

Abstract
Osteosarcoma (OS) is characterized by aggressive features including invasiveness and high incidence of metastasis. OS patients with metastases are difficult to treat and suffer from a poor prognosis. DPY30 (protein dpy- 30 homolog) is a key component of SET1/MLL family of H3K4 methyltransferases, which is implicated in the progression of multiple cancers. However, the potential functional engagement of DPY30 in OS remains to be unveiled. The objective of this study is to investigate the potential roles of DPY30 in the regulation of malignant phenotypes of OS cells. We examined DPY30 expression from a published dataset (GSE28424) as well as in OS tissues and adjacent normal tissues from OS patients. The association of DPY30 expression level and clinicopathologic parameters was assessed by Chi-square test. The role of DPY30 in regulating the malignant phenotype of OS cells and tumorigenesis was examined by in vitro functional assays and xenograft mouse model. We reported an upregulation of DPY30 in OS tumor tissues in both published dataset and clinical samples. A high level of DPY30 expression was associated with larger tumor size and more metastasis in OS patients, as well as poor overall survival. DPY30 knockdown in OS cells significantly impairs proliferation, migration and invasion, but induced cellular apoptosis. We further demonstrated that the agonist of PI3K/AKT pathway can rescue the inhibitory effects of DPY30 knockdown in OS cells. Together, our data indicate that DPY30 functions as an oncogene to promote the malignancy of OS cells possibly through PI3K/AKT pathway. The dependency of OS cells on DPY30 overexpression is a targetable vulnerability in OS cells.
Key words: Osteosarcoma, DPY30, PI3K/AKT signal pathway
Introduction
Osteosarcoma (OS) is one of the most common tumors frequently diagnosed in children and adolescents.1At present, surgery in combination with chemotherapy remains as the mainstay for OS treatment. Although novel strategies such as immunotherapy and targeted therapy have been developed to tackle metastatic OS, the treatment outcome is limited by its high malignancy.2 The 5-year overall survival of OS patients with optimal surgery is about 70%,3 which increases to 8 % after post-operative neoadjuvant chemotherapy.4 However, the 5-year survival rate of OS patients with distant metastasis is as low as 30%,5 and metastasis accounts for the leading cause of death in OS patients.6,7 Dissecting the molecular mechanisms underlying the malignant progression of OS could provide insights into the formulation of effective treatment strategies.
Histones are the structural proteins involved in the genome organization and epigenetic regulation.8 Epigenetic perturbations caused by the deregulation of histone methyltransferases or demethylases are regarded as important factors in cancer progression. 9,1. Protein dpy-30 homolog (DPY30), one of the integral subunits of SET1/MLL H3K4 methyltransferase complex, is involved in the regulation of gene expression and cell differentiation by modulating H3K4 methylation.11-13 The deregulation of DPY30 has been reported in different cancers.13,14 Previous studies demonstrated that DPY30 is highly expressed in cholangiocarcinoma,15 cervical cancer,16 ovarian cancer13 and gastric cancer,17 suggesting its oncogenic role in tumorigenesis. However, its potential contribution to the malignant progression of OS cells remains to be explored. Given that DPY30 promotes the epithelial-mesenchymal transition (EMT), migration and invasion in different cancer cells13,16 and the fact that OS cells are highly invasive,6,7 we postulate that DPY30-mediated EMT process underpins the differentiation of OS cells and contributes to its malignant progression.
In this study, we investigated the potential role of DPY30 in malignant phenotypes of OS cells, using cell line model and xenograft tumorigenesis model. We reported a high-level expression of DPY30 in OS tumor tissues, which was associated with larger tumor size and more metastasis in OS patients. Silencing DPY30 suppressed the proliferation and invasion and dampened the mesenchymal transition of OS cells. The oncogenic role of DPY30 seemed to be mediated by PI3K/AKT pathway. Together, our data indicate that DPY30 promotes the malignancy of OS cells through PI3K/AKT pathway, suggesting the possibility of targeting DPY30 as an anti-cancer strategy in OS treatment.
Methods and Materials
Data retrieval
The microarray data were downloaded from the GEO database (GSE28424), which contains 19 OS cell lines and four normal bone cell lines. The differentially expressed genes (DEGs) were analyzed using the Limma package in Bioconductor (http://mirrors. nju.edu.cn/bioconductor/3.15/bioc/html/ limma.html).
Clinical samples
A total number of 44 patients diagnosed with OS were recruited at Department of Orthopedics, Yantaishan Hospital, Yantai City, Shandong Province, China. Inclusion criteria: patients above 20 years-old, patients without record of chronic disease; patients were diagnosed with OS, which was confirmed by two independent pathologists. Exclusion criteria: patients with chronic diseases, patients diagnosed with other cancers, patients who had gone previous treatment. OS tumor tissues and the adjacent normal tissues were collected by surgery. The tissues were snap-frozen in liquid nitrogen and stored in -80°C until further use. This study was approved by the Institute Research Ethics Committee of Yantaishan Hospital (no. 2018036).
