

Abstract
Subterranean estuaries are biogeochemically active coastal sites resulting from the underground mixing of fresh aquifer groundwater and seawater. In these systems, microbial activity can largely transform the chemical elements that may reach the sea through submarine groundwater discharge (SGD), but little is known about the microorganisms thriving in these land‐sea transition zones. We present the first spatially‐resolved characterization of the bacterial assemblages along a coastal aquifer in the NW Mediterranean, considering the entire subsurface salinity gradient. Combining bulk heterotrophic activity measurements, flow cytometry, microscopy and 16S rRNA gene sequencing we find large variations in prokaryotic abundances, cell size, activity and diversity at both the horizontal and vertical scales that reflect the pronounced physicochemical gradients. The parts of the transect most influenced by freshwater were characterized by smaller cells and lower prokaryotic abundances and heterotrophic production, but some activity hotspots were found at deep low‐oxygen saline groundwater sites enriched in nitrite and ammonium. Diverse, heterogeneous and highly endemic communities dominated by Proteobacteria, Patescibacteria, Desulfobacterota and Bacteroidota were observed throughout the aquifer, pointing to clearly differentiated prokaryotic niches across these transition zones and little microbial connectivity between groundwater and Mediterranean seawater habitats. Finally, experimental manipulations unveiled large increases in community heterotrophic activity driven by fast growth of some rare and site‐specific groundwater Proteobacteria. Our results indicate that prokaryotic communities within subterranean estuaries are highly heterogeneous in terms of biomass, activity and diversity, suggesting that their role in transforming nutrients will also vary spatially within these terrestrial–marine transition zones.
Keywords: aquatic prokaryotic communities, coastal aquifer, microbial activity, mixing zone, Patescibacteria, seawater intrusion, submarine groundwater discharge, subterranean estuaries, terrestrial–marine interface
1. INTRODUCTION
Coastal aquifers are located at the intersection between the land and the ocean and constitute a subterranean hydrological connection between groundwater and seawater. This land‐ocean interaction is crucial both from a terrestrial and a marine perspective: it affects the availability of fresh groundwater in coastal areas (threatened by seawater intrusion) and impacts coastal ecosystems through the discharge of groundwater and associated chemicals to the coastal ocean, the so‐called submarine groundwater discharge (SGD; Burnett et al., 2003; Robinson et al., 2018). SGD has indeed been recognized as a major source of chemicals (e.g., nutrients, metals) to the coastal ocean (e.g., Kwon et al., 2014), having important implications for ocean biogeochemistry, marine biota and coastal ecosystem services (Alorda‐Kleinglass et al., 2021; Lecher & Mackey, 2018; Santos et al., 2021). Before discharging to the ocean, fresh meteoric groundwater can mix with seawater intruded in the coastal aquifer, forming a zone of pronounced gradients in physicochemical conditions such as salinity, pH, temperature or oxygen, the so‐called subterranean estuary (Duque et al., 2020; Moore, 1999). Subterranean estuaries are characterized by important biogeochemical transformations, which are often mediated by microorganisms that control the cycling and fate of the chemical constituents in groundwater (e.g., nutrients, metals, carbon) (Moore, 2010). Microbial communities inhabiting subterranean estuaries thus determine the chemical quality of groundwater and control the magnitude of SGD‐associated chemical fluxes. However, the scarcity of microbial studies in subterranean estuaries, and coastal aquifers in general, prevents a good understanding of the relevance of these microbial reactors in modulating SGD‐derived chemical fluxes to the sea (Archana et al., 2021; Ruiz‐González et al., 2021).
Chemical fluxes associated to SGD, and thus the microbial processes controlling them, are of particular relevance in semi‐arid and oligotrophic seas such as Mediterranean Sea, where any nutrient input may have disproportionate effects in coastal ecosystems (Herut et al., 1999; Ludwig et al., 2009; Rodellas et al., 2015). For example, in the Mediterranean Sea, recent estimates unveiled that SGD‐derived nutrient inputs (inorganic nitrogen, phosphorus and silica) can be larger than those from rivers or atmospheric deposition (Rodellas et al., 2015). Although the magnitude of SGD fluxes to coastal areas has been widely studied over the last decade, establishing that coastal aquifers are important sources of metals, pollutants and nutrients worldwide (Cho et al., 2018; Tovar‐Sánchez et al., 2014) the role of microorganisms in determining SGD has mostly been inferred from indirect chemical evidences pointing to biotic transformations of elements (Goyetche et al., 2022; Huettel et al., 2014; Rodellas et al., 2018; Weinstein et al., 2011).
In the Mediterranean, only two studies explicitly addressed the composition of coastal aquifer microbial communities, reporting pronounced diversity variations along a vertical salinity gradient (Héry et al., 2014; Sola et al., 2020), and suggesting that microbial activity may modify the chemical composition of the groundwater within the aquifer. However, they sampled only different depths within a single well, thus disregarding the longitudinal heterogeneity of microbial assemblages at varying distances from the shore. Since groundwater flow rates are usually on the order of millimeters to meters per day (Santos et al., 2021), even small distances along the subterranean estuary may result into largely heterogeneous microbial communities influenced not only by the changing physicochemical properties but also by varying degrees of connectivity with the inland aquifer, the terrestrial surface, and the sea (Ruiz‐González et al., 2021). Understanding the role of these communities thus requires spatially‐resolved subterranean samplings, but these are logistically challenging and hence most microbial studies on coastal aquifers have been restricted to few sampling sites (Ruiz‐González et al., 2021). Moreover, despite that the number of microbial studies in coastal aquifers has increased in recent years (Archana et al., 2021; Ruiz‐González et al., 2021), most have been based on amplicon sequencing and thus very little is known regarding basic features such as the cell abundance and size, metabolic status or activity rates of coastal groundwater microbiota.
Here, we aimed to explore the spatial patterns and drivers of the microbial communities in a subterranean estuary located in the NW Mediterranean, considering both the vertical and the longitudinal scales. Profiting from a unique sampling site in an alluvial aquifer where a series of installed piezometers allow sampling the entire groundwater salinity gradient at different depths, we studied the taxonomic composition and activity of the bacterial communities through a combination of bulk prokaryotic activity measurements, flow cytometry, microscopy and 16S rRNA gene sequencing, and compared them with variations in physicochemical conditions. We hypothesized that the pronounced physicochemical gradients and the slow groundwater velocities promote the establishment of spatially heterogeneous communities adapted to specific subsurface niches, but that the bidirectional movement of water caused by the dynamic interplay between groundwater discharge and seawater intrusion (Folch et al., 2020; Martínez‐Pérez et al., 2022; Palacios et al., 2020) might result in a certain connectivity between those communities closer to the terrestrial surface or the sea. We finally investigated experimentally how predictable may be the responses of the studied microbial communities, assessing whether a similar manipulation of the physicochemical environment of assemblages sampled at different portions of the aquifer led to site‐specific shifts in activity and taxonomic composition.
2. MATERIALS AND METHODS
2.1. Study site, sampling and physicochemical parameters
The study was conducted on 11 July 2018 at an experimental site in an alluvial aquifer connected to the Mediterranean Sea. The site is located near the mouth of the ephemeral stream of Argentona, 30 km northeast from Barcelona (NW Mediterranean), in a Mediterranean temperate climate characterized by sporadic rainfall episodes, mostly in spring and fall.
