

Abstract
Pharmacogenomic (PGx) testing has emerged as a compelling strategy that clinicians can use to inform antidepressant medication selection and dosing, but the clinical efficacy of this strategy has been questioned. We systematically reviewed and meta‐analyzed clinical trials for an association between the use of PGx‐guided antidepressant therapy and depressive symptom remission in patients with major depressive disorder (MDD). We included prospective, controlled clinical trials published in English up to July 12, 2022. Data extraction and synthesis adhered to the 2020 Preferred Reporting Items for Systematic Reviews and Meta‐Analyses guidelines. Each trial was assessed for risk of bias and a random‐effects model was used to estimate pooled risk ratios. Thirteen trials comprising 4,767 patients were analyzed, including 10 randomized controlled trials, and three open label trials. Across all included trials, those that received PGx‐guided antidepressant therapy (n = 2,395) were 1.41 (95% confidence interval (CI) = 1.15–1.74, P = 0.001) more likely to achieve remission compared with those that received unguided antidepressant therapy (n = 2,372). Pooled risk ratios for randomized controlled trials and open label trials were 1.46 (95% CI: 1.13–1.88) and 1.26 (95% CI = 0.84–1.88), respectively. These results suggest that PGx‐guided antidepressant therapy is associated with a modest but significant increase in depressive symptom remission in adults with MDD. Efforts to address the heterogeneity in PGx test composition (i.e., genes and alleles tested) and accompanying prescribing recommendations across trials will likely reduce the uncertainty about the efficacy of PGx‐guided antidepressant therapy in the literature.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Clinical trials have reported mixed findings regarding the efficacy of pharmacogenomic (PGx) guided antidepressant therapy among adults with major depressive disorder (MDD), resulting in uncertainty about the use of PGx testing in practice.
WHAT QUESTION DID THIS STUDY ADDRESS?
Does the whole of the clinical trial evidence support the efficacy of PGx‐guided antidepressant therapy in MDD?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This is the most comprehensive systematic review and meta‐analysis of the clinical trial evidence on PGx‐guided antidepressant therapy in MDD. The results suggest a modest but significantly favorable effect of PGx‐guided antidepressant therapy on depressive symptom remission.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
PGx testing merits consideration by healthcare providers when treating adults with MDD, particularly patients with moderate to severe depression and a history of inadequate response or intolerability to at least one antidepressant medication.
Pharmacological treatment of major depressive disorder (MDD) is largely a trial‐and‐error process, with <50% of individuals responding to their first medication and even fewer achieving remission. 1 This process can lead to treatment‐resistant depression which results in significantly greater healthcare resource utilization. 2 It is therefore critical to develop and implement clinical tools to aid in personalizing medication selection and dosing for patients with MDD.
Pharmacogenomic (PGx) testing is one objective tool that has been trialed for decades, but the paucity of prospective, clinical efficacy data has led to controversy surrounding whether this tool is ready for everyday use in the clinic. PGx testing utilizes associations between genetic variation and pharmacokinetic (e.g., drug levels in the blood) and clinical (i.e., efficacy and tolerability) outcomes to inform medication selection and dosing based on an individual's genome. Expert groups, such as the Clinical Pharmacogenetics Implementation Consortium (CPIC) and Dutch Pharmacogenomics Working Group, as well as drug regulators (e.g., US Food and Drug Administration (FDA)) have provided PGx‐based dosing guidelines intended for use by clinicians during decision making with regard to specific medications. 3 , 4 , 5 In the context of MDD, the bulk of current evidence and guidelines focus on variation in two drug metabolizing genes (CYP2C19 and CYP2D6) to inform selection and dosing of antidepressant therapy. 4 , 5 , 6 , 7 Notably, other genes (e.g., SLC6A4 and HTR2A) have been implicated as biomarkers in antidepressant medication response and are included on many commercial PGx test panels, despite limited clinical validity and efficacy. 8 , 9 , 10 , 11 , 12 , 13
Clinical trials have evaluated the use of PGx testing to inform antidepressant treatment and two meta‐analyses have been completed for a portion of these trials. 14 , 15 The first meta‐analysis, by Rosenblat and colleagues, included one open label trial and 4 randomized controlled trials (RCTs) and estimated a pooled risk ratio for depressive symptom remission of 1.74 in favor of PGx‐guided treatment. 15 Similarly, Bousman et al. estimated a pooled risk ratio of 1.71 among five RCTs. 14 Since the publication of these seminal meta‐analyses, several new trials have been published justifying a need to perform an updated meta‐analysis. Herein, we systematically reviewed the literature for prospective, controlled clinical trials that evaluated the efficacy of PGx testing as a strategy to inform antidepressant treatment and conducted a meta‐analysis to determine if this strategy is associated with depressive symptom remission.
METHODS
Search strategy
The systematic review followed the 2020 Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) reporting recommendations. 16 The following search string was used in PubMed to identify clinical trials published up to July 12, 2022, that assessed the efficacy of PGx‐guided antidepressant therapy: ((major depressive disorder OR mood disorder) AND (antidepressant) AND (pharmacogenetic OR pharmacogenomic) AND (prospective OR randomized OR randomized OR open label OR open‐label) AND (trial)). Bibliographies of all research articles were hand‐searched for additional references not identified in our primary search.
Selection criteria and data extraction
Two reviewers (authors L.C.B. and J.S.) independently assessed all articles identified by the search strategy for eligibility using the following criteria: (i) prospective, controlled trial examining the efficacy of PGx‐guided antidepressant therapy, (ii) only adult participants were included (aged ≥18), (iii) remission data was reported or available by request from the corresponding author of the trial, and (iv) the trial was published in English and full text was available. Articles for which a consensus on eligibility between the two reviewers was not obtained were evaluated by a third reviewer (author C.B.).
