

Abstract
Chronic inflammation represents a major threat to human health since long‐term systemic inflammation is known to affect distinct tissues and organs. Recently, solid evidence demonstrated that chronic inflammation affects hematopoiesis; however, how chronic inflammation affects hematopoietic stem cells (HSCs) on the mechanistic level is poorly understood. Here, we employ a mouse model of chronic multifocal osteomyelitis (CMO) to assess the effects of a spontaneously developed inflammatory condition on HSCs. We demonstrate that hematopoietic and nonhematopoietic compartments in CMO BM contribute to HSC expansion and impair their function. Remarkably, our results suggest that the typical features of murine multifocal osteomyelitis and the HSC phenotype are mechanistically decoupled. We show that the CMO environment imprints a myeloid gene signature and imposes a pro‐inflammatory profile on HSCs. We identify IL‐6 and the Jak/Stat3 signaling pathway as critical mediators. However, while IL‐6 and Stat3 blockage reduce HSC numbers in CMO mice, only inhibition of Stat3 activity significantly rescues their fitness. Our data emphasize the detrimental effects of chronic inflammation on stem cell function, opening new venues for treatment.
Keywords: chronic inflammation, chronic multifocal osteomyelitis, hematopoietic stem cells, IL‐6/Jak/Stat3, niche
Subject Categories: Immunology, Signal Transduction, Stem Cells & Regenerative Medicine
Chronic inflammation induces HSC expansion and reduces HSC fitness by hyperactivating the IL‐6/Jak/Stat3 signaling pathway. These effects of chronic inflammation on HSCs are mitigated upon inhibition of Stat3.

Introduction
Chronic inflammation is a hallmark of autoimmune and autoinflammatory disorders and is characterized by a low‐grade and persistent increase in cytokines and chemokines. During the last years, compelling evidence linked sterile chronic inflammation with disease risk and mortality (Emerging Risk Factors et al, 2010, Dregan et al, 2014, Proctor et al, 2015, Straub & Schradin, 2016). However, the underlying mechanisms of how chronic inflammation affects hematopoiesis and the most primitive hematopoietic stem cells (HSCs) remain poorly understood.
HSCs are a rare population of cells whose activity is regulated by a plethora of cell intrinsic and extrinsic factors (Blank et al, 2008; Rossi et al, 2012; Pietras et al, 2015; Pinho & Frenette, 2019). While cell intrinsic factors have been extensively studied, the role of cell extrinsic factors remains rather elusive. Cell extrinsic factors are provided by specialized BM cells located in close proximity to HSCs and referred to as the BM niche. The BM niche consists of distinct cell types, which provide an optimal environment for HSCs and can influence HSC activity by secreting factors such as neurotransmitters, cytokines, chemokines, hormones, and growth factors (Zhang et al, 2003; Katayama et al, 2006; Sacchetti et al, 2007; Mendez‐Ferrer et al, 2008, 2010; Chow et al, 2011; Ding et al, 2012). Interestingly, while some cytokines provide pro‐survival signals to HSCs and impose quiescence, excessive exposure to inflammatory cytokines may lead to impaired self‐renewal and increased differentiation, thus compromising HSC function (Essers et al, 2009; Baldridge et al, 2010; Chen et al, 2010; Boettcher et al, 2012; Kunisaki et al, 2013; Helbling et al, 2019).
During the last decade, several studies attempted to decipher how chronic inflammation affects HSCs. For instance, mice challenged with pathogens, polyinosinic:polycytidylic acid (pI:pC) or lipopolysaccharide (LPS) exhibited functional defects in HSCs (Baldridge et al, 2010; Esplin et al, 2011; Zhao et al, 2013; Walter et al, 2015; Matatall et al, 2016; Isringhausen et al, 2021; Bogeska et al, 2022). However, all these models are based on the artificial administration of biological agents or factors and provided a limited understanding on how sterile chronic inflammation affects HSCs. In a non‐infection context, genetic and collagen‐induced mouse models of rheumatoid arthritis reported that HSCs displayed skewing toward the myeloid lineage, but surprisingly, their quiescence and long‐term repopulating abilities were not affected (Ma et al, 2009; Oduro Jr et al, 2012; Hernandez et al, 2020). On the contrary, dysregulated HSC activity was recently reported in a mouse model of spondyloarthritis, a chronic inflammatory disease (Regan‐Komito et al, 2020). Possibly, the contradictory results on the effect of chronic inflammation on HSCs can be explained by the distinct, and sometimes controversial, murine models employed to induce chronic inflammation, and thus, further studies investigating the effect of sterile chronic inflammation on the functionality of HSCs are needed.
Chronic multifocal osteomyelitis (CMO) is a murine autoinflammatory disorder that resembles a human condition known as chronic recurrent multifocal osteomyelitis (CRMO; Giedion et al, 1972). CMO mice carry a missense mutation, L98P, in the gene proline‐serine–threonine phosphatase‐interacting protein 2 (Pstpip2), and consequently lack Pstpip2 protein (Ferguson et al, 2006; Chitu et al, 2009). CMO mice exhibit a gradual increase in pro‐inflammatory cytokines and suffer from chronic inflammation (Chitu et al, 2009). Up to 9–10 weeks old, CMO mice are healthy and asymptomatic; however, they develop visible signs of inflammation and exhibit a 100% penetrance phenotype by the age of 14–15 weeks. The inflammatory signs are manifested as a progressive disease characterized by swollen and deformed paws, tail kinks, splenomegaly, bone lesions, and inflamed ears (Byrd et al, 1991; Hentunen et al, 2000; Ferguson et al, 2006; Chitu et al, 2009, 2012; Lukens et al, 2014a). The pathology is driven by the expansion of the granulocytic compartment, which contributes to the production of high levels of interleukin 1β (IL‐1β; Chitu et al, 2009; Drobek et al, 2015). Importantly, IL‐1β signaling pathway was shown to be crucial for disease development, as in the absence of interleukin‐1 receptor type 1 (IL‐1RI), the disease was abolished (Cassel et al, 2014; Lukens et al, 2014a).
Here, we asked how sterile chronic inflammation affects HSC long‐term activity. By using CMO mice we observed that chronic inflammation exerts detrimental effects on HSCs, inducing an expansion of the HSC pool and reducing HSC fitness. In contrast to the critical role of IL‐1β in disease progression, inactivation of the IL‐1β/MyD88 signaling pathway was not enough to prevent defects in the HSC compartment. Instead, we demonstrate that IL‐6 and the Jak/Stat3 signaling pathway mediate HSC alterations. However, while both IL‐6 and Stat3 blockage significantly reduce HSC numbers in CMO mice, only inhibition of Stat3 activity is able to rescue HSC function. Altogether, our observations show that sterile chronic inflammation, in addition to the typical features of chronic multifocal osteomyelitis, has detrimental effects on HSCs and provides new insights into the mechanisms driving these phenotypes.
Results
Increased number and impaired fitness of HSCs in the BM of CMO mice
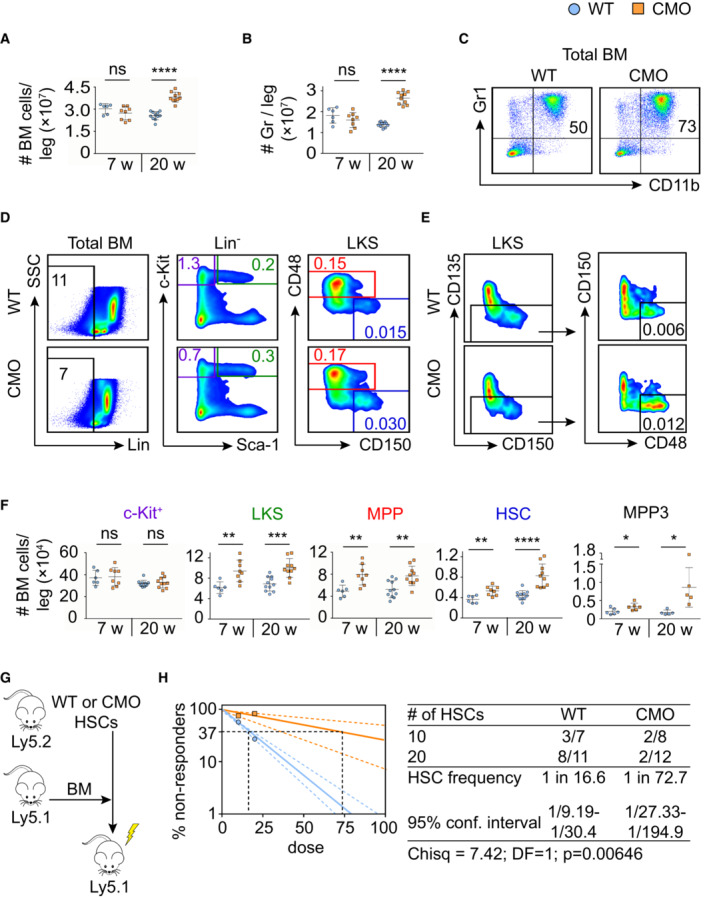
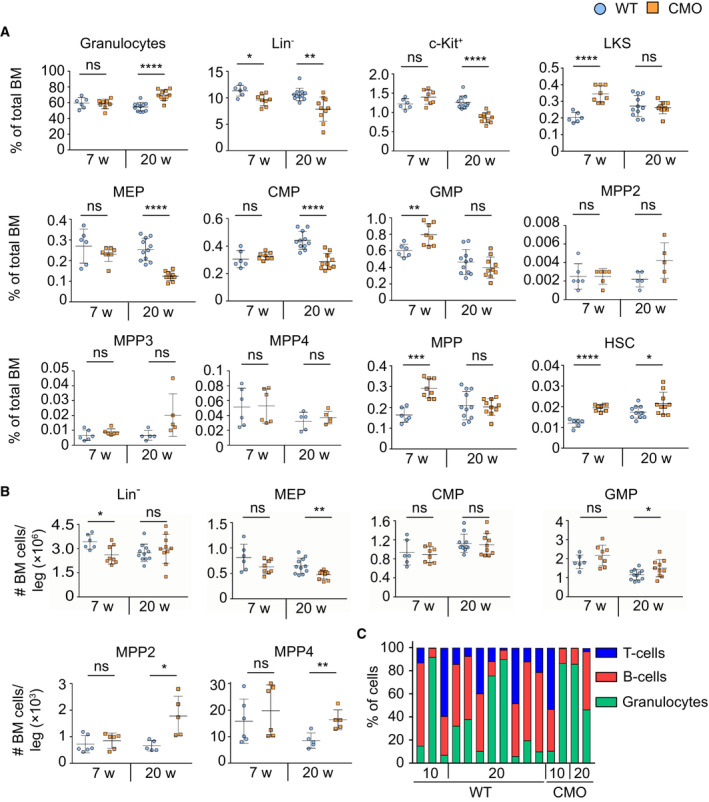
Since we and others previously reported that CMO mice suffer from sterile chronic inflammation and exhibit increased cytokine levels (Chitu et al, 2009, 2012; Cassel et al, 2014; Lukens et al, 2014a; Drobek et al, 2015), we employed this mouse model to investigate the effects of chronic inflammation on the BM composition, and in particular the HSC pool, defined here as Lin− c‐kit+ Sca‐1− CD48− CD150+ (Kiel et al, 2005). We observed that 7‐week‐old CMO mice, which are asymptomatic, showed a similar number of BM cells as WT controls, while 20‐week‐old mice, which are symptomatic, exhibited a significant increase in BM cellularity (Fig 1A), mainly driven by increased granulocytic counts (Figs 1B and C, and EV1A). Myeloid progenitor populations in CMO mice demonstrated a decrease in the number of megakaryocyte‐erythroid progenitors (MEPs), while the number of granulocyte‐monocyte progenitors (GMPs) was expanded (Fig EV1B); nevertheless, this bias did not affect the total number of c‐Kit+ myeloid progenitors (Fig 1D–F). Further, we detected an increased number of Lin− c‐Kit+ Sca‐1+ cells (LKS), LKS CD48+ CD150− multipotent progenitors (MPPs), including the earliest myeloid precursor MPP3, and HSCs (Fig 1D–F). Strikingly, these stem cell‐enriched populations were expanded already in asymptomatic CMO mice, and later persisted as the disease progressed (Fig 1F). As expected, in terms of percentages, the expansion of LKS, MPPs, and HSCs was masked in older mice due to the increased BM cellularity and was only detected in asymptomatic CMO mice, which did not suffer from increased BM cellularity yet (Fig EV1A).
Figure 1. Expansion and reduced fitness of HSCs in the BM of CMO mice.
-
A, BAbsolute number of BM cells (A) and granulocytes (B). Y‐axes indicate the number of cells per leg. X‐axes indicate the age of the mice: 7 or 20 weeks old (w). Each symbol indicates values for one biological replicate (mouse). At least six mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (****P < 0.0001, ns, not significant).
-
C, DRepresentative flow cytometry plots from 20‐week‐old WT and CMO BM. Plots illustrate the percentage of granulocytes defined as Gr1+ CD11b+ (C) and distinct BM subpopulations (D). Color boxes indicate the following populations: Lin− cells (black), Lin− c‐Kit+ Sca‐1− (c‐Kit+, purple), Lin− c‐Kit+ Sca‐1+ (LKS, green), Lin− c‐Kit+ Sca‐1+ CD48+ CD150− (MPPs, red), and Lin− c‐Kit+ Sca‐1+ CD48− CD150+ (HSCs, blue). Numbers indicate the percentage of cells in total BM.