The Argentona experimental site was established in 2015 with the construction of a series of piezometers installed perpendicularly to the shoreline spanning a distance of 100 m from the seashore (Figure S1). Most piezometers are gathered in nests consisting of three piezometers in which a 2 m screened area allows collecting water from depths ranging from 6 to 22 mbs (metres below the surface, Figure 1a, Table 1) that cover the whole salinity gradient of the subterranean estuary. The aquifer is dominated by quaternary sediments formed by layers of gravels, sands and clays and overlies a granitic basement (Folch et al., 2020; Martínez‐Pérez et al., 2022). The groundwater table is located at ~3 mbs. Fresh groundwater flows seaward mostly in the aquifer upper part (<15 mbs), whilst the deep area (>20 mbs) is the most affected by a seawater intrusion extending >150 m inland (Figure 1a). There is thus a transition zone between terrestrial and marine groundwater from ~15 to ~20 mbs, where the strongest vertical physicochemical gradients occur. Although these are the general hydrogeological patterns of the aquifer, there are horizontal layers of silts and clays that act as impermeable units, resulting in a system that behaves as a multilayered aquifer (e.g., at some points, brackish levels are observed beneath high salinity levels; Martínez‐Pérez et al., 2022). In addition, there is another upper zone of interaction between freshwater and seawater due to wave action and tidal pumping that drives the circulation of seawater across the beachface, generating a seawater recirculation cell (or upper saline plume, Figure 1a). Some of the main chemical reactions occurring within the aquifer have been recently inferred through the chemical identification of the freshwater and saline end‐members and the spatial distribution of their mixing ratios (Goyetche et al., 2022; Martínez‐Pérez et al., 2022), suggesting that there might be significant denitrification in the mixing zone and iron and manganese reduction in the saline portion of the aquifer.
FIGURE 1.

(a) Schematic representation of the experimental site in Argentona showing the location of the piezometers (black bars) and the screened intervals (grey) of each piezometer (grey areas). Piezometer samples are referred to in the text as “groundwater” samples (n = 10) and are distinguished from the beach porewater and the seawater samples. N1 and N2 refer to two nests which consist of three piezometers screened at different depths. The red dots indicate the location of the four sites from which groundwater for the experimental incubations was collected. (b) Spatial variability in the measured physicochemical parameters. The vertical grey lines indicate the position of the piezometers, and the horizontal dotted line indicates the soil surface. The symbols indicate whether the samples belong to the aquifer (Groundwater), to beach porewater obtained next to the shoreline (Porewater) or to surface seawater (Sea). Cond, conductivity; DO, dissolved oxygen; Temp, temperature
TABLE 1.
Sample name, sampling method and depth (considering the screened interval in each piezometer), distance to the shoreline, and categorization of groundwater types depending on salinity (see also Figure 1a)
| Sample ID | Sampling method | Sampling depth (m) | Screened interval (m) | Shore distance (m) | Aquifer water | Salinity |
|---|---|---|---|---|---|---|
| N2_15 | Installed piezometer | 11 | 10–12 | 97.9 | Fresh | 0.5 |
| N2_20 | Installed piezometer | 16 | 15–17 | 99.8 | Brackish | 19.4 |
| N2_25 | Installed piezometer | 21 | 20–22 | 98.3 | Marine | 30.2 |
| N1_15 | Installed piezometer | 13.5 | 12.5–14.5 | 62.9 | Fresh | 3.3 |
| N1_20 | Installed piezometer | 19.5 | 18.4–20.4 | 60.2 | Marine | 33.6 |
| N1_25 | Installed piezometer | 21.8 | 20.9–22.8 | 62.3 | Marine | 27.1 |
| PP15 | Installed piezometer | 6 | 2–13 | 52.4 | Fresh | 1.1 |
| PP20_Top | Installed piezometer | 10 | 3–18 | 39.5 | Brackish | 8.5 |
| PP20_Bottom | Installed piezometer | 15 | 3–18 | 39.5 | Brackish | 25.3 |
| PP18 | Installed piezometer | 15 | 13–15 | 37 | Brackish | 6.3 |
| Beach_Porewater | Manual piezometer | 1 | 1 | 2 | 35.2 | |
| Sea | Manual collection | 0.5 | – | 5 (offshore) | 36.4 |
In total, 12 samples were collected along the aquifer‐sea continuum, 4 of which were also used for the experimental incubations (see below, Figure 1a, Table 1). Samples from piezometers (hereafter groundwater samples, n = 10) were collected using a submersible pump placed in the middle of the screened part (Figure 1a), which ranged from 6 and 22 mbs (Table 1). In the case of the PP20, two samples (10 m depth ‐top‐ and 15 m depth ‐bottom‐) were taken given that the screened area spans the entire piezometer, as in PP15 (Figure 1a). Piezometers were purged discarding at least three times the piezometer volume to ensure that the collected sample represented the aquifer groundwater. In situ temperature, dissolved oxygen, pH, water electrical conductivity and salinity were measured with a multiparameter probe (YSI) by pumping groundwater through a flow cell. Additionally, 1 m deep beach porewater sample was collected at 2 m from the shoreline, using a direct‐push well‐point piezometer and extracted using a hand‐held vacuum pump. Surface seawater was collected near the shoreline using a submersible pump.
For each water sample, an aliquot for inorganic nutrients (nitrate, nitrite, ammonium, phosphate and silicate) was collected in 10 ml‐polyethylene vials by filtering 10 ml of water through 0.45 μm nylon syringe filters and kept refrigerated until storage at −20°C once in the laboratory. The analyses were performed at the Chemistry Service of the Institut de Ciències del Mar (ICM‐CSIC) using colorimetric method with a SEAL Autoanalyser 3 (AAC)HR(SEAL Analytica), coupled to a Jasco FP2020 for the determination of ammonia. Samples for microbial analyses were collected in acid‐rinsed 10 L‐polycarbonate carboys after prefiltering by 200 μm to remove large particles and were kept refrigerated and in the dark until processing in the laboratory, between 4–6 h after collection.
2.2. Experimental incubations
Four selected groundwater samples of low prokaryotic biomass and activity (two fresh ‐N2_15, PP15‐ and two brackish ‐PP18, PP20_Top‐, see Figure 1a) were incubated in acid‐rinsed autoclaved 2 L polycarbonate bottles for 2.5 days in the dark and at close to in situ temperature, with no carbon or nutrient addition, to explore how these microbial communities react to a similar manipulation. This experimental setting changed some environmental conditions (e.g., limitation of the interaction with aquifer solids, different oxygen concentration, etc.), yet it was not intended to mimic natural changes potentially encountered by aquifer assemblages, but rather to address whether some prokaryotes within the aquifer can react quickly to changes in the environment and whether the pool of responding taxa differs across sites and represents rare or abundant microbes. Samples for all the microbial parameters (see below) were collected after the incubation period.
2.3. Prokaryotic abundances, cell size and heterotrophic activity
Free‐living prokaryotic abundances, average cell sizes, and the number of cells with high nucleic acid content (HNA cells) were determined by flow cytometry. Duplicate 1.8 ml samples preserved with 1% paraformaldehyde +0.05 glutaraldehyde (final conc.) and stored at −80°C until analysis with a Becton‐Dickinson FACSCalibur flow cytometer staining cells with SYBR Green I. Prokaryotic cell size was estimated using the relationship between the average bacterial size and the average fluorescence of the SYBR Green I stained prokaryotes relative to that of standard beads (Gasol & Del Giorgio, 2000), applying an in‐house calibration between Syto13 and SybrGreen. Bulk prokaryotic heterotrophic production was estimated using the 3H‐leucine incorporation method (Kirchman et al., 1985), incubating three aliquots (1.2 ml) and one trichloroacetic acid (TCA)‐killed control (5% final concentration) with 3H‐leucine (160 Ci mmol, 20 nM final conc.) for about 2.5–3.0 h in the dark at in situ temperature. The incorporation was stopped by adding cold TCA (5% final conc.), samples were processed by the centrifugation method of Smith and Azam (1992) and the amount of the incorporated radioisotope was quantified with a Beckman scintillation counter.
