

SUMMARY
Inadequate potassium (K+) consumption correlates with increased mortality and poor cardiovascular outcomes. Potassium effects on blood pressure have been described previously; however, whether or not low K+ independently affects kidney disease progression remains unclear. Here, we demonstrate that dietary K+ deficiency causes direct kidney injury. Effects depend on reduced blood K+ and are kidney specific. In response to reduced K+, the channel Kir4.2 mediates altered proximal tubule (PT) basolateral K+ flux, causing intracellular acidosis and activation of the enzyme glutaminase and the ammoniagenesis pathway. Deletion of either Kir4.2 or glutaminase protects from low-K+ injury. Reduced K+ also mediates injury and fibrosis in a model of aldosteronism. These results demonstrate that the PT epithelium, like the distal nephron, is K+ sensitive, with reduced blood K+ causing direct PT injury. Kir4.2 and glutaminase are essential mediators of this injury process, and we identify their potential for future targeting in the treatment of chronic kidney disease.
Graphical Abstract
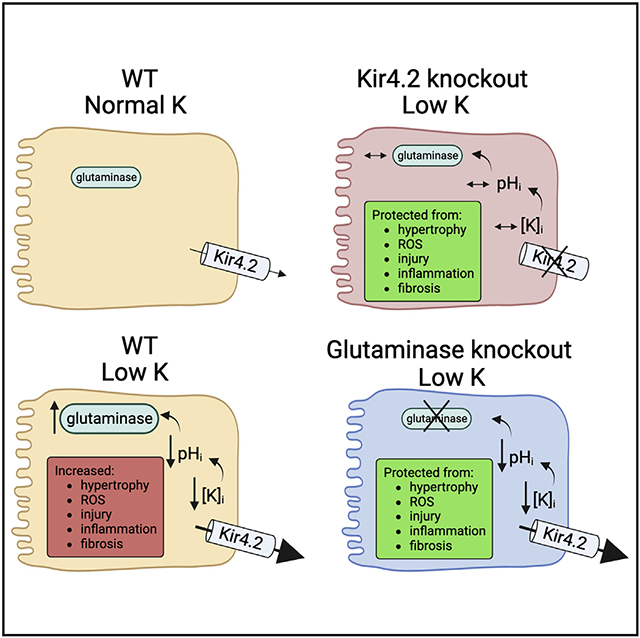
In brief
Terker et al. demonstrate that dietary K+ deficiency causes kidney-specific injury and inflammation. Effects are mediated by the proximal tubule basolateral K+ channel Kir4.2, which promotes ammoniagenesis in response to reduced blood K+ levels. Deletion of this channel or the enzyme glutaminase protect animals from low-K+-mediated kidney injury.
INTRODUCTION
Modern dietary trends include an electrolyte profile that is relatively high in sodium (Na+) yet low in potassium (K+).1 A diet with a high Na+-to-K+ ratio has repeatedly been linked to poor cardiovascular outcomes including hypertension, stroke, and cardiovascular mortality.2-5 Primacy has often been attributed to Na+, and most mechanistic and clinical studies correlate effects of high Na+ intake on cardiovascular health. Clinical practice also reflects this focus on Na+ consumption with periodically updated Na+ consumption recommendations for patients guided by research findings. While effects of reduced dietary K+ have received less attention, there is strong evidence that low K+ intake has a prominent role in the development of cardiovascular disease.
More recent clinical studies show that low K+ consumption correlates with increased incidence and progression of chronic kidney disease, as well as higher rates of graft failure in kidney transplant recipients,6 but mechanistic details are lacking.5,7 Proximal tubule (PT) cells are the predominant epithelial targets of kidney injury as they constitute the bulk of kidney mass and perform the majority of reabsorptive functions. Their fate dictates renal recovery after injury.8 Along the distal convoluted tubule (DCT), prior work has demonstrated direct effects of extracellular K+ on modulating distal epithelial cell function and Na+ reabsorption.9-11 Low blood K+ has been linked to renal hypertrophy and PT damage, but whether these effects result directly from reduced peritubular K+ influencing the proximal epithelium remains unanswered. If true, such a mechanism would have important implications for individuals with chronic kidney disease (CKD). As standard practice is to recommend dietary K+ restriction for patients with CKD, current approaches could be unintentionally contributing to kidney disease progression.
Previous descriptions of the distal nephron epithelium as a K+-sensitive segment show that altered basolateral K+ flux is mediated by the inwardly rectifying K+ (Kir) channel Kir4.1, which forms functional heterotetramers with Kir5.1.9 While Kir4.1 expression is confined to the distal nephron, the related Kir4.2 is expressed along the PT basolateral membrane and also heterotetramerizes with Kir5.1 to form functional channels. Along the PT, Kir4.2 regulates basolateral membrane potential and intracellular pH (pHi).12 In a model of systemic acidosis, its deletion inhibited ammoniagenesis and caused a proximal renal tubular acidosis, though its role in a low-K+ state has not been previously investigated.
Along with acidosis, low K+ is one of the most potent stimuli for renal ammoniagenesis.13,14 Ammoniagenesis is a metabolic process initiated by the enzyme glutaminase. Low-K+-induced ammoniagenesis is thought to contribute to the generation of metabolic alkalosis as suggested by increased urinary acid excretion following K+ depletion.14 We hypothesized that chronically elevated glutaminase activity in a setting of reduced dietary K+ intake may induce an excessive metabolic load on the kidney, contributing to cell stress and kidney injury.
In this study, we tested the hypothesis that low dietary K+ causes direct kidney injury. We show that injury is largely kidney specific, sparing other organs. Using a combination of in vivo and in vitro approaches, we further demonstrate that effects are mediated via PT basolateral K+ efflux through Kir4.2 in the setting of extracellular K+ reductions. The resulting intracellular K+ reduction, in turn, causes intracellular acidosis, a potent activator of glutaminase activity. Finally, we demonstrate that in the absence of glutaminase function, PT cells are protected from low-K+-mediated injury both in culture and in vivo.
RESULTS
A high-salt/low-K+ diet causes kidney injury and inflammation via reduced blood K+
We initially determined effects of high-salt intake on kidney inflammation, injury, and fibrosis. Animals that consumed a high-salt diet with normal K+ (HS/NK) for 4 weeks did not have significant differences in total kidney mRNA abundance of inflammatory, injury, or fibrosis transcripts compared with mice that were maintained on normal salt and NK (NS/NK) (Figure 1A). Differences in fibrosis were also not observed at the protein level as determined by Sirius red staining (Figures 1B and S1A). As most people consume a diet that is high in Na+ and low in K+, we modeled effects of this diet by comparing mice consuming a HS/NK diet with those fed a high-salt and K+-deficient (HS/0K) diet. Animals fed HS/0K had increased total kidney mRNA abundance of multiple inflammatory, injury, fibrotic, and immune cell transcripts compared with those animals on HS/NK (Figures 1C-1E and S1B). This was accompanied by increased immune cell infiltration as determined by flow cytometry and immunostaining (Figures 1F and S1C-S1E). Immunostaining also confirmed increases in injury markers kidney injury molecule 1 (Kim-1) and neutrophil gelatinase-associated lipocalin (NGAL) and fibrosis as observed through Sirius red staining (Figures 1G, 1H, and S1F). Quantification of tubule injury revealed that epithelial damage was diffuse throughout all anatomical regions, with the highest rates of injury observed in the cortex (Figure S1G). While the HS/0K diet also increased cytokine transcript abundance in circulating leukocytes, quantitative PCR analysis from heart, liver, colon, spleen, and skeletal muscle did not demonstrate similar increases in mRNA abundance of inflammatory or fibrotic transcripts, suggesting that solid organ effects were largely restricted to the kidney (Figure S2). To confirm that low dietary K+ intake alone, in the absence of high salt, was sufficient to cause kidney injury, we compared the effects of NS/NK and NS/0K intakes in mice. Dietary K+ restriction, without high salt, still increased total kidney mRNA abundance of inflammatory, injury, and fibrotic transcripts and caused increased immune cell kidney infiltration as determined by flow cytometry (Figure S3; Table S1).