-
EGating strategy to identify MPP3, defined as Lin− c‐Kit+ Sca‐1+ CD135− CD150− CD48+. Numbers indicate the percentage of MPP3 in total BM.
-
FQuantification of the absolute number of distinct stem and progenitor populations in BM. Each symbol indicates values for one biological replicate (mouse). At least five mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant).
-
GTransplantation scheme of 20‐week‐old WT and CMO HSCs.
-
HFrequency of functional WT (blue) and CMO (orange) HSCs measured by limiting dilution competitive repopulation unit assays and calculated using ELDA online software based on Poisson distribution statistics (Chi‐square test; Chisq = 7.42; P = 0.00646). Graph shows the curve fit of the log fraction of nonresponding mice (solid lines) and confidence intervals (dashed lines) versus the number of mice tested. Logarithmic plot; X‐axis indicates the dose of transplanted cells and Y‐axis percentages of negative responders. Reconstitution was evaluated in blood of recipient mice 16 weeks after transplantation. A responder mouse was defined as engraftment ≥ 0.5% Ly5.2+ cells and contribution ≥ 0.5% in at least two out of three lineages (granulocytes, B‐cells, and T‐cells).
Figure EV1. CMO BM exhibits increased cellularity and expansion of HSPCs in a time‐dependent fashion.
-
A, BPercentage (A) and absolute number (B) of distinct BM subpopulations. Each symbol represents one biological replicate (mouse). Blue round symbols indicate values for WT mice and square orange symbols indicate values for CMO mice. X‐axes indicate the age of the mice: 7 or 20 weeks old (w). Data represent mean ± s.d. At least six mice were used per group in two separate experiments. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant).
-
CTri‐lineage reconstitution upon transplantation of WT and CMO HSCs. The x‐axis indicates the dose of donor cells. The y‐axis indicates the percentage of cells defined as Gr1+ CD11b+ myeloid cells (green), B220+ B‐cells (red), and CD3+ T‐cells (blue). Each column represents values for one mouse.
Since the number of phenotypically defined HSCs is increased in the BM of CMO mice (Fig 1F), we next investigated whether this phenotypical expansion affected their functionality. Limiting dilution transplantation assays were performed using 20‐week‐old donors as indicated in Fig 1G. Analysis of peripheral blood (PB) of recipient mice 16 weeks post‐transplantation showed that the engraftment of CMO HSCs presented similar lineage distribution as the WT HSCs (Fig EV1C). However, the long‐term repopulating ability of CMO HSCs was impaired in comparison to WT HSCs (Fig 1H). Altogether, these results indicate that in CMO mice BM HSCs are expanded and their function compromised, suggesting that chronic inflammation might affect HSC properties.
Both the hematopoietic and the nonhematopoietic compartment in CMO BM exert detrimental effects on HSCs
Since chronic inflammation in CMO mice is mediated by hematopoietic cells and by nonhematopoietic niche cells (Chitu et al, 2009, 2012), we next investigated which of these compartments executes detrimental effects on HSCs. First, to investigate whether CMO hematopoietic cells were able to affect the functional properties of HSCs, we assessed the function of WT HSCs that co‐habited with WT or CMO hematopoietic cells. As depicted in Fig 2A, WT or CMO BM (Ly5.2) cells isolated from 20‐week‐old mice were mixed with WT BM (Ly5.1) in a 10:1 ratio and transplanted into lethally irradiated primary recipient mice (Ly5.1). Twelve weeks post‐transplantation donor Ly5.1 HSCs, which co‐habited with either WT or CMO donor hematopoietic cells, were isolated and re‐transplanted into lethally irradiated secondary recipients (Ly5.2). Sixteen weeks post‐transplantation, engraftment of WT HSCs that co‐habited with WT or CMO hematopoietic cells was determined by flow cytometric analysis. We observed that CMO hematopoietic cells negatively affected the long‐term repopulating capacity of WT HSCs in comparison to the effect that WT hematopoietic cells had on WT HSCs (Figs 2B and EV2A). Next, we investigated whether the nonhematopoietic BM niche was also able to alter HSC properties. Thus, WT BM cells (Ly5.1) were transplanted into 10‐week‐old WT or CMO congenic mice (Ly5.2) in which hematopoietic cells were ablated by lethal irradiation, while the radioresistant niche cells were preserved (Sugrue et al, 2013; Fig 2C). Twelve weeks after transplantation, Ly5.1 WT donor cells (total BM or HSCs) exposed to either WT or CMO BM niche, were isolated and re‐transplanted into lethally irradiated secondary recipients (Ly5.2). We observed that WT BM cells exposed to the CMO BM niche engrafted significantly worse in secondary recipients than WT BM cells exposed to WT BM niche (Figs 2D and E, and EV2B and C), while their capacity to reconstitute B‐cells, T‐cells, and myeloid cells was not affected (Fig EV2D). Similarly, limiting dilution transplantation assays demonstrated that WT HSCs exposed to CMO BM niche had significantly impaired engraftment in secondary recipients compared with WT HSCs exposed to WT BM niche (Fig 2F), while preserving their ability to reconstitute B, T, and myeloid cells (Fig EV2E). Altogether, these results indicate that both inflammatory signals coming from hematopoietic cells and the nonhematopoietic BM niche exert detrimental effects on HSC fitness.
Figure 2. Hematopoietic cells and nonhematopoietic BM niche exert detrimental effects on HSCs.
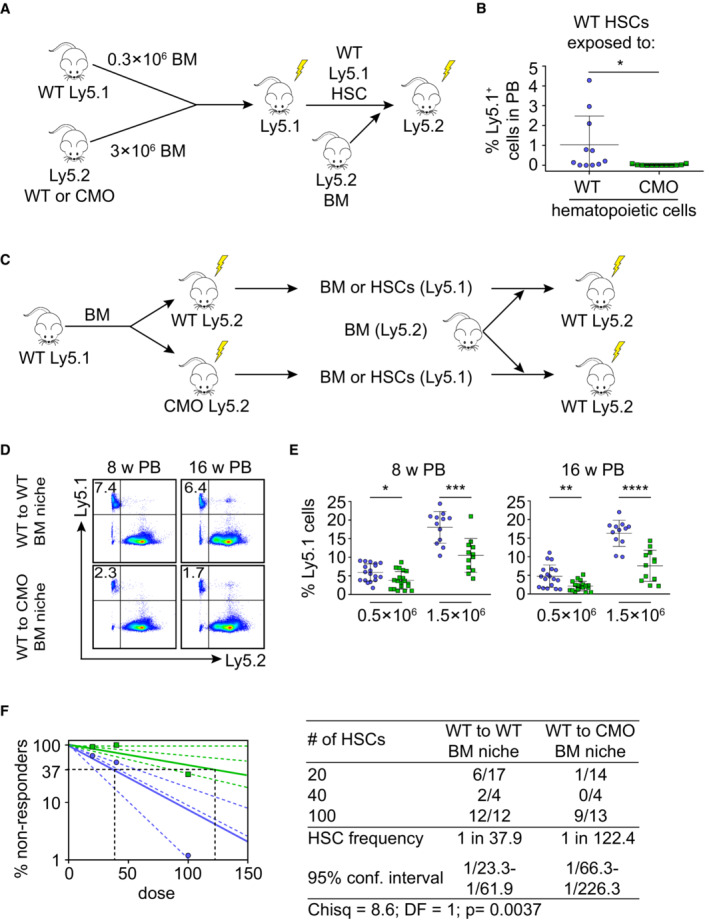
- Schematic representation of the experimental setup. 0.3 × 106 WT BM Ly5.1 cells were mixed with 3 × 106 BM Ly5.2 cells isolated from WT or CMO mice and transplanted into lethally irradiated Ly5.1 WT primary recipient mice. Twelve weeks after transplantation, donor Ly5.1 HSCs were sorted and re‐transplanted into lethally irradiated Ly5.2 secondary recipients in the presence of Ly5.2 BM support.
- Percentage of Ly5.1+ cells derived from WT HSCs exposed to WT (blue) or CMO (green) hematopoietic cells. Each symbol indicates values for one biological replicate (mouse). At least 11 mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05).
- Schematic representation of experimental setup. 3 × 106 Ly5.1 WT BM cells were transplanted into lethally irradiated WT or CMO primary recipient mice (Ly5.2). 12 weeks after transplantation, Ly5.1 WT donor BM or HSCs cells were isolated and re‐transplanted into lethally irradiated secondary recipients (Ly5.2) in the presence of support BM (Ly5.2).
- Representative flow cytometry plots from secondary recipient mice transplanted with 0.5 × 106 WT BM exposed to WT (WT to WT) or to CMO (WT to CMO) BM niche. Numbers indicate the percentage of WT donor‐derived Ly5.1+ cells in peripheral blood (PB) 8 and 16 weeks (w) after transplantation.
- Quantification of panel (D) Y‐axes show the contribution of WT BM Ly5.1+ cells after exposure to WT (blue) or to CMO (green) BM niche. X‐axes indicate the dose of transplanted cells. Each symbol indicates values for one biological replicate (mouse). At least 12 mice were used per group in three separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
- Frequency of functional HSCs after exposure to WT (blue) or to CMO (green) BM niche. Logarithmic plot shows the percentage of nonresponder recipient mice. Responders exhibited engraftment of ≥ 0.01% Ly5.1+ cells and contribution ≥ 0.5% to at least one out of three lineages (granulocytes, B‐cells, and T‐cells) at week 16 post‐transplantation. X‐axis indicates the dose of transplanted cells. Graph shows the curve fit of the log fraction of nonresponding mice (solid lines) and confidence intervals (dashed lines) versus the number of mice tested. Right table shows the number of responders and the total number of recipients transplanted per cell dose. Frequencies of HSCs were calculated using ELDA online software based on Poisson distribution statistics (Chi‐square test; Chisq = 8.6; P = 0.0037).
Figure EV2. CMO hematopoietic cells and the CMO BM niche exert detrimental effects on HSCs.
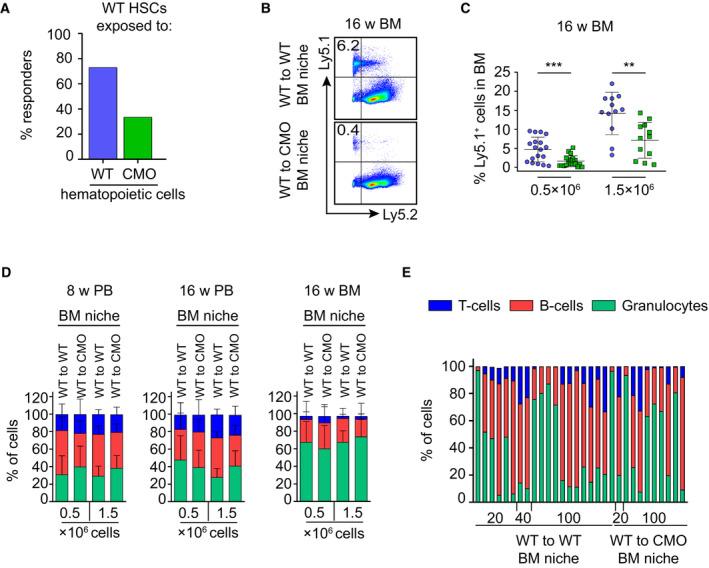
- Percentage of responder recipient mice that received WT HSCs, which co‐habited with WT or CMO hematopoietic cells. A recipient was considered as a positive responder when engraftment was ≥ 0.01% Ly5.1+ cells and contribution ≥ 0.5% in at least one out of three lineages (granulocytes, B‐cells, and T‐cells) at week 16 post‐transplantation. Percentage of Ly5.1+ donor‐derived cells in blood of secondary recipients.
- Representative flow cytometry plots from secondary recipient mice transplanted with 0.5 × 106 WT BM exposed to WT (WT to WT) or CMO (WT to CMO) BM niche. Y‐axis shows Ly5.1+ cells and x‐axis Ly5.2+ cells in BM of recipient mice 16 weeks after transplantation. Numbers indicate the percentage of WT donor‐derived Ly5.1+ cells.
- Quantification of panel (B). X‐axis indicates the dose of transplanted cells. At least 12 animals were used per group in three separate experiments. Values for WT BM exposed to WT BM niche are indicated with a blue symbol, and values for WT BM exposed to CMO BM niche are indicated in a green symbol. Each symbol represents one biological replicate (mouse). Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (**P < 0.01, ***P < 0.001).
- Tri‐lineage reconstitution from secondary transplantation of WT BM exposed to WT (WT to WT) or CMO (WT to CMO) BM niche. Graphics indicate analysis of peripheral blood (PB) 8 and 16 weeks after transplantation, and BM 16 weeks after transplantation. Each column indicates mean ± s.d. (at least 12 mice were used per group in three separate experiments). Y‐axes indicate the percentage of donor‐derived Ly5.1+ Gr1+ CD11b+ granulocytes (green), B220+ B‐cells (red), and CD3+ T‐cells (blue). The x‐axes indicate the number of donor cells.
- Tri‐lineage reconstitution 16 weeks after transplantation in blood from secondary recipients transplanted with WT HSCs exposed to WT (WT to WT) or CMO (WT to CMO) BM niche. Each column represents values for one biological replicate (mouse). Y‐axes indicate the percentage of donor‐derived Ly5.1+ Gr1+ CD11b+ granulocytes (green), B220+ B‐cells (red), and CD3+ T‐cells (blue). The x‐axes indicate the number of donor cells.
