

Abstract
Abstract
Cardiac mechanical afterload induces an intrinsic autoregulatory increase in myocyte Ca2+ dynamics and contractility to enhance contraction (known as the Anrep effect or slow force response). Our prior work has implicated both nitric oxide (NO) produced by NO synthase 1 (NOS1) and calcium/calmodulin‐dependent protein kinase II (CaMKII) activity as required mediators of this form of mechano‐chemo‐transduction. To test whether a single S‐nitrosylation site on CaMKIIδ (Cys290) mediates enhanced sarcoplasmic reticulum Ca2+ leak and afterload‐induced increases in sarcoplasmic reticulum (SR) Ca2+ uptake and release, we created a novel CRISPR‐based CaMKIIδ knock‐in (KI) mouse with a Cys to Ala mutation at C290. These CaMKIIδ‐C290A‐KI mice exhibited normal cardiac morphometry and function, as well as basal myocyte Ca2+ transients (CaTs) and β‐adrenergic responses. However, the NO donor S‐nitrosoglutathione caused an acute increased Ca2+ spark frequency in wild‐type (WT) myocytes that was absent in the CaMKIIδ‐C290A‐KI myocytes. Using our cell‐in‐gel system to exert multiaxial three‐dimensional mechanical afterload on myocytes during contraction, we found that WT myocytes exhibited an afterload‐induced increase in Ca2+ sparks and Ca2+ transient amplitude and rate of decline. These afterload‐induced effects were prevented in both cardiac‐specific CaMKIIδ knockout and point mutant CaMKIIδ‐C290A‐KI myocytes. We conclude that CaMKIIδ activation by S‐nitrosylation at the C290 site is essential in mediating the intrinsic afterload‐induced enhancement of myocyte SR Ca2+ uptake, release and Ca2+ transient amplitude (the Anrep effect). The data also indicate that NOS1 activation is upstream of S‐nitrosylation at C290 of CaMKII, and that this molecular mechano‐chemo‐transduction pathway is beneficial in allowing the heart to increase contractility to limit the reduction in stroke volume when aortic pressure (afterload) is elevated.
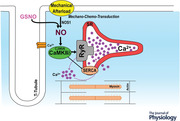
Key points
A novel CRISPR‐based CaMKIIδ knock‐in mouse was created in which kinase activation by S‐nitrosylation at Cys290 (C290A) is prevented.
How afterload affects Ca2+ signalling was measured in cardiac myocytes that were embedded in a hydrogel that imposes a three‐dimensional afterload.
This mechanical afterload induced an increase in Ca2+ transient amplitude and decay in wild‐type myocytes, but not in cardiac‐specific CaMKIIδ knockout or C290A knock‐in myocytes.
The CaMKIIδ‐C290 S‐nitrosylation site is essential for the afterload‐induced enhancement of Ca2+ transient amplitude and Ca2+ sparks.
Keywords: CaMKII, cardiac myocyte, mechano‐chemo‐transduction, post‐translational modifications, S‐nitrosylation
Abstract figure legend CaMKII S‐nitrosylation at Cys‐290 mediates mechano‐chemo‐transduction and afterload‐induced increases in Ca2+ transient amplitude and Ca2+ sparks.
Introduction
The heart exhibits two classical mechanisms of intrinsic autoregulation to increase contractile force: 1) upon elevated preload (high end‐diastolic volume or sarcomere length; Frank–Starling effect) and 2) upon elevated afterload that raises the force against which the heart must perform work to eject blood (high aortic pressure or aortic valve resistance; Anrep effect). The Frank–Starling effect is instantaneous and is induced by altered structure of the myofilament lattice and myofilament Ca2+ sensitivity, but the molecular details remain incompletely resolved (Ait‐Mou et al., 2016; Allen & Kentish, 1985; de Tombe et al., 2010; Gordon et al., 1966; Kampourakis & Irving, 2021). The Anrep effect or slow force response (SFR) takes several minutes to fully develop upon an abrupt increase in cardiac afterload or sarcomere length, respectively, and involves intracellular signalling and increased Ca2+ transients, but again the molecular biochemical mechanisms are unresolved for this aspect of mechano‐chemo‐transduction in the heart (Cingolani et al., 2013; Dowrick et al., 2019). As the Anrep effect or SFR is studied experimentally, it is hard to isolate the effect of afterload, because the acute increase in afterload or sarcomere length also alters preload.
To isolate afterload effects, we embed resting isolated myocytes in a hydrogel, so that the three‐dimensional afterload is only sensed when the myocyte actively contracts (Hegyi et al., 2021; Jian et al., 2014; Shimkunas et al., 2021). In this system we have demonstrated an afterload‐induced increase in Ca2+ transient amplitude and spontaneous SR Ca2+ release events (Ca2+ sparks) that was nitric oxide (NO) dependent, required NO synthase 1 (NOS1) and was suppressed by inhibition of Ca2+/calmodulin‐dependent protein kinase II (CaMKII) by KN‐93 (Jian et al., 2014). It was recently shown that increasing afterload (with preload relatively controlled) caused an increase in left ventricular contractility that was blunted in hearts from CaMKII‐knockout mice (Reil et al., 2020), suggesting that CaMKII activity is directly involved in the Anrep effect. The CaMKII signalling pathway has also been implicated in these long‐term maladaptive processes involved in pathological hypertrophy and heart failure (Anderson et al., 2011; Hegyi et al., 2019).
CaMKII has four main isoforms, but the predominant isoform in cardiac myocytes is CaMKIIδ (Anderson et al., 2011). In cardiac myocytes, CaMKII is dynamically regulated by the periodic rises in intracellular [Ca2+] ([Ca2+]i) during each beat that couples to kinase activation (Bers, 2002), with activation increased by frequency and Ca2+ transient amplitude (Erickson et al., 2011). In addition, CaMKIIδ is subject to autophosphorylation (at Thr287) (Hudmon & Schulman, 2002) that is known to promote autonomous activity or memory, even after [Ca2+]i declines. Additional post‐translational modifications (PTMs) within the same CaMKIIδ regulatory domain (Fig. 1A ) via oxidation (at M281/M282) (Erickson et al., 2008), O‐GlcNAcylation (at S280) (Erickson et al., 2013) and S‐nitrosylation at C290 (Coultrap & Bayer, 2014; Erickson et al., 2015) can likewise promote this autonomous CaMKII activity. Notably, nitrosylation of C290 can promote autonomous, pathologically sustained kinase activity (molecular memory) leading to arrhythmogenic SR Ca2+ release, while S‐nitrosylation at C273 limits the ability of Ca–CaM to activate CaMKII (Curran et al., 2014; Erickson et al., 2015).
Figure 1. CaMKIIδ‐C290A knock‐in mouse model.
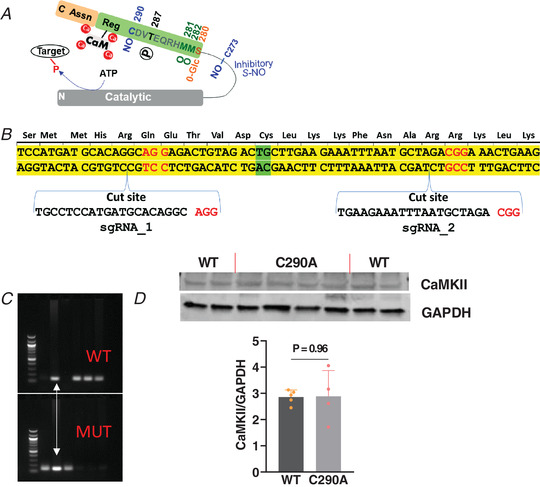
A, schematic representation of a CaMKII monomer highlighting amino acids in the regulatory domain (Reg) that have been shown to promote autonomous activation via autophosphorylation (P), S‐nitrosylation (NO), oxidation (O) and O‐GlcNAcylation (O‐Glc). B, homology‐directed repair knock‐in schematic representation of Cys290 to Ala mouse generation with exon 11 in yellow and PAM sequence in red. C, PCR showing genotype results for WT, MUT and HET (white arrow) mice. D, western blot of CaMKII expression levels in WT and mutant C290A mice. N animal = 5 and 4, respectively. Unpaired t test.
