

Abstract
Interferon regulatory factor (IRF) 3 and IRF7 are master regulators of type I interferon (IFN‐I)‐dependent antiviral innate immunity. Upon viral infection, a positive feedback loop is formed, wherein IRF7 promotes further induction of IFN‐I in the later stage. Thus, it is critical to maintain a suitably low level of IRF7 to avoid the hyperproduction of IFN‐I. In this study, we find that early expression of IFN‐I‐dependent STAT1 promotes the expression of XAF1 and that XAF1 is associated specifically with IRF7 and inhibits the activity of XIAP. XAF1‐knockout and XIAP‐transgenic mice display resistance to viral infection, and this resistance is accompanied by increases in IFN‐I production and IRF7 stability. Mechanistically, we find that the XAF1‐XIAP axis controls the activity of KLHL22, an adaptor of the BTB‐CUL3‐RBX1 E3 ligase complex through a ubiquitin‐dependent pathway. CUL3‐KLHL22 directly targets IRF7 and catalyzes its K48‐linked ubiquitination and proteasomal degradation. These findings reveal unexpected functions of the XAF1‐XIAP axis and KLHL22 in the regulation of IRF7 stability and highlight an important target for antiviral innate immunity.
Keywords: IFN‐I, IRF7, KLHL22, ubiquitination, XAF1‐XIAP axis
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
A XAF1‐XIAP‐KLHL22 axis functions as a major regulator to control IRF7 protein abundance and helps maintaining proper IFN‐I production.

Introduction
Interferon regulatory factors (IRFs), a family of transcription factors, play important roles in immune defense by regulating interferon (IFN) expression and the development of various immunocytes (Lohoff & Mak, 2005). Nine IRF family members have been identified in mammals based on homologous structures, and these IRFs form a regulatory network that mediates the regulation of IFNs through diverse functions and interactions with other molecules (Yanai et al, 2012). The functions and partner interactions of IRFs depend on their structures. A DNA‐binding domain (DBD) is an intrinsic characteristic of IRFs and is required for binding with interferon‐stimulated response elements (ISREs). In addition, the IRF‐association domain (IAD) plays a major regulatory role in mediating IRF dimerization and interactions with other transcription factors (Yanai et al, 2012).
The first identified IRFs, IRF1 and IRF2, exhibit opposite functions during IFN regulation, with IRF1 upregulating the expression of type I and type II IFN (IFN‐I and IFN‐γ) and IRF2 suppressing their expression (Matsuyama et al, 1993; Song et al, 2021). IRF4 and IRF8, identified later, were found to exhibit physiological functions similar to those of IRF2; however, the functions of IRF4 and IRF8 are multifaceted, as they exhibit opposite functions in different complexes (Nelson et al, 1993; Brass et al, 1996; Shaffer et al, 2009). IRF9 contributes to the interaction between IFN‐I signaling and the p53 pathway (Munoz‐Fontela et al, 2016). Among the IRFs, IRF3, IRF5, and IRF7 are the major drivers of IFN‐I regulation in antiviral immunity. When viruses invade cells, host pattern recognition receptors (PRRs), including Toll‐like receptors and intracellular sensors, respond to viral components, activating kinases involved in IRF activation (McNab et al, 2015; Jefferies, 2019). IRF3 is constitutively expressed allowing quick responses to risk signals. Activated IRF3 is phosphorylated and translocated in dimer form to the nucleus where it promotes the transcription of IFN‐I genes, especially Ifnb, to defend against viral infection at an early stage. Synthesized IFN‐I binds with the IFN‐alpha/beta receptor (IFNAR), which subsequently results in Irf7 transcription and thereby induces increased IFN‐I, especially interferon‐alpha (Ifna), expression. This process contributes to the clearance of viruses and danger signaling to uninfected cells (Honda et al, 2006a). In the late stage of viral infection, IRF7 induces increased IFN‐I production, forming a positive feedback loop to produce IFN‐I more rapidly and efficiently (Sato et al, 2000b; Honda et al, 2005). Therefore, it is generally believed that IRF7 shows a greater ability to induce IFN‐I expression than IRF3.
Although IFN‐I is broadly considered a critical factor in resistance to viral infection (Lin & Young, 2014), persistent expression of IFN‐I results in chronic inflammatory diseases such as systemic lupus erythematosus (SLE) (Dusheiko, 1997; Ronnblom & Leonard, 2019). Therefore, it is possible that the cessation of IFN‐I signaling is necessary to allow the immune system to rest after an infection has been cleared. Given the essential roles of IRFs in IFN‐I regulation, the investigation into the mechanism(s) by which IRFs, especially IRF3 and IRF7, are regulated to prevent hyper‐reaction is necessary. Posttranslational modification (PTM) is a fast and effective regulatory mechanism that triggers protein activity or confers molecular stability. Previous studies have indicated that IRF3 and IRF7 activation depend on PTM, which is essential for IFN‐I induction (Ning et al, 2011). Indeed, ubiquitination and phosphorylation have been identified as the most frequent types of IRF3 and IRF7 modifications, which promote their activation or suppression (Higgs & Jefferies, 2008). Evidence indicates that polyubiquitination is a critical modification that suppresses the activity of IRF3 and IRF7. The E3 ubiquitin ligase TRIM21 contributes to IRF3 and IRF7 stability by regulating their K48‐linked polyubiquitination, which leads to their degradation in a proteasome‐ or autophagy‐dependent manner. By contrast, SUMOylation of IRF3 and IRF7 by TRIM28 contributes to their stability by blocking the polyubiquitylation site, preventing their degradation (Jefferies, 2019). Peptidylprolyl isomerase (Pin1) has also been reported to bind with IRF3 phosphorylated at the Ser339‐Pro340 motif to mediate protein polyubiquitination and proteasome‐dependent degradation (Saitoh et al, 2006). E3 ligases may be hijacked by infecting viruses (Yu et al, 2005; Yu & Hayward, 2010); however, their involvement in viral infection is unclear, and studies on possible E3 ligase promotion of IRF7 ubiquitination and degradation in innate immune cells are rare.
In this study, we found that the mRNA level of IFN‐I gradually decreased in bone marrow‐derived macrophages (BMDMs) 6 h after viral infection, but Irf7 mRNA transcription continuously increased. Using RNA sequencing (RNA‐seq) and mass spectrometry (MS), we identified a critical molecule, XIAP‐associated factor‐1 (XAF1), that interacts with IRF7 and whose mRNA abundance continuously increased along with that of IRF7 upon IFN‐β stimulation. XAF1 deficiency clearly enhanced the expression of IFN‐I and suppressed the onset of viral infection in vivo. Our experiments also demonstrated that the protein stability of IRF7 is negatively regulated by XAF1, which suppresses K48‐linked polyubiquitination and thus inhibits proteasome‐dependent degradation. XAF1 did not exhibit any E3 ubiquitin ligase activity but did act as a natural antagonist of XIAP. Notably, we observed similar IRF7‐stabilizing and IFN‐I‐inducing phenotypes in XAF1‐knockout (KO) and XIAP‐transgenic (XIAPTg) BMDMs. Nevertheless, the data revealed that XIAP inhibited the ubiquitination and degradation of IRF7 in vitro. We next confirmed that KLHL22 (an adaptor of BTB‐CUL3‐RBX1 (BCR)) is an E3 ligase that directly targets IRF7, catalyzes the K48‐linked ubiquitination of IRF7 to trigger proteasomal degradation, and significantly inhibits the production of IFN‐I. The activity of KLHL22 was inhibited by XIAP, as XIAP altered the stability of KLHL22 through polyubiquitination‐associated proteasomal degradation. Our findings showed that the mechanism of IFN‐I production involves an essential negative feedback loop. Targeting this loop may facilitate the development of therapeutic drugs for viral infection or chronic inflammatory diseases mediated by IFN‐I.
Results
IFN‐I‐inducing XAF1 is potentially involved in IRF7 ubiquitination
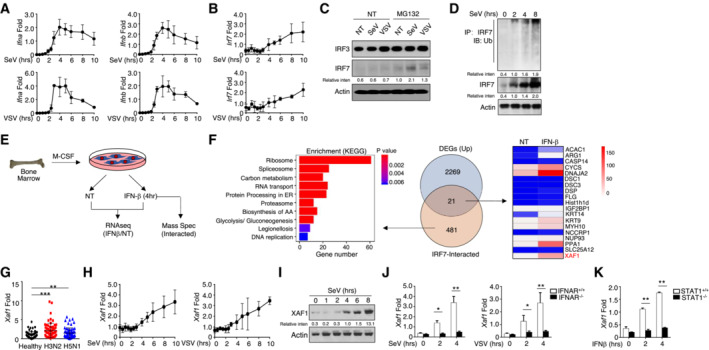
A low level of IRF7 expression is typically observed; however, IRF7 is intensively induced by IFN‐I‐mediated signaling. Although IRF7 is involved in the induction of IFN‐I in the late phase of viral infections, no sustained increase in the production of IFN‐I was observed. To clarify the relationship between IFN‐I production and IRF7 dynamics, we challenged wild‐type (WT) BMDMs with Sendai virus (SeV) or vesicular stomatitis virus (VSV). The mRNA levels of Ifna and Ifnb increased from 4 to 6 h after viral infection; they gradually decreased thereafter (Fig 1A). By contrast, the transcriptional abundance of Irf7 continuously increased even at 10 h after infection (Fig 1B). Paradoxically, we found that viral infection did not significantly induce increases in IRF7 protein levels (Fig 1C). Hence, we speculated that IRF7 might undergo proteolysis‐dependent degradation at the late stage of virus challenge. An increase in the level of IRF7 was observed after treatment with MG132, suggesting that proteasome‐dependent degradation may be involved in regulating the protein level of IRF7 (Fig 1C). Ubiquitin is conjugated to target proteins by E3 ubiquitin ligases and controls the stability of the target. As expected, the ubiquitination of IRF7 gradually increased upon SeV stimulation (Fig 1D).
Figure 1. IFN‐I‐induced XAF1 expression is negatively correlated with the Irf7 mRNA level.
-
A, BmRNA levels of IFN‐I (A) and IRF7 (B) in virus‐stimulated WT BMDMs were measured by qRT–PCR at the indicated time points.
-
CWT BMDMs were stimulated with SeV or VSV‐WT (MOI = 1) for 9 h and treated with MG132 2 h before harvest. The indicated proteins in whole‐cell lysates were detected by IB.
-
DWT BMDMs were stimulated with SeV for the indicated time course, and whole‐cell lysates were subjected to IP using an anti‐IRF7 antibody. Cell lysates were also subjected to direct IB, and actin was used as a loading control.
-
ESchematic of the experimental procedure for RNA‐seq and MS.
-
FLeft panel, KEGG analysis of IRF7‐interacting proteins as determined by MS; middle panel, Venn diagrams illustrating the overlap between DEGs identified through analysis of the RNA‐seq and MS results; right panel, heatmap showing overlapping DEGs.
-
GAbundant XAF1 mRNA expression was induced by different influenza viruses at an MOI of 1 for 6 h in PBMCs obtained from healthy donors (n = 64) as measured by qRT–PCR.
-
H, IThe mRNA (H) and protein (I) levels of XAF1 in virus‐induced WT BMDMs were measured throughout the indicated time course by qRT–PCR and IB, respectively.
-
J, KqRT–PCR analysis of XAF1 mRNA in BMDMs derived from IFNAR‐KO mice (J) and STAT1‐KO mice (K).