A custom data extraction template was used by the two primary reviewers (authors L.C.B. and J.S.) to summarize the selected articles. Extracted information included authors, year, study design, sample size, trial eligibility criteria, trial duration, sample characteristics (i.e., age, sex, and ancestry), remission measure used, and the gene composition of the PGx test used in the trial. When information was missing or incomplete for an eligible study, a request for additional information was made to the corresponding author of the trial.
Study risk of bias assessment
The Cochrane Risk of Bias Tools were utilized to assess bias in the RCTs (RoB2) and open label, controlled studies (ROBINS I) included in the analysis. 17
Statistical analysis
Data were analyzed using Review Manager version 5.4 (Revman) and the Major package in Jamovi version 2.2.5 (Jamovi). 18 , 19 The risk ratio (RR) was used as the effect size estimator and was calculated by contrasting the counts of remission, defined as a Hamilton Depression Rating Scale‐17 (HDRS‐17) score ≤7, the Clinical Global Impression scale ≤2, or the Patient Health Questionnaire ≤5 within pharmacogenomic‐guided and unguided treatment groups. Remission was selected as the outcome of interest because it is the goal of antidepressant therapy, all included trials assessed remission, and it is associated with a lower risk of depressive episode relapse and recurrence. The pooled RR was calculated using a random effects model using the inverse variance method. To determine the influence of any given study each of the eligible studies were omitted and the meta‐analyses were then re‐run to determine their influence on the pooled effect size following the recommendations of Viechtbauer and Cheung. 20 Because six of the included trials evaluated the GeneSight test, we also omitted these trials and re‐ran the meta‐analysis to determine whether these trials biased the overall results. Random‐effect meta‐regressions were conducted to assess the potential moderating effect of mean age of trial participants, sex (percentage of female subjects in the trial), ancestry (percentage of individuals of European background in the trial), weeks to primary end point measurement, average number of prior antidepressant treatments, and average severity of depression at study inclusion. Heterogeneity in effect sizes among trials was tested using the chi‐square statistic (with P < 0.10 indicating significant heterogeneity), and its magnitude was quantified using the I‐squared statistic, which is an index that describes the proportion of the total variation in the study effect size estimates that is due to heterogeneity and is independent of the number of studies included in the meta‐analysis and the metric of the effect size. Publication bias was evaluated using a funnel plot and Egger's regression test for funnel plot asymmetry. 21
RESULTS
The systematic review and selection process is summarized in Figure 1 . Five RCTs from our previous meta‐analysis were included in this study. 14 Newly found articles through July 15, 2022, included 145 from our Boolean search and 10 from hand searches of bibliographies. After removal of duplicates, the abstracts of 150 articles were screened and 132 were excluded for not meeting our eligibility criteria, resulting in 24 articles for retrieval and full text review. Among these 24 articles, 16 did not meet eligibility criteria, leaving a total of 8 new trials in addition to the five previously analyzed RCTs. 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33
Figure 1.
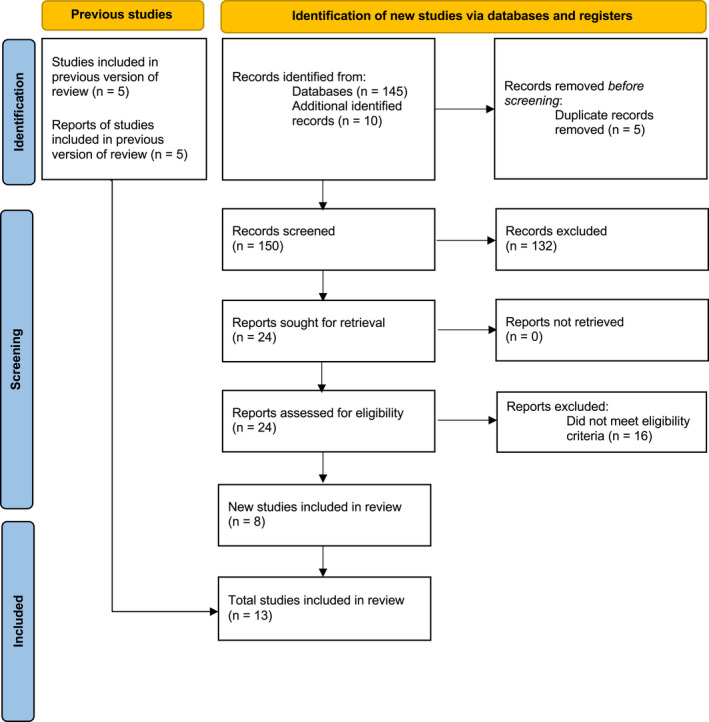
Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) diagram. Of 150 records screened, and five previous studies analyzed, our search resulted in a total of 13 new studies for inclusion in the analysis.
Characteristics of the 13 meta‐analyzed trials are summarized in Table 1 . For the meta‐analysis, a total of 4,767 (range 44–1944) adults participated across the included trials. Eligibility criteria across the trials varied. All but two trials exclusively enrolled participants with a primary diagnosis of MDD. 27 , 32 Ten of the 13 trials used a single (participant) or double (participant and rater) blinded RCT design and the remaining three trials used a controlled open label design. The majority (9/13, 69%) of the trials measured their primary end point at 8‐weeks post‐baseline (range 8–24 weeks) and all but the McCarthy et al. 32 and Oslin et al. 33 trials utilized the HDRS‐17 as their measure of remission. The genes tested varied across the trials but all included CYP2C19 and CYP2D6. Genetic testing was performed by commercial laboratories for all except the Shan et al. trial. 30 Ten of the 13 trials were preregistered at clinicaltrials.gov or similar registry and appeared to adhere to the initial protocol. 23 , 24 , 25 , 26 , 27 , 29 , 31 , 32 , 33 , 34 , 35 , 36
Table 1.
