

Abstract
In recent years, multiple disciplines have focused on mitochondrial biology and contributed to understanding its relevance towards adult-onset neurodegenerative disorders. These are complex dynamic organelles that have a variety of functions in ensuring cellular health and homeostasis. The plethora of mitochondrial functionalities confers them an intrinsic susceptibility to internal and external stressors (such as mutation accumulation or environmental toxins), particularly so in long-lived postmitotic cells such as neurons. Thus, it is reasonable to postulate an involvement of mitochondria in aging-associated neurological disorders, notably neurodegenerative pathologies including Alzheimer’s disease and Parkinson’s disease. On the other hand, biological effects resulting from neurodegeneration can in turn affect mitochondrial health and function, promoting a feedback loop further contributing to the progression of neuronal dysfunction and cellular death. This review examines state-of-the-art knowledge, focus on current research exploring mitochondrial health as a contributing factor to neuroregeneration, and the development of therapeutic approaches aimed at restoring mitochondrial homeostasis in a pathological setting.
Key Words: Alzheimer’s disease, axon, energy homeostasis, glymphatic system, mitochondria, mitostasis, neurodegeneration, neuroregeneration, Parkinson’s disease, therapeutical strategies
Introduction
Mitochondria are ubiquitous cytoplasmic organelles that form tubular structures, organized in a cell-spanning network. As dynamic organelles, mitochondria are under constant fission, fusion, mitophagy, and biogenesis (Figure 1). Mitochondrial fission is a cell division process that directs mitochondrial contents, creates heterogeneity, and contributes to the eradication of defective mitochondria (Zhou et al., 2017). Mitochondrial health and function are ensured by coordinated biogenesis and mitophagy (Ho et al., 2017).
Figure 1.
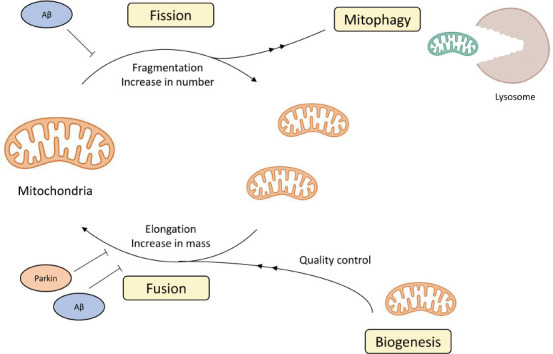
The complicated lives of mitochondria.
The figure illustrates the lifecycle of mitochondria and the effect of disease-specific protein interaction on mitochondria dynamics. Orange mitochondria represent ones, and green mitochondria represent damaged ones (namely from oxidative damage). Blue circles represent the Alzheimer’s disease-related peptide Aβ and orange circle represents the Parkinson’s disease-related Parkin. Aβ: Amyloid beta.
In addition to their widely described role as the primary cellular adenosine 5′ triphosphate (ATP) producers, mitochondria control important cellular events, such as Ca2+ homeostasis, reactive oxygen species (ROS) signaling, ion channel regulation, or exocytosis (Rowland and Voeltz, 2012; Trigo et al., 2022). Mitochondrial fusion allows for the exchange of mitochondrial material and contributes to the regulation of ROS and calcium buffering. In fact, cellular homeostasis is directly overseen by mitochondria via modulation of intracellular calcium levels, together with the endoplasmic reticulum or in response to stress, by activating the unfolded protein response, primarily accomplished through ROS signaling and immune response modulation (Singer and Chandel, 2019; Trigo et al., 2022). Mitochondrial ATP synthesis occurs in a well-characterized and described manner, along the mitochondrial electron transport chain complex (Zhao et al., 2019); however, a by-product of this process is the production of ROS. Classically associated with cellular damage and deleterious effects (Brand, 2016), recent studies have established important signaling roles for ROS in a physiological and healthy state (D’Autréaux and Toledano, 2007). Indeed, these are described to be crucial messengers in several signaling pathways (De Giusti et al., 2013), with ROS signal transfer happening via oxidative modification of proteins, such as receptors, kinases, phosphatases, caspases, and transcription factors (De Giusti et al., 2013).
On the other hand, due to its plethora of cellular roles, mitochondrial stress has important links to apoptotic, necrotic, and apoptosis-necrosis hybrid cell death mechanisms (Fricker et al., 2018), playing an important role in the development and progression of neurodegenerative diseases (Naumova and Šachl, 2020). For a long time, this topic has stimulated the interest of physicians and scientists, and the role of mitochondria in apoptosis and the toxic effects of ROS production have been widely studied (Kausar et al., 2018); however, the fact that most clinical attempts to modulate these processes on their own have failed mandates a re-assessment of pathophysiology. This review explores the various roles and therapeutic potential of mitochondria in neurological disorders, with a focus on novel research aiming to promote mitochondria homeostasis to restore neuronal function in pathological settings.
Search Strategy and Selection Criteria
This review article used PubMed of the National Institute of Health (NIH), National Library of Medicine (https://pubmed.ncbi.nlm.nih.gov/), and Google Scholar (https://scholar.google.com/), assessed between April and June 2022. Search strategy and selection criteria used combinations of keywords such as mitochondria; homeostasis; mitostasis; neurodegeneration; neuroregeneration; Alzheimer’s disease; Parkinson’s disease; glymphatic system. There was no limit on the year of publication, affiliation, authorship, or journal, but there was a preference for more recent research.