2.4. DNA extraction and characterization of prokaryotic communities
Prokaryotic communities from all sites were characterized by Illumina sequencing of the 16S rRNA gene. Between 2.3 and 10 L of water (depending on the expected prokaryotic abundance based on previous exploratory samplings and flow cytometry data) were filtered onto 0.2 μm polycarbonate membrane filters (47 mm, Merck Millipore) using a peristaltic pump. The DNA was extracted from the filters using the standard phenol‐chloroform protocol with slight modifications (Salazar, Cornejo‐Castillo, Benitez‐Barrios, et al., 2015). The V3–V4 region of the 16S rRNA gene was amplified with the general bacterial primers 341F and 805R (Herlemann et al., 2011) and sequenced in an Illumina MiSeq platform following a paired‐end approach at the RTLGenomics facility (https://rtlgenomics.com/). These primers were selected because previous studies have shown that they are amongst the most efficient in recovering some groups of bacteria that are common in groundwaters, such as the phylum Patescibacteria (Peura et al., 2012; Ruiz‐González et al., 2021) and have been used in recent groundwater microbial surveys (e.g., Herrmann et al., 2019).
To identify amplicon sequence variants (ASVs), 16S rRNA amplicon reads were analysed through the dada2 pipeline version 1.16.0 (Callahan et al., 2016) on r version 4.0.3 (R Core Team, 2018), which resolves ASVs by modelling the errors in Illumina‐sequenced amplicon reads. Reads were trimmed to 260 bases and the reverse ones to 190 bases according to their quality scores, and primers were removed using cutadapt version 3.5 (Martin, 2011). To retain more rare taxa along the subterranean estuary, samples were “pseudo”‐pooled for the dada() step. Paired‐ends were merged after inference of amplicon variants and chimeras were removed with removeBimeraDenovo(). Taxonomic assignment of the different ASVs was performed using the function assignTaxonomy() against SILVA version 138 through the RDP naive Bayesian classifier method described in Wang et al. (2007). ASVs assigned to chloroplasts or mitochondria were removed for subsequent analyses. The ASV table was randomly subsampled down to the minimum number of reads per sample (10,655 reads) using the rrarefy function in the vegan package (Oksanen et al., 2015) for comparing richness and diversity across samples. All raw sequences used in this study are publicly available at the European Nucleotide Archive (PRJEB52186).
2.5. Quantification of ribosome‐containing bacterial groups through catalysed reporter deposition fluorescence in situ hybridization (CARD‐FISH)
DNA sequencing does not differentiate between living, dormant or dead cells, so we used CARD‐FISH (Pernthaler et al., 2002) to quantify the potentially viable cells by targeting those that contained intact ribosomes (or enough ribosomes to be detected). In all samples except the beach porewater, 10 or 50 ml of water (depending on the expected prokaryotic abundance) were fixed with paraformaldehyde (1% final concentration) at 4°C in the dark for the determination of the in situ abundances of different bacterial groups. Fixed samples were filtered through 0.22‐μm polycarbonate filters (GTTP, 25 mm, Millipore) and stored at 20°C until processing. We used 10 horseradish peroxidase‐probes based on some of the most abundant groups detected by sequencing: Beta42a and Gam42a for Beta‐ and Gammaproteobacteria (Manz et al., 1992), Alf968 for Alphaproteobacteria (Neef, 1997), CF319 for clades belonging to the Bacteroidetes group (Manz et al., 1996), HGC96a for Actinobacteria (Roller et al., 1994) OD1‐289 for Parcubacteria clades within Patescibacteria (Gong et al., 2014), Delta495a for most Deltaproteobacteria (Loy et al., 2002) and Syn405 for the marine cyanobacteria Synechococcus (West et al., 2001). Eub338‐II‐III were also applied to quantify most Eubacteria (Daims et al., 1999).
Prior to hybridization, cells were permeabilized with lysozyme and achromopeptidase. Hybridizations were carried out at 35°C overnight (except 2 h at 46°C for OD1) and hybridization conditions were established adding different proportions of formamide (30% for Actinobacteria and OD1, 45% for Alphaproteobacteria, 50% for Delta495a, 55% for the rest of probes). Counterstaining of CARD‐FISH filters was done with 4,6‐diamidino‐2‐phenylindole (DAPI), and the abundances of total prokaryotes and the targeted bacterial groups were estimated with an automated epifluorescence microscope Axio Imager.Z2m connected to a Zeiss camera (AxioCam MRm, Carl Zeiss MicroImaging) at 630× magnification, counting a minimum of 55 fields of view per filter with the automated image analysis software ACMEtool3 (version 2013‐04‐07, M. Zeder, technobiology GmbH2014). Due to the small size of OD1 cells, their abundance was estimated manually with an Olympus BX61 epifluorescence microscope counting a minimum of 10 fields (550–1200 DAPI‐stained cells).
2.6. Statistical analysis
Differences between prokaryotic communities were visualized using nonmetric multidimensional scaling (NMDS), metaMDS function, R vegan package (Oksanen et al., 2015) based on Bray‐Curtis distances. Differences in taxonomic composition among types of water were tested using analysis of similarity (ANOSIM R Vegan). The role of individual physicochemical variables in explaining changes in prokaryotic community structure was assessed by means of Mantel linear correlations (R Vegan). Distance matrices were constructed considering all samples (n = 12) or only groundwater samples (n = 10) computing the Euclidean distances of each single variable and were correlated to the Bray‐Curtis dissimilarities between prokaryotic communities. Prokaryotic taxonomic richness (the number of ASV per sample) and diversity (Shannon index) were calculated using the rarefied ASV table. Statistically significant differences in prokaryotic abundance, size, heterotrophic production or the abundances of the different groups between the types of water were determined by means of one‐way ANOVA followed by Tukey's post hoc tests. Spearman correlations were used to explore the variation in microbial parameters in relation to physicochemical variables (R ggpubr) and the results of the correlation analysis were presented in a heatmap (R ComplexHeatmap). All analyses were run using r software version 4.0.3 (R Core Team, 2018).
3. RESULTS
3.1. Physicochemical conditions
Temperature, salinity, dissolved oxygen, pH and inorganic nutrient concentrations varied largely both longitudinally and vertically along the subterranean estuary (Figure 1b), following the main hydrogeological patterns of the site (i.e., fresh groundwater flowing seaward at the upper part of the aquifer; seawater intrusion in the deep aquifer area, Figure 1a). Groundwater collected from the shallowest piezometers (6–13.5 m) was characterized by fresh and/or slightly brackish groundwater (salinity range 0.5–3.3) showing the highest pH values (c. 7.2), intermediate oxygen (0.7–1.6 mg/L) and phosphate (1.3–2.3 μM) concentrations, and the highest concentrations of nitrate and silicate (610–1100 and 340–400 μM, respectively, Figure 1b). On the contrary, the deepest part of the aquifer (21–22 m) harboured the most saline groundwater (27–34) and showed the lowest pH (6.1–6.6), oxygen (0.5–0.8 mg/L), phosphate (0.6–1.8 μM) and nitrate (0.7–6.1 μM) concentrations, and the highest concentrations of ammonium and nitrite (5–30 and 0.5–1.7 μM, respectively). Brackish groundwater (6.3–25.3) occupied the intermediate depths (10–16 m) within the aquifer and was characterized by intermediate levels of most parameters except for an enrichment in dissolved oxygen in N2_20 and PP18, and in phosphate in PP20 (Figure 1b). Compared to surface seawater, groundwater samples were significantly colder, more acidic, depleted in oxygen and highly enriched in inorganic nitrogen, phosphate and silicate. For most of the analysed parameters, the beach porewater sample was very similar to the seawater (Figure 1b), consistent with the shoreface circulation of seawater due to wave‐setup (Figure 1a). According to their salinities, groundwater samples are classified here‐in‐after in three clusters as fresh (<4), brackish (4–27) and saline (>27, see Table 1).
