

Abstract
Microglia are activated in response to a number of different pathological states within the CNS including injury, ischemia, and infection. Microglial activation results in their production of pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α. While release of these factors is typically intended to prevent further damage to CNS tissue, they may also be toxic to neurons and other glial cells. Mounting evidence indicates that chronic microglial activation may also contribute to the development and progression of neurodegenerative disorders. Unfortunately, determining the role of pro-inflammatory cytokines in these disorders has been complicated by their dual roles in neuroprotection and neurodegeneration. The purpose of this review is to summarize current understanding of the involvement of cytokines in neurodegenerative disorders and their potential signaling mechanisms in this context. Taken together, recent findings suggest that microglial activation and pro-inflammatory cytokines merit interest as targets in the treatment of neurodegenerative disorders.
Keywords: Microglia, Pro-inflammatory cytokines, Neurodegeneration, Tumor necrosis factor-α, Interleukin-1, Interleukin-6
1. Introduction
Cytokines comprise a group of small polypeptides (8–30 kDa) possessing tremendous diversity in their potential actions. Most cytokines act at very low concentrations (picomolar to nanomolar) and signal in either an autocrine or paracrine fashion to modulate local cellular activities including survival, growth, and differentiation. Cytokines are also rapidly upregulated in response to disease, injury, and infection and serve an important role in tissue repair in these acute pathologic states. These peptides have typically been classified as either pro-inflammatory or anti-inflammatory based on their actions in peripheral tissues. Recently, chronic microglial production of pro-inflammatory cytokines including interleukins (IL-1 and IL-6), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) has received considerable attention for its role in neurodegenerative disorders. The purpose of this review is to outline current evidence regarding how these cytokines may contribute to the process of neurodegeneration and their potential as therapeutic targets in a wide range of central nervous system (CNS) diseases.
2. Microglia: culprits of cytokine production in neurodegenerative diseases
Microglia are the unique resident immune cells of the CNS acting as primary mediators of inflammation. Although microglial density is region-specific, they comprise between 5 and 20% of all cells in the human brain, accounting for approximately 20% of the glial population [98,136]. Within healthy CNS tissue, microglia possess a unique ramified morphology with a small, round soma and numerous branching processes. Although long considered to be in a “resting” state, recent evidence indicates that ramified microglia have critical physiologic roles including determination of neuronal fate, migration, axonal growth, and synaptic remodeling during brain and spinal cord development. The critical contributions of microglia to CNS maturation can be attributed to their actions in phagocytosis of cellular debris, release of a variety of cell signaling factors including neurotrophins and extracellular matrix components, and direct contact with neurons [13,70,94,96,179].
Microglia also possess the necessary machinery and characteristics to act as sensors for disruption in normal homeostasis within the mature CNS [69,158]. Microglia are rapidly activated following a number of pathologic events including altered neuronal function, infection, injury, ischemia, and inflammation (Fig. 1). Activation results in a transition in microglial morphology to an ameboid state facilitating the migration of these cells to the site of insult [41]. Microglial response to CNS pathology also results in initiation of a number of immune functions including phagocytosis, antigen processing and presentation, and production of both cytotoxic and neurotrophic factors [41,180]. Mediators of cytotoxicity released from activated microglia include reactive oxygen and nitrogen species (superoxide anion, nitric oxide), arachidonic acid metabolites (eicosanoids), excitotoxic glutamate, quinolic acid, and histamine (Fig. 1) [128]. Release of these factors results from stimulation of microglia with lipopolysaccharide (LPS), amyloid protein, and high concentrations of IFN-γ [117,120]. Alternatively, microglia may also act as sources of neurotrophic factors including nerve growth factor (NGF), brain derived neurotrophic factor (BDNF), and neurotrophin-4/5 (NT-4/5) [13,127]. Microglia-mediated neuroprotection and neurogenesis has been shown to occur due to exposure to IL-4 and low levels of IFN-γ [23]. These findings indicate that microglial actions may be dependent on the nature of the activating stimulus. To this end, recent studies have shown that microglial phagocytosis of invading bacteria is associated with their release of pro-inflammatory factors whereas clearance of apoptotic debris is associated with production of anti-inflammatory factors [71,113]. While short-term microglial activity is generally accepted to serve a neuroprotective role, chronic activation has been implicated as a potential mechanism in neurodegenerative disorders. A special emphasis has recently been placed on microglial release of pro-inflammatory cytokines including IL-1, IL-6, TNF-α, and IFN-γ and their roles in neuronal degeneration.
Fig. 1.
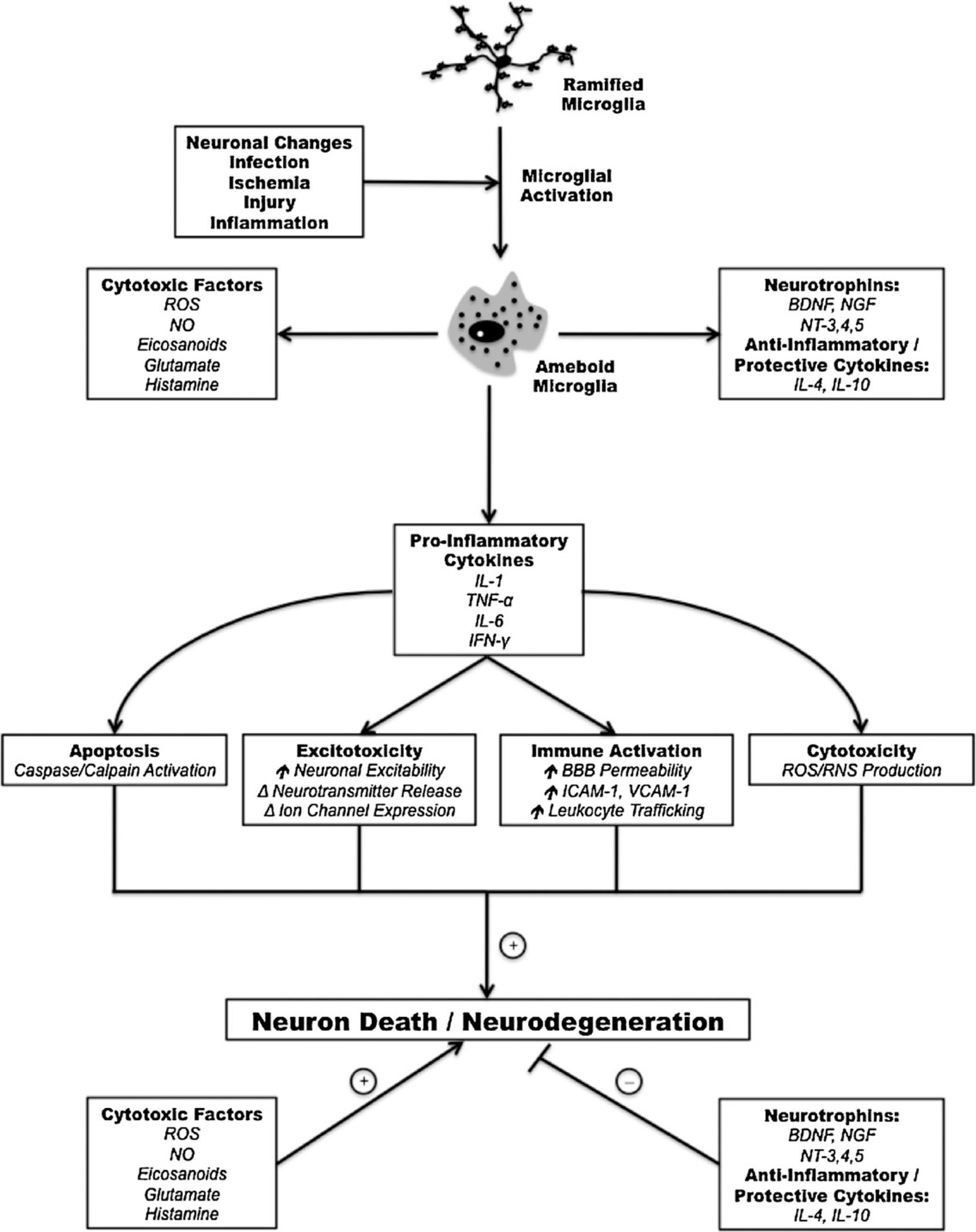
Common mechanisms by which microglial activation and subsequent pro-inflammatory cytokine release may contribute to neurodegenerative pathology.
It is important to note, however, that microglia are not the sole producers of cytokines following CNS insult. Astrocytes have also been implicated in the generation of pro-inflammatory mediators involved in neurodegenerative disorders [103,109,175]. Cytokines other than those classically described as pro-inflammatory may also have a role in the development of these disorders. For example, studies have identified IL-33 as having a potential role in Alzheimer’s disease. Chapuis et al. and other groups have demonstrated that multiple polymorphisms in the gene encoding IL-33 may confer increased risk of Alzheimer’s disease [29,188]. While these are both important topics deserving of attention, the focus of this review will remain on the role of classical pro-inflammatory cytokines (namely TNF-α, IL-1, and IL-6) in neurodegeneration.
3. Evidence for cytokine involvement in neurodegenerative disorders: an undeniable case?
3.1. Alzheimer’s disease
Alzheimer’s disease (AD) is the most common form of dementia in the elderly resulting in a progressive decline in a number of cognitive functions including short-term memory. AD is characterized by the formation of two characteristic lesions: extracellular β-amyloid deposits forming senile plaques and intracellular neurofibrillary tangles made up of the microtubule associated protein tau. A strong link between inflammation, primarily mediated by pro-inflammatory cytokines, and AD has been established both in clinical data and bench research. Indirect evidence suggests that initiation of strong local inflammatory responses within the brain following traumatic injury may be a significant risk factor for the development of AD [63,112]. Recent findings also suggest that AD may be associated with a more widespread inflammatory state characterized by increased peripheral blood levels of IL-1β, IL-6, TNF-α, TGF-β, and IL-18 [162]. To this end, systemic inflammatory activation following lipopolysaccharide (LPS) administration appears to exacerbate disease features in multiple animal models of AD [100].
Recent studies have provided further evidence supporting an association between microglial cytokine production and AD. In particular, in vitro activation of microglia from Alzheimer’s patients and non-demented controls with Aβ peptide results in their release of pro-inflammatory cytokines including IL-1β, IL-6, and TNF-α in a dose-dependent manner [110,141]. A number of post-mortem investigations have also demonstrated the presence of these proinflammatory cytokines in close proximity to AD lesion sites [44,64,83]. In addition, genetic analysis indicates an apparent link between disease development and specific polymorphisms in the genes encoding TNF-α and IL-1β [42,43].