Mitochondria in Neurodegeneration
AgeDisruption of mitochondrial health and function is deeply related to the etiology of several chronic neurodegenerative diseases. Among them, the genetic optic neuropathy optic atrophy type 1 (OPA1) is caused by mutations in the OPA1 gene, which codes for a mitochondrial dynamin-related GTPase involved in mitochondrial architecture, fusion, and metabolism. Another example is mutations in the mitofusin 2 (MFN2) gene, an essential regulator of mitochondrial fusion, which result in Charcot-Marie-Tooth disease (Chen et al., 2005). Moreover, at least 11 different missense mutations in mitochondrial DNA (mtDNA) in select genes encoding subunits in enzyme complexes I, III, and IV, have been linked to Leber’s hereditary optic neuropathy, a neurodegenerative disease that causes optic nerve atrophy and leads to blindness in young adults (Brown et al., 1992). Of note, inherited mitochondrial disorders are mostly caused by mutations in the polymerase (POLG1) gene, which encodes the mtDNA polymerase catalytic subunit, resulting in a series of neurodegenerative illnesses, including childhood myocerebrohepatopathy spectrum diseases, Alpers’ syndrome, ataxia neuropathy spectrum disorders, myoclonus epilepsy myopathy sensory ataxia, and chronic progressive external ophthalmoplegia (Wong et al., 2008).
Furthermore, a causal relation has been established between aging-related neurodegenerative diseases and mitochondria (Matsui et al., 2021). While the evidence of mitochondrial enrolment in more common human neurodegenerative diseases, such as dementia, was initially considered mostly circumstantial (Martin, 2010), recent studies appear to establish a solid association between mitochondrial dynamics and Alzheimer’s disease (AD) and Parkinson’s disease dementia (Grimm and Eckert, 2017; Gómez-Suaga et al., 2018; Wong et al., 2020; Misrani et al., 2021; Sharma et al., 2021b), the two most common neurodegenerative diseases (Nussbaum and Ellis, 2003) (Figure 2).
Figure 2.
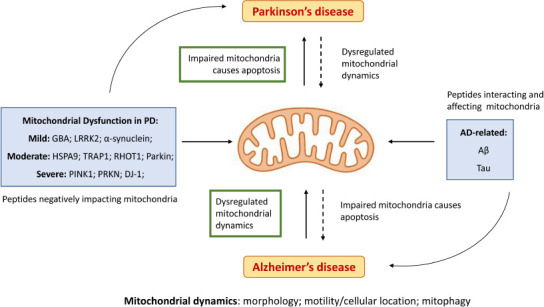
Importance of mitochondria homeostasis in AD and PD.
The most promising mitostasis therapeutic targets in neurodegeneration are highlighted in green. Dashed arrows represent lesser impacts in degeneration pathways and solid arrows represent main action mechanisms. Blue boxes represent the known clinical representations of PD and AD in mitochondria with a causational hypothesis in the literature (Cheng and Bai, 2018). AD: Alzheimer’s disease; Aβ: amyloid β peptide; DJ-1: protein/nucleic acid deglycase DJ-1; GBA: glucocerebrosidase; HSPA9: stress-70 protein, mitochondrial; LRRK2: leucine-rich repeat kinase 2; PD: Parkinson’s disease; PINK1: mitochondrial serine/threonine-protein kinase; PRKN: E3 ubiquitin-protein ligase parkin; RHOT1: mitochondrial Rho GTPase 1; tau: microtubule-associated tau protein; TRAP1: heat shock protein 75 kDa, mitochondrial.
Mitostasis alterations in Alzheimer’s disease
AD is the most common form of dementia in the elderly population, and although its prevalence is rising in lockstep with rising life expectancy in modern societies, the mechanisms of neuronal degeneration in AD are still somewhat unclear and current knowledge is partially lacking. In fact, neurodegeneration has been hypothesized to be caused by a cumulative number of factors, including alterations in protein metabolism and processing, excitotoxicity, oxidative stress, and, more recently, mitochondrial dysfunction (Nussbaum and Ellis, 2003; Guo et al., 2013; Henriques et al., 2015).
There are, however, solid data establishing a correlation between the processing of the amyloidogenic peptide amyloid beta (Aβ), the principal component of AD amyloid plaques (Gandy et al., 1993; da Cruz e Silva et al., 2004; Henriques et al., 2015), and mitochondria health and homeostasis. The amyloid precursor protein (APP) (Gandy et al., 1993; da Cruz e Silva et al., 2004; Henriques et al., 2015) has a mitochondrial targeting sequence (Pavlov et al., 2009) and interacts with mitochondrial import proteins when overexpressed in vitro, preventing the mitochondrial import and promoting bioenergetic deficits (Wilkins and Swerdlow, 2017). APP variants were described to associate with mitochondria in the AD brain (Pavlov et al., 2009), namely by interacting with outer mitochondrial membrane translocases, such as translocase of outer mitochondrial membrane 40 (TOMM40) and translocase of inner mitochondrial membrane 23 (TIMM23) (Cha et al., 2012; Eldeeb et al., 2020). The brain of AD patients further shows evidence of mitochondrial dysfunction, namely increased ROS production and abnormalities in levels and activity of the respiratory chain subunit, mirrored by elevated mitochondrial levels of APP (Sharma et al., 2021a). Of note, the APP-derived Aβ interacts with the mitochondrial matrix protein Aβ-binding alcohol dehydrogenase, which contributes to Aβ toxicity, believed to act via mitochondrial dysfunction and oxidative stress (Yao et al., 2011; Figure 2).
The peptide Aβ has been described to affect mitochondria in several ways, in the context of AD (Reddy, 2009; Cai and Tammineni, 2017; Cheng and Bai, 2018; Perez Ortiz and Swerdlow, 2019; Tang et al., 2019; Wong et al., 2020; Sharma et al., 2021a). The amyloidogenic Aβ builds-up in mitochondrial membranes, causing structural and functional damage and preventing neurons from functioning normally (Reddy, 2009); additionally, oligomeric Aβ has been found to increase intracellular Ca2+ levels and to promote its excessive accumulation in mitochondria (Reddy, 2009). Aβ has also been described to physiologically bind mitochondria in the cerebral cortex of AD mouse models (Eckert et al., 2010) and to interact with mitochondria in the human brain cortex (Calvo-Rodriguez and Bacskai, 2021). In both human and mutant APP transgenic models, deletion of mitochondrial peptidyl-prolyl cis-trans isomerase F (PPIF) gene has a neuroprotective effect in response to oxidative and Aβ-induced stress (Akhter et al., 2017).