CMO mice lacking MyD88 adaptor protein retain HSC expansion and reduced fitness
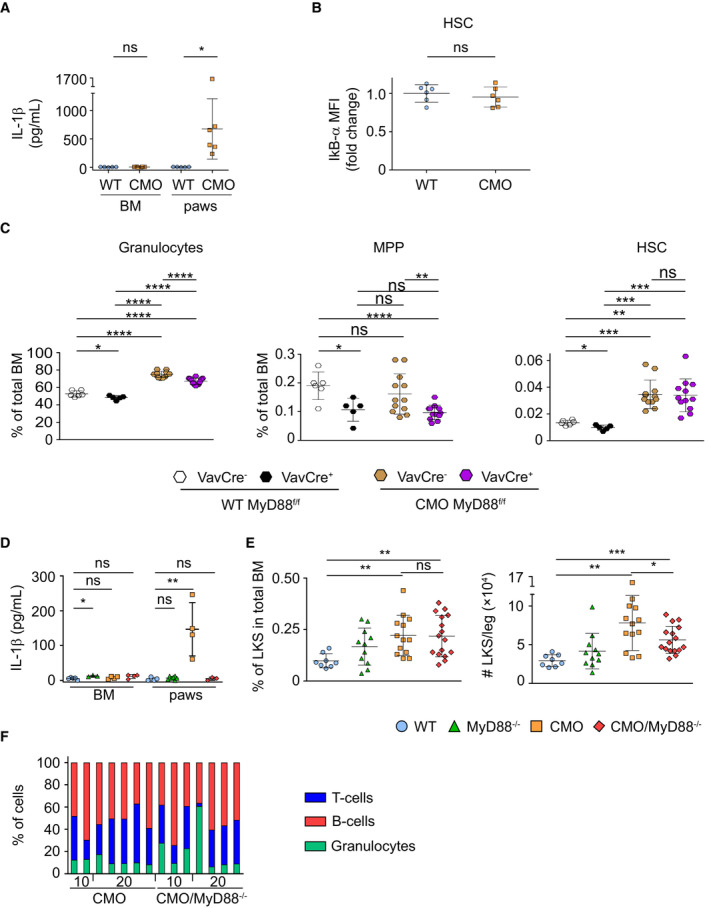
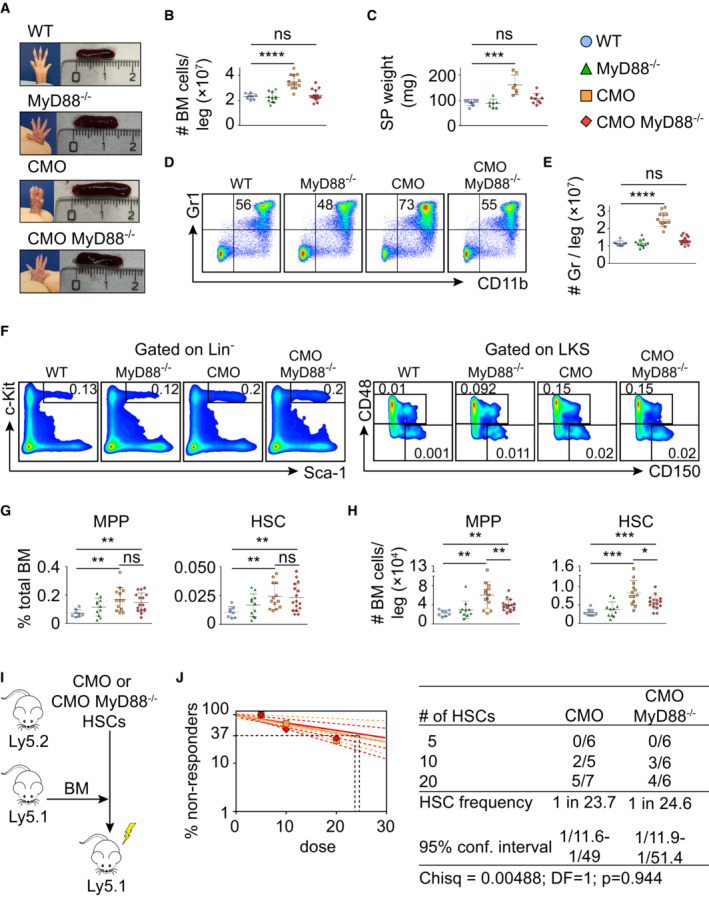
We and others have shown that IL‐1β, a cytokine suggested to impair HSC functionality (Pietras et al, 2016), is the main driving force behind the inflammatory phenotype of CMO mice (Cassel et al, 2014; Lukens et al, 2014a; Drobek et al, 2015). Remarkably, increased IL‐1β levels can be detected in paws, but not in BM, of CMO mice (Fig EV3A), and accordingly, WT and CMO HSCs exhibited similar levels of IκB‐α, a negative modulator of the NF‐κΒ signaling pathway (Fig EV3B). Thus, we investigated whether deletion of myeloid differentiation primary response 88 (MyD88) adaptor protein, an essential part of the IL‐1β signaling pathway, would be sufficient to prevent the CMO phenotype, including the expansion of the HSC pool. First, we investigated whether ablation of MyD88 specifically in the hematopoietic compartment would be sufficient to observe the rescue, and thus, CMO mice were crossed to MyD88flox/flox VAV‐iCre transgenic mice. Interestingly, we observed that CMO MyD88flox/flox VAV‐iCre+ mice, lacking expression of MyD88 specifically in hematopoietic cells, retained all characteristics of CMO mice, including swollen paws (Fig 3A), increased BM cellularity (Fig 3B), expansion of granulocytes (Figs 3C and D, and EV3C), and increased number of MPPs and HSCs (Figs 3E and F, and EV3C). Next, we crossed CMO mice with whole‐body MyD88 knockout (KO) mice and observed that CMO MyD88 KO mice did not present swollen paws (Fig 4A). Further, ablation of MyD88 in CMO mice prevented the increased BM cellularity (Fig 4B), splenomegaly (Fig 4A and C), and the hyperproduction of granulocytes observed in CMO mice (Fig 4D and E). In line with these findings, CMO MyD88 KO mice did not exhibit increased levels of IL‐1β (Fig EV3D). However, we observed that the populations enriched for stem cells (LKS, MPP, and HSC) remained expanded in the BM of CMO MyD88 KO mice compared with WT BM (Figs 4F–H and EV3E). Further, we observed that CMO MyD88 KO HSC retained the functional impairment exhibited by CMO HSCs (Figs 4I and J, and EV3F). Altogether, these experiments indicate that the IL‐1β/MyD88 signaling pathway in hematopoietic and nonhematopoietic cells is dispensable to induce expansion and impair the fitness of HSCs, suggesting that other factors, rather than IL‐1β, mediate HSC defects. Further, our data indicate that MyD88‐mediated signaling in the BM niche, but not in the hematopoietic cells, is crucial to induce the increase in BM cellularity, splenomegaly, and the hyperproduction of granulocytes observed in CMO mice.
Figure EV3. Phenotypic analysis of CMO mice and CMO mice lacking MyD88.
-
AY‐axis indicates IL‐1β (pg/ml) in BM and paws isolated from WT (blue symbols) and symptomatic CMO (orange symbols) mice. Each symbol represents one biological replicate (mouse). At least five mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, ns, not significant).
-
BIkB‐α levels in HSCs isolated from WT or CMO mice. Y‐axis indicates IkB‐α mean fluorescence intensity (MFI). Values are normalized to the average of WT levels. Each symbol represents one biological replicate (mouse). At least six mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (ns, not significant).
-
CPercentage of distinct BM subpopulations in distinct murine lines. In this figure, each symbol represents one mouse (biological replicates). At least five mice were used per group in three separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant).
-
DIL‐1β levels in BM and paws. Y‐axis indicated IL‐1β (pg/ml) in BM and paws isolated from WT (blue symbols) and symptomatic CMO (orange symbols) mice. Each symbol represents one biological replicate (mouse). At least four mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ns, not significant).
-
EPercentage (left panel) and absolute number (right panel) of LKS in BM. Each symbol represents one biological replicate (mouse). At least eight mice were used per group in three separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant).
-
FTri‐lineage reconstitution upon transplantation of CMO and CMO MyD88−/− HSCs. The x‐axis indicates the dose of donor cells. The y‐axis indicates the percentage of cells defined as Gr1+ CD11b+ myeloid cells (green), B220+ B‐cells (red), and CD3+ T‐cells (blue). Each column represents values for one mouse.
Figure 3. CMO mice lacking MyD88 in hematopoietic cells retain HSC expansion in BM.
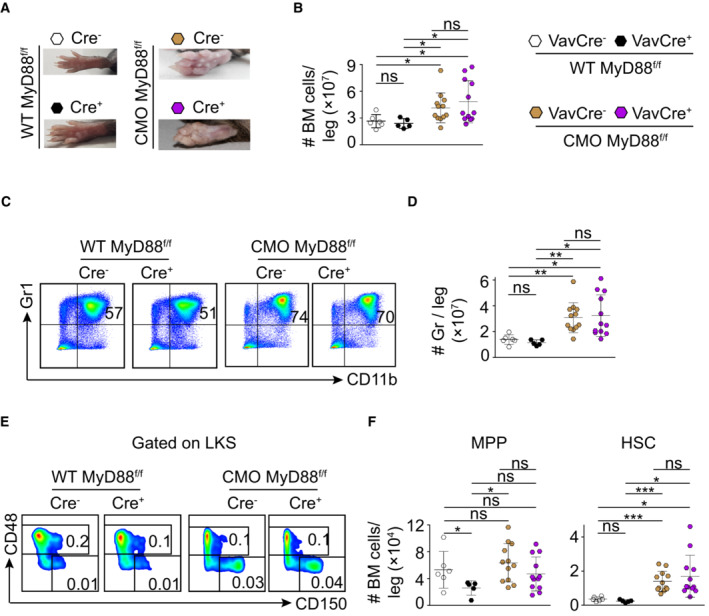
- Picture of representative paws from WT MyD88flox/flox VAV‐iCre− (white symbol), WT MyD88flox/flox VAV‐iCre+ (black symbol), CMO MyD88flox/flox VAV‐iCre− (brown symbol), and CMO MyD88flox/flox VAV‐iCre+ (purple symbol) mice.
- Absolute number of BM cells per leg.
- Representative flow cytometry plots from BM cells isolated from the indicated mouse genotypes. Y‐axes indicate Gr1 expression and x‐axes CD11b expression. Numbers indicate the percentage of Gr1+ CD11b+ granulocytes.
- Quantification of the absolute number of granulocytes per leg.
- Representative flow cytometry plots from indicated mouse genotypes. Y‐axes indicate CD48 expression and x‐axes CD150 expression in the LKS population. Numbers indicate the percentage of MPPs (upper box) and HSCs (lower box) in total BM.
- Quantification of the absolute number of MPPs and HSCs per leg. Y‐axes indicate the number of MPPs (left panel) and HSCs (right panel) per leg.
Data information: In this figure, each symbol indicates values for one biological replicate (mouse). At least five mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant).
Figure 4. CMO/MyD88−/− double‐mutant mice retain HSC expansion and functional defects.
-
ARepresentative paw and spleen pictures from each experimental group: WT, MyD88−/−, CMO, and CMO/MyD88−/− mice.
-
BAbsolute number of BM cells per leg. Each symbol represents a biological replicate (mouse). At least eight mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (****P < 0.0001, ns, not significant).
-
CSpleen weight (mg). Each symbol indicates values for one biological replicate (mouse). At least seven mice were used per group in two separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (***P < 0.001, ns, not significant).
-
DRepresentative flow cytometry plots from WT, MyD88−/−, CMO, and CMO/MyD88−/− mice. Plots illustrate the expression of Gr1 (Y‐axes) and CD11b (X‐axes) in BM. Numbers indicate the percentage of Gr1+ CD11b+ granulocytes.
-
EQuantification of the absolute number of granulocytes per leg. Each symbol indicates values for one biological replicate (mouse). At least eight mice were used per group in three separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (****P < 0.0001, ns, not significant).
-
FRepresentative flow cytometry plots from WT, MyD88−/−, CMO, and CMO/MyD88−/− mice. Left panels: Y‐axes indicate c‐Kit expression and x‐axes Sca‐1 expression in lineage− cells. Box indicates LKS population. Right panels: y‐axes indicate CD48 expression and x‐axes CD150 expression in the LKS population. Boxes represent MPPs (upper box), and HSCs (lower box). Numbers indicate the percentage of total BM.
-
G, HPercentage (G) and absolute number (H) of MPPs and HSCs per leg. Blue symbols indicate WT, green MyD88−/−, orange CMO, and red CMO/MyD88−/− double‐mutant mice. Each symbol represents a 20‐week‐old mouse (biological replicates). At least eight mice were used per group in three separate experiments. Data represent mean ± s.d. Statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant).
-
ITransplantation scheme of CMO and CMO MyD88−/− HSCs.