Unfortunately, animal models of AD have not proven unambiguous in promoting our understanding of the involvement of microglial cytokines in disease progression. Early work in Tg2575 transgenic mice over-producing Aβ peptide was unsuccessful in demonstrating mRNA expression of pro-inflammatory cytokines in hippocampal and cortical tissues up to 14 months of age utilizing a ribonuclease protection assay. The same study reported the presence of IL-1β reactive astrocytes in close apposition to Aβ plaques, but did not investigate microglial cytokine expression [121]. In comparison, other laboratories have shown the presence of cortical microglia expressing IL-1β, IL-6 and TNF-α proteins in similar mouse models [12,77]. Although the degree of cytokine expression in animal models is still debated, these studies have provided clues to their degenerative effects in AD. Deletion of the tumor necrosis factor receptor 1 (TNFR1) in APP23 transgenic mice results in decreased Aβ production and plaque formation and reverses impairments in cognitive performance [75]. Together, these studies suggest an important role for pro-inflammatory cytokines in AD, but further work is needed to understand their actions in disease pathology.
3.2. Parkinson’s disease
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder and is caused by the progressive loss of dopaminergic (DA) neurons from the substantia nigra pars compacta (SNpc) that normally innervate the striatum. The pathological hallmark of PD is intracellular accumulation of α-synuclein leading to the formation of Lewy bodies. PD may result in a number of different presenting symptoms including resting tremor, bradykinesia, cogwheel rigidity, and postural instability. Epidemiologic findings from a number of studies suggest that inflammation may be involved in the pathogenesis of PD. For example, long-term use of the non-steroidal anti-inflammatory drug (NSAID) ibuprofen appears to reduce the risk for developing PD [144]. This result may be explained, in part, by post-mortem analysis of cerebrospinal fluid (CSF) and brain demonstrating elevated protein levels of pro-inflammatory cytokines in PD patients [16,122,123]. In addition, transgenic mice engineered to over-express α-synuclein show increased DA neuronal susceptibility following stereotaxic injections of LPS within the nigra as compared to wild-type controls [55]. Together, these data demonstrate a role for inflammatory events in the progressive neurodegeneration associated with PD.
McGeer et al. were the first to demonstrate microglial activation in the SNpc of PD brains [118]. Additional work has confirmed these findings and also found that microglial reactivity may be more widespread than simply the SNpc: increased numbers of activated microglia were found in the hippocampus, putamen, cingulate cortex, and transentorhinal cortex of patients with PD [87]. Post-mortem findings such as these unfortunately are unable to determine when microglial activation first occurs in disease pathology. Recent PET imaging studies using an isoqunoline, [11C](R)-PK11195, which binds to the peripheral benzodiazepine receptor expressed by activated microglia/macrophages, have provided evidence for their reactivity in early-stage PD patients [45,132]. The same study also noted a correlation between microglial activation and loss of dopaminergic terminals in the midbrain. Western blot analysis and enzyme-linked immunosorbent assay (ELISA) suggest that microglial activation in the PD brain results in increased expression of pro-inflammatory cytokines [147]. Indeed, elevated production of IL-1β, IL-6, and TNF-α were noted in microglia activated by α-synuclein in vitro [160].
Although the mechanisms by which microglial-derived cytokines participate in the progressive neurodegeneration in PD are largely unknown, basic science has begun address this significant issue. Dopaminergic neurons appear to be exquisitely sensitive to TNF-α insult in vitro [119]. A role for TNF-α has also been suggested in multiple animal models of PD. Increased expression of TNF-α is noted following administration of 6-hydroxydopamine (6-OHDA) or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [124,156]. In addition, disruption of TNF-α signaling by at least two methods appears to reduce toxicity of these compounds to DA neurons. Mice lacking the TNF-α receptor show decreased loss of TH-immunoreactivity after MPTP administration [156]. Lentiviral therapy with a dominant-negative TNF two weeks after 6-OHDA lesion also prevented progressive degeneration of DA neurons and microglial activation, further indicating a role for cytokine signaling in models of PD [72]. Similarly, IFN-γ deficiency in mice prevents paraquat-induced changes in locomotor behaviors and DA neuron survival [105]. Although these findings collectively indicate that pro-inflammatory cytokines may have a detrimental role in the progression of PD, further studies are needed to determine how they may participate in early-stage pathogenesis.
3.3. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is an adult-onset disorder characterized by the selective degeneration of both upper and lower motor neurons leading to development of muscle weakness, spasticity, atrophy, dysarthria, and dysphagia. ALS is rapidly progressive, often resulting in death approximately three years on average after initial diagnosis [17]. Although the vast majority of cases of ALS occur without a known cause, at least 10–15% of patients with this condition have an inherited form referred to as familial ALS (fALS). The most common form of fALS is caused by a gain of function mutation in the gene encoding the Cu2+/Zn2+ superoxide dismutase enzyme (SOD1) [17,40]. An association between inflammation and ALS has not been fully established, but existing clues provide some evidence of inflammatory activation in this disease. For example, elevated levels of the pro-inflammatory cytokines IL-6 and TNF-α have been described in the CSF, serum, and skin of patients with ALS [131,149,167]. Subsequent imaging studies measuring [11C](R)-PK11195 binding to the peripheral benzodiazepine receptor site of activated microglia/macrophages suggest microglial activation in this disorder may be prevalent throughout the CNS [45]. Increased microglial reactivity was noted in the spinal cord, motor cortex, pons, dorsolateral prefrontal cortex, and thalamus of ALS patients when compared to healthy controls [173]. In fact, increased binding of PK11195 to the peripheral benzodiazepine receptor of activated microglia in the motor cortex was significantly associated with clinical signs of upper motor neuron lesion, suggesting activated microglia may play a significant role in disease severity.
Experimental findings have also added to our understanding of the involvement of microglia and pro-inflammatory cytokines in ALS. Pramatarova et al. demonstrated that neuron-specific transgenic expression of mutant SOD1 (mSOD1) did not result in altered locomotor function [137]. In contrast, non-specific expression of mSOD1 causes severe motor impairment, suggesting that glial cells may play an important role in ALS pathology [92]. Microglial activation in the G93A mSOD1 model was first noted in the ventral horn of the spinal cord during the late pre-symptomatic phase (80 days) [5]. IL-1α and IL-1β were also increased in the pre-symptomatic phase and reached even higher levels when significant hindlimb paralysis developed (120 days) [79]. The existence of both microglial activation and pro-inflammatory cytokine production before disease symptomatology indicates these biochemical changes may be directly involved in motor neuron degeneration. In support of this, mRNA expression of apoptosis-associated proteins including caspases, TNFR1, and Fas is significantly increased during the symptomatic phase in mSOD1 mice [78,79]. Our group and others have also demonstrated in vitro sensitivity of primary motor neurons and hybrid motoneuron cell lines to cytokine-induced apoptosis [38,153]. Further work is needed to fully determine the mechanisms by which pro-inflammatory cytokines and motor neuron apoptosis contribute to the disease process in ALS.
4. Modulatory role of the sympathetic nervous system in pro-inflammatory cytokine release
Interestingly, the sympathetic nervous system (SNS) plays an important role in the modulation of pro-inflammatory cytokine production by microglia. Following activation by acute stressors, the SNS releases the catecholamines norepinephrine (NE) and epinephrine (Epi) which are thought to act directly on both microglia and peripheral immune cells to regulate their activity [47]. mRNA expression of multiple adrenergic receptors (ARs) including α1, α2, β1, and β2-ARs has been demonstrated in vitro in microglia isolated from the rat forebrain [125]. Subsequent studies using selective agonists indicated that β1 and β2 adrenergic receptors present on microglia are functional, responding in a classical manner to activation by increases in intracellular cyclic AMP (cAMP) [164]. While accumulation of intracellular cAMP in peripheral blood mononuclear cells (PBMCs) following catecholamine stimulation has classically been thought to prevent their production of pro-inflammatory cytokines, the microglial response to NE and Epi appears to depend on the environmental context [47]. For example, multiple studies indicate that activation of β-adrenergic receptors attenuates production of pro-inflammatory mediators including TNF-α, IL-6, IL-12, and NO in cultured microglia exposed to LPS [27,50,138]. Conversely, β-AR stimulation in the absence of LPS activates microglia in vitro resulting in their release of IL-1β [164,171]. Similar responses have also been reported in vivo following exposure to acute stressors where NE release was associated with increased CNS production of IL-1β and IL-6 [14,90]. In both of these studies, microglial activation and pro-inflammatory cytokine release was blocked by administration of β-AR antagonists further indicating a pivotal role for adrenergic receptors in the response of microglia to catecholamine stimulation. Together, these findings suggest that sympathetic activation and cytokine release may be tightly coupled depending on the cellular and environmental context.
Unfortunately, the role of the interaction between the sympathetic nervous system and microglial activity in neurodegenerative disorders has been largely overlooked. Despite evidence indicating that catecholamines may facilitate microglial activation, anti-inflammatory effects of β-AR stimulation have been shown in animal models of excitotoxicity, AD, and PD [54,60,139]. These findings have been attributed to the differential effects of β-AR activation on inhibiting systemic inflammatory responses when compared to local stimulation of CNS microglia [57,157]. Further work is necessary to determine the particular environmental conditions that may promote catecholamine-induced activation of microglia in neurodegenerative disorders. In addition, future studies should focus on understanding how increased cytokine levels in the diseased brain may modulate activity of the SNS. For example, both IL-1 and TNF-α have been shown to inhibit release of norepinephrine in the rat myenteric plexus [84,85]. Elenkov et al. have also shown that TNF-α acts at pre-synaptic terminals to suppress the release of NE from the rat median eminence [46]. However, our knowledge concerning cytokine modulation of the adrenergic system is still incomplete. This is a particularly important field of study especially as it pertains to changes in SNS signaling due to cytokine dysregulation in neurodegenerative disorders.