As such, although targeting mitochondrial dysfunction is becoming an increasingly plausible therapeutic option in AD, a realistic application has not yet been clearly established (Figure 2).
Mitostasis alterations in Parkinson’s disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting over six million people worldwide. A mostly sporadic progressive degenerative disease with unknown etiology, impairment of mitochondrial respiration has long been linked with Parkinsonism (Langston et al., 1983), and further studies have characterized the impact of mitochondria on PD onset and progression (Grimm and Eckert, 2017; Gómez-Suaga et al., 2018; Park et al., 2018; Magalhaes et al., 2021; Prasuhn et al., 2021). A current promising therapeutic avenue has focused on restoring mitochondrial dysfunction; however, trials aimed at improving mitochondrial function in PD have not yet been successful (Park et al., 2018; Figure 2).
Impairment of mitochondrial biogenesis, mitophagy, and trafficking, disruption of the electron transport chain, increased ROS production, or Ca2+ imbalance, can all contribute to PD-associated mitochondrial dysfunction (Park et al., 2018; Grünewald et al., 2019). If protective mechanisms fail, a negative cycle will eventually elicit all these dysfunctions, leading to cellular impairment and, eventually, neuronal death, and the role of mitochondria in regulating cell death via apoptosis, Ca2+ homeostasis, cell division control, and growth have indeed all been described to be altered in PD (Bose and Beal, 2016).
As previously described, ROS are physiologically produced by mitochondria at very low levels, playing signaling roles in the cell. Physiologically generated ROS are thus regulated by mitochondrial antioxidants, such as the manganese superoxide dismutase (MnSOD/SOD2) and glutathione, that ensure oxidative species are kept at signaling levels, preventing their increase to pathological concentrations (Prasuhn et al., 2021). However, these antioxidants can be overwhelmed by excessive ROS production, resulting in uncontrolled oxidative stress, eventually leading to macromolecule oxidative damage. High energy requiring cells such as neurons are particularly dependent on mitochondrial respiration and thus must tightly balance ROS production to ensure it does not reach pathological levels, which could contribute to neurodegeneration. Subsequently, a decrease in glutathione in the substantia nigra pars compacta has been theorized as an early modifiable event for PD (Prasuhn et al., 2021).
Protein inhibitor of activated STAT2 (PIAS2) is an E3-type small ubiquitin-like modifier (SUMO) ligase, stabilizing protein interaction and mediating protein SUMOylation. Increased levels of PIAS2 have been linked to cognitive and motor impairments in PD mouse models, associated with decreased mitophagy and the consequent accumulation of senescent mitochondria, as well as oxidative stress, and modulating this pathway has been shown to prevent neuronal damage and cognitive and motor impairments (Magalhaes et al., 2021). Being a transcriptional coregulator of several paramount cellular pathways, namely the STAT pathway and the p53 pathways (Rabellino et al., 2012), PIAS2 is one of the new therapeutic targets being explored in neurodegenerative disorders.
Mitochondria as Neuronal Health Promotors
The nervous system is particularly reliable on mitochondria metabolism and homeostasis, since it is the most energy-dependent system, and more so during the process of aging, when metabolism and homeostasis are physiologically altered, including glucose hypometabolism and reduction in mitochondrial electron transport chain activity (reviewed elsewhere (Trigo et al., 2022)). Consequently, several therapeutic strategies have been developed to restore and maintain mitochondria health, not only in response to neuronal degeneration on its own, but also to promote physiological, healthy aging (Murphy and Hartley, 2018). A promising approach aims to restore mitochondrial trafficking and dynamics.
Mitochondrial dynamics, the cycle of constant mitochondria fusion and fission, is a highly complex and balanced process, where mitochondria localization and trafficking are controlled by a wide array of molecular mediators (Sabouny and Shutt, 2020), including the interaction with the endoplasmic reticulum (Chan, 2020). These characteristics are not surprising, as mitochondria movement and dynamics are widely known to be essential for neuronal health, growth, and regeneration (Trigo et al., 2019), and the cohesiveness and location of the intricate mitochondrial network are of paramount relevance for healthy cellular metabolism, particularly in the long neuronal axons (Wai and Langer, 2016). Mitochondria are highly dynamic organelles, constantly relocating and dispersing and re-entering the network, which is of central importance to ensure that neuronal energetic requirements are met, from the cellular body to the axon, dendrites and, ultimately, the synapse (Parrado-Fernández et al., 2018).
Recent studies further emphasized the importance of mitochondria in the context of PD in particular, as most genetic mutations associated with this disease have been associated with mitochondria location (such as mitochondria-endoplasmic reticulum contact sites) (Cutillo et al., 2020), with an impact on mitochondria dynamics (Seager et al., 2020). Recent research has reiterated the impact of mitochondrial biogenesis (Ge et al., 2020; Kumar et al., 2020), where it was demonstrated that defective mitophagy in Parkin-knockout dopaminergic neurons is a consequence primarily of defects in mitochondrial biogenesis, which results in mitochondrial dysfunction. This work auspiciously also demonstrates that modulating Parkin activity can modulate mitochondrial biogenesis, but although promising, further work is required before this therapeutic strategy can be translated to a clinical setting. Mitochondrial biogenesis is a complicated activity, akin to cellular division, that requires the coordination of several types of cellular machinery, confounded by the fact that mitochondria possess their own genetic material (mtDNA); its replication, by cell-independent machinery, is also an important factor worth considering (Chandra et al., 2019a, b).