Data information: All data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. The data are presented as the means ± SDs. The significance of differences was determined by t‐tests. *P < 0.05, **P < 0.01, and ***P < 0.005.
Please see Appendix Fig S1 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
To identify the critical molecules involved in IRF7 ubiquitination, we analyzed interferon‐stimulated genes (ISGs), which interacted with IRF7 in BMDMs after IFN‐β stimulation (Fig 1E and Table EV2). A total of 481 IRF7‐interacting proteins were identified from the MS data, some of which are involved in the proteasome pathway, as revealed by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Fig 1F). We also assessed the major differentially expressed genes (DEGs) on the basis of mRNA abundance in WT BMDMs with or without IFN‐β stimulation. Overall, 21 genes exhibited overlap between the DEGs and the IRF7‐interacting proteins. Among these genes, we identified XAF1, a natural antagonist of XIAP, as a candidate regulator of IRF7 protein stability (Fig 1F). Significant upregulation of XAF1 expression was detected in peripheral blood mononuclear cells (PBMCs) obtained from healthy donors challenged with either the H3N2 or H5N1 virus compared with those from noninfected donors (Fig 1G). Consistent with these findings, viral infection was found to trigger increased XAF1 expression at both the transcriptional and translational levels (Fig 1H and I). In addition, Xaf1 clearly acted as an ISG that relied on IFN‐I‐mediated signaling in the absence of XAF1 induction in IFNAR‐ or STAT1‐deficient BMDMs challenged with virus or IFN‐β (Fig 1J and K). These data indicate that the XAF1‐mediated pathway potentially regulates IRF7 stability and restricts the production of IFN‐I in the late period of virus infection.
XAF1 deficiency specifically promotes IFN‐I production and enhances antiviral innate immunity
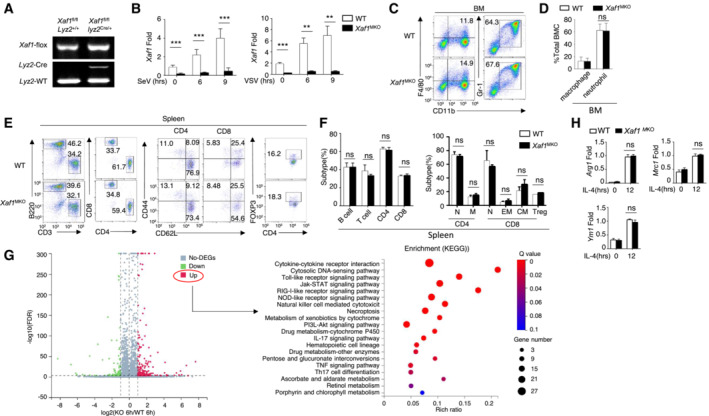
To clarify the role of XAF1 in regulating the antiviral response, we first performed a luciferase reporter assay to evaluate the effect of XAF1 on the production of IFN‐β. As shown in Fig 2A, elevated XAF1 expression notably reduced IFN‐β transcriptional activity upon virus challenge. We then crossed Xaf1‐floxed mice with Lyz2‐Cre mice to generate myeloid cell‐specific XAF1‐KO (XAF1MKO) mice (Fig EV1A). Real‐time quantitative PCR (qPCR) confirmed the loss of XAF1 expression in the BMDMs of the XAF1MKO mice (Fig EV1B). The XAF1MKO mice were born at the expected Mendelian ratios and exhibited a normal survival ratio. We did not observe any obvious abnormalities in the development of macrophages or neutrophils (Fig EV1C and D). Additionally, compared with WT mice, XAF1MKO mice exhibited no significant difference in the frequency of T cells or B cells in the spleen (Fig EV1E and F). The frequency of regulatory T cells among CD4+ splenic T cells in the XAF1MKO mice was comparable to that in the WT mice (Fig EV1E and F). To elucidate the function of XAF1 in regulating inflammatory responses, we monitored the major lipopolysaccharide (LPS)‐induced changes in mRNA abundance in the transcriptome. We identified 188 DEGs upregulated in XAF1‐deficient BMDMs compared with BMDMs from WT littermates. KEGG analysis indicated that the major signaling pathway altered in XAF1‐deficient BMDMs was associated with the TLR signaling pathway and the IFN‐I signaling pathway (Fig EV1G). As expected, XAF1‐deficient BMDMs showed significantly enhanced IFN‐I production in response to SeV, VSV, or herpes simplex virus type‐1 (HSV‐1; Fig 2B). The XAF1‐deficient BMDMs also exhibited significantly inhibited GFP‐tagged VSV replication, as determined by fluorescence microscopy and flow cytometry analyses (Fig 2C). Additionally, XAF1 exhibited similar performance in modulating IFN‐I production induced by the TLR agonists LPS, polyinosinic‐polycytidylic acid (polyI:C) and CpG oligodeoxynucleotides (Fig 2D). A series of ISGs, such as Ifit3, Irf1, Mx2, and Slfn8, were upregulated in XAF1‐deficient BMDMs upon LPS or polyI:C stimulation (Fig 2E). However, other proinflammatory cytokines, including Tnfa, Il6, and Il10, were not affected by the depletion of XAF1 (Fig 2F). In addition, the levels of M2 macrophage markers induced by IL‐4 were comparable between WT and XAF1‐deficient BMDMs (Fig EV1H). In addition to exerting effects in BMDMs, SeV‐ or HSV‐induced IFN‐I was significantly enhanced in XAF1‐deficient mouse embryonic fibroblasts (MEFs; Fig 2G). These data confirm that XAF1 acts as a negative regulator in antiviral immunity by suppressing IFN‐I induction.
Figure 2. XAF1 negatively regulates IFN‐I induction.
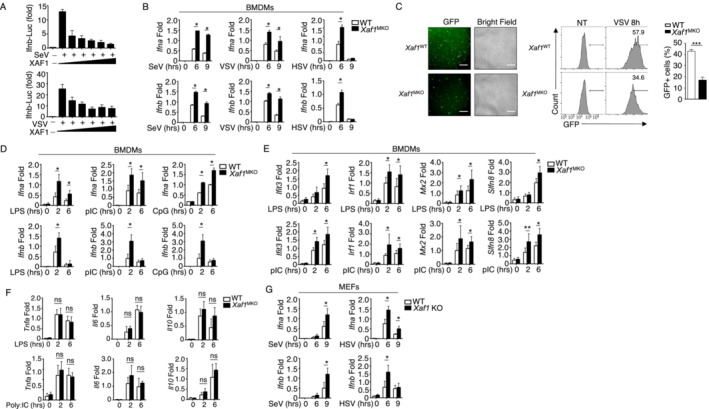
- HEK293T cells were transfected with IFN‐β luciferase reporters and XAF1‐expressing plasmids and stimulated with SeV or VSV‐WT (MOI = 1). The readouts were normalized to Renilla luciferase activity and are presented as the fold change relative to activity in untransfected cells.
- qRT–PCR analysis of IFN‐I induction in WT and XAF1‐deficient BMDMs stimulated with different viruses.
- WT or XAF1‐deficient BMDMs were infected with GFP‐expressing VSV (VSV‐GFP) at an MOI of 0.1 for 8 h. The data are presented as representative images and FACS data, showing the infected (GFP+) and total (bright field) cells. Scale bar, 1,000 μm.
- qRT–PCR analysis of the mRNA levels of IFN‐I in WT and XAF1‐deficient BMDMs stimulated with distinct TLR agonists.
- Some of the LPS‐ or polyI:C‐induced ISGs in WT and XAF1‐deficient BMDMs were measured by qPCR.
- mRNA levels of the indicated cytokines in WT and XAF1‐deficient BMDMs activated by distinct TLR agonists.
- IFN‐I expression in WT and XAF1‐KO MEFs stimulated with SeV or HSV, evaluated by qPCR.
Data information: All data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. The data are presented as the means ± SDs. The significance of differences was determined by t‐tests. *P < 0.05, **P < 0.01, and ***P < 0.005.
Figure EV1. XAF1 deficiency in myeloid cells had no effect on the development of peripheral immune cells.
-
AGenotyping PCR of WT (XAF1fl/flLyz2+/+) and myeloid‐conditional XAF1‐KO (XAF1MKO, XAF1fl/flLyz2+/Cre) mice.
-
BqRT–PCR analysis showing clear ablation of XAF1 in the BMDMs of XAF1MKO mice.
-
C, DFlow cytometry analysis of macrophages (CD11b+F4/80+) and neutrophils (CD11b+Ly6G+) in bone marrow (BM) from 6‐ to 8‐week‐old WT and XAF1MKO mice (n = 3).
-
E, FFlow cytometry analysis of the frequencies of distinct immune cells in the spleen (left), naïve and memory T cells (middle), and Treg cells (right) from WT or XAF1MKO mice (n = 3).
-
GVolcano plot illustrating the upregulated and downregulated DEGs in XAF1‐deficient BMDMs compared with WT littermates after 6 h of LPS stimulation. The KEGG analysis showed that the upregulated DEGs were enriched in biological processes.
-
HqRT–PCR analysis of ARG1, MRC1, and YM1 in IL‐4‐treated M2 macrophages in WT and XAF1‐deficient BMDMs.
Data information: All data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. Data are represented as the means ± SDs. The significance of differences was determined by a t‐test. ns, not significant.
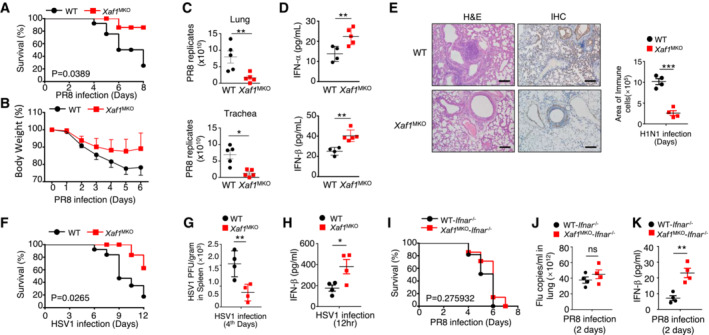
Mice with XAF1‐deficient myeloid cells are resistant to viral infection
To assess the function of XAF1 in the antiviral innate immune response, we infected WT and XAF1MKO mice with the H1N1 strain PR8 via intranasal (i.n.) administration at a 50% tissue culture infectious dose (TCID50). Mouse body weight and survival ratio were monitored for up to 8 days. Compared with the WT mice, the XAF1MKO mice showed an increased survival rate (Fig 3A) and reduced loss of body weight (Fig 3B). A lower viral load in the lungs and tracheas was observed in the XAF1MKO mice, suggesting that these mice were substantially more resistant to PR8 infection (Fig 3C). Enzyme‐linked immunosorbent assay (ELISA) revealed elevated IFN‐I induction, which potentially contributed to the viral resistance of the XAF1MKO mice (Fig 3D). Consistent with these findings, hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) indicated that the XAF1MKO mice displayed significantly attenuated immune cell infiltration and lung injury (Fig 3E). In addition to differing from the WT mice in response to the RNA virus, the XAF1MKO mice exhibited less susceptibility to infection by the HSV‐1 DNA virus (Fig 3F and G) due to enhanced production of IFN‐β (Fig 3H). To confirm the role of excessive IFN‐I levels in the antiviral response, we crossed XAF1MKO mice with IFNAR‐deficient mice. In mice with the IFNAR‐deficient background, the virus resistance of the XAF1MKO mice was absent, as indicated by survival ratios and viral loads similar to those of the WT group, although IFN‐β production was still increased (Fig 3I–K). Previous studies have indicated that inflammatory bowel disease is associated with the induction of proinflammatory cytokines, including tumor necrosis factor (TNF), interleukin (IL)‐6, and IL‐23. Therefore, we evaluated whether XAF1 deficiency contributes to the onset of colitis in mice. We used dextran sulfate sodium (DSS) to construct models of acute colitis; among these models, WT and XAF1MKO mice exhibited similar body weight loss, stool consistency index values, hemorrhage scores, and colon lengths. These results indicated that XAF1 deficiency is not essential for proinflammatory cytokine induction (Fig EV2A–D). In addition, the results collectively indicated an essential and specific role of XAF1 in regulating IFN signaling in macrophages, which contributed to the regulation of antiviral innate immunity but not an inflammatory disease in vivo.