5. Pro-inflammatory cytokine signaling in neurodegeneration: muddy waters of neurodegeneration vs. neuroprotection?
5.1. TNF-α
TNF-α exists either as a 26 kDa membrane-bound protein or a 17 kDa soluble form generated by the activity by a metalloprotease known as TNF-α converting enzyme (TACE; ADAM17). Although microglia have been implicated as the major producers of TNF-α in neurodegenerative disorders and CNS insult, numerous studies have demonstrated that astrocytes and neurons may also be a source of this pro-inflammatory cytokine [19,101]. TNF-α signaling occurs via two distinct receptors termed TNFR1 (also known as Tnfrsf1a/p55) expressed almost ubiquitously among cell types and TNFR2 (Tnfrsf1b/p75) which is present in lower levels in specific neuronal subtypes and glial cells in the brain [32,166]. TNFR1 appears to have important roles in promoting neuronal death through activation of pro-apoptotic proteins or survival by increasing nuclear factor-n B (NF-κB) activity. Upon TNF binding, dissociation of a silencer protein allows the intracellular death domain of TNFR1 to interact with the TNF receptor-associated death domain adaptor protein (TRADD) [67]. Depending on the cellular context, TRADD–TNFR1 association allows for recruitment of distinct signaling complexes. Complex I is composed of TRADD, TNF receptor associated proteins 2/5 (TRAF2/5), cellular inhibitor-of-apoptosis proteins 1/2 (c-IAP 1/2), ubiquitin-conjugating enzyme 13 (Ubc 13), and receptor interacting protein (RIP) [182]. Association of these signaling proteins promotes lysine 63 (Lys 63)-linked polyubiquitination of RIP via the actions of TRAF2/5 and c-IAP1/2 [114,178,186]. Polyubiquitinated RIP can in turn activate IκB kinase (IKK) either through direct interaction with its regulatory subunit NF-κB essential modulator (NOME) or via recruitment of the transforming growth factor β-activated kinase 1 (TAK1) complex [15,91,184]. Phosphorylation of IκB by IKK allows for subsequent nuclear translocation of NF-κB and transcription of its target genes including anti-apoptotic proteins (e.g., IAPs, Bcl-2, Bcl-xL) and pro-inflammatory factors (e.g., cytokines, chemokines, COX-2) [2]. Complex I may also initiate transcription of pro-inflammatory molecules and enzymes by activation of JNK and p38 kinases [66,177]. Together, these results indicate that formation of complex 1 following TNFR1 binding appears to play an important role in inflammation and cell survival.
TNFR1 may also signal via an alternate protein complex (Complex II) to initiate apoptotic cell death. Complex II is formed by the association of TNFR1 and TRADD with Fas-associated death domain protein (FADD) [11]. This allows the N-terminal death effector domain (DED) of FADD to interact with similar domains on the pro-forms of caspase-8 and caspase-10, leading to their activation [31,151]. Activated initiator caspases such as caspase-8 and caspase-10 may then initiate apoptosis directly by cleavage of caspase-3 or indirectly through activation of Bax (a pro-apoptotic member of the Bcl-2 family) and eventual release of cytochrome c from mitochondria [190]. In this manner, signaling via complex II after ligand binding of TNFR1 results in apoptotic cell death seemingly counteracting the NF-κB- and kinase-mediated effects of complex I. Recent findings indicate that activation of NF-κB is sufficient to prevent TNF-α-induced apoptosis [106]. However, the conditions which may promote anti- or pro-apoptotic signaling through complex I and complex II, respectively, remain largely unknown. Further insight into this issue may provide novel targets for preventing neuronal death in neurodegenerative disorders.
Signaling through TNFR2 shares a number of common similarities with the cascades initiated by TNFR1. Specifically, TNFR2 acts via a number of different signaling pathways to increase NF-κB-mediated transcription of anti-apoptotic and pro-inflammatory gene targets. TNFR2 responds preferentially to binding by membrane-bound TNF-α and does not possess a death domain [62]. Instead, TNFR2 is capable of directly interacting with TRAF2 to promote formation of a protein complex comprised of TRAF1/2 and c-IAP1/2 leading to RIP-dependent activation of NF-κB and JNK/p38 kinases in a manner similar to that described above for TNFR1 [142]. Additionally, TNFR2 stimulation also appears to mediate cytoplasmic accumulation of NF-κB-inducing kinase (NIK) through interactions between the TRAF/c-IAP protein complex and TRAF3. NIK buildup provides an alternate means to promote activity of IKK and thus NF-κB [174]. Neuronal TNFR2 is also capable of increasing NF-κB-mediated transcription by activation of the phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway [68]. These findings suggest that TNFR2 signaling mediates at least part of the protective and regulatory effects of TNF-α in the CNS.
5.2. IL-1
The IL-1 family consists of several different proteins including IL-1α, IL-1β, and an endogenous IL-1 receptor antagonist (IL-1RA). IL-1α is found primarily as a membrane-bound protein that is thought to participate mainly in paracrine and autocrine signaling, whereas IL-1β is typically found in a soluble, secreted form. Both IL-1α and IL-1β are synthesized in pro forms which are then cleaved by proteases (i.e. caspase-1 for IL-1β) to generate mature proteins [129]. Signaling by both IL-1α and IL-1β is mediated by the type I IL-1 receptor (IL-1RI). An additional receptor, the type II IL-1 receptor (IL-1RII), is thought to act as a decoy receptor that does not participate in active IL-1 signaling [18]. Ligand binding of IL-1RI results in a conformational change that allows for its association with an accessory receptor chain known as IL-1 receptor accessory protein (IL-1RAcP). The high affinity complex formed by IL-1RI and IL-1RAcP can then recruit a number of adaptor proteins to the receptor including the myeloid differentiation response gene 88 (MyD88) and tumor necrosis factor-associated factor 6 (TRAF6) leading to activation of a number of different kinases including IL-1 receptor associated kinases 1 and 4 (IRAK1, IRAK4), transforming growth factor β-activated kinase (TAK), and members of the mitogen activated protein kinase (MAPK) cascade (p38, JNK, and ERK1/2) [20,35,116,126,146,161]. Together, the actions of these kinases facilitate the transcription of a wide variety of IL-1-responsive genes via the actions of NF-κB and the AP-1 transcription factor complex consisting of members of the Fos, Jun, and ATF families of transcription factors [130,181]. IL-1RI may also promote the hydrolysis of sphingomyelin by neutral sphingomyelinase (nSMase) in the neuronal membrane to produce ceramide [81]. Cellular accumulation of cermide may result in activation of apoptotic pathways, activity of Src kinases, and alterations in neuronal electrophysiology [21,30,39].
5.3. IL-6
IL-6 is characterized as a 26 kDa glycoprotein, which was formerly known by a variety of names including B-cell differentiation factor, T-cell differentiation factor, hybridoma/plasmacytoma growth factor, and hepatocyte stimulating factor [36]. IL-6 binds and activates a receptor protein complex comprised of one non-signaling, membrane-associated α subunit (IL-6R) and two gp130 subunits responsible for signal transduction [111,152]. Interestingly, a soluble form of IL-6R (sIL-6R) may also be formed by alternative RNA splicing or protease cleavage and subsequently associate with cell surface gp130 to participate in a process referred to as IL-6 transsignaling [26]. Ligand binding of this protein complex results in homodimerization of gp130 and activates multiple signaling mechanisms. First, gp130 homodimerization facilitates activation of members of the Janus kinase family including JAK1, JAK2, and TYK2. These kinases may then act to phosphorylate signal transducer and activator of transcription (STAT) proteins, particularly STAT1 and STAT3 [10,89]. Upon phosphorylation, STAT1 and STAT3 undergo nuclear translocation and promote transcription of their target genes. STAT1 is thought to be an important regulator of signaling by interferons (IFNs) involved in innate immune responses including type I (IFN-α, IFN-β, and IFN-ω) and type II (IFN-γ) IFNs [7,104]. In contrast, STAT3 mediates the cell survival and proliferation-promoting effects of IL-6 by modulating expression of genes involved in cell division/progression of the cell cycle (cyclin D1, c-myc, c-fos) and suppression of apoptosis (survivin, c-IAP2, Bcl-xL, Bcl-2) [1,80]. Activated IL-6R–gp130 receptor complexes may also initiate signaling by MAPKs. In this case, the MAPK cascade is initiated by tyrosine phosphorylation of the Src homology region 2 domain-containing phosphatase-2 (SHP-2). SHP-2 subsequently activates the Ras/Raf/MAPK signaling pathway leading to activation of multiple members of the MAPK family involved in both cellular survival and stress responses including classical extracellular signal-regulated kinases 1/2 (ERK1/2), p38 MAPK, and JNK [37,148,163]. Finally, IL-6 can also promote PI3K/Akt signaling. Akt may in turn promote cell survival by acting on a number of substrates including transcription factors (forkhead/FOXO), cell cycle regulators (CDK2), and pro-apoptotic (Bax, Bad, caspase-9) and anti-apoptotic (Bcl-2) proteins [22,107,115].
5.4. Common pro-inflammatory cytokine signaling pathways involved in neurodegeneration
The signaling pathways outlined above illustrate only a small portion of the complexity in the actions of pro-inflammatory cytokines. Although a clear link has been established between these cytokines and neurodegeneration, their signaling mechanisms appear to involve a balance between promoting cell survival, apoptosis, and pro-inflammatory responses. Although the factors that may disrupt this normal equilibrium remain largely unknown, this may partly explain the conflicting data surrounding the role of pro-inflammatory cytokines in neuronal degeneration: IL-1, IL-6, and TNF-α have often been described as having neuroprotective effects both in vivo and in vitro [24]. However, recent findings have elucidated a number of pro-inflammatory cytokine actions that may directly contribute to neuronal degeneration. Interestingly, many of these degenerative mechanisms may be commonly activated by multiple pro-inflammatory cytokines (Fig. 1). One example of this is cytokine modulation of neuronal excitability and neurotransmitter release. Recent evidence indicates that IL-1 may mediate increased neuronal excitability through direct interaction of its receptor complex with N-methyl-D-aspartate (NMDA) receptors and inhibition of Ca2+-induced K+ channels [58,189]. In addition, increased microglial production of nitric oxide (NO) following cytokine exposure may also contribute to neuronal hyperexcitability by initiating Ca2+-dependent release of glutamate from astrocytes [8,145]. Pro-inflammatory cytokines may also disrupt neuronal ionic balance by altering activity and expression on ion channels and ionotropic neurotransmitter receptors [76,183,191]. Finally, cytokine exposure has been shown to modulate neuronal release of multiple neurotransmitters including GABA, dopamine, acetylcholine, and serotonin both in cell culture and tissue preparations [69]. Recent evidence indicates that intracerebroventricular administration of LPS may also increase frequency of spike wave discharges in a genetic rat model of absence epilepsy providing further evidence for pro-inflammatory cytokine modulation of neuronal excitability [95]. These effects may result in excitotoxic injury to neurons. Pro-inflammatory cytokines also share the ability to induce apoptosis in neurons and glial cells. As discussed above, TNF-α may directly mediate apoptotic death by interaction with TNFR1. The actions of IL-1 in inducing apoptosis are less clearly defined, but appear to be dependent on the presence or absence of additional cytokines and signaling molecules. For example, IL-1 has been shown to promote cell death when combined with either IFN-γ or TNF-α in primary human neuron cultures [28,82]. Pro-inflammatory cytokines can also contribute to damage of neurons and glia by promoting trafficking of peripheral immune cells into the CNS. Entry of immune cells into the CNS may be facilitated by cytokine-mediated increases in blood–brain-barrier permeability or enhanced movement of leukocytes into the CNS by increasing expression of cell adhesion molecules essential for extravasation (e.g., intracellular adhesion molecule 1 [ICAM-1], vascular cell adhesion molecule 1 [VCAM-1]) and trafficking [6,51,52]. Pro-inflammatory cytokines also promote increased production of factors which are toxic to neurons and glial cells including reactive oxygen species (ROS) and NO.