In the present study, we use wild‐type (WT), cardiac‐specific CaMKIIδ ablation mice (CaMKIIδ‐cKO), as well as novel CRISPR‐based CaMKIIδ‐C290A knock‐in (KI) mice to test the hypotheses that the afterload‐induced enhancement of Ca2+ transients in intact adult ventricular myocytes requires the CaMKIIδ isoform specifically, and more specifically that the single S‐nitrosylation site (C290) on CaMKIIδ is necessary. We used myocytes that were either mechanically unloaded or embedded in the cell‐in‐gel system to impose a mechanical afterload (Jian et al., 2014), and monitored Ca2+ handling using confocal line‐scan imaging. We conclude that the CaMKIIδ‐C290 S‐nitrosylation site is essential for the afterload‐induced enhancement of Ca2+ transient amplitude, involving both increased SR Ca2+ uptake and sensitization of SR Ca2+ release via ryanodine receptors (RyR).
Methods
Ethical approval
All animal handling and laboratory procedures were conducted in compliance with the NIH guidelines for animal research and with approval of the Institutional Animal Care and Use Committee at the University of California, Davis (protocol no. 21572).
CaMKIIδ C290A knock‐in mouse model
CaMKIIδ‐C290A‐KI mice were generated via the UC Davis Mouse Biology Program, to ablate one of the nitrosylation sites on CaMKIIδ. To replace the WT C290 residue codon ‘TGC’ with a ‘GCC’ Ala residue at position 290, we used CRISPR/Cas9 homology‐directed repair to knock‐in the Ala codon into the CaMKIIδ mouse locus in place of that for Cys (Fig. 1B ). We took advantage of an offset oligo (5′‐ATGACTTCTGCTTTCAGTAGCTTTCACCTCACCAGATCAACCCTCCAGCCCTCCCACAACAATGTCAAATTTCTTACCTTCAGTTTCCGTCTAGCATTAAATTTCTTCAAGGCGTCTACAGTCTCCTGCCTGTGCATCATGGAGGCAACAGTAGAGCGTTGCTAA‐3′) that is complementary to the target strand and distal to the protospacer adjacent motif (PAM). Additionally, we used the following sgRNAs 5′‐TGCCTCCATGATGCACAGGCAGG‐3′ and 5′‐TGAAGAAATTTAATGCTAGACGG‐3′, which cleaves the genome upstream and downstream from the engineered mutation, knocking in via a semi‐long ssODN method to ensure highest possible success.
The mice were genotyped, and CaMKIIδ mRNA was sequenced to confirm the amino acid substitution in the C290A knock‐in. To determine mouse genotype, polymerase chain reaction (PCR) was performed on genomic DNA from 0.4 cm lengths of mouse tails obtained from neonatal mice. All PCR reagents were from Quantabio (Beverly, MA, USA). Thirty‐five cycles were performed on the samples using ProFlex PCR System (Thermo Fisher Scientific, Waltham, MA, USA) as follows: denaturation at 95°C for 0.5 min, annealing at 60°C for 0.5 min, extension at 72°C for 0.5 min, final extension cycle at 72°C for 5 min, and a soak cycle at 4°C. Reaction products were analysed on 2% agarose gels in which the following bands were expected: WT (+/+) allele: 137 bp; knockout (−/−) allele: 137 bp; heterotypic (+/−) alleles: both bands. Forward primer: CTACTGTTGCCTCCATGATGCA; reporter 1: CTGTAGACTGCTTGAAGAAAT; reverse primer: CTTTGCCATGACTTCTGCTTTCA; reporter 2: ACTGTAGACGCCTTGAAG.
Conventional echocardiography and Doppler imaging
Transthoracic echocardiography was performed in mice anaesthetized by isoflurane inhalation (1.5%), which was later individually adjusted (1–3%) to achieve a stable heart rate of 400−500 beats/min to avoid fusion of the waves. Core body temperature was carefully monitored and maintained at 37°C during the entire procedure. Transthoracic echocardiography was performed using a VisualSonics Vevo 2100 system equipped with a 40 MHz transducer (Fujifilm VisualSonics, Toronto, ON, Canada). Systolic function (cardiac contractile function) indices were obtained from short‐axis M‐mode scans at the midventricular level. Apical four‐chamber views were assessed for diastolic function measurements using pulsed‐wave and tissue Doppler imaging at the level of the mitral valve. Measurements were collected at baseline conditions for both C290A and WT mice. All parameters were measured at least three times, and means are presented.
Cardiomyocyte isolation
Adult mice between 10 and 12 weeks old were used including WT (The Jackson Laboratory, Bar Harbor, ME, USA, stock no. 000664), CaMKIIδ cardiac‐specific KOs, and the novel CaMKIIδ C290A mice with a Cys to Ala substitution at position 290. The animals were kept at standard temperature, humidity and lighting. Food and drinking water were provided ad libitum. For the enzymatic isolation of left ventricular cardiomyocytes, the perfusion system was filled with minimal essential medium (MEM) solution and cleared of any bubbles by fast forwarding the motor. The perfusion system water bath was set to 37°C and a rate of ∼5 ml/min. The top section of the perfusion system was cleared of MEM, then filled with MEM + enzyme (50 ml MEM + 60 mg collagenase type 2 (Worthington Biochemical Corp., Lakewood, NJ, USA, lot number: 41A20883) and 1.3 mg protease (Sigma‐Aldrich, St Louis, MO, USA, P5147, CAS number: 9036‐06‐0)). The mice were injected subcutaneously with diluted 0.3 ml heparin and anaesthetized with 5% isoflurane. After sedation, mice were moved onto an absorbent pad with the nose/mouth in a nose cone which continuously delivers 2% isoflurane. The mice were taped to the absorbent pad and reactions to noxious stimuli checked. When areflexia was achieved, hearts were excised and cleaned in cold wash solution (100 ml MEM + 0.1 ml heparin), with any extra tissue or fat cut off. Mechanism of death was exsanguination while the mice were unconscious. The entire heart was then cannulated in cold wash solution on ice. Hearts were retrogradely perfused on constant flow ∼5 ml/min, 37°C. When adequately digested, a cut was made below the atria to drop the ventricles into a small dish with stopping solution (mouse 2.25 ml 10× MEM + 250 μl fetal bovine serum). Ventricular myocytes were dispersed mechanically and filtered through a nylon mesh and allowed to sediment for ∼10 min. The sedimentation was repeated three times using increasing [Ca2+] from 0.125 to 0.25 then 0.5 mM. Finally, ventricular myocytes were kept in Tyrode's solution (0.5 mM [Ca2+]) at room temperature until use.
Fluorescence measurement of [Ca2+] using Fluo‐4
[Ca2+]i was measured using a single wavelength calcium indicator, Fluo‐4 AM (10 μM, Thermo Fisher Scientific, cat. no.: F14201, lot number: 2146860). Freshly isolated cardiomyocytes were loaded with Fluo‐4 AM and Pluronic acid (0.02%, Thermo Fisher Scientific, cat. no.: P3000MP, lot number: 1990297). The indicator was loaded for 30 min at room temperature followed by wash and de‐esterification for 30 min. Supernatant was removed and replaced three times with normal Tyrode's solution (NT) containing 145 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 5.5 mM glucose, 10 mM HEPES (pH 7.4, 23°C) – while increasing [Ca2+] with each wash to a final of 1.8 mM. All loading was done at room temperature.
Cell‐in‐gel system
Elastic gel matrix was made of a polyvinyl alcohol (PVA) hydrogel system composed of underivatized PVA (98 kDa) and a tetravalent boronate–polyethylene glycol cross‐linker, all custom made (Onofiok et al., 2010). Freshly isolated cardiomyocytes were loaded with Fluo‐4 AM as described above and suspended in a 7% PVA solution; then, a 7.5% cross‐linker solution was added in equal volume. Optionally, submicrometre fluorescent beads were embedded in the gel to track displacement. Upon incubation for 15 min at room temperature, the boronate group cross‐links the PVA hydrogel, embedding the cell in the 3D gel matrix. The boronate group also cross‐links the cis‐diols of the cell surface glycans to PVA, thereby tethering the cell surface to the gel. The optically transparent gel allows for Ca2+ signals to be observed while the cell‐in‐gel system is perfused. This gel system helps investigate mechano‐chemo‐transduction signalling pathways by activating intrinsic mechanical mechanisms in contracting myocytes.