Studies included in systematic review and meta‐analysis
| PMID | Title | Author and year | Trial registration | Study design | Sample size (n) | Primary end point (weeks) | Age, years | Female (%) | Ancestry (% European) | Eligibility | Baseline depressive symptom score/number of failed medications | Scale (remission definition) | Target genes | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 23,047,243 | Using a PGx algorithm to guide the treatment of depression | Hall‐Flavin 2012 | N/A | Open‐label, controlled | 44 | 8 | PGx = 43; TAU = 42 | PGx = 54; TAU = 54 | “The subjects almost exclusively identified themselves to be of European ancestry.” | 25–70 years MDD by DSM‐IV HAM‐D17 ≥ 14 baseline |
QIDS‐C16 PGx = 16.0; TAU = 15.4 PGx = 4.4; TAU = 4.4 |
HAM‐D17 (not reported) | CYP2D6, CYP2C19, CYP1A2, HTR2A, SLC6A4 | [22] |
| 24,018,772 | Utility of integrated PGx testing to support the treatment of MDD in a psychiatric outpatient setting | Hall‐Flavin 2013 | NCT01610063 | Open‐label, controlled | 165 | 8 | PGx = 41; TAU = 44 | PGx = 69; TAU = 77 | “…almost exclusively European ancestry of the participants, …” | 18–72 years MDD by DSM‐IV HAM‐D17 ≥ 14 baseline |
HAM‐D17 PGx = 23.0; TAU = 22.5 PGx = 3.6; TAU = 4.7 |
HAM‐D17 (≤ 7) | CYP2D6, CYP2C19, CYP1A2, HTR2A, SLC6A4 | [23] |
| 24,229,738 | A prospective, randomized, double‐blind study assessing the clinical impact of integrated pharmacogenomic testing for MDD | Winner 2013 | NCT01261364 | Blinded (participant & rater) RCT | 49 | 10 | PGx = 51; TAU = 48 | PGx = 69; TAU = 92 |
PGx = 96; TAU = 100 |
Age ≥ 18 years depression based on interview HAM‐D17 score ≥ 14 |
PGx = NA; TAU = NA PGx = 2.9; TAU = 2.7 |
HAM‐D17 (≤7) | CYP2D6, CYP2C19, CYP1A2, SLC6A4, HTR2A | [24] |
| 26,243,841 | Improved antidepressant remission in major depression via a Pharmacokinetic Pathway Polygene Pharmacogenetic Report | Singh 2015 | ACTRN12613001135707 | Blinded (participant & rater) RCT | 128 | 12 | PGx = 44; TAU = 44 | PGx = 58; TAU = 61 | PGx = 100; TAU = 100 | Age ≥ 18 years MDD by DSM‐V HAM‐D17 score ≥ 18 |
HAM‐D17 PGx = 24.8 TAU = 24.7 PGx = NA; TAU = NA |
HAM‐D17 (≤ 7) | CYP2C19, CYP2D6, UGT1A1 ABCB1, ABCC1 | [25] |
| 28,705,252 | Efficacy of prospective PGx testing in the treatment of MDD: results of a randomized, double‐blind clinical trial | Perez 2017 | NCT02529462 | Blinded (participant) RCT | 280 | 12 | PGx = 52; TAU = 51 | PGx = 64; TAU = 63 |
PGx = 91; TAU = 94 |
Age ≥ 18 years MDD by DSM‐IV‐TR CGI‐S ≥ 4 new to treatment or inadequately controlled |
HAM‐D17 PGx = 19.5; TAU = 19.0 PGx = NA; TAU = NA |
HAM‐D17 (≤7) | CYP1A2, CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4, ABCB1, AKT1, BDNF, CACNG2, CES1, COMT, CRHR1, DDIT4, DRD3, EPHX1, FCHSD1, GRIK2, GRIK4, HLA‐A, HTR1A, HTR2A, HTR2C, LPHN3, NEFM, OPRM1, RGS4, RPTOR, SLC6A4, UGT2B15 | [26] |
| 28,992,526 | Improved efficacy with targeted PGx‐guided treatment of patients with depression and anxiety: A RCT demonstrating clinical utility | Bradley 2018 | NCT02878928 | Blinded (participant & rater) RCT | 93 | 8 | PGx = 48; TAU = 47 | PGx = 73; TAU = 72 |
PGx = 63; TAU = 63 |
19–87 years MDD and/or anxiety by DSM‐V New to treatment OR inadequately controlled |
HAM‐D17 PGx = 20; TAU = 20 PGx = NA; TAU = N/A |
HAM‐D17 (≤ 7) | CYP1A2, CYp2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5 SLC6A4, COMT, HTR2A, MTHFR | [27] |
| 30,466,219 | A PGx‐based antidepressant treatment for patients with MDD: Results from an 8‐week, Randomized, Single‐blinded Clinical Trial | Han 2018 | Not reported | Blinded (participant & rater) RCT | 100 | 8 | PGx = 44; TAU = 44 | PGx = 77; TAU = 73 |
PGx = 0; TAU = 0 |
Age > 20 years MDD by DSM‐V ≥ CGI‐3 OR intolerance to current medication |
QIDS‐C16 PGx = 24.5; TAU = 23.1 PGx = 2.5; TAU = 2.1 |