6. Conclusions concerning pro-inflammatory cytokines as a therapeutic target in neurodegenerative disorders: bullseye or bust?
Given the mounting evidence for their role in neurodegenerative disorders and the host of mechanisms by which they may cause neuronal degeneration, pro-inflammatory cytokines have appropriately received considerable attention as therapeutic targets in neurodegenerative disorders. Caution must be taken in this approach because of the multiple roles these cytokines may have in both neurodegeneration and neuroprotection. Nonetheless, both experimental and clinical evidence suggest that inhibiting inflammatory responses (and more specifically pro-inflammatory cytokines) may be a viable option in the treatment of neurodegenerative disorders. The earliest evidence to this point stems from epidemiological reports suggesting the use of some non-aspirin NSAIDs may reduce the risk of developing both AD and PD [49,53,56]. Unfortunately, these findings remain controversial and several studies indicate that NSAIDs may not be effective in preventing disease progression in patients with existing AD [3,135,140]. This may indicate another challenge in using drugs targeting inflammatory responses because efficacy of treatment may depend on therapy initiation long before overt clinical signs of disease are evident. This would require considerable progress in our current repertoire of biomakers for neurodegenerative disease.
Our group and others have focused on the use of endogenously produced anti-inflammatory substances in the treatment of CNS injury and other neurodegenerative disorders. Some of these agents, such as estrogen and melatonin used in our laboratory, may have multiple actions in preventing both inflammation and downstream effects of pro-inflammatory cytokines including production of ROS and induction of apoptosis [25,88,143,150,154,155]. Unfortunately, evidence for the clinical efficacy of these drugs against neurodegeneration is lacking. Still other groups have placed an emphasis on inhibiting microglial activation in the treatment of neurodegenerative disorders. Minocycline, originally developed as a broad spectrum tetracycline antibiotic, has generated interest because of its ability to prevent activation of microglia [65]. In particular, minocycline has been shown to inhibit expression of pro-inflammatory cytokines (IL-1β, TNF-α) by microglia [99,168]. Minocycline has shown particular promise in reversing functional defects in animal models of AD, PD, ALS, and spinal cord injury (SCI) [33,97,102,185]. A number of clinical trials are currently underway to test the efficacy of minocycline in neurodegeneration. Unfortunately, a phase III multi-center randomized trial in ALS patients demonstrated that minocycline treatment may worsen disease progression, calling into question the translational potential of this drug [61].
Recent investigations have focused on targeting individual cytokines in neurodegeneration. IL-1 has received particular attention because a recombinant form of its endogenous inhibitor, IL-1RA, is well-tolerated and has been clinically approved for use in rheumatoid arthritis [59]. A recent phase II clinical trial demonstrated that recombinant human IL-1RA may improve clinical outcome in stroke patients with cortical infarcts [48]. In addition, IL-1RA exposure demonstrated protective effects against CNS injury and neuron damage resulting from NMDA-mediated excitotoxicity [4,176]. Interestingly, AD patients have been shown to have decreased CSF levels of IL-1RA, indicating that IL-1RA therapy could have particular benefit in these patients [165]. Several inhibitors of TNF-α signaling have also been designed which may be useful as therapeutics. Pilot studies and case reports indicate that perispinal administration of etanercept, which inhibits TNF-α by acting as a decoy receptor, may have efficacy in the treatment of AD patients. In particular, etanercept treatment was associated with rapid improvement in language and visuospatial/executive tasks [169,170]. The rate of improvement in these patients indicates that etanercept may enhance cognitive function by reversing changes in neuronal excitability/activity associated with TNF-α exposure (as described above), although this has not been investigated. Further work is necessary to confirm findings from earlier studies using etanercept in large, well-designed clinical trials.
As previously discussed, the interaction between the SNS and microglial cytokine production may also be a viable target in the treatment of neurodegenerative disorders. Most of the studies in this field have focused on the effects of inhibiting peripheral immune responses on neurodegeneration. Mounting evidence suggests that agonists of β-adrenergic receptors may exert anti-inflammatory effects in animal models of CNS degeneration [34,54,60,139]. These findings appear to be contradictory to studies demonstrating that β-AR activation in microglia promotes their production of pro-inflammatory cytokines. β-AR agonists may instead exert their anti-inflammatory effects systemically by blocking activation of peripheral immune cells. In fact, increased intercellular levels of cAMP following β-AR stimulation has been shown to attenuate cytokine production by PBMCs [47]. Other studies have focused on the use of phosphodiestarase (PDE) inhibitors that act to prevent degradation of cAMP in peripheral immune cells as a method to prevent pro-inflammatory cytokine production. Hasko et al. have demonstrated that adminstration of both rolipram (a selective PDEIV inhibitor) and amrinone (selective PDEIII inhibitor) inhibit production of the pro-inflammatory mediators IL-12, IFN-γ, TNF-α, and NO to prevent immune activation in a mouse model of LPS-mediated endotoxemia [74]. Interestingly, rolipram has also been shown to promote neuroprotection in animal models of spinal cord injury and multiple sclerosis through its immunomodulatory effects [9,86,133,134]. Adenosine signaling has also been implicated as a potential target to modulate the systemic inflammatory response [73]. Reports indicate that adenosine may protect against cardiac ischemia by inhibiting peripheral production of pro-inflammatory cytokines through its actions at A1 and A2 receptors [93,108,159,187]. Interestingly, mice lacking the adenosine A1 receptor show increased expression of pro-inflammatory IL-1β in peripheral immune cells [172]. These animals also have increased disease severity in experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis [172]. Thus, the adenosine A1 receptor appears to be an important modulator of immune system function and activation. Taken together, these data suggest that interaction between the SNS and pro-inflammatory cytokine production both centrally and peripherally may be a viable target in the treatment of neurodegenerative disorders. However, further studies are needed to fully understand the effects of SNS activity on local microglial and peripheral responses as well as the contribution of the peripheral immune system to neurodegeneration.
Together, evidence from clinical and translational studies targeting the general inflammatory response and pro-inflammatory cytokines in neurodegenerative disorders suggest that this approach may have merit. However, it must be noted that inflammation often serves a protective role in the CNS. Future studies should place particular emphasis on the conditions that promote cytokine-mediated neuronal degeneration over survival. Defining specific changes in the CNS environment responsible for enhanced detrimental effects of pro-inflammatory cytokines may also reveal new targets in the treatment of neurodegenerative disorders. Inflammation should also be carefully viewed as only one contributor to the mechanisms underlying neurodegeneration. Design of therapeutics targeting only the inflammatory component may be short-sighted. Instead, drugs attenuating multiple mechanisms of neuronal loss may have greater promise in the treatment of neurodegenerative diseases. Nonetheless, the volume of evidence linking pro-inflammatory cytokine involvement to neurodegeneration makes this an exciting field of study with particular clinical relevance.
Acknowledgements
Completion of our projects was made possible by funding from the National Institutes of Health (NIH) and National Institute of Neurological Disorders and Stroke (NINDS): (NS-31622, NS-38146, and NS-41088) and the State of South Carolina Spinal Cord Injury Research Fund.