Confocal imaging of Ca2+ signals
Ca2+ transients and diastolic Ca2+ events (sparks and waves) were detected via line‐scan imaging with a laser scanning confocal microscope – Bio‐Rad Laboratories (Hercules, CA, USA) Radiance 2100, equipped with a ×40 oil immersion objective lens, at 6 ms/line. Fluo‐4 was excited at a wavelength of 488 nm of the argon laser, and the emitted fluorescence was collected through a 500−530 nm bandpass emission filter. To measure systolic Ca2+ transients, intact cardiomyocytes were plated on laminin‐coated coverslips or loaded in‐gel as described above and paced at 0.5 Hz in a field stimulation chamber, until Ca2+ cycling reached steady state. Fluo‐4 fluorescence was recorded for at least five beats during steady state. To assess SR Ca2+ load, 20 mM caffeine in NT was applied to activate RyRs and rapidly deplete the SR Ca2+ content, such that the amplitude indicates the SR Ca2+ load. The exponential decay rate constant of the Ca2+ transient induced by rapid caffeine application reflects Ca2+ extrusion via sodium–calcium exchange (NCX) (Guo et al., 2007). ImageJ was used for image processing and analysis; Ca2+ sparks (measured from ∼10 s after stimulation stopped) were detected using the SparkMaster plugin (Picht et al., 2007) first, and then verified by human inspection. Scanning speed (in lines per second) and pixel size were adjusted according to the microscope setting during image acquisition, and criteria values were set to 3.8 as recommended (Picht et al., 2007). Based on non‐cellular fluorescent regions in the image, the ‘background’ is determined. Images with Ca2+ waves, which can confound spark frequency measures, were also excluded from Ca2+ spark analysis. Fluorescence intensities were background‐subtracted and normalized to the corrected baseline.
Statistical analysis and presentation of data
Data are presented as means ± SD, as indicated. Normality of the data was assessed by the Shapiro–Wilk test. To determine statistical significance of differences in normally distributed data, paired Student's t test, unpaired Student's t test, or one‐way ANOVA was used as indicated. For data that were not normally distributed, a non‐parametric test was used. In cellular experiments, we performed hierarchical statistical analyses (nested tests) to account for inter‐subject variability and dependence between samples. The specific statistical test used is indicated in each figure legend. GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA) was used for data analysis. P < 0.05 was considered statistically significant.
Results
To determine the role of CaMKIIδ‐C290 in mediating mechano‐chemo‐transduction, we generated a C290A‐KI mouse. Figure 1A illustrates five PTM sites in the highly conserved regulatory region of CaMKII, immediately adjacent to the calmodulin (CaM) binding site. These include O‐GlcNAcylation (S280), oxidation (MM281/282), autophosphorylation (T287) and S‐nitrosylation (C290), each of which can promote autonomous kinase activity after Ca–CaM dissociation and memory (Coultrap & Bayer, 2014; Erickson et al., 2008, 2013, 2015; Howe et al., 2004). S‐nitrosylation at C273 has also been shown to suppress CaMKIIδ activation by Ca–CaM (Erickson et al., 2015).
Figure 1B shows the design of the CRISPR strategy and PAM sites used to replace the endogenous cysteine in CaMKIIδ with a serine. The mice were genotyped, and CaMKIIδ mRNA was sequenced (see Methods) to confirm the amino acid substitution in the C290A‐KI. Figure 1C shows blots for WT and C290A mutant, allowing identification of homozygous WT and C290A mice, as well as heterozygous mice expressing both WT and mutant alleles. The C290A‐KI mice all expressed similar basal CaMKII protein levels (Fig. 1D ).
The new CaMKIIδ‐C290A mice were born at expected Mendelian genotype ratios and exhibited normal growth and maturation. CaMKIIδ‐C290A mice vs. WT littermates at the age we studied (10–12 weeks) exhibited unaltered baseline heart rate and cardiac contractile function on echocardiography (Fig. 2A and B ), and normal heart and body weights (Fig. 2C ). Thus, the new C290 nitrosylation resistant mutant mice exhibited no obvious baseline phenotype differences from the WT mice.
Figure 2. CaMKIIδ‐C290A knock‐in mouse model.
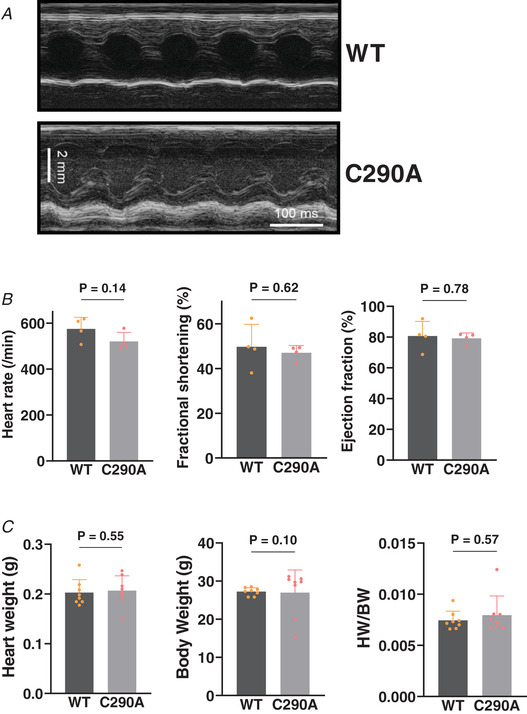
A, representative echocardiogram of the LV in a WT and C290A mouse. B, unaltered heart rate and echocardiographic fractional shortening and ejection fraction in C290A mice. N animal = 4. Unpaired t test. C, normal heart weight, body weight and their ratio in CaMKIIδ C290A mice at baseline. N animal = 8. Mann–Whitney test.
CaMKIIδ‐C290A myocytes exhibit normal Ca transients and β‐adrenergic receptor responses
First, we assessed the effects of CaMKII‐C290A mutation on basal myocyte Ca2+ handling. Figure 3A illustrates the confocal line‐scan Ca2+ imaging protocol. Mouse cardiomyocytes were paced at 0.5 Hz for 30 s, and the last five stabilized Ca2+ transients (CaTs; Fig. 3A ) used for quantitative analysis. After pacing, myocytes were at rest for 1 min during which spontaneous Ca2+ sparks were recorded, and then 20 mM caffeine was rapidly applied to release all SR Ca2+ and assess the SR Ca2+ load. CaMKIIδ‐C290A mice exhibited unaltered Ca2+ transient amplitude, time to peak [Ca2+]i and time constant (τ) in comparison to their WT littermates (Fig. 3B ).
Figure 3. Ca2+ transients in novel C290A mice.
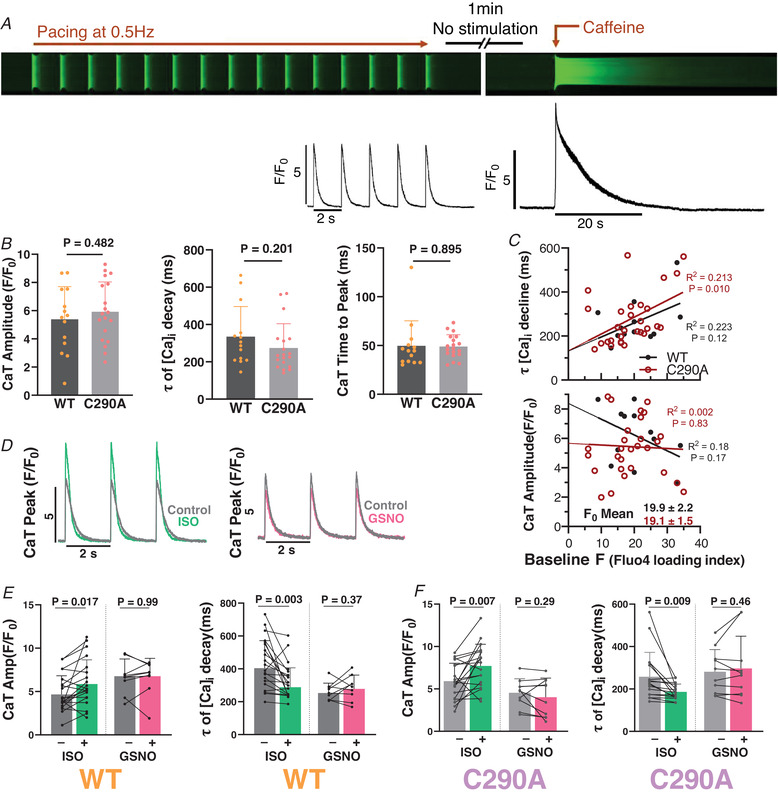
A, experimental protocol and representative longitudinal line scan with pseudo‐colour insets. B, mean ± SD values of baseline Ca2+ transient (CaT) data for wild‐type (WT) mice in comparison with their C290A littermates under load‐free conditions. n cell/N animal = 14/5, 19/5 respectively. Nested t test. C, influence of Fluo‐4 loading level on tau (τ) of [Ca2+]i decline and CaT amplitude. Simple linear regression. D, CaT peaks of exemplar C290A cardiomyocytes under control conditions and with 100 nM isoproterenol (ISO; left), or with 150 μM S‐nitrosoglutathione (GSNO; right) all stimulated at 0.5 Hz. E and F, summary data for CaT amplitude and τ in WT and C290A littermates. n cell/N animal = 20/6, 9/5, 19/5 and 10/4, respectively. Paired t test.