Cell culture
Human OS cell lines (U2OS, HOS, Saos2 and MG63) were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). OS cells were grown in DMEM (Invitrogen, Waltham, MA, USA) supplemented with 10% FBS (ExCell Bio, Shanghai, China) and 1% penicillin-streptomycin (HyClone, Logan, UT, USA). These cells were cultured in a humidified incubator with containing 5% CO2 at 37°C. Human immortalized osteoblasts (hFOB 1.19) were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China), and cultured in DMEM/F12 (Invitrogen) containing 0.3 mg/mL G418 (TargetMol, Boston, MA, USA) at 34°C. The cells were routinely checked for mycoplasma contamination.
Lentivirus-mediated stable knockdown of DPY30
The knockdown of endogenous DPY30 in U2OS and HOS cells was conducted using a lentiviral vector-based shRNA technique. The shRNA sequences targeting human DPY30 in this study were: 5′-GACCACCAAATCCCATTGAAT-3′ (shDPY30#1), 5′- GCACAGTTTGAAGATCGAAAC-3′(shDPY30#2) and 5′- GACAACCAAA TCCCATTGAAT-3′(shDPY30#3). The shRNA control (shCtrl) sequence was 5′-GGCTACGTCCAGGAGCGCACC- 3′. All the oligonucleotides (synthesized by RiboBio Co. Ltd., Guangzhou, China) were annealed and inserted into the pLKO.1 lentiviral vector. Lentiviruses were produced by transfecting HEK293 T cells with the lentivirus and packing plasmids (Lentiviral Packaging-Lentiviral Packaging Kit, Sigma) using polyethylenimine transfection method. The viral supernatant was collected by centrifuging the culture medium for 10 min at 1000 x g. To generate stable shRNA-mediated knockdown of DPY30 in U2OS and HOS cells, 1×105 cells were seeded in a 24-well plate. When cells reached at 60% confluence, viral transduction was performed with lentiviral supernatant at a MOI (multiplicity of infection) of 5. 48 h after transduction, the infected cells were selected with 1.0 μg/mL puromycin for two weeks to eliminate the uninfected cells before further experiment.
Western blot
Cells cultured in 6-well plates (density, 3×105 cells/mL) were lysed with RIPA lysis containing protease inhibitor cocktail (Thermo Fisher Scientific, MA, USA) on ice for 30 min, followed by centrifugation at 4°C and 12,000 x g for 5 min. The supernatant was collected and the supernatant protein concentration was quantified by a BCA Protein assay kit (Beyotime Biotechnology, Shanghai, China). The protein sample was mixed with sampleloading buffer, followed by boiling for 5 min at 100 °C. 10 μg protein was used for SDS-PAGE for protein separation and transferred to a poly-vinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). The membranes were blocked in 5% skim milk at room temperature for 1 h, followed by overnight incubation with primary antibodies at 4°C. The bound antibodies were detected using secondary antibody conjugated with horseradish peroxidase, and the protein bands were visualized using an enhanced chemiluminescence system (ProteinSimple, San Jose, CA, USA). The protein bands were then analyzed using Quantity One software Version 4.0.1 (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Experiments were performed in triplicate. The following antibodies (Abcam, Madison, WI, USA) were used in this study: GAPDH (Ab9485, 1:2000), PI3K (ab133595, 1:1000), p-PI3K (Y607) (ab182651, 1:1000), p-AKT (Ser474) (ab38513, 1:1000), AKT (ab131168, 1:1000), DPY30 (Ab187690, 1:1500), E-cadherin (ab40772, 1:1000), N-cadherin (ab76011, 1:1500).
Quantitative real-time PCR
Total RNA from the cells and tissue samples was extracted using Trizol reagent (Invitrogen, CA, USA). Reverse transcription was performed according to the guidelines using PrimeScript™ RT reagent Kit with gDNA Eraser (RR047A, Takara, Dalian, China). Quantitative real-time PCR was performed in a 7500 Real Time PCR System (Applied Biosystems, CA, USA) using TB Green™ Premix Ex Taq™ II kit (RR820A, Takara, Dalian, China). The PCR cycling condition used: 95°C 5 min, 40 cycles of 95°C 15 sec, 60°C 30 sec and 72°C 45 sec. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control for normalization. All experiment was performed from three independent biological samples. The primer sequences used in this study (Shanghai Sangon Biotechnology Co., Ltd., Shanghai, China): DPY30: 5′-AACGCAGGTTGCAGAAAATCCT-3′ and 5′-TCTGATCCAGGTAGCACGAG- 3′. GAPDH: 5′-CCACCCATGGCAAATTCC ATGGCA-3′ and 5′-TCTAGACGGCAGGT CAGGTCCACC-3′.