Figure 3. XAF1MKO mice exhibit resistance to viral infection.
-
A, BSurvival rate (A) and body weight loss (B) were monitored for 8 days (n = 10).
-
CViral titers in the lung and trachea were quantified by TCID50 assay on day 2 (n = 5).
-
DELISA of IFN‐I levels in the sera of WT and XAF1MKO mice infected with H1N1 strain PR8 on day 2 (n = 3).
-
EH&E‐stained lung tissue sections 2 days after infection. Inflammation scores are presented in a bar graph (n = 4). Scale bar, 200 μm.
-
F–HSurvival rate (F, n = 12) and viral titer (G, n = 4) in the spleen and IFN‐β in the serum (H, n = 4) of WT and XAF1MKO mice intravenously injected with HSV‐1 (3 × 106 PFU per mouse).
-
I–KWT and XAF1MKO mice crossed with IFNAR−/− mice were i.n. infected with a sublethal dose (0.1 HA) of PR8. The survival rate (I, n = 10), viral titer in the lung (J, n = 4), and serum IFN‐β level (K, n = 4) were monitored for the indicated times.
Data information: All data are representative of at least three biologically independent experiments. The data are presented as the means ± SDs. The significance of differences was determined by t‐tests. *P < 0.05, **P < 0.01, and ***P < 0.005.
Figure EV2. No difference in the colitis model or activation of the MAPK and NF‐κB pathways was observed between WT and XAF1MKO mice.
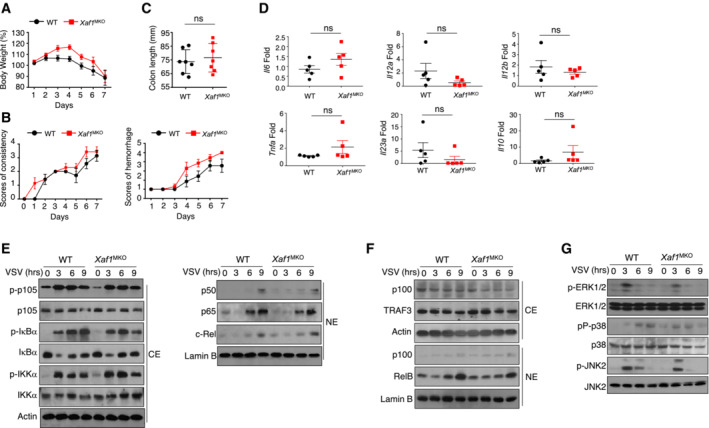
-
A, BBody weight loss (A) and stool diarrhea and hematochezia scores (B) in WT and XAF1MKO mice of the DSS‐induced colitis model.
-
C, DColon length (C) and proinflammatory cytokine production (D) of DSS‐treated WT and XAF1MKO mice on day 8.
-
E, FThe indicated proteins in cytoplasmic (CE) and nuclear extracts (NE) of WT and XAF1‐deficient BMDMs were detected by IB.
-
GThe activation of MAPKs in total cell lysates was measured by IB.
Data information: The data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. Data are represented as the means ± SDs. The significance of differences was determined by a t‐test.
Source data are available online for this figure.
XAF1 regulates IFN signaling by promoting IRF7 degradation
To clarify the mechanism by which XAF1 controls IFN‐I production, we first examined the activation of the TBK1‐IRF3 axis after viral infection. Immunoblot assays revealed comparable phosphorylation levels of TBK1 and IRF3 in BMDMs derived from WT and XAF1MKO mice upon virus challenge (Fig 4A and B). Furthermore, no difference in IRF3 dimerization was found between VSV‐stimulated BMDMs with XAF1 and those deficient in XAF1 (Fig 4C). Upregulation of IFN‐I production in XAF1‐deficient BMDMs was not due to enhanced binding of IRF3 to interferon stimulation response elements (ISREs), as revealed by chromatin immunoprecipitation (ChIP) assays (Fig 4D). The p38 MAPK pathway and canonical/noncanonical NF‐κB pathway are critical for the expression of IFN‐I and proinflammatory cytokines (Mikkelsen et al, 2009; Wang et al, 2010; Bais et al, 2019). However, our data indicated that neither the MAPK pathway nor the canonical/noncanonical NF‐κB pathway differed between XAF1‐deficient BMDMs and WT BMDMs upon viral infection (Fig EV2E–G). These data demonstrate that the upregulation of IFN‐I production mediated by XAF1 deficiency was independent of the MAPK, NF‐κB, and TBK1‐IRF3 pathways.
Figure 4. XAF1 regulates IRF7 stability.
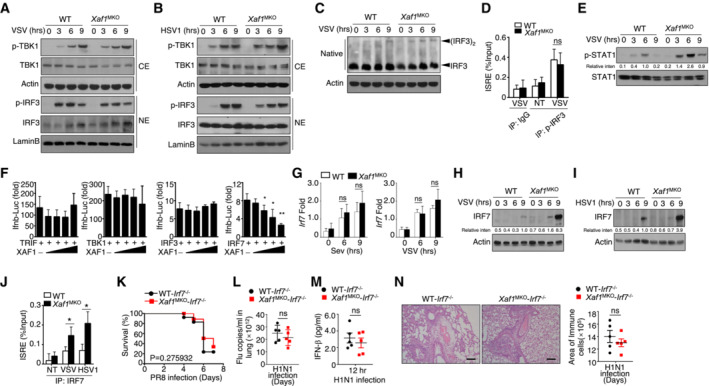
-
A, BVirus‐induced phosphorylation of TBK1 and IRF3 in cytoplasmic (CE) and nuclear (NE) extracts of WT and XAF1‐deficient BMDMs was measured by IB.
-
CMonomeric and dimeric IRF3 expression (top blot) in whole‐cell lysates of WT and XAF1‐deficient BMDMs stimulated with VSV‐WT (MOI = 1) was measured by IB.
-
DChIP assays were performed, and the results were quantified by qRT–PCR to detect IRF3 binding to an ISRE in WT and XAF1‐deficient BMDMs activated by VSV‐WT (MOI = 1) for 9 h.
-
EIB was performed to determine the VSV‐induced phosphorylation level of STAT1.
-
FHEK293T cells were transfected with IFN‐β luciferase reporters in the presence (+) or absence (−) of the indicated XAF1 expression plasmids. Luciferase assays were used to determine the fold change in expression with respect to the empty vector group 36 h after transfection.
-
GqRT–PCR analysis of Irf7 mRNA in WT and XAF1‐deficient BMDMs stimulated with distinct virus infections.
-
H, IIB analysis of VSV‐ or HSV‐1‐induced IRF7 expression as measured in whole‐cell lysates of WT and XAF1‐deficient BMDMs.
-
JChIP assays were performed and quantified by qRT–PCR to detect IRF7 binding to an ISRE in WT and XAF1‐deficient BMDMs stimulated with VSV‐WT (MOI = 1) or HSV‐1.
-
K–NWT and XAF1MKO mice crossed with mice with an IRF7−/− background were i.n. infected with a sublethal dose (0.1 hyaluronic acid [HA]) of the H1N1 strain PR8. The survival rate (K, n = 15), viral titer in the lung (L, n = 5), serum IFN‐β level (M, n = 5), and intensity of H&E‐stained lung tissue sections (N, n = 5) were determined at the indicated time points. Scale bar, 200 μm.
Data information: All data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. The data are presented as the means ± SDs. The significance of differences was determined by t‐tests. *P < 0.05, **P < 0.01, and ***P < 0.005.
Please see Appendix Fig S2 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
As the expression of multiple ISGs was elevated, we speculated that the IFN signaling pathway might be affected by XAF1 deficiency. Evidence has indicated that the phosphorylation of STAT1, which responds to IFN‐I signaling, plays an important role in ISG induction. As expected, phosphorylation of STAT1 after VSV stimulation was significantly increased in XAF1‐deficient BMDMs compared to that in WT BMDMs (Fig 4E). In addition to IRF3, IRF7 is a master regulator of IFN‐I induction in antiviral immune responses. A luciferase assay implied that XAF1 expression suppressed the activity of IRF7 and IFN‐β transcription in a dose‐dependent manner but had no effect on the activity of TRIF, TBK1, or IRF3 (Fig 4F). Although the mRNA level of Irf7 was comparable between WT and XAF1‐deficient BMDMs (Fig 4G), greater IRF7 protein accumulation was detected in the XAF1‐deficient BMDMs challenged with the virus (Fig 4H and I). Consistent with these findings, a slight increase in IRF7 enrichment at an ISRE was found in XAF1‐deficient BMDMs (Fig 4J). To confirm the critical role of IRF7 in the XAF1‐mediated antiviral response, we generated XAF1MKO‐IRF7 double‐KO mice. In the mice with an IRF7‐deficient background, the virus resistance of the XAF1MKO mice was absent; these mice showed a pathological reaction similar to that of the WT mice (Fig 4K–N). Taken together, our data suggest that XAF1 repressed the expression of IFN‐I by regulating IRF7 protein stability without affecting the activity of NF‐κB, MAPKs, or TBK1‐IRF3.
XAF1 interacts with IRF7 and promotes its ubiquitination
To verify the mechanism mediating IRF7 degradation, we treated WT and XAF1‐deficient BMDMs with a proteasome inhibitor (MG132) and lysosome inhibitor (chloroquine, CQ). The results indicated that both the proteasome and lysosomes were involved in IRF7 degradation in the WT BMDMs (Fig 5A). Substrate ubiquitination typically causes substrate degradation. Indeed, polyubiquitination, especially K48‐linked ubiquitination of IRF7, was clearly reduced in the XAF1‐deficient BMDMs compared with the WT BMDMs after VSV stimulation (Fig 5B and C). To confirm the function of XAF1 regarding the IRF family, we cotransfected XAF1 and distinct IRFs into HEK293T cells. Coimmunoprecipitation (co‐IP) assays demonstrated that XAF1 was physically associated with IRF7 and IRF1 but not with IRF3 or IRF2 (Fig 5D). In addition, we observed a strong endogenous interaction between XAF1 and IRF7 in VSV‐stimulated BMDMs (Fig 5E). IRF7 contains five critical domains, including a DBD in the N‐terminal region as well as a constitutive domain (CAD), a virus‐activated domain (VAD), an inhibitory domain (ID), and a signal response domain (SRD) in the C‐terminal region. We next analyzed the IRF7‐ and XAF1‐interacting domains in transfected HEK293T cells. Co‐IP results revealed that the association between IRF7 and XAF1 was dependent on the ID of IRF7 (Fig 5F). XAF1 contains 7 zinc finger (ZF) domains, and truncation assays revealed that 4–6 of these ZF domains contributed to the interaction of XAF1 with IRF7 (Fig 5G). Using a deletion mutant of each ZF domain, we found that ZF6 played the dominant role in the interaction of IRF7 and XAF1 (Fig 5H). Furthermore, ZF6 of XAF1 played a critical role in suppressing IRF7‐mediated IFN‐I production, as demonstrated by the qPCR assay of reconstituted XAF1‐deficient MEFs (Fig 5I).