Some initial WT myocyte experiments exhibited unusually long twitch [Ca]i decline τ values, which we suspected might be attributed to excess Fluo‐4 loading (assessed as basal resting F 0; Fig. 3C and Supporting Fig. 1C ), which perforce would increase intracellular Ca2+ buffering, slow [Ca2+]i decline and limit Ca2+ transient amplitude. All‐cell data were analysed as τ vs. baseline F 0 (Supporting Fig. 1B and C ), and we excluded from Fig. 3 the group of WT myocytes with uniquely high indicator loading (F 0 > 40). In doing so, the mean F 0 level of indicator loading was well‐matched between WT and C290A myocytes (19.9 ± 2.2 and 19.1 ± 1.5; Fig. 3C ). The unaltered Ca2+ transients in C290A vs. WT myocytes are consistent with the unaltered echocardiographic differences (Fig. 2B ).
Both C290A‐KI and WT mouse myocytes showed a classical response towards the β‐adrenergic receptor (β‐AR) agonist, isoproterenol (ISO; 100 nM), which was added 5 min before pacing was restarted. ISO significantly increased both the CaT amplitude (Fig. 3D–F ) and rate of Ca2+ transient decline. In contrast, acute exposure to 150 μM S‐nitrosoglutathione (GSNO; an NO donor) for 5 min did not alter Ca2+ transient amplitude or kinetics of [Ca2+]i decline in either WT or C290A mouse myocytes (Fig. 3E and F ).
Figure 4 shows Ca2+ spark analysis for myocytes during the post‐stimulation period indicated in the protocol (10–60 s after stimulation was stopped). In WT myocytes, both ISO and GSNO treatment significantly increased Ca2+ spark frequency, and to similar extents (Fig. 4D ). However, in C290A myocytes, GSNO failed to increase Ca2+ spark frequency, in contrast to a similar ISO‐induced increase in Ca2+ spark frequency in C290A as in WT myocytes (Fig. 4D and E ). There was also no difference observed in SR Ca load with these treatments across the groups, but our ability to detect small differences in SR Ca load may be limited because peak [Ca2+]i may be near indicator saturation. These data from intact myocytes are consistent with GSNO‐induced CaMKII nitrosylation and activation with consequent RyR sensitization, and show that this effect is abolished by the single point mutation of a known S‐nitrosylation site (C290A) in CaMKIIδ.
Figure 4. Ca2+ spark measurements in WT vs. C290A mice.
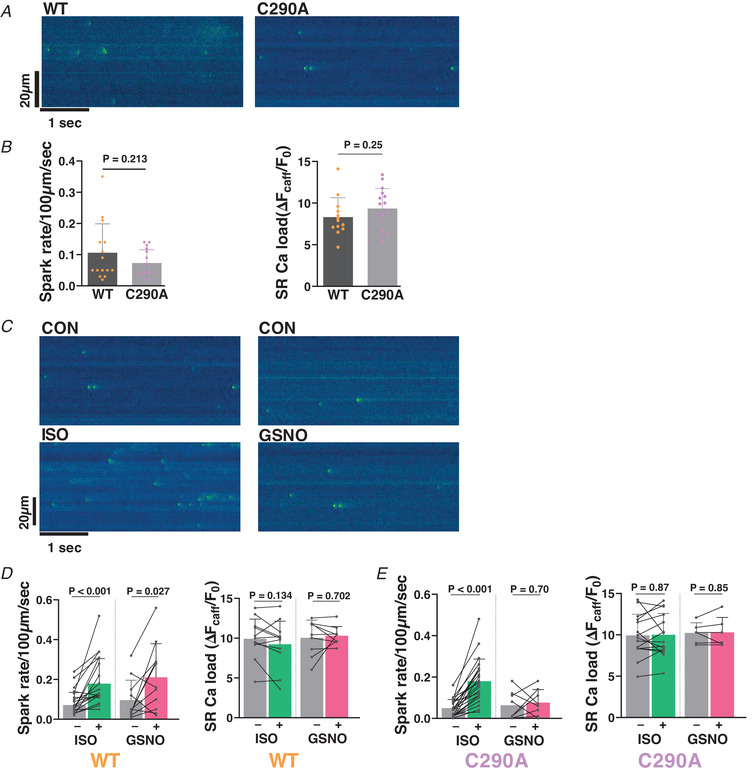
A, representative spontaneous Ca2+ sparks seen in the pseudo‐coloured confocal linescan images of C290A cardiomyocytes and their wild‐type (WT) littermates. B, mean ± SD values of Ca2+ spark rate under load‐free conditions in C290A vs. WT cardiomyocytes. n cell/N animal = 15/5, 16/5, 13/5 and 16/5, respectively. Nested t test. C, representative pseudo‐coloured confocal line‐scans from quiescent C290A cardiomyocytes under control conditions and with 100 nM isoproterenol (ISO, left), or with 150 μM S‐nitrosoglutathione (GSNO, right). D and E, mean ± SD Ca2+ spark rate data are shown for WT (D) and C290A (E) cardiomyocytes in response to ISO (left) and GSNO (right). n cell/N animal = 18/6, 11/4, 12/5 and 10/4 for WT and 22/6, 8/3, 15/4 and 6/3 for C290A, respectively. Paired t test.
Collectively, our data demonstrate that the CaMKIIδ‐C290A‐KI mice displayed normal physiological myocyte function, similar to their WT littermates. However, the GSNO‐induced increase in arrhythmogenic SR Ca2+ leak seen in WT myocytes was suppressed in myocytes harbouring the point mutation in CaMKIIδ (C290A).
Afterload‐induced increase in diastolic SR calcium leak requires CaMKIIδ S‐nitrosylation
We next tested whether the myocyte‐intrinsic S‐nitrosylation effect seen with GSNO was also induced by increased mechanical afterload in WT but suppressed in the C290A myocytes. We used a cell‐in‐gel system, in which myocytes encounter a mechanical afterload as they contract against a viscoelastic hydrogel (Hegyi et al., 2021; Jian et al., 2014; Shimkunas et al., 2021). Our group previously showed that NOS1 and CaMKII were both involved in mediating afterload‐induced increases in intracellular CaTs and Ca2+ sparks in intact ventricular cardiomyocytes from WT mice (Jian et al., 2014). Figure 5A shows that WT cardiomyocytes contracting in the gel (vs. load‐free) displayed enhanced systolic CaT amplitudes (Fig. 5D ), faster [Ca2+]i decline (Fig. 5E ), faster time to peak [Ca2+]i (Fig. 5F ), reduced fractional shortening (Fig. 5G ) and a robust increase in diastolic Ca2+ sparks (Fig. 6). The increase in CaTs and Ca2+ sparks in cardiomyocytes under afterload in‐gel are consistent with an increase in RyR sensitivity, and the faster CaT decay would be most consistent with enhanced SR Ca2+ uptake. While altered myofilament Ca2+ buffering at higher force might complicate this, that would tend to reduce peak [Ca]i, which was not observed. As a control, we tested whether individual gel‐forming components might alter cardiomyocyte function. We preincubated cardiomyocytes with PVA or cross‐linker alone, followed by perfusion with normal Tyrode's solution and contraction measurements. There was no difference in contraction measured as sarcomere length shortening for either PVA or cross‐linker treatment – without mixing to form hydrogel (Fig. 5H ). Previous in‐gel studies used NOS1 knockout mice and the useful CaMKII inhibitor KN‐93 to implicate NOS1 and CaMKII in the cell‐in‐gel afterload effects on Ca2+ handling (Jian et al., 2014). Indeed, that confirmed the role of the NOS isoform as the NO source in the mechano‐chemo‐transduction pathway, but KN‐93 is an imperfectly selective inhibitor of CaMKII.
Figure 5. Influence of afterload on myocyte Ca2+ transients.