Immunohistochemistry
Immunohistochemical staining was performed using rabbit anti-human DPY30 polyclonal antibody (Nouvs) and anti-human Ki-67 antibody (Abcam) in OS tissues and the para-carcinoma tissues collected OS patients (n=88), as well as in the xenograft tumor tissues for mouse model (n=12). The tissues were fixed in 4% paraformaldehyde overnight. After deparaffinization and rehydration, 4-μm sections of formalin-fixed paraffin-embedded (FFPE) tumor tissues treated with 0.3% hydrogen peroxide for 30 min to inhibit endogenous peroxidase activity, and blocked with 10% normal donkey serum (NDS) and 1% BSA in 1×phosphate buffered saline (PBS) for 1 h at room temperature. Slides were then incubated with primary antibody (1:500 dilutions in the blocking buffer) overnight at 4°C. After washing with TBST buffer, the slides were further incubated with secondary antibody (HRP-conjugated, 1:2000 dilutions) in blocking buffer for 2 h at room temperature. Signal development was performed using SABC-HRP Kit (Vector Laboratories, Burlingame, CA, USA) for 10 min, and slides were counterstained with hematoxylin staining buffer (Sigma-Aldrich, Darmstadt, Germany) for 5 min. The images were captured under Leica AM6000 microscope (Leica, Wetzlar, Germany). Antibody IgG isotype (Abcam) was used as negative controls (Supplementary Figure 1). For each OS tumor sample or xenograft tissue, the number of positively stained cells in 5 sections was quantified.
Figure 1.
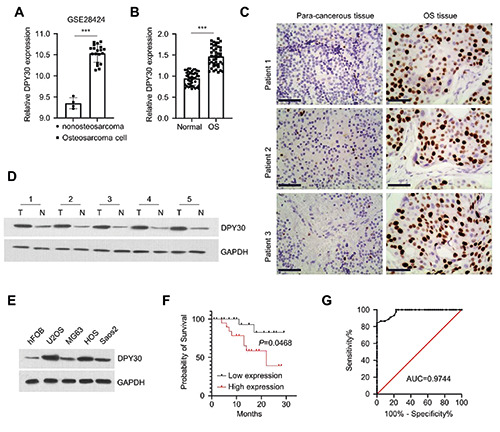
The expression of DPY30 in clinical OS samples and cell lines. A) The expression of DPY30 from the GEO dataset (GSE28424; including 19 osteosarcoma cell lines and 4 normal bone cell lines). B)The expression of DPY30 in the OS tumor tissues and para-carcinoma normal tissues (n=44) were assessed by RT-qPCR.(C) Immunohistochemical (IHC) staining demonstrated the overexpression of DPY30 in OS tumor tissues in comparison to para-carcinoma normal tissues. D) DPY30 protein expression was determined by Western blot in 5 pairs of OS tumor tissues and para-carcinoma normal tissues. E) DPY30 protein expression was determined by Western blot in 4 different OS cells lines (U2OS, HOS, Saos2 and MG63) and human osteoblast cell line (hFOB). F) Kaplan Meier curve and log-rank test were used to compare the overall survival in 44 OS patients. G) Receiver operative characteristics (ROC) curve analysis of DPY30 in the prognosis of OS patients. ***p<0.001.
CCK-8 proliferation assay
Cell proliferation assay was assessed using Cell Counting Kit- 8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s protocol. Cells were seeded in to 96-well plates at a density of 1000 cells/well and cultured for 0, 24, 48, and 72 h; 10 μL of CCK8 reagents was added to the cell culture at indicated time point 1 h incubation. The absorbance (OD value) in each well was captured at 450 nm on a microplate reader.
EdU incorporation assay
Cell in DNA synthesis phase was determined with BeyoClick EdU Cell Proliferation Kit (Beyotime Biotechnology, Beijing, China). Cells cultured in 96-well plates were pulsed with 1 x EdU solution in cell culture medium for 3 h. The medium was discarded and cells were fixed with 3.7% formaldehyde in PBS for 10 min. The fixed cells were washed twice with PBS. Cells were permeabilized with 0.5% Triton® X-100 in PBS for 15 min. After permeabilization, cells were then incubated with 1 x Click-iT® reaction cocktail for 30 min. The cells were washed twice with PBS and further stained by 1 μM DAPI. Cellular images were recorded using Leica AM6000 microscope, and the results were analyzed with Image J Pro Plus 6 software (NIH, Bethesda, MD, USA). The number of EdU-positive cells from 5 randomly selected fields was counted.
Cell migration and invasion assay
The effect of DPY30 knockdown on OS cell migration and invasion was determined by a Transwell assay as previously described.18 Briefly, OS cells were suspended in 100 μL of serumfree DMEM and seeded at the density of 2×104 cells/well in the upper chambers of Transwell plate (24-well plate, 8-mm pore size, Corning, NY, USA). The transwell upper chamber (Corning) without Matrigel (BD Biosciences, MA, USA) was used for migration assay, while transwell upper chamber coated with Matrigel was used for invasion assay. Then, 500 μL of DMEM containing 10% FBS was added to the lower chambers. After culturing for 24 h, cells were fixed with 4% paraformaldehyde at room temperature for 30 min and stained with 0.1% crystal violet (Sigma, Germany) for 20 min. The non-migrating cells in the upper chambers were removed with a cotton swab. Cells were photographed under Leica AM6000 microscope and the number of invading cells was recorded in five random microscopic fields.