Figure 5. XAF1 promotes the ubiquitination of IRF7.
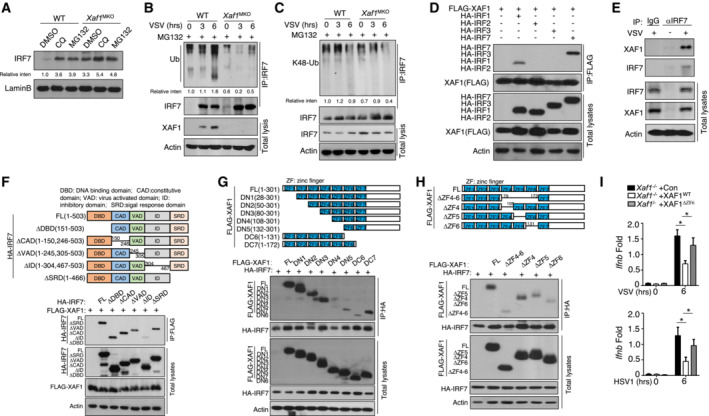
-
AAfter treatment with CQ or MG132, IRF7 in nuclear extracts of WT and XAF1‐deficient BMDMs was detected by IB.
-
B, CWT and XAF1‐deficient BMDMs were infected with VSV‐WT (MOI = 1) and treated with MG132 for 2 h before harvest. Whole‐cell lysates (WLs) were subjected to IP using an anti‐IRF7 antibody, and the indicated proteins were detected by IB.
-
DHEK293T cells were cotransfected with XAF1 and distinct IRF‐expressing plasmids. IB HA was performed followed by IP with an anti‐FLAG antibody.
-
EThe interaction between XAF1 and IRF7 was assessed in WT BMDMs stimulated by VSV‐WT (MOI = 1). WLs were subjected to IP using an anti‐IRF7 or anti‐IgG antibody and then to IB and detected with anti‐XAF1 antibody.
-
FThe associations between XAF1 and various IRF7 truncation mutants were detected through IP and IB of the transfected HEK293T cells.
-
G, HThe associations between IRF7 and various XAF1 truncation mutants were detected through IP and IB.
-
IqRT–PCR analysis of the indicated genes in XAF1‐deficient MEFs expressing XAF1WT and XAF1ΔZF6 and subjected to the indicated virus stimulation.
Data information: Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. The data are presented as the means ± SDs and are representative of at least three biologically independent experiments. The statistical analyses revealed variations among the experimental replicates. Two‐tailed unpaired t‐tests were performed. *P < 0.05.
Please see Appendix Fig S3 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
XIAP inhibits the ubiquitination of IRF7 and promotes IFN‐I induction
XAF1 acts as a natural antagonist to suppress XIAP activity; thus, XAF1 deficiency potentially increases XIAP activity. Because XAF1 failed to show E3 ligase activity, we tested whether XIAP is involved in regulating IRF7 stability (Fig 6A). We found no significant difference in the mRNA abundance of XIAP between XAF1‐deficient and WT BMDMs upon IFN‐β stimulation (Fig 6B). When cotransfected with critical IFN‐I inducers, XIAP specifically enhanced IRF7‐mediated IFN‐β induction but had no effect on IRF3, TRIF, TBK1, MAVS or RIG‐I (Fig 6C). To further evaluate the function of XIAP in IFN‐I induction, we generated Xiap Tg mice to potentially mimic the phenotype of XAF1‐KO mice. Compared with WT mice, Xiap Tg mice exhibited normal growth and survival without any obvious difference in the frequency of T cells or B cells in the spleen (Fig EV3A and B) or inguinal lymph nodes (Fig EV3C and D). Furthermore, the frequency of regulatory T cells among CD4+ splenic T cells and the development of T cells in the thymus in Xiap Tg mice were comparable to those in WT mice (Fig EV3A–F). The innate immune system of Xiap Tg mice exhibited no abnormalities in the development of macrophages or neutrophils, which were similar to those in WT mice (Fig EV3G and H).
Figure 6. XIAP enhances IRF7 stability and promotes IFN‐I induction.
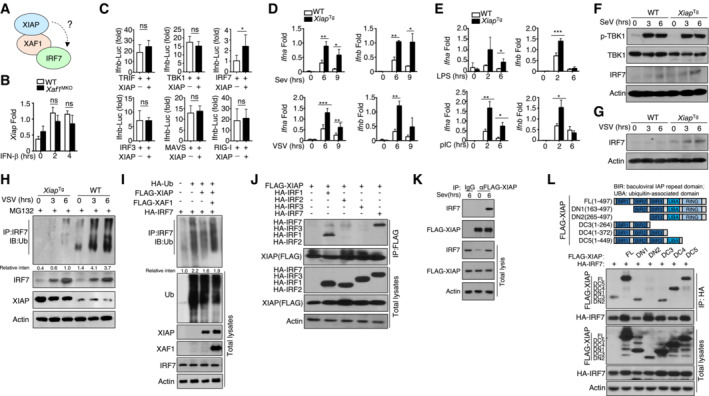
-
AModel diagram showing the potential regulatory mechanisms involving XAF1, XIAP, and IRF7.
-
BqRT–PCR analysis of XIAP expression in WT and XAF1‐deficient BMDMs stimulated with IFN‐β.
-
CHEK293T cells were transfected with IFN‐β luciferase reporters in the presence (+) or absence (−) of the indicated XIAP expression plasmids. Luciferase assays were performed, and the results are reported as fold changes with respect to the empty vector group 36 h after transfection.
-
D, EqRT–PCR analysis of IFN‐I expression in WT or XIAPTg BMDMs stimulated with distinct viruses (D) or TLR agonists (E).
-
F, GThe virus‐induced expression of IRF7 in WT and XIAPTg BMDMs was measured by IB.
-
HWT and XIAPTg BMDMs were infected with VSV‐WT (MOI = 1) and treated with MG132 2 h before harvest. Whole‐cell lysates were subjected to IP using an anti‐IRF7 antibody.
-
IHEK293T cells were transfected with FLAG‐tagged ubiquitin and the indicated expression plasmids. The ubiquitination level of IRF7 was determined by IB, and total cell lysates were also subjected to direct IB.
-
JHEK293T cells were cotransfected with XAF1 and distinct IRF‐expressing plasmids. IBHA was performed, followed by IP with an anti‐FLAG antibody.
-
KThe interaction between XIAP and IRF7 was assessed in XIAP‐overexpressing MEFs activated by SeV. Whole‐cell lysates (WLs) were subjected to IP using an anti‐FLAG‐XIAP or anti‐IgG antibody and then subjected to IB and detected with the anti‐IRF7 antibody.
-
LThe associations between IRF7 and various XIAP truncation mutants were detected through IP and IB.
Data information: The data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. The data are presented as the means ± SDs. The significance of a difference was determined by t‐tests. *P < 0.05, **P < 0.01 and ***P < 0.005.
Please see Appendix Fig S4 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
Figure EV3. XIAP is not required for the development of peripheral immune cells.
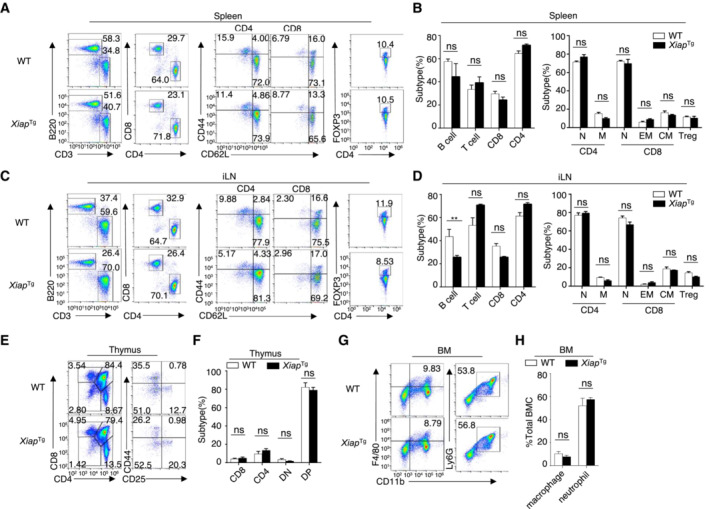
-
A–FFlow cytometry analysis of the absolute numbers of different immune cells in the spleen (A, B, upper), inguinal lymph nodes (C, D, middle), and thymus (E, F, lower) from WT and XAF1MKO mice (n = 3).
-
G, HFlow cytometry analysis of macrophages (CD11b+F4/80+) and neutrophils (CD11b+Ly6G+) in bone marrow (BM) from 6‐ to 8‐week‐old WT and XAF1MKO mice (n = 3).
Data information: The data are representative of at least three biologically independent experiments. Data are represented as the means ± SDs. The significance of differences was determined by t‐tests. **P < 0.01.
Consistent with the effects of XAF1 deficiency, the expression of IFN‐I in XIAPTg BMDMs was significantly higher than that in WT BMDMs after viral infection (Fig 6D) or stimulation with a Toll‐like receptor agonist (Fig 6E). Overexpression of XIAP clearly promoted virus‐induced IRF7 expression without affecting the phosphorylation of TBK1 (Fig 6F and G). After treatment with MG132, an increase in the ubiquitination level of IRF7 was found in XIAPTg BMDMs stimulated with VSV compared with VSV‐stimulated WT BMDMs (Fig 6H). In transfected HEK293T cells, XIAP obviously repressed IRF7 ubiquitination, and this effect was disrupted by XAF1 (Fig 6I). Similar to XAF1, XIAP selectively interacted with IRF7 and IRF1 (Fig 6J), implying that XIAP and XAF1 might form a complex to regulate IRF7. In addition, an endogenous co‐IP assay further confirmed the interaction between XIAP and IRF7 in MEFs stimulated with SeV (Fig 6K). XIAP was previously shown to have three BIR domains and a ubiquitin‐associated (UBA) domain, which allows XIAP to bind to ubiquitin, and a RING domain. Co‐IP results indicated that the BIR2 domain is important for the interaction between XIAP and IRF7 (Fig 6L). These data indicate that XIAP acts as a positive regulator of IRF7 stability but not as the direct E3 ligase targeting IRF7 for degradation.
CUL3‐KLHL22 directly ubiquitinates IRF7 and thereby inhibits IFN‐I induction
To identify the E3 ligase that directly regulates IRF7 degradation, we used MS to analyze the ubiquitination‐related proteins that interact with IRF7 (Fig 7A). Among the molecules, four E3‐ligase‐related proteins (PRPF19, KLHL22, TRIM28, and DDB1) significantly increased the ubiquitination level of IRF7 in cotransfected HEK293T cells (Fig 7B). In contrast to the other three proteins, KLHL22, a substrate‐specific adaptor of the BTB‐CUL3‐RBX1 E3 ubiquitin ligase complex, decreased the protein level of IRF7, and this effect was reversed by MG132 treatment (Fig EV4A). Additionally, KLHL22 significantly decreased IRF7‐mediated IFN‐β transcriptional activity in a dose‐dependent manner (Fig 7C and D). These data suggest that CUL3‐KLHL22 is a potential E3 ligase that regulates the protein level and ubiquitination of IRF7.
Figure 7. CUL3‐KLHL22 directly promotes the ubiquitination and degradation of IRF7.