Figure 1. Reduced dietary K+ causes kidney injury.

(A) Total kidney transcript abundance of inflammatory, injury, and fibrosis genes in animals that consumed an NS/NK diet compared with those that consumed an HS/NK diet for 4 weeks.
(B) Effects of the same dietary treatments as (A) on kidney fibrosis as determined by Sirius red staining. Quantification presented in Figure S1A.
(C–E) Total transcript kidney transcript abundance of (C) inflammatory cytokines, (D) injury markers, and (E) fibrosis genes in animals that consumed an HS/NK diet compared with an HS/0K diet for 3 weeks.
(F) Kidney abundance of leukocyte subsets as determined by flow cytometry in animals that consumed an NS/NK, HS/NK, or HS/0K diet for 3 weeks.
(G) Effects of HS/NK compared with HS/0K diet on Kim1 and NGAL expression as determined by immunohistochemical staining on kidney sections.
(H) Effects of the same dietary treatments as (G) on kidney fibrosis as determined by Sirius red.
Quantifications for (G) and (H) presented in Figure S1F. n = 5 per group, except n = 10 for HS/0K in (H). *p < 0.05 by unpaired t test or Mann-Whitney; †p < 0.05 by Kruskal-Wallis test followed by Dunn’s multiple comparison test versus HS/NK. Scale bar: 50 μm. Normality was determined by Shapiro-Wilk test. Error bars indicate SEM.
To determine at what level of dietary K+ deleterious effects occur, a dietary K+ titration was performed to produce graded changes in blood K+ levels (Figure 2A; Table S2A). Blood K+ was inversely correlated with changes in kidney weight, renal transcript abundance of inflammatory cytokines, injury markers, and fibrosis markers, and abundance of immune cell subsets (Figures 2B-2D). Kim1 mRNA expression began to increase when blood K+ decreased below 3.5 mM, although changes in other transcripts generally required slightly lower blood K+ levels (Figure S4A). Therefore, all subsequent experiments were performed using diets containing high salt and 0.04% K+ (HS/0.04K), which corresponded to a blood K+ level of 2.6 mM.
Figure 2. Effects of changes in blood K+ on kidney injury.

(A–D) Effects of dietary K+ titration on (A) blood K+, (B) kidney weight, (C) total kidney mRNA abundance of inflammatory, injury, and fibrosis transcripts, and (D) kidney leukocyte subset abundance determined by flow cytometry. All animals were on 6% NaCl along with designated K+ content and were treated for 3 weeks.
(E) Blood K+ levels in mice consuming an HS/0.04K diet on normal water compared with those on HS/0.04K diet with water supplemented with amiloride. Animals were treated for 3 weeks.
(F–H) Total kidney abundance of (F) inflammatory cytokines, (G) injury markers, and (H) fibrotic transcripts in mice treated with HS/0.04K diet alone or with amiloride.
n = 5 per group except in (B), where n = 9 for 0K and 10 for 0.8K, and (E), where n = 4 per group. †p < 0.05 by one-way ANOVA with Dunnett’s multiple comparison versus 0.8% K+. #p < 0.05 by Kruskal-Wallis test. For (C) and (D), all genes and cell subsets were positive by #. *p < 0.05 by unpaired t test or Mann-Whitney test. Normality was determined by Shapiro-Wilk test. Error bars indicate SEM.
Additional physiological data are presented in Tables S2A and S2B.
To confirm that blood K+ itself, and not reduced dietary K+ consumption, was the nephrotoxic signal, we next used the K+-sparing diuretic amiloride to influence blood K+ independent of diet. Amiloride treatment maintained normokalemia in mice consuming an HS/0.04K diet compared with the HS/0.04K diet alone (Figures 2E, S4B, and S4C; Table S2B). Amiloride treatment also prevented increased total kidney transcript abundance of inflammatory, injury, and fibrosis markers (Figures 2F-2H), supportive of the hypothesis that blood K+ mediates kidney injury.
Aldosterone-induced renal fibrosis and injury requires reductions in blood K+
Aldosterone is known to cause kidney injury.15 It also causes hypokalemia, and while the kidney damage has traditionally been attributed to effects mediated by dietary Na+, our data demonstrating a relationship between blood K+ and injury led us to hypothesize that low K+ may be mediating some of the observed effects. To test this hypothesis, we treated mice with aldosterone and an HS/NK diet, and, as expected, aldosterone-treated animals had reduced blood K+ levels (Figure 3A; Table S3). They also had increased total kidney mRNA abundance of inflammatory, injury, and fibrotic transcripts (Figures 3B-3D). Increased kidney fibrosis was also observed (Figures 3E and S5A). Maintaining blood K+ in the physiological range with either a high-K+ diet or amiloride while also treating with aldosterone reduced or prevented the increase in nearly all of these parameters (Figures 3B-3E). Importantly, an abundance of the alpha subunit of the epithelial Na+channel (αENaC), a known aldosterone target gene, remained elevated despite normokalemia (Figure S5B).
Figure 3. Aldosterone injury effects are mediated by K+.

(A) Blood K+ levels in animals that underwent a sham procedure, aldosterone infusion only, aldosterone infusion + high K+ diet, or aldosterone infusion + amiloride treatment.
(B–D) Total kidney abundance of (B) inflammatory cytokines, (C) injury markers, and (D) fibrotic transcripts in mice treated as in (A).
(E) Effects of treatments as described in (A) on kidney fibrosis as determined by Sirius red.
Quantification for (E) presented in Figure S5A. n = 5 per group for all except for the aldo group, where n = 4. †p < 0.05 by one-way ANOVA with Dunnett’s multiple comparison versus sham. Scale bar: 50 μm. Normality was determined by Shapiro-Wilk test. Error bars indicate SEM.
Additional physiological data are presented in Table S3.
Low K+ exerts deleterious effects on the PT through Kir4.2 and reduced pHi
A consequence of reduced extracellular K+ is a reduction in pHi.16 To confirm this effect in PT cells in culture, we cultured human renal proximal tubule epithelial cells (hRPTECs) in K+-deplete medium in vitro. Low-K+ conditions caused a rapid reduction in pHi that corrected over time (Figure 4A). To determine if this intracellular acid load contributed to low-K+ injury in vivo, we alkali-loaded animals in conjunction with an HS/0.04K diet and determined effects on the kidney. Despite similar blood K+ levels, animals that were co-treated with NaHCO3 were more alkalotic and had reduced kidney weight and reduced mRNA transcript abundance of inflammatory, injury, and fibrosis genes compared with mice co-treated with NaCl-supplemented drinking water (Figures 4B-4H; Table S4).
Figure 4. Effects of alkali supplementation on low K+ injury.

(A) Time course of pHi measurements in hRPTECs after changing culture conditions from normal-K+ to low-K+ medium. 1/RFU on the y axis is proportional to pHi.
(B–E) Effects of NaHCO3 supplementation on (B) blood K+ levels, (C) blood Cl− levels, (D) blood HCO3− levels, and (E) kidney weight in animals consuming an HS/0.04K diet for 3 weeks.