Nonetheless, restoring mitochondria homeostasis is an exceptionally promising future therapeutic strategy in PD. Dysfunctional mitochondrial ETC complex and increased oxidative stress are deeply interconnected with the vast majority of mitochondrial dysfunction in PD and, as such, therapeutic tools aiming to influence both aspects of mitochondrial homeostasis and maintenance have been investigated in recent years, particularly derivates from coenzyme Q10 (Negida et al., 2016; Zhu et al., 2017).
Mitochondrial complex I dysfunction increases oxidative stress and consequent oxidative damage (Rak et al., 2016), which is prevented by coenzyme Q10. Its mechanism of action is to bypass mitochondrial complex I dysfunction, via the Q-cycle (sequential oxidation and reduction of the electron carrier Coenzyme Q in the mitochondria), which directly decreases the extent of oxidative stress (Wu et al., 2016). However, although promising in animal studies, current clinical trials have not yet proven to be successful (Negida et al., 2016; Zhu et al., 2017; Attia and Maklad, 2018). However, a promising result from Drosophila models, not yet explored in mammalian models, observed Vitamin K2 (long-chain menaquinone 7) to act in a similar manner to coenzyme Q10 (Vos et al., 2012).
Nicotinamide (Vitamin B3) and its derivates are also being explored as strategies to restore ATP production. In this line of investigation, Nicotinamide has been observed to normalize redox levels, with initial studies reporting that it may also affect metabolism-regulating sirtuins (Prasuhn et al., 2021). Nicotinamide metabolism is highly relevant for mitochondrial complex I and could be used to rescue ETC disturbances (Lehmann et al., 2017).
Predictive techniques, tailored patient profiles, targeted prevention, and customization of medical services are crucial for overall medical care. In mitochondrial-related pathologies, due to the lack of causal medications and cures for individuals, these alterations may act as a marker for predicting and measuring disease progress (Koklesova et al., 2022). For clinical application, the ideas here discussed are suited for the perspectives of personalized medicine. Recent studies discussed how specific mitochondrial impairments may lay grounds for patient stratification strategies, implemented in the context of 3P medicine (predictive; preventive, and personalized) (Koklesova et al., 2021).
The usefulness of this observation in the translation for clinical and bedside care is recently evidenced and discussed in the literature (Koklesova et al., 2021, 2022).
Movement to and from the central nervous system: the role of mitochondria
The brain plays a unique role in iron metabolism due to the blood-brain barrier which restricts its access to plasma iron (Mills et al., 2010), and understanding the process of iron release into the brain and transport regulation could help explain the iron accumulation in the brain observed in various neurodegenerative disorders (Singh et al., 2014; Duck and Connor, 2016). Ferrosenescence primarily affects the nervous system and is linked to excessive iron ion accumulation in the brain (Sfera et al., 2018).
The major functional forms of iron, heme and iron-sulphur clusters, are synthesized in the mitochondria (Ma et al., 2006), and mitochondrial iron homeostasis must be regulated to minimize ROS production (Xu et al., 2010). Recent studies in yeast and mammal models suggest a relationship between impaired mitochondrial iron homeostasis and aging (Doulias et al., 2008; Irazusta et al., 2010; Xu et al., 2010; Hughes et al., 2020). Current research describes the disruption of mitochondrial iron transport during aging, resulting in accumulation of mitochondrial iron and destruction of mitochondrial structural components (Buffenstein et al., 2008; Ward and Cloonan, 2019).
Iron content varies between brain regions and cell types, with autopsy analyses showing the total iron concentration in the substantia nigra and globus pallidus of the basal ganglia to rise with age (Walker et al., 2016).
While the blood-brain barrier regulates the movement of cells and molecules from circulation into and from the central nervous system, the brain nonetheless also requires alternative clearance means to clear away metabolic debris and physiological by-products. Since it lacks a conventional lymphatic system, the relevance of this waste removal process is more evident when considering that waste accumulation is a significant contributor to neurodegenerative disease origin and progression (Trigo et al., 2022).
The glymphatic system is a newly described structure, through which subarachnoid cerebral-spinal fluid enters the brain interstitium along the outside of penetrating arteries (para-arterial influx) and mixes with interstitial fluid (Iliff et al., 2012); the resulting fluid then passes through the interstitium, where it is drained via para-venous pathways to the meningeal lymphatic vessels, where it reaches the cervical lymphatics (Iliff et al., 2012). Extracellular proteins, such as the amyloidogenic Aβ peptide, tau, or phosphorylated tau, are cleared from the interstitium by this fluid movement through the brain (Iliff et al., 2012; Tarasoff-Conway et al., 2015). The role played by this system is especially important in the deeper areas of the brain, where interstitial solutes are incapable to directly reach the blood-brain barrier, and thus require an alternative clearance pathway (Silva et al., 2021). The glymphatic system is highly dependent on the astrocytic water channel aquaporin-4, evidenced by reduced cerebral-spinal fluid influx and solute clearance in the parenchymal interstitium in aquaporin-4-knock out animal models (Silva et al., 2021). Glymphatic dysfunction has been hypothesized to particularly contribute to aging-associated brain dysfunction due to increased Aβ accumulation (Kress et al., 2014), and impaired glymphatic clearance has in fact been described in AD animal models, associated with Aβ deposition (Peng et al., 2016); considering the link between Aβ deposition and metabolic and mitochondrial impairment (Reddy and Beal, 2008), an obvious research hypothesis would be to explore the correlation between mitochondria function and glymphatic clearance in the context of neurodegenerative disorders, but this link is yet to be convincingly established.