-
JFrequency of functional CMO (orange) and CMO MyD88−/− (red) HSCs measured by limiting dilution competitive repopulation unit assays and calculated using ELDA online software based on Poisson distribution statistics (Chi‐square test; Chisq = 0.00488; P = 0.944). Graph shows the curve fit of the log fraction of nonresponding mice (solid lines) and confidence intervals (dashed lines) versus the number of mice tested. Logarithmic plot; X‐axis indicates the dose of transplanted cells and Y‐axis percentages of negative responders. Reconstitution was evaluated in blood of recipient mice 16 weeks after transplantation. A responder mouse was defined as engraftment ≥ 0.5% Ly5.2+ cells and contribution ≥ 0.5% in at least two out of three lineages (granulocytes, B‐cells, and T‐cells).
The CMO BM niche imprints a myeloid and pro‐inflammatory profile in HSCs
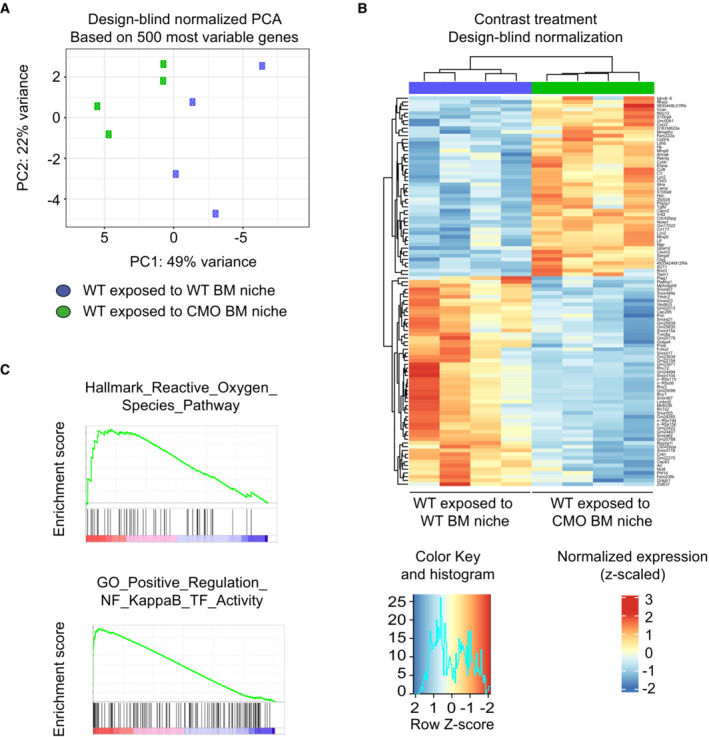
To better understand how the CMO niche affects the functionality of HSCs, we performed gene expression profile analysis of WT HSCs exposed to WT or CMO BM niche (Fig 2C). WT HSCs transplanted into lethally irradiated 10‐week‐old WT or CMO recipients were sorted 12 weeks after transplantation and subjected to RNA sequencing (RNA‐seq). The principal component analysis demonstrated a certain degree of variability among samples (Fig EV4A); however, unsupervised cluster analysis gathered HSCs exposed to WT BM niche to one clade and HSCs exposed to CMO BM niche to another clade (Fig EV4B). We identified 104 genes differentially expressed between WT HSCs exposed to WT or CMO BM niche (P‐value < 0.05, log2 fold change > 0.585, FDR < 0.01; Fig 5A; Dataset EV1). In particular, 48 genes were upregulated and 56 genes were downregulated in WT HSCs exposed to CMO niche. Further, enrichment analysis showed top relevant pathways upregulated in WT HSCs exposed to CMO niche (Figs 5B–E and EV4C). Among those, several pathways related to myeloid inflammatory responses and myeloid differentiation/activation were identified, and particularly interesting was the upregulation of a myeloid immunity signature and of the IL‐6/Jak/Stat3 signaling pathway (Fig 5F). Altogether, these data suggest that the CMO BM niche can affect HSC fitness by activating an inflammatory response and imposing expression of myeloid markers in HSCs.
Figure EV4. RNA‐seq analysis of WT HSCs exposed to WT recipient mice (WT exposed to WT BM niche) or WT HSC exposed to CMO recipient mice (WT exposed to CMO BM niche).
-
APrincipal component analysis of four samples of WT HSC exposed to WT recipient mice (WT exposed to WT BM niche, blue symbols) and four samples of WT HSC exposed to CMO recipient mice (WT exposed to CMO BM niche, green symbols).
-
BHeatmap of unsupervised hierarchical cluster analysis of genes differentially expressed in WT HSC exposed to WT or CMO recipient mice (P < 0.05, log2 fold change > 0.5). Data is normalized to z‐scores for each gene. Red color indicates increased and blue color decreased gene expression in comparison to the universal mean for each gene.
-
CRepresentative enrichment plots showing upregulated pathways in WT HSC exposed to CMO recipient mice from MSigDB Hallmark gene set v.7 and MSigDB GO Biological Process gene set v.7.
Figure 5. CMO BM niche induces activation of pathways involved in myeloid differentiation and inflammation in HSCs.
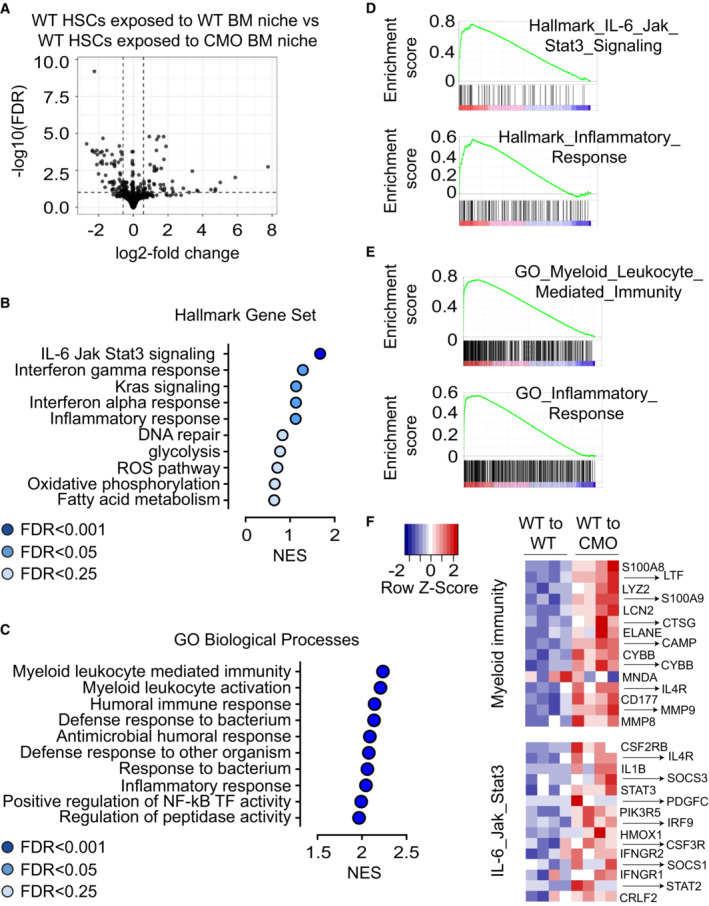
-
AVolcano plot showing the differentially expressed genes in WT HSCs exposed to WT BM niche compared with WT HSCs exposed to CMO BM niche.
-
B, CGene Set Enrichment Analysis (GSEA) showing top relevant pathways upregulated in WT HSCs exposed to CMO BM niche compared with WT HSCs exposed to WT BM niche. Data were generated using (B) MSigBD Hallmark gene set v.7 (ranked according to NES values, FDR < 0.25) and (C) and MSigDB GO Biological Processes gene set v.7 (ranked according to NES values, FDR < 0.00001).
-
D, ERepresentative enrichment plots showing (D) two upregulated pathways in WT HSC exposed to CMO recipient mice from MSigDB Hallmark gene set v.7 and (E) two from MSigDB GO Biological Process gene set v.7.
-
FHeatmaps of unsupervised hierarchical cluster analysis of the top 15 genes differentially expressed in the Hallmark myeloid immunity signature (upper heatmap) and top 15 genes differentially expressed in the GO IL‐6/Jak/Stat3 signaling pathway (lower heatmap) in WT HSC exposed to WT or CMO recipient mice (P < 0.05, log2 fold change > 0.5). Data is normalized to z‐scores for each gene. Red color indicates increased and blue color decreased gene expression in comparison to the universal mean for each gene.
CMO mice exhibit increased levels of IL‐6, elevated pStat3 in HSCs, and increased HSC survival
Since the RNA‐seq analysis indicated that the IL‐6/Jak/Stat3 signaling pathway was upregulated in WT HSC exposed to the CMO BM niche, we first assessed IL‐6 levels in WT and CMO mice, as well as in transplanted WT and CMO recipients. We observed that the amount of IL‐6 was significantly higher in serum and BM of CMO mice compared with WT (Fig 6A), and a similar tendency was observed in CMO mice upon transplantation (Fig EV5A). Thus, we next investigated the effect of IL‐6 stimulation on the levels of pStat3 in WT HSC and observed that IL‐6 stimulation upregulated pStat3 levels in WT HSCs (Fig 6B). On the contrary, IL‐1β, which was reported to be elevated in CMO mice and responsible for the CMO inflammatory phenotype (Cassel et al, 2014; Lukens et al, 2014a; Drobek et al, 2015), failed to trigger Stat3 phosphorylation in HSCs (Fig 6B). Next, we verified the RNA‐seq results by phospho‐flow analysis. We observed a significant upregulation of pStat3 levels in WT HSCs exposed to CMO BM niche in comparison to WT HSCs exposed to WT BM niche (Fig 6C and D), and accordingly, we observed upregulation of pStat3 target genes in WT LKS cells isolated from CMO recipients (Fig EV5B). Further, we investigated whether pStat3 was similarly increased in CMO mice. In line with the results in the transplantation setting (Fig 6C and D), HSCs from CMO mice exhibited significantly higher pStat3 levels than WT control HSCs (Fig 6E and F). We also assessed whether the increase in pStat3 levels was exclusive to the HSC pool in CMO mice. Western blot and phospho‐flow analysis using c‐Kit+ BM cells demonstrated that pStat3 was also increased in c‐Kit+ myeloid progenitors (Fig EV5C and D). Since the IL‐6/Jak/Stat3 signaling pathway favors cell survival (Gilbert & Hemann, 2010, 2012), we next assessed HSC viability in WT and CMO mice. Indeed, we observed that the percentage of Annexin V+ apoptotic cells was lower in stem and progenitor cells (including LKS, MPP, and HSC) from CMO mice in comparison to WT controls (Fig 6G and H). Altogether, these data suggest that the augmented levels of IL‐6 detected in CMO mice may contribute to the increased pStat3 levels in CMO HSCs and promote HSC survival.
Figure 6. CMO HSCs exhibit increased IL‐6/Jak/Stat3 signaling and reduced apoptosis.
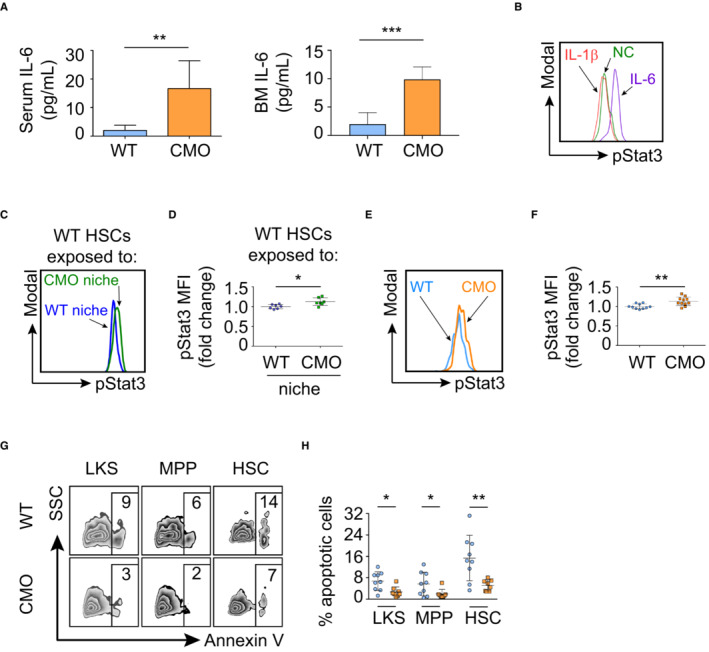
-
AIL‐6 levels in serum and BM of WT (n = 6) and CMO (n = 8) symptomatic mice. Y‐axis indicates the amount of IL‐6 (pg/ml). Data represent mean ± s.d.
-
BRepresentative flow cytometry histograms of pStat3 signal in WT HSCs stimulated with IL‐1β (red), IL‐6 (purple), or nonstimulated control (NC, green).
-
CRepresentative flow cytometry histograms of pStat3 signal from WT HSCs exposed to WT (blue) or to CMO (green) niche.
-
DQuantification of panel (C). Y‐axis indicates pStat3 mean fluorescence intensity (MFI). Values are normalized to the average of the WT. Each symbol indicates values for one biological replicate (mouse). At least seven mice were used per group in three separate experiments. Data represent mean ± s.d.
-
ERepresentative flow cytometry histograms of pStat3 signal from WT (blue) and CMO (orange) HSCs.
-
FQuantification of panel (E). Y‐axis indicates pStat3 mean fluorescence intensity (MFI). Values are normalized to the average of the WT. Each symbol represents one biological replicate, either a WT mouse (blue) or a CMO mouse (orange). At least 10 mice were used per group in three separate experiments. Data represent mean ± s.d.