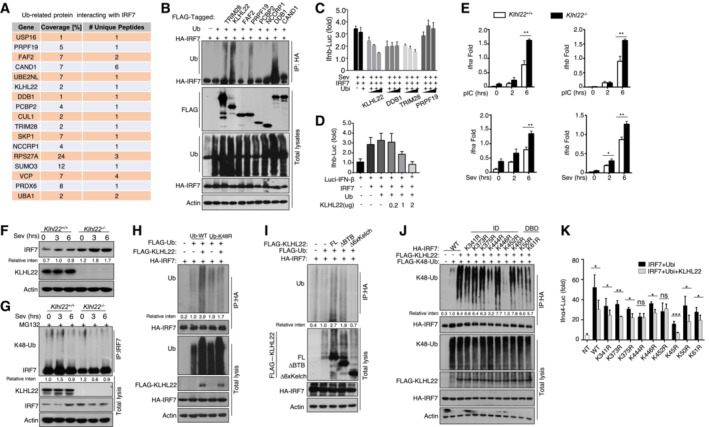
-
AThe list of ubiquitination (Ub)‐related proteins that interact with IRF7 according to the MS results.
-
BHEK293T cells were transfected with IRF7 and the indicated ubiquitin‐related expression plasmid. IB was performed to detect FLAG, followed by IP with an anti‐hyaluronic acid (HA) antibody.
-
C, DHEK293T cells were transfected with IFN‐β luciferase reporters and the indicated expression plasmids. Luciferase assays were performed to determine the fold changes with respect to the empty vector group.
-
EqRT–PCR analysis of IFN‐I in WT and KLHL22‐KO MEFs stimulated with polyI:C or SeV. The data are presented as the fold change relative to the Actb mRNA level.
-
FImmunoblot analysis of SeV‐induced IRF7 in whole‐cell lysates (WLs) of WT or KLHL22‐KO MEFs.
-
GAfter treatment with MG132, IRF7 was isolated by IP from WLs of KLHL22‐KO MEFs and subjected to IB using anti‐K48‐ubiquitin. Total cell lysates were also subjected to direct IB.
-
HHEK293T cells were transfected with IRF7 and FLAG‐K48R‐ubiquitin or FLAG‐ubiquitin in the presence (+) or absence (−) of KLHL22 expression plasmids. HA‐tagged IRF7 was isolated by IP, and the ubiquitination level was then measured by IB.
-
IHEK293T cells were transfected with IRF7 and distinct KLHL22 truncation mutants. IB of FLAG was performed, followed by IP with an anti‐HA antibody.
-
JHEK293T cells were transfected with KLHL22, FLAG‐K48‐ubiquitin, and various IRF7 point mutants. After treatment with MG132, IB of K48‐Ub was performed, followed by IP with an anti‐HA antibody.
-
KHEK293T cells were transfected with IFN‐α4 luciferase reporters and the indicated expression plasmids. Luciferase assays were performed to determine fold changes with respect to the empty vector group.
Data information: The data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. The data are presented as the means ± SDs. The significance of differences was determined by t‐test. *P < 0.05, **P < 0.01, and ***P < 0.005.
Please see Appendix Fig S5 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
Figure EV4. KLHL22 mediates K48‐linked ubiquitination of IRF7 and promotes its degradation.
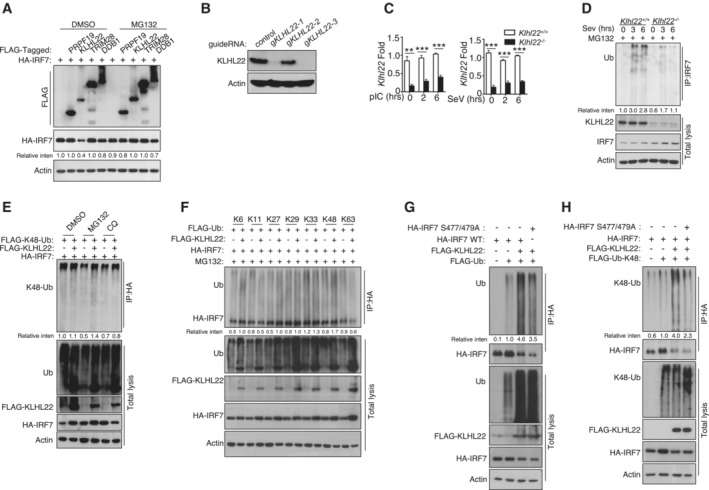
-
AHEK293T cells were transfected with IRF7 and the indicated expression plasmids. After treatment with MG132, the indicated proteins were detected by IB.
-
B, CIB and qRT–PCR analysis showing specific ablation of KLHL22 in KLHL22‐KO MEFs generated by CRISPR.
-
DAfter treatment with MG132, IRF7 was isolated by IP from whole‐cell lysates (WLs) of KLHL22‐KO MEFs and subjected to IB using anti‐ubiquitin. Total cell lysates were also subjected to direct IB.
-
EHEK293T cells were transfected with IRF7 and FLAG‐K48‐ubiquitin in the presence (+) or absence (−) of KLHL22 expression plasmids. After pretreatment with MG132 or CQ, HA‐tagged IRF7 was isolated by IP, and the ubiquitination level was then measured by IB.
-
FHEK293T cells were transfected with multiple ubiquitin mutants (mutations at K6, K11, K27, K29, K33, K48, and K63) and the indicated expression plasmids. HA‐tagged IRF7 was isolated by IP, and the ubiquitination level was then measured by IB.
-
G, HHEK293T cells were transfected with HA‐tagged IRF7 and the inactive mutant variants (S477 and 479A) whose phosphorylation sites at serine 477 and 479 were mutated from serine to alanine. HA‐tagged IRF7 was isolated by IP, and the ubiquitination level was then measured by IB.
Data information: Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. Data are represented as the means ± SDs. The data are representative of at least three biologically independent experiments.
Please see Appendix Fig S7 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
To further explore the role of KLHL22 in controlling the production of IFN‐I, we generated KLHL22‐KO MEFs by using CRISPR–Cas9 and single guide RNAs (sgRNAs; Fig EV4B and C). Notably, we observed a substantial increase in IFN‐I production in KLHL22‐KO MEFs compared with control MEFs upon polyI:C or SeV stimulation (Fig 7E). Moreover, an elevated IRF7 protein level was detected in KLHL22‐deficient MEFs, even in the early stage of stimulation (Fig 7F). IRF7 degradation relied on CUL3‐KLHL22‐mediated ubiquitination, especially K48‐linked ubiquitination (Figs 7G, and EV4D and E), and this degradation was blocked by the use of mutated K48R ubiquitin (Figs 7H and EV4F). Moreover, as suggested by the obvious reduction in IRF7 ubiquitination when the phosphorylation sites on serine 477 and 479 were mutated to alanine, KLHL22‐mediated ubiquitin‐dependent degradation of IRF7 was not wholly dependent on direct IRF7 phosphorylation (Fig EV4G and H). KLHL22 possesses a BTB domain with which CUL3 interacts and a six‐repeat Kelch domain that enables substrate recognition (Chen et al, 2018). The distinct truncation of KLHL22 indicated that the six‐repeat Kelch domain is more important than the BTB domain in promoting IRF7 ubiquitination (Fig 7I). Furthermore, recombinant CUL3‐RBX1‐KLHL22 proteins were sufficient to promote IRF7 ubiquitination in vitro (Fig EV5A and B). Co‐IP assays further showed that the interaction of IRF7 with KLHL22 was dependent on its inhibitor domain (Fig EV5C). KLHL22‐induced ubiquitination sites on IRF7 were mainly located in the ID and DBD, as suggested by a significant reduction in IRF7 ubiquitination when these two domains were retained (Fig EV5D). We next generated a series of point mutants in the ID and DBD of IRF7, replacing lysine (K) residues with arginine (R) residues. Some of the mutations moderately inhibited KLHL22‐mediated ubiquitination (Fig EV5E), but only the K444R and K452R IRF7 mutants, which exhibited more obvious reductions in KLHL22‐mediated K48‐linked ubiquitination (Fig 7J), were resistant to the effects of KLHL22‐mediated IFN‐I impairment (Fig 7K). In summary, we identified CUL3‐KLHL22 as an E3 ligase that mediates K48‐linked ubiquitination and proteasomal degradation of IRF7 and that is involved in IFN‐I induction.
Figure EV5. KLHL22 interacts with IRF7 and promotes its ubiquitination.
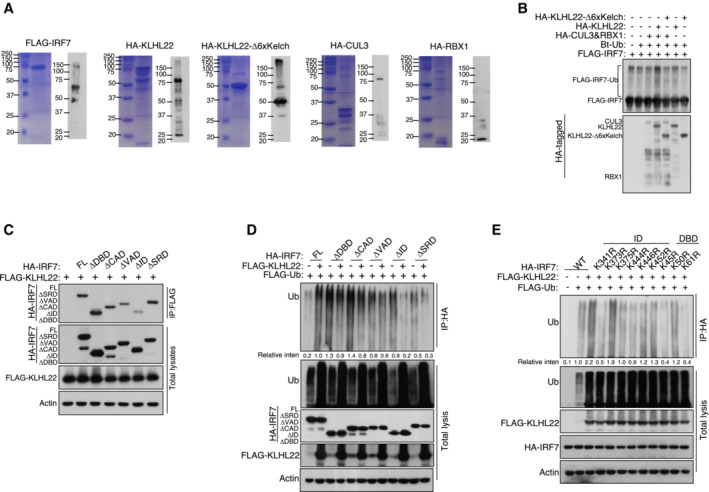
- Purification of recombinant proteins was performed by Coomassie blue staining and IB.
- CUL3‐KLHL22 E3 ligase catalyzed IRF7 ubiquitination in a cell‐free system. Ubiquitin and recombinant FLAG‐IRF7 were incubated with recombinant CUL3–RBX1 and KLHL22 or KLHL22‐Δ6xKelch.
- The associations between KLHL22 and various truncations of IRF7 were detected through the indicated IP and IB analyses.
- HEK293T cells were transfected with KLHL22 and IRF7 variants. IB of FLAG was performed followed by IP with an anti‐HA antibody.
- HEK293T cells were transfected with KLHL22, FLAG‐ubiquitin, and various IRF7 point mutants. IB of FLAG was performed, followed by IP with an anti‐HA antibody. The data are representative of at least three biologically independent experiments.
Data information: Please see Appendix Fig S8 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
XIAP promotes the ubiquitination of KLHL22 and suppresses its effect on IFN‐I production
To determine whether XIAP participates in CUL3‐KLHL22‐mediated IRF7 ubiquitination, we cotransfected these molecules into HEK293T cells. As shown in Fig 8A, CUL3‐KLHL22‐mediated ubiquitination of IRF7 was inhibited by XIAP in a dose‐dependent manner. Notably, XIAP promoted the polyubiquitination of KLHL22 in vitro (Fig 8B) and moderately reduced the KLHL22 protein level upon virus challenge (Fig 8C). Under MG132 treatment, XIAP overexpression enhanced the total and K48‐linked ubiquitination levels of KLHL22 (Fig 8D). To examine whether XIAP functions as an upstream regulator of KLHL22, we overexpressed XIAP in KLHL22‐KO MEFs using a retrovirus and confirmed the results by immunoblotting (IB; Fig 8E). The qPCR data revealed that XIAP overexpression did not promote further IFN‐I production in the absence of KLHL22 (Fig 8F), indicating that KLHL22 was the critical downstream mediator in XIAP‐regulated IFN‐I signaling. Overall, these data demonstrate that IRF7 ubiquitination was directly mediated by KLHL22 and that this process was suppressed by XIAP, which regulated the ubiquitination of KLHL22. As an antagonist of XIAP, XAF1 stabilized KLHL22 and attenuated IFN‐I production. Our findings not only provide essential insight into the mechanism by which IRF7 regulates protein stability but also suggest the potential advantages of antiviral drugs that target XAF1, XIAP, and/or KLHL22.