Cell apoptosis assay
Cell apoptosis was performed using PI and fluorescein isothiocynate (FITC)-conjugated Annexin V staining kit (Beyotime Biotechnology, Beijing, China). Cells were trypsinized and re-suspended in annexin V binding buffer. One μL annexin V-FITC and 1 μL PI reagent were added to 500 μL cell suspension with 5x105 cells in annexin-V staining buffer. The staining mixture was incubated for 30 min in the dark. Stained cells were centrifuged and washed twice with staining buffer and re-suspended in 500 μL PBS. The percentage of apoptotic cells was detected by BD FACS CantoTM II Flow Cytometer (BD Biosciences). The data were analyzed using FlowJo 5.0 software (FlowJo LLC, OR, USA). Both the early apoptotic (annexin-V positive only) and late apoptotic (Annexin V and PI double positive) cells were counted to estimate the apoptotic index.
In vivo tumorigenesis assay
The protocol for the xenograft tumorigenesis mouse model was performed according to a previous study.19 A total number of 12 male immunodeficient BALB/c nude mice (30-40 gram, 6 weeks) were housed in a pathogen-free animal house with a 12-h light/dark cycle. The mice were randomly assigned into two groups (6 mice each): i) sh-NC group (injected with HOS cells infected with sh-NC), ii) sh-DPY30 (injected with HOS cells infected with sh-DPY30). 2×106 cells in 0.25 mL PBS was subcutaneously injected into the flank of each mouse. Tumor volume was monitored every 7 days using a caliper. Tumor volume was calculated by formula V= ¼ 1/2 (A·B·C), where A denotes the major tumor axis and B is the minor tumor axis. Four weeks after tumor cell injection, all the mice were euthanized by CO2 asphyxiation. The euthanizing chamber was connected to a CO2 cylinder with a flow rate to displace 40% of chamber volume per minute. Mice were asphyxiated for 15 min until no movement was observed. Death was assured by cervical dislocation, and no leg flex or heartbeat was observed. The tumors of terminally dead mice were removed for weight measurement and fixed until further IHC staining analysis. All animal procedures were approved by the Animal Care and Use Ethical Committee of Yantaishan Hospital (202106003).
Statistical analysis
All in vitro experiments were conducted for three independent times. Data were presented as mean ± standard error of mean (SEM), and statistical analyses were performed with GraphPad Prism 7.0 software (GraphPad Software, Inc., San Diego, CA, USA). The significance of difference was assessed using unpaired two-tailed Student’s t-test (for two groups) or one-way ANOVA and Tukey’s post-hoc test (for more than two groups). Chi-square test was employed to assess the association between DPY30 expression level and the clinical parameters in OS patients. P <0.05 indicated a significant difference.
Results
DPY30 expression is upregulated in OS tissues and cell lines
To examine the expression of DPY30 in OS tissues, we retrieved a published dataset from the GEO database (http://www.ncbi.nlm.nih.gov/geo/; GSE28424; including 19 OS cell lines and 4 normal bone cell lines). The expression level of DPY30 mRNA was significantly higher in OS cell lines when compared to the normal bone cells lines (Figure 1A). In clinical samples collected from OS patients (n=44), DPY30 also showed a significant upregulation in OS tumor tissues in comparison to paracarcinoma tissues, as measured by RT-qPCR (Figure 1B), immunohistochemistry (Figure 1C), and Western blot (Figure 1D). In addition, the upregulation of DPY30 was confirmed by Western blot in 4 different OS cells lines (U2OS, HOS, Saos2 and MG63), with human osteoblast cell line (hFOB) as the control (Figure 1E).
Correlation between DPY30 and clinicopathological characteristics of OS
Meanwhile, we also analyzed the correlation of DPY30 expression levels with clinicopathological parameters in OS patients. OS patients (n=44) were divided into DPY30 highexpression and low-expression groups based on the median DPY30 expression value determined by RT-qPCR. As shown in Table 1, high DPY30 expression was associated with larger tumor size (p=0.0014) and more distant metastases (p=0.0395), but had no significant correlations with the age, gender, entering stage and differentiation state (P >0.05). In addition, Kaplan Meier curve and analysis of 44 OS patients showed that a high level of DPY30 expression was associated with a poorer overall survival (Figure 1F). Receiver operative characteristics (ROC) curve analysis further indicates that DPY30 could be a potential marker for the prognosis in OS patients (Figure 1G). Overall, these data suggest that DPY30 overexpression was associated the malignancy and the poor prognosis in OS patients.
DPY30 knockdown suppresses OS cell proliferation and induces apoptosis
To investigate the functional role of DPY30 in OS cells, OS cell lines (U2OS and HOS) with stable shRNA-mediated knockdown were established with lentiviral system. Three shRNAs were used for DPY30 silencing and sh-DPY30#2 showed the optimal knockdown efficiency (Figure 2A). Cells with sh-DPY30#2 knockdown were selected for the subsequent experiments. CCK-8 proliferation assay showed that in both U2OS and HOS cell lines, DPY30 knockdown significantly impaired cell growth of OS cells (Figure 2B). The impaired cell proliferation was further validated by EdU incorporation assay, which showed a reduced cell number with DNA synthesis after DPY30 knockdown (Figure 2C). Further, flow cytometric analysis revealed that DPY30 silencing caused an increase of apoptotic events in both OS cell lines (Figure 2D).
