

SUMMARY
The precise and accurate identification and quantification of transcriptional start sites (TSSs) is key to understanding the control of transcription. The core promoter consists of the TSS and proximal non‐coding sequences, which are critical in transcriptional regulation. Therefore, the accurate identification of TSSs is important for understanding the molecular regulation of transcription. Existing protocols for TSS identification are challenging and expensive, leaving high‐quality data available for a small subset of organisms. This sparsity of data impairs study of TSS usage across tissues or in an evolutionary context. To address these shortcomings, we developed Smart‐Seq2 Rolling Circle to Concatemeric Consensus (Smar2C2), which identifies and quantifies TSSs and transcription termination sites. Smar2C2 incorporates unique molecular identifiers that allowed for the identification of as many as 70 million sites, with no known upper limit. We have also generated TSS data sets from as little as 40 pg of total RNA, which was the smallest input tested. In this study, we used Smar2C2 to identify TSSs in Glycine max (soybean), Oryza sativa (rice), Sorghum bicolor (sorghum), Triticum aestivum (wheat) and Zea mays (maize) across multiple tissues. This wide panel of plant TSSs facilitated the identification of evolutionarily conserved features, such as novel patterns in the dinucleotides that compose the initiator element (Inr), that correlated with promoter expression levels across all species examined. We also discovered sequence variations in known promoter motifs that are positioned reliably close to the TSS, such as differences in the TATA box and in the Inr that may prove significant to our understanding and control of transcription initiation. Smar2C2 allows for the easy study of these critical sequences, providing a tool to facilitate discovery.
Keywords: transcription start site, promoter, cis‐regulatory elements, template switching reverse transcriptase, rolling circle amplification, technical advance
Significance Statement
We have developed a technique called Smar2C2 and used it to identify transcription start sites in maize, rice, sorghum, soybean and wheat. This wide panel of plant TSSs facilitated the identification of evolutionarily conserved features, such as patterns in the dinucleotides that compose the initiator element and differences in the TATA‐box sequence motif, that may prove significant to understanding and controlling transcription initiation.
INTRODUCTION
The accurate genome‐wide discovery and quantification of transcriptional start sites (TSSs) is needed to understand the control of transcription initiation and regulation. The core promoter is located directly upstream of the 5′ DNA sense strand, and non‐coding sequences directly downstream of the TSS are critical in determining transcriptional regulation. These sequences directly flanking the TSS serve as binding sites for the transcription pre‐initiation complex (PIC), which contains RNA polymerase II and associated general transcription factors (Hampsey, 1998). Although core promoters may be sufficient to initiate basal levels of transcription (Kadonaga, 2012), they still colocalize with accessible chromatin and histone modifications associated with transcription initiation (Felsenfeld, 1992). Although it is unclear exactly which sequences are needed to form a functional core promoter, in eukaryotes they often contain a TATA box, an initiator element (Inr) and a B recognition element. In addition, other common sequences such as the downstream promoter element, the CAAT box and the GC box are located proximally to the TSS, and likewise regulate transcription (Brázda et al., 2021). These core promoter sequence features alone are likely to be insufficient for basal transcription and, to interact correctly with the core promoter, they must be precisely positioned relative to the TSS. For this reason, the identification of active core promoter elements and proximal cis‐regulatory elements are contingent on the annotation of the TSS.
Examining promoter sequences for sites of protein binding may not be sufficient, as even the most ubiquitous promoter motifs are only found in a subset of identified promoters. For example, the Inr is the most common sequence feature of promoters in eukaryotes, but is only present in 53.3% of eukaryotic promoters (Brázda et al., 2021). Additionally, the TATA box, perhaps the most well‐known example of a sequence associated with a core promoter sequence, is only found in 24.4% of eukaryotic promoters (Brázda et al., 2021). These known sequences also possess significant heterogeneity in actual combinations of known motifs detected in promoters, although the function of this remains somewhat unclear (Yamamoto et al., 2009). The lack of consistent promoter motifs has led to speculation that DNA secondary structures are likely to underpin transcription at many promoters (Bansal et al., 2014). Indeed, secondary DNA structures such as G‐quadruplexes (Huppert & Balasubramanian, 2007), cytosine‐rich i‐motifs (Assi et al., 2018) and cruciforms (Miura et al., 2018) can all influence core promoter function. However, DNA secondary structure identification is not as simple as motif or repeat identification (Lee et al., 2018), making accurate and widespread TSS annotation critical for their discovery.
Beyond understanding the nature of core promoters, better annotation of TSSs will help detect the use of alternative TSSs. Although there are still a lot of unknown variables surrounding TSSs, Arabidopsis thaliana alternative TSSs have been shown to change the inclusion of upstream open reading frames in response to exposure to blue light and regulate RNA expression via non‐sense‐mediated decay (Kurihara et al., 2018). In Zea mays (maize), alternative TSSs were shown to generate proteins that exclude different domains and coding regions of several genes, leading to altered final products (Mejía‐Guerra et al., 2015). On an evolutionary timescale it has been shown that new promoters can arise from the inclusion of a new internal exon, providing a pathway for the rapid generation of new alternative TSSs (Fiszbein et al., 2019). Alternative TSSs have been implicated in processes including circadian rhythms (Kurihara et al., 2018) and carbon sensing (Wiese et al., 2004). However, the technical limitations surrounding accurate TSS detection impede the identification of alternative TSS utilization, leaving their regulatory importance relatively unknown.
Although variation in core promoter sequence motif and nucleotide composition is well established, it is unknown what specific combination of these core elements is required for transcript initiation. Understanding the minimum motif composition needed for promoter activity, a true ‘minimal promoter’ without tissue‐specific effects, would greatly benefit plant synthetic biology, specifically advancing the design of artificial promoters. If a true minimal promoter sequence was known, it could be combined with any number of cis‐regulatory elements to precisely tailor transcription to a desired spatiotemporal context. Recently, Jores et al. (2021) synthesized 79 838 annotated promoters to express a reporter in protoplasts and transient expression systems, which yielded valuable information with regards to broad promoter sequence trends. However, this approach was dependent on annotations derived from RNA‐seq, which although effective for quantifying expression and identifying isoforms, is less accurate when compared with empirical TSS data (Adiconis et al., 2018; Mejía‐Guerra et al., 2015). The accurate identification of TSSs in the genome is a critical early step needed to dissect and generate a reliable minimal synthetic promoter, an invaluable breakthrough that would impact fields ranging from stress‐tolerant crops (Hou et al., 2012) to pharmaceutical production in planta (Lomonossoff & D'Aoust, 2016).
Despite the value of accurate TSS annotation, empirical TSS data are rarely generated and are lacking for many species. Several methods can measure TSSs, including Cap Analysis of Gene Expression (CAGE), which is the most widely used method and is experimentally robust (Shiraki et al., 2003). Although CAGE can accurately and precisely annotate TSSs in a wide range of organisms, it is technically difficult to perform, requires a large volume of input RNA, cannot determine polymerase chain reaction (PCR) duplication, and is time intensive (Adiconis et al., 2018). One common approach to label TSSs has been the use of template‐switching reverse transcriptases (TSRTs) (Adiconis et al., 2018). TSRT deposits a few ectopic cytosines after reaching the 5′ end of a template sequence (Zhu et al., 2001), allowing researchers to attach a template‐switching oligo directly at the TSS. RNA Annotation and Mapping of Promoters for Analysis of Gene Expression (RAMPAGE) (Batut et al., 2013), full‐length cDNA sequencing like single‐cell tagged reverse transcription (STRT) (Islam et al., 2012) and recently the Survey of Transcription Initiation at Promoter Elements (STRIPE‐seq) (Policastro et al., 2021) all utilize TSRTs to identify TSSs, each with unique advantages. We have attempted to utilize TSRT technology to create a rapid and technically simple technique that can generate a large number of accurate PCR de‐duplicated TSSs from extremely low levels of input RNA.
Here, we present Smar2C2, a technique that builds and improves on previous work using TSRT and rolling circle amplification to measure TSSs and transcription termination sites (TTSs) in a wide panel of plant species and tissues. Smar2C2 is experimentally simple, rapid and capable of measuring a large number of unique TSSs, all while requiring extremely low levels of input RNA. We use Smar2C2 to annotate TSSs from five angiosperm species and 10 tissues, facilitating the identification of evolutionarily conserved plant promoter features.