(F–H) Total kidney abundance of (F) inflammatory cytokines, (G) injury markers, and (H) fibrotic transcripts in mice treated as in (B).
n = at least 9 per group in (A), n = 5 and 4 per group in (B), n = 5 per group for (C) and (D), n = 10 and 9 for (E), and n = 10 per group for (F)–(H). †p < 0.05 by one-way Kruskal-Wallis test with Dunn’s multiple comparison versus time = 0. *p < 0.05 by unpaired t test or Mann-Whitney test. Normality was determined by Shapiro-Wilk test. Error bars indicate SEM.
Additional physiological data are presented in Table S4.
These results suggested that secondary development of intracellular acidosis was required for low-K+-mediated PT injury. The well-established role of Kir4.1 mediating K+ signaling on distal epithelia led us to hypothesize a similar function for the PT-specific K+ channel Kir4.2. Kir4.2 is known to have an important role in setting the PT basolateral membrane potential,12 suggesting it may serve as a PT K+ sensor. To determine if Kir4.2 mediates effects of low-K+ injury on the PT, we fed Kir4.2 knockout (Kir4.2−/−) animals and wild-type (WT) controls an HS/0.04K diet. This exacerbated the previously described type II renal tubular acidosis (RTA) in Kir4.2 knockouts, causing profound hypokalemia and systemic acidosis (Figures 5A, S6A, and S6B; Table S5). Plasma aldosterone levels were reduced in both genotypes after consuming the HS/0.04K diet for 8 days (Figure S6C). Rapid weight loss in knockout animals limited the duration of experiments, so data were collected after 3 and 8 days of treatment. Despite the lower blood K+, Kir4.2−/− mice failed to undergo renal hypertrophy, as demonstrated by reduced kidney weight compared with WT controls (Figure 5B). WT mice demonstrated increased abundance of phosphorylated S6 kinase 1 after 8 days, consistent with activation of the mammalian target of rapamycin complex 1 (mTORC1) (Figure S6D). Such changes were not evident in Kir4.2−/− mice. While 3 days of HS/0.04K diet did not dramatically affect expression of inflammatory, injury, or fibrosis transcripts (data not shown), Kir4.2−/− mice had lower abundances of these transcripts after 8 days of treatment compared with WT animals (Figure 5C). Flow cytometry data showed increased kidney abundance of macrophages in WT animals following 8 days of the HS/0.04K diet, though this effect was absent in Kir4.2−/− mice (Figure 5D). Furthermore, transcript abundance and blood K+ displayed a linear or exponential relationship for several genes including interleukin-6 (IL-6), IL-1β, tumor necrosis factor α (TNF-α), CCL2, Kim1, NGAL, Col1a1, Col3a1, and α-smooth muscle actin (α-SMA) in WT animals (Figures 5E-5G and S6E-S6J). In Kir4.2−/− animals, this analysis revealed the absence of such a relationship for IL-6, IL-1β, CCL2, Kim1, Col1a1, Col3a1, and α-SMA. While slopes were statistically different for IL-6, Kim1, Col1a1, Col3a1, and α-SMA, similar conclusions could not be drawn for TNF-α, IL-1β, CCL2, and NGAL, though knockout abundances were reduced relative to WT controls for all genes (Figures S6E-S6J).
Figure 5. Effects of Kir4.2 deletion on low-K+- and aldosterone-mediated kidney injury.

(A) Blood K+ levels in WT and Kir4.2−/− animals following 3 and 8 days of treatment with an HS/0.04K diet.
(B) Relationship between kidney weight and blood K+ in WT and Kir4.2−/− mice.
(C) Total kidney abundance of inflammatory, injury, and fibrosis transcripts in WT and Kir4.2−/− animals after treatment with an HS/0.04K diet for 8 days.
(D) Kidney macrophage abundance in WT and Kir4.2−/− animals after treatment with an HS/0.04K diet for 3 and 8 days as determined by flow cytometry.
(E–G) Relationships between total kidney transcript abundance of (E) IL-6, (F) Kim1, and (G) Col1a1 and blood K+ levels in WT and Kir4.2−/− animals.
(H) Blood K+ levels in WT and Kir4.2−/− animals following 7 days of aldosterone infusion.
(I–K) Total kidney abundance of (I) inflammatory cytokines, (J) injury markers, and (K) fibrosis transcripts in mice treated as in (H).
n ≥ 5 for all. *p < 0.05 by unpaired t test or Mann-Whitney. For (B), p < 0.05 for test of difference in elevations of two lines and p = 0.06 for test of difference in slopes. For (E)–(G), regression analysis for WT animals demonstrated a negative correlation, while this was not the case for knockout mice; p < 0.05 for a difference in slopes between genotypes. Normality was determined by Shapiro-Wilk test. Error bars indicate SEM.
Additional physiological data are presented in Tables S5 and S6.
These data suggest that Kir4.2 is required to sense and respond to plasma K+ changes along the PT. This led us to hypothesize that if low K+ mediates aldosterone effects on injury and fibrosis through the proximal epithelium (Figure 3), then Kir4.2 deletion will afford protection in settings of aldosterone excess. While Kir4.2−/− animals had lower blood K+ levels following aldosterone infusion, their renal abundance of inflammatory, injury, and fibrosis transcripts were substantially reduced when compared with WT controls, confirming this hypothesis (Figures 5H-5K; Table S6).
Renal glutaminase is required for low-K+-mediated kidney injury
Low K+ is known to have potent metabolic effects on the PT epithelium, including activation of the ammoniagenesis pathway.13 We confirmed increased kidney transcript abundance of glutaminase in WT mice after consumption of an HS/0K diet (Figure S7A) and subsequently demonstrated increased protein abundance in PT cells via co-staining with lotus tetragonolobus lectin (LTL) (Figure 6A). Reduced injury in Kir4.2−/− animals following consumption of an HS/0.04K diet was associated with an absence of increased renal glutaminase abundance compared with WT controls (Figures 6B and S7B). Differences in glutamate dehydrogenase abundance between genotypes were not detected (Figure S7C). To determine if increased glutaminase activity contributes to low-K+ injury, we next cultured hRPTECs in normal- or low-K+ conditions. Low-K+ medium increased protein abundance of the PT injury marker Kim1. Importantly, this effect was absent in the presence of the glutaminase inhibitor BPTES (Figure 6C). Consistent with these data, low-K+ conditions also modestly increased reactive oxygen species (ROS) production as determined through measurements with the compound H2DCFDA (Figure S8A). In the presence of BPTES, changes in ROS production were not detectable (Figure S8B).
Figure 6. Kidney-specific glutaminase deletion protects against low-K+ kidney injury.

(A) Representative immunofluorescence co-staining for glutaminase and the PT marker LTL in animals treated as in (A).
(B) Kidney glutaminase abundance in WT and Kir4.2−/− animals at baseline and following treatment with an HS/0.04K diet for 8 days.
(C) Effects of glutaminase inhibition on low-K+-mediated increases in Kim1 abundance in hRPTECs in vitro.
(D) Total kidney glutaminase protein abundance in control and KS GLS−/− animals.
(E) Blood K+ and HCO3− levels from control and KS GLS−/− animals after treatment with an HS/0.04K diet for 3 weeks.
(F) Kidney weight from control and KS GLS−/− animals after treatment with an HS/0.04K diet for 3 weeks.
(G) Kidney abundance of leukocyte subsets as determined by flow cytometry in animals treated as in (B).
(H–J) Total kidney abundance of (H) inflammatory cytokines, (I) fibrotic transcripts, and (J) injury markers in animals treated as in (E).
(K) Kidney total Kim1 protein abundance in mice treated as in (E).
n ≥ 5 per group for all panels except n = 3 per group in (C) and n = 4 for K+ in (E). *p < 0.05 by unpaired t test or Mann-Whitney. **p < 0.05 for interaction by two-way ANOVA. Scale bar: 50 μm. Normality was determined by Shapiro-Wilk test. Error bars indicate SEM.