3.2. Spatial variations in prokaryotic abundances, heterotrophic production and cell size
The physicochemical gradients coincided with pronounced variations in the assessed microbial parameters (Figure 2). With some exceptions, groundwater communities were characterized by lower prokaryotic abundances and activity and smaller cell sizes than surface seawater assemblages. Considering only the piezometer samples, the abundance, activity and average cell size increased following the groundwater salinity gradient from fresh to saline. Prokaryotic abundance varied more than fivefold, ranging from 2 × 104 cells/ml in N2_15 to >1.2 × 105 cells/ml in the deep saline site N2_25 (Figure 2a). The communities from the two deepest saline sites (N2_25 and N1_25) showed the highest prokaryotic heterotrophic production, even surpassing that in the surface seawater sample. Low heterotrophic production rates were detected in most other groundwater sites (range 1.4–5.4 [mean 4.7] pmol 3H‐Leu L/h, Figure 2b). The proportion of prokaryotic cells with high nucleic acids content (HNA cells), indicative of larger genomes and usually associated to copiotrophic bacterial taxa (Vila‐Costa et al., 2012), also increased from the freshest (40% of total cells) to the most saline (90%) samples (Figure 2c), as did the average cell sizes (Figure 2d, range 0.06–0.09 μm3, Figure 2d).
FIGURE 2.
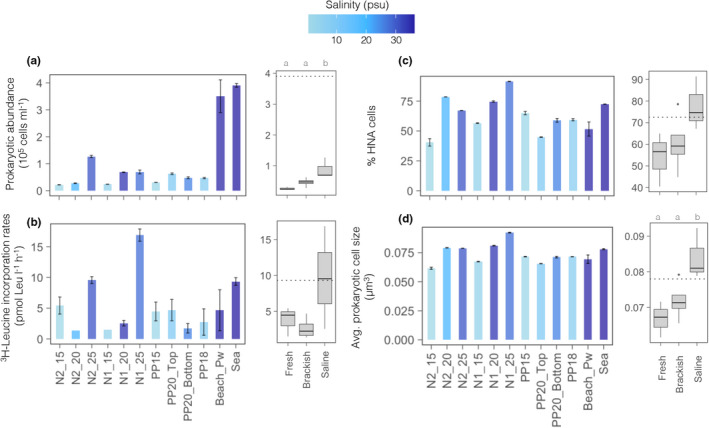
(a) Variations throughout the studied transect in total prokaryotic abundances, (b) prokaryotic heterotrophic production measured as 3H‐leucine incorporation rates, (c) percentage of HNA cells and (d) average cell size of the prokaryotic cells. Error bars indicate standard deviation of duplicate or triplicate samples. The colour indicates the salinity of each sample. In each case, the boxplots show variations in these same properties between the different water types (fresh, brackish, saline) within the aquifer. The dotted line indicates the value corresponding to the seawater sample. Different letters indicate significant differences between water types (Tukey's post hoc text, p < .05). Site acronyms as in Figure 1a
3.3. Taxonomic composition of bacterial communities across the subterranean estuary
Sequencing of the 16S rRNA gene resulted in 429,876 quality sequences, which clustered into 9579 ASVs. Excluding the experimental incubations, 351,966 reads (9154 ASVs) were detected. The number of ASV per sample varied between 431 and 1440, and the Shannon diversity index ranged from 3.4 to 6.6 (Table 2). This variability in richness and diversity was not statistically related to any of the measured physicochemical nor other prokaryotic variables. Between 0.05% and 11% of community sequences were classified as Archaea (mostly Nanoarchaeota, Table 2) even though the selected primers capture mostly bacteria. The ASV accumulation curve did not reach a clear asymptote when pooling all samples, suggesting that more sampling sites or a higher sequencing depth would be needed capture the diversity hidden in this subterranean estuary (Figure S2a). However, individual species rarefaction curves indicate a good local coverage of the local ASV richness with the sequencing depth used (Figure S2b).
TABLE 2.
Number of amplicon sequence variants (ASVs), Shannon diversity index, % of archaeal reads and number of phyla detected across the studied prokaryotic communities. The rarefied ASV table was used for these analyses
| Sample ID | Number of ASVs | Shannon index | % Archaea | Number of phyla |
|---|---|---|---|---|
| N2_15 | 830 | 5.3 | 1.5 | 41 |
| N2_20 | 856 | 5.6 | 6.1 | 41 |
| N2_25 | 1030 | 5.5 | 1.8 | 40 |
| N1_15 | 858 | 5.9 | 11.2 | 39 |
| N1_20 | 624 | 3.4 | 0.3 | 33 |
| N1_25 | 431 | 4.8 | 0.1 | 29 |
| PP15 | 1440 | 6.5 | 5.8 | 43 |
| PP20_Top | 1308 | 6.6 | 6.4 | 46 |
| PP20_Bottom | 934 | 5.9 | 3.9 | 42 |
| PP18 | 697 | 5.1 | 0.7 | 31 |
| Beach_Porewater | 1256 | 5.7 | 0.1 | 22 |
| Sea | 728 | 5.3 | 0.2 | 24 |
The NMDS ordination of prokaryotic communities along the subterranean estuary showed a clear segregation of communities between fresh, brackish and saline groundwater sites, and these in turn differed largely from the seawater and beach porewater communities (ANOSIM R = 0.75, p < .001, Figure 3a). In terms of taxonomic composition, 63 phyla were detected within the 10 groundwater samples. Communities were dominated by Proteobacteria (28% of groundwater reads), followed by Patescibacteria (12%), Desulfobacterota (10%) and Bacteroidota (8%). Despite the clear clustering of sites depicted by the NMDS, we observed a highly heterogeneous distribution of the main phyla or classes across the studied samples, with groups such as Desulfobacterota and Firmicutes showing high local relative abundances at single locations, reaching up to 63 and 49% of the community at the deep saline groundwater sites N1_20 and N1_25, respectively (Figure 3b). Other groups displayed more clear abundance variations depending on groundwater salinity (Figure 3c). Within Proteobacteria, the gammaproteobacterial order Burkholderiales (formerly considered Betaproteobacteria) prevailed in fresh groundwater, whereas other Gamma‐ and Alphaproteobacteria were enriched in brackish groundwater sites. The phyla Nitrospirota and Planctomycetota decreased pronouncedly from fresh‐ towards saline groundwaters, and Bacteroidota and Campylobacterota showed the opposite pattern. Verrucomicrobiota and Patescibateria were showed higher relative abundances in fresh and brackish sites compared to the deep saline groundwater (Figure 3c).
FIGURE 3.