RESULTS
Design of Smar2C2
Smar2C2 incorporates barcoded primers and rolling circle replication to generate sequencing libraries with large numbers of uniquely barcoded TSS reads. Following first‐strand cDNA synthesis from RNA using a poly‐dT primer and a TSRT (Figure 1a,b), Smar2C2 uses a template‐switching oligo that incorporates a unique barcode attached to the 5′ mRNA end that is used to generate the full‐length cDNA second strand (Figure 1c,d). This gives each RNA molecule a unique molecular identifier (UMI), facilitating downstream PCR duplicate removal.
Figure 1.
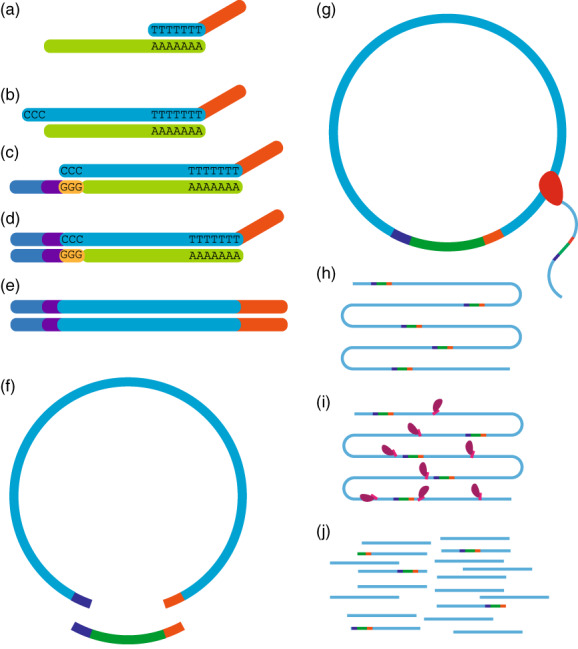
Design overview of Smar2C2.
(a) cDNA is generated with a template‐switching reverse transcriptase using extracted RNA (light green) and a poly‐dT primer (light blue) with an adaptor (orange) (b). (c) A template‐switching oligo containing an adaptor (blue) and unique molecular identifier (UMI) (purple) is bound to the deposited cytosines and used to add the second adaptor and UMI to the cDNA (d). The final construct (e) is circularized using a linker (dark green) (f) and amplified using rolling circle amplification (g). Rolling circle amplification generates a linear strand of repeating segments (h), and Tn5 is used to generate a final library for sequencing (i). This places the transcription start site (TSS), identifying adaptor and UMI in variable locations within the read (j), allowing for them to be sequenced and extracted bioinformatically.
Following this minimal amplification, using in situ PCR sequences attached by the template‐switching oligo and the initial poly‐dT primer used in cDNA synthesis, the final sequence (Figure 1e) is circularized using a linker and the sample is treated with a wide range of exonucleases to remove any residual linear products (Figure 1f). Circularized DNA is then amplified using rolling circle amplification (Figure 1g), which generates long linear concatemers of DNA composed of repeating segments of the original cDNA input (Figure 1h). This concatemer is subsequently fragmentated for library preparation. Although any library preparation can be used, for convenience we opted to use Tn5 to fragment libraries and insert sequencing adaptors (Figure 1i). The library is then sequenced and reads containing the TSS adaptor are identified by their proximity to the adaptor attached via the CCC deposited by the TSRT (Figure 1j) (see Experimental procedures).
We used Smar2C2 to identify TSSs in Glycine max (soybean), Oryza sativa (rice), Sorghum bicolor (sorghum), Triticum aestivum (wheat) and Zea mays (maize), in both leaf and root tissue taken from 4‐week‐old seedlings. For all species and tissues, two biological replicates were generated. Additionally, to assess the efficacy of TSS identification we included a Spike‐in RNA Variant Control (SIRV) in the soybean data set, which contains known RNA fragments. In the maize leaf data set, 51.1% of all read pairs (145.9 million reads) contained a TSS, of which 16.1% (approx. 8.2% of total reads) contained less than 27 bp of genomic DNA and were removed from the downstream analysis. A total of 73.5% (105.8 million reads) of the TSS reads were uniquely mapped to the genome, with 9.46% (12.4 million reads) TSS reads multimapped and discarded from further analysis. Aligned reads were then de‐duplicated using the UMI attached to each read. In total, 53.0% (56.7 million reads) of the reads contained a unique UMI. This resulted in an average of 4.80 UMIs per TSS position. In total, 19.94% of the sequenced reads contained a TSS adaptor, were mapped successfully to the genome and contained a unique UMI. In the maize shoot, this resulted in 56 764 592 individual TSS reads being identified. This high number of reads is beyond what was required for previous applications, and should allow for the detection of more subtle TSSs.
Soybean leaf libraries were also subjected to a series of 10‐fold serial dilutions following RNA extraction, with samples containing 40 ng, 4 ng, 400 pg and 40 pg sequenced to test the minimum input of RNA alongside the standard input of 400 ng. All input levels generated successful libraries, with no detectable loss in library complexity. Technical replicates had a high degree of similarity, with R2 values of greater than 0.93 for all standard 400‐ng input libraries, and the reduction of input RNA levels only reduced the R2 values slightly (Figure S1).
Individual TSSs can be analyzed as a single position, but it is often useful to group TSSs that occur in a small region into transcription start regions (TSRs). Aligned reads were assigned to a single base pair (bp) TSS as well as a cluster of start sites called the TSR using tsrchitect (Raborn et al., 2017). TSRs were assigned to genes that were either overlapping or contained an annotation 1000 bp downstream of the TSR; any TSR outside this range was defined as distal. Highly transcribed genes contain small falsely annotated TSRs within genes that are the result of infrequent aberrant transcripts, decayed RNA or other technical artifacts. These artifacts are present in other validated TSS data sets (Mejía‐Guerra et al., 2015; Policastro et al., 2021), as well as in the spike‐in control, and are often removed by setting a threshold based on a percentage of promoter proximal reads from which to consider reads (Policastro et al., 2021). However, as a result of the high read depth, we risked either identifying a large number of aberrant TSSs inside of highly transcribed genes with a low threshold or eliminating TSSs that appear valid in more lowly transcribed genes when using a high threshold. Therefore, we excluded TSRs that do not contain at least 10% of the total reads for any individual gene. This compromise allowed us to consider the local TSS environment when deciding which reads should be considered in further analysis.
In relation to other 5′ RNA‐sequencing methods, the time and material costs associated with Smar2C2 are closest to the previously reported STRT (Table S1) (Adiconis et al., 2018), with a low cost of library prep relative to other techniques and roughly 10 h of benchwork, spread across 2 days. This is unsurprising given that, of the techniques assessed, Smar2C2 shares many common steps with STRT, deviating at the circularization of full‐length cDNA. Although with Smar2C2 not all reads sequenced contain a TSS, this technique is able to compensate for some of the costs in library preparation.
Validation of Smar2C2 accuracy
As an initial form of unbiased validation, an RNA spike‐in control (Lexogen SIRV‐Set 2) was added to a soybean sample. These synthetic RNA isoforms contained TSSs with a high degree of complexity, such as alternative TSSs, overlapping transcripts and antisense transcripts (Figure S2). Comparing the known TSSs with those revealed by Smar2C2, we were able to evaluate the accuracy of Smar2C2. We found a high degree of accuracy between the known TSSs and those found via Smar2C2, and when using the same thresholds to identify TSRs as described above, which require at least 10% of the total reads from the final transcript, 99.1% of reads were within one base pair of the annotated TSS. Finally, the primary transcription start site (pTSS), defined as the single‐nucleotide position within a TSR containing the highest read count, within each TSR was directly on the annotated TSS for every spike‐in RNA. Most TSS reads not positioned around an annotated TSS appeared at locations within the annotated gene with only a single read, which is consistent with rare aberrant transcripts and degradation products. These results show that when Smar2C2 is assessed against a known control it accurately annotates TSSs with a high degree of single‐nucleotide accuracy.