Figure 8. XIAP induces the ubiquitination of KLHL22.
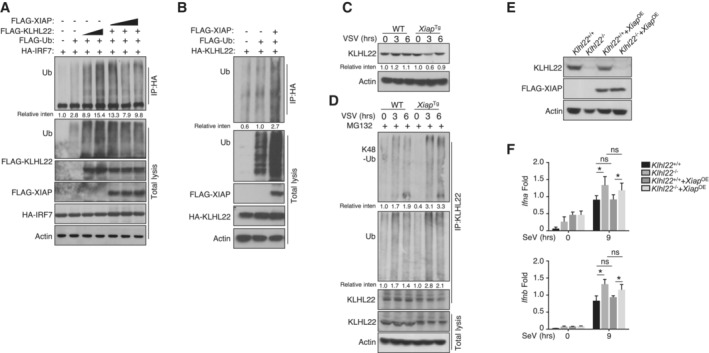
- HEK293T cells were transfected with FLAG‐tagged ubiquitin and the indicated expression plasmids. The ubiquitination levels of IRF7 were measured by IB. Cell lysates were subjected to direct IB.
- HEK293T cells were transfected with KLHL22 in the presence (+) or absence (−) of XIAP. IB of FLAG was performed followed by IP with an anti‐hyaluronic acid (HA) antibody.
- IB of VSV‐induced KLHL22 expression in whole‐cell lysates (WLs) of WT and Xiap Tg mice.
- After treatment with MG132, KLHL22 was isolated by IP from WLs of WT and Xiap Tg mice and subjected to IB using an anti‐ubiquitin or anti‐K48‐ubiquitin antibody. Protein lysates were also subjected to direct IB.
- IB showed the ablation of KLHL22 and the overexpression of FLAG‐XIAP in KLHL22‐KO MEFs expressing XIAP (KLHL22−/−XIAPOE).
- qRT–PCR analysis of IFN‐I in WT and KLHL22‐KO MEFs expressing XIAP and subjected to SeV stimulation.
Data information: The data are representative of at least three biologically independent experiments. Data from the qPCR assay are presented as the fold change relative to the Actin mRNA level. The data are presented as the means ± SDs. The significance of differences was determined by t‐tests. *P < 0.05.
Please see Appendix Fig S6 for information regarding replicates, quantification, and statistical evaluation for biochemical data in this figure.
Source data are available online for this figure.
Discussion
IRFs, especially IRF3 and IRF7, function as direct inducers of IFN‐I. When host cells undergo viral infection, IRF3‐ and IRF7‐mediated signaling cascades result in the induction of ISGs and suppress a series of biological processes (Thompson et al, 2011; Reikine et al, 2014). During the early stage of viral infection, inducible IFN‐I binds to IFNARs and activates Janus kinase (JAK) signaling cascades, which lead to the phosphorylation and activation of STAT1/2. Together with IRF9, phosphorylated STAT1/2 binds to the ISRE region on the promoters of various ISGs. As an ISG, IRF7 promotes further transcription of IFN‐I (Marie et al, 1998; Sato et al, 1998; Honda et al, 2006b). In the late stage of viral infection, this process is a common mechanism to protect uninfected cells from viral invasion. In addition to its association with a viral infection, IRF7 is strongly associated with the onset of SLE, as suggested by assays of single‐nucleotide polymorphisms (SNPs) (International Consortium for Systemic Lupus Erythematosus et al, 2008; Okabe et al, 2008; Salloum et al, 2010). IRF7 plays multiple roles in viral infections and inflammatory diseases, and a negative feedback mechanism to control IRF7 stability and activation is essential to maintain immune homeostasis. However, whether a specific ISG suppresses the protein expression of IRF7 and, if so, via what mechanism, remains unclear. This lack of knowledge has prevented the development of antiviral or anti‐inflammatory drugs that target IRF7.
Consistent with our findings, those of other studies have demonstrated that MG132 treatment or mutation of certain E3 ligases, such as CUL1 (a component of the E3 ligase complex SKP1‐CUL1‐F‐box) and TRIM21/Ro52, contributes to the stability of IRF7 and promote its ubiquitination (Sato et al, 2000a; Prakash & Levy, 2006). RTA‐associated ubiquitin ligase (RAUL), a component of HECT E3 ubiquitin ligase, has been found to promote the ubiquitination of the transcription factor TIP120B for proteolysis. Similar to TRIM21, RAUL regulates the stability of both IRF3 and IRF7. This ligase broadly contributes to the inhibition of IRF3/7 to maintain suitable IFN‐I production levels under standard physiological conditions or after viral infection. Some viruses escape immune surveillance by hijacking RAUL to promote the degradation of IRF3 and IRF7 (Yu & Hayward, 2010). In contrast to RAUL, there are no reports that XAF1, an ISG, is hijacked by pathogens; moreover, its function is crucial for IFN‐I signaling in uninfected and resting innate immune cells to prevent the continuous expression of IFN‐I. These observations suggest that XAF1 may be a potential antiviral drug because it maintains an effective and proper amount of IFN‐I.
In this study, we clarified how PTM, especially ubiquitination, regulates IRF7 stability to repress IFN‐I production. Indeed, the protein level of IRF7 can be reduced at the translational stage. OASL1, an ISG, has been demonstrated to inhibit the translation of IRF7, which negatively regulates IFN‐I production (Lee et al, 2013). This finding may also explain the observation that highly abundant Irf7 mRNA did not consistently lead to a high IRF7 protein level. Indeed, our data revealed an interesting mechanism by which IFN‐I receptor signaling and an ISG negative feedback loop regulate endogenous IRF7 protein levels. In addition to influencing viral infection, IFN‐I displays an antitumor function and the ability to induce chronic inflammatory diseases such as SLE. The XAF1‐XIAP‐KLHL22 axis functions as a major regulator that controls IRF7 protein abundance and helps to maintain a proper level of this protein. XAF1 has been shown to regulate apoptosis and promote antitumor activity. Furthermore, KLHL22 plays a critical role in mediating the activation of mTORC1 and may be an oncogene, making it a potential therapeutic target in breast cancer (Chen et al, 2018). Therefore, in future, it will be interesting to determine whether XAF1‐XIAP‐KLHL22‐mediated downregulation of IFN‐I in innate immune cells is involved in promoting tumorigenesis.
In summary, we determined that the stability of the IRF7 protein is regulated by CUL3‐KLHL22 through the K48‐linked polyubiquitin‐dependent proteasomal pathway. In addition, the balance of XAFI and XIAP serves to dynamically adjust the activity and stability of KLHL22, indirectly controlling the production of IRF7. The mRNA level of Irf7 is continuously elevated during virus challenge, from the early to late stages, but no permanently induced transcription of IFN‐I was observed, which indicates that negative feedback signaling protects against tissue damage due to cytokine hyperexpression. Previous studies have shown that XAF1 forms a complex with IRF1 to promote IRF1‐mediated transcription of proapoptotic genes (Jeong et al, 2018). IRF1 was originally recognized as a transcription factor that induces ISG expression (Yarilina et al, 2008). It would also be useful to examine the role of IRF1 in IFN‐I induction and its relationship with KLHL22. Overall, our study provides novel ideas for drug targets that repress viral infection and/or IFN‐I‐mediated autoimmune diseases.
Materials and Methods
Mice and cell lines
Xaf1 fl/fl mice were produced by Shanghai Model Organisms Center, Inc. To generate macrophage conditional Xaf1 MKO (Xaf1 f/f Lyz2‐Cre) mice or Xaf1 −/− mice, the Xaf1‐floxed mice were crossed with Lyz2‐Cre (004781) or EIIa‐Cre (003724) mice (all from Jackson Laboratory, C57BL/6 background), respectively. The Ifnar1 −/− mice were a gift from Prof. Shu Zhu (Zhejiang University), and the Irf7 −/− mice were a gift from Prof. Xiao‐peng Qi (Shandong University). For experiments, conditional KO mice were compared with their littermate WT (Xaf1 fl/fl ) mice, which served as controls. In the animal studies, the WT and multiple KO mice were randomly grouped. The experimental data were collected in a blinded manner, and each individual was identified with an ear tag. Mice were kept in specific pathogen‐free (SPF) conditions, and all animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Zhejiang University. The gene‐specific PCR primers used for genotyping are shown in Table EV1.
For MEFs, heterozygous mice were bred to obtain Xaf1 −/− and WT embryos from the same pregnant female mice. Embryos were removed from mice at embryonic days (E) 13.5–14.5 and were subjected to cutting and enzymatic disaggregation after the removal of blood and liver tissue to generate single MEFs.
Isolation of PBMCs
PBMCs were isolated from anticoagulated blood samples from healthy donors (College of Animal Sciences Zhejiang University and Life Sciences Institute, Zhejiang University), by using lymphocyte separation media (MULTISCIENCES) and density‐gradient centrifugation. The use of PBMCs was in compliance with institutional guidelines and approved protocols of Zhejiang University. Written informed consent was obtained from all adult subjects.
Antibodies and reagents
Antibodies targeting IkBa (C‐21, 1:1,000), p65 (C‐20, 1:1,000), Lamin B (C‐20, 1:1,000), ERK (K‐23, 1:2,000), phospho‐ERK (E‐4, 1:1,000), JNK2 (C‐17, 1:1,000), p38 (H‐147, 1:1,000), p105/p50 (C‐19, 1:1,000), TBK1 (108A429, 1:1,000), IRF3 (SC‐9082, 1:1,000), STAT1 (sc‐592, 1:1,000), RelB (C‐19; 1:1,000), and c‐Rel (sc‐71, 1:1,000), ubiquitin (P4D1; 1:1,000), and control rabbit IgG (sc‐2027) were purchased from Santa Cruz Biotechnology. Antibodies targeting phospho‐IκBα (Ser32, 14D4, 1:1,000), phospho‐JNK (Thr180/Tyr185, #9251, 1:1,000), phospho‐p38 (Thr180/Tyr182, 3D7, 1:1,000), phospho‐p105 (Ser933, 18E6, 1:1,000), phospho‐TBK1 (Ser172, D52C2, 1:1,000), phospho‐IRF3 (Ser396, 4D4G, 1:1,000), phospho‐STAT1 (Ser727, 9177 S, 1:1,000), p100/p52 (4,882; 1:1,000), and K48‐linkage‐specific polyubiquitin (D9D5, 1:8,000) were purchased from Cell Signaling Technology Inc. Anti‐actin (C‐4; 1:10,000), anti‐HA (12CA5), anti‐FLAG (M2), horseradish peroxidase (HRP)–conjugated anti‐HA (3F10), and anti‐FLAG (M2) were purchased from Sigma–Aldrich. IRF7 (ET1‐89610) was purchased from HUABIO. KLHL22 (16214‐1‐AP) was purchased from Proteintech. Xaf1 (GTX51339) was purchased from GeneTax. Other fluorescence‐labeled antibodies are listed in the section on flow cytometry and cell sorting.
LPS (derived from Escherichia coli strain 0127: B8) and CpG (2216) were purchased from Sigma–Aldrich. R848 and polyI:C were purchased from Amersham. MG132 (N‐carbobenzyloxy‐L‐leucyl‐L‐leucyl‐L‐leucinal) was applied for 2 h (at 10 μM for HEK293T cells) or 1 h (at 5 μM for BMDMs).