HAM‐D17 (≤ 7) | CYP1A2, CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4, ABCB1, AKT1, BDNF, CACNG2, CES1, COMT, CRHR1, DDIT4, DRD3, EPHX1, FCHSD1, GRIK2, GRIK4, HLA‐A, HTR1A, HTR2A, HTR2C, LPHN3, NEFM, OPRM1, RGS4, RPTOR, SLC6A4, UGT2B15 | [28] |
| 30,677,646 | Impact of pharmacogenomics on clinical outcomes in MDD in the GUIDED trial: A large, patient‐ and rater‐blinded, randomized, controlled study | Greden 2019 | NCT02109939 | Blinded (participant & rater) RCT | 1,167 | 8 | PGx = 47; TAU = 48 | PGx = 72; TAU = 70 |
PGx = 79; TAU = 82 |
Age ≥ 18 years MDD by ≥11 on QIDS‐C16 Inadequate response |
HAM‐D17 PGx = 21.1; TAU = 21.4 PGx = 3.5; TAU = 3.5 |
HAM‐D17 (≤7) | CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP2B6 SLC6A4, HTR2A | [29] |
| 31,572,113 | Preliminary Clinical Investigation of Combinatorial PGx Testing for the Optimized Treatment of Depression: A Randomized Single‐Blind Study | Shan 2019 | Not reported | Blinded (participant & rater) RCT | 71 | 8 | PGx = 27; TAU = 29 | PGx = 61; TAU = 65 |
PGx = 0; TAU = 0 |
Age 18–51 years MDD by DSM‐V HAM‐D17 ≥ 17 baseline Tx naive or free from medication for 2 weeks |
HAM‐D17 PGx = 21 TAU = 20.9 PGx = NA; TAU = NA |
HAM‐D17 (≤ 7) | CYP2C19, CYP2D6, CYP1A2 SLC6A4, HTR2A | [30] |
| 32,383,277 | Randomized, controlled, participant‐ and rater‐blind trial of PGx test‐guided treatment vs. treatment as usual for MDD | Perlis 2020 | NCT02634177 | Blinded (participant & rater) RCT | 296 | 8 | PGx = 48; TAU = 48 | PGx = 71; TAU = 73 |
PGx = 74; TAU = 72 |
Age ≥ 18 years MDD by DSM‐V > 18 SIGH‐D‐17 |
SIGH‐D‐17 PGx = 22.5; TAU = 22.1 PGx = 1.43 TAU = 1.52 |
SIGH‐D (≤ 7) | CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A4/5 SLC6A4, CACANA1C, ANK3, 5HT2C, MC4R, DRD2, COMT, ADRA2A, MTHFR, BDNF, OPRM1, GRIK1 | [31] |
| 33,938,307 | A prospective study to determine the clinical utility of PGx testing of veterans with treatment‐resistant depression | McCarthy 2021 | NCT04469322 | Blinded (participant) RCT | 102 | 8 | PGx = 52.5; TAU = 50.3 | PGx = 21; TAU = 24 |
PGx = 66; TAU = 69 |
Age ≥ 18 years veterans, MDD or Bipolar Depression Failed at least 1 medication |
QIDS‐SR PGx = 13.9; TAU = 13 PGx = NA; TAU = NA |
CGI (≤ 2) | CYP1A2, CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4, CYP3A5, DRD2, HLA‐B, 5HTTLPR, HTR2A, HTR2C, POLG, SLC6A4, UGT1A4 | [32] |
| 35,288,545 | Clinical utility of combinatorial PGx testing in depression: A Canadian patient‐ and rater‐blinded, RCT | Tiwari 2022 | NCT02466477 | Blinded (participant & rater) RCT | 308 | 8 | PGx1 = 40; PGx2 = 41; TAU = 42 | PGx1 = 66; PGx2 = 65; TAU = 63 | PGx1 = 80; PGx2 = 83; TAU = 89 | Age ≥ 18 years MDD by DSM‐IV QIDS‐C16 ≥ 11 Inadequate response to at least 1 medication |
HAM‐D17 PGx 1 = 21.3; PGx2 = 21.5; TAU = 21.4 PGx 1 = 4; PGx2 = 3.7; TAU = 3 |
HAM‐D17 (≤ 7) | CYP1A2, CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4 CNRI, GCG, HTR2A, HCRTR2, MC4R, NDUFS1, NPY, SLC6A4 | [34] |
| 35,819,423 |
Effect of PGx Testing for Drug‐Gene Interactions on Medication Selection and Remission of Symptoms in MDD The PRIME Care Randomized Clinical Trial |
Oslin 2022 | NCT03170362 | Open‐Label, controlled | 1944 | 24 | PGx = 48; TAU = 47 | PGx = 24; TAU = 27 |
PGx = 67 TAU = 70 |
Age 18–80 years, veterans, MDD, History of at least 1 treatment episode, plan to start new episode of antidepressant monotherapy, PHQ‐9 > 9. |
PHQ‐9 PGx = 17.5; TAU = 17.5 PGx = NA; TAU = NA |
PHQ‐9 (≤5) | CYP2D6, CYP2C19, CYP2C9, CYP2B6, CYP1A, CYP3A4, UDT1A4, UDT2B15, SLC6A4, HTR2A, HLA‐A, HLA‐B | [33] |
DSM‐IV, Diagnostic and Statistical Manual of Mental Disorders Fourth Edition; HAMD, Hamilton Rating Scale for Depression; MDD, major depression disorder; N/A, not applicable; PGx, pharmacogenomic; RCT, randomized controlled trial.