In fact, only recently have the pathological implications of glymphatic system dysfunction started to be characterized. Evidence is starting to accumulate suggesting a close association between mitochondrial and glymphatic system function. The primarily described glymphatic role of waste removal is intimately related with metabolic activity, and initial observations evidence pathological mitochondrial alterations in the central nervous system, namely in astrocytes, associated with cerebral clearance in dementia patients with idiopathic normal pressure hydrocephalus, a syndrome characterized by increased cerebral-spinal fluid pressure (Tan et al., 2021). This effect appears to be related to mitostasis, as reduced mitochondria-endoplasmic reticulum distance in neuronal soma was reported (Hasan-Olive et al., 2019; Tan et al., 2021; Salehpour et al., 2022), but, again, causality has not yet been established.
Although still a recently described process with many unknowns, the glymphatic system is nonetheless starting to be studied in the context of brain-associated pathologies, and its therapeutic potential, particularly when combined with a focus on mitochondria function, is significant. Photobiomodulation therapy, which promotes mitochondrial ATP metabolism and regulates the production and dissemination of ROS (de Freitas and Hamblin, 2016), has been explored to restore and promote glymphatic function, and is already considered a promising neurodegenerative tool (Salehpour et al., 2022). In other pathologies of the central nervous system, such as ischemic stroke, the glymphatic system is being explored not only as a therapeutic target but also as a diagnostic and prognostic asset (Lv et al., 2021).
Future Perspectives
In sum, developing therapeutic approaches aiming not only to restore neuronal metabolism, but also acting on mitochondrial biogenesis, trafficking, or dynamics, are a rising concern in the clinical treatment of neurodegenerative diseases. Predictive techniques, tailored patient profiles, targeted prevention, and customization of medical services are crucial for overall medical care. Mitochondrial-related alterations may act as biomarkers to anticipate pathology onset and assess disease progression (Koklesova et al., 2022). For clinical applications, the ideas here discussed are indicated for personalized medicine approaches: recent studies have explored how specific mitochondrial impairments may lay grounds for patient stratification strategies, implemented in the context of 3P medicine (predictive; preventive, and personalized) (Koklesova et al., 2021).
Moreover, current research is constantly unraveling new functions and capacities for mitochondria hereto unimaginable (for instance a capacity to act as light lenses in cone photoreceptors (Ball et al., 2022)). These and future findings could lead to the identification of new pathological links, that only now are starting to be considered. Moreover, although not currently capable of restoring the healthy basal state on its own, this approach can also be a stepping stone for drug co-delivery systems, integrating several tactics for neuronal health.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
Funding: This work was supported by a grant from the Fundação para a Ciência e Tecnologia of the Ministério da Educação e Ciência (2020.02006.CEECIND), iBiMED, University of Aveiro and the Fundação para a Ciência e Tecnologia of the Ministério da Educação e Ciência (to DT).
References
- 1.Akhter F, Chen D, Yan SF, Yan SS. Mitochondrial perturbation in Alzheimer's disease and diabetes. Prog Mol Biol Transl Sci. 2017;146:341–361. doi: 10.1016/bs.pmbts.2016.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attia HN, Maklad YA. Neuroprotective effects of coenzyme Q10 on paraquat-induced Parkinson's disease in experimental animals. Behav Pharmacol. 2018;29:79–86. doi: 10.1097/FBP.0000000000000342. [DOI] [PubMed] [Google Scholar]
- 3.Ball JM, Chen S, Li W. Mitochondria in cone photoreceptors act as microlenses to enhance photon delivery and confer directional sensitivity to light. Sci Adv. 2022;8:45. doi: 10.1126/sciadv.abn2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bose A, Beal MF. Mitochondrial dysfunction in Parkinson's disease. J Neurochem 139. 2016;Suppl:216–231. doi: 10.1111/jnc.13731. [DOI] [PubMed] [Google Scholar]
- 5.Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Brown MD, Voljavec AS, Lott MT, MacDonald I, Wallace DC. Leber's hereditary optic neuropathy: a model for mitochondrial neurodegenerative diseases. FASEB J. 1992;6:2791–2799. doi: 10.1096/fasebj.6.10.1634041. [DOI] [PubMed] [Google Scholar]
- 7.Buffenstein R, Edrey YH, Yang T, Mele J. The oxidative stress theory of aging: embattled or invincible?Insights from non-traditional model organisms. Age (Dordr) 2008;30:99–109. doi: 10.1007/s11357-008-9058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Q, Tammineni P. Mitochondrial aspects of synaptic dysfunction in Alzheimer's disease. J Alzheimers Dis. 2017;57:1087–1103. doi: 10.3233/JAD-160726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calvo-Rodriguez M, Bacskai BJ. Mitochondria and calcium in Alzheimer's disease: from cell signaling to neuronal cell death. Trends Neurosci. 2021;44:136–151. doi: 10.1016/j.tins.2020.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Cha MY, Han SH, Son SM, Hong HS, Choi YJ, Byun J, Mook-Jung I. Mitochondria-specific accumulation of amyloid βinduces mitochondrial dysfunction leading to apoptotic cell death. PLoS One. 2012;7:e34929. doi: 10.1371/journal.pone.0034929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. 2020;15:235–259. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 12.Chandra G, Shenoi RA, Anand R, Rajamma U, Mohanakumar KP. Reinforcing mitochondrial functions in aging brain: An insight into Parkinson's disease therapeutics. J Chem Neuroanat. 2019a;95:29–42. doi: 10.1016/j.jchemneu.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Chandra R, Calarco CA, Lobo MK. Differential mitochondrial morphology in ventral striatal projection neuron subtypes. J Neurosci Res. 2019b;97:1579–1589. doi: 10.1002/jnr.24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Kurokawa J, Kass RS. Phosphorylation of the A-kinase-anchoring protein Yotiao contributes to protein kinase A regulation of a heart potassium channel. J Biol Chem. 2005;280:31347–31352. doi: 10.1074/jbc.M505191200. [DOI] [PubMed] [Google Scholar]