DPY30 knockdown impairs the invasion and migration capacities of OS cells
High invasiveness is an aggressive malignant behavior of OS cells.20 Therefore, we further explored whether DPY30 regulates the invasion and migration ability of OS cells in vitro. We found that DPY30 knockdown significantly impaired the migratory and invasive abilities of U2OS and HOS cells (Figure 3 A,B). EMT is a key cellular event driving the migratory phenotype in cancer cells.13,16 We therefore examined the expression of EMT markers after DPY30 knockdown. Western blot assays showed that DPY30 knockdown can reduced the expression of N-cadherin (mesenchymal marker), while the expression of E-cadherin (epithelial marker) significantly increased (Figure 3C). Together, these findings suggest that DPY30 is required for the high invasiveness of OS cells by maintain the mesenchymal status of OS cells.
DPY30 knockdown suppresses the PI3K/AKT pathway in OS cells
Previous studies have reported that the activation of PI3K/AKT pathway plays a crucial role in the malignant progression of OS.21-24 We therefore determined the effect of DPY30 on the activation of PI3K/AKT pathway in OS cells. Western blot analysis showed that the phosphorylation levels of PI3K and AKT were significantly attenuated upon DPY30 knockdown, while the levels of total PI3K and AKT proteins were not changed (Figure 4A). To further confirm the implication of PI3K/AKT signaling in the role of DPY30, we applied PI3K agonist (740 Y-P), which rescued the phosphorylation level of PI3K and AKT upon DPY30 knockdown (Figure 4B). CCK8 assay demonstrated that the inhibitory effect of DPY30 knockdown on cell proliferation was partially abrogated upon the treatment of 740 Y-P (Figure 4C). Apoptosis induction by DPY30 knockdown was largely suppressed by 740 Y-P (Figure 4D). Similarly, 740 Y-P also rescued cellular migration and invasion upon DPY30 knockdown (Figure 4 E,F). These results imply that the activity of PI3K/AKT pathway underlies the functional role of DPY30 in OS cells.
Figure 2.
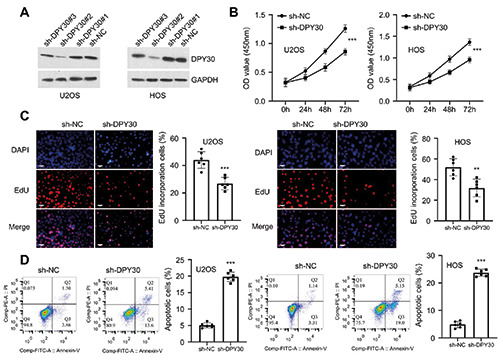
DPY30 knockdown suppresses the malignancy in OS cells. A) The knockdown efficiencies of three sh-RNAs targeting DPY30 were determined by Western blot in U2OS and HOS cells. B) CCK-8 proliferation assay in U2OS and HOS cells upon DPY30 knockdown. C) EdU incorporation assay in U2OS and HOS cells upon DPY30 knockdown. D) Flow cytometry analysis of apoptotic events in U2OS and HOS cells upon DPY30 knockdown. **p<0.01; ***p<0.001.
Table 1.
Correlations of DPY30 expression with clinicopathologic features in osteosarcoma patients.
| Clinical pathological parameters | n | DPY30 expression | p | |
|---|---|---|---|---|
| High expression group (n=22) | Low expression group (n=22) | |||
| Age (years) | 0.22 | |||
| <20 | 26 | 11 | 15 | |
| >20 | 18 | 11 | 7 | |
| Gender | 0.1118 | |||
| Male | 29 | 12 | 17 | |
| Female | 15 | 10 | 5 | |
| Tumor size | 0.0009 | |||
| <8 cm | 23 | 6 | 17 | |
| ≥8 cm | 21 | 16 | 5 | |
| Distant metastasis | 0.026 | |||
| Yes | 29 | 18 | 11 | |
| No | 15 | 4 | 11 | |
| Entering stage | 0.7569 | |||
| I+II | 17 | 9 | 8 | |
| III | 27 | 13 | 14 | |
| Differentiation state | 0.0658 | |||
| High | 18 | 6 | 12 | |
| Low | 26 | 16 | 10 | |
Figure 3.
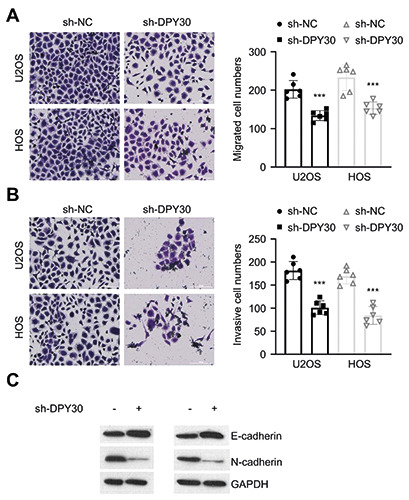
DPY30 knockdown suppresses the invasion and migration capacities of OS cells. In vitro migration (A) and invasion (B) assay in U2OS and HOS cells upon DPY30 knockdown. (C) Protein levels of E-cadherin and N-cadherin were determined by Western blot in U2OS and HOS cells upon DPY30 knockdown. **p<0.01; ***p<0.001.