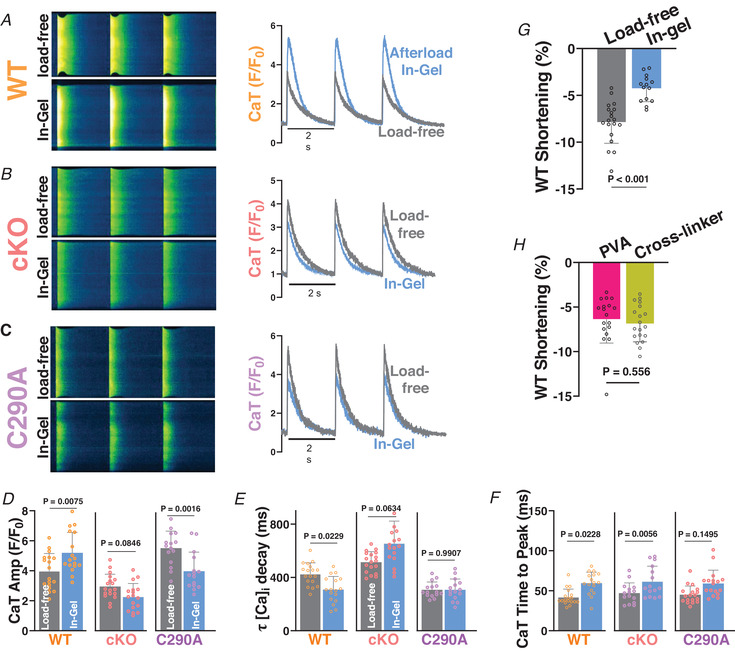
A–C, representative confocal pseudo‐coloured line scan images of systolic Ca2+ transients (left) obtained using Fluo‐4 imaging and measured Ca2+ transients (CaT; right) in load‐free control and cell‐in‐gel for WT (A), CaMKIIδ‐cKO (B) and CaMKIIδ‐C290A (C) mice. D–F, influence of load (In‐Gel) on CaT Amp (D), on τ decay (E) and on time to peak (F) for WT, C290A and KO. Load‐free and In‐Gel n cell/N animal = 18/4 and 17/4 for WT; 18/4 and 17/4 for cKO; and 15/3 and 15/3 for C290A, respectively. Nested t test. G, fractional shortening (%) in load‐free and in‐gel conditions. n cell/N animal = 19/5 and 14/4. Nested t test. H, effect of PVA alone or Crosslinker alone on shortening. n cell/N animal = 18/4 and 19/4. Nested t test.
Figure 6. Ca2+ sparks and waves in load‐free or afterloaded (in‐gel) myocytes.
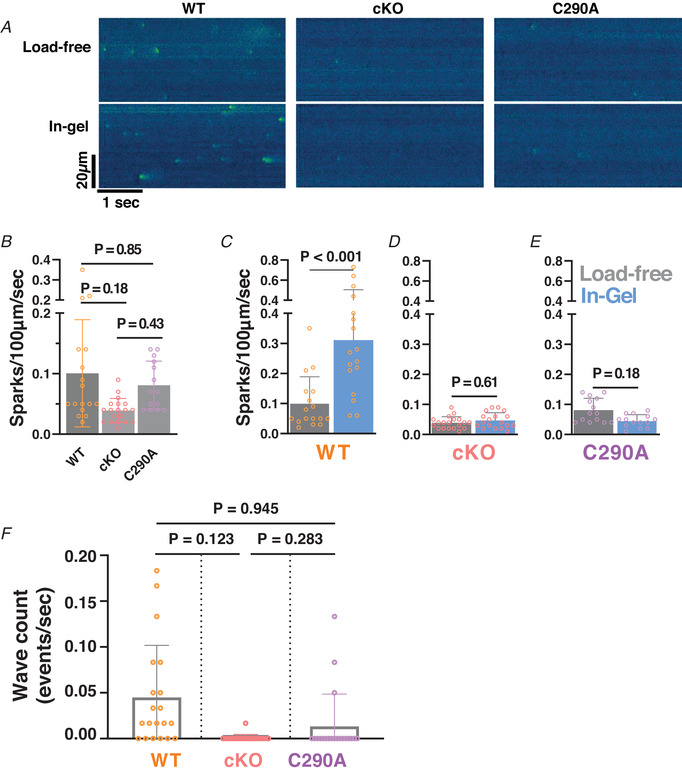
A, representative spontaneous Ca2+ sparks seen in the pseudo‐coloured confocal linescan images of cardiomyocytes with and without load in wild‐type (WT, left), cardiac specific CaMKIIδ‐KO (cKO, middle) and CaMKIIδ nitrosylation resistant (C290A, right) mice. B, mean ± SD values of resting Ca2+ spark rate in WT, cKO and C290A cardiomyocytes. n cell/N animal = 17/4, 19/4 and 14/3, respectively. Nested one‐way ANOVA. C–E, mean ± SD values of Ca2+ spark rate in WT, cKO and C290A cardiomyocytes under load‐free conditions (grey) and in‐gel (blue). n cell/N animal = 17/4, 17/4, 19/4, 18/4, 14/3, 14/3, respectively. Nested t tests. F, wave frequency in WT, cKO and C290A myocytes (records in which waves occurred were excluded from spark analysis). n cell/N animal = 20/4, 20/4 and 20/3, respectively. Nested one‐way ANOVA.
Here, we used cardiac specific CaMKIIδ knockout mice (cKO) to more explicitly test whether CaMKIIδ, in particular, is required for mediating the afterload‐induced effects in Ca2+ handling. Figure 5B, D and E shows that compared to WT myocytes in the same conditions, the afterload‐induced increase in CaT amplitude and rate of [Ca]i decline were completely prevented in the CaMKIIδ‐cKO myocytes. Moreover, in the absence of CaMKIIδ, CaT amplitude became smaller and slower under afterload (Fig. 5D–E ). Thus, the CaMKIIδ isoform is specifically required for this intrinsic afterload‐induced increase in Ca2+ transients.
Furthermore, the new CaMKIIδ‐C290A‐KI myocytes allowed a more explicit test of whether this single cysteine (C290) on CaMKIIδ is critical for the afterload‐induced effects on Ca2+ transients. Figure 5C–F shows that contrary to their WT littermates, C290A myocytes contracting in the gel (vs. load‐free contractions) displayed a decrease in CaT amplitude (Fig. 5D ) and no change in the τ of twitch [Ca]i decline (Fig. 5E ). Thus, this specific S‐nitrosylation target on CaMKIIδ‐C290 is essential to the afterload‐induced increase in CaTs and accelerated SR Ca2+ uptake.
Figure 6A–B shows that CaMKII‐cKO and WT myocytes exhibited mean Ca2+ spark frequencies of 0.03 and 0.1 sparks/100 μm/s, respectively (P = 0.18, nested one‐way ANOVA, multiple comparison test). Given the activating effect of CaMKII on diastolic RyR2, a difference might have been expected (Guo et al., 2006; Wehrens et al., 2004), but in resting myocytes CaMKII activation is typically low (Erickson et al., 2011). More importantly, Fig. 6C and D exhibits a pronounced afterload increase in Ca2+ spark frequency in WT myocytes that was completely absent in the CaMKIIδ‐cKO myocytes. The C290A‐KI myocytes had a baseline Ca2+ spark frequency that was roughly in‐between the WT and CaMKIIδ‐cKO myocytes. But as with the CaMKIIδ‐cKO, the point‐mutant C290A (Fig. 6E ) abolished the afterload‐induced Ca2+ spark frequency observed in WT myocytes. In fact, there tended to be a slight decrease in Ca2+ spark frequency in C290A myocytes, which could possibly be explained by potential S‐nitrosylation of the CaMKIIδ inhibitory S‐nitrosylation site (C273) that cannot be opposed by the activating effect of C290 nitrosylation in the C290A mouse. In the above Ca2+ spark frequency analysis, sweeps were excluded from spark analysis if propagating Ca2+ waves occurred because such waves would tend to reduce SR Ca2+ content and hence also reduce overall probability of Ca2+ sparks. The WT myocytes under afterload conditions exhibited many more arrhythmogenic Ca2+ waves than were seen in either CaMKIIδ‐cKO or C290A‐KI myocytes (Fig. 6F ).
Taken together, these mechano‐chemo‐transduction studies point to NOS1‐mediated S‐nitrosylation of CaMKIIδ‐C290 as the critical mediator of the intrinsic afterload‐induced increase in cardiac Ca2+ transient enhancement that allows the heart to contract more strongly in response to greater circulatory afterload. Indeed, C290 was required for not only the afterload‐induced increase in Ca2+ transient amplitude and faster SR Ca2+ uptake but also for the afterload‐induced increase in arrhythmogenic Ca2+ waves. Ergo, S‐nitrosylation of CaMKIIδ C290 is necessary for the mediation of afterload‐induced increase in Ca2+ transients, but also potentially arrhythmogenic Ca2+ sparks and waves.