- 15.Cheng Y, Bai F. The association of tau with mitochondrial dysfunction in Alzheimer's disease. Front Neurosci. 2018;12:2014–2019. doi: 10.3389/fnins.2018.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cutillo G, Simon DK, Eleuteri S. VPS35 and the mitochondria: Connecting the dots in Parkinson's disease pathophysiology. Neurobiol Dis. 2020;145:105056. doi: 10.1016/j.nbd.2020.105056. [DOI] [PubMed] [Google Scholar]
- 17.D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 18.da Cruz e Silva OAB, Fardilha M, Henriques AG, Rebelo S, Vieira S, da Cruz e Silva EF. Signal transduction therapeutics: relevance for Alzheimer's disease. J Mol Neurosci. 2004;23:123–142. doi: 10.1385/JMN:23:1-2:123. [DOI] [PubMed] [Google Scholar]
- 19.de Freitas LF, Hamblin MR. Proposed mechanisms of photobiomodulation or low-level light therapy. IEEE J Sel Top Quantum Electron. 2016;22:7000417. doi: 10.1109/JSTQE.2016.2561201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Giusti V, Caldiz C, Ennis I, Pérez N, Cingolani H, Aiello E. Mitochondrial reactive oxygen species (ROS) as signaling molecules of intracellular pathways triggered by the cardiac renin-angiotensin II-aldosterone system (RAAS) Front Physiol. 2013;4:126. doi: 10.3389/fphys.2013.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doulias PT, Vlachou C, Boudouri C, Kanavaros P, Siamopoulos KC, Galaris D. Flow cytometric estimation of “labile iron pool” in human white blood cells reveals a positive association with ageing. Free Radic Res. 2008;42:253–259. doi: 10.1080/10715760801911649. [DOI] [PubMed] [Google Scholar]
- 22.Duck KA, Connor JR. Iron uptake and transport across physiological barriers. BioMetals. 2016;29:573–591. doi: 10.1007/s10534-016-9952-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckert A, Schulz KL, Rhein V, Götz J. Convergence of amyloid-βand tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010;41:107–114. doi: 10.1007/s12035-010-8109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eldeeb MA, Ragheb MA, Esmaili M. How does protein degradation regulate TOM machinery-dependent mitochondrial import? Curr Genet. 2020;66:501–505. doi: 10.1007/s00294-020-01056-0. [DOI] [PubMed] [Google Scholar]
- 25.Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal cell death. Physiol Rev. 2018;98:813–880. doi: 10.1152/physrev.00011.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gandy SE, Caporaso GL, Buxbaum JD, de Cruz Silva O, Iverfeldt K, Nordstedt C, Suzuki T, Czernik AJ, Nairn AC, Greengard P. Protein phosphorylation regulates relative utilization of processing pathways for Alzheimer beta/A4 amyloid precursor protein. Ann N Y Acad Sci. 1993;695:117–121. doi: 10.1111/j.1749-6632.1993.tb23038.x. [DOI] [PubMed] [Google Scholar]
- 27.Ge P, Dawson VL, Dawson TM. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol Neurodegener. 2020;15:20. doi: 10.1186/s13024-020-00367-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gómez-Suaga P, Bravo-San Pedro JM, González-Polo RA, Fuentes JM, Niso-Santano M. ER-mitochondria signaling in Parkinson's disease review-article. Cell Death Dis. 2018;9:337. doi: 10.1038/s41419-017-0079-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017;143:418–431. doi: 10.1111/jnc.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grünewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson's disease. Prog Neurobiol. 2019;177:73–93. doi: 10.1016/j.pneurobio.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Guo CY, Sun L, Chen XP, Zhang DS. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasan-Olive MM, Enger R, Hansson HA, Nagelhus EA, Eide PK. Pathological mitochondria in neurons and perivascular astrocytic endfeet of idiopathic normal pressure hydrocephalus patients. Fluids Barriers CNS. 2019;16:39. doi: 10.1186/s12987-019-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henriques AG, Oliveira JM, Carvalho LP, da Cruz e Silva OAB. Aβinfluences cytoskeletal signaling cascades with consequences to Alzheimer's disease. Mol Neurobiol. 2015;52:1391–1407. doi: 10.1007/s12035-014-8913-4. [DOI] [PubMed] [Google Scholar]
- 34.Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya E V, Figueroa ME, Passegué E. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543:205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes CE, Coody TK, Jeong M-Y, Berg JA, Winge DR, Hughes AL. Cysteine toxicity drives age-related mitochondrial decline by altering iron homeostasis. Cell. 2020;180:296–310. doi: 10.1016/j.cell.2019.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irazusta V, Obis E, Moreno-Cermeño A, Cabiscol E, Ros J, Tamarit J. Yeast frataxin mutants display decreased superoxide dismutase activity crucial to promote protein oxidative damage. Free Radic Biol Med. 2010;48:411–420. doi: 10.1016/j.freeradbiomed.