Abbreviations:
- ILs
interleukins
- TNF-α
tumor necrosis factor-α
- IFNs
interferons
- CNS
central nervous system
- LPS
lipopolysaccharide
- NGF
nerve growth factor
- BDNF
brain derived neurotrophic factor
- NT-4/5
neurotrophin-4/5
- AD
Alzheimer’s disease
- TGF-β
transforming growth factor β
- IL-18
interleukin-18
- Aβ
amyloid beta
- APP
amyloid precursor protein
- TNFR1
tumor necrosis factor receptor 1
- PD
Parkinson’s disease
- SNpc
substantia nigra pars compacta
- DA
dopamine
- NSAID
non-steroidal anti-inflammatory drug
- CSF
cerebrospinal fluid
- ELISA
enzyme-linked immunosorbent assay
- 6-OHDA
6-hydroxydopamine
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- ALS
amyotrophic lateral sclerosis
- fALS
familial amyotrophic lateral sclerosis
- SOD1
superoxide dismutase
- TACE
TNF-α converting enzyme
- TRADD
TNF receptor-associated death domain adaptor protein
- TRAFs
TNF receptor associated proteins
- c-IAPs
cellular inhibitor-of-apoptosis proteins
- Ubc 13
ubiquitin-conjugating enzyme 13
- RIP
receptor interacting protein
- NFκB
nuclear factor kappa B
- IκB
inhibitor of κB
- IKK
IκB kinase
- NOME
NF-κB essential modulator
- TAK1
transforming growth factor β-activated kinase 1
- COX-2
cyclooxygenase-2
- FADD
Fas-associated death domain protein
- DED
death effector domain of FADD
- JNK
c-Jun N-terminal kinase
- NIK
NFκB-inducing kinase
- PI3K
phosphatidylinositol-3-kinase
- IL-1RA
IL-1 receptor antagonist
- IL-1RI
type I IL-1 receptor
- IL-1RII
type II IL-1 receptor
- IL-1RAcP
IL-1 receptor accessory protein
- MyD88
myeloid differentiation response gene 88
- IRAKs
IL-1 receptor associated kinases
- MAPKs
mitogen activated protein kinases
- ERKs
extracellular signal-regulated kinases
- AP-1
activator protein 1
- ATFs
activating transcription factors
- nSMase
neutral sphingomyelinase
- IL-6R
interleukin-6 receptor
- sIL-6R
soluble IL-6 receptor
- JAK
Janus kinase
- STAT
signal transducer and activator of transcription proteins
- SHP-2
Src homology region 2 domain-containing phosphatase-2
- FOXO
forkhead box proteins
- CDK
cyclin dependent kinase
- NMDA
N-methyl-D-aspartate
- NO
nitric oxide
- GABA
gamma-aminobutyric acid
- ICAM-1
intracellular adhesion molecule 1
- VCAM-1
vascular cell adhesion molecule 1
- ROS
reactive oxygen species
- SCI
spinal cord injury
- SNS
sympathetic nervous system
- NE
norephinephrine
- Epi
epinephrine
- ARs
adrenergic receptors
- cAMP
cyclic adenosine monophosphate
- PBMCs
peripheral blood mononuclear cells
- PDE
phosphodiestarase
- EAE
experimental autoimmune encephalomyelitis
References
- [1].Aggarwal BB, Sethi G, Ahn KS, Sandur SK, Pandey MK, Kunnumakkara AB, Sung B, Ichikawa H, Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution, Ann. N. Y. Acad. Sci. 1091 (2006) 151–169. [DOI] [PubMed] [Google Scholar]
- [2].Ahn KS, Aggarwal BB, Transcription factor NF-kappaB: a sensor for smoke and stress signals, Ann. N. Y. Acad. Sci. 1056 (2005) 218–233. [DOI] [PubMed] [Google Scholar]
- [3].Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ, Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial, JAMA 289 (2003) 2819–2826. [DOI] [PubMed] [Google Scholar]
- [4].Akuzawa S, Kazui T, Shi E, Yamashita K, Bashar AH, Terada H, Interleukin-1 receptor antagonist attenuates the severity of spinal cord ischemic injury in rabbits, J. Vasc. Surg. 48 (2008) 694–700. [DOI] [PubMed] [Google Scholar]
- [5].Alexianu ME, Kozovska M, Appel SH, Immune reactivity in a mouse model of familial ALS correlates with disease progression, Neurology 57 (2001) 1282–1289. [DOI] [PubMed] [Google Scholar]
- [6].Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, Raine CS, Brosnan CF, John GR, IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program, J. Immunol. 177 (2006) 5574–5584. [DOI] [PubMed] [Google Scholar]
- [7].Arulampalam V, Kolosenko I, Hjortsberg L, Bjorklund AC, Grander D, Tamm KP, Activation of STAT1 is required for interferon-alpha-mediated cell death, Exp. Cell Res. 317 (2011) 9–19. [DOI] [PubMed] [Google Scholar]
- [8].Bal-Price A, Moneer Z, Brown GC, Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes, Glia 40 (2002) 312–323. [DOI] [PubMed] [Google Scholar]
- [9].Beaumont E, Whitaker CM, Burke DA, Hetman M, Onifer SM, Effects of rolipram on adult rat oligodendrocytes and functional recovery after contusive cervical spinal cord injury, Neuroscience 163 (2009) 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bellido T, Borba VZ, Roberson P, Manolagas SC, Activation of the Janus kinase/STAT (signal transducer and activator of transcription) signal transduction pathway by interleukin-6-type cytokines promotes osteoblast differentiation, Endocrinology 138 (1997) 3666–3676. [DOI] [PubMed] [Google Scholar]
- [11].Bender LM, Morgan MJ, Thomas LR, Liu ZG, Thorburn A, The adaptor protein TRADD activates distinct mechanisms of apoptosis from the nucleus and the cytoplasm, Cell Death Differ. 12 (2005) 473–481. [DOI] [PubMed] [Google Scholar]
- [12].Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR, Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice, Neurobiol. Aging 20 (1999) 581–589. [DOI] [PubMed] [Google Scholar]
- [13].Bessis A, Bechade C, Bernard D, Roumier A, Microglial control of neuronal death and synaptic properties, Glia 55 (2007) 233–238. [DOI] [PubMed] [Google Scholar]
- [14].Blandino P Jr., Barnum CJ, Deak T, The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress, J. Neuroimmunol. 173 (2006) 87–95. [DOI] [PubMed] [Google Scholar]
- [15].Blonska M, Shambharkar PB, Kobayashi M, Zhang D, Sakurai H, Su B, Lin X, TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation, J. Biol. Chem. 280 (2005) 43056–43063. [DOI] [PubMed] [Google Scholar]
- [16].Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P, Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients, Neurosci. Lett. 202 (1995) 17–20. [DOI] [PubMed] [Google Scholar]
- [17].Boillee S, Vande Velde C, Cleveland DW, ALS: a disease of motor neurons and their nonneuronal neighbors, Neuron 52 (2006) 39–59. [DOI] [PubMed] [Google Scholar]
- [18].Bourke E, Cassetti A, Villa A, Fadlon E, Colotta F, Mantovani A, IL-1 beta scavenging by the type II IL-1 decoy receptor in human neutrophils, J. Immunol. 170 (2003) 5999–6005. [DOI] [PubMed] [Google Scholar]
- [19].Breder CD, Hazuka C, Ghayur T, Klug C, Huginin M, Yasuda K, Teng M, Saper CB, Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration, Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 11393–11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brikos C, Wait R, Begum S, O’Neill LA, Saklatvala J , Mass spectrometric analysis of the endogenous type I interleukin-1 (IL-1) receptor signaling complex formed after IL-1 binding identifies IL-1RAcP, MyD88, and IRAK-4 as the stable components, Mol. Cell. Proteomics 6 (2007) 1551–1559. [DOI] [PubMed] [Google Scholar]
- [21].Brugg B, Michel PP, Agid Y, Ruberg M, Ceramide induces apoptosis in cultured mesencephalic neurons, J. Neurochem. 66 (1996) 733–739. [DOI] [PubMed] [Google Scholar]
- [22].Brunet A, Datta SR, Greenberg ME, Transcription-dependent and - independent control of neuronal survival by the PI3K-Akt signaling pathway, Curr. Opin. Neurobiol. 11 (2001) 297–305. [DOI] [PubMed] [Google Scholar]
- [23].Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M, Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells, Mol. Cell. Neurosci. 31 (2006) 149–160. [DOI] [PubMed] [Google Scholar]
- [24].Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW, Gahring LC, Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways, J. Immunol. 163 (1999) 3963–3968. [PubMed] [Google Scholar]
- [25].Carroll JC, Pike CJ, Selective estrogen receptor modulators differentially regulate Alzheimer-like changes in female 3xTg-AD mice, Endocrinology 149 (2008) 2607–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J, Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils, Blood 110 (2007) 1748–1755. [DOI] [PubMed] [Google Scholar]
- [27].Chang JY, Liu LZ, Catecholamines inhibit microglial nitric oxide production, Brain Res. Bull. 52 (2000) 525–530. [DOI] [PubMed] [Google Scholar]
- [28].Chao CC, Hu S, Ehrlich L, Peterson PK, Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors, Brain Behav. Immun. 9 (1995) 355–365. [DOI] [PubMed] [Google Scholar]
- [29].Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, Hubans C, Maurage CA, Huot L, Bensemain F, Laumet G, Ayral AM, Fievet N, Hauw JJ, DeKosky ST, Lemoine Y, Iwatsubo T, Wavrant-Devrieze F, Dartigues JF, Tzourio C, Buee L, Pasquier F, Berr C, Mann D, Lendon C, Alperovitch A, Kamboh MI, Amouyel P, Lambert JC, Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer’s disease, Mol. Psychiatry 14 (2009) 1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chik CL, Li B, Negishi T, Karpinski E, Ho AK, Ceramide inhibits L-type calcium channel currents in rat pinealocytes, Endocrinology 140 (1999) 5682–5690. [DOI] [PubMed] [Google Scholar]
- [31].Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM, FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis, Cell 81 (1995) 505–512. [DOI] [PubMed] [Google Scholar]
- [32].Choi SJ, Lee KH, Park HS, Kim SK, Koh CM, Park JY, Differential expression, shedding, cytokine regulation and function of TNFR1 and TNFR2 in human fetal astrocytes, Yonsei Med. J. 46 (2005) 818–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Choi Y, Kim HS, Shin KY, Kim EM, Kim M, Park CH, Jeong YH, Yoo J, Lee JP, Chang KA, Kim S, Suh YH, Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models, Neuropsychopharmacology 32 (2007) 2393–2404. [DOI] [PubMed] [Google Scholar]
- [34].Christiansen SH, Selige J, Dunkern T, Rassov A, Leist M, Combined anti-inflammatory effects of beta(2)-adrenergic agonists and PDE4 inhibitors on astrocytes by upregulation of intracellular cAMP, Neurochem. Int. (2011), Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- [35].Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD, Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation, Mol. Cell. Biol. 28 (2008) 3538–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Coulie PG, Vanhecke A, Van Damme J, Cayphas S, Poupart P, De Wit L, Content J, High-affinity binding sites for human 26-kDa protein (interleukin 6, B cell stimulatory factor-2, human hybridoma plasmacytoma growth factor, interferon-beta 2), different from those of type I interferon (alpha, beta), on lymphoblastoid cells, Eur. J. Immunol. 17 (1987) 1435–1440. [DOI] [PubMed] [Google Scholar]
- [37].Cunnick JM, Meng S, Ren Y, Desponts C, Wang HG, Djeu JY, Wu J, Regulation of the mitogen-activated protein kinase signaling pathway by SHP2, J. Biol. Chem. 277 (2002) 9498–9504. [DOI] [PubMed] [Google Scholar]