Additional physiological data are presented in Table S7.
To determine if glutaminase inhibition protects against low-K+ injury in vivo, we generated kidney-specific glutaminase knockout animals (KS GLS−/−) using the Pax8-LC1 Cre system (Figures 6D and S9). Following consumption of an HS/0.04K diet, blood K+ and HCO3− did not differ between KS GLS−/− mice and controls (Figure 6E; Table S7). However, KS GLS−/− kidneys did not show the same degree of hypertrophy and contained a reduced inflammatory infiltrate compared with WT animals, as we detected fewer kidney CD45+ cells and reduced myeloid and macrophage subsets in knockouts (Figures 6F and 6G). KS GLS−/− animals had reductions in total kidney transcript abundance of TNF-α, IL-1β, IL-6, NGAL, Col1a1, and Col1a3 (Figures 6H-6J). Kim1 transcript trended toward reduced abundance in KS GLS−/− mice, and immunoblotting indicated reduced Kim1 protein abundance in knockouts compared with WT controls (Figures 6J and 6K).
DISCUSSION
Reductions in dietary K+ intake strongly correlate with poor cardiovascular health outcomes. Mechanistic descriptions of K+ affecting the distal nephron epithelium provide a partial explanation for this, but whether or not K+ directly affects PT cell function remains unknown. Herein, we demonstrated that low dietary K+ injures PT cells. Injurious effects of K+ on the kidney occur directly through reduced ambient K+ levels and are continuous throughout the range of blood K+, with lower levels correlating with increased injury. Low K+ did not have inflammatory or profibrotic effects on other solid organs, but rather it demonstrated kidney specificity. Data obtained through a combination of in vitro and in vivo studies support a model for injury dependence upon increased basolateral K+ efflux, via Kir4.2, and the development of intracellular acidosis (Figure 7). Intracellular acidosis activates glutaminase to drive renal ammoniagenesis, and our findings show that this increased enzymatic activity contributes to kidney injury.
Figure 7. Model of low-K+ kidney injury.

In WT animals, low K+ causes increased basolateral K+ efflux through Kir4.2, reducing intracellular K+ (K+i) and pHi. This increases glutamine uptake along the basolateral membrane and its catabolism through GLS, producing ammonia, which is secreted into the lumen via NHE3. Glutamate dehudrogenase (GLUD) catalyzes conversion to α-ketoglutarate (α-KG). Metabolic flux through this pathway is increased under low-K+ conditions, causing increased kidney hypertrophy, inflammation, injury, and fibrosis. Kir4.2 deletion prevents alterations in basolateral K+ flux under low-K+ conditions. Kir4.2−/− mice do not develop intracellular acidosis or increased glutamine catabolism and are protected from kidney hypertrophy and injury. While GLS knockout mice do develop an intracellular acidosis under low-K+ conditions, the absence of GLS prevents increased glutamine catabolism, affording renoprotection. Error bars indicate SEM.
Our findings are reminiscent of hypokalemic nephropathy, a relatively uncommon clinical entity in which patients present with severe hypokalemia and injury of the renal epithelium.17,18 The mechanisms we have identified in this study are likely relevant to this process in patients. While targeting the pathway described here might be a viable treatment approach in this setting, our findings of increased Kim1 mRNA in animals with an average blood K+ level of 3.3 mM after 3 weeks of treatment suggest that less severe reductions in dietary and blood K+ can have deleterious kidney effects chronically. Long-term damaging effects of reduced dietary K+ may be more pervasive among the general population and could explain the association between reduced urine K+ and CKD incidence and progression.2,3,5,19 If prevalent in humans, this mechanistic relationship has important implications for the clinical care of patients with CKD. Dietary K+ restriction is a common component of the clinical approach to treating CKD. It is typically instituted to avoid negative cardiac effects of hyperkalemia, but overly aggressive K+ restriction could be harmful to the kidneys in the long term. While findings from several studies have supported this hypothesis,20,21 others have not.22,23 Moreover, clinical studies have been observational, making mechanistic inferences difficult. This hypothesis is currently being tested in a clinical trial in which patients with CKD stages 3 and 4 are being supplemented with KCl to determine effects on disease progression.24,25 Future studies should determine what level of K+ intake is optimal not only for patients at various stages of CKD but also for the general population to maintain cardiovascular health and preserve kidney function.
High aldosterone levels are known to contribute to renal fibrosis but also potently reduce blood K+ levels through effects on the distal nephron. Prior reports from a DOCA/salt model demonstrated that renal hypertrophy was dependent upon reductions in plasma K+ levels.26 Our data are consistent with this finding and also implicate reduced K+ in the renal fibrosis and injury observed in states of aldosteronism. Expression of the mineralocorticoid receptor (MR) begins along the thick ascending limb and extends distally to the aldosterone-sensitive distal nephron (ASDN).27 Along the ASDN, mineralocorticoid effects are enhanced through the actions of the enzyme 11β hydroxysteroid dehydrogenase type 2. Given the lack of MR expression along the PT, aldosterone-induced PT epithelial injury is likely through an indirect mechanism. Along the distal nephron, low K+ is a secondary mediatory of NaCl cotransporter (NCC) activation in the setting of hyperaldosteronism. NCC-mediated Na+ transport is not increased directly through DCT MR activity but rather through changes in basolateral membrane potential affected by blood K+.28,29 The current data support an analogous role for reduced K+ causing PT injury through altered Kir4.2-mediated basolateral K+ flux. While low K+ has direct effects along both proximal and distal segments, the majority of renal epithelial mass comprises the PT, suggesting that kidney inflammatory and injury effects are dominated by proximal epithelium. However, an accurate assignment of proximal and distal contributions to the observed phenotype are the subject of ongoing investigation.
This integral role of Kir4.2 in both dietary and aldosterone-mediated low K+ injury highlights its importance in PT responses to changes in plasma K+. Along the DCT, it has been proposed that Kir4.1 acts as a “K+ sensor,” mediating changes in membrane potential that occur secondary to altered plasma K+.9 Bignon et al. had proposed that Kir4.2 serves a similar K+-sensing role along the PT, and our data are supportive of this hypothesis.12 Given that the kidney comprises mostly PT, Kir4.2 may be the dominant pathway by which the kidney responds to blood K+ changes to maintain systemic electrolyte homeostasis. The worsened hypokalemia observed in mice lacking Kir4.2 further suggests this.
Reductions in basolateral K+ along the proximal epithelium stimulate ammoniagenesis. The initial enzymatic step of ammonia production is catalyzed by glutaminase, which converts glutamine to glutamate, and therefore renal ammonia production requires increased kidney glutamine uptake.13 The stimulus for glutaminase activation is likely the reduced pHi following exposure to low extracellular K+ that we demonstrated in cultured PT epithelial cells. These findings are supported by previous reports,30 though others were unable to demonstrate this phenomenon,31 likely due to differences in cell lines that were studied. While additional studies are required to define a causative role for pHi, this is consistent with prior data showing that acidosis activates glutaminase32 and our finding that Kir4.2 deletion, which prevents the development of intracellular acidosis, also prevents increased glutaminase abundance and kidney injury. However, our measured reduction in pHi was largely transient, raising the possibility that while altered pHi may initiate the downstream sequence of molecular events, it may not serve to sustain the signal chronically. Before such a role for reduced cytosolic pH can be ruled out, it needs to be clearly demonstrated that PT pHi completely returns to baseline in vivo chronically in an animal model of low K+.