Risk of bias
Table 2 summarizes the risk of bias assessments for the 10 RCTs and three open label trials. Among the RCTs, selection and attrition biases were judged to be low. However, performance bias was judged as high for all RCTs. Although participants in all RCTs were blinded to the results, the physicians were not. In addition, two RCTs did not blind raters of the primary outcome, resulting in a high risk of detection bias. 26 , 32 High risk of reporting bias was also noted for the Bradley et al. 27 trial, given that remission status was only reported for a subset (n = 93) of participants with moderate to severe depressive symptoms (HDRS ≥25). Last, with the exception of the Shan et al. 30 and Oslin et al. 33 trials, all studies were industry sponsored, a study attribute that has been linked to more favorable outcomes in drug and device studies. For the three open label trials, pre‐intervention selection, at‐intervention information, and post‐intervention confounding biases were judged to be low. However, post‐intervention information and industry biases were judged as high for two out of three trials. Moderate risk of post‐intervention reporting bias was noted for the Hall‐Flavin et al. 2012 trial, 22 whereas pre‐intervention confounding and post‐intervention selection biases were judged as moderate for the Hall‐Flavin et al. 2013 trial. 23
Table 2.
Risk of bias RCT and open label studies
| Authors | Random sequence generation (selection bias) | Allocation concealment (selection bias) | Blinding of participants, personnel (performance bias) | Blinding of outcome assessment (detection bias) | Incomplete outcome data (attrition bias) | Selective outcome reporting (reporting bias) | Other sources of bias | Ref |
|---|---|---|---|---|---|---|---|---|
| Randomized controlled trials: RoB2 | ||||||||
| Winner et al. 2013 | Low: “A basic randomization scheme was utilized that did not balance for age, gender, ethnicity” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [24] |
| Singh 2015 | Low: “… via computerized randomization only half [of subjects] had this information analyzed” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [25] |
| Pérez et al. 2017 | Low: “Randomization was stratified by center with a 1:1 ratio for intervention and control group, using a computer‐ generated random list” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded Raters not blinded | High: Remission assessed by an unblinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [26] |
| Bradley et al. 2018 | Low: “… randomized by disease and severity and allocated in a 1:1 ratio…” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | High: Results for remission were reported in a subset of the sample | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [27] |
| Han et al. 2018 | Low: “All recruited patients were randomly allocated either to PGATx or treatment as usual (TAU)” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded Rater blinding unknown | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [28] |
| Greden et al. 2019 | Low: “Eligible patients were randomized 1:1 to TAU or the guided‐care (intervention) arm.” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [29] |
| Shan et al. 2019 | Low: “They were randomly allocated to guided and unguided groups” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician | [30] |
| Perlis et al. 2020 | Low: “Subjects were randomized 1:1 to either AGT or TAU treatment conditions” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded Raters blinded | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [31] |
| Tiwari et al 2022 | Low: “Patients were randomized 1:1:1 to one of three treatment arms, including two intervention arms and a TAU arm” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded Raters blinded | Low: Remission assessed by a blinded rater | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [32] |
| McCarthy et al. 2021 | Low: “Subjects were randomly assigned according to a predetermined schedule to either the PGX or TAU” | Low: Central allocation method used | High: Patients were blind to study group Treating clinician not blinded Raters not blinded | High: Remission assessed by unblinded treating clinician | Low: Balanced loss to follow up | Low: Results of all baseline measures were reported | High: Recruitment bias Patients were recruited by treating clinician Industry bias Industry sponsored | [34] |
| Authors | Bias due to Confounding (pre‐intervention confounding bias) | Bias in selection of participants into the study (pre‐intervention selection bias) | Bias in classification of intervention (at‐intervention information bias) | Bias due to deviations from intended interventions (post‐intervention confounding bias) | Bias due to missing data (post‐intervention selection bias) | Bias in measurement of outcomes (post‐intervention information bias) | Bias in the selection of reported results (post‐intervention reporting bias) | Notes |
|---|---|---|---|---|---|---|---|---|
| Open‐Label Trials: ROBINS‐I | ||||||||
| Hall‐Flavin et al. 2012 |
Low: Potential confounders were assessed and an appropriate analysis method was employed |
Low: Participants were selected based on characteristics observed prior to the intervention |
Low: Classification of intervention status was done prospectively |
Low: No indication of deviations noted. |
Low: Missing data present but balanced between guided and unguided arms. “seven were excluded from the analysis due to missing data (four in the unguided treatment group and three in the guided treatment group)” |
High: Raters were aware of the participant's intervention group |
Moderate: Trial was not preregistered and several secondary analyses performed |
In‐Kind Support from Industry |
| Hall‐Flavin et al. 2013 |
Moderate: Potential confounders were assessed but not included in the outcome analysis |
Low: Participants were selected based on characteristics observed prior to the intervention |
Low: Classification of intervention status was done prospectively |
Low: No indication of deviations noted |
Moderate: Missing data present and unbalanced between guided and unguided arms. “20 in the unguided group and 42 in the guided group” |
High: Raters were aware of the participant's intervention group |
Low: Reported results align with the trial's preregistration |
In‐Kind Support from Industry |
| Oslin et al. 2022 |
Moderate: Potential confounders were assessed but not included in the outcome analysis |
Low: Participants were selected based on characteristics observed prior to the intervention |
Low: Classification of intervention status was done prospectively |
Low: No indication of deviations noted |
Low: To account for data not being missing completely at random, the models for the repeated remission rates were extended to consider inverse‐probability–weighted selection models (valid under missing at random assumption) and pattern mixture models |
Low: Raters were blinded to participant's intervention group |
Low: Reported results align with the trial's preregistration |
Commercial test but not supported by industry |
PGx, pharmacogenetics; RCT, randomized controlled trial.