- [38].Das A, Smith JA, Gibson C, Varma AK, Ray SK, Banik NL, Estrogen receptor agonists and estrogen attenuate TNF-alpha-induced apoptosis in VSC4.1 motoneurons, J. Endocrinol. 208 (2011) 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Davis CN, Tabarean I, Gaidarova S, Behrens MM, Bartfai T, IL-1beta induces a MyD88-dependent and ceramide-mediated activation of Src in anterior hypothalamic neurons, J. Neurochem. 98 (2006) 1379–1389. [DOI] [PubMed] [Google Scholar]
- [40].Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. , Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase, Science 261 (1993) 1047–1051. [DOI] [PubMed] [Google Scholar]
- [41].Dheen ST, Kaur C, Ling EA, Microglial activation and its implications in the brain diseases, Curr. Med. Chem. 14 (2007) 1189–1197. [DOI] [PubMed] [Google Scholar]
- [42].Di Bona D, Candore G, Franceschi C, Licastro F, Colonna-Romano G, Camma C, Lio D, Caruso C, Systematic review by meta-analyses on the possible role of TNF-alpha polymorphisms in association with Alzheimer’s disease, Brain Res. Rev. 61 (2009) 60–68. [DOI] [PubMed] [Google Scholar]
- [43].Di Bona D, Plaia A, Vasto S, Cavallone L, Lescai F, Franceschi C, Licastro F, Colonna-Romano G, Lio D, Candore G, Caruso C, Association between the interleukin-1beta polymorphisms and Alzheimer’s disease: a systematic review and meta-analysis, Brain Res Rev 59 (2008) 155–163. [DOI] [PubMed] [Google Scholar]
- [44].Dickson DW, The pathogenesis of senile plaques, J. Neuropathol. Exp. Neurol. 56 (1997) 321–339. [DOI] [PubMed] [Google Scholar]
- [45].Doorduin J, de Vries EF, Dierckx RA, Klein HC, PET imaging of the peripheral benzodiazepine receptor: monitoring disease progression and therapy response in neurodegenerative disorders, Curr. Pharm. Des. 14 (2008) 3297–3315. [DOI] [PubMed] [Google Scholar]
- [46].Elenkov IJ, Kovacs K, Duda E, Stark E, Vizi ES, Presynaptic inhibitory effect of TNF-alpha on the release of noradrenaline in isolated median eminence, J. Neuroimmunol. 41 (1992) 117–120. [DOI] [PubMed] [Google Scholar]
- [47].Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES, The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system, Pharmacol. Rev. 52 (2000) 595–638. [PubMed] [Google Scholar]
- [48].Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ, A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients, J. Neurol. Neurosurg. Psychiatry 76 (2005) 1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Etminan M, Gill S, Samii A, Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies, BMJ 327 (2003) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Farber K, Pannasch U, Kettenmann H, Dopamine and noradrenaline control distinct functions in rodent microglial cells, Mol. Cell. Neurosci. 29 (2005) 128–138. [DOI] [PubMed] [Google Scholar]
- [51].Ferrari CC, Depino AM, Prada F, Muraro N, Campbell S, Podhajcer O, Perry VH, Anthony DC, Pitossi FJ, Reversible demyelination, blood–brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain, Am. J. Pathol. 165 (2004) 1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Forster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D, Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood–brain barrier, J. Physiol. 586 (2008) 1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gagne JJ, Power MC, Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis, Neurology 74 (2010) 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Galea E, Heneka MT, Dello Russo C, Feinstein DL, Intrinsic regulation of brain inflammatory responses, Cell. Mol. Neurobiol. 23 (2003) 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM, Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration, J. Neurosci. 28 (2008) 7687–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gao X, Chen H, Schwarzschild MA, Ascherio A, Use of ibuprofen and risk of Parkinson disease, Neurology 76 (2011) 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Garcia-Bueno B, Caso JR, Leza JC, Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms, Neurosci. Biobehav. Rev. 32 (2008) 1136–1151. [DOI] [PubMed] [Google Scholar]
- [58].Gardoni F, Boraso M, Zianni E, Corsini E, Galli CL, Cattabeni F, Marinovich M, Di Luca M, Viviani B, Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1beta and NMDA stimulation, J. Neuroinflamm. 8 (2011) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Geyer M, Muller-Ladner U, Actual status of antiinterleukin-1 therapies in rheumatic diseases, Curr. Opin. Rheumatol. 22 (2010) 246–251. [DOI] [PubMed] [Google Scholar]
- [60].Gleeson LC, Ryan KJ, Griffin EW, Connor TJ, Harkin A, The beta2-adrenoceptor agonist clenbuterol elicits neuroprotective, anti-inflammatory and neurotrophic actions in the kainic acid model of excitotoxicity, Brain Behav. Immun. 24 (2010) 1354–1361. [DOI] [PubMed] [Google Scholar]
- [61].Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H, Neville HE, Boylan K, Mozaffar T, Belsh JM, Ravits J, Bedlack RS, Graves MC, McCluskey LF, Barohn RJ, Tandan R, Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial, Lancet Neurol. 6 (2007) 1045–1053. [DOI] [PubMed] [Google Scholar]
- [62].Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P, The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor, Cell 83 (1995) 793–802. [DOI] [PubMed] [Google Scholar]
- [63].Griffin WS, Sheng JG, Gentleman SM, Graham DI, Mrak RE, Roberts GW, Microglial interleukin-1 alpha expression in human head injury: correlations with neuronal and neuritic beta-amyloid precursor protein expression, Neurosci. Lett. 176 (1994) 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Griffin WS, Sheng JG, Roberts GW, Mrak RE, Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution, J. Neuropathol. Exp. Neurol. 54 (1995) 276–281. [DOI] [PubMed] [Google Scholar]
- [65].Guasti L, Richardson D, Jhaveri M, Eldeeb K, Barrett D, Elphick MR, Alexander SP, Kendall D, Michael GJ, Chapman V, Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain, Mol. Pain 5 (2009) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Guicciardi ME, Gores GJ, AIP1: a new player in TNF signaling, J. Clin. Invest. 111 (2003) 1813–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gururaja TL, Yung S, Ding R, Huang J, Zhou X, McLaughlin J, Daniel-Issakani S, Singh R, Cooper RD, Payan DG, Masuda ES, Kinoshita T, A class of small molecules that inhibit TNFalpha-induced survival and death pathways via prevention of interactions between TNFalphaRI, TRADD, and RIP1, Chem. Biol. 14 (2007) 1105–1118. [DOI] [PubMed] [Google Scholar]
- [68].Gustin JA, Ozes ON, Akca H, Pincheira R, Mayo LD, Li Q, Guzman JR, Korgaonkar CK, Donner DB, Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappa B activation, J. Biol. Chem. 279 (2004) 1615–1620. [DOI] [PubMed] [Google Scholar]
- [69].Hanisch UK, Microglia as a source and target of cytokines, Glia 40 (2002) 140–155. [DOI] [PubMed] [Google Scholar]
- [70].Hanisch UK, Kettenmann H, Microglia: active sensor and versatile effector cells in the normal and pathologic brain, Nat. Neurosci. 10 (2007) 1387–1394. [DOI] [PubMed] [Google Scholar]
- [71].Hanisch UK, Prinz M, Angstwurm K, Hausler KG, Kann O, Kettenmann H, Weber JR, The protein tyrosine kinase inhibitor AG126 prevents the massive microglial cytokine induction by pneumococcal cell walls, Eur. J. Immunol. 31 (2001) 2104–2115. [DOI] [PubMed] [Google Scholar]
- [72].Harms AS, Barnum CJ, Ruhn KA, Varghese S, Trevino I, Blesch A, Tansey MG, Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson’s disease, Mol. Ther. 19 (2011) 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hasko G, Pacher P, Vizi ES, Illes P, Adenosine receptor signaling in the brain immune system, Trends Pharmacol. Sci. 26 (2005) 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hasko G, Szabo C, Nemeth ZH, Salzman AL, Vizi ES, Suppression of IL-12 production by phosphodiesterase inhibition in murine endotoxemia is IL-10 independent, Eur. J. Immunol. 28 (1998) 468–472. [DOI] [PubMed] [Google Scholar]
- [75].He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y, Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice, J. Cell Biol. 178 (2007) 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].He XH, Zang Y, Chen X, Pang RP, Xu JT, Zhou X, Wei XH, Li YY, Xin WJ, Qin ZH, Liu XG, TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury, Pain 151 (2010) 266–279. [DOI] [PubMed] [Google Scholar]
- [77].Heneka MT, O’Banion MK, Terwel D, Kummer MP, Neuroinflammatory processes in Alzheimer’s disease, J. Neural Transm. 117 (2010) 919–947. [DOI] [PubMed] [Google Scholar]
- [78].Hensley K, Fedynyshyn J, Ferrell S, Floyd RA, Gordon B, Grammas P, Hamdheydari L, Mhatre M, Mou S, Pye QN, Stewart C, West M, West S, Williamson KS, Message and protein-level elevation of tumor necrosis factor alpha (TNF alpha) and TNF alpha-modulating cytokines in spinal cords of the G93A–SOD1 mouse model for amyotrophic lateral sclerosis, Neurobiol. Dis. 14 (2003) 74–80. [DOI] [PubMed] [Google Scholar]
- [79].Hensley K, Floyd RA, Gordon B, Mou S, Pye QN, Stewart C, West M, Williamson K, Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A–SOD1 mouse model of amyotrophic lateral sclerosis, J. Neurochem. 82 (2002) 365–374. [DOI] [PubMed] [Google Scholar]
- [80].Hirano T, Ishihara K, Hibi M, Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors, Oncogene 19 (2000) 2548–2556. [DOI] [PubMed] [Google Scholar]
- [81].Hofmeister R, Wiegmann K, Korherr C, Bernardo K, Kronke M, Falk W, Activation of acid sphingomyelinase by interleukin-1 (IL-1) requires the IL-1 receptor accessory protein, J. Biol. Chem. 272 (1997) 27730–27736. [DOI] [PubMed] [Google Scholar]
- [82].Hu S, Peterson PK, Chao CC, Cytokine-mediated neuronal apoptosis, Neurochem. Int. 30 (1997) 427–431. [DOI] [PubMed] [Google Scholar]
- [83].Hull M, Berger M, Volk B, Bauer J, Occurrence of interleukin-6 in cortical plaques of Alzheimer’s disease patients may precede transformation of diffuse into neuritic plaques, Ann. N. Y. Acad. Sci. 777 (1996) 205–212. [DOI] [PubMed] [Google Scholar]
- [84].Hurst S, Collins SM, Interleukin-1 beta modulation of norepinephrine release from rat myenteric nerves, Am. J. Physiol. 264 (1993) G30–G35. [DOI] [PubMed] [Google Scholar]
- [85].Hurst SM, Collins SM, Mechanism underlying tumor necrosis factor-alpha suppression of norepinephrine release from rat myenteric plexus, Am. J. Physiol. 266 (1994) G1123–G1129. [DOI] [PubMed] [Google Scholar]
- [86].Iannotti CA, Clark M, Horn KP, van Rooijen N, Silver J, Steinmetz MP, A combination immunomodulatory treatment promotes neuroprotection and locomotor recovery after contusion SCI, Exp. Neurol. 230 (2011) 3–15. [DOI] [PubMed] [Google Scholar]