Major downstream products of ammoniagenesis are ammonia itself, glucose, lipids, protein, and carbon dioxide.13 This increased metabolic demand is associated with the well-established phenomenon of low-K+-induced renal hypertrophy. Decades ago it was demonstrated that alkali loading in a rat model of hypokalemic nephropathy reduced tubulointerstitial fibrosis, ammoniagenesis, and renal hypertrophy, implicating acid-base changes in the pathogenesis.17,33,34 It was subsequently proposed that ammonia drove the development of tubulointerstitial fibrosis through its role in promoting activation of the innate immune. We confirmed a protective effect of bicarbonate and observed increased kidney abundance of multiple leukocyte subsets following dietary K+ restriction with the most dramatic changes in macrophage abundance. Increased kidney macrophage abundance was significantly prevented in both Kir4.2−/− and KS GLS−/− animals, which is consistent with these prior findings, suggesting a role for ammoniagenesis in promoting kidney inflammation. Prior work has also clearly demonstrated increased renal ROS production in animals treated with a low-K+ diet.35,36 While these reports propose that ROS production partially serves a homeostatic function in the renal response to K+ depletion, it is highly likely these effects co-exist with the well-described toxic effects of oxidative stress. Our cell data support this hypothesis in that they show increased ROS production in PT cells following low K+ culture, consistent with prior reports in Madin-Darby canine kidney (MDCK) cells.36 Moreover, increased ROS production is dependent on glutaminase activity, as its inhibition with BPTES prevented this effect.
The injury patterns observed with low-K+ diet and aldosterone are specific, and it cannot be assumed that identified protective pathways will also confer protection in other types of kidney injury. We have demonstrated that low K+ contributes to injury in our models, and the results suggest that higher K+ intake may be protective from CKD progression more generally. Wang et al. demonstrated this in a CKD model of reduced nephron mass and proposed that the effects were mediated by reduced inflammation.37
Future work will investigate if Kir4.2 or glutaminase inhibition protects in other animal models of kidney injury. Ion channels and enzymes are two of the most common pharmacological targets, and glutaminase has been widely investigated as a drug target in the cancer field.38 The efficacy of Na+-glucose co-transporter 2 inhibitors suggest that targeting other metabolic pathways in PT epithelial cells could be an effective approach for the treatment or prevention of CKD. As our results highlight the importance of both Kir4.2 and glutaminase function in mediating PT injury, these insights provide opportunities to intervene.
Limitations of the study
While multiple physiological parameters were measured in this study, we do not present comprehensive blood pressure analyses. Previous reports demonstrate combining high-Na+ and reduced-K+ intake raises blood pressure in mouse models.10,39,40 Because direct effects of low K+ on renal epithelial cells and elevated renal perfusion pressures likely contribute to kidney damage in our models, future studies will be required to assign proportional contributions for these two parameters. Similarly, we demonstrate that the HS/0.04K diet reduces plasma aldosterone, suggesting that it is not a major cause of the observed phenotype, yet a clear determination of its role and those of other endocrine mediators requires further investigation. It is also important to note we do not present measurements of Kir4.2 function. While our data demonstrate that its presence is required to observe the complete injury phenotype, we are unable to conclude which aspect of its function mediates the effects. Lastly, experiments were carried out on male animals aged 8–12 weeks, so additional studies are required to determine if effects are generalizable to both sexes and aged mice.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andrew Terker (andrew.s.terker@vumc.org).
Materials availability
Cell lines and animal models plasmids generated in this study are available from the lead contact.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal experiments were performed in accordance with the guidelines and with the approval of the Institutional Animal Care and Use Committee of Vanderbilt University Medical Center (M2200012-00). All animals used were male, aged 8–12 weeks. C57Bl/6 mice for experiments using only wild-type (WT) animals were purchased from Jackson labs and were used at 9 weeks of age. Kir4.2 knockout mice were generated by J. Denton. The Kir4.2 knockout mouse strain was designed by Sigma-Aldrich using CRISPR/Cas9. In vitro transcribed (IVT) sgRNA (50 ng/ul) and SpCas9 mRNA (100 ng/ul) (Sigma-Aldrich) were injected into the cytoplasm of C57BL/6N mouse zygotes (Envigo) by the Vanderbilt Genome Editing Resource (Vanderbilt University). The strain was backcrossed five generations to C57Bl6/N prior to use. Kidney-specific glutaminase knockouts animals were generated by crossing glutaminase (GLS1) floxed mice, purchased from Jackson Labs, with Pax8-LC1 Cre mice, which have been previously reported.28 Recombination was induced at 6 weeks of age by treating animals with a diet containing doxycycline (200 mg/kg diet) for two weeks. Age-matched littermates (8–12 weeks old) were used for experiments. All mice were genotyped by PCR on ear or tail snip DNA before and after experiments. Primers used for Kir4.2 knockout genotyping include F: GGTAGGAGATTAACACCATACTG and R: GAGAGTCCACTTTCATAATGCAG. Those used for glutaminase knockouts include: Pax8: F: CCA TGT CTA GAC TGG ACA AGA and R: CTC CAG GCC ACA TAT GAT TAG, LC1: F: TGGGCGGCATGGTGCAAGTT and R: CGGTGCTAACCAGCGTTTTC, GLS floxed allele: F: GGCCTGCTTAATGTTTCCTG and R: GGCATATCCCTGAGTTCGAG.
Cell culture
Human renal proximal tubule epithelial cells (hRPTECs) were purchased from the ATCC and cultured according to the supplier’s recommendations. For pH experiments, cells were plated in a black 96-well plate with clear bottoms (Thermo) at a density of 2 × 104 per well. The following day, cells were changed to either normal K+ (4 mM potassium gluconate) or K+ deficient (0 mM potassium gluconate) medium that was prepared in our lab (omitting Phenol red) according to prior reports.10 Cells were cultured in these conditions for amount of time indicated in Figure 4A. 30 min prior to reading, pHrodo green (ThermoFisher) was added to the wells according to the manufacturer’s protocol. The plate was then read at an Ex/Em of 509/533 nm on a SpectraMax iD3 (Molecular Devices). For reactive oxygen species (ROS) detection, the cell-permeant 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCFDA) was used similar to previous reports.41 Cells were plated as above and changed to either normal or K+ deficient medium as above followed by overnight culture. The following day, H2DCFDA (Thermo Fisher) was added at a final concentration of 5 μM for 15 min followed by washing and reading on a SpectraMax iD3 (Molecular Devices) at an Ex/Em of 495/520 nm. BPTES (Selleck Chemicals) was added to culture medium at a final concentration of 10 μM where indicated.
For detection of Kim1, hRPTECs cells were maintained according to the supplier’s recommendations. Cells were plated in 12 well plates (Thermo) at a density of 105 cells per well. Low K+ medium was prepared in our lab according to previous reports,10 and supplemented with 10% FBS. Final [K+] was 1.5 mM for low K+ medium and K+ gluconate was added to a final [K+] of 5.5 mM for normal K+ medium. BPTES (Selleck Chemicals) was added to culture medium at a final concentration of 10 μM where indicated. Cells were collected the following day in lysis buffer42 and centrifuged at 4°C for 15 min at 10,000 x g. Subsequent analysis was performed using protein lysate as described under ‘Western blot’ in the following section.
METHOD DETAILS
Diet experiments
All custom diets were purchased from Envigo (Indianapolis, In). Our standard mouse chow was used for Figures 1 and S3, which contained 1.21% K+ and 0.99% NaCl (same NaCl content was used for NS/0K diet). High salt/normal K+ (HS/NK) diets contained 6% NaCl with 0.8% K+. All other high salt diets contained 6% NaCl with K+ content indicated in the figures and corresponding figure legends. Amiloride was supplemented in drinking water at a concentration of 50 mg/L. For NaHCO3 experiments, NaCl or NaHCO3 was supplemented in drinking water at 0.28 M concentration with 2% sucrose. All other studies were carried out for three weeks except for those involving Kir4.2−/− animals, which were performed for either three or eight days as indicated in figures and corresponding legends.