-
GRepresentative flow cytometry plots from WT (upper plots) and CMO (lower plots) BM cells. Numbers indicate the percentage of Annexin V+ cells in distinct BM populations.
-
HQuantification of panel (G). Y‐axis indicates the percentage of Annexin V+ cells and x‐axis the distinct BM subpopulations. Each symbol represents one biological replicate (WT mice in blue and CMO mice in orange). At least eight mice were used per group in three separate experiments. Data represent mean ± s.d.
Data information: In this figure, statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ***P < 0.001).
Figure EV5. Analysis and inhibition of IL‐6 and Stat3 in vivo .
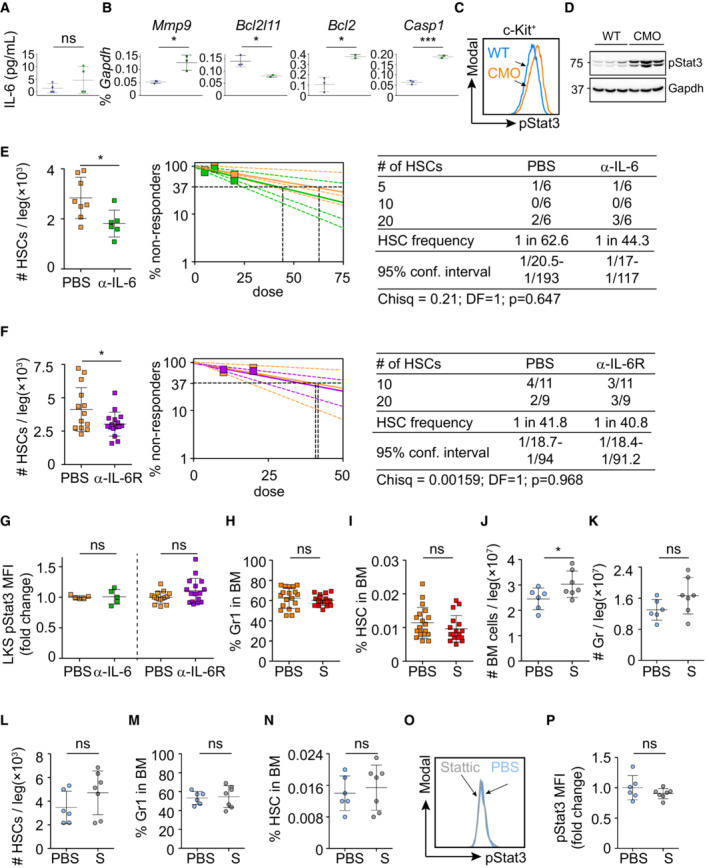
-
AIL‐6 levels in BM of WT (n = 4) and CMO (n = 4) recipient mice upon transplantation of WT BM cells. Y‐axis indicates the amount of IL‐6 (pg/ml).
-
BQuantitative RT–PCR in WT LKS cells exposed to WT (blue symbols) or CMO (green symbols) BM niche. The y‐axes represent Mmp9, Bcl2l11, Bcl2, and Casp1 expression relative to Gapdh.
-
CRepresentative flow cytometry histograms of pStat3 signal from WT (blue) and CMO (orange) c‐Kit+ cells.
-
DWestern blot analysis for pStat3 in WT and CMO c‐Kit+ cells. Gapdh expression was used as a loading control. Positions of m.w. standards are indicated (KDa). (E and F right panels) HSCs are measured by limiting dilution competitive repopulation unit assays and calculated using ELDA online software based on Poisson distribution statistics. Graphs show the curve fit of the log fraction of nonresponding mice (solid lines) and confidence intervals (dashed lines) versus the number of mice tested. Logarithmic plot; X‐axis indicates the dose of transplanted cells and Y‐axis percentages of negative responders. Reconstitution was evaluated in blood of recipient mice 16 weeks after transplantation. A responder mouse was defined as engraftment ≥ 0.5% Ly5.2+ cells and contribution ≥ 0.5% in at least two out of three lineages (granulocytes, B‐cells, and T‐cells).
-
ENumber of phenotypically enumerated HSCs per leg (left panel) and frequency of functional HSCs (right panel) in CMO mice treated with PBS control (orange) or IL‐6‐blocking antibody (green).
-
FNumber of phenotypically enumerated HSCs per leg (left panel) and frequency of functional HSCs (right panel) in CMO mice treated with PBS control (orange) or IL‐6 receptor‐blocking compound (purple).
-
GpStat3 levels in LKS cells isolated from CMO mice treated as indicated. Y‐axis indicates pStat3 mean fluorescence intensity (MFI). Values are normalized to the average of PBS treatment.
-
H, IPercentage of granulocytes (H) and HSCs (I) in BM of CMO mice treated with PBS control or Stattic.
-
J–LAbsolute number of cells (J), granulocytes (K), and HSCs (L) per leg isolated from WT mice treated with PBS control (blue) or Stattic (S; gray).
-
M, NPercentage of granulocytes (M) and HSCs (N) in BM isolated from WT mice treated with PBS control (blue) or Stattic (S; gray).
-
ORepresentative flow cytometry histograms of pStat3 signal in LKS cells from WT mice treated with PBS control (blue) or Stattic (gray).
-
PQuantification of panel (K). Y‐axis indicates pStat3 mean fluorescence intensity (MFI). Values are normalized to the average of the WT. In this figure, each symbol represents value for one biological replicate. Data represent mean ± s.d. Unless otherwise indicated, statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, ns, not significant).
pStat3 inhibition prevents HSC expansion and restores HSC fitness in CMO mice
Since our data pointed at IL‐6 as a contributing cytokine to HSC defects, we next investigated whether blockage of IL‐6 activity in CMO mice would be sufficient to prevent the detrimental effects in HSCs. Thus, CMO mice were treated with IL‐6‐ or IL‐6 receptor‐blocking antibodies, and the effects on HSC numbers and activity were determined. We observed that IL‐6‐ and IL‐6 receptor‐blocking antibodies reduced the number of HSCs in CMO mice in comparison to the vehicle control‐treated animals (Fig EV5E and F). However, limiting dilution transplantation assays demonstrated that neither of the approaches was sufficient to rescue the CMO HSC functional defects (Fig EV5E and F). In line with the lack of a functional rescue, we observed that Stat3 phosphorylation was not reduced upon antibody treatment (Fig EV5G). Since the Jak/Stat3 signaling pathway can be activated by other cytokines (McLemore et al, 2001; Yu et al, 2009; Nguyen et al, 2015), we next investigated whether pStat3 directly contributes to the expansion and reduced fitness of HSCs. CMO mice were treated with PBS control or a Stat3 inhibitor, referred to as Stattic (Li et al, 2020), which inhibits Stat3 phosphorylation (Fig 7A). As expected, Stattic‐treated CMO mice developed signs of inflammation such as swollen paws (Fig 7B), symptoms mainly driven by IL‐1β, a cytokine not affected by Stattic administration. Similarly, Stattic‐treated CMO mice retained the increased BM cellularity and expansion of granulocytes (Figs 7C and D, and EV5H). However, Stattic‐treated CMO mice had significantly fewer HSCs in their BM than the PBS‐treated CMO control group (Figs 7E and F, and EV5I). Further, limiting dilution transplantation assays demonstrated improved CMO HSC activity upon Stat3 inhibition in comparison to PBS treatment (Fig 7G and H). Phospho‐flow analysis verified a decrease in pStat3 levels in the LKS compartment in Stattic‐treated CMO mice (Fig 7I and J). Of note, in WT mice, Stattic treatment did not affect the number of HSCs and pStat3 levels (Fig EV5J–P). Altogether, our data indicate that despite the possible contribution of IL‐6 to the CMO HSC detrimental effects, specific blockage of this cytokine/receptor was not sufficient to rescue HSC functional defects, while inhibition of the downstream Jak/Stat3 signaling pathway reduced HSC numbers and improved HSC fitness in CMO mice.
Figure 7. Stat3 inhibition prevents the expansion of CMO HSCs and rescues HSC fitness.
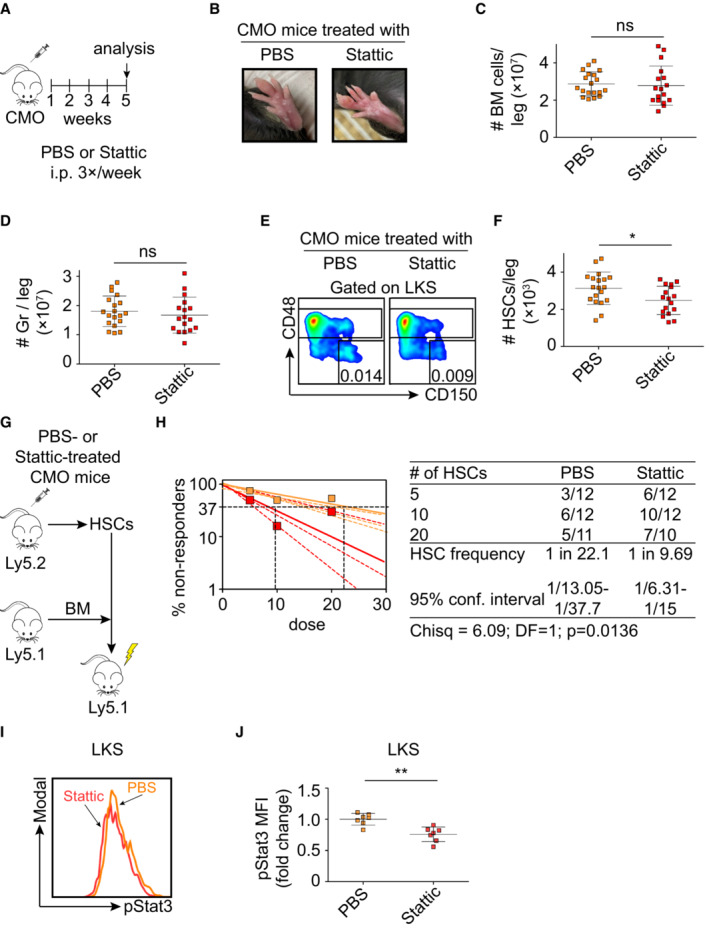
-
AScheme of Stattic treatment.
-
BPictures of representative paws from CMO mice treated with PBS control or Stattic. Pictures were taken on the last day of the experiment before sacrificing the mice.
-
C, DAbsolute number of BM cells (C) and granulocytes (D) in CMO mice. Y‐axes indicate the number of cells per leg. X‐axes indicate PBS (orange) or Stattic treatment (red). Each symbol indicates values for one biological replicate (mouse). At least 17 mice were used per group in three separate experiments. Data represent mean ± s.d.
-
ERepresentative flow cytometry plots from PBS‐ (left plot) and Stattic‐treated (right plot) CMO mice.
-
FQuantification of the absolute number of HSCs per leg. Each symbol represents one biological replicate (PBS‐treated CMO mouse, orange, and Stattic‐treated CMO mouse, red). Data represent mean ± s.d. At least 17 mice were used per group in three separate experiments.
-
GSchematic representation of the experimental design.
-
HFrequency of functional HSCs in CMO mice after treatment with Static (red) or PBS control (orange). Graph shows the curve fit of the log fraction of nonresponding mice (solid lines) and confidence intervals (dashed lines) versus the number of mice tested. Logarithmic plot shows the percentage of nonresponder recipient mice. Responders exhibited engraftment of ≥ 0.5% Ly5.1+ cells and contribution ≥ 0.5% in at least two out of three lineages (granulocytes, B‐cells, and T‐cells) at week 16 post‐transplantation. X‐axis indicates the dose of transplanted cells. Right table shows the number of responders and the total number of recipients transplanted per cell dose. Frequencies of HSCs were calculated using ELDA online software based on Poisson distribution statistics (Chi‐square test; Chisq = 6.09; P = 0.0136).
-
IRepresentative flow cytometry histograms of pStat3 signal from PBS‐ or Stattic‐treated CMO mice.
-
JQuantification of panel (I). Y‐axis indicates pStat3 mean fluorescence intensity (MFI). Values are normalized to the average of the PBS‐treated CMO mice. Each symbol represents values for a biological replicate (mouse). At least seven mice were used per group in two separate experiments. Data represent mean ± s.d.
Data information: In this figure statistical significance was assessed using two‐tailed Student's t‐tests (*P < 0.05, **P < 0.01, ns, not significant), unless otherwise indicated.
Discussion
The effects of infection and chronic inflammation on HSCs have mostly been studied in WT mice treated with particular stimuli, such as Mycobacterium avium (Baldridge et al, 2010; Matatall et al, 2016), pI:pC (Walter et al, 2015; Bogeska et al, 2022), viruses (Isringhausen et al, 2021), LPS (Esplin et al, 2011; Zhao et al, 2013), IL‐1 (Pietras et al, 2016), IFNα (Essers et al, 2009), or collagen (Hernandez et al, 2020). These studies reported that the effects of chronic inflammation on HSCs can vary from detrimental to considerably mild. Here, we employed a model of a progressive autoinflammatory disorder based on mice harboring a naturally occurring mutation that leads to sterile chronic inflammation. Thus, we were able to bypass the artificial setup of externally inducing inflammation and assess the consequences of a spontaneously developed inflammatory condition. This approach gave us a unique insight into the interactions of the inflammatory BM niche, immune cells, and HSCs. We showed that both hematopoietic cells and BM niche contributed to the expansion and functional impairment of HSCs. In line with our study, the KRNxG7 mouse model of rheumatoid arthritis also displayed myeloid skewing (Oduro Jr et al, 2012); however, KRNxG7 mice did not exhibit defects in HSCs (Ma et al, 2009). We speculate that this difference can be explained by the particular inflammatory profile of each disease model.