Meta‐analysis
Random‐effects pooled RRs and forest plots for the 10 RCTs and three open label trials are shown in Figure 2 . The pooled RR was 1.26 (95% CI = 0.84–1.88, P = 0.26) for open label trials and 1.46 (95% CI = 1.13–1.88, P = 0.003) for the RCTs. The pooled RR for all trials (RCTs + open label) was 1.41 (95% CI = 1.15–1.74, P = 0.001). None of the trials were identified as potential influential studies. Regardless of which trial was omitted from the meta‐analysis, the pooled RR remained statistically significant and overlapped with the 95% CI of the pooled RR from the meta‐analysis of all 10 RCTs (Table 3 ). Likewise, removal of the six trials that evaluated the GeneSight test did not significantly alter the pooled RR (RR = 1.46, 95% CI = 1.02–2.09, P = 0.04). Meta‐regression analyses of the 13 trials did not show a moderating effect for age (Beta = −0.012, P = 0.485), sex (Beta = −0.004, P = 0.517), ancestry (Beta = −0.001, P = 0.928), nor weeks to primary end point measurement (Beta = −0.019, P = 0.394; Table S1 ). However, moderating effects were detected for the average number of prior antidepressant treatments (Beta = 0.229, P = 0.026) among seven trials with available data and depressive symptom severity at study inclusion (Beta = 0.115, P = 0.036) among 12 trials with available data (Table S1 ). As the average number of prior antidepressant treatments and the average baseline depressive symptom severity increased so did the risk ratio in favor of PGx‐guided antidepressant treatment. Examination of funnel plot asymmetry and Egger's regression analysis showed no indication of publication bias (Z = 1.26, P = 0.205; Figure S1 ).
Figure 2.
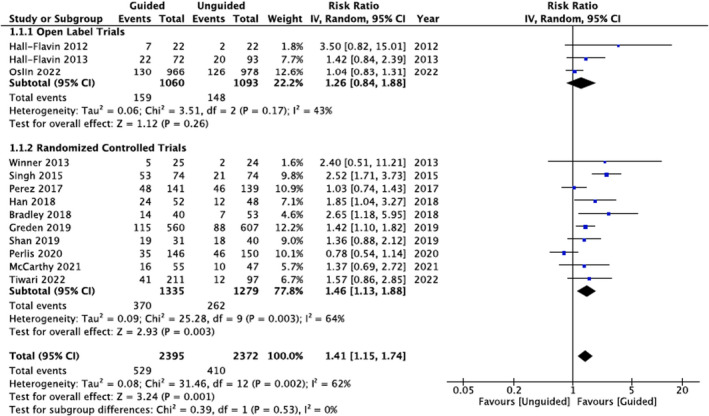
Meta‐analysis of PGx RCTs, open label, and combined. Pooled risk ratios were calculated using random‐effects models. CI, confidence interval; PGx, Pharmacogenomic; RCTs, randomized controlled trials.
Table 3.
Random‐effects meta‐analyses after omission of each of the 13 prospective, controlled clinical trials
| Omitted study | Pooled risk ratio | 95% CI |
|---|---|---|
| Singh 2015 | 1.32 | 1.06–1.65 |
| Bradley 2018 | 1.40 | 1.08–1.81 |
| Han 2018 | 1.43 | 1.09–1.88 |
| Winner 2013 | 1.45 | 1.11–1.88 |
| Tiwari 2022 | 1.46 | 1.10–1.92 |
| Shan 2019 | 1.48 | 1.11–1.98 |
| McCarthy 2021 | 1.48 | 1.12–1.94 |
| Greden 2019 | 1.49 | 1.09–2.03 |
| Perez 2017 | 1.55 | 1.17–2.04 |
| Perlis 2020 | 1.58 | 1.26–1.98 |
| Hall‐Flavin 2012 | 1.39 | 1.13–1.71 |
| Hall‐Flavin 2013 | 1.42 | 1.13–1.78 |
| Oslin 2022 | 1.48 | 1.18–1.86 |
| Total | 1.41 | 1.15–1.74 |
CI, confidence interval.
DISCUSSION
We identified and systematically reviewed 13 prospective, controlled trials that examined the effect of PGx‐guided antidepressant treatment on depressive symptom remission among individuals with MDD. Pooled RRs from these trials suggest a modest but significantly favorable effect of PGx testing on depressive symptom remission. Individuals that received PGx‐guided antidepressant treatment were 41% (95% CI = 15–74%) more likely to achieve depressive symptom remission relative to their counterparts who received treatment as usual. Notably, this effect is lower than that reported in the two previous and smaller meta‐analyses that showed PGx‐guided antidepressant treatment increased the likelihood of achieving depressive symptom remission by 71% and 74%. 14 , 15
Despite this attenuation of the initially reported impact of PGx‐guided antidepressant treatment on depressive symptom remission, the results continue to support the notion that PGx testing merits consideration by healthcare providers when treating adults with MDD. This may be particularly relevant to individuals with moderate to severe depression and a history of inadequate response or intolerability to psychotropic medications, characteristics that significantly enhanced the efficacy of PGx‐guided antidepressant treatment.