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 38.Kausar S, Wang F, Cui H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells. 2018;7:274. doi: 10.3390/cells7120274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koklesova L, Samec M, Liskova A, Zhai K, Büsselberg D, Giordano FA, Kubatka P, Golunitschaja O. Mitochondrial impairments in aetiopathology of multifactorial diseases: common origin but individual outcomes in context of 3P medicine. EPMA J. 2021;12:27–40. doi: 10.1007/s13167-021-00237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koklesova L, Mazurakova A, Samec M, Kudela E, Biringer K, Kubatka P, Golubnitschaja O. Mitochondrial health quality control: measurements and interpretation in the framework of predictive, preventive, and personalized medicine. EPMA J. 2022;13:177–193. doi: 10.1007/s13167-022-00281-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar M, Acevedo-Cintrón J, Jhaldiyal A, Wang H, Andrabi SA, Eacker S, Karuppagounder SS, Brahmachari S, Chen R, Kim H, Ko HS, Dawson VL, Dawson TM. Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human dopamine neurons. Stem Cell Reports. 2020;15:629–645. doi: 10.1016/j.stemcr.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 44.Lehmann S, Loh SHY, Martins LM. Enhancing NAD+salvage metabolism is neuroprotective in a PINK1 model of Parkinson's disease. Biol Open. 2017;6:141–147. doi: 10.1242/bio.022186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lv T, Zhao B, Hu Q, Zhang X. The glymphatic system: a novel therapeutic target for stroke treatment. Front Aging Neurosci. 2021;13:689098. doi: 10.3389/fnagi.2021.689098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma Y, De Groot H, Liu Z, Hider RC, Petrat F. Chelation and determination of labile iron in primary hepatocytes by pyridinone fluorescent probes. Biochem J. 2006;395:49–55. doi: 10.1042/BJ20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magalhaes J, Tresse E, Ejlerskov P, Hu E, Liu Y, Marin A, Montalant A, Satriano L, Rundsten CF, Carlsen EMM, Rydbirk R, Sharifi-Zarchi A, Andersen JB, Aznar S, Brudek T, Khodosevich K, Prinz M, Perrier JM, Sharma M, Gasser T, et al. PIAS2-mediated blockade of IFN-βsignaling: a basis for sporadic Parkinson disease dementia. Mol Psychiatry. 2021;26:6083–6099. doi: 10.1038/s41380-021-01207-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin LJ. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals. 2010;3:839–915. doi: 10.3390/ph3040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsui H, Ito J, Matsui N, Uechi T, Onodera O, Kakita A. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson's disease. Nat Commun. 2021;12:3101. doi: 10.1038/s41467-021-23452-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mills E, Dong XP, Wang F, Xu H. Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med Chem. 2010;2:51–64. doi: 10.4155/fmc.09.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Misrani A, Tabassum S, Yang L. Mitochondrial dysfunction and oxidative stress in Alzheimer's disease. Front Aging Neurosci. 2021;13:617588. doi: 10.3389/fnagi.2021.617588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018;17:865–886. doi: 10.1038/nrd.2018.174. [DOI] [PubMed] [Google Scholar]
- 53.Naumova N, Šachl R. Regulation of cell death by mitochondrial transport systems of calcium and Bcl-2 proteins. Membranes (Basel) 2020;10:299. doi: 10.3390/membranes10100299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Negida A, Menshawy A, Ashal G El, Elfouly Y, Hani Y, Hegazy Y, El Ghonimy S, Fouda S, Rashad Y. Coenzyme Q10 for patients with Parkinson's disease: a systematic review and meta-analysis. CNS Neurol Disord Drug Targets. 2016;15:45–53. doi: 10.2174/1871527314666150821103306. [DOI] [PubMed] [Google Scholar]
- 55.Nussbaum RL, Ellis CE. Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 56.Park JS, Davis RL, Sue CM. Mitochondrial dysfunction in Parkinson's disease: new mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep. 2018;18:21. doi: 10.1007/s11910-018-0829-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parrado-Fernández C, Schneider B, Ankarcrona M, Conti MM, Cookson MR, Kivipelto M, Cedazo-Mínguez Á, Sandebring-Matton A. Reduction of PINK1 or DJ-1 impair mitochondrial motility in neurites and alter ER-mitochondria contacts. J Cell Mol Med. 2018;22:5439–5449. doi: 10.1111/jcmm.13815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pavlov PF, Petersen CH, Glaser E, Ankarcrona M. Mitochondrial accumulation of APP and Aβ: Significance for Alzheimer disease pathogenesis. J Cell Mol Med. 2009;13:4137–4145. doi: 10.1111/j.1582-4934.2009.00892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, Regan S, Kasper T, Peng S, Ding F, Benveniste H, Nedergaard M, Deane R. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiol Dis. 2016;93:215–225. doi: 10.1016/j.nbd.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in Alzheimer's disease: role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol. 2019;176:3489–3507. doi: 10.1111/bph.14585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prasuhn J, Davis RL, Kumar KR. Targeting mitochondrial impairment in Parkinson's disease: challenges and opportunities. Front Cell Dev Biol. 2021;8:615461. doi: 10.3389/fcell.2020.615461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rabellino A, Carter B, Konstantinidou G, Wu SY, Rimessi A, Byers LA, Heymach JV, Girard L, Chiang CM, Teruya-Feldstein J, Scaglioni PP. The SUMO E3-ligase PIAS1 regulates the tumor suppressor PML and its oncogenic counterpart PML-RARA. Cancer Res. 2012;72:2275–2284. doi: 10.1158/0008-5472.CAN-11-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rak M, Bénit PP, Chrétien D, Bouchereau J, Schiff M, El-Khoury R, Tzagoloff A, Rustin P. Mitochondrial cytochrome c oxidase deficiency. Clin Sci. 2016;130:393–407. doi: 10.1042/CS20150707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reddy PH. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp Neurol. 2009;218:286–292. doi: 10.1016/j.expneurol.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–625. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sabouny R, Shutt TE. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem Sci. 2020;45:564–577. doi: 10.1016/j.tibs.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 68.Salehpour F, Khademi M, Bragin DE, Diduro JO. Photobiomodulation therapy and the glymphatic system: promising applications for augmenting the brain lymphatic drainage system. Int J Mol Sci. 2022;23:2975. doi: 10.3390/ijms23062975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seager R, Lee L, Henley JM, Wilkinson KA. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 2020;4:NS20200008. doi: 10.1042/NS20200008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sfera A, Bullock K, Price A, Inderias L, Osorio C. Ferrosenescence: the iron age of neurodegeneration? Mech Ageing Dev. 2018;174:63–75. doi: 10.1016/j.mad.2017.11.012. [DOI] [PubMed] [Google Scholar]