DPY30 knockdown suppresses the tumorigenesis of OS cells in vivo
To further confirm the oncogenic role of DPY30, we injected HOS cells with sh-NC or sh-DPY30 into BALB/c nude mice as the xenograft tumorigenesis model. We found that the average tumor volume (Figure 5A) and tumor weight (Figure 5B) were significantly decreased in the DPY30 knockdown group. IHC staining further demonstrated that the number of Ki-67 (cell proliferation marker) and DPY30-positive cells in the xenograft tissue section was reduced upon DPY30 knockdown (Figure 5C). Collectively, these observations suggest that DPY30 knockdown can inhibit the growth of OS cells in vivo.
Discussion
OS is a common type of aggressive bone tumor that mainly originates from mesenchymal tissues.25,26 The 5-year overall survival rate is about 70% in OS patients without metastasis.27,28 However, in the patients with metastasis, the 5-year overall survival rate drops to as low as 30%. This is particularly relevant in patients who had developed lung metastasis before diagnosis.1,29,30 In this study, we found that DPY30 is upregulated in OS tumor tissues and OS cell lines. Silencing DPY30 in OS cells suppressed the cell proliferation, migration and invasion, while induced apoptosis. Importantly, in xenograft mouse model, OS cells with DPY30 knockdown showed retarded tumorigenesis. The above data support the oncogenic role of DPY30 in OS cells.
Figure 4.
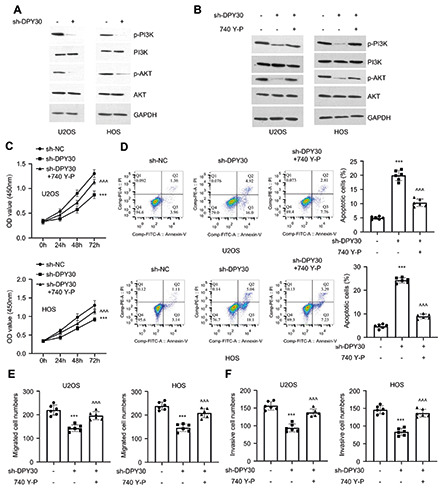
PI3K/AKT signaling pathway underlies the effect of DPY30 in OS cells. A) The phosphorylation levels of PI3K and AKT in U2OS and HOS cells upon DPY30 knockdown. B) The phosphorylation levels of PI3K and AKT in U2OS and HOS cells upon DPY30 knockdown with or without the treatment of PI3K agonist (740 Y-P). C) CCk-8 proliferation assay, cellular apoptosis (D), migration (E) and invasion (F) assay in U2OS and HOS cells upon DPY30 knockdown with or without the treatment of PI3K agonist (740 Y-P). **p<0.01; ***p<0.001; ^^p<0.01; ^^^p<0.001; *compared to sh-NC; ^compared to sh-DPY30.
As the integral core subunits of SET1/MLL complex, DPY30 knockdown can induces G2/M phase cell cycle arrest,31 which is consistent with our observation that DPY30 knockdown inhibits the proliferation of OS cells. Additionally, our results indicate that DPY30 knockdown induced the apoptosis. Moreover, high DPY30 expression was associated with a poorer overall survival in OS patients. These data were consistent with the role of DPY30 in promoting cell survival and predicting the prognosis in other cancers, such as ovarian cancer and cholangiocarcinoma.15,32
Furthermore, DPY30 knockdown impaired the migration and invasion in OS cells, and high level of DPY30 expression was associated with more distant metastases in OS patients. High invasiveness is an aggressive malignant behavior of OS cells,20 and EMT is a crucial cellular event orchestrating the migratory phenotype in cancer cells.13,16 Consistently, we showed that DPY30 silencing attenuated the mesenchymal phenotype in OS cells, as revealed by the reduction of N-cadherin expression. These data are consistent with the previous reports that DPY30 regulates EMT process in cervical squamous carcinoma and epithelial ovarian cancer.13,16 Together, these data highlight the crucial role of DPY30 in regulating the migratory phenotype of OS cells, which suggests that the upregulation of SPY30 may contribute to the metastasis in OS patients.
Previous studies showed that the constitutive activation in PI3K/AKT pathway contributes to the epigenetic changes in cancer progression.33,34 In this study, we demonstrated that DPY30 knockdown suppressed the activation of PI3K/AKT signaling pathway, which underlies the functional role of DPY30. Indeed, the activation of PI3K/AKT pathway is involved in the progression of various cancers including ovarian,35 breast cancer,35 glioblastomas, 36 endometrial carcinoma.37 There is growing body of evidence that the PI3K/Akt pathway promotes cell proliferation and metabolic reprogramming to support metastasis.33,34 In OS, PI3K/AKT pathway not only supports the initiation and progression of tumor, but also contributes to the metabolic reprogramming and cellular transformation required for migration and invasion. 38,39 In addition, PI3K/AKT pathway can crosstalk with other oncogenic signals such as WNT signaling to dictate cellular changes such as EMT.40 One of the remaining questions of our study is how DPY30 regulates the activity of PI3K/AKT signaling? A genome-wide approach such as RNA-seq, ChIP-seq or ATAC-seq may shed light on how the histone H3K4 methyltransferase component DPY30 impinges on the oncogenic PI3K/AKT signaling.