In addition to synthetic controls, we assessed the validity of the TSSs discovered on a genome‐wide scale. We compared the Smar2C2 TSSs against the well‐validated CAGE technique using TSS data generated in the maize shoot from a previous publication (Mejía‐Guerra et al., 2015). For Smar2C2 pTSSs that are located upstream of the same gene as the CAGE reads, the CAGE reads tend to align directly on the annotated pTSS (Figure 2a,b). The distribution of TSS reads surrounding annotated TSSs in CAGE and Smar2C2 is also similar, with a strong peak centered on the existing annotation (Figure S3). Although the number of reads distal to any existing gene annotation is higher than that seen in other existing TSS data sets, it is consistent with published CAGE data reprocessed using the same pipeline (Figure 2c) (Mejía‐Guerra et al., 2015). Of the 18 398 peaks identified by CAGE in the maize shoot, 13 716 (74.5%) were also identified via Smar2C2. In contrast, CAGE identified only 21.5% of the peaks identified in Smar2C2. One reason for these differences is that CAGE uses a randomer instead of a poly‐dT primer and generates 27‐bp mappable fragments of cDNA instead of the longer Smar2C2 fragments, which average 79 bp but can be >140 bp in length. Additionally, although the same tissue and genotype was sampled for the CAGE and Smar2C2 data sets, the CAGE sample was harvested from more juvenile plants, which will also contribute to differences between the two data sets. Finally, CAGE lacks any UMI for de‐duplication, which may cause PCR duplicates to form false TSSs. We theorized that false TSSs arising from PCR duplicates should be more common at short peaks located farther from other clusters of TSSs. To test this, we omitted the de‐duplication step of our bioinformatic pipeline and examined the change in short TSSs. Of the TSRs under 10 bp, we observed an increase from 81.0% to 85.1% in TSRs of 1 bp in length, whereas all other TSRs showed a decrease overall (Table S2). The TSRs that appeared in CAGE but were absent in Smar2C2 were more likely to be one nucleotide in length, with 80.0% of discordant peaks being one nucleotide long whereas 61.1% of concordant peaks were one nucleotide long. The higher proportion of single‐nucleotide peaks that become enriched PCR duplicates if not removed suggests that some of the difference in the data sets arises from the technical limitations of CAGE.
Figure 2.
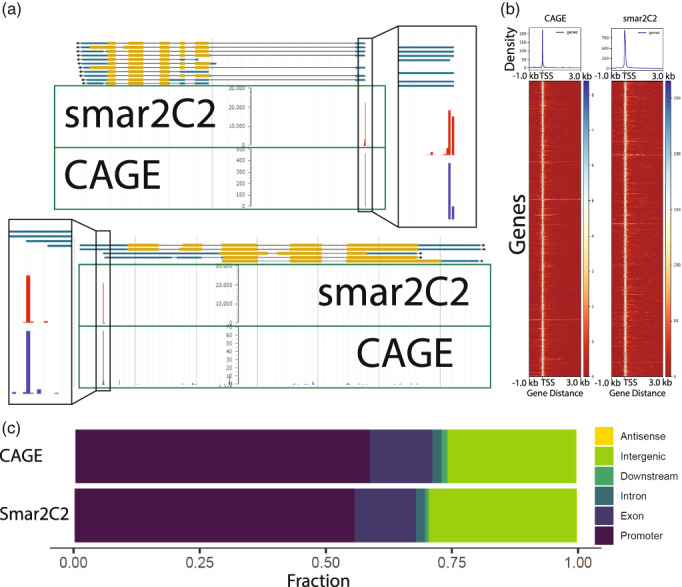
Smar2C2 overlap with CAGE.
(a) Browser tracks showing the overlap between Smar2C2 and existing CAGE data in Zea mays (maize) at a single base‐pair resolution relative to existing annotations. The box indicates a magnified image of the browser track to highlight single base‐pair resolution. (b) A heat map of CAGE reads and Smar2C2 reads centered on the genic transcription start site (TSS) identified by Smar2C2 shows that when CAGE reads are present at a Smar2C2 TSS they show a high degree of precise and concentrated overlap. (c) A comparison of the location of TSS reads in CAGE and Smar2C2 using the same processing pipeline.
To further investigate the validity of the Smar2C2 TSSs, we examined them for the chromatin states expected at TSSs (Figure 3a). For active promoters, accessible chromatin is expected directly upstream of the TSS, to facilitate transcription factor binding and PIC formation (Klemm et al., 2019). Accessible chromatin was measured with an assay for transposase‐accessible chromatin with high‐throughput sequencing (ATAC‐seq) (Lu et al., 2017), which revealed a strong enrichment directly upstream of the Smar2C2 pTSS locations (Figure 3b). Different histone modifications are correlated with transcription initiation and transcription elongation directly downstream of the TSS. Specifically, H3K4me3 and H3K56ac are well‐known histone modifications associated with transcription initiation that are found directly downstream of the TSS. These stand in contrast to H3K4me1 and H3K36me3, histone modifications associated with transcriptional elongation that are found further downstream of TSSs within the gene body in plant genomes (Mendieta et al., 2021; Ricci et al., 2019; Roudier et al., 2011; Zhang et al., 2009). Comparing previously published libraries from chromatin immunoprecipitation assays with sequencing (ChIP‐seq) (Ricci et al., 2019) with transcription‐associated histone modifications to the Smar2C2 pTSS coordinates, we observe that, as expected, H3K4me3 and H3K56ac are highly enriched directly downstream of Smar2C2 pTSSs, whereas H3K4me1 and H3K36me3 are enriched further downstream of Smar2C2 pTSSs in the gene bodies (Figure 3b). Across the pTSSs discovered around annotated genes, 85.5% displayed predicted histone modifications associated with transcription initiation, whereas 5.7% displayed unexpected relative locations of histone modifications associated with transcription initiation. The remaining 8.8% were not located within 1000 bp of the domains enriched for histone modifications associated with transcription initiation or elongation. Although it is possible that this discordance arises from differences between experimental set‐ups, the most likely explanation is the high sensitivity of Smar2C2 relative to ChIP‐seq. Together with the strong overlap with CAGE, as well as the correlation of chromatin states surrounding the TSS, we are confident that Smar2C2 accurately identifies TSSs.
Figure 3.
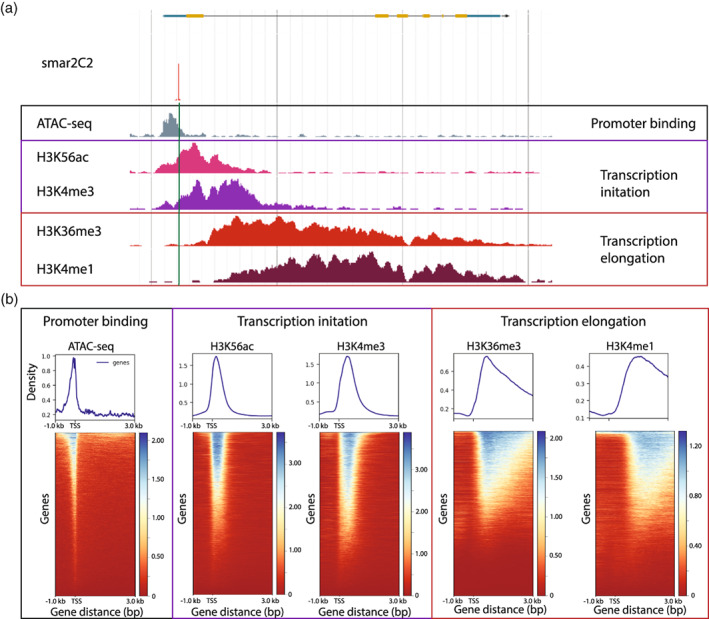
Validation of Smar2C2 TSS using epigenomic data.
(a) The expected orientation of epigenomic data relative to the transcription start site (TSS) in plants with accessible chromatin identified via ATAC‐seq (gray) present upstream in the promoter, histone modifications of transcription initiation H3K56ac (pink) and H3K4me3 (purple) directly downstream, and histone modifications of transcription elongation H3K36me3 (red) and H3K4me1 (burgundy) further downstream in the gene body. (b) Heat maps of epigenomic and CAGE data centered on the primary TSS identified with Smar2C2 show that these histone modification patterns are consistent across the entire genome, with genes ranked by the volume of ChIP data.
To examine the Smar2C2 TSSs further, we compared our results with previous work focused on improving annotations using chromatin modification (Mendieta et al., 2021). In addition to supporting the identified Smar2C2 TSSs, histone modifications associated with transcription initiation and elongation can also be used to identify misannotated and unannotated genes in the genome (Figure 4). In total, 1396 of the Smar2C2 TSSs are proximal to mis‐annotated genes identified via ChIP‐seq (Mendieta et al., 2021). Additionally, 1189 TSSs are distal to known genes that overlap novel genes discovered using histone modification data, providing a potential method of novel gene identification (Mendieta et al., 2021). Smar2C2 libraries also possess the potential to be used for annotation in a similar manner to standard RNA‐seq data, providing a possible improvement to existing genome annotation pipelines.