(a) Nonmetric multidimensional scaling (NMDS) plots based on Bray–Curtis distances between the studied prokaryotic communities, colour‐coded by the different water types (freshwater, brackish, saline groundwater). The size of the symbols is proportional to salinity values. Normalized stress values are shown. (b) Taxonomic composition of the studied prokaryotic communities. Groundwater samples are organized by water type (fresh, brackish, saline) and ordered by increasing conductivity. The classification was performed at the Phylum level except in the case of Proteobacteria and Patescibacteria where the main classes and even orders were distinguished. Prot, Proteobacteria; Patesc, Patescibacteria; Gam, Gammaproteobacteria. Only the most abundant phyla (>0.1% of total sequences) are shown. (c) Variations in the relative contribution of the different phyla between the different types of water considering only the aquifer samples (n = 10). Gammaproteobacteria does not include the order Burkholderiales, which is presented separately. The dashed and dotted lines represent the contribution of each group in the beach porewater and seawater samples, respectively, for comparison
The surface marine community had higher proportion of Bacteroidota, Alphaproteobacteria and Cyanobacteria compared to aquifer sites (Figure 3b,c). Marine Cyanobacteria were still detected in the beach porewater (5%) but not in aquifer samples. Indeed, the porewater composition in terms of dominant phyla was generally similar to that of the surface seawater community but showed higher proportions of Alpha‐ and Gammaproteobacteria.
3.4. Microscopy quantification of ribosome‐containing bacterial groups
Probes for some of the major bacterial phyla or classes detected (excluding Firmicutes and Planctomycetes) were used to identity ribosome‐containing (i.e., potentially more active or viable) bacteria. Eubacteria‐labelled cells ranged from 14% to 44% of total DAPI counts within the aquifer, and increased from fresh‐ to saline groundwater (Figure 4a,b). 50% of the cells were identified as Eubacteria in the surface marine assemblage. Within Eubacteria, we observed large variations in the abundance of the different groups, although in general the groups that comprised the highest proportions of ribosome‐containing cells were Alphaproteobacteria, Deltaproteobacteria, Bacteroidetes and Betaproteobacteria (Figure 4c). Alphaproteobacteria prevailed in fresh groundwater whereas Gammaproteobacteria, Bacteroidetes, Actinobacteria and Patescibacteria increased their contribution towards the deep saline groundwater. Deltaproteobacteria showed minimum relative abundances in brackish waters, and the rest of groups showed no clear pattern along the salinity gradient (Figure 4c).
FIGURE 4.
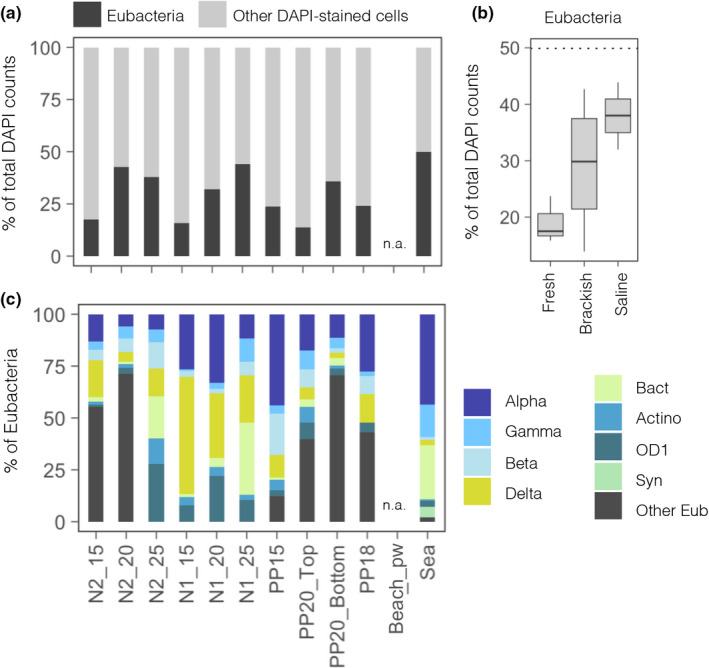
Spatial variations in the abundances of the different bacterial groups detected by CARD‐FISH. Proportion of cells (of total DAPI counts) hybridized with the probes for Eubacteria (Eub) (a) across individual samples and (b) between the different types of water considering only the aquifer samples (n = 10). The dotted line represents the % of Eubacteria in the surface seawater sample, for comparison. (c) Proportion of the different targeted groups expressed as % of total Eubacteria‐positive cells. n.a., not available. Site acronyms as in Figure 1. Alpha‐, Gamma‐, Beta‐ and Deltaproteobacteria (Alph, Gam, Bet, Del), Bacteroidetes (Bact), Actinobacteria (Act) Parcubacteria (OD1) and Synechococcus (Syn)
3.5. Environmental drivers of microbial communities along the subterranean estuary
The observed differences in the overall taxonomic structure (assessed by 16S rRNA gene sequencing) of the groundwater microbial communities (n = 10) were significantly correlated with differences in some of the measured environmental variables, mainly pH and salinity (Mantel R = .44 and 0.41, p < .005), nitrate (Mantel R = 0.33, p < .01) and ammonium (Mantel R = 0.31, p < .02).
When considering individual microbial parameters or the abundances of specific bacterial groups, nitrite concentration and/or salinity were among the variables positively correlating with changes in the abundance of ribosome‐containing Eubacteria, Actinobacteria, Bacteroidetes or Gammaproteobacteria, but also with variations in total prokaryotic abundance, average cell size, and % HNA cells (Figure 5). Conversely, nitrate concentration and pH were the variables correlating negatively with most groups as well as the other microbial properties (Figure 5). Salinity, nitrate and pH also explained variations in the amplicon‐based abundance of groups such as Desulfobacterota and Campylobacterota (Figure 5), whereas other groups were linked to variations in specific parameters such as ammonium (Actinobacteriota and Planctomycetota), dissolved oxygen (Alphaproteobacteria), or nitrite and silicate (Bacteroidota).
FIGURE 5.
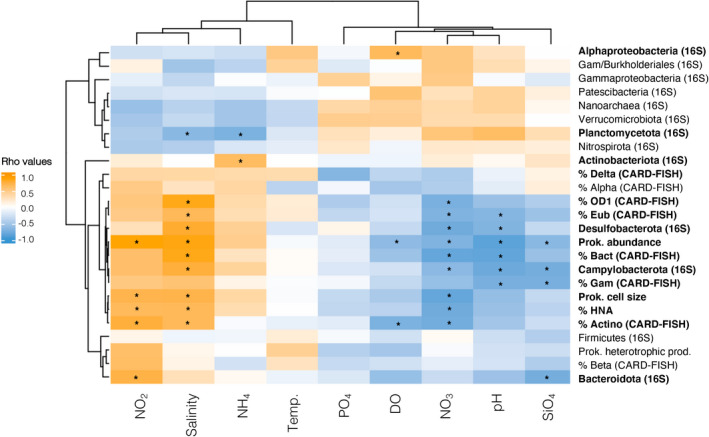
Hierarchically clustered heatmap of correlations between different microbial parameters and the measured physicochemical conditions, considering only the aquifer samples (n = 10). Prok., prokaryotic. The colour gradient indicates the Spearman correlation coefficients (Rho values). CARD‐FISH group relative abundances represent % of total DAPI counts, and amplicon‐based abundances (16S) represent % of community sequences. 16S‐derived Gammaproteobacteria excludes Burkholderiales. The asterisks (*) indicate significant relationships (p < .05), also highlighted in bold
3.6. Connectivity of microbial communities along the aquifer‐sea gradient
In order to explore whether the studied microbial communities were connected by the bidirectional movement of groundwater and seawater, we used the sequence data to estimate the ubiquity of the different ASVs as well as their presence in the adjacent seawater. We first explored the individual dynamics of the 20 most abundant ASVs within the aquifer. Together, these 20 ASVs accounted for 2.5% to 55% of the local sequences across aquifer samples, but only 0.2% and 1.6% in the beach porewater and seawater communities, respectively (Figure 6a). These ASVs belonged to eight different phyla (Table S1) and displayed very different habitat preferences and niche breadths, either appearing in only one or two sites (such as some ASVs belonging to Zetaproteobacteria, Firmicutes, Burkholderiales) or showing presence across most part of the salinity gradient (Alphaproteobacterial ASVs). Only two out of the 20 ASVs, which belonged to the alphaproteobacterial genera Sphingobium and Sphingomonas, were also detected in the beach porewater and/or the surface seawater assemblages (Figure 6b, Table S1).