- [87].Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y, Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains, Acta Neuropathol. 106 (2003) 518–526. [DOI] [PubMed] [Google Scholar]
- [88].Ionov M, Burchell V, Klajnert B, Bryszewska M, Abramov AY, Mechanism of neuroprotection of melatonin against beta-amyloid neurotoxicity, Neuroscience 180 (2011) 229–237. [DOI] [PubMed] [Google Scholar]
- [89].Jenab S, Quinones-Jenab V, The effects of interleukin-6, leukemia inhibitory factor and interferon-gamma on STAT DNA binding and c-fos mRNA levels in cortical astrocytes and C6 glioma cells, Neuro Endocrinol. Lett. 23 (2002) 325–328. [PubMed] [Google Scholar]
- [90].Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M, Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines, Neuroscience 135 (2005) 1295–1307. [DOI] [PubMed] [Google Scholar]
- [91].Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ, TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains, Mol. Cell 15 (2004) 535–548. [DOI] [PubMed] [Google Scholar]
- [92].Kato S, Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences, Acta Neuropathol 115 (2008) 97–114. [DOI] [PubMed] [Google Scholar]
- [93].Ke JJ, Yu FX, Rao Y, Wang YL, Adenosine postconditioning protects against myocardial ischemia-reperfusion injury though modulate production of TNF-alpha and prevents activation of transcription factor NF-kappaB, Mol. Biol. Rep. 38 (2011) 531–538. [DOI] [PubMed] [Google Scholar]
- [94].Kettenmann H, Hanisch UK, Noda M, Verkhratsky A, Physiology of microglia, Physiol. Rev. 91 (2011) 461–553. [DOI] [PubMed] [Google Scholar]
- [95].Kovacs Z, Czurko A, Kekesi KA, Juhasz G, Intracerebroventricularly administered lipopolysaccharide enhances spike-wave discharges in freely moving WAG/Rij rats, Brain Res. Bull. 85 (2011) 410–416. [DOI] [PubMed] [Google Scholar]
- [96].Kreutzberg GW, Microglia: a sensor for pathological events in the CNS, Trends Neurosci. 19 (1996) 312–318. [DOI] [PubMed] [Google Scholar]
- [97].Kriz J, Nguyen MD, Julien JP, Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis, Neurobiol. Dis. 10 (2002) 268–278. [DOI] [PubMed] [Google Scholar]
- [98].Lawson LJ, Perry VH, Dri P, Gordon S, Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain, Neuroscience 39 (1990) 151–170. [DOI] [PubMed] [Google Scholar]
- [99].Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR, Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation, Pain 115 (2005) 71–83. [DOI] [PubMed] [Google Scholar]
- [100].Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D, LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice, J Neuroinflammation 7 (2010) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW, Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta, J. Immunol. 150 (1993) 2659–2667. [PubMed] [Google Scholar]
- [102].Lee SM, Yune TY, Kim SJ, Park DW, Lee YK, Kim YC, Oh YJ, Markelonis GJ, Oh TH, Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat, J. Neurotrauma 20 (2003) 1017–1027. [DOI] [PubMed] [Google Scholar]
- [103].Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Hoi Yu AC, Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease, Curr. Alzheimer Res. 8 (2011) 67–80. [DOI] [PubMed] [Google Scholar]
- [104].Lin W, Lin Y, Interferon-gamma inhibits central nervous system myelination through both STAT1-dependent and STAT1-independent pathways, J. Neurosci. Res. 88 (2010) 2569–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Litteljohn D, Mangano E, Shukla N, Hayley S, Interferon-gamma deficiency modifies the motor and co-morbid behavioral pathology and neurochemical changes provoked by the pesticide paraquat, Neuroscience 164 (2009) 1894–1906. [DOI] [PubMed] [Google Scholar]
- [106].Liu ZG, Hsu H, Goeddel DV, Karin M, Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death, Cell 87 (1996) 565–576. [DOI] [PubMed] [Google Scholar]
- [107].LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA, Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations, Drug Resist. Updat. 11 (2008) 32–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lozza G, Conti A, Ongini E, Monopoli A, Cardioprotective effects of adenosine A1 and A2A receptor agonists in the isolated rat heart, Pharmacol. Res. 35 (1997) 57–64. [DOI] [PubMed] [Google Scholar]
- [109].Lu Y, Zhu L, Gao YJ, Pain-related aversion induces astrocytic reaction and proinflammatory cytokine expression in the anterior cingulate cortex in rats, Brain Res. Bull. 84 (2011) 178–182. [DOI] [PubMed] [Google Scholar]
- [110].Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM Jr., Brachova L, Yan SD, Walker DG, Shen Y, Rogers J, Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro, Glia 35 (2001) 72–79. [DOI] [PubMed] [Google Scholar]
- [111].Lupardus PJ, Skiniotis G, Rice AJ, Thomas C, Fischer S, Walz T, Garcia KC, Structural snapshots of full-length Jak1, a transmembrane gp130/IL-6/IL6-Ralpha cytokine receptor complex, and the receptor-Jak1 holocomplex, Structure 19 (2011) 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Lye TC, Shores EA, Traumatic brain injury as a risk factor for Alzheimer’s disease: a review, Neuropsychol. Rev. 10 (2000) 115–129. [DOI] [PubMed] [Google Scholar]
- [113].Magnus T, Chan A, Grauer O, Toyka KV, Gold R, Microglial phagocytosis of apoptotic inflammatory T cells leads to down-regulation of microglial immune activation, J. Immunol. 167 (2001) 5004–5010. [DOI] [PubMed] [Google Scholar]
- [114].Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG, Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 11778–11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Manning BD, Cantley LC, AKT/PKB signaling: navigating downstream, Cell 129 (2007) 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].McDermott EP, O’Neill LA, Ras participates in the activation of p38 MAPK by interleukin-1 by associating with IRAK, IRAK2, TRAF6, and TAK-1, J. Biol. Chem. 277 (2002) 7808–7815. [DOI] [PubMed] [Google Scholar]
- [117].McDonald DR, Brunden KR, Landreth GE, Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia, J. Neurosci. 17 (1997) 2284–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].McGeer PL, Itagaki S, Boyes BE, McGeer EG, Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains, Neurology 38 (1988) 1285–1291. [DOI] [PubMed] [Google Scholar]
- [119].McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM, Tumor necrosis factor alpha is toxic to embryonic mesencephalic dopamine neurons, Exp. Neurol. 169 (2001) 219–230. [DOI] [PubMed] [Google Scholar]
- [120].Meda L, Cassatella MA, Szendrei GI, Otvos L Jr., Baron P, Villalba M, Ferrari D, Rossi F, Activation of microglial cells by beta-amyloid protein and interferon-gamma, Nature 374 (1995) 647–650. [DOI] [PubMed] [Google Scholar]
- [121].Mehlhorn G, Hollborn M, Schliebs R, Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology, Int. J. Dev. Neurosci. 18 (2000) 423–431. [DOI] [PubMed] [Google Scholar]
- [122].Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T, Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease, Neurosci. Lett. 211 (1996) 13–16. [DOI] [PubMed] [Google Scholar]
- [123].Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T, Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients, Neurosci. Lett. 165 (1994) 208–210. [DOI] [PubMed] [Google Scholar]
- [124].Mogi M, Togari A, Tanaka K, Ogawa N, Ichinose H, Nagatsu T, Increase in level of tumor necrosis factor (TNF)-alpha in 6-hydroxydopamine-lesioned striatum in rats without influence of systemic L-DOPA on the TNF-alpha induction, Neurosci. Lett. 268 (1999) 101–104. [DOI] [PubMed] [Google Scholar]
- [125].Mori K, Ozaki E, Zhang B, Yang L, Yokoyama A, Takeda I, Maeda N, Sakanaka M, Tanaka J, Effects of norepinephrine on rat cultured microglial cells that express alpha1, alpha2, beta1 and beta2 adrenergic receptors, Neuropharmacology 43 (2002) 1026–1034. [DOI] [PubMed] [Google Scholar]
- [126].Muroi M, Tanamoto K, TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-kappaB, J. Leukoc. Biol. 83 (2008) 702–707. [DOI] [PubMed] [Google Scholar]
- [127].Nakajima K, Kikuchi Y, Ikoma E, Honda S, Ishikawa M, Liu Y, Kohsaka S, Neurotrophins regulate the function of cultured microglia, Glia 24 (1998) 272–289. [DOI] [PubMed] [Google Scholar]
- [128].Nakajima K, Kohsaka S, Microglia: activation and their significance in the central nervous system, J. Biochem. 130 (2001) 169–175. [DOI] [PubMed] [Google Scholar]
- [129].Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA, IL-1beta processing in host defense: beyond the inflammasomes, PLoS Pathog. 6 (2010) e1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV, Toll-like receptors in neurodegeneration, Brain Res Rev 59 (2009) 278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ono S, Hu J, Shimizu N, Imai T, Nakagawa H, Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis, J. Neurol. Sci. 187 (2001) 27–34. [DOI] [PubMed] [Google Scholar]
- [132].Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T, Microglial activation and dopamine terminal loss in early Parkinson’s disease, Ann. Neurol. 57 (2005) 168–175. [DOI] [PubMed] [Google Scholar]
- [133].Paintlia AS, Paintlia MK, Singh I, Singh AK, Combined medication of lovastatin with rolipram suppresses severity of experimental autoimmune encephalomyelitis, Exp. Neurol. 214 (2008) 168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Paintlia AS, Paintlia MK, Singh I, Skoff RB, Singh AK, Combination therapy of lovastatin and rolipram provides neuroprotection and promotes neurorepair in inflammatory demyelination model of multiple sclerosis, Glia 57 (2009) 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Pasqualetti P, Bonomini C, Dal Forno G, Paulon L, Sinforiani E, Marra C, Zanetti O, Rossini PM, A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease, Aging Clin. Exp. Res. 21 (2009) 102–110. [DOI] [PubMed] [Google Scholar]
- [136].Polazzi E, Monti B, Microglia and neuroprotection: from in vitro studies to therapeutic applications, Prog. Neurobiol. 92 (2010) 293–315. [DOI] [PubMed] [Google Scholar]
- [137].Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA, Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment, J. Neurosci. 21 (2001) 3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Prinz M, Hausler KG, Kettenmann H, Hanisch U, Beta-adrenergic receptor stimulation selectively inhibits IL-12p40 release in microglia, Brain Res. 899 (2001) 264–270. [DOI] [PubMed] [Google Scholar]