Biosafety
Experiments with H3N2, H5N1, and H1N1 (PR8) virus were carried out in a biosafety level 3 facility, and these viruses were obtained from the Center for Disease Control and Prevention of Zhejiang province. All animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Zhejiang University.
Plasmids
FLAG‐tagged ubiquitin, K6, K11, K27, K29, K33, K48, and K63 plasmids in the pRK5 vector and TRIF, TBK1, MAVS, RIG‐I, IRF1, IRF2, and IRF3 plasmids were provided by S.‐c. Sun. XAF1, XIAP, IRF7, and KLHL22 were cloned into the expression vector for HA‐tagged pcDNA3.1 or FLAG‐tagged pRK5. IRF7 truncated variants (1–237, 1–410, 132–457, and 238–457) were subcloned into HA‐tagged pcDNA3.1 vector by PCR. Truncated XAF1 variants (1–131, 1–172, 28–301, 50–301, 80–301, 108–301, and 132–301) and XIAP variants (1–264, 1–372, 1–449, 163–497, and 265–497) were subcloned into FLAG‐tagged PRK5 vectors. The domain‐deleted plasmids of XAF1 (ΔZF4, ΔZF5, ΔZF6, ΔZF4‐6) and XIAP (ΔBIR2, ΔBIR3, ΔBIR2‐3) were all subcloned into the FLAG‐tagged PRK5 vector.
LentiCRISPR v2 plasmids were provided by Dr. Li Shen; we then replaced puromycin with a GFP tag to obtain the lentiCRISPR v2‐GFP plasmid. pMD2.G and psPAX2 plasmids were provided by Prof. Yichuan Xiao.
Macrophage preparation and stimulation
WT, Xaf1 MKO, or Xiap Tg mice were sacrificed and soaked in 70% ethanol for a few minutes. Then, the tibia and fibula were removed, the muscle and connective tissue were excised, and the bone marrow (BM) cells were collected. BM cells were cultured with DMEM plus 20% FBS and 30% L929 cell culture supernatant. Three days later, the culture media were changed. Mature BMDMs were collected in PBS containing 2.5 mM EDTA and 5% FBS after 5 days. After starvation in DMEM containing 1% FBS for 8 h, the cells were subjected to experiments.
Starvation‐treated BMDMs were stimulated with LPS (1 μg/ml for IB experiments and 100 ng/ml for cytokine induction experiments), pIC (20 μg/ml), CpG (5 μM), R848 (1 μg/ml), SeV, HSV, or VSV (multiplicity of infection, MOI = 1), or VSV‐GFP (MOI = 0.1). Total and subcellular extracts were prepared for IB assays, and total RNA was prepared for qRT–PCR assays.
ELISA and real‐time quantitative PCR (RT–qPCR)
Samples for ELISA were subjected to a commercial system from eBioscience for analysis. For RT–qPCR, total RNA was extracted using TRIzol reagent (Life Technology) and subjected to cDNA synthesis; cDNA was then used with iQTM SYBR Green Supermix (Bio‐Rad) for qRT–PCR analysis, which was performed in an iCycler sequence detection system (Bio‐Rad). The quantitative results were normalized to Actb. The gene‐specific PCR primers (all for mouse genes) are listed in Table EV1.
Immunoblot and immunoprecipitation
The prepared cell lysates or subcellular extracts were subjected to IB or IP immediately. For IP in primary cells, IRF7 was pulled down by using an IRF7 antibody (anti‐IRF7). For IP in HEK293 cells that overexpressed the indicated tagged vectors, IP was performed with an anti‐FLAG M2/anti‐HA antibody. Subsequently, immunoprecipitated samples were enriched with suitable agarose beads. Elution from microbeads was subjected to SDS–polyacrylamide gel electrophoresis (SDS–PAGE). The separated proteins were transferred onto nitrocellulose (NC) membranes (GE) after electrophoresis. For IB, the NC membrane was blocked with 5% nonfat milk and incubated with a specific primary antibody and HRP‐conjugated secondary antibody successively. Signals were detected by enhanced chemiluminescence (Amersham Biosciences).
Luciferase reporter gene assays
HEK293 cells (2 × 105) were transfected by calcium phosphate precipitation with a firefly luciferase reporter driven by the IFN‐β or IFNα4 promoter, along with the mentioned cDNA expression vectors and the control Renilla luciferase reporter. After transfection for approximately 36 h, the cells were lysed with 1× Passive Lysis Buffer and violent vibration for 15 min and then collected for dual‐luciferase assays (Promega). The activity of firefly luciferase was normalized to that of Renilla luciferase.
Generation of KLHL22‐KO MEFs
KLHL22‐KO stable MEF lines were generated using a lentiviral system. CRISPR guide sequences targeting the second exon of Klhl22 were designed by http://crispr.mit.edu and cloned into lentiCRISPR v2‐GFP vectors. The sequences were as follows: sgKLHL22_1_mouse: 5′ GGATCCTCTTTGACGTTGTCC 3′; sgKLHL22_2_mouse: 5′ GATCGGATTCTGCTAGCTGCA 3′; and sgKLHL22_3_mouse: 5′ GGGAGTGCTGTGTGCTTCGAT 3′. HEK293T cells were used to package the virus. HEK293T cells in 6‐well plates were transfected with the guide RNA plasmids indicated above, together with pMD2.G and psPAX2. After 36 h of transfection, supernatants containing the virus were collected to infect wild‐type MEFs supplemented with 8 μg/ml polybrene for 24 h. GFP‐positive cells were sorted into 96‐well plates and validated by western blotting.
Ubiquitination assays
BMDMs or MEFs were pretreated with or without MG132 for 2 h and then lysed with Nonidet P‐40 lysis buffer (50 mM Tris–HCl, pH 7.5, 120 mM NaCl, 1% Nonidet P‐40, 1 mM EDTA, and 1 mM DTT) containing 6 M urea and protease inhibitors. IRF7 was isolated by IP with antibodies targeting IRF7. The ubiquitinated proteins were detected by IB using an anti‐ubiquitin (Santa, P4D1) or anti‐K48‐linkage‐specific polyubiquitin (D9D5) antibody. For transfection, HEK293T cells were pretreated with or without MG132 or chloroquine and lysed on ice for 15 min with the lysis buffer mentioned above. Then, the samples were centrifuged at 13,300 g. For 5 min at 4°C. Supernatants were then diluted to 1:3 with Nonidet P‐40 lysis buffer containing protease but not 6 M urea. For the ubiquitination assay of HA‐tagged IRF7 or KLHL22, anti‐HA antibodies were added to diluted lysates for overnight binding. Protein A beads were then incubated with lysates at 4°C for 1 h to enrich anti‐HA antibodies. Immunoprecipitates were washed three times with Nonidet P‐40 lysis buffer containing 0.1% DTT and analyzed by IB.
For in vitro ubiquitination assays, 500 ng of HA‐CUL3 and HA‐RBX1 mixed with 2 μg of HA‐KLHL22 or HA–KLHL22‐Δ6 × Kelch purified from E. coli BL21 cells was used. The E3 ligase complex was mixed with 2 μg of FLAG‐IRF7 purified from E. coli BL21 cells, 1.2 μg of biotinylated‐Ub, 600 ng of E1 (UBE1), 800 ng of E2 (UBCH5B), 5 mM Mg‐ATP, and 10 × ubiquitin reaction buffer and incubated at 37°C for 1 h. The above reagents, except for the annotated reagents, were purchased from Enzo Life Sciences. Reactions were terminated by boiling at 100°C with SDS sample buffer for 10 min and analyzed by IB.
Purification of recombinant proteins
Recombinant human FLAG‐IRF7, HA‐KLHL22, HA‐KLHL22‐Δ6 × Kelch, HA‐CUL3, and HA‐RBX1 were amplified by PCR and inserted into the pGFP‐2 × Strep expression vector. All recombinant proteins were expressed in 400 ml of E. coli BL21 cells and cultured at 37°C until the optical density reached 0.6. For induction, 0.2 mM IPTG was added to the culture overnight at 16°C. Harvested cells were resuspended in lysis buffer (50 mM Tris–HCl at pH 7.5, 500 mM NaCl, 1 mM EDTA·2Na, 1% Triton X‐100, 20 mM β‐ME, and 1 mM PMSF) and lysed by sonication (40% output, 16 cycles of 15‐s on and 60‐s off). After centrifugation at 35,000 g for 20 min, the soluble fractions of the lysates were loaded onto a gravity flow column filled with Strep‐Tactin beads (NUPTEC). After extensive washes with washing buffer (20 mM Tris–HCl at pH 7.5, 500 mM NaCl, 1 mM EDTA·2Na, 1% Triton X‐100, 20 mM β‐ME, and 0.1 mM PMSF), the bound proteins were eluted with the same buffer containing 10 mM desthiobiotin. The proteins were then dialyzed overnight at 4°C against 20 mM Tris at pH 7.5, 150 mM NaCl, 10% glycerol, and 1 mM DTT. After dialysis, the purified proteins were concentrated with Amicon Ultra Centrifugal Filters (Millipore) and stored at −80°C. The purity of the proteins was examined by SDS–PAGE and Coomassie Blue staining, and their concentrations were determined by the Bradford assay in combination with Coomassie Blue staining.
Fluorescence microscopy
Macrophages were seeded into 12‐well plates containing 70% alcohol‐pretreated slides for starvation overnight; they were infected with VSV‐GFP in serum‐free medium for 1 h, and this medium was replaced with growth medium for another 11 h. Cells treated with or without stimulation were fixed with 4% paraformaldehyde (PFA) for 20 min. Then, the cells were washed with PBS three times. The infected (GFP+) cells were visualized under a fluorescence microscope and quantified by flow cytometry.
Flow cytometry
Single‐cell suspensions from bone marrow, spleen, or draining lymph nodes were subjected to flow cytometry using CytoFlex (Beckman Coulter) and the following fluorescence‐labeled antibodies from eBioscience: PB‐conjugated anti‐CD4; PE‐conjugated anti‐F4/80; PerCP5.5‐conjugated anti‐CD8 and anti‐Ly6G; APC‐conjugated anti‐CD62L and CD3; APC‐CY7‐conjugated anti‐CD11b; FITC‐conjugated anti‐CD44 and Foxp3; and PE‐conjugated anti‐B220.
Virus infection
For the H1N1 (PR8) infection model, age‐matched (6–8 weeks of age) mice were housed in microisolator cages in a Biosafety Level 3 facility. Mice were anesthetized with dry ice and received 1 × 106 TCID50 PR8 (obtained from the Center for Disease Control and Prevention of Zhejiang province) in 40 μl phosphate‐buffered saline dripped into the nasal cavity. The infected mice were monitored to determine changes in body weight and survival rate for 8 days. Samples of sera, lung, trachea, and spleen from the infected mice were collected immediately after sacrifice on day 2 and day 5. The sera were used for measuring IFNs by ELISA, and the tissues were homogenized to determine the various copies. In addition, a fraction of the lung in the same area was placed into formalin and processed for histology. For the model of HSV‐1 infection, the virus (100 μl) was administered by intravenous inoculation of the lateral tail vein; mice were monitored to determine the survival rate for up to 12 days. Sera and spleen samples were collected to measure IFN production or virus load. For the in vivo and in vitro infections with VSV, SeV, or HSV‐1, the MOI was set to 1.