FIGURE 6.

(a) Cumulative contribution to total community sequences of the 20 most abundant ASVs found within the aquifer. (b) Individual read counts (considering the rarefied ASV table to allow comparisons among samples) of the 20 most abundant ASVs across the aquifer‐sea continuum, coloured by the assigned taxonomy. The classification was performed at the Phylum level except in the case of Proteobacteria and Patescibacteria where some main classes and even orders were distinguished (see Table S1 for taxonomic classification of the shown ASVs down to the genus level). Prot, Proteobacteria; Patesc, Patescibacteria; Gam, Gammaproteobacteria
Between 41% and 94% of the ASVs found in each groundwater sample were not detected in any other site of the subterranean estuary (Figure 7a). These unique ASVs represented between 21% to 71% of the local community sequences suggesting that several of these unique taxa were locally abundant. No ASV was found to be present across all studied communities, in accordance with the fact that most ASVs were rare and present in very few of the studied sites (Figure S3a,b). Consequently, we did not find the often reported positive correlation between the mean abundance of the different ASVs and their occurrence (i.e., number of sites in which they were detected, Figure S3c).
FIGURE 7.

(a) Percentage of unique ASVs (i.e., ASVs detected exclusively in one sample) and their contribution to total sequences in each of the studied communities. (b) Contribution of ASVs detected exclusively in the aquifer (light blue), or in the seawater sample with no presence in the aquifer (dark blue). Shared ASVs were those present in both the sea and in any aquifer sample (black). (c) Contribution of ASVs with different niche preferences across the study site, identified based on the type of water where they showed their maximum relative abundance. Groundwater samples are organized by water type (fresh, brackish, saline) and are ordered by increasing conductivity, from the least (N2_15) to the most (N1_20) saline
We then categorized ASVs depending on whether they were found exclusively in aquifer samples or whether they were detected in the sea but not in groundwater samples (Figure 7b). This showed that between 93%–100% of sequences of aquifer communities belonged to ASVs exclusively found within the aquifer. Nonetheless, there were 28 ASVs present in the aquifer that accounted for c. 20% of the surface marine community (Figure 7b). Of these 28 shared ASVs, 15 had higher individual abundances in the aquifer than in the sea, meaning that they could be aquifer ASVs that had been dispersed to the sea, and together represented 2.5% of the surface marine community (Figure S4a). These included several Alphaproteobacterial genera such as Sphingomonas, Sphingobium, Erythrobacter or Brevundimonas, as well as other genera like Sedimenticola (Gammaproteobacteria), Desulforporinus (Firmicutes), Bacteroides, Sediminibacterium and Cloacibacterium (Bacteroidetes) or Acidovorax (Burkholderiales). The remaining 13 ASVs showed the opposite pattern, being more abundant in the sea than in the aquifer, and did not represent more than 0.6% of the local aquifer communities (Figure S4b). These comprised mostly typical marine taxa such as Synechococcus, SAR11 and several Rhodobacterales and Gammaproteobacteria (details not shown).
Finally, the preferred habitat of each ASV within the aquifer‐sea continuum was explored by categorizing ASVs depending on the habitat where they showed their maximum abundances (Figure 7c). We observed that the different aquifer habitats (fresh, brackish, saline groundwater) harboured very few sequences of ASVs with preference for other habitats, with the exception of the brackish site N2_20 and PP18 in which c. 25% belonged to ASVs with maximum abundances in fresh and saline groundwater (Figure 7c). The beach porewater community showed no presence of any of the aquifer prevailing taxa but 12% of the reads were of marine origin.
3.7. Growth of groundwater ASVs during experimental incubations
The changes in conditions caused by experimental bottle enclosure of two fresh and two brackish groundwater communities resulted in large increases in total prokaryotic abundance and activity, the proportion of HNA cells and the average cell size (Figure 8a–d), despite their low in situ abundances and activity. After incubations, all four communities were dominated by Gammaproteobacteria (>50%–90% of sequences), followed by Alphaproteobacteria (5%–32%) and/or Betaproteobacteria in fresh groundwater communities (18%–25%) (Figure 8e). These groups also comprised most of the detected Eubacterial cells, but larger proportions of Alphaproteobacteria were detected compared to the sequence data (Figure 8f).
FIGURE 8.
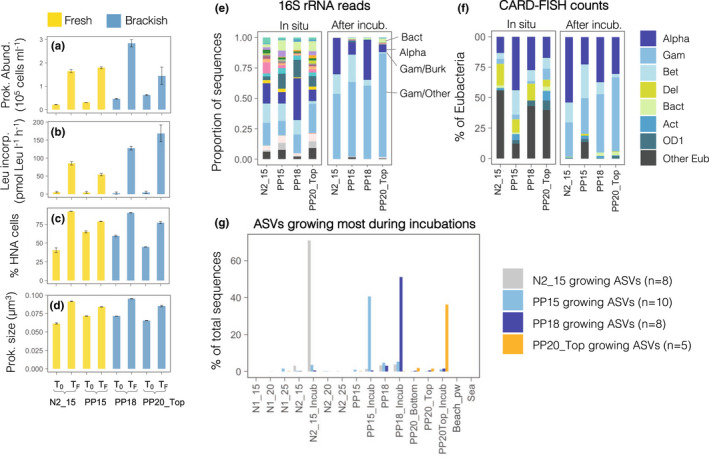
Changes in groundwater prokaryotic communities during experimental bottle enclosure. (a) Variations and relative increase in prokaryotic abundance, (b) heterotrophic production measured as 3H‐leucine incorporation rates, (c) percentage of HNA cells and (d) average cell size of the prokaryotic cells during ∼2.5‐day experimental enclosure of fresh and brackish groundwater from four selected sites. Error bars indicate standard deviation of duplicate or triplicate samples. (e, f) Taxonomic composition of the communities after the incubations assessed by (e) 16S rRNA gene sequencing and (f) CARD‐FISH. CARD‐FISH probe acronyms as in Figure 4. (g) Increase in abundance of the ASVs growing most during the experimental incubations. Note that in all cases these growing ASVs were rare in the original communities and throughout the aquifer
We also assessed whether the ASVs growing most during incubations differed depending on the original groundwater communities, and whether these growing ASVs were rare or abundant at other sites throughout the aquifer (Figure 8g). To do so, we identified those ASVs that showed >10‐fold increases in relative abundance during incubation and that were abundant in the final communities (>1% of local sequences). Between five and 10 ASVs met these criteria, which accounted for 35%–70% of the four communities after incubations but never represented more than 5% in the original assemblages, meaning that they were rare in situ. The pool of growing ASVs also appeared to be highly specific from each community, as they comprised very few sequences across all other sites in the aquifer (Figure 8g).