Aldosterone infusion
Aldosterone was infused as previously reported.28 For studies involving only wild-type mice (Figure 3), aldosterone was dissolved in polyethylene glycol 200 and loaded into an osmotic minipump (model 1004, Alzet), implanted subcutaneously, and infused at a rate of 240 μg/kg/day for three weeks. Sham animals underwent an identical procedure, but without pump implantation. Animals were then fed indicated diets, which were either HS/NK (6% NaCl, 0.8% K+) or HS/HK (6% NaCl, 5% K+). Amiloride was supplemented in drinking water at a dose of 50 mg/L. Experiments involving Kir4.2−/− were performed for seven days using aldosterone infused via osmotic minipump (model 1007D, Alzet) at a rate of 180 μg/kg/day. Both WT and knockouts were maintained on HS/NK for the duration of the knockout studies.
Quantitative Real-Time PCR
Indicated organs or peripheral blood buffy coat (obtained from cardiac puncture blood samples after a 2000 x g microfuge spin) was snap frozen in Trizol reagent (Invitrogen) and RNA was subsequently isolated according to the manufacturer’s protocol. SuperScript IV First-Strand Synthesis System kit (Invitrogen) was used to synthesize cDNA from equal amounts of total RNA from each sample. Quantitative RT-PCR was performed using TaqMan real-time PCR (7900HT, Applied Biosystems). The Master Mix and all gene probes were purchased from Applied Biosystems. The probes used are listed in the key resources table. Relative quantification of specific PCR products was determined by the 2−ΔΔCT method. Data were normalized to RPS18.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Kim1 | Novus | Cat# AF-1817 |
| NGAL | Novus | Cat# AF-1857 |
| CD68 | Novus | Cat# 125212 |
| CD3 | abcam | Cat# MCA1477 |
| Glutaminase | Bio-Rad | Cat# 56750 |
| Glutamate dehydrogenase | Cell signaling | Cat# 12793 |
| Kir4.2 | Cell signaling | Cat# APC-058 |
| LTL-fluorescein | Alomone | Cat# FL-1321-2 |
| β-actin | Vector | Cat# A1978 |
| GAPDH | Millipore | Cat# HRP-60004 |
| S6 kinase (total) | Proteintech | Cat# 9202 |
| phospho S6 kinase (T389) | Cell signaling | Cat# 9205 |
| AKT (total) | Cell signaling | Cat# 4691 |
| phospho AKT (S473) | Cell signaling | Cat# 4060 |
| CD45-BV785 | Cell signaling | Cat# 103149 |
| CD11b-PerCP5.5 | Biolegends | Cat# 45-0112-82 |
| Ly6G-APC | Biolegends | Cat# 127614 |
| F4/80-PE-Cy7 | Biolegends | Cat# 123114 |
| CD3-FITC | Biolegends | Cat# 100204 |
| CD8-APC-Cy7 | Biolegends | Cat# 100714 |
| Chemicals, peptides, and recombinant proteins | ||
| pHrodo green | ThermoFisher | Cat# P35373 |
| H2DCFDA | ThermoFisher | Cat# D399 |
| BPTES | Selleck | Cat# S7753 |
| Amiloride | Millipore Sigma | Cat# 1019701 |
| Aldosterone | Millipore Sigma | Cat# A9477 |
| DNAse I | BioRad | Cat #7326828 |
| Collagenase D | Roche | Cat# 11088858001 |
| Picrosirius red | MilliporeSigma | Cat# 365548 |
| Critical commercial assays | ||
| Superscript IV First Strand Synthesis System Kit | ThermoFisher | Cat# 18091050 |
| Aldosterone ELISA | IBL America | Cat# IB79134 |
| Ammonium assay | Pointe Scientific | Cat# 23-666-102 |
| Creatinine companion | EXOCELL | Cat# 1012 |
| BCA Protein Assay | ThermoFisher | Cat# 23225 |
| Experimental models: Cell lines | ||
| Renal proximal tubule epithelial cells | ATCC | PCS-400-010 |
| Experimental models: Organisms/strains | ||
| Kir4.2 knockout animals | Vanderbilt University | N/A |
| Glutaminase floxed animals | Jackson Labs | 017956 |
| Pax8-LC1 transgenic animals | Dr. Leslie Gewin | N/A |
| Oligonucleotides | ||
| See Tables S8 and S9 | N/A | |
| Software and algorithms | ||
| Graphpad Prism v9 | Graphpad Software, LLC | https://www.graphpad.com/scientific-software/prism/ |
| QuPath | Relman and Schwartz18 | https://qupath.github.io/ |
Flow cytometry
Following animal euthanasia, kidneys were minced on ice by hand with a razor blade and incubated for one hour in collagenase D (Roche) and DNAse I (Biorad) at 37° with intermittent agitation. Suspension was then put through a 40μM filter and washed with HBBS supplemented with 3% fetal bovine serum (FBS). Lysate was then centrifuged for 10 min at 1000 rpm after which the supernatant was aspirated and pellet was resuspended in 1 mL RBC lysis buffer. After a 4 min incubation, 1 mL PBS with 0.5% FBS was added and cells were centrifuged again for 10 min at 1000 rpm. The supernatant was removed and pellet was resuspended in Zombie Violet (Biolegends), spun again for 10 min at 1000 rpm, and resuspended in staining antibody cocktail for 60 min (antibodies listed in key resources table). Cells were then washed again, resuspended, and flow cytometry was performed on a NovoCyte flow cytometer (Agilent). Cell debris and dead cells were excluded based on scatter signals and Zombie staining with gating determined by antibody signal (antibodies listed in key resources table). Novoexpress software was used for analysis.
Immunohistochemistry
Immunostaining was performed as reported previously.42,43 Following euthanasia, kidneys were removed and incubated at room temperature overnight in fixative containing 3.7% formaldehyde, 10 mM sodium m-periodate, 40 mM phosphate buffer, and 1% acetic acid. The fixed kidney was dehydrated through a graded series of ethanols, embedded in paraffin, sectioned (5 μm), and mounted on glass slides. Antibodies used are listed in Key Resources Table. For each experiment, all sections were mounted onto a single slide and stained together to reduce variability. Whole sections for each animal were analyzed using QuPath software.44 Threshold for positivity was determined using software and the algorithm was applied uniformly to all sections on the slide.
Immunofluorescence
Following euthanasia, kidneys were removed and processed as described under the ‘immunohistochemistry’ section. Immunofluorescence was performed as described previously.45 Briefly, the deparaffinized sections underwent antigen retrieval with citrate buffer by microwave heat for 15 min, and then were blocked with 10% normal goat serum for 1 h at room temperature. Antibodies used for staining were all diluted to a final concentration of 1:50 in phosphate buffered saline and sections were incubated overnight. Sections were viewed and imaged with a Nikon TE300 fluorescence microscope and spot-cam digital camera (Diagnostic Instruments).
Picrosirius red staining
Picrosirius red staining (MilliporeSigma, 365548) was performed according to the protocol provided by the manufacturer as previously reported.42 Quantification was performed using QuPath software as described under the immunohistochemistry section.
Tubular injury score
Analysis was performed as reported previously.41 In a blinded fashion, percentage of tubules that displayed cell necrosis, loss of the brush border, cast formation, and tubular dilatation was scored according to the following: 0, none; 1, ≤10%; 2, 11%–25%; 3, 26%–45%; 4, 46%–75%; 5, >76%.