CMO mice exhibit increased levels of pro‐inflammatory mediators, such as MIP‐1α, IL‐1α, IL‐1β, IL‐6, IFNγ, MCP‐1, G‐CSF, and M‐CSF (Chitu et al, 2009, 2012; Cassel et al, 2014; Lukens et al, 2014a; Drobek et al, 2015). Remarkably, the whole CMO phenotype reported so far was dependent on the hyperproduction of IL‐1β, and the CMO defects could be fully rescued by the inactivation of the IL‐1β signaling pathway (Cassel et al, 2014; Lukens et al, 2014a). Interestingly, previous studies showed the effect of IL‐1 on HSCs (Heimfeld et al, 1991; Orelio et al, 2008) and Pietras et al (2016) demonstrated that chronic exposure of mice to IL‐1β compromised the self‐renewal ability of murine HSCs. Nevertheless, in sharp contrast to the critical role of IL‐1β in the development of the CMO phenotype, here we observed that the effects on HSCs occur in an IL‐1β independent fashion. On one hand, we demonstrated that hematopoietic cell‐specific deletion of MyD88 adaptor protein, an essential molecule in the IL‐1β signaling pathway, had no effect on the CMO disease onset nor on the expansion of HSCs. On the other hand, CMO mice with whole‐body MyD88 deletion did not develop signs of the autoinflammatory disorder; however, these mice still exhibited an expansion of the HSC compartment. These observations indicate that signals originating in the nonhematopoietic BM niche drive HSC expansion under chronic inflammatory conditions, although strikingly, independently of the pro‐inflammatory cytokine IL‐1β.
IL‐6 is a multifunctional cytokine whose production is enhanced during inflammation. Here, we demonstrated that IL‐6 levels are elevated in CMO mice and that IL‐6 can directly act on HSCs and increase the levels of pStat3, promoting myelopoiesis, survival, and expansion of the HSC pool. Nevertheless, inhibition of IL‐6 activity, either by using IL‐6‐ or IL‐6 receptor‐blocking antibodies, although able to reduce HSC numbers in CMO mice, was not sufficient to restore the functional defects in HSCs. These observations suggest that additional factors may contribute to HSC alterations under chronic inflammatory conditions, and future studies should address this point. Remarkably, an additional aspect that could contribute to HSC alterations in CMO mice is the microbiota. It was previously published that CMO mice were protected from developing an inflammatory phenotype when fed with a diet high in fat and cholesterol and that these effects were mediated by changes in the intestinal microbiota (Lukens et al, 2014b). In fact, a large body of evidence supports the role of microbiota in the pathogenesis of inflammatory disorders (Clarke et al, 2010; Maloy & Powrie, 2011; Koh & Kim, 2017; Tajik et al, 2020), and thus, future research should determine whether the microbiome composition can also affect the HSC pool under chronic inflammatory conditions.
We demonstrated that the nonhematopoietic compartment in the BM can imprint a pro‐inflammatory profile in HSCs exposed to a chronic inflammatory context. Using RNA‐seq in WT HSCs exposed to the CMO niche, we reported the activation of IFNα and IFNγ responses, NF‐κΒ activity, and elevated ROS pathway, which was previously reported to regulate HSC activity (Ito et al, 2006; Baldridge et al, 2010; King & Goodell, 2011; Mirantes et al, 2014; Poulos et al, 2016; Nakagawa et al, 2018). Particularly interesting was the activation of the IL‐6/Jak/Stat3 signaling pathway, since we and others reported its critical role in stressed‐induced myelopoiesis (Maeda et al, 2005, 2009; Danek et al, 2020), a hallmark of the CMO phenotype. As mentioned above, blocking IL‐6 activity was not sufficient to restore the HSC defects in CMO mice; however, the Jak/Stat3 signaling pathway can be activated by distinct cytokines (McLemore et al, 2001; Yu et al, 2009; Nguyen et al, 2015). Accordingly, using Stat3 inhibitors, which are currently in clinical trials (Beebe et al, 2018), we demonstrated that Stat3 blockage was sufficient to prevent HSC expansion and rescue HSC fitness in CMO mice, enforcing the idea that in addition to IL‐6, other factors activating the Jak/Stat3 signaling pathway contribute to detrimental effects observed under chronic inflammatory conditions in HSCs. In fact, we also observed that upon transplantation of WT BM cells into CMO recipients, the levels of IL‐6 were not elevated in all CMO recipients, supporting the notion that additional cytokines are involved in activating the Jak/Stat3 signaling pathway. Thus, future studies should determine which additional factors in the CMO mouse model contribute to the activation of this signaling pathway in HSCs.
Technically, we need to consider that we employed lethal irradiation of WT and CMO recipient mice to perform BM transplantation studies and determine the role of the niche on WT HSCs. Thus, a potential pitfall of this approach is that the irradiation may affect cellular components from the niche or induce a certain degree of inflammation that could affect the BM reconstitution upon transplantation (Cao et al, 2011; Green et al, 2013; Severe et al, 2019). Future experiments assessing the role of WT or CMO BM niche on HSCs may benefit from nonmyeloablative strategies to condition the recipient mice (Hsieh et al, 2007; Du et al, 2018).
CMO mice share many features with CRMO patients, including elevated levels of cytokines and chemokines in serum such as IL‐1β and IL‐6 (Chitu et al, 2009; Hofmann et al, 2016). Patients diagnosed with CRMO are mostly children, as the median age of diagnosis is 10 years old. CMRO is a type of nonbacterial osteomyelitis, characterized by bone pain and fever. Its etiology is unknown, but it is unlikely caused by infections or autoimmunity. Current treatment consists of nonsteroidal anti‐inflammatory drugs (NSAIDs), corticosteroids, and disease‐modifying anti‐rheumatic drugs (DMARDs; Hofmann et al, 2017). Our data indicate that the CMO phenotype is mediated by two distinct mechanisms; (i) the IL‐1β/MyD88 signaling pathway, responsible for the swollen paws, tail nodes, increased BM cellularity, and elevated number of granulocytes, and (ii) the Jak/Stat3 signaling pathway, in part activated by IL‐6, and responsible for HSC alterations. We observed that by treating CMO mice with Stattic, a Stat3 inhibitor, we were able to prevent HSC expansion, a condition reported in the context of chronic inflammation in humans (Baylis et al, 2013; Cook et al, 2019; Bick et al, 2020; Caiado et al, 2021), and to rescue HSC fitness. Remarkably, Stattic administration did not affect the BM composition, including the HSC pool, in WT mice. Interestingly, the distinct effects of Stattic in WT and CMO mice could be explained by the different levels of Stat3 activation in WT and CMO HSCs, which would point to a therapeutic window. Thus, our data open new venues of intervention for particular autoinflammatory disorders in which a combination of anti‐inflammatory drugs and Stat3 inhibitors would aim at reducing inflammation and preserve HSC activities, respectively.
Materials and Methods
Animal models
Three types of CMO mice were used in this study: Pstpip2 wt/wt (WT controls), Pstpip2 cmo/cmo (experimental group lacking the Pstpip2 protein and referred to as CMO mice), and Pstpip2 wt/cmo (used for colony maintenance). CMO mice were 4‐ to 7‐weeks‐old when they are still asymptomatic. CMO symptomatic mice were 20‐ to 25‐weeks‐old and exhibited swollen paws, tail kinks, and inflamed ears. CMO mice were crossed to whole‐body MyD88−/− mice and generated Pstpip2 wt/wt MyD88 −/− (control), Pstpip2 cmo/cmo MyD88 −/− (CMO mice lacking MyD88 adaptor protein) and Pstpip2 wt/cmo MyD88 −/− (used for colony maintenance). Finally, CMO mice were crossed to MyD88flox/flox VAV‐iCre mice generating CMO MyD88flox/flox VAV‐iCre− and CMO MyD88flox/flox VAV‐iCre+ mice. Mice were maintained in the animal facility of the Institute of Molecular Genetics of the CAS. All experiments were approved by the ethical committee of the institute.
Flow cytometric analysis and HSC sorting
WT and CMO mice were sacrificed by cervical dislocation, femurs and tibias were isolated, and crunched using pestle and mortar, or in the case of the spleen, processed with syringe pistons. After obtaining single‐cell suspensions, red blood cells were lysed. Cells were then labeled with fluorescence‐conjugated antibodies and analyzed on LSRII or Symphony instruments (BD Biosciences, San Jose, CA, USA). Antibodies that were used for phenotypic analysis: Gr1 APC (RB6‐8C5), CD11b FITC (M1/70), anti‐mouse Lineage Cocktail Pacific Blue (including CD3 (17A2); Gr1 (RB6‐8C5); CD11b (M1/70); CD45R/B220 (RA3‐6B2); TER‐119 (Ter‐119)), c‐Kit PE (2B8), Sca‐1 APC (D7), FcγRII/III Pe‐Cy7 (93), CD34 FITC (RAM 34), CD48 FITC (HM48‐1), CD150 Pe‐Cy7 (TC15‐12F12.2). Transplanted mice were analyzed using Ly5.1 FITC (A20), Ly5.2 PE‐Cy7 (104), Gr1 APC (RB6‐8C5), CD11b APC (M1/70), CD45/B220 APC, CD45/B220 PE (RA3‐6B2), and CD3ε PE (145‐2C11). Analysis of the pStat3 and IkB‐α levels was done using anti‐mouse Lineage Cocktail Pacific Blue (including CD3 (17A2); Gr1 (RB6‐8C5); CD11b (M1/70); CD45R/B220 (RA3‐6B2); TER‐119 (Ter‐119)), c‐Kit APC (2B8), Sca‐1 BV650 (D7), CD48 FITC (HM48‐1), CD150 Pe‐Cy7 (TC15‐12F12.2). Preparation of BM HSCs for sorting and transplantation was a two‐step process. First, the Lin+ fraction of the BM cells was labeled using biotinylated lineage markers: CD45/B220 (RA3‐6B2), CD3 (145‐2c11), Ter119 (TER‐119), Gr1 (RB6‐8C5), and CD11b (M1/70). These cells were further labeled with anti‐biotin magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) and depleted on a MACS separator (Miltenyi Biotec) according to the manufacturer's protocol. Second, the Lin− fraction of the BM was labeled with the following antibodies: c‐Kit (2B8), Sca‐1 APC (E13‐161.7), CD48 FITC (HM48‐1), CD150 Pe‐Cy7 (TC15‐12F12.2), and streptavidin‐eFluor450. Influx instrument (BD Biosciences) was employed to sort HSCs according to the following sorting strategy (Danek et al, 2020): Lin−, c‐Kit+, Sca‐1+, CD48− CD150+. For secondary transplantations, biotinylated Ly5.1 or Ly5.2 were added to antibody premixes. In all flow cytometric analyses, except pStat3 analysis in fixed cells, Hoechst 33258 was added to cell suspensions to exclude dead cells. Antibodies were purchased from BD Biosciences, eBioscience (San Diego, CA, USA), BioLegend (San Diego, CA, USA), or Exbio (Praha, Vestec, Czech Republic). Data were obtained using Diva software (BD Biosciences) and analyzed using FlowJo software (Tree Star Incorporation, Ashland, OR, USA).
HSC limiting dilution transplantation assays
Donor strains used in these assays expressed Ly5.2, and congenic C57BL/6 mice used as recipients expressed Ly5.1. Unless otherwise indicated, mice employed were 8–12 weeks old. Up to three different doses of HSCs (5, 10, and 20), defined as LKS CD48− CD150+ were sorted and intravenously transplanted together with 5 × 105 WT BM (Ly5.1+) support cells. Recipient mice were lethally irradiated with 6 Gy prior to transplantation. Peripheral blood (PB) and BM were analyzed 16 weeks post‐transplantation. Cells were stained with antibodies against Ly5.1 and Ly5.2 to distinguish the donor‐derived from support cells. Additionally, lineage‐specific antibodies (B220, CD3, CD11b, and Gr1), were employed to assess the reconstitution of B‐cells, T‐cells, and myeloid cells, respectively. A recipient mouse was defined as positive if the engraftment of donor cells was ≥ 0.5% Ly5.2+ cells contributing to at least two out of the three lineages. The frequency of HSCs was calculated with ELDA online software (Hu & Smyth, 2009) using Poisson statistics and the method of maximum likelihood to the proportion of negative recipients in a limiting dilution setting.
Exposure of WT BM to WT or CMO hematopoietic cells in vivo
Twenty‐week‐old C57BL/6 (Ly5.2+) WT or CMO whole BM was isolated and mixed with C57BL/6 (Ly5.1+) whole BM in a ratio of 10:1 and transplanted intravenously into lethally irradiated recipient C57BL/6 (Ly5.1+) mice. Twelve weeks later, donor Ly5.1+ HSCs, defined as LKS CD48− CD150+ were isolated and transplanted into lethally irradiated secondary C57BL/6 (Ly5.2+) recipient mice. A single dose of 20 HSCs was used, together with 0.5 × 106 of support cells (Ly5.2). PB and BM of recipient mice were analyzed 16 weeks post‐transplantation. Cells were stained with Ly5.1 and Ly5.2 antibodies to distinguish donor and support cells as well with B220, CD3, CD11b, and Gr1 to determine tri‐lineage reconstitution. A recipient mouse was defined as positive if the engraftment of donor cells was ≥ 0.01% Ly5.1+ cells contributing to at least one out of the three lineages. Unless otherwise indicated, mice employed were 8–12 weeks old.