Figure 4.
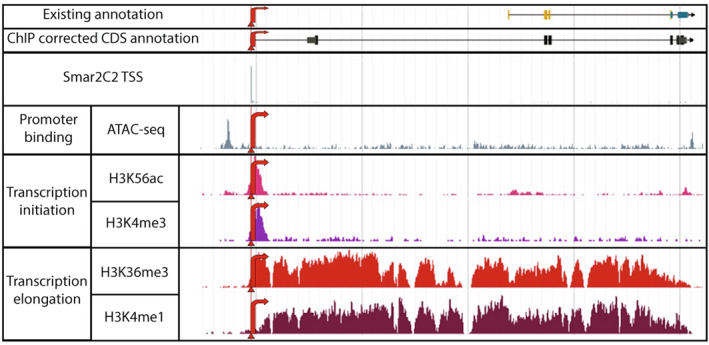
Smar2C2 transcription start site (TSS) compared against ChiP‐corrected annotation.
Previous work has established that genome annotations can be improved by using histone modification ChIP‐seq data to predict the location of TSSs within the genome that might differ significantly from the existing annotation. Smar2C2 TSSs can be used to corroborate the corrected annotations. An example browser shot of the existing annotation, the corrected annotation predicted via ChIP‐seq data and the relevant ChIP‐seq data tracks is shown here. The triangles represent the single base‐pair TSS, as determined by Smar2C2, whereas the red arrows indicate the direction of transcription.
Applications of precise TSS identification
As a result of the simplicity of the protocol, we generated, with replicates, high‐quality TSS data for all the aforementioned species in both leaf and root tissue. We examined these new TSSs and explored the positional enrichment of sequence motifs relative to them. Previous work has established that classical core promoter sequences like the TATA box and Inr are present in maize and Arabidopsis with precise positional requirements (Andersson & Sandelin, 2020; Mejía‐Guerra et al., 2015). By capitalizing on the ease of execution and the increased sequencing depth of this technique we can add validity to the annotation of core promoters while expanding our understanding in a comparative evolutionary context across closely related species. We detected anywhere between 57 000 and 165 000 TSRs, which was largely dependent on the total depth of sequencing for each individual library and the species being sampled (Table S3). Although the total number of TSSs and TSRs discovered between species varied, the percentage of TSSs discovered divided by the total library size remained within 1.5% of other tissues and replicates, indicating that the total number of TSSs discovered is consistent within species and across tissues. The number of reads located proximal to known genes was fairly consistent, with between 69 and 74% of the generated TSRs located next to and transcribing into existing gene annotations. The one exception was wheat, where only 49% of shoot TSRs and 54% of root TSRs were located proximal to existing annotations. It is unclear whether these differences are associated with the available gene annotations and genome assemblies or are caused by unknown biological factors.
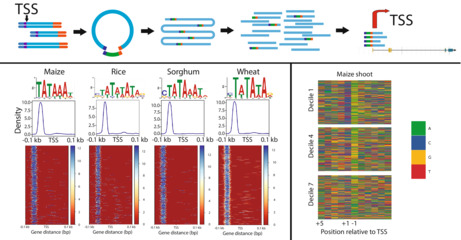
To explore the general sequence patterns surrounding the TSS, sequence logos were created using tsrexplorer (Policastro et al., 2021) by looking at the information content of individual nucleotides relative to the Smar2C2 pTSS (Figure 5a). The information content for individual nucleotides at the top expression decile appeared similar between all the species surveyed. Each species displayed a clear sequence preference directly on the pTSS, likely displaying the Inr sequence consisting of a pyrimidine purine dinucleotide (Hoskins et al., 2011). The TATA box was also visible from the −34 to −28 nucleotides relative to the pTSS, showing a strong enrichment for TA nucleotides characteristic of the classic motif (Haberle & Stark, 2018). Lastly, the information content of CG nucleotides in the region directly between the TATA box and the Inr sequences from nucleotides −11 to −24 was higher than that in the surrounding regions, excluding the Inr, possibly indicating the presence of a previously described Y‐patch motif (Jores et al., 2021). As pTSS strength decreases, the information content at each of these major areas tends to decrease as well, usually around the TATA box and Inr. However, other areas can also gain information. For example, in rice and sorghum roots the information content of the 5′ untranslated region (5′ UTR) increases greatly, showing a GTAC pattern in lowly expressed genes (Figure S4). Highlighting the importance of accurate TSS annotation, this motif detection is extremely dependent on pTSS position, with even single nucleotide shifts in either direction eliminating the observed nucleotide enrichment.
Figure 5.
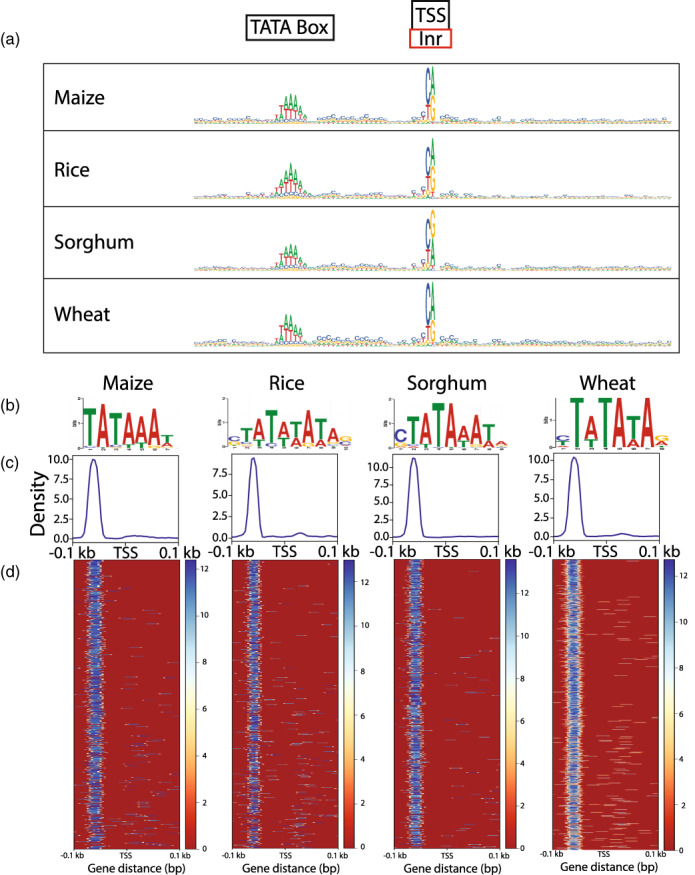
TATA‐box motifs identified by Smar2C2.
Sequence logos show sequence patterns surrounding the transcription start site (TSS), including possible initiator element (Inr) and TATA‐box sequences (a). The TATA‐box motifs discovered close to the TSS display precise positional enrichment relative to the TSS (c), and are found in the classic region from positions −28 to −35 of the promoter (c, d). TATA‐box motifs discovered in the highest expression decile show some sequence deviation from the classic TATA‐box motif, with Oryza sativa (rice), Sorghum bicolor (sorghum) and Triticum aestivum (wheat) displaying enrichment for a C preceding the more classic TA‐rich motif (b).
With precise location data we can also begin to examine small sequence variation in core promoter motifs. The TATA‐box sequence, which is usually located in the region between nucleotides −34 and −28, was compared across several species as well as across expression levels (Figure 5a). The precise location information, which only varied by one nucleotide in either direction, lets us examine only TA‐rich motifs that fall within the predicted region. This allowed for increased resolution and the removal of noise resulting from similar motifs that were not located within the specific region (Figure 5c,d). The canonical TATA‐box motif in eukaryotes is TATAWAW, and the annotated motifs discovered in these plants seem to mostly follow this trend, although there was some variability in the newly discovered motif (Figure 5b). For example, except for maize, all plant genes in the highest expression decile seemed to favor a CTATAWAW motif. As expression levels decrease, plant genes had more variation in the motifs that are detected near the TATA box. The TATA box is a major contributor to core promoter strength (Jores et al., 2021) and these observed differences in sequence motifs may impact core promoter activity, so an understanding the diversity of these motifs is the first step towards comprehending their function.