- 71.Sharma C, Kim S, Nam Y, Jung UJ, Kim SR. Mitochondrial dysfunction as a driver of cognitive impairment in alzheimer's disease. Int J Mol Sci. 2021a;22:4850. doi: 10.3390/ijms22094850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma VK, Singh TG, Mehta V. Stressed mitochondria: a target to intrude alzheimer's disease. Mitochondrion. 2021b;59:48–57. doi: 10.1016/j.mito.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 73.Silva I, Silva J, Ferreira R, Trigo D. Glymphatic system, AQP4, and their implications in Alzheimer's disease. Neurol Res Pract. 2021;3:5. doi: 10.1186/s42466-021-00102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singer BD, Chandel NS. Immunometabolism of pro-repair cells. J Clin Invest. 2019;129:2597–2607. doi: 10.1172/JCI124613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singh N, Haldar S, Tripathi AK, Horback K, Wong J, Sharma D, Beserra A, Suda S, Anbalagan C, Dev S, Mukhopadhyay CK, Singh A. Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities. Antioxidants Redox Signal. 2014;20:1324–1363. doi: 10.1089/ars.2012.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan C, Wang X, Wang Y, Wang C, Tang Z, Zhang Z, Liu J, Xiao G. The pathogenesis based on the glymphatic system, diagnosis, and treatment of idiopathic normal pressure hydrocephalus. Clin Interv Aging. 2021;16:139–153. doi: 10.2147/CIA.S290709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang J, Oliveros A, Jang MH. Dysfunctional mitochondrial bioenergetics and synaptic degeneration in Alzheimer disease. Int Neurourol J. 2019;23:S5–10. doi: 10.5213/inj.1938036.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Ménard J, Zetterberg H, Wisniewski T, de Leon MJ. Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Trigo D, Goncalves MB, Corcoran JPT. The regulation of mitochondrial dynamics in neurite outgrowth by retinoic acid receptor βsignaling. FASEB J. 2019;33:7225–7235. doi: 10.1096/fj.201802097R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trigo D, Avelar C, Fernandes M, Sá J, Cruz e Silva O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022;596:1095–1110. doi: 10.1002/1873-3468.14298. [DOI] [PubMed] [Google Scholar]
- 81.Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B, Meganathan R, Morais VA, Verstreken P. Vitamin K 2 is a mitochondrial electron carrier that rescues Pink1 deficiency. Science. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- 82.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 83.Walker T, Michaelides C, Ekonomou A, Geraki K, Parkes HG, Suessmilch M, Herlihy AH, Crum WR, So PW. Dissociation between iron accumulation and ferritin upregulation in the aged substantia nigra: attenuation by dietary restriction. Aging (Albany NY) 2016;8:2488–2508. doi: 10.18632/aging.101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ward DM, Cloonan SM. Mitochondrial iron in human health and disease. Annu Rev Physiol. 2019;81:453–482. doi: 10.1146/annurev-physiol-020518-114742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilkins HM, Swerdlow RH. Amyloid precursor protein processing and bioenergetics. Brain Res Bull. 2017;133:71–79. doi: 10.1016/j.brainresbull.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wong KY, Roy J, Fung ML, Heng BC, Zhang C, Lim LW. Relationships between mitochondrial dysfunction and neurotransmission failure in Alzheimer's disease. Aging Dis. 2020;11:1291–1316. doi: 10.14336/AD.2019.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wong LJ, Naviaux RK, Brunetti-Pierri N, Zhang Q, Schmitt ES, Truong C, Milone M, Cohen BH, Wical B, Ganesh J, Basinger AA, Burton BK, Swoboda K, Gilbert DL, Vanderver A, Saneto RP, Maranda B, Arnold G, Abdenur JE, Waters PJ, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008;29:E150–172. doi: 10.1002/humu.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu M, Gu J, Guo R, Huang Y, Yang M. Structure of mammalian respiratory supercomplex I1III2IV1. Cell. 2016;167:1598–1609. doi: 10.1016/j.cell.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 89.Xu J, Marzetti E, Seo AY, Kim JS, Prolla TA, Leeuwenburgh C. The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech Ageing Dev. 2010;131:487–493. doi: 10.1016/j.mad.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yao J, Du H, Yan S, Fang F, Wang C, Lue LF, Guo L, Chen D, Stern DM, Gunn Moore FJ, Chen JX, Arancio O, Yan SS Du. Inhibition of amyloid-β(Aβ) peptide-binding alcohol dehydrogenase-Aβinteraction reduces Aβaccumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review) Int J Mol Med. 2019;44:3–15. doi: 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou TJ, Zhang SL, He CY, Zhuang QY, Han PY, Jiang SW, Yao H, Huang YJ, Ling WH, Lin YC, Lin ZN. Downregulation of mitochondrial cyclooxygenase-2 inhibits the stemness of nasopharyngeal carcinoma by decreasing the activity of dynamin-related protein 1. Theranostics. 2017;7:1389–1406. doi: 10.7150/thno.17647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhu ZG, Sun MX, Zhang WL, Wang WW, Jin YM, Xie CL. The efficacy and safety of coenzyme Q10 in Parkinson's disease: a meta-analysis of randomized controlled trials. Neurol Sci. 2017;38:215–224. doi: 10.1007/s10072-016-2757-9. [DOI] [PubMed] [Google Scholar]