Discussion
The mechanisms by which mechanical stress feeds back on contraction itself, Ca2+ cycling and electrophysiology involve complex systems of mechano‐chemo‐transduction that have been gaining more attention in the cardiovascular field as interactions among these individual fields have become better appreciated (Cingolani et al., 2013; Dowrick et al., 2019; Hegyi et al., 2021; Izu et al., 2020; Jian et al., 2014; Kampourakis & Irving, 2021; Petroff et al., 2001; Prosser et al., 2011; Reil et al., 2020; Shimkunas et al., 2021; Shin et al., 2010; Toischer et al., 2008, 2010). Here we focus on how mechanical afterload enhances Ca2+ transients, mediating a myocyte‐intrinsic form of autoregulation that can boost contraction (Shimkunas et al., 2021) and help the heart pump against an elevated afterload in the Anrep effect. Our cell‐in‐gel system mimics in vivo mechanical conditions in two ways. First, it imposes multiaxial 3D mechanical stress selectively during contraction (afterload), and it also tethers cell surface proteins to the gel and thereby engages endogenous cell‐surface signalling system responses to both normal and shear stress. In WT mice and a genetic mouse model of familial hypertrophic cardiomyopathy, selective inhibition of either NOS1 or CaMKII prevented the afterload‐induced rise in spontaneous Ca2+ activities (Jian et al., 2014), highlighting the involvement of both nitrosylation and CaMKII in this form of mechano‐chemo‐transduction. We also know from previous studies that CaMKII can be S‐nitrosylated at C290 (in CaMKIIδ), and that this promotes autonomous CaMKII activation (Coultrap & Bayer, 2014; Erickson et al., 2015; Pereira et al., 2017) that can enhance RyR sensitivity and SR Ca2+ uptake, and other contractile, electrophysiological and nuclear signalling processes (Anderson et al., 2011; Hegyi et al., 2019).
Our new CRISPR‐based knock‐in mouse, which selectively prevents S‐nitrosylation at a single amino acid on CaMKIIδ (C290A), enables molecular dissection of afterload‐induced enhancement of Ca2+ signalling in adult ventricular myocytes with otherwise normal CaMKIIδ function. These mice exhibit no overt phenotype (vs. WT littermates) but show suppressed ability of the exogenous NO‐donor GSNO to promote myocyte RyR opening in the form of spontaneous diastolic SR Ca2+ release events (Ca2+ sparks). Moreover, with only endogenous NOS signalling available, this single amino acid substitution in CaMKIIδ in C290A mice abolished the afterload‐induced enhancement of such Ca2+ sparks (Fig. 6A and E ). Previous work had attributed afterload‐induced Ca2+ sparks to NOS1‐induced NO production using NOS1 knockout mice and CaMKII activity using the imperfect CaMKII inhibitor KN‐93 (Jian et al., 2014). Our results here highlight that the afterload‐induced enhancement of Ca2+ sparks requires cardiac myocyte CaMKIIδ and specifically amino acid C290 therein, based on results in CaMKIIδ‐cKO and ‐C290A, respectively. Moreover, we conclude that this process involves NOS1‐dependent S‐nitrosylation of CaMKIIδ at C290 to enhance CaMKII activity, which is known to promote SR Ca2+ release channel activity via phosphorylation at RyR2‐S2814 and enhanced Ca2+ sensitivity of the RyR channel (Guo et al., 2006; Uchinoumi et al., 2016; van Oort et al., 2010; Wehrens et al., 2004). Notably, this RyR sensitization would also increase fractional SR Ca2+ release, but that effect would diminish over several beats as the larger Ca2+ transient would drive higher Ca2+ efflux from the myocyte, resulting in reduced SR Ca content that can substantially limit steady state enhancement of Ca2+ transients (Ai et al., 2005; Diaz et al., 2005). In addition, this RyR sensitization can be proarrhythmic by increasing the propensity for Ca2+ waves that can induce triggered beats. Thus, the higher RyR Ca2+ sensitivity by itself is likely insufficient to explain the marked afterload‐induced increase in steady state Ca2+ transients.
Mechanical afterload also induced a robust acceleration of SR Ca2+ uptake rate, based on the kinetics of Ca2+ transient decline, which would enhance SR Ca2+ content and better sustain the enhanced Ca2+ transient amplitudes as observed in Fig. 5D . Consistent with the Ca2+ spark results, the acceleration of twitch [Ca2+]i decline and larger Ca2+ transient amplitudes were also prevented in both the CaMKIIδ‐cKO and C290A KI myocytes (Fig. 5D and E ). This suggests that CaMKIIδ S‐nitrosylation at C290 is a critical mediator of at least these two SR targets that synergize and contribute to the afterload‐induced increase in Ca2+ transient amplitude and decay kinetics.
The RyR effects via the NOS1–CaMKII–RyR pathway have now been well‐studied and may be critical in mediating β‐AR effects at the RyR (Curran et al., 2014; Gutierrez et al., 2013; Pereira et al., 2017, 2013). CaMKIIδ also phosphorylates phospholamban (PLN) at Thr17 and enhances SERCA2 function, akin to β‐AR activation and PKA‐dependent phosphorylation of PLN at Ser16, which is quantitatively dominant in acute β‐AR responses. Higher myocyte pacing rates gradually increase PLN phosphorylation at the Thr17 CaMKII site over several minutes (Hagemann et al., 2000; Huke & Bers, 2007), but the time course and quantitative extent of that phosphorylation do not match well with the observed frequency‐dependent acceleration of SR Ca2+ uptake (Huke & Bers, 2007). Thus, while CaMKIIδ C290 is critical for the afterload enhanced SR Ca2+ uptake rate, the detailed molecular mechanism may require additional study. In a parallel initial electrophysiological study in rabbit ventricular myocytes in the same cell‐in‐gel system, we found that afterload induced an increase in both L‐type Ca2+ current (I Ca) and action potential duration (Hegyi et al., 2021). Both of those effects would enhance myocyte Ca2+ loading and contribute to the enhanced Ca transient amplitude observed, which can intrinsically accelerate the τ of [Ca2+]i decline (Bers & Berlin, 1995). The involvement of CaMKIIδ or C290 in these afterload‐induced electrophysiological changes has not yet been tested, but since CaMKII alters the gating of Ca2+, Na+ and K+ currents (Anderson et al., 2011; Hegyi et al., 2019), follow‐up studies may clarify whether CaMKIIδ and C290 are essential for some of those afterload‐induced electrophysiological effects.
CaMKIIδ has two known sites for nitrosylation, C290 and C273. While S‐nitrosylation of C290 on CaMKIIδ promotes autonomous kinase activity, S‐nitrosylation of C273 suppresses activation by Ca–CaM (Erickson et al., 2015). Given the role of nitrosylation in mechano‐chemo‐transduction, the differential effect of nitrosylation at these two sites suggests an added layer of complexity, as activating the inhibitory C273 site could complicate the physiological mechano‐chemo‐transduction process. Additionally, we cannot rule out other possible post‐translational modifications at these S‐nitrosylation sites, including glutathionylation and oxidation. We also have not addressed the upstream afterload‐induced signal transduction mechanism, which has not yet been elucidated, but candidates could include integrin and dystrophin linked signalling.
An increase in afterload (higher gel stiffness; up to10 kPa) leads to progressively increasing myocyte Ca2+ transient amplitude, which limits the reduction of fractional shortening as afterload increases (Shimkunas et al., 2021). This would translate to a relatively preserved stroke volume with increased afterload that is suppressed by genetic ablation of NOS1 (Jian et al., 2014), CaMKIIδ or the replacement of WT CaMKIIδ with CaMKIIδ‐C290A‐KI (Fig. 5). It would be of interest to test whether cardiac output decreases more in C290A mice than in WT in response to acute or chronic pressure‐overload, as reported for CaMKIIδ‐KO mice (Reil et al., 2020). Mechanical preload has also been shown to promote increases in Ca2+ spark frequency, involving NADPH oxidase (NOX2) that produces reactive oxygen species (ROS) (Prosser et al., 2011), so it is possible that there are somewhat separate but interacting signalling pathways in mediating intrinsic afterload‐ and preload‐induced changes in myocyte Ca2+ handling. NOX2 can be activated by NO signalling or CaMKII activation (Girouard et al., 2009; Lu et al., 2020), suggesting a complex interplay between these two pathways that merits further study regarding the specific role of CaMKII oxidation in mechano‐chemo‐transduction.