Limitations
Several caveats should be considered when interpreting the results of this study. First, the clinical generalizability of the findings is limited. Although two of the included trials exclusively enrolled individuals of Chinese ancestry and two trials were predominantly male subjects, most of the individuals included across the 13 trials were female adults aged 40–50 years with a European background. As such, uncertainty remains about the efficacy of PGx‐guided antidepressant treatment in diverse settings and emphasizes the critical need for trial evidence that is more representative of real‐world clinical populations. Second, prescribing physicians in all the included trials were not blinded to intervention allocation, which may have introduced a high risk of performance bias as well as attention and ancillary treatment biases. Innovative trial designs are needed to address these biases. One example is being employed in an ongoing Australian PGx trial (ACTRN12621000181808), in which prescribing recommendations are provided to all healthcare providers in an identical format, without reference to the participant's genetic results. Recommendations for participants allocated to the PGx‐guided group are tailored to their CYP2C19 and CYP2D6 genotypes, whereas the treatment as usual group receives recommendations based on current Australian antidepressant prescribing guidelines. Third, we are unable to determine which gene or combination of genes are driving the observed effect. Although all the included trials tested for genetic variants in CYP2C19 and CYP2D6, they also tested for variants in several other genes (e.g., SLC6A4 and HTR2A) that are often used by commercial laboratories to derive antidepressant prescribing recommendations, despite the absence of dosing guidelines for these genes. Complicating this matter further is that many of the PGx tests used in the included trials utilize proprietary combinatorial or “black box” algorithms that can lead to discordant recommendations among tests and with current PGx‐based dosing guidelines. 36 As a result, one could argue that the true impact of PGx‐guided antidepressant treatment on depressive symptom remission is likely being attenuated by such heterogeneity. As standardization efforts, such as the Standardizing Laboratory Practices in Pharmacogenomics initiative, develop and regulations related to PGx testing tighten, we expect this heterogeneity to lessen. 37 , 38 Finally, not all studies reported side effects in an evaluable way, precluding us from assessing the pooled effect of PGx‐guided antidepressant treatment on side effects in this population.
CONCLUSION
The gap between the availability of PGx testing and its clinical use is beginning to narrow but uncertainty about its efficacy for improving antidepressant treatment and depressive symptom remission continues to be a considerable constraint on widespread adoption in psychiatry. The findings of this systematic review and meta‐analysis, address this constraint and suggest that healthcare providers that use PGx‐guided antidepressant prescribing should expect modest but significant increases in depressive symptom remission in adults with MDD.
FUNDING
No financial support was provided for this study.
CONFLICTS OF INTEREST
L.C.B. is Founder and Principal Consultant for Great Scott! Consulting LLC, a PGx consulting company, and stockholder of Myriad Genetics. J.D.S. is an employee of Tempus Labs. D.J.M. reports to have been a co‐investigator on two pharmacogenomic studies where genetic test kits were provided as in‐kind contribution by Myriad Neuroscience. He has not received any payments or any equity, stocks, or options from any pharmacogenomic companies. C.B. is Founder and CEO of Sequence2Script Inc. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
L.C.B., J.D.S., K.B., A.M., D.J.M., and C.A.B. wrote the manuscript. L.C.B. and C.A.B. designed the research. J.D.S., L.C.B., K.B., and A.M. performed the research. C.A.B. analyzed the data.
REGISTRATION AND PROTOCOL
This review and meta‐analysis were registered in PROSPERO (registration number: CRD314807) and followed PRISMA guidelines. 16
Supporting information
Figure S1
Table S1
ACKNOWLEDGMENTS
None.
[Correction added on 16 November 2022, after first online publication: Affiliations linked with the authors ‘Daniel Müller’ and ‘Chad Bousman’ has been corrected in this version.]
References
- 1. Warden, D. , Rush, A.J. , Trivedi, M.H. , Fava, M. & Wisniewski, S.R. The STAR*D project results: a comprehensive review of findings. Curr. Psychiat. Rep. 9, 449–459 (2007). [DOI] [PubMed] [Google Scholar]
- 2. Sussman, M. , O'sullivan, A.K. , Shah, A. , Olfson, M. & Menzin, J. Economic burden of treatment‐resistant depression on the U.S. Health care system. J. Manag. Care Spec. Pharm. 25, 823–835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Relling, M.V. , Klein, T.E. , Gammal, R.S. , Whirl‐Carrillo, M. , Hoffman, J.M. & Caudle, K.E. The clinical pharmacogenetics implementation consortium: 10 years later. Clin. Pharmacol. Ther. 107, 171–175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swen, J. , Nijenhuis, M. , Boer, A. et al. Pharmacogenetics: from bench to byte—an update of guidelines. Clin. Pharmacol. Ther. 89, 662–673 (2011). [DOI] [PubMed] [Google Scholar]
- 5. FDA . Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/drugs/science‐and‐research‐drugs/table‐pharmacogenomic‐biomarkers‐drug‐labeling. Accessed April 27, 2022.
- 6. Hicks, J.K. et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin. Pharmacol. Ther. 98, 127–134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hicks, J. et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 102, 37–44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bousman, C. , Bengesser, S. , Aitchison, K. et al. Review and consensus on pharmacogenomic testing in psychiatry. Pharmacopsychiatry 54, 5–17 (2020). [DOI] [PubMed] [Google Scholar]
- 9. Bousman, C.A. & Hopwood, M. Commercial pharmacogenetic‐based decision‐support tools in psychiatry. Lancet Psychiatry 3, 585–590 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Maruf, A.A. , Fan, M. , Arnold, P.D. , Müller, D.J. , Aitchison, K.J. & Bousman, C.A. Pharmacogenetic Testing Options Relevant to Psychiatry in Canada: options de tests pharmacogénétiques pertinents en psychiatrie au Canada. Can. J. Psychiatry 65, 521–530 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stein, K. , Maruf, A.A. , Müller, D.J. , Bishop, J.R. & Bousman, C.A. Serotonin transporter genetic variation and antidepressant response and tolerability: a systematic review and meta‐analysis. J Personalized Med. 11, 1334 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wan, Y. , Zhai, X. , Tan, H. , Ai, Y. & Zhao, L. Associations between the 1438A/G, 102T/C, and rs7997012G/a polymorphisms of HTR2A and the safety and efficacy of antidepressants in depression: a meta‐analysis. Pharmacogenomics J. 21, 200–215 (2021). [DOI] [PubMed] [Google Scholar]
- 13. Ren, F. , Ma, Y. , Zhu, X. , Guo, R. , Wang, J. & He, L. Pharmacogenetic association of bi‐ and triallelic polymorphisms of SLC6A4 with antidepressant response in major depressive disorder. J. Affect Dis. 273, 254–264 (2020). [DOI] [PubMed] [Google Scholar]
- 14. Bousman, C.A. , Arandjelovic, K. , Mancuso, S.G. , Eyre, H.A. & Dunlop, B.W. Pharmacogenetic tests and depressive symptom remission: a meta‐analysis of randomized controlled trials. Pharmacogenomics 20, 37–47 (2019). [DOI] [PubMed] [Google Scholar]
- 15. Rosenblat, J.D. , Lee, Y. & McIntyre, R.S. The effect of pharmacogenomic testing on response and remission rates in the acute treatment of major depressive disorder: a meta‐analysis. J Affect Disorders. 241, 484–491 (2018). [DOI] [PubMed] [Google Scholar]
- 16. Page, M.J. , McKenzie, J.E. , Bossuyt, P.M. et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 372, n71 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Higgins, J.P.T. , Altman, D.G. , Gotzsche, P.C. et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 343, d5928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collaboration TC . Review Manager (RevMan) V5.4. Cochrane Reviews; 2022.