Exposure of WT BM to WT or CMO BM niche in vivo
C57BL/6 (Ly5.1+) mouse was used as a donor, and C57BL/6 (Ly5.2+) WT and CMO mice were used as recipients. A single dose of 3 × 106 donor cells was injected intravenously into lethally irradiated (6 Gy) recipients. Twelve weeks later, donor Ly5.1+ cells were isolated and transplanted into lethally irradiated secondary C57BL/6 (Ly5.2+) recipients. Two doses were used for secondary transplantation: 0.5 × 106 or 1.5 × 106 were transplanted per recipient mouse, together with 0.5 × 106 competitor (Ly5.2+) BM cells. PB was analyzed 8 and 16 weeks post‐transplantation, and BM was analyzed 16 weeks post‐transplantation. Cells were stained with Ly5.1 and Ly5.2 antibodies to distinguish donor and support cells as well with B220, CD3, CD11b, and Gr1 to determine tri‐lineage reconstitution. Separately, WT Ly5.1+ HSCs, defined as LKS CD48− CD150+ were isolated from primary recipients and transplanted into secondary C57BL/6 (Ly5.2+) recipients. Three different doses of HSCs (20, 40, and 100) were sorted and transplanted intravenously, together with 0.5 × 106 WT BM (Ly5.2+) support cells. PB and BM of recipient mice were analyzed 16 weeks post‐transplantation. Cells were stained with Ly5.1 and Ly5.2 antibodies to distinguish donor and support cells as well with B220, CD3, CD11b, and Gr1 to determine tri‐lineage reconstitution. A recipient mouse was defined as positive if the engraftment of donor cells was ≥ 0.01% Ly5.1+ cells contributing to at least one out of the three lineages. Unless otherwise indicated, mice employed were 8–12 weeks old.
RNA sequencing and data analysis
HSCs, defined as Lin− c‐Kit+ Sca‐1+ CD48− CD150+, were sorted using an Influx instrument (BD Biosciences), lysed with Tri Reagent RT (Molecular Research Center, Cincinnati, OH, USA), and RNA was extracted with RNEasy MinElute Cleanup Kit (Qiagen, Venlo, Netherlands). Libraries (300 pg mRNA per sample) were prepared with SMARTer® Stranded Total RNA‐Seq—Pico Input Mammalian library preparation kit v2 (Takara). Library size distribution was evaluated on the Agilent 2100 Bioanalyzer using the High Sensitivity DNA Kit (Agilent). Libraries were sequenced on the Illumina NextSeq® 500 instrument using 75 bp single‐end high output configuration. Read quality was assessed by FastQC. For subsequent read processing, a bioinformatic pipeline nf‐core/rnaseq version 1.3 was used. Individual steps included removing sequencing adaptors with Trim Galore, mapping to reference genome GRCm38 (Ensembl annotation version 94) with HISAT2, and quantifying expression on gene level with featureCounts. Counts mapped per gene served as input for differential expression analysis using DESeq2 R Bioconductor package. Prior to the analysis, genes not expressed in at least two samples were discarded. Shrunken log2‐fold changes using the adaptive shrinkage estimator were used for differential expression analysis. Genes exhibiting minimal absolute log2‐fold change value of 0.585 (1.5 fold change) and statistical significance (adjusted P‐value < 0.1) between compared groups of samples were considered as differentially expressed for subsequent interpretation and visualization.
ELISA assays
IL‐6 ELISA
Serum was collected from 20‐ to 28‐week‐old WT and CMO mice and the levels of IL‐6 were measured using the Invitrogen™ eBioscience™ Mouse IL‐6 ELISA Ready‐SET‐Go!™ Kit, catalog number (88‐7064‐22). Manufacture instructions were followed. IL‐6 levels in BM were measured by crunching the femur and tibiae in 600 μl PBS. Samples were spun, supernatants were transferred to new tubes, and total protein concentration was determined using Pierce™ BCA Protein Assay Kit, catalog number 23225. Sixty microgram of total protein was used for ELISA (Mouse IL‐6 Quantikine ELISA Kit, catalog number M6000B).
IL‐1β ELISA
Femur and tibiae (WT, CMO, MyD88xCMO, CMO MyD88‐flox VavCre, age 16 w‐37 w) were flushed with 500 μl of buffer mix (2× Lysis Buffer and Complete™ ULTRA Tablets, Mini, EASYpack Protease Inhibitor Cocktail). Next, samples were spun, supernatants transferred to new tubes and total protein concentration determined as indicated above. One hundred microgram total protein was used for ELISA (IL‐1 beta Mouse Uncoated ELISA Kit, Invitrogen, catalog number 88‐7013). Hind paws were homogenized with a mixer in RIPA buffer supplemented with protease inhibitor cocktail (Protease Inhibitor Cocktail Set III, EDTA‐Free—Calbiochem, Millipore, catalog number 539134). Total protein concentration was measured as mentioned above. Sixty microgram of total protein was used for ELISA (IL‐1 beta Mouse Uncoated ELISA Kit, Invitrogen, catalog number 88‐7013).
Intracellular flow cytometry analysis
Whole BM cells (4 × 106 cells/sample) were stained for cell‐surface markers. Samples were then fixed using BD Fixation/Permeabilization Solution Kit (catalog #: 554714) and further permeabilized using Phosflow Perm Buffer III (catalog #: 558050). Finally, they were stained overnight with pStat3 PE (13A3‐1) or IkB‐α (3D6C02) antibodies (Biolegend) and analyzed on FACS.
RNA isolation, cDNA preparation, and quantitative RT–PCR
RNA from murine LKS cells was extracted with Tri Reagent RT (Molecular Research Center) and treated with DNaseI (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Briefly, cDNA was prepared using SuperScript II Reverse Transcriptase (Thermo Fisher Scientific). Quantitative RT–PCR was performed using a LightCycler® 480 SYBR Green I Master mix and samples were run on a LightCycler® 480 Instrument II (both Roche Molecular Systems, Pleasanton, CA, USA). For each sample, transcript levels of tested genes were normalized to Gapdh. Primer sequences used for quantitative RT–PCR are listed below:
| Gene | Orientation | Organism | Sequence (5′–3′) |
|---|---|---|---|
| Mmp9 | F | Human | ACGGTTGGTACTGGAAGTTC |
| R | Human | CCAACTTATCCAGACTCCTGG | |
| Bcl2l11 | F | Mouse | CGCAGATCTTCAGGTTCCTC |
| R | Mouse | ACAAACCCCAAGTCCTCCTT | |
| Bcl2 | F | Mouse | GGTCTTCAGAGACAGCCAGG |
| R | Mouse | GATCCAGGATAACGGAGGCT | |
| Casp1 | F | Mouse | AGTCCTGGAAATGTGCCATC |
| R | Mouse | TCAGCTCCATCAGCTGAAAC | |
| Gapdh | F | Mouse | AACTTTGGCATTGTGGAAGG |
| R | Mouse | ATCCACAGTCTTCTGGGTGG |
Stattic treatment
Stattic (Stat3 inhibitor) was purchased from MedChemExpress (catalog #: HY‐13818). WT and CMO mice were injected intraperitoneally with PBS control or Stattic (20 mg/kg). A total of 13 injections were administered, with the schedule three times per week, for 4 weeks, followed by a final injection the day before the analysis.
IL‐6‐blocking antibody and IL‐6 receptor‐blocking antibody (MR16‐1) treatment
Ultra‐LEAF™ Purified anti‐mouse IL‐6 antibody (MP5‐20F3, Biolegend), an IL‐6‐blocking antibody, was administered intraperitoneally three times per week (Monday to Friday, every other day) for 4 weeks. MR16‐1 (rat anti‐mouse interleukin‐6 receptor monoclonal antibody) was a kind gift from Chugai Pharmaceutical, Tokyo, Japan. The specificity and blocking ability of this monoclonal antibody were verified in previous reports (Katsume et al, 2002; Okazaki et al, 2002). CMO mice received an intraperitoneal injection of 2 mg of MR16‐1 the first week, followed by weekly administration of 0.5 mg of MR16‐1 for 4 weeks. PBS was administered on the same schedule as a negative control. Animals were sacrificed and analyzed 2 days after the last injection (in case of Ultra‐LEAF™ Purified anti‐mouse IL‐6 antibody) or 5 days after the last injection (in case of MR16‐1 treatment).
Western blotting analysis
WT and CMO BM were isolated and enriched for c‐Kit+ fraction using MACS separator. The cells were then lysed in SDS–PAGE sample buffer and centrifuged at 120,000 × g to remove insoluble material. The samples were then separated using reducing SDS–PAGE (10% polyacrylamide gel). Following separation, samples were transferred to PVDF membrane and stained with anti‐pStat3 and anti‐Gapdh antibodies.
Apoptosis assays
Apoptosis was assessed as previously reported (Zjablovskaja et al, 2017). Briefly, WT and CMO BM cells were stained as indicated above for LKS, MPP, and HSC labeling in combination with Annexin V (BD Biosciences). Samples were analyzed on a Symphony instrument (BD Biosciences).
Statistical analysis
Statistical significance for indicated datasets was determined using a two‐sided, unpaired Student's t‐test. P‐values < 0.05 were considered statistically significant. Scatter dot plots depict the mean with error bars representing standard deviation (s.d.). Frequency of functional HSCs was measured by limiting dilution competitive repopulation unit assays and calculated using ELDA online software based on Poisson distribution statistics.
Author contributions
Srdjan Grusanovic: Formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Petr Danek: Data curation; investigation; visualization; methodology. Maria Kuzmina: Formal analysis; investigation; visualization; methodology. Miroslava K Adamcova: Formal analysis; investigation; methodology. Monika Burocziova: Formal analysis; investigation; visualization; methodology. Romana Mikyskova: Investigation; methodology. Karolina Vanickova: Investigation; methodology. Sladjana Kosanovic: Investigation; methodology. Jana Pokorna: Investigation; methodology. Milan Reinis: Supervision. Tomas Brdicka: Supervision; investigation; methodology. Meritxell Alberich‐Jorda: Conceptualization; formal analysis; supervision; funding acquisition; investigation; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
PDF+
Acknowledgements
The study was supported by GACR (Czech Science Foundation) grant 20‐03380S, by the National Institute for Cancer Research (Programme EXCELES, ID Project No. LX22NPO5102)—Funded by the European Union—Next Generation EU, and by institutional funding from the IMG CAS (RVO 68378050) to MA‐J, and by a GA UK fellowship (project No. 1372219) from Charles University in Prague to SG. This project received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement No. 860002. This research was partially funded by the Czech Health Research Council grant No. NV18‐05‐00562 to MR. The flow cytometry data presented in this paper were produced at the Flow Cytometry Core Facility, IMG CAS, Prague, Czech Republic. The authors would like to thank Olena Sapega for technical assistance and Chugai Pharmaceutical Co., Ltd. and Dr. Tadamitsu Kishimoto for providing MR16‐1.
EMBO Reports (2023) 24: e54729.
Data availability
The RNA‐sequencing data from this publication have been deposited to the Array Express database (https://www.ebi.ac.uk/biostudies/arrayexpress) and assigned the identifier E‐MTAB‐10956.