4. DISCUSSION
In the last decades, there have been significant advances in the understanding of SGD, mainly related to the characterization of its magnitude and chemical fluxes to the coastal ocean, its drivers and its biogeochemical and societal implications (Alorda‐Kleinglass et al., 2021; Lecher & Mackey, 2018; Santos et al., 2021; Taniguchi et al., 2019). However, major gaps in knowledge remain about the important microbially‐mediated chemical transformations occurring in the coastal aquifer before the solutes reach the ocean, mostly due to the scarcity of microbial studies targeting coastal aquifers (Ruiz‐González et al., 2021; Seibert et al., 2020). By sampling the microbial assemblages inhabiting the different compartments within a well‐characterized subterranean estuary, here we have shown a large spatial (vertical and longitudinal) heterogeneity in these communities, as we had hypothesized based on the pronounced physicochemical gradients, yet very little connectivity between local aquifer microbial communities. Our results point to clearly differentiated microbial niches across this land‐sea transition zone and provide one of the most detailed microbial descriptions of a coastal aquifer conducted so far.
4.1. Increased abundance, activity and larger size of prokaryotic cells towards the saline portions of the subterranean estuary
Unlike in surface aquatic ecosystems, there are almost no reports on the abundance, activity, or cell size of the microorganisms in coastal aquifers because most studies have focused mainly on the taxonomic characterization of communities (Ruiz‐González et al., 2021). Our study shows that fresher portions of the aquifer were characterized by smaller cells, lower prokaryotic abundance and lower heterotrophic production than the deeper saline groundwater (Figure 2). Fresh groundwater prokaryotic abundances were 13‐ to 18‐fold lower than those in surface seawater, ranging from 2 × 104 to 3 × 104 cells/ml. These low values fall in between those reported for pristine inland fresh groundwater (102–106 cells/ml, Griebler & Lueders, 2009) and are lower than those found by the two studies reporting prokaryotic abundances on the fresh parts of a sandy beach aquifer (6 × 105–1.3 × 106 cells/ml, Santoro et al., 2008) and of a coastal alluvial aquifer (2 × 105–>6 × 107 cells/ml, Velasco Ayuso, Acebes, et al., 2009, Velasco Ayuso, Guerrero, et al., 2009). However, a recent seasonal comparison of two shallow beach tidal subterranean estuaries showed even lower prokaryotic abundances, yet only at certain times of the year (e.g., in winter, <7 × 103 cells/ml, Calvo‐Martin et al., 2022).
The low biomass, activity and cell size of Argentona fresh groundwater prokaryotes agrees with the scarcity of dissolved organic carbon (DOC) typical of inland freshwater aquifers (Hofmann & Griebler, 2018), which often leads to miniaturization of prokaryotes in response to stress or starvation, becoming inactive or dormant (Jones & Lennon, 2010; Velimirov, 2001). In turn, the higher bacterial biomass, size, and heterotrophic activity found in the saline portion of the Argentona site could imply a source of organic carbon coming from the sea. Although we did not quantify DOC concentrations in the present study, a similar sampling conducted 1 year later (July 2019) showed low DOC concentrations, with fresh groundwater carrying on average less DOC (average 30 μM) than the brackish and saline groundwater sites (average 57 and 47 μM, respectively, C. Ruiz‐González). However, precisely N1_25 and N2_25 had very low DOC concentrations (<30 μM) despite showing the highest prokaryotic abundances and activity (details not shown). It is nonetheless possible that other organic carbon sources exist, such as the in situ production of DOC by autotrophic microorganisms, as suggested for the deep ocean and other organic carbon limited communities (Sebastián et al., 2018, 2019) and supported by accumulating evidence about the relevance of dark carbon fixation in groundwater systems (Overholt et al., 2022). Alternatively, differences in DOC quality and lability could also explain the observed variations in prokaryotic abundance or activity regardless of its bulk concentrations (e.g., Calvo‐Martin et al., 2022), but to our knowledge little is known about the origin, lability and composition of the DOC pool along subterranean estuaries.
In accordance with the trend of increasing activity towards saline groundwater, the percentage of Eubacterial‐hybridized cells (i.e., bacteria containing ribosomes) increased from the fresh (average 19% of DAPI‐stained cells) to the saline portions of the aquifer (average 38%) (Figure 4). These values are smaller than those commonly found in surface freshwater or marine ecosystems (50%–98%, Alonso‐Sáez et al., 2007; Ruiz‐González et al., 2013), even in very oligotrophic habitats such as the bathypelagic ocean (e.g., 38%–49%, Varela et al., 2007). This agrees with the observation that a large fraction of communities from inland aquifers are locally inactive (Griebler & Lueders, 2009). Within the aquifer, the abundance of Eubacteria correlated positively with the average cell size and the percentage of HNA prokaryotes (Pearson's R = .64 and .66, respectively, p < .05), but not with the prokaryotic heterotrophic production, suggesting that not all ribosome‐containing cells are equally active.
4.2. High spatial heterogeneity of dominant microbial groups along the subterranean estuary
In terms of microbial community composition, we found that Proteobacteria (mostly Alpha‐ and Gammaproteobacteria), Desulfobacteriota (formerly included within Deltaproteobacteria) and Bacteroidota were among the dominant phyla, in agreement with most available studies on subterranean estuaries (Archana et al., 2021; Ruiz‐González et al., 2021). However, Patescibacteria was the second most dominant phylum reaching up to 23% of local sequences. Patescibacteria (also known as candidate phyla radiation) includes a high diversity of recently discovered bacterial groups with unusually small cell sizes that are widespread in inland groundwaters (Brown et al., 2015; Herrmann et al., 2019; Luef et al., 2015), but which have rarely been reported in high abundances in coastal aquifers. The ubiquity and abundance of Patescibacteria in the Argentona site may be because most commonly used primers in groundwater studies fail to retrieve these groups, whereas the ones used here capture a higher diversity within Patescibacteria (Peura et al., 2012; Ruiz‐González et al., 2021). Patescibacteria are characterized by reduced genomes and hence limited metabolic capacities, and seem to be well adapted to the oligotrophic and relatively stable groundwater ecosystems, where many probably thrive in symbiosis with other microorganisms (Brown et al., 2015; He et al., 2021; Luef et al., 2015; Nelson & Stegen, 2015; Tian et al., 2020). The ecological role of these ultrasmall groups is still not fully understood, but their abundance in the Argentona site suggests that these groups may also be relevant players in coastal groundwater habitats.
Applying CARD‐FISH, we observed that groups such as Alphaproteobacteria and Deltaproteobacteria (recently reclassified into several phyla including Desulfobacterota) appeared as some of the most abundant among the ribosome‐containing cells, although Betaproteobacteria, Bacteroidetes and Gammaproteobacteria also dominated at some sites (Figure 4). We also detected minute free‐living cells labelled with the OD1 probe that targets Parcubacterial clades within Patescibacteria, which accounted for up to 28% of the ribosome‐containing cells in some brackish or marine groundwater communities. Their detection as free‐living cells agrees with other studies suggesting that not all members within this group may be symbiotic (Beam et al., 2020; Herrmann et al., 2019).
The CARD‐FISH probes used in this study are most probably biased against some of the groundwater diversity, because their design was based on sequences available in public databases, which only recently have started to include groundwater taxa (Ruiz‐González et al., 2021). In any case, our findings support that many of the main groups detected by sequencing were also potentially functional in these communities. Further studies aimed at distinguishing between the active and the inactive groundwater microbial components will be needed, because the inactive taxa can obscure any links between the environment, the taxonomic structure of the communities and their role in ecosystems, particularly when DNA sequencing is used (Niño‐García et al., 2016a, 2016b; Wisnoski et al., 2020).