We examined the sequences directly surrounding (±1 bp) the measured site of transcription initiation, as dinucleotide ratios surrounding the TSSs have been described in other eukaryotes as pyrimidine–purine combinations (Nepal et al., 2013; Policastro et al., 2021). Analysis across all four grass species show an enrichment of C/T at the −1 position directly upstream of the pTSS and A/G at the +1 position directly downstream of the pTSS (Figures 6a and S5). As expected, all possible pyrimidine–purine dinucleotide pairings were the four most represented dinucleotides in the four grasses studied, and, although they were not evenly distributed, these dinucleotides had similar frequencies across expression deciles. The most highly expressed genes were enriched most commonly for CA, followed by CG, TG and TA, which are all enriched relative to the other dinucleotide frequencies. In the top decile of expression, CA shows a 6.12‐fold enrichment over background genomic dinucleotide frequencies, whereas the top decile of CG shows a 4.8‐fold increase over background genomic frequencies. All four common dinucleotides trended downwards as the expression of the pTSS decreases, approaching random distributions at lower levels of expression (Figures 6b and S6). Importantly, these uneven distributions of possible dinucleotide ratios are conserved across all four grass species examined. It is possible that there are more complicated Inr motifs beyond simple dinucleotides, like those reported in fruit flies (Haberle & Stark, 2018). As expression decreases, the dinucleotide TA quickly becomes no more common than any other dinucleotide. However, TA does appear to be a part of the only longer motif (AAACCCTAG) that was significantly enriched at the pTSS in all four grass species promoters. In every species except wheat, the motif is enriched on a specific single nucleotide that places TA as the dinucleotide pair flanking the pTSS (Figure S7). This motif is very similar to the canonical binding site of MYB‐related transcription factors (O'Malley et al., 2016), but how this motif functions in a core promoter is unknown (Figure S7). The sequence patterns surrounding the TSSs identified by Smar2C2 provide additional validity to Smar2C2, and also highlight the uses for accurate TSS data.
Figure 6.
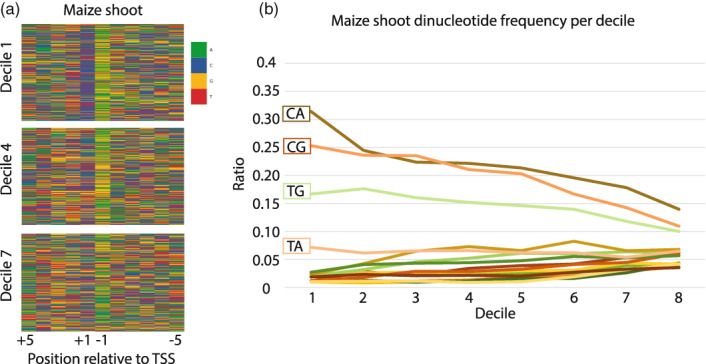
Nucleotide trends directly flanking the transcription start site (TSS).
(a) A nucleotide heat map in Zea mays (maize) centered on the 10 nucleotides flanking the TSS shows a clear sequence bias, with C/T being more present at the +1 nucleotide, directly upstream of the TSS, and A/G being more present at the −1 nucleotide, directly downstream of the TSS. This general pattern is more prevalent at higher expression deciles, becoming less apparent as the expression levels decrease. (b) These sequence patterns can also be examined as dinucleotide ratios flanking the TSS. Although the pattern of C/T upstream and A/G downstream is consistent, there is clearly a significant bias towards CA, and then CG followed closely by TG. These patterns are most apparent at the highest expression deciles and become less pronounced as expression decreases.
DISCUSSION
There are several well‐vetted methods for the identification and annotation of TSSs in all domains of life. However, despite the ability to generate these data sets and investigate the fundamental nature of TSSs with regards to transcription, barring a few model organisms, the overall availability of data remains sparse. CAGE is a proven technique that can reliably identify TSSs, but it is difficult to perform, is confounded by PCR duplicates, time and cost, and requires a large volume of input of RNA. As a result of these limitations, many improvements have been made on various aspects of CAGE, usually revolving around improving the ease of execution and decreasing the input requirements (Adiconis et al., 2018; Cumbie et al., 2015; Salimullah et al., 2011; Yamashita et al., 2011). Other attempts to measure TSSs have focused around technologies like TSRTs, which have had obvious utility and advantages since their use in the generation of full‐length cDNAs (Baran‐Gale et al., 2018; Zhu et al., 2001) and have been used successfully to identify TSSs previously (Batut et al., 2013; Islam et al., 2012). Techniques like STRT have used the TSRT to generate full‐length cDNA with adaptors attached on either end of the read for single‐cell applications, but then rely on enzymatic digestion and adaptor ligation to generate libraries that can be used with high‐throughput sequencing (Islam et al., 2012). However, this is only able to generate a limited number of TSSs from a single sample and does not increase with increased sample input (Adiconis et al., 2018). Through Smar2C2, we have built upon these existing techniques to generate a TSS profiling method that combines the ease of full‐length cDNA generation with quantifiable high‐throughput sequencing to generate high numbers of TSS reads.
The initial use of TSRT alongside a poly‐dT primer allows for easy generation of cDNA from extremely low levels of RNA input. TSRTs have been widely used in low‐input protocols and single‐cell cDNA generation, and poly‐dT primers are easy to use and exclude the abundant non‐polyadenylated RNA. Smar2C2 generated TSS data using as little as 40 pg of total input RNA while showing minimal losses in data quality. Although not unique to Smar2C2, the TSRT allows for the use of an adaptor that contains a unique molecular identifier, allowing the removal of PCR duplicates, unlike CAGE.
We have also demonstrated that Smar2C2 measures valid, accurate and precise TSSs through many orthogonal validation approaches. Not only does Smar2C2 show concordance with CAGE, but it also correctly identifies TSSs from complex spike‐in controls. Moreover, Smar2C2 TSSs displayed all the predicted histone modifications associated with transcription initiation. Although Smar2C2 lacks efficiency in terms of the total number of library reads that identifies a TSS, it is very efficient in terms of reagent and time costs. The ability to generate a large number of TSSs from a single sample using low inputs in a short period of time can compensate for the costs of sequencing the library.
To demonstrate the utility of Smar2C2 genome‐wide TSS measurements, we measured the TSSs of five agronomically important crops. This allowed for the comparison of core promoter features both within species and across species. We characterized the TSS dinucleotide sequences, as well as upstream and downstream sequence motifs. With precise TSS data, we identified short sequence characteristics that would otherwise be undetectable, improving the annotation of core promoter motifs and detecting previously unreported patterns in TATA box and Inr sequences. Many of these core promoter features were conserved across all species examined.
By combining widely used methods of sequence analysis with precise TSS positioning we can begin to discover patterns and information that would otherwise be unavailable. When entire sequences are examined for motif enrichment it can be difficult to separate what is a binding site for a functional regulatory element and what is unconserved noise present in any non‐coding sequence, and this problem becomes more drastic as sequence patterns become shorter or more flexible while retaining function. By using positional data we can eliminate large portions of potential sequences for consideration, allowing for a fine‐tuned examination of regions that we know to be important for core promoter function.
The improved resolution and novel sequence patterns in core promoter sequences revealed by the precise TSS annotation advance the basic understanding of promoter function, as well as the biotechnology that manipulates the promoters. Some attempts have been made to analyze the sequence motifs present in a subset of promoters, such as genes that changed expression in response to heat shock, and use that information to generate a synthetic promoter that activates under the desired conditions (Maruyama et al., 2017). Better knowledge of core promoter sequences will facilitate the identification of the minimal sequences required for transcriptional initiation, or a true ‘minimal promoter’. Having a bona fide plant minimal promoter will improve our understanding of transcription, while expanding the ability to express plant synthetic constructs with tight spatiotemporal precision. In addition to tailoring the expression of agronomically important genes, more specific promoters benefit diverse fields, including plant‐produced biopharmaceuticals (Lomonossoff & D'Aoust, 2016). The precise control of expression can also enable the implementation of synthetic genetic circuits in plants (McCarthy & Medford, 2020), expanding expression patterns to include possible AND, IF, and OR Boolean operators. Plant synthetic biology is still in its infancy, and a strong understanding of transcriptional initiation will be critical in unlocking new avenues of discovery and advancement.