DSS‐induced colitis
To determine changes in mortality, DSS (molecular mass, 36,000 to 50,000 Da; MP Biomedicals) was added to drinking water at 3.5% (w/v) for 6 days followed by 2 days of regular water. Body weight and the presence of diarrhea and hematochezia were recorded every day for up to 8 days, and the mice were then sacrificed to enable colon length measurement, histological analysis, and immune cell isolation from the mucosa.
Histopathology
The lung fraction was removed from WT or Xaf1 MKO mice, fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned, stained with H&E, and then examined by light microscopy to determine histological changes. Two different investigators evaluated and determined the inflammation scores of randomly numbered slides.
ChIP–qPCR assay
ChIP assays were performed with macrophages stimulated for 6 h with VSV or HSV‐1. The cells were fixed with 1% formaldehyde and sonicated as previously described (Nelson et al, 2006). Lysates were subjected to IP with the appropriate antibodies, and the precipitated DNA was then purified by Qiaquick columns (Qiagen) and quantified by qPCR using a pair of primers that amplified the target region of the indicated promoter. The precipitated DNA is presented as the frequency of the total input DNA.
LC–MS/MS analysis
To identify proteins that interact with IRF7, macrophages were seeded into 10‐cm dishes and starved overnight. Cells were treated with IFN‐β for 4 h before lysis. Lysates were collected, and IRF7 was pulled down for IP. Samples were resolved by SDS–PAGE, and the whole lane was analyzed by MS. All proteome samples were analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) using an EASY nLC3000 connected to a Thermo Fisher Scientific Q Exactive HF‐X MS system. MS data files were analyzed using MaxQuant software with the integrated Andromeda search engine. Data were compared against the human UniProt database (20,367 entries, release‐2019_04; SwissProt) supplemented with commonly observed contaminants. The MS data detailing putative IRF7‐interacting proteins and ubiquitination‐related proteins that interact with IRF7 are shown in Table EV2.
RNA‐seq analysis
RNA samples were collected from WT and Xaf1MKO BMDMs with or without IFN‐β/LPS stimulation. RNA was purified with the Qiagen RNeasy Mini Kit according to the manufacturer's instructions and subjected to RNA‐seq analysis. RNA sequencing was performed by the Life Science Institute Sequencing and Microarray Facility using an Illumina sequencer. The raw reads were aligned to the mouse reference genome (mm10) using Tophat2 RNA‐sequencing alignment software. The mapping rate was 70% overall across all the samples in the dataset.
DEGs were identified in the count data using the R package DESeq2. Overlapping genes were analyzed for Gene Ontology biological processes using DAVID. DEGs (fold change of ≥ 1.5 or ≤ 1.5) of LPS‐activated BMDMs compared with nontreated BMDMs were identified. Overlapping genes were archived for network analysis. A network of interactions was constructed using STRING 10. KEGG pathway enrichment analysis was performed, and significantly enriched terms based on low P‐values and false discovery rates were used for further analysis.
Statistical analysis
Data are presented as the means ± SDs. The two‐tailed Student's t‐test was used to compare the differences between the two groups. One‐way analysis of variance (ANOVA) was applied, and the Bonferroni correction was used for multiple comparisons. The Gehan‐Breslow‐Wilcoxon test was used for the animal survival assay. P‐values of < 0.05 were considered significant, and the level of significance is indicated as * (P < 0.05), ** (P < 0.01), or *** (P < 0.001). All statistical analyses were performed using Prism 6 software.
Author contributions
Bao‐qin Liu: Investigation; methodology; writing – original draft; writing – review and editing. Rong‐bei Liu: Investigation; methodology; writing – original draft; writing – review and editing. Wen‐ping Li: Investigation; methodology; writing – original draft. Xin‐tao Mao: Resources; data curation; investigation; methodology. Yi‐ning Li: Investigation. Tao Huang: Investigation. Hao‐li Wang: Investigation. Hao‐tian Chen: Investigation. Jiang‐yan Zhong: Validation; investigation; visualization. Bing Yang: Data curation; methodology. Ren‐jie Chai: Visualization; methodology. Qian Cao: Resources; validation; methodology. Jin Jin: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing. Yi‐yuan Li: Conceptualization; supervision; funding acquisition; investigation; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank the core facilities of the Life Sciences Institute of Zhejiang University for technical assistance. This study was supported by the distinguished Young Scientist Fund of the Natural Science Foundation of China (82125016), Zhejiang Provincial Natural Science Foundation of China (LR19C080001, LQ21H030013), Jiangsu Science and Technology Project (Social Development) (BE2019669), National Natural Science Foundation of China (82071046, 82100540), and Key R&D Program of Zhejiang Province (2020C03075).
EMBO Reports (2023) 24: e55387.
Data availability
The raw RNA‐sequence data reported in this paper were deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2017) at the Beijing Institute of Genomics (BIG) Data Center (Nucleic Acids Res 2021), BIG, Chinese Academy of Sciences, under the accession number BioProject PRJCA007172. The link is https://ngdc.cncb.ac.cn/gsa/browse/CRA005357.
References
- Bais SS, Ratra Y, Khan NA, Pandey R, Kushawaha PK, Tomar S, Medigeshi G, Singh A, Basak S (2019) Chandipura virus utilizes the prosurvival function of RelA NF‐κB for its propagation. J Virol 93: e00081‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Kehrli E, Eisenbeis CF, Storb U, Singh H (1996) Pip, a lymphoid‐restricted IRF, contains regulatory domain that is important for autoinhibition and ternary complex formation with the Ets factor PU.1. Genes Dev 10: 2335–2347 [DOI] [PubMed] [Google Scholar]
- Chen J, Ou Y, Yang Y, Li W, Xu Y, Xie Y, Liu Y (2018) KLHL22 activates amino‐acid‐dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature 557: 585–589 [DOI] [PubMed] [Google Scholar]
- Dusheiko G (1997) Side effects of alpha interferon in chronic hepatitis C. Hepatology 26: 112S–121S [DOI] [PubMed] [Google Scholar]
- Higgs R, Jefferies CA (2008) Targeting IRFs by ubiquitination: regulating antiviral responses. Biochem Soc Trans 36: 453–458 [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T (2006a) Type I inteferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25: 349–360 [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T (2006b) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25: 349–360 [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida NJN (2005) IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 434: 772–777 [DOI] [PubMed] [Google Scholar]
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN) , Harley JB, Alarcon‐Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD et al (2008) Genome‐wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40: 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies CA (2019) Regulating IRFs in IFN driven disease. Front Immunol 10: 325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S‐I, Kim J‐W, Ko K‐P, Ryu B‐K, Lee M‐G, Kim H‐J, Chi S‐G (2018) XAF1 forms a positive feedback loop with IRF‐1 to drive apoptotic stress response and suppress tumorigenesis. Cell Death Dis 9: 806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kim B, Oh GT, Kim YJ (2013) OASL1 inhibits translation of the type I interferon‐regulating transcription factor IRF7. Nat Immunol 14: 346–355 [DOI] [PubMed] [Google Scholar]
- Lin FC, Young HA (2014) Interferons: success in anti‐viral immunotherapy. Cytokine Growth Factor Rev 25: 369–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohoff M, Mak TW (2005) Roles of interferon‐regulatory factors in T‐helper‐cell differentiation. Nat Rev Immunol 5: 125–135 [DOI] [PubMed] [Google Scholar]
- Marie I, Durbin JE, Levy DE (1998) Differential viral induction of distinct interferon‐alpha genes by positive feedback through interferon regulatory factor‐7. EMBO J 17: 6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A et al (1993) Targeted disruption of IRF‐1 or IRF‐2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75: 83–97 [PubMed] [Google Scholar]
- McNab F, Mayer‐Barber K, Sher A, Wack A, O'Garra A (2015) Type I interferons in infectious disease. Nat Rev Immunol 15: 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, Arthur JSC, Flavell RA, Ghosh S, Paludan SR (2009) RIG‐I‐mEediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells. J Biol Chem 284: 10774–10782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz‐Fontela C, Mandinova A, Aaronson SA, Lee SW (2016) Emerging roles of p53 and other tumour‐suppressor genes in immune regulation. Nat Rev Immunol 16: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185 [DOI] [PubMed] [Google Scholar]
- Nelson N, Marks MS, Driggers PH, Ozato K (1993) Interferon consensus sequence‐binding protein, a member of the interferon regulatory factor family, suppresses interferon‐induced gene transcription. Mol Cell Biol 13: 588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning S, Pagano J, Barber GNJG (2011) IRF7: activation, regulation, modification and function. Immunity 12: 399–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, Kawane K, Nagata S (2008) IFN regulatory factor (IRF) 3/7‐dependent and ‐independent gene induction by mammalian DNA that escapes degradation. Eur J Immunol 38: 3150–3158 [DOI] [PubMed] [Google Scholar]
- Prakash A, Levy DE (2006) Regulation of IRF7 through cell type‐specific protein stability. Biochem Biophys Res Commun 342: 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikine S, Nguyen JB, Modis Y (2014) Pattern recognition and signaling mechanisms of RIG‐I and MDA5. Front Immunol 5: 342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnblom L, Leonard D (2019) Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med 6: e000270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Tun‐Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S (2006) Negative regulation of interferon‐regulatory factor 3‐dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol 7: 598–605 [DOI] [PubMed] [Google Scholar]
- Salloum R, Franek BS, Kariuki SN, Rhee L, Mikolaitis RA, Jolly M, Utset TO, Niewold TB (2010) Genetic variation at the Irf7/Phrf1 locus is associated with autoantibody profile and serum interferon alpha activity in lupus patients. J Invest Med 58: 661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N (1998) Positive feedback regulation of type I IFN genes by the IFN‐inducible transcription factor IRF‐7. FEBS Lett 441: 106–110 [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N et al (2000a) Distinct and essential roles of transcription factors IRF‐3 and IRF‐7 in response to viruses for IFN‐alpha/beta gene induction. Immunity 13: 539–548 [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka NJI (2000b) Distinct and essential roles of transcription factors IRF‐3 and IRF‐7 in response to viruses for IFN‐α/β gene induction. Immunity 13: 539–548 [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Emre NC, Romesser PB, Staudt LM (2009) IRF4: immunity. malignancy! therapy? Clin Cancer Res 15: 2954–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R, Gao Y, Dozmorov I, Malladi V, Saha I, McDaniel MM, Parameswaran S, Liang C, Arana C, Zhang B et al (2021) IRF1 governs the differential interferon‐stimulated gene responses in human monocytes and macrophages by regulating chromatin accessibility. Cell Rep 34: 108891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MR, Kaminski JJ, Kurt‐Jones EA, Fitzgerald KA (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3: 920–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Basagoudanavar SH, Wang X, Hopewell E, Albrecht R, García‐Sastre A, Balachandran S, Beg AA (2010) NF‐κB RelA subunit is crucial for early IFN‐β expression and resistance to RNA virus replication. J Immunol 185: 1720–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai H, Negishi H, Taniguchi T (2012) The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Onco Targets Ther 1: 1376–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarilina A, Park‐Min K‐H, Antoniv T, Hu X, Ivashkiv LB (2008) TNF activates an IRF1‐dependent autocrine loop leading to sustained expression of chemokines and STAT1‐dependent type I interferon–response genes. Nat Immunol 9: 378–387 [DOI] [PubMed] [Google Scholar]
- Yu Y, Hayward GS (2010) The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33: 863–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang SE, Hayward GS (2005) The KSHV immediate‐early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome‐mediated degradation. Immunity 22: 59–70 [DOI] [PubMed] [Google Scholar]