Blood electrolyte measurements
Blood electrolytes were measured on samples obtained by cardiac puncture with an iSTAT analyzer using Chem8+ cartridges (Abbott, Abbot Park, Il) or with a Diamond Diagnostics (Holliston, Ma) Carelyte Plus unit.
Western Blot
Kidneys were snap frozen in liquid nitrogen at the time of euthanasia and subsequently transferred to −80°C for storage. For tissue lysate preparation, tissue was homogenized with a Tissue-Tearor homogenizer (Biospec Products), in lysis buffer as previously reported.42 Lysate was then centrifuged at 6000 rpm for 15 min. Protein concentration was measured by BCA protein assay (Pierce) followed by gel electrophoresis on a 4–20% Bis-Tris gel (Biorad).
Plasma aldosterone measurement
Performed as previously reported,10 according to ELISA kit manufacturer’s protocol (IBL America).
Urine ammonia measurement
Spot urines were collected, urine was diluted 1:50, and urine ammonia was measured according to the manufacturer’s protocol (Pointe Scientific). Values were normalized to urine creatinine which was measured as previously reported using the creatinine companion kit (Exocell).42
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM. Statistical tests used are indicated in figure legends. Normality was determined using the Shapiro-Wilk test. Nonparametric analyses were used if distribution was not normal. Comparisons were made with unpaired Student’s t-test, Mann-Whitney test, Kruskal-Wallis test, one-way ANOVA, or two-way ANOVA with or without repeated measures as appropriate and indicated in the figure legends. Corrections for multiple comparisons were performed as indicated in figure legends.
Supplementary Material
Highlights.
K+ deficiency causes kidney-specific injury and inflammation
Injury effects are dependent on the proximal K+ channel Kir4.2
Kir4.2 mediates activation of the ammoniagenesis pathway under low-K+ conditions
Kidney deletion of glutaminase prevents low-K+-mediated kidney injury
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. Eric Figueroa for assistance with generation of the Kir4.2 knockout animals. These studies were supported by NIH grants DP5OD033412 (A.S.T.), DK51265, DK95785, DK62794, DK7569, and P30DK114809 (R.C.H. and M.-z.Z.); VA Merit Award 00507969 (R.C.H.); and the Vanderbilt Center for Kidney Disease and Vanderbilt Diabetes Research and Training Center (DK020593) pilot and feasibility grant (J.S.D.). A.S.T. was also supported by a postdoctoral fellowship from the American Heart Association. J.P.A. is a Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program Scholar.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111840.
REFERENCES
- 1.Adrogué HJ, and Madias NE (2007). Sodium and potassium in the pathogenesis of hypertension. N. Engl. J. Med 356, 1966–1978. 10.1056/NEJMra064486. [DOI] [PubMed] [Google Scholar]
- 2.Neal B, Wu Y, Feng X, Zhang R, Zhang Y, Shi J, Zhang J, Tian M, Huang L, Li Z, et al. (2021). Effect of salt substitution on cardiovascular events and death. N. Engl. J. Med 385, 1067–1077. 10.1056/NEJMoa2105675. [DOI] [PubMed] [Google Scholar]
- 3.O’Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, Yan H, Lee SF, Mony P, Devanath A, et al. (2014). Urinary sodium and potassium excretion, mortality, and cardiovascular events. N. Engl. J. Med 371, 612–623. 10.1056/NEJMoa1311889. [DOI] [PubMed] [Google Scholar]
- 4.Yang Q, Liu T, Kuklina EV, Flanders WD, Hong Y, Gillespie C, Chang MH, Gwinn M, Dowling N, Khoury MJ, and Hu FB (2011). Sodium and potassium intake and mortality among US adults: prospective data from the Third National Health and Nutrition Examination Survey. Arch. Intern. Med 171, 1183–1191. 10.1001/archin-ternmed.2011.257. [DOI] [PubMed] [Google Scholar]
- 5.Elfassy T, Zhang L, Raij L, Bibbins-Domingo K, Lewis CE, Allen NB, Liu KJ, Peralta CA, Odden MC, and Zeki Al Hazzouri A (2020). Results of the CARDIA study suggest that higher dietary potassium may be kidney protective. Kidney Int. 98, 187–194. 10.1016/j.kint.2020.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenga MF, Kieneker LM, Soedamah-Muthu SS, van den Berg E, Deetman PE, Navis GJ, Gans RO, Gaillard CA, Bakker SJ, and Joosten MM (2016). Urinary potassium excretion, renal ammoniagenesis, and risk of graft failure and mortality in renal transplant recipients. Am. J. Clin. Nutr 104, 1703–1711. 10.3945/ajcn.116.134056. [DOI] [PubMed] [Google Scholar]
- 7.Kim HW, Park JT, Yoo TH, Lee J, Chung W, Lee KB, Chae DW, Ahn C, Kang SW, Choi KH, et al. (2019). Urinary potassium excretion and progression of CKD. Clin. J. Am. Soc. Nephrol 14, 330–340. 10.2215/CJN.07820618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang-Panesso M, Kadyrov FF, Lalli M, Wu H, Ikeda S, Kefaloyianni E, Abdelmageed MM, Herrlich A, Kobayashi A, and Humphreys BD (2019). FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J. Clin. Invest 129, 5501–5517. 10.1172/JCI125519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, and Wang WH (2017). Potassium sensing by renal distal tubules requires Kir4.1. J. Am. Soc. Nephrol 28, 1814–1825. 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, et al. (2015). Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 21, 39–50. 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, and Ellison DH (2016). Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 89, 127–134. 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bignon Y, Pinelli L, Frachon N, Lahuna O, Figueres L, Houillier P, Lourdel S, Teulon J, and Paulais M (2020). Defective bicarbonate reabsorption in Kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion. Kidney Int. 97, 304–315. 10.1016/j.kint.2019.09.028. [DOI] [PubMed] [Google Scholar]
- 13.Kamm DE, and Strope GL (1973). Glutamine and glutamate metabolism in renal cortex from potassium-depleted rats. Am. J. Physiol 224, 1241–1248. 10.1152/ajplegacy.1973.224.6.1241. [DOI] [PubMed] [Google Scholar]
- 14.Tannen RL (1977). Relationship of renal ammonia production and potassium homeostasis. Kidney Int. 11, 453–465. 10.1038/ki.1977.63. [DOI] [PubMed] [Google Scholar]
- 15.Del Vecchio L, Procaccio M, Viganò S, and Cusi D (2007). Mechanisms of disease: the role of aldosterone in kidney damage and clinical benefits of its blockade. Nat. Clin. Pract. Nephrol 3, 42–49. 10.1038/ncpneph0362. [DOI] [PubMed] [Google Scholar]
- 16.Adler S, and Fraley DS (1977). Potassium and intracellular pH. Kidney Int. 11, 433–442. 10.1038/ki.1977.61. [DOI] [PubMed] [Google Scholar]
- 17.Tolins JP, Hostetter MK, and Hostetter TH (1987). Hypokalemic nephropathy in the rat. Role of ammonia in chronic tubular injury. J. Clin. Invest 79, 1447–1458. 10.1172/JCI112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Relman AS, and Schwartz WB (1956). The nephropathy of potassium depletion; a clinical and pathological entity. N. Engl. J. Med 255, 195–203. 10.1056/NEJM195608022550501. [DOI] [PubMed] [Google Scholar]