Many additional analyses are enabled by Smar2C2 that were not outlined in this report and are beyond the scope of this particular project. For example, Smar2C2 precisely measured TTSs, which can be analyzed separately or examined when they occur on a read pair with their corresponding TSS. As a result of the paired‐end nature of these data this allows for an examination of the coupling of alternative TSSs, alternative splicing, and alternative TTSs. Additionally, we discovered many TSSs and TSRs distal to existing gene annotations. It is unclear what these reads correspond to, but they provide an exciting opportunity to discover transcripts absent in current annotations. Traditional annotations often cannot empirically define TSSs, instead estimating TSSs based on annotations from related organisms or RNA‐seq. This has been shown to be potentially inaccurate (Mendieta et al., 2021), and lacks the resolution critical for the discovery of motifs outlined in this paper. Smar2C2 can both add novel TSS annotations and refine known gene TSS annotation.
In summation, we have demonstrated that Smar2C2 allows for the rapid accurate placement of a previously unobtainable number of TSSs while using extremely low RNA inputs. Each individual TSS is tagged with a UMI, ensuring that every mapped read originated from a unique transcript. Lastly, we have demonstrated with artificial transcripts, chromatin data, and predicted sequences that our TSSs are extremely accurate, with single‐nucleotide resolution. Smar2C2 is simple to execute, quick and inexpensive. The advantages of Smar2C2 should aid more widespread measurements of TSSs, improving the understanding of promoter function, transcriptional regulation, cis‐regulatory element identification and genome annotations.
EXPERIMENTAL PROCEDURES
Plant growth and tissue preparation
Plants were grown in soil (Sungro Horticulture Professional Growing Mix; Sungro, https://www.sungro.com) mixed with Marathon 1% Granular Greenhouse and Nursery Insecticide (https://www.ohp.com/Products/marathon_1g.php) and Gnatrol WDG Larvicide (https://nufarm.com/usturf/product/gnatrol‐wdg/). Plants were grown at 21°C with a long‐day photoperiod (16 h light/8 h dark) at 60% humidity. Shoot and root tissue were harvested from plants 4 weeks after planting.
RNA extraction
RNA was extracted from plants using the Monarch Total RNA Miniprep Kit with the Tough‐To‐Lyse protocol (New England Biolabs, https://international.neb.com). Tissue was dissected from the plant and flash frozen in liquid nitrogen. Tissue was then ground in a porcelain mortar and pestle for 3 min. The sample was then suspended in 800 μl of 1× DNA/RNA protection reagent and centrifuged at 16 000 g for 2 min to pellet the debris. Then 800 μl of the supernatant was transferred to a fresh Eppendorf tube, and an equal volume of RNA Lysis Buffer (800 μl) was added. The sample was then vortexed briefly and 800 μl of the sample was transferred to the gDNA removal column. The sample was centrifuged for 30 sec at 16 000 g and the flow through was recovered. Then 800 μl of RNase free 200‐proof pure ethanol was then added to the flow through and mixed thoroughly by pipetting. After that, 800 μl of the mixture was transferred to an RNA purification column and centrifuged for 30 seconds at 16 000 g . The column was then washed with 500 ul of RNA wash buffer and the flow through was discarded. Then 5 μl of DNase I was combined with 75 μl of DNase I reaction buffer (New England Biolabs) per sample, and the mixture was carefully pipetted directly on top of the column matrix. The sample was allowed to incubate for 15 min at room temperature (18–23°C). Then 500 μl of RNA priming buffer was added and the solution was centrifuged for 30 sec at 16 000 g . The sample was washed with 500 μl of RNA wash buffer and spun for 30 sec, followed by a second wash of 500 μl and a 2‐minute spin at 16 000 g . The wash buffer was then discarded and the sample was re‐spun for 1 min at 16 000 g to ensure no ethanol contamination. Next, 50 μl for nuclease‐free water was added and the sample was spun for 30 sec at 16 000 g . The sample was then placed on ice and the total double‐stranded RNA (dsRNA) was quantified using the Qubit RNA broad range assay kit. All reagents and materials used for these steps were used exclusively for RNA extraction and are kept sealed and separate to ensure no RNase contamination. All work stations, gloves, mortar and pestle, and nitrogen storage vessels were thoroughly cleaned with ethanol and RNaseZap.
cDNA generation
A 400‐ng portion of the sample was added to a strip tube and incubated with 2 μl of 10 μm ISPCR poly‐dT primer and 1 μl of 10 mm dNTP. The protocol was also tested with 40 ng, 4 ng, 400 pg and 40 pg of input RNA generated via serial dilution. Nuclease‐free water was added to the sample to bring it to 6 μl and the sample was mixed via pipetting. The sample was incubated at 72°C for 3 min and then immediately placed on ice for 5 min. Following this, 2 μl of 5× First‐Strand buffer, 1 μl of 20 mm DTT, 0.5 μl of 100 μm ISPCR TSO‐UMI oligo (IDT, Coralville, IA, USA) and 0.5 μl of SMARTscribe Reverse Transcriptase (Takara Bio, Kusatsu, Japan) were added and mixed gently with pipetting. The sample was incubated for 90 min at 42°C and then 75°C before being placed on ice. Then, 13 μl of Q5 High‐Fidelity 2× Master Mix (New England Biolabs), 2 μl of 5000 U/ml RNase H (New England Biolabs) and 2 μl of 10 μm ISPCR oligo (IDT) were then added to the solution. The sample was then incubated for 37°C for 15 min, 95°C for 1 min, 65°C for 10 min and 98°C for 45 sec, followed by 6 cycles of 98°C for 10 sec, 63°C for 30 sec and 72°C for 3 min. The samples were then purified using the Monarch PCR and DNA cleanup kit with a binding buffer ratio of 5:1 and eluted in 10 μl of water. All reagents and materials used for these steps were used exclusively for RNA extraction and are kept sealed and separate to ensure no RNase contamination. All work stations, gloves, mortar and pestle, and nitrogen storage vessels were thoroughly cleaned with ethanol and RNaseZap.
Linker formation
Combine 25 μl KAPA HiFi HotStart ReadyMix (2×), 1 μl 10 μm forward splint, 1 μl 10 μm reverse splint and 23 μl of water. Incubate at 95°C for 3 min, 98°C for 1 min, 62°C for 1 min and 72°C for 6 min. The Zymo Select‐a‐Size cleanup kit (Zymo Research, Irvine, CA, USA) was used, with 85 μl of 100% ethanol added to 500 μl of select‐a‐size DNA binding buffer. Final concentrations were determined using the Qubit dsDNA broad range kit (Thermo Fisher Scientific, Waltham, MA, USA).
Circularization and rolling circle
A 200‐ng portion of splint DNA and 10 μl of NEBuilder HiFi DNA Assembly (New England Biolabs) were added to 10 μl of the sample solution. The solution is then incubated for 60 min at 50°C. Add 5 μl of NEBuffer2 (New England Biolabs), 1 μl of exonuclease I (New England Biolabs), 1 μl of lambda exonuclease (New England Biolabs), 0.5 μl of exonuclease III (New England Biolabs) and 21.5 μl of water. Incubate the sample for 37°C for 60 min followed by 80°C for 20 min. The samples were then purified using the Monarch PCR and DNA cleanup kit with a binding buffer ratio of 5:1 and eluted in 20 μl of water. Following this, add 19 μl of water, 2.5 μl of 10 mm dNTP (New England Biolabs), 2.5 μl of Exo‐Resistant Random Primer (Thermo Fisher Scientific), 5 μl of 10X phi29 buffer and 1 μl of phi29 DNA polymerase (New England Biolabs) to the solution. Incubate for 4 h at 30°C followed by 10 min at 65°C. Purify using the Monarch PCR and DNA cleanup kit with a binding buffer ratio of 5:1 and elute in 19 μl of water.
Tn5 fragmentation and library preparation
Add 20 μl of 2× TD buffer and 2 μl of loaded Tn5 to 19 μl of sample. Incubate for 30 minutes at 37°C. Purify using the Monarch PCR and DNA cleanup kit (New England Biolabs) with a binding buffer ratio of 5:1 and elute in 10 μl of water. Add 1.25 μl of 25 μm barcoded primer 1 (IDT), 1.25 μl of 25 μm barcoded primer 2 (IDT) and 12.5 μl of Q5 High‐Fidelity 2× Master Mix (New England Biolabs) to the 10 μl of sample and incubate for 72°C for 5 min, 98°C for two minutes, followed by six cycles of 98°C for 10 sec, 63°C for 30 sec and 72°C for 90 sec. Purify using the Monarch PCR and DNA cleanup kit with a binding buffer ratio of 5:1 and quantify the final concentration using the Qubit dsDNA broad range kit (Thermo Fisher Scientific).