In summary, our study revealed an oncogenic role of DPY30 in OS progression, which may serve as an anti-cancer target and prognostic marker. A high level of DPY30 in OS cells is required to support malignant phenotypes such hyper-proliferation, migration and invasion. We further demonstrated that DPY30 expression is necessary for the in vivo tumorigenesis in mouse model. Future work is needed to clarify how DPY30 regulates PI3K/AKT signaling in OS cells.
Figure 5.
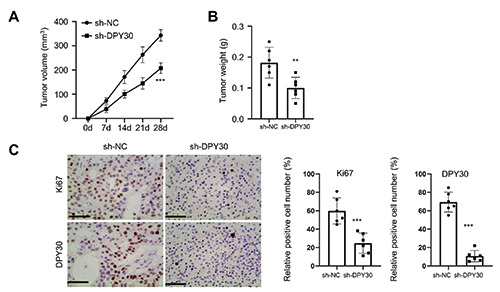
DPY30 silencing suppresses tumorigenesis in mouse model. HOS cells with sh-NC and sh-DPY30 were injected into nude mice. The tumor volume (A) and weight (B) were determined at day 28 between the two groups (n=6 in each group). C) Immunohistochemical (IHC) staining of DPY30 and Ki-67 in the xenograft tumor tissues from sh-NC and sh-DPY30 groups (n=6 in each group, 5 sections were analyzed from each xenograft tumor tissue). **p<0.01; ***p<0.001.
Funding Statement
Funding: This work was supported by the Shandong Province Medical and Health Technology Development Plan Project (202104070255); the Shandong Province Key Research and Development Program (2018GSF118186); the Yantai Key Research and DevelopmentPlan Project (2017WS114), the General program of Shandong Natural Science Foundation (ZR2022MH199).
References
- 1.Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer 2014;14:722-35. [DOI] [PubMed] [Google Scholar]
- 2.Gill J, Gorlick R. Advancing therapy for osteosarcoma. Nat Rev Clin Oncol 2021;18:609-24. [DOI] [PubMed] [Google Scholar]
- 3.Smeland S, Bielack SS, Whelan J, Bernstein M, Hogendoorn P, Krailo MD, et al. Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 2019;109:36-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kane GM, Cadoo KA, Walsh EM, Emerson R, Dervan P, O’Keane C, et al. Perioperative chemotherapy in the treatment of osteosarcoma: a 26-year single institution review. Clin Sarcoma Res 2015;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim W, Han I, Lee JS, Cho HS, Park JW, Kim H-S. Postmetastasis survival in high-grade extremity osteosarcoma: A retrospective analysis of prognostic factors in 126 patients. J Surg Oncol 2018;117:1223-31. [DOI] [PubMed] [Google Scholar]
- 6.McGuire JJ, Nerlakanti N, Lo CH, Tauro M, Utset-Ward TJ, Reed DR, et al. Histone deacetylase inhibition prevents the growth of primary and metastatic osteosarcoma. Int J Cancer 2020;147:2811-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mc Auley G, Jagannathan J, O’Regan K, Krajewski KM, Hornick JL, Butrynski J, et al. Extraskeletal osteosarcoma: Spectrum of imaging findings. Am J Roentgenol 2012;198:W31-W7. [DOI] [PubMed] [Google Scholar]
- 8.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012;13:343-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 2011;11:726-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li N, Xue W, Yuan H, Dong B, Ding Y, Liu Y, et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J Clin Invest 2017;127:1284-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 2012;81:65-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang H, Shukla A, Wang X, Chen W-y, Bernstein BE, Roeder RG. Role for dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 2011;144:513-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Zhang S, Li A, Zhang A, Zhang S, Chen L. DPY30 is required for the enhanced proliferation, motility and epithelial- mesenchymal transition of epithelial ovarian cancer cells. Int J Mol Med 2018;42:3065-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z, Augustin J, Chang C, Hu J, Shah K, Chang CW, et al. The DPY30 subunit in SET1/MLL complexes regulates the proliferation and differentiation of hematopoietic progenitor cells. Blood 2014;124:2025-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong ZF, Zhang WQ, Wang SJ, Li SY, Shang J, Liu F, Shen DY. Upregulation of DPY30 promotes cell proliferation and predicts a poor prognosis in cholangiocarcinoma. Biomed Pharmacother 2020;123:109766. [DOI] [PubMed] [Google Scholar]
- 16.He FX, Zhang LL, Jin PF, Liu DD, Li AH. DPY30 regulates cervical squamous cell carcinoma by mediating epithelialmesenchymal transition (EMT). Onco Targets Ther 2019;12:7139-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YJ, Han ME, Baek SJ, Kim SY, Oh SO. Roles of DPY30 in the proliferation and motility of gastric cancer cells. PLoS One 2015;10:e0131863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Mai W, Chen M, Hu J, Zhuo Z, Lei X, et al. Arenobufagin inhibits prostate cancer epithelial-mesenchymal transition and metastasis by down-regulating β-catenin. Pharmacol Res 2017;123:130-42. [DOI] [PubMed] [Google Scholar]