- 19.Swift SL, Drexler Y, Sotres-Alvarez D, Raij L, Llabre MM, Schneiderman N, Horn LV, Lash JP, Mossavar-Rahmani Y, and Elfassy T (2022). Associations of sodium and potassium intake with chronic kidney disease in a prospective cohort study: findings from the Hispanic Community Health Study/Study of Latinos, 2008-2017. BMC Nephrol. 23, 133. 10.1186/s12882-022-02754-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazzaferro S, de Martini N, Cannata-Andía J, Cozzolino M, Messa P, Rotondi S, Tartaglione L, and Pasquali M; On Behalf Of The Era-Edta Ckd-Mbd Working Group (2021). Focus on the possible role of dietary sodium, potassium, phosphate, magnesium, and calcium on CKD progression. J. Clin. Med 10, 958. 10.3390/jcm10050958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wieërs MLAJ, Mulder J, Rotmans JI, and Hoorn EJ (2022). Potassium and the kidney: a reciprocal relationship with clinical relevance. Pediatr. Nephrol 37, 2245–2254. 10.1007/s00467-022-05494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hundemer GL, Sood MM, Ramsay T, and Akbari A (2022). Urinary potassium excretion and progression from advanced CKD to kidney failure. Can. J. Kidney Health Dis 9. 10.1177/20543581221084501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He J, Mills KT, Appel LJ, Yang W, Chen J, Lee BT, Rosas SE, Porter A, Makos G, Weir MR, et al. (2016). Urinary sodium and potassium excretion and CKD progression. J. Am. Soc. Nephrol 27, 1202–1212. 10.1681/ASN.2015010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terker A, Saritas T, and McDonough A (2022). The highs and lows of potassium intake in chronic kidney disease - does one size fit all? J. Am. Soc. Nephrol 33, 1638–1640. 10.1681/ASN.2022070743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gritter M, Wouda RD, Yeung SMH, Wieers MLA, Geurts F, de Ridder MAJ, Ramakers CRB, Vogt L, de Borst MH, Rotmans JI, et al. (2022). Effects of short-term potassium chloride supplementation in patients with CKD. J. Am. Soc. Nephrol 10.1681/ASN.2022020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, Domenighetti AA, Pedrazzini T, and Burnier M (2005). Potassium supplementation reduces cardiac and renal hypertrophy independent of blood pressure in DOCA/salt mice. Hypertension 46, 547–554. 10.1161/01.HYP.0000178572.63064.73. [DOI] [PubMed] [Google Scholar]
- 27.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, and Ellison DH (1998). 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J. Am. Soc. Nephrol 9, 1347–1358. 10.1681/ASN.V981347. [DOI] [PubMed] [Google Scholar]
- 28.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang CL, and Ellison DH (2016). Direct and indirect mineralocorticoid effects determine distal salt transport. J. Am. Soc. Nephrol 27, 2436–2445. 10.1681/ASN.2015070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czogalla J, Vohra T, Penton D, Kirschmann M, Craigie E, and Loffing J (2016). The mineralocorticoid receptor(MR) regulates ENaC but not NCC in mice with random MR deletion. Pflugers Arch. 468, 849–858. 10.1007/s00424-016-1798-5. [DOI] [PubMed] [Google Scholar]
- 30.Amemiya M, Tabei K, Kusano E, Asano Y, and Alpern RJ (1999). Incubation of OKP cells in low-K+ media increases NHE3 activity after early decrease in intracellular pH. Am. J. Physiol 276, C711–C716. 10.1152/ajpcell.1999.276.3.C711. [DOI] [PubMed] [Google Scholar]
- 31.Abu Hossain S, Chaudhry FA, Zahedi K, Siddiqui F, and Amlal H (2011). Cellular and molecular basis of increased ammoniagenesis in potassium deprivation. Am. J. Physiol. Renal Physiol 301, F969–F978. 10.1152/ajprenal.00010.2011. [DOI] [PubMed] [Google Scholar]
- 32.Tong J, Harrison G, and Curthoys NP (1986). The effect of metabolic acidosis on the synthesis and turnover of rat renal phosphate-dependent glutaminase. Biochem. J 233, 139–144. 10.1042/bj2330139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark EC, Nath KA, Hostetter MK, and Hostetter TH (1990). Role of ammonia in tubulointerstitial injury. Miner. Electrolyte Metab 16, 315–321. [PubMed] [Google Scholar]
- 34.Nath KA, Hostetter MK, and Hostetter TH (1991). Increased ammoniagenesis as a determinant of progressive renal injury. Am. J. Kidney Dis 17, 654–657. 10.1016/s0272-6386(12)80344-1. [DOI] [PubMed] [Google Scholar]
- 35.Babilonia E, Wei Y, Sterling H, Kaminski P, Wolin M, and Wang WH (2005). Superoxide anions are involved in mediating the effect of low K intake on c-Src expression and renal K secretion in the cortical collecting duct. J. Biol. Chem 280, 10790–10796. 10.1074/jbc.M414610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X, Yin W, Doi SQ, Robinson SW, Takeyasu K, and Fan X (2003). Stimulation of Na, K-ATPase by low potassium requires reactive oxygen species. Am. J. Physiol. Cell Physiol 285, C319–C326. 10.1152/ajpcell.00536.2002. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Soltero L, Zhang P, Huang XR, Lan HY, and Adrogue HJ (2007). Renal inflammation is modulated by potassium in chronic kidney disease: possible role of Smad7. Am. J. Physiol. Renal Physiol 293, F1123–F1130. 10.1152/ajprenal.00104.2007. [DOI] [PubMed] [Google Scholar]
- 38.Pavlova NN, and Thompson CB (2016). The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47. 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyd-Shiwarski CR, Weaver CJ, Beacham RT, Shiwarski DJ, Connolly KA, Nkashama LJ, Mutchler SM, Griffiths SE, Knoell SA, Sebastiani RS, et al. (2020). Effects of extreme potassium stress on blood pressure and renal tubular sodium transport. Am. J. Physiol. Renal Physiol 318, F1341–F1356. 10.1152/ajprenal.00527.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vitzthum H, Seniuk A, Schulte LH, Müller ML, Hetz H, and Ehmke H (2014). Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. J. Physiol 592, 1139–1157. 10.1113/jphysiol.2013.266924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sasaki K, Terker AS, Tang J, Cao S, Arroyo JP, Niu A, Wang S, Fan X, Zhang Y, Bennett SR, et al. (2022). Macrophage interferon regulatory factor 4 deletion ameliorates aristolochic acid nephropathy via reduced migration and increased apoptosis. JCI Insight 7, e150723. 10.1172/jci.insight.150723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasaki K, Terker AS, Pan Y, Li Z, Cao S, Wang Y, Niu A, Wang S, Fan X, Zhang MZ, and Harris RC (2021). Deletion of myeloid interferon regulatory factor 4 (Irf4) in mouse model protects against kidney fibrosis after ischemic injury by decreased macrophage recruitment and activation. J. Am. Soc. Nephrol 32, 1037–1052. 10.1681/ASN.2020071010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terker AS, Sasaki K, Arroyo JP, Niu A, Wang S, Fan X, Zhang Y, Nwosisi S, Zhang MZ, and Harris RC (2021). Activation of hypoxia-sensing pathways promotes renal ischemic preconditioning following myocardial infarction. Am. J. Physiol. Renal Physiol 320, F569–F577. 10.1152/ajprenal.00476.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: open source software for digital pathology image analysis. Sci. Rep 7, 16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan Y, Cao S, Tang J, Arroyo JP, Terker AS, Wang Y, Niu A, Fan X, Wang S, Zhang Y, et al. (2022). Cyclooxygenase-2 in adipose tissue macrophages limits adipose tissue dysfunction in obese mice. J. Clin. Invest 132, e152391. 10.1172/JCI152391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.