- [139].Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, Wilson B, Lu RB, Hong JS, Flood PM, beta2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway, J. Immunol. 186 (2011) 4443–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P, et al. , Clinical trial of indomethacin in Alzheimer’s disease, Neurology 43 (1993) 1609–1611. [DOI] [PubMed] [Google Scholar]
- [141].Rogers J, Lue LF, Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease, Neurochem. Int. 39 (2001) 333–340. [DOI] [PubMed] [Google Scholar]
- [142].Rothe M, Sarma V, Dixit VM, Goeddel DV, TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40, Science 269 (1995) 1424–1427. [DOI] [PubMed] [Google Scholar]
- [143].Samantaray S, Sribnick EA, Das A, Knaryan VH, Matzelle DD, Yallapragada AV, Reiter RJ, Ray SK, Banik NL, Melatonin attenuates calpain upregulation, axonal damage and neuronal death in spinal cord injury in rats, J. Pineal Res. 44 (2008) 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Samii A, Etminan M, Wiens MO, Jafari S, NSAID use and the risk of Parkinson’s disease: systematic review and meta-analysis of observational studies, Drugs Aging 26 (2009) 769–779. [DOI] [PubMed] [Google Scholar]
- [145].Santello M, Bezzi P, Volterra A, TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus, Neuron 69 (2011) 988–1001. [DOI] [PubMed] [Google Scholar]
- [146].Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S, Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling, J. Immunol. 171 (2003) 4304–4310. [DOI] [PubMed] [Google Scholar]
- [147].Sawada M, Imamura K, Nagatsu T, Role of cytokines in inflammatory process in Parkinson’s disease, J. Neural Transm. Suppl. (2006) 373–381. [DOI] [PubMed] [Google Scholar]
- [148].Schiemann WP, Bartoe JL, Nathanson NM, Box 3-independent signaling mechanisms are involved in leukemia inhibitory factor receptor alpha- and gp130-mediated stimulation of mitogen-activated protein kinase. Evidence for participation of multiple signaling pathways which converge at Ras, J. Biol. Chem. 272 (1997) 16631–16636. [DOI] [PubMed] [Google Scholar]
- [149].Sekizawa T, Openshaw H, Ohbo K, Sugamura K, Itoyama Y, Niland JC, Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases, J. Neurol. Sci. 154 (1998) 194–199. [DOI] [PubMed] [Google Scholar]
- [150].Shang XL, Zhao JH, Cao YP, Xue YX, Effects of synaptic plasticity regulated by 17beta-estradiol on learning and memory in rats with Alzheimer’s disease, Neurosci. Bull. 26 (2010) 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Sheikh MS, Huang Y, Death receptor activation complexes: it takes two to activate TNF receptor 1, Cell Cycle 2 (2003) 550–552. [PubMed] [Google Scholar]
- [152].Skiniotis G, Boulanger MJ, Garcia KC, Walz T, Signaling conformations of the tall cytokine receptor gp130 when in complex with IL-6 and IL-6 receptor, Nat. Struct. Mol. Biol. 12 (2005) 545–551. [DOI] [PubMed] [Google Scholar]
- [153].Smith JA, Zhang R, Varma AK, Das A, Ray SK, Banik NL, Estrogen partially down-regulates PTEN to prevent apoptosis in VSC4.1 motoneurons following exposure to IFN-gamma, Brain Res. 1301 (2009) 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Sribnick EA, Matzelle DD, Ray SK, Banik NL, Estrogen treatment of spinal cord injury attenuates calpain activation and apoptosis, J. Neurosci. Res. 84 (2006) 1064–1075. [DOI] [PubMed] [Google Scholar]
- [155].Sribnick EA, Samantaray S, Das A, Smith J, Matzelle DD, Ray SK, Banik NL, Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats, J. Neurosci. Res. 88 (2010) 1738–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP, Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease, FASEB J. 16 (2002) 1474–1476. [DOI] [PubMed] [Google Scholar]
- [157].Sternberg EM, Neuroendocrine regulation of autoimmune/inflammatory disease, J. Endocrinol. 169 (2001) 429–435. [DOI] [PubMed] [Google Scholar]
- [158].Stoll G, Jander S, The role of microglia and macrophages in the pathophysiology of the CNS, Prog. Neurobiol. 58 (1999) 233–247. [DOI] [PubMed] [Google Scholar]
- [159].Stone TW, Ceruti S, Abbracchio MP, Adenosine receptors and neurological disease: neuroprotection and neurodegeneration, Handb. Exp. Pharmacol. (2009) 535–587. [DOI] [PubMed] [Google Scholar]
- [160].Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ, Synuclein activates microglia in a model of Parkinson’s disease, Neurobiol. Aging 29 (2008) 1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Suzuki K, Hino M, Kutsuna H, Hato F, Sakamoto C, Takahashi T, Tatsumi N, Kitagawa S, Selective activation of p38 mitogen-activated protein kinase cascade in human neutrophils stimulated by IL-1beta, J. Immunol. 167 (2001) 5940–5947. [DOI] [PubMed] [Google Scholar]
- [162].Swardfager W, Lanctot K, Rothenburg L, Wong A, Cappell J, Herrmann N, A meta-analysis of cytokines in Alzheimer’s disease, Biol. Psychiatry 68 (2010) 930–941. [DOI] [PubMed] [Google Scholar]
- [163].Takahashi-Tezuka M, Yoshida Y, Fukada T, Ohtani T, Yamanaka Y, Nishida K, Nakajima K, Hibi M, Hirano T, Gab1 acts as an adapter molecule linking the cytokine receptor gp130 to ERK mitogen-activated protein kinase, Mol. Cell. Biol. 18 (1998) 4109–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Tanaka KF, Kashima H, Suzuki H, Ono K, Sawada M, Existence of functional beta1- and beta2-adrenergic receptors on microglia, J. Neurosci. Res. 70 (2002) 232–237. [DOI] [PubMed] [Google Scholar]
- [165].Tarkowski E, Liljeroth AM, Nilsson A, Minthon L, Blennow K, Decreased levels of intrathecal interleukin 1 receptor antagonist in Alzheimer’s disease, Dement. Geriatr. Cogn. Disord. 12 (2001) 314–317. [DOI] [PubMed] [Google Scholar]
- [166].Tartaglia LA, Goeddel DV, Two TNF receptors, Immunol. Today 13 (1992) 151–153. [DOI] [PubMed] [Google Scholar]
- [167].Terenghi F, Allaria S, Nobile-Orazio E, Circulating levels of cytokines and their modulation by intravenous immunoglobulin in multifocal motor neuropathy, J. Peripher. Nerv. Syst. 11 (2006) 67–71. [DOI] [PubMed] [Google Scholar]
- [168].Tikka TM, Koistinaho JE, Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia, J. Immunol. 166 (2001) 7527–7533. [DOI] [PubMed] [Google Scholar]
- [169].Tobinick EL, Gross H, Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration, J Neuroinflammation 5 (2008) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Tobinick EL, Gross H, Rapid improvement in verbal fluency and aphasia following perispinal etanercept in Alzheimer’s disease, BMC Neurol. 8 (2008) 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Tomozawa Y, Yabuuchi K, Inoue T, Satoh M, Participation of cAMP and cAMP-dependent protein kinase in beta-adrenoceptor-mediated interleukin-1 beta mRNA induction in cultured microglia, Neurosci. Res. 22 (1995) 399–409. [DOI] [PubMed] [Google Scholar]
- [172].Tsutsui S, Schnermann J, Noorbakhsh F, Henry S, Yong VW, Winston BW, Warren K, Power C, A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis, J. Neurosci. 24 (2004) 1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB, Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study, Neurobiol. Dis. 15 (2004) 601–609. [DOI] [PubMed] [Google Scholar]
- [174].Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M, Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling, Nat. Immunol. 9 (2008) 1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].van Neerven S, Nemes A, Imholz P, Regen T, Denecke B, Johann S, Beyer C, Hanisch UK, Mey J, Inflammatory cytokine release of astrocytes in vitro is reduced by all-trans retinoic acid, J. Neuroimmunol. 229 (2010) 169–179. [DOI] [PubMed] [Google Scholar]
- [176].Vogt C, Hailer NP, Ghadban C, Korf HW, Dehghani F, Successful inhibition of excitotoxic neuronal damage and microglial activation after delayed application of interleukin-1 receptor antagonist, J. Neurosci. Res. 86 (2008) 3314–3321. [DOI] [PubMed] [Google Scholar]
- [177].Wajant H, Scheurich P, Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling, Int. J. Biochem. Cell Biol. 33 (2001) 19–32. [DOI] [PubMed] [Google Scholar]
- [178].Wajant H, Scheurich P, TNFR1-induced activation of the classical NF-kappaB pathway, FEBS J. 278 (2011) 862–876. [DOI] [PubMed] [Google Scholar]
- [179].Wakselman S, Bechade C, Roumier A, Bernard D, Triller A, Bessis A, Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor, J. Neurosci. 28 (2008) 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Walter L, Neumann H, Role of microglia in neuronal degeneration and regeneration, Semin. Immunopathol. 31 (2009) 513–525. [DOI] [PubMed] [Google Scholar]
- [181].Weber A, Wasiliew P, Kracht M, Interleukin-1 (IL-1) pathway, Sci. Signal. 3 (2010) cm1. [DOI] [PubMed] [Google Scholar]
- [182].Wertz IE, Dixit VM, Ubiquitin-mediated regulation of TNFR1 signaling, Cytokine Growth Factor Rev. 19 (2008) 313–324. [DOI] [PubMed] [Google Scholar]
- [183].Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poirier C, Mattson MP, Geiger JD, Haughey NJ, Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors, J. Neurochem. 109 (2009) 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD, Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected], Nat. Cell Biol. 8 (2006) 398–406. [DOI] [PubMed] [Google Scholar]
- [185].Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S, Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease, J. Neurosci. 22 (2002) 1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Xia ZP, Chen ZJ, TRAF2: a double-edged sword? Sci. STKE 2005 (2005) pe7. [DOI] [PubMed] [Google Scholar]
- [187].Yu FX, Ke JJ, Fu Y, Liao B, Shi YK, Protective effects of adenosine in rabbit sinoatrial node ischemia-reperfusion model in vivo: control of arrhythmia by hyperpolarization-activated cyclic nucleotide-gated (HCN)4 channels, Mol. Biol. Rep. 38 (2011) 1723–1731. [DOI] [PubMed] [Google Scholar]
- [188].Yu JT, Song JH, Wang ND, Wu ZC, Zhang Q, Zhang N, Zhang W, Xuan SY, Tan L, Implication of IL-33 gene polymorphism in Chinese patients with Alzheimer’s disease, Neurobiol. Aging (2010), Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- [189].Zhang R, Yamada J, Hayashi Y, Wu Z, Koyama S, Nakanishi H, Inhibition of NMDA-induced outward currents by interleukin-1beta in hippocampal neurons, Biochem. Biophys. Res. Commun. 372 (2008) 816–820. [DOI] [PubMed] [Google Scholar]
- [190].Zhao Y, Li S, Childs EE, Kuharsky DK, Yin XM, Activation of prodeath Bcl-2 family proteins and mitochondria apoptosis pathway in tumor necrosis factor-alpha-induced liver injury, J. Biol. Chem. 276 (2001) 27432–27440. [DOI] [PubMed] [Google Scholar]
- [191].Zhou C, Ye HH, Wang SQ, Chai Z, Interleukin-1beta regulation of N-type Ca2+ channels in cortical neurons, Neurosci. Lett. 403 (2006) 181–185. [DOI] [PubMed] [Google Scholar]