References
- Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA (2010) Quiescent haematopoietic stem cells are activated by IFN‐gamma in response to chronic infection. Nature 465: 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylis D, Bartlett DB, Patel HP, Roberts HC (2013) Understanding how we age: insights into inflammaging. Longev Healthspan 2: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe JD, Liu JY, Zhang JT (2018) Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol Ther 191: 74–91 [DOI] [PubMed] [Google Scholar]
- Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, Szeto MD, Liao X, Leventhal MJ, Nasser J et al (2020) Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586: 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank U, Karlsson G, Karlsson S (2008) Signaling pathways governing stem‐cell fate. Blood 111: 492–503 [DOI] [PubMed] [Google Scholar]
- Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, Heikenwalder M, Manz MG (2012) Cutting edge: LPS‐induced emergency myelopoiesis depends on TLR4‐expressing nonhematopoietic cells. J Immunol 188: 5824–5828 [DOI] [PubMed] [Google Scholar]
- Bogeska R, Mikecin AM, Kaschutnig P, Fawaz M, Buchler‐Schaff M, Le D, Ganuza M, Vollmer A, Paffenholz SV, Asada N et al (2022) Inflammatory exposure drives long‐lived impairment of hematopoietic stem cell self‐renewal activity and accelerated aging. Cell Stem Cell 29: e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd L, Grossmann M, Potter M, Shen‐Ong GL (1991) Chronic multifocal osteomyelitis, a new recessive mutation on chromosome 18 of the mouse. Genomics 11: 794–798 [DOI] [PubMed] [Google Scholar]
- Caiado F, Pietras EM, Manz MG (2021) Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J Exp Med 218: e20201541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Wu X, Frassica D, Yu B, Pang L, Xian L, Wan M, Lei W, Armour M, Tryggestad E et al (2011) Irradiation induces bone injury by damaging bone marrow microenvironment for stem cells. Proc Natl Acad Sci USA 108: 1609–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel SL, Janczy JR, Bing X, Wilson SP, Olivier AK, Otero JE, Iwakura Y, Shayakhmetov DM, Bassuk AG, Abu‐Amer Y et al (2014) Inflammasome‐independent IL‐1beta mediates autoinflammatory disease in Pstpip2‐deficient mice. Proc Natl Acad Sci USA 111: 1072–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu Y, Zheng P (2010) Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J Clin Invest 120: 4091–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Ferguson PJ, de Bruijn R, Schlueter AJ, Ochoa LA, Waldschmidt TJ, Yeung YG, Stanley ER (2009) Primed innate immunity leads to autoinflammatory disease in PSTPIP2‐deficient cmo mice. Blood 114: 2497–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Nacu V, Charles JF, Henne WM, McMahon HT, Nandi S, Ketchum H, Harris R, Nakamura MC, Stanley ER (2012) PSTPIP2 deficiency in mice causes osteopenia and increased differentiation of multipotent myeloid precursors into osteoclasts. Blood 120: 3126–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Lucas D, Hidalgo A, Mendez‐Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N et al (2011) Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 208: 261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16: 228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EK, Izukawa T, Young S, Rosen G, Jamali M, Zhang L, Johnson D, Bain E, Hilland J, Ferrone CK et al (2019) Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv 3: 2482–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danek P, Kardosova M, Janeckova L, Karkoulia E, Vanickova K, Fabisik M, Lozano‐Asencio C, Benoukraf T, Tirado‐Magallanes R, Zhou Q et al (2020) Beta‐catenin‐TCF/LEF signaling promotes steady‐state and emergency granulopoiesis via G‐CSF receptor upregulation. Blood 136: 2574–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G, Morrison SJ (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481: 457–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dregan A, Charlton J, Chowienczyk P, Gulliford MC (2014) Chronic inflammatory disorders and risk of type 2 diabetes mellitus, coronary heart disease, and stroke: a population‐based cohort study. Circulation 130: 837–844 [DOI] [PubMed] [Google Scholar]
- Drobek A, Kralova J, Skopcova T, Kucova M, Novak P, Angelisova P, Otahal P, Alberich‐Jorda M, Brdicka T (2015) PSTPIP2, a protein associated with autoinflammatory disease, interacts with inhibitory enzymes SHIP1 and Csk. J Immunol 195: 3416–3426 [DOI] [PubMed] [Google Scholar]
- Du W, Liu W, Mizukawa B, Shang X, Sipple J, Wunderlich M, Geiger H, Davies S, Mulloy J, Pang Q et al (2018) A non‐myeloablative conditioning approach for long‐term engraftment of human and mouse hematopoietic stem cells. Leukemia 32: 2041–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerging Risk Factors C, Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J (2010) C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. Lancet 375: 132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, Humphrey MB, Yang Q, Borghesi LA, Kincade PW (2011) Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol 186: 5367–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MAG, Offner S, Blanco‐Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A (2009) IFN alpha activates dormant haematopoietic stem cells in vivo. Nature 458: 904–908 [DOI] [PubMed] [Google Scholar]
- Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldschmidt TJ, Tygrett LT, Schlueter AJ, El‐Shanti H (2006) A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone 38: 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedion A, Holthusen W, Masel LF, Vischer D (1972) Subacute and chronic "symmetrical" osteomyelitis. Ann Radiol (Paris) 15: 329–342 [PubMed] [Google Scholar]
- Gilbert LA, Hemann MT (2010) DNA damage‐mediated induction of a chemoresistant niche. Cell 143: 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Hemann MT (2012) Context‐specific roles for paracrine IL‐6 in lymphomagenesis. Genes Dev 26: 1758–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DE, Adler BJ, Chan ME, Lennon JJ, Acerbo AS, Miller LM, Rubin CT (2013) Altered composition of bone as triggered by irradiation facilitates the rapid erosion of the matrix by both cellular and physicochemical processes. PLoS ONE 8: e64952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimfeld S, Hudak S, Weissman I, Rennick D (1991) The in vitro response of phenotypically defined mouse stem cells and myeloerythroid progenitors to single or multiple growth factors. Proc Natl Acad Sci USA 88: 9902–9906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbling PM, Pineiro‐Yanez E, Gerosa R, Boettcher S, Al‐Shahrour F, Manz MG, Nombela‐Arrieta C (2019) Global transcriptomic profiling of the bone marrow stromal microenvironment during postnatal development, aging, and inflammation. Cell Rep 29: e4 [DOI] [PubMed] [Google Scholar]
- Hentunen TA, Choi SJ, Boyce BF, Dallas MR, Dallas SL, Shen‐Ong GL, Roodman GD (2000) A murine model of inflammatory bone disease. Bone 26: 183–188 [DOI] [PubMed] [Google Scholar]
- Hernandez G, Mills TS, Rabe JL, Chavez JS, Kuldanek S, Kirkpatrick G, Noetzli L, Jubair WK, Zanche M, Myers JR et al (2020) Pro‐inflammatory cytokine blockade attenuates myeloid expansion in a murine model of rheumatoid arthritis. Haematologica 105: 585–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann SR, Kubasch AS, Range U, Laass MW, Morbach H, Girschick HJ, Hedrich CM (2016) Serum biomarkers for the diagnosis and monitoring of chronic recurrent multifocal osteomyelitis (CRMO). Rheumatol Int 36: 769–779 [DOI] [PubMed] [Google Scholar]
- Hofmann SR, Kapplusch F, Girschick HJ, Morbach H, Pablik J, Ferguson PJ, Hedrich CM (2017) Chronic recurrent multifocal osteomyelitis (CRMO): presentation, pathogenesis, and treatment. Curr Osteoporos Rep 15: 542–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh MM, Langemeijer S, Wynter A, Phang OA, Kang EM, Tisdale JF (2007) Low‐dose parenteral busulfan provides an extended window for the infusion of hematopoietic stem cells in murine hosts. Exp Hematol 35: 1415–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK (2009) ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347: 70–78 [DOI] [PubMed] [Google Scholar]
- Isringhausen S, Mun Y, Kovtonyuk L, Krautler NJ, Suessbier U, Gomariz A, Spaltro G, Helbling PM, Wong HC, Nagasawa T et al (2021) Chronic viral infections persistently alter marrow stroma and impair hematopoietic stem cell fitness. J Exp Med 218: e20192070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y et al (2006) Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 12: 446–451 [DOI] [PubMed] [Google Scholar]
- Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS (2006) Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124: 407–421 [DOI] [PubMed] [Google Scholar]
- Katsume A, Saito H, Yamada Y, Yorozu K, Ueda O, Akamatsu K, Nishimoto N, Kishimoto T, Yoshizaki K, Ohsugi Y (2002) Anti‐interleukin 6 (IL‐6) receptor antibody suppresses Castleman's disease like symptoms emerged in IL‐6 transgenic mice. Cytokine 20: 304–311 [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ (2005) SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121: 1109–1121 [DOI] [PubMed] [Google Scholar]
- King KY, Goodell MA (2011) Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol 11: 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JH, Kim WU (2017) Dysregulation of gut microbiota and chronic inflammatory disease: from epithelial defense to host immunity. Exp Mol Med 49: e337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K et al (2013) Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502: 637–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Song Y, Li P, Li M, Wang H, Xu T, Yu X, Yu Y, Tai Y, Chen P et al (2020) Downregulation of RIG‐I mediated by ITGB3/c‐SRC/STAT3 signaling confers resistance to interferon‐alpha‐induced apoptosis in tumor‐repopulating cells of melanoma. J Immunother Cancer 8: e000111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, Gross JM, Calabrese C, Iwakura Y, Lamkanfi M, Vogel P, Kanneganti TD (2014a) Critical role for inflammasome‐independent IL‐1beta production in osteomyelitis. Proc Natl Acad Sci USA 111: 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, Bandi SR, Calabrese CR, Vande Walle L, Lamkanfi M et al (2014b) Dietary modulation of the microbiome affects autoinflammatory disease. Nature 516: 246–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YD, Park C, Zhao H, Oduro KA Jr, Tu X, Long F, Allen PM, Teitelbaum SL, Choi K (2009) Defects in osteoblast function but no changes in long‐term repopulating potential of hematopoietic stem cells in a mouse chronic inflammatory arthritis model. Blood 114: 4402–4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Baba Y, Nagai Y, Miyazaki K, Malykhin A, Nakamura K, Kincade PW, Sakaguchi N, Coggeshall KM (2005) IL‐6 blocks a discrete early step in lymphopoiesis. Blood 106: 879–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Malykhin A, Teague‐Weber BN, Sun XH, Farris AD, Coggeshall KM (2009) Interleukin‐6 aborts lymphopoiesis and elevates production of myeloid cells in systemic lupus erythematosus‐prone B6.Sle1.Yaa animals. Blood 113: 4534–4540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F (2011) Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474: 298–306 [DOI] [PubMed] [Google Scholar]
- Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, King KY (2016) Chronic infection depletes hematopoietic stem cells through stress‐induced terminal differentiation. Cell Rep 17: 2584–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLemore ML, Grewal S, Liu F, Archambault A, Poursine‐Laurent J, Haug J, Link DC (2001) STAT‐3 activation is required for normal G‐CSF‐dependent proliferation and granulocytic differentiation. Immunity 14: 193–204 [DOI] [PubMed] [Google Scholar]
- Mendez‐Ferrer S, Lucas D, Battista M, Frenette PS (2008) Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452: 442–447 [DOI] [PubMed] [Google Scholar]
- Mendez‐Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN, Frenette PS (2010) Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466: 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirantes C, Passegue E, Pietras EM (2014) Pro‐inflammatory cytokines: Emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res 329: 248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa MM, Chen H, Rathinam CV (2018) Constitutive activation of NF‐kappaB pathway in hematopoietic stem cells causes loss of quiescence and deregulated transcription factor networks. Front Cell Dev Biol 6: 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PM, Putoczki TL, Ernst M (2015) STAT3‐activating cytokines: a therapeutic opportunity for inflammatory bowel disease? J Interferon Cytokine Res 35: 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oduro KA Jr, Liu F, Tan Q, Kim CK, Lubman O, Fremont D, Mills JC, Choi K (2012) Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood 120: 2203–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki M, Yamada Y, Nishimoto N, Yoshizaki K, Mihara M (2002) Characterization of anti‐mouse interleukin‐6 receptor antibody. Immunol Lett 84: 231–240 [DOI] [PubMed] [Google Scholar]
- Orelio C, Haak E, Peeters M, Dzierzak E (2008) Interleukin‐1‐mediated hematopoietic cell regulation in the aorta‐gonad‐mesonephros region of the mouse embryo. Blood 112: 4895–4904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Reynaud D, Kang YA, Carlin D, Calero‐Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, Passegue E (2015) Functionally distinct subsets of lineage‐biased multipotent progenitors control blood production in Normal and regenerative conditions. Cell Stem Cell 17: 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Mirantes‐Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B et al (2016) Chronic interleukin‐1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self‐renewal. Nat Cell Biol 18: 607–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Frenette PS (2019) Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol 20: 303–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos MG, Ramalingam P, Gutkin MC, Kleppe M, Ginsberg M, Crowley MJP, Elemento O, Levine RL, Rafii S, Kitajewski J et al (2016) Endothelial‐specific inhibition of NF‐kappaB enhances functional haematopoiesis. Nat Commun 7: 13829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor MJ, McMillan DC, Horgan PG, Fletcher CD, Talwar D, Morrison DS (2015) Systemic inflammation predicts all‐cause mortality: a Glasgow inflammation outcome study. PLoS ONE 10: e0116206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan‐Komito D, Swann JW, Demetriou P, Cohen ES, Horwood NJ, Sansom SN, Griseri T (2020) GM‐CSF drives dysregulated hematopoietic stem cell activity and pathogenic extramedullary myelopoiesis in experimental spondyloarthritis. Nat Commun 11: 155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, Mayle A, Goodell MA (2012) Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell 11: 302–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M et al (2007) Self‐renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131: 324–336 [DOI] [PubMed] [Google Scholar]
- Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, Kokkaliaris KD, Rhee C, Lee D, Scadden EW et al (2019) Stress‐induced changes in bone marrow stromal cell populations revealed through single‐cell protein expression mapping. Cell Stem Cell 25: 570–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RH, Schradin C (2016) Chronic inflammatory systemic diseases: an evolutionary trade‐off between acutely beneficial but chronically harmful programs. Evol Med Public Health 2016: 37–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue T, Lowndes NF, Ceredig R (2013) Mesenchymal stromal cells: radio‐resistant members of the bone marrow. Immunol Cell Biol 91: 5–11 [DOI] [PubMed] [Google Scholar]
- Tajik N, Frech M, Schulz O, Schalter F, Lucas S, Azizov V, Durholz K, Steffen F, Omata Y, Rings A et al (2020) Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nat Commun 11: 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P et al (2015) Exit from dormancy provokes DNA‐damage‐induced attrition in haematopoietic stem cells. Nature 520: 549–552 [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9: 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ et al (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425: 836–841 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ling F, Wang HC, Sun XH (2013) Chronic TLR signaling impairs the long‐term repopulating potential of hematopoietic stem cells of wild type but not Id1 deficient mice. PLoS ONE 8: e55552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zjablovskaja P, Kardosova M, Danek P, Angelisova P, Benoukraf T, Wurm AA, Kalina T, Sian S, Balastik M, Delwel R et al (2017) EVI2B is a C/EBPalpha target gene required for granulocytic differentiation and functionality of hematopoietic progenitors. Cell Death Differ 24: 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]