- 19. Project TJ . Jamovi. 2022. https://www.jamovi.org
- 20. Viechtbauer, W. & Cheung, M.W.‐L. Outlier and influence diagnostics for meta‐analysis. Res. Synth. Methods 1, 112–125 (2010). [DOI] [PubMed] [Google Scholar]
- 21. Egger, M. , Smith, G.D. , Schneider, M. & Minder, C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 315, 629–634 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hall‐Flavin, D.K. et al. Using a pharmacogenomic algorithm to guide the treatment of depression. Transl. Psychiatry 2, e172 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hall‐Flavin, D.K. et al. Utility of integrated pharmacogenomic testing to support the treatment of major depressive disorder in a psychiatric outpatient setting. Pharmacogenet. Genom. 23, 535–548 (2013). [DOI] [PubMed] [Google Scholar]
- 24. Winner, J.G. , Altar, C.A. , Carhart, J.M. , Allen, J.D. & DeChairo, B.M. A prospective, randomized, double‐blind study assessing the clinical impact of integrated pharmacogenomic testing for major depressive disorder. Discov. Med. 16, 219–227 (2013). [PubMed] [Google Scholar]
- 25. Singh, A.B. Improved antidepressant remission in major depression via a pharmacokinetic pathway polygene pharmacogenetic report. Clin. Psychopharm. Neu. 13, 150–156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pérez, V. , Salavert, A. , Espadaler, J. et al. Efficacy of prospective pharmacogenetic testing in the treatment of major depressive disorder: results of a randomized, double‐blind clinical trial. BMC Psychiatry 17, 250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bradley, P. et al. Improved efficacy with targeted pharmacogenetic‐guided treatment of patients with depression and anxiety: a randomized clinical trial demonstrating clinical utility. J. Psychiatr. Res. 96, 100–107 (2018). [DOI] [PubMed] [Google Scholar]
- 28. Han, C. et al. A pharmacogenomic‐based antidepressant treatment for patients with major depressive disorder: results from an 8‐week, randomized, Single‐blinded Clinical Trial. Clin. Psychopharm. Neu. 16, 469–480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Greden, J.F. et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: a large, patient‐ and rater‐blinded, randomized, controlled study. J. Psychiatr. Res. 111, 59–67 (2019). [DOI] [PubMed] [Google Scholar]
- 30. Shan, X. et al. Preliminary clinical investigation of combinatorial pharmacogenomic testing for the optimized treatment of depression: a randomized single‐blind study. Front. Neurosci. 13, 960 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Perlis, R.H. , Dowd, D. , Fava, M. , Lencz, T. & Krause, D.S. Randomized, controlled, participant‐ and rater‐blind trial of pharmacogenomic test‐guided treatment versus treatment as usual for major depressive disorder. Depress. Anxiety 37, 834–841 (2020). [DOI] [PubMed] [Google Scholar]
- 32. McCarthy, M.J. , Chen, Y. , Demodena, A. et al. A prospective study to determine the clinical utility of pharmacogenetic testing of veterans with treatment‐resistant depression. J. Psychopharmacol. 35, 992–1002 (2021). [DOI] [PubMed] [Google Scholar]
- 33. Oslin, D.W. , Lynch, K.G. , Shih, M. et al. Effect of pharmacogenomic testing for drug‐gene interactions of medication selection and remission of symptoms in major depressive disorder: the PRIME care randomized clinical trial. JAMA 328, 151–161 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tiwari, A.K. et al. Clinical utility of combinatorial pharmacogenomic testing in depression: a Canadian patient‐ and rater‐blinded, randomized, controlled trial. Transl. Psychiatry 12, 101 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lundh, A. , Lexchin, J. , Mintzes, B. , Schroll, J.B. & Bero, L. Industry sponsorship and research outcome. Cochrane Database Syst. Rev. 2017, MR000033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bousman, C.A. & Dunlop, B.W. Genotype, phenotype, and medication recommendation agreement among commercial pharmacogenetic‐based decision support tools. Pharmacogenomics J. 18, 613–622 (2018). [DOI] [PubMed] [Google Scholar]
- 37. Blazy, C. , Ellingrod, V. & Ward, K. Variability between clinical pharmacogenetics implementation consortium (CPIC®) guidelines and a commercial pharmacogenetics Laboratory in Genotype to phenotype interpretations for patients utilizing psychotropics. Front. Pharmacol. 13, 939313 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. American Society of Pharmacovigilance . Standardizing Laboratory Practices in Pharmacogenomics. https://www.stopadr.org/stripe. Accessed April 29, 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1