Post‐translational modifications in CaMKIIδ’s critical regulatory domain by autophosphorylation (T287), O‐GlcNAcylation (S280), oxidation (MM281/282) and S‐nitrosylation (C290) each promotes autonomous or chronic CaMKII activity that has heretofore been implicated in pathological consequences in many pathways (Anderson et al., 2011; Coultrap & Bayer, 2014; Erickson et al., 2008, 2013, 2015; Hegyi et al., 2019). Here, we have identified a potentially important beneficial adaptive effect of S‐nitrosylation at CaMKIIδ‐C290 that may critically mediate the intrinsic autoregulation mechanism known as the Anrep effect (or slow force response) which boosts the heart's ability to contract when it encounters a mechanical afterload such as elevated arterial pressure or aortic valvular resistance that is independent of neuronal input. Furthermore, this S‐nitrosylation of CaMKIIδ‐C290 may represent a novel therapeutic strategy to enhance myocardial function.
Additional information
Competing interests
None.
Author contributions
C.C.A., C.Y.K., Y.C., D.M.B. and J.B. conceived the conceptual design of the study. C.C.A. and J.M.H. performed data acquisition and analysis. C.C.A., C.Y.K., J.M.H., E.Y.S., S.B., Y.C., D.M.B. and J.B. performed visualization and interpretation of results. C.C.A. drafted the manuscript, C.C.A., C.Y.K., J.M.H., E.Y.S., S.B., Y.C., D.M.B. and J.B. edited and revised it critically for important intellectual content. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the National Institutes of Health (NIH) R01‐HL030077 (D.M. Bers), P01‐HL141084 (D.M. Bers), R01‐HL141460 (Y. Chen‐Izu), R01‐HL142282 (D.M. Bers and J. Bossuyt).
Supporting information
Statistical Summary Document
Peer Review History
Figure S1
Figure S2
Acknowledgements
We thank Dr Zhong Jian for preparing and providing the PVA hydrogel kit.
Biography
Chidera Alim is a post‐doctoral scholar at the University of California, Davis. She earned her PhD in Physiology at UC Davis, after graduating from the illustrious Howard University where she received her BSc in Biology. Her research interests surround the spatiotemporal regulation of the calcium/calmodulin‐dependent protein kinase II (CaMKII), its post‐translational modifications and its role in mechano‐chemo‐transduction. Her current research is focused on understanding the regulation and function of CaMKII in cardiovascular health and disease, in order to advance the treatment of heart failure and cardiac arrhythmia.

Handling Editors: Bjorn Knollmann & Michael Shattock
The peer review history is available in the Supporting information section of this article (https://doi.org/10.1113/JP283427#support‐information‐section).
This is an Editor's Choice article from the 15 November 2022 issue.
Contributor Information
Donald M. Bers, Email: dmbers@ucdavis.edu.
Julie Bossuyt, Email: jbossuyt@ucdavis.edu.
Data availability statement
Data are available on request from the corresponding authors: D.B. and J.B.
References
- Ai, X. , Curran, J. W. , Shannon, T. R. , Bers, D. M. , & Pogwizd, S. M. (2005). Ca2+/calmodulin‐dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circulation Research, 97(12), 1314–1322. [DOI] [PubMed] [Google Scholar]
- Ait‐Mou, Y. , Hsu, K. , Farman, G. P. , Kumar, M. , Greaser, M. L. , Irving, T. C. , & de Tombe, P. P. (2016). Titin strain contributes to the Frank‐Starling law of the heart by structural rearrangements of both thin‐ and thick‐filament proteins. Proceedings of the National Academy of Sciences, USA, 113(8), 2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, D. G. , & Kentish, J. C. (1985). The cellular basis of the length‐tension relation in cardiac muscle. Journal of Molecular and Cellular Cardiology, 17(9), 821–840. [DOI] [PubMed] [Google Scholar]
- Anderson, M. E. , Brown, J. H. , & Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. Journal of Molecular and Cellular Cardiology, 51(4), 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers, D. M. (2002). Cardiac excitation‐contraction coupling. Nature, 415(6868), 198–205. [DOI] [PubMed] [Google Scholar]
- Bers, D. M. , & Berlin, J. R. (1995). Kinetics of [Ca]i decline in cardiac myocytes depend on peak [Ca]i. American Journal of Physiology, 268(1), C271‐C277. [DOI] [PubMed] [Google Scholar]
- Cingolani, H. E. , Perez, N. G. , Cingolani, O. H. , & Ennis, I. L. (2013). The Anrep effect: 100 years later. American Journal of Physiology. Heart and Circulatory Physiology, 304(2), H175‐H182. [DOI] [PubMed] [Google Scholar]
- Coultrap, S. J. , & Bayer, K. U. (2014). Nitric oxide induces Ca2+‐independent activity of the Ca2+/calmodulin‐dependent protein kinase II (CaMKII). Journal of Biological Chemistry, 289(28), 19458–19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, J. , Tang, L. , Roof, S. R. , Velmurugan, S. , Millard, A. , Shonts, S. , Wang, H. , Santiago, D. , Ahmad, U. , Perryman, M. , Bers, D. M. , Mohler, P. J. , Ziolo, M. T. , & Shannon, T. R. (2014). Nitric oxide‐dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS One, 9(2), e87495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tombe, P. P. , Mateja, R. D. , Tachampa, K. , Ait Mou, Y. , Farman, G. P. , & Irving, T. C. (2010). Myofilament length dependent activation. Journal of Molecular and Cellular Cardiology, 48(5), 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, M. E. , Graham, H. K. , O'Neill, S. C. , Trafford, A. W. , & Eisner, D. A. (2005). The control of sarcoplasmic reticulum Ca content in cardiac muscle. Cell Calcium, 38(3–4), 391–396. [DOI] [PubMed] [Google Scholar]
- Dowrick, J. M. , Tran, K. , Loiselle, D. S. , Nielsen, P. M. F. , Taberner, A. J. , Han, J. C. , & Ward, M. L. (2019). The slow force response to stretch: Controversy and contradictions. Acta Physiologica, 226(1), e13250. [DOI] [PubMed] [Google Scholar]
- Erickson, J. R. , Joiner, M. L. , Guan, X. , Kutschke, W. , Yang, J. , Oddis, C. V. , Bartlett, R. K. , Lowe, J. S. , O'Donnell, S. E. , Aykin‐Burns, N. , Zimmerman, M. C. , Zimmerman, K. , Ham, A. J. , Weiss, R. M. , Spitz, D. R. , Shea, M. A. , Colbran, R. J. , Mohler, P. J. , & Anderson, M. E. (2008). A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell, 133(3), 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, J. R. , Nichols, C. B. , Uchinoumi, H. , Stein, M. L. , Bossuyt, J. , & Bers, D. M. (2015). S‐nitrosylation induces both autonomous activation and inhibition of calcium/calmodulin‐dependent protein kinase II δ. Journal of Biological Chemistry, 290(42), 25646–25656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, J. R. , Patel, R. , Ferguson, A. , Bossuyt, J. , & Bers, D. M. (2011). Fluorescence resonance energy transfer‐based sensor Camui provides new insight into mechanisms of calcium/calmodulin‐dependent protein kinase II activation in intact cardiomyocytes. Circulation Research, 109(7), 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, J. R. , Pereira, L. , Wang, L. , Han, G. , Ferguson, A. , Dao, K. , Copeland, R. J. , Despa, F. , Hart, G. W. , Ripplinger, C. M. , & Bers, D. M. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O‐linked glycosylation. Nature, 502(7471), 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard, H. , Wang, G. , Gallo, E. F. , Anrather, J. , Zhou, P. , Pickel, V. M. , & Iadecola, C. (2009). NMDA receptor activation increases free radical production through nitric oxide and NOX2. Journal of Neuroscience, 29(8), 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A. M. , Huxley, A. F. , & Julian, F. J. (1966). The variation in isometric tension with sarcomere length in vertebrate muscle fibres. Journal of Physiology, 184(1), 170–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, T. , Ai, X. , Shannon, T. R. , Pogwizd, S. M. , & Bers, D. M. (2007). Intra‐sarcoplasmic reticulum free [Ca2+] and buffering in arrhythmogenic failing rabbit heart. Circulation Research, 101(8), 802–810. [DOI] [PubMed] [Google Scholar]