Sequencing
Libraries were sequenced using the NovaSeq 6000 platform (Illumina, https://illumina.com), with each full RNA input replicate receiving between 83 and 230 million paired‐end reads, and serial dilution samples receiving between 11 and 41 million paired‐end reads (Table S3).
TSS identification
cutadapt 3.4 (Martin, 2011) was used to quality trim reads, identify and remove generic sections of the TSS adaptor (AAGCAGTGGTATCAACGCAGAGTAC) and filter out reads that did not contain the TSS adaptor. umi‐tools 1.0.1 (Smith et al., 2017) was then used to extract the sequence containing the UMI (NNNNNNNNNNNNATGGG) and place it in the fasta header. Sequences were then mapped using star 2.7.9a (Dobin et al., 2013), removing all filters for variation in read length size for the variability caused by the adaptor location within the read. Output bam files were then indexed using samtools 1.14 (Li et al., 2009). Duplicate reads were then removed using ‘dedup’ in umi‐tools.
Transcription start sites and TSRs were identified using tsrchitect 1.2.0 (Raborn et al., 2017), requiring a tag‐count threshold of 25 reads and a cluster distance of 20 nucleotides. A custom script (www.github.com/aem11309/smar2C2) was then used to identify TSSs within 1000 bp of an annotated gene. The total number of reads for each individual gene was then calculated and TSRs that did not contain at least 10% of the reads for any given gene were removed to prevent background reads from highly transcribed genes to be annotated as TSSs. The primary TSS was identified from each TSR as the mostly highly transcribed single‐nucleotide position.
Motifs were discovered de novo, compared against existing databases and positional enrichment was determined using meme‐suite (Bailey et al., 2015). Dinucleotide frequencies and sequence logo motifs were generated using tsrexplorer (Policastro et al., 2021). Data from previous studies including ChiP‐seq (Mendieta et al., 2021; Ricci et al., 2019) and ATAC‐seq (Lu et al., 2017) validation heat maps were generated using deeptools (Ramírez et al., 2014). bedtools 2.30.0 and emboss 6.6.0 were used for general sequence manipulation (Quinlan, 2014; Rice et al., 2000).
AUTHOR CONTRIBUTIONS
AEM, CV and RJS conceived and designed the experiments. AEM performed the experiments. AEM and JPM analyzed the data. AEM, JPM and RJS wrote the article.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest associated with this work.
OPEN RESEARCH BADGES
This article has earned Open Data and Open Materials badges. All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE197144. All plants used in this study are commonly available lines with no transformations or other modifications.
Supporting information
Figure S1. Smar2C2 CPM values are consistent across replicates and at low input.
Figure S2. Smar2C2 identified transcription start sites relative to SIRV6 spike‐in control annotations.
Figure S3. TSS read density surrounding annotated transcription start sites in CAGE and Smar2C2.
Figure S4. Sequence logos across expression deciles display potential motifs.
Figure S5. Nucleotide patterns surrounding the transcription start site are more prominent at high expression levels across multiple species.
Figure S6. Dinucleotide patterns flanking the transcription start site and across expression deciles are conserved across multiple species.
Figure S7. Motifs centered on transcription start sites resemble known MYB‐related transcription factor binding sites.
Table S1. Transcription start site library time and cost compared with previously reported values.
Table S2. Transcription start region size distribution with and without UMI de‐duplication.
Table S3. Final mapped de‐duplicated transcription start sites compared with initial sequencing read depth.
ACKNOWLEDGEMENTS
We would like to acknowledge the Georgia Genomics & Bioinformatics Core as well as the Georgia Advanced Computing Resource Center for research support. JPM was supported by the National Institute of General Medical Sciences of the National Institutes of Health (T32GM007103). This research was supported by grants from the National Science Foundation (IOS‐1546867 and IOS‐1856627) to RJS.
DATA AVAILABILITY STATEMENT
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE197144. All data used in this article are available and can be accessed under this accession.
REFERENCES
- Adiconis, X. , Haber, A.L. , Simmons, S.K. , Levy Moonshine, A. , Ji, Z. , Busby, M.A. et al. (2018) Comprehensive comparative analysis of 5′‐end RNA‐sequencing methods. Nature Methods, 15(7), 505–511. 10.1038/s41592-018-0014-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, R. & Sandelin, A. (2020) Determinants of enhancer and promoter activities of regulatory elements. Nature Reviews Genetics, 21(2), 71–87. 10.1038/s41576-019-0173-8 [DOI] [PubMed] [Google Scholar]
- Assi, H.A. , Garavís, M. , González, C. & Damha, M.J. (2018) I‐motif DNA: structural features and significance to cell biology. Nucleic Acids Research, 46(16), 8038–8056. 10.1093/nar/gky735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, T.L. , Johnson, J. , Grant, C.E. & Noble, W.S. (2015) The MEME suite. Nucleic Acids Research, 43(W1), W39–W49. 10.1093/nar/gkv416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal, M. , Kumar, A. & Yella, V.R. (2014) Role of DNA sequence based structural features of promoters in transcription initiation and gene expression. Current Opinion in Structural Biology, 25, 77–85. 10.1016/j.sbi.2014.01.007 [DOI] [PubMed] [Google Scholar]
- Baran‐Gale, J. , Chandra, T. & Kirschner, K. (2018) Experimental design for single‐cell RNA sequencing. Briefings in Functional Genomics, 17(4), 233–239. 10.1093/bfgp/elx035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batut, P. , Dobin, A. , Plessy, C. , Carninci, P. & Gingeras, T.R. (2013) High‐fidelity promoter profiling reveals widespread alternative promoter usage and transposon‐driven developmental gene expression. Genome Research, 23(1), 169–180. 10.1101/gr.139618.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brázda, V. , Bartas, M. & Bowater, R.P. (2021) Evolution of diverse strategies for promoter regulation. Trends in Genetics, 37(8), 730–744. 10.1016/j.tig.2021.04.003 [DOI] [PubMed] [Google Scholar]
- Cumbie, J.S. , Ivanchenko, M.G. & Megraw, M. (2015) NanoCAGE‐XL and CapFilter: an approach to genome wide identification of high confidence transcription start sites. BMC Genomics, 16(1), 597. 10.1186/s12864-015-1670-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C.A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. et al. (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29(1), 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld, G. (1992) Chromatin as an essential part of the transcriptional mechanim. Nature, 355(6357), 219–224. [DOI] [PubMed] [Google Scholar]
- Fiszbein, A. , Krick, K.S. , Begg, B.E. & Burge, C.B. (2019) Exon‐mediated activation of transcription starts. Cell, 179, 1551–1565.e17. 10.1016/j.cell.2019.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle, V. & Stark, A. (2018) Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews Molecular Cell Biology, 19(10), 621–637. 10.1038/s41580-018-0028-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampsey, M. (1998) Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiology and Molecular Biology Reviews, 62(2), 465–503. 10.1128/mmbr.62.2.465-503.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins, R.A. , Landolin, J.M. , Brown, J.B. , Sandler, J.E. , Takahashi, H. , Lassmann, T. et al. (2011) Genome‐wide analysis of promoter architecture in Drosophila melanogaster. Genome Research, 21(2), 182–192. 10.1101/gr.112466.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, L. , Chen, L. , Wang, J. , Xu, D. , Dai, L. , Zhang, H. et al. (2012) Construction of stress responsive synthetic promoters and analysis of their activity in transgenic Arabidopsis thaliana. Plant Molecular Biology Reporter, 30(6), 1496–1506. 10.1007/s11105-012-0464-0 [DOI] [Google Scholar]
- Huppert, J.L. & Balasubramanian, S. (2007) G‐quadruplexes in promoters throughout the human genome. Nucleic Acids Research, 35(2), 406–413. 10.1093/nar/gkl1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, S. , Kjällquist, U. , Moliner, A. , Zajac, P. , Fan, J.B. , Lönnerberg, P. et al. (2012) Highly multiplexed and strand‐specific single‐cell RNA 5′ end sequencing. Nature Protocols, 7(5), 813–828. 10.1038/nprot.2012.022 [DOI] [PubMed] [Google Scholar]