- 19.Xue Y, Guo Y, Liu N, Deng Z, Jian Y, Cai H, et al. MicroRNA- 22-3p targeted regulating transcription factor 7-like 2 (TCF7L2) constrains the Wnt/β-catenin pathway and malignant behavior in osteosarcoma. Bioengineered 2022;13:9135-9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan TM, Roberts RD, Lizardo MM. Understanding and modeling metastasis biology to improve therapeutic strategies for combating osteosarcoma progression. Front Oncol 2020;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He J, Zhang W, Zhou X, Yan W, Wang Z. Aloin induced apoptosis by enhancing autophagic flux through the PI3K/AKT axis in osteosarcoma. Chin Med 2021;16:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Shen X, Ma M, Liu W, Yang W, Wang P, et al. ZIP10 drives osteosarcoma proliferation and chemoresistance through ITGA10-mediated activation of the PI3K/AKT pathway. J Exp Clin Cancer Res 2021;40:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang H-y, Guo J-q, Sang B-t, Cheng J-n, Wu X-m. PDGFRβ modulates aerobic glycolysis in osteosarcoma HOS cells via the PI3K/AKT/mTOR/c-Myc pathway. Biochem Cell Biol 2022;100:75-84. [DOI] [PubMed] [Google Scholar]
- 24.Shi Z, Wang K, Xing Y, Yang X. CircNRIP1 encapsulated by bone marrow mesenchymal stem cell-derived extracellular vesicles aggravates osteosarcoma by modulating the miR-532-3p/AKT3/PI3K/AKT axis. Front Oncol 2021;11:658139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer 2009;125:229-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004. Cancer 2009;115:1531-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins M, Wilhelm M, Conyers R, Herschtal A, Whelan J, Bielack S, et al. Benefits and adverse events in younger versus older patients receiving neoadjuvant chemotherapy for osteosarcoma: findings from a meta-analysis. J Clin Oncol 2013;31:2303-12. [DOI] [PubMed] [Google Scholar]
- 28.Bernthal NM, Federman N, Eilber FR, Nelson SD, Eckardt JJ, Eilber FC, et al. Long-term results (> 25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 2012;118:5888-93. [DOI] [PubMed] [Google Scholar]
- 29.Gill J, Ahluwalia MK, Geller D, Gorlick R. New targets and approaches in osteosarcoma. Pharmacol Ther 2013;137:89-99. [DOI] [PubMed] [Google Scholar]
- 30.Klein MJ, Siegal GP. Osteosarcoma: Anatomic and histologic variants. Am J Clin Pathol 2006;125:555-81. [DOI] [PubMed] [Google Scholar]
- 31.Hong Z-F, Zhang W-Q, Wang S-J, Li S-Y, Shang J, Liu F, et al. Upregulation of DPY30 promotes cell proliferation and predicts a poor prognosis in cholangiocarcinoma. Biomed Pharmacother 2020;123:109766. [DOI] [PubMed] [Google Scholar]
- 32.Liu Q, Yang H, Hua H. Overexpression of miR-493-3p suppresses ovarian cancer cell proliferation, migration and invasion through downregulating DPY30. Reprod Biol 2022;22:100610. [DOI] [PubMed] [Google Scholar]
- 33.Yang Q, Jiang W, Hou P. Emerging role of PI3K/AKT in tumor-related epigenetic regulation. Semin Cancer Biol 2019;59:112-24. [DOI] [PubMed] [Google Scholar]
- 34.Spangle JM, Roberts TM, Zhao JJ. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim Biophys Acta Rev Cancer 2017;1868:123-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res 2005;11:2875-8. [DOI] [PubMed] [Google Scholar]
- 36.Knobbe CB, Trampe-Kieslich A, Reifenberger G. Genetic alteration and expression of the phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathol Appl Neurobiol 2005;31:486-90. [DOI] [PubMed] [Google Scholar]
- 37.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res 2005;65:10669-73. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Yu XH, Yan YG, Wang C, Wang WJ. PI3K/Akt signaling in osteosarcoma. Clin Chim Acta 2015;444:182-92. [DOI] [PubMed] [Google Scholar]
- 39.Soghli N, Ferns GA, Sadeghsoltani F, Qujeq D, Yousefi T, Vaghari-Tabari M. MicroRNAs and osteosarcoma: Potential targets for inhibiting metastasis and increasing chemosensitivity. Biochem Pharmacol 2022;201:115094. [DOI] [PubMed] [Google Scholar]
- 40.Zhang A, He S, Sun X, Ding L, Bao X, Wang N. Wnt5a promotes migration of human osteosarcoma cells by triggering a phosphatidylinositol-3 kinase/Akt signals. Cancer Cell Int 2014;14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]