- Guo, T. , Zhang, T. , Mestril, R. , & Bers, D. M. (2006). Ca2+/calmodulin‐dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circulation Research, 99(4), 398–406. [DOI] [PubMed] [Google Scholar]
- Gutierrez, D. A. , Fernandez‐Tenorio, M. , Ogrodnik, J. , & Niggli, E. (2013). NO‐dependent CaMKII activation during beta‐adrenergic stimulation of cardiac muscle. Cardiovascular Research, 100(3), 392–401. [DOI] [PubMed] [Google Scholar]
- Hagemann, D. , Kuschel, M. , Kuramochi, T. , Zhu, W. , Cheng, H. , & Xiao, R. P. (2000). Frequency‐encoding Thr17 phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. Journal of Biological Chemistry, 275(29), 22532–22536. [DOI] [PubMed] [Google Scholar]
- Hegyi, B. , Bers, D. M. , & Bossuyt, J. (2019). CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. Journal of Molecular and Cellular Cardiology, 127, 246–259. [DOI] [PubMed] [Google Scholar]
- Hegyi, B. , Shimkunas, R. , Jian, Z. , Izu, L. T. , Bers, D. M. , & Chen‐Izu, Y. (2021). Mechanoelectric coupling and arrhythmogenesis in cardiomyocytes contracting under mechanical afterload in a 3D viscoelastic hydrogel. Proceedings of the National Academy of Sciences, USA, 118(31), e2108484118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, C. J. , Lahair, M. M. , McCubrey, J. A. , & Franklin, R. A. (2004). Redox regulation of the calcium/calmodulin‐dependent protein kinases. Journal of Biological Chemistry, 279(43), 44573–44581. [DOI] [PubMed] [Google Scholar]
- Hudmon, A. , & Schulman, H. (2002). Structure‐function of the multifunctional Ca2+/calmodulin‐dependent protein kinase II. Biochemical Journal, 364(3), 593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huke, S. , & Bers, D. M. (2007). Temporal dissociation of frequency‐dependent acceleration of relaxation and protein phosphorylation by CaMKII. Journal of Molecular and Cellular Cardiology, 42(3), 590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izu, L. T. , Kohl, P. , Boyden, P. A. , Miura, M. , Banyasz, T. , Chiamvimonvat, N. , Trayanova, N. , Bers, D. M. , & Chen‐Izu, Y. (2020). Mechano‐electric and mechano‐chemo‐transduction in cardiomyocytes. Journal of Physiology, 598(7), 1285–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian, Z. , Han, H. , Zhang, T. , Puglisi, J. , Izu, L. T. , Shaw, J. A. , Onofiok, E. , Erickson, J. R. , Chen, Y. J. , Horvath, B. , Shimkunas, R. , Xiao, W. , Li, Y. , Pan, T. , Chan, J. , Banyasz, T. , Tardiff, J. C. , Chiamvimonvat, N. , Bers, D. M. , Lam, K. S. , & Chen‐Izu, Y. (2014). Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Science Signaling, 7(317), ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampourakis, T. , & Irving, M. (2021). The regulatory light chain mediates inactivation of myosin motors during active shortening of cardiac muscle. Nature Communication, 12(1), 5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, S. , Liao, Z. , Lu, X. , Katschinski, D. M. , Mercola, M. , Chen, J. , Heller Brown, J. , Molkentin, J. D. , Bossuyt, J. , & Bers, D. M. (2020). Hyperglycemia acutely increases cytosolic reactive oxygen species via o‐linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circulation Research, 126(10), e80–e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onofiok, E. , Lam, K. S. , & Luo, J. (2010). Three‐dimensional cell adhesion matrix.
- Pereira, L. , Bare, D. J. , Galice, S. , Shannon, T. R. , & Bers, D. M. (2017). β‐Adrenergic induced SR Ca2+ leak is mediated by an Epac‐NOS pathway. Journal of Molecular and Cellular Cardiology, 108, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, L. , Cheng, H. , Lao, D. H. , Na, L. , van Oort, R. J. , Brown, J. H. , Wehrens, X. H. , Chen, J. , & Bers, D. M. (2013). Epac2 mediates cardiac beta1‐adrenergic‐dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation, 127(8), 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroff, M. G. , Kim, S. H. , Pepe, S. , Dessy, C. , Marbán, E. , Balligand, J. L. , & Sollott, S. J. (2001). Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nature Cell Biology, 3(10), 867–873. [DOI] [PubMed] [Google Scholar]
- Picht, E. , Zima, A. V. , Blatter, L. A. , & Bers, D. M. (2007). SparkMaster: Automated calcium spark analysis with ImageJ. American Journal of Physiology Cell Physiology, 293(3), C1073‐C1081. [DOI] [PubMed] [Google Scholar]
- Prosser, B. L. , Ward, C. W. , & Lederer, W. J. (2011). X‐ROS signaling: Rapid mechano‐chemo transduction in heart. Science, 333(6048), 1440–1445. [DOI] [PubMed] [Google Scholar]
- Reil, J. C. , Reil, G. H. , Kovacs, A. , Sequeira, V. , Waddingham, M. T. , Lodi, M. , Herwig, M. , Ghaderi, S. , Kreusser, M. M. , Papp, Z. , Voigt, N. , Dobrev, D. , Meyhofer, S. , Langer, H. F. , Maier, L. S. , Linz, D. , Mugge, A. , Hohl, M. , Steendijk, P. , & Hamdani, N. (2020). CaMKII activity contributes to homeometric autoregulation of the heart: A novel mechanism for the Anrep effect. Journal of Physiology, 598(15), 3129–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimkunas, R. , Hegyi, B. , Jian, Z. , Shaw, J. A. , Kazemi‐Lari, M. A. , Mitra, D. , Leach, J. K. , Li, X. , Jaradeh, M. , Balardi, N. , Chen, Y. J. , Escobar, A. L. , Baker, A. J. , Bossuyt, J. , Banyasz, T. , Chiamvimonvat, N. , Lam, K. S. , Bers, D. M. , Izu, L. T. , & Chen‐Izu, Y. (2021). Mechanical load regulates excitation‐Ca2+ signaling‐contraction in cardiomyocyte. Circulation Research, 128(6), 772–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, S. H. , Hung, C. L. , Uno, H. , Hassanein, A. H. , Verma, A. , Bourgoun, M. , Køber, L. , Ghali, J. K. , Velazquez, E. J. , Califf, R. M. , Pfeffer, M. A. , & Solomon, S. D. ; Valsartan in Acute Myocardial Infarction Trial (VALIANT) Investigators (2010). Mechanical dyssynchrony after myocardial infarction in patients with left ventricular dysfunction, heart failure, or both. Circulation, 121(9), 1096–1103. [DOI] [PubMed] [Google Scholar]
- Toischer, K. , Kögler, H. , Tenderich, G. , Grebe, C. , Seidler, T. , Van, P. N. , Jung, K. , Knöll, R. , Körfer, R. , & Hasenfuss, G. (2008). Elevated afterload, neuroendocrine stimulation, and human heart failure increase BNP levels and inhibit preload‐dependent SERCA upregulation. Circulation Heart Failure, 1(4), 265–271. [DOI] [PubMed] [Google Scholar]
- Toischer, K. , Rokita, A. G. , Unsöld, B. , Zhu, W. , Kararigas, G. , Sossalla, S. , Reuter, S. P. , Becker, A. , Teucher, N. , Seidler, T. , Grebe, C. , Preuss, L. , Gupta, S. N. , Schmidt, K. , Lehnart, S. E. , Krüger, M. , Linke, W. A. , Backs, J. , Regitz‐Zagrosek, V. , …, Hasenfuss, G. (2010). Differential cardiac remodeling in preload versus afterload. Circulation, 122(10), 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchinoumi, H. , Yang, Y. , Oda, T. , Li, N. , Alsina, K. M. , Puglisi, J. L. , Chen‐Izu, Y. , Cornea, R. L. , Wehrens, X. H. T. , & Bers, D. M. (2016). CaMKII‐dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. Journal of Molecular and Cellular Cardiology, 98, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oort, R. J. , McCauley, M. D. , Dixit, S. S. , Pereira, L. , Yang, Y. , Respress, J. L. , Wang, Q. , De Almeida, A. C. , Skapura, D. G. , Anderson, M. E. , Bers, D. M. , & Wehrens, X. H. (2010). Ryanodine receptor phosphorylation by calcium/calmodulin‐dependent protein kinase II promotes life‐threatening ventricular arrhythmias in mice with heart failure. Circulation, 122(25), 2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens, X. H. T. , Lehnart, S. E. , Reiken, S. R. , & Marks, A. R. (2004). Ca2+/calmodulin‐dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circulation Research, 94(6), e61–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Summary Document
Peer Review History
Figure S1
Figure S2
Data Availability Statement
Data are available on request from the corresponding authors: D.B. and J.B.