- Jores, T. , Tonnies, J. , Wrightsman, T. , Buckler, E.S. , Cuperus, J.T. , Fields, S. et al. (2021) Synthetic promoter designs enabled by a comprehensive analysis of plant core promoters. Nature Plants, 7(6), 842–855. 10.1038/s41477-021-00932-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadonaga, J.T. (2012) Perspectives on the RNA polymerase II core promoter. In Wiley interdisciplinary reviews: developmental biology (vol. 1, issue 1), 1, 40–51. 10.1002/wdev.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm, S.L. , Shipony, Z. & Greenleaf, W.J. (2019) Chromatin accessibility and the regulatory epigenome. In. Nature Reviews Genetics, 20(4), 207–220. 10.1038/s41576-018-0089-8 [DOI] [PubMed] [Google Scholar]
- Kurihara, Y. , Makita, Y. , Kawashima, M. , Fujita, T. , Iwasaki, S. & Matsui, M. (2018) Transcripts from downstream alternative transcription start sites evade uORF‐mediated inhibition of gene expression in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America, 115(30), 7831–7836. 10.1073/pnas.1804971115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, N.K. , Li, X. & Wang, D. (2018) A comprehensive survey on genetic algorithms for DNA motif prediction. Information Sciences, 466, 25–43. 10.1016/j.ins.2018.07.004 [DOI] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonossoff, G.P. & D'Aoust, M.A. (2016) Plant‐produced biopharmaceuticals: a case of technical developments driving clinical deployment. In. Science, 353(6305), 1237–1240. 10.1126/science.aaf6638 [DOI] [PubMed] [Google Scholar]
- Lu, Z. , Hofmeister, B.T. , Vollmers, C. , DuBois, R.M. & Schmitz, R.J. (2017) Combining ATAC‐seq with nuclei sorting for discovery of cis‐regulatory regions in plant genomes. Nucleic Acids Research, 45(6), e41. 10.1093/nar/gkw1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011) Cutadapt removesadapter sequences from high-throughput sequencing reads. EMBnet.Journal, 17(1). 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Maruyama, K. , Ogata, T. , Kanamori, N. , Yoshiwara, K. , Goto, S. , Yamamoto, Y.Y. et al. (2017) Design of an optimal promoter involved in the heat‐induced transcriptional pathway in Arabidopsis, soybean, rice and maize. Plant Journal, 89(4), 671–680. 10.1111/tpj.13420 [DOI] [PubMed] [Google Scholar]
- McCarthy, D.M. & Medford, J.I. (2020) Quantitative and Predictive Genetic Parts for Plant Synthetic Biology. Frontiers in Plant Science, 11, 512–526. 10.3389/fpls.2020.512526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejía‐Guerra, M.K. , Li, W. , Galeano, N.F. , Vidal, M. , Gray, J. , Doseff, A.I. et al. (2015) Core promoter plasticity between maize tissues and genotypes contrasts with predominance of sharp transcription initiation sites. The Plant Cell, 27(12), 3309–3320. 10.1105/tpc.15.00630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendieta, J.P. , Marand, A.P. , Ricci, W.A. , Zhang, X. & Schmitz, R.J. (2021) Leveraging histone modifications to improve genome annotations. G3: genes, genomes, Genetics, 11(10), jkab263. 10.1093/g3journal/jkab263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, O. , Ogake, T. & Ohyama, T. (2018) Requirement or exclusion of inverted repeat sequences with cruciform‐forming potential in Escherichia coli revealed by genome‐wide analyses. Current Genetics, 64(4), 945–958. 10.1007/s00294-018-0815-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal, C. , Hadzhiev, Y. , Previti, C. , Haberle, V. , Li, N. , Takahashi, H. et al. (2013) Dynamic regulation of the transcription initiation landscape at single nucleotide resolution during vertebrate embryogenesis. Genome Research, 23(11), 1938–1950. 10.1101/gr.153692.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley, R.C. , Huang, S.S.C. , Song, L. , Lewsey, M.G. , Bartlett, A. , Nery, J.R. et al. (2016) Cistrome and Epicistrome features shape the regulatory DNA landscape. Cell, 165(5), 1280–1292. 10.1016/j.cell.2016.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Policastro, R.A. , Mcdonald, D.J. , Brendel, V.P. & Zentner, G.E. (2021) Flexible analysis of TSS mapping data and detection of TSS shifts with TSRexploreR. NAR Genomics and Bioinformatics, 3(2), lqab051. 10.1093/nargab/lqab051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A.R. (2014) BEDTools: the swiss‐Army tool for genome feature analysis. Current Protocols in Bioinformatics, 2014, 11.12.1–11.1234. 10.1002/0471250953.bi1112s47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raborn, R.T. , Sridharan, K. and Brendel, V.P. (2017). TSRchitect: Promoter identification from large‐scale TSS profiling data . 10.18129/B9.bioc.TSRchitect [DOI]
- Ramírez, F. , Dündar, F. , Diehl, S. , Grüning, B.A. & Manke, T. (2014) DeepTools: A flexible platform for exploring deep‐sequencing data. Nucleic Acids Research, 42(W1), W187–W191. 10.1093/nar/gku365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci, W.A. , Lu, Z. , Ji, L. , Marand, A.P. , Ethridge, C.L. , Murphy, N.G. et al. (2019) Widespread long‐range cis‐regulatory elements in the maize genome. Nature Plants, 5(12), 1237–1249. 10.1038/s41477-019-0547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, P. , Longden, L. & Bleasby, A. (2000) EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics, 16(6), 276–277. 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- Roudier, F. , Ahmed, I. , Bérard, C. , Sarazin, A. , Mary‐Huard, T. , Cortijo, S. et al. (2011) Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO Journal, 30(10), 1928–1938. 10.1038/emboj.2011.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimullah, M. , Mizuho, S. , Plessy, C. & Carninci, P. (2011) NanoCAGE: a high‐resolution technique to discover and interrogate cell transcriptomes. Cold Spring Harbor Protocols, 6(1), 96–110. 10.1101/pdb.prot5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki, T. , Kondo, S. , Katayama, S. , Waki, K. , Kasukawa, T. , Kawaji, H. et al. (2003) Cap analysis gene expression for high‐throughput analysis of transcriptional starting point and identification of promoter usage. Proceedings of the National Academy of Sciences of the United States of America, 100(26), 15776–15781. 10.1073/pnas.2136655100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T. , Heger, A. & Sudbery, I. (2017) UMI‐tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Research, 27(3), 491–499. 10.1101/gr.209601.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese, A. , Elzinga, N. , Wobbes, B. & Srneekens, S. (2004) A conserved upstream open reading frame mediates sucrose‐induced repression of translation. Plant Cell, 16(7), 1717–1729. 10.1105/tpc.019349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, Y.Y. , Yoshitsugu, T. , Sakurai, T. , Seki, M. , Shinozaki, K. & Obokata, J. (2009) Heterogeneity of Arabidopsis core promoters revealed by high‐density TSS analysis. Plant Journal, 60(2), 350–362. 10.1111/j.1365-313X.2009.03958.x [DOI] [PubMed] [Google Scholar]
- Yamashita, R. , Sathira, N.P. , Kanai, A. , Tanimoto, K. , Arauchi, T. , Tanaka, Y. et al. (2011) Genome‐wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Research, 21(5), 775–789. 10.1101/gr.110254.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Bernatavichute, Y.V. , Cokus, S. , Pellegrini, M. & Jacobsen, S.E. (2009) Genome‐wide analysis of mono‐, di‐ and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biology, 10(6), R62. 10.1186/gb-2009-10-6-r62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y.Y. , Machleder, E.M. , Chenchik, A. , Li, R. & Siebert, P.D. (2001) Reverse transcriptase template switching: a SMART™ approach for full‐length cDNA library construction. In. BioTechniques, 30(4), 892–897. 10.2144/01304pf02 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Smar2C2 CPM values are consistent across replicates and at low input.
Figure S2. Smar2C2 identified transcription start sites relative to SIRV6 spike‐in control annotations.
Figure S3. TSS read density surrounding annotated transcription start sites in CAGE and Smar2C2.
Figure S4. Sequence logos across expression deciles display potential motifs.
Figure S5. Nucleotide patterns surrounding the transcription start site are more prominent at high expression levels across multiple species.
Figure S6. Dinucleotide patterns flanking the transcription start site and across expression deciles are conserved across multiple species.
Figure S7. Motifs centered on transcription start sites resemble known MYB‐related transcription factor binding sites.
Table S1. Transcription start site library time and cost compared with previously reported values.
Table S2. Transcription start region size distribution with and without UMI de‐duplication.
Table S3. Final mapped de‐duplicated transcription start sites compared with initial sequencing read depth.
Data Availability Statement
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE197144. All data used in this article are available and can be accessed under this accession.