4.3. Salinity as a key driver of aquifer prokaryotic communities
We observed a clear clustering of aquifer microbial communities based mostly on groundwater salinity, with brackish assemblages located between fresh and saline groundwater communities (Figure 3a). At the Argentona site, fresh groundwater flows seawards mainly in the upper part of the aquifer, whereas the deep aquifer area is influenced by the mixing between intruded seawater and meteoric water. The depth and extent of the saltwater wedge varies depending on the season and on meteorologic and oceanographic episodic events (e.g., storms, precipitation, Folch et al., 2020; Palacios et al., 2020). This causes steep gradients in salinity, oxygen, pH and redox conditions both at the horizontal and the vertical scale, but also in inorganic nutrient concentrations, which are differentially delivered by fresh or marine water. All these variables are well‐known drivers of aquatic bacterial communities and have been related to the compositional variations of prokaryotic communities from other coastal aquifers in the Mediterranean region (Héry et al., 2014; Sola et al., 2020) and elsewhere (Archana et al., 2021; Calvo‐Martin et al., 2022; Ruiz‐González et al., 2021).
Salinity, in particular, is considered one of the key drivers of prokaryote community assembly across natural ecosystems (Lozupone & Knight, 2007). However, unlike in surface estuaries where salinity variations cause a gradual succession of taxa (Bouvier & del Giorgio, 2002; Herlemann et al., 2011; Troussellier et al., 2002), in the Argentona site we found abrupt changes in taxonomic composition even between groundwater samples with similar salinity values (e.g., the three deepest saline sites were dominated by totally different phyla, Figure 3b). This points to a large effect of other drivers in shaping bacterial communities, including not only other physicochemical parameters (e.g., oxygen content, availability of electron donors and acceptors), but also the geological and hydrogeological properties of the aquifer, such as the lithology, grain size or organic matter content of aquifer solids, porosity, hydraulic conductivities, etc. These properties determine the degree of water‐solid interaction, the characteristics of groundwater flow (e.g., groundwater velocity and transit times), connectivity between different parts of the aquifer and the coastal ocean, as well as the potential retention and supply of carbon and nutrients. Indeed, the highest prokaryotic activity, the largest cell size and the highest proportion of HNA cells in the subterranean estuary were found in N1_25, probably caused by the specific hydrogeological characteristics of this sample since the screened interval of this piezometer is the only one located in an altered granite layer, with lower hydraulic conductivities resulting in more isolated communities (Folch et al., 2020).
The dominance of Desulfobacterota and of Firmicutes in the deep saline sites N1_20 and N1_25, respectively, is remarkable (Figure 3b). Members of Desulfobacterota include sulphate‐reducing groups that have been identified across multiple aquatic and terrestrial ecosystems, but often with preference to anoxic conditions (Waite et al., 2020). Many use sulphate, sulphite, thiosulphate, elemental sulphur or iron as terminal electron acceptors, and some can reduce elemental metals, mainly iron. The abundant detection of large ribosome‐containing Deltaproteobacteria through CARD‐FISH suggests that these sulphate‐reducing bacteria are probably active, being key players in the deepest and most saline part, in accordance with expected increases in sulphate reduction towards higher salinities within subterranean estuaries (Santoro, 2010). Firmicutes, which also include sulphate‐reducing groups and can produce endospores to resist extreme conditions (Filippidou et al., 2016), have also been identified as common microbial components of groundwater, usually increasing their contribution when salinity increases (Sang et al., 2018; Sola et al., 2020). The presence of metal‐ and sulphate‐reducing taxa indicates a possible depletion of other electron acceptors, which is consistent with the measured low oxygen and NO3 − concentrations and with chemical evidences of iron and manganese reduction in the deep saline groundwater at the Argentona site (Goyetche et al., 2022; Martínez‐Pérez et al., 2022).
Other prokaryotic groups showed more gradual changes along the salinity gradient, being more prevalent in the fresher portions of the aquifer (Burkholderiales, Nitrospirota, Planctomycetota), or preferring brackish (Gamma‐ and Alphaproteobacteria) or saline sites (Bacteroidota and Campylobacterota). This means that hydrological changes, such as variations in the extent of the saline intrusion (Folch et al., 2020; Palacios et al., 2020) or the occurrence of extreme precipitation events (Diego‐Feliu et al., 2022), may trigger major changes in the physicochemical parameters of the subterranean estuary and thus impact communities and their associated metabolic functions. For example, increases in salinity have been associated with decreased denitrification, nitrification and anammox (anaerobic ammonium oxidation, Jiao et al., 2018; Santoro, 2010). This pattern agrees with the lowest abundances found in Argentona saline groundwater of potentially nitrate‐oxidizing bacteria such as Nitrospinota and Nitrospirota, or of anammox bacteria such as the genus Ca. Brocadia (Planctomycetes). On the contrary, increases in salinity is expected to favor metabolisms such as dissimilatory nitrogen reduction to ammonium (DNRA, Santoro, 2010). DNRA can be carried out by a high diversity of bacterial groups, including Proteobacteria, Planctomycetes, Bacteroidetes, Firmicutes and Campylobacterota (Giblin et al., 2013; Hanson et al., 2013; Mohan et al., 2004), in agreement with the higher concentrations of Argentona saline groundwaters and the increased abundances of some of these groups. Given the high nitrate concentrations entering the Argentona subterranean estuary, mostly derived from agricultural activities (Martínez‐Pérez et al., 2022; Rufí‐Salís et al., 2019), it is expected that these bacterial communities play a major role on the subsurface nitrogen cycle. All this suggests that the spatial distribution of prokaryotic groups observed here will vary seasonally following the temporal variations in aquifer physicochemistry, but whether such seasonal changes will be due to a spatial redistribution of the same microbial taxa or to the growth of new groups peaking at different moments of the year remains unknown. Moreover, there are multiple evidences that a large fraction of the groundwater microbial biomass and activity is concentrated on aquifer surfaces, and that these communities differ taxonomically and functionally from free‐living cells (Archana et al., 2021; Ruiz‐González et al., 2021), so it is likely that our data, based only in groundwater samples, are providing an incomplete view of the microbial complexity hidden within the Argentona aquifer.
Other important components of groundwater microbial communities are the lithoautotrophs, which fix carbon dioxide and meet their energy requirements by oxidising inorganic electron donors (Griebler & Lueders, 2009). Several phyla detected in the Argentona site are known to contain groups capable of chemolithoautotrophic metabolisms, such as Nitrospira and Nitrospina, Ca. Brocadia (Planctomycetota), Sulfurimonas (Campylobacterota), Mariprofundus (Zetaproteobacteria) or Thiobacillus (Burkholderiales). Sulfurimonas and Mariprofundus, obligate chemolitoautotrophic sulphur‐ and iron‐oxidizing bacteria, respectively, were also found in a coastal Mediterranean carbonate aquifer (Héry et al., 2014). All this would agree with accumulating evidence from inland aquifers suggesting that chemolitoautotrophy is more important than previously believed in subsurface ecosystems (Anantharaman et al., 2016; Jewell et al., 2016; Kumar et al., 2018; Overholt et al., 2022), and the fact that the above‐mentioned autotrophic groups dominated in different portions of the subterranean estuary suggests that inorganic carbon fixation might occur along the entire salinity gradient. However, to our knowledge, the relative contribution of autotrophic metabolisms to total carbon cycling in subterranean estuaries has never been addressed.
4.4. High endemicity and low connectivity between microbial communities along the groundwater‐seawater continuum
The hydrological connectivity with surrounding ecosystems is also a relevant driver of the distribution of prokaryotic communities and of their role in subterranean estuaries (Ruiz‐González et al., 2021). Several studies have reported compositional changes in coastal aquifer microbial communities caused by seasonal changes in groundwater table (Chen et al., 2020; Menning et al., 2018), waves, tides (McAllister et al., 2015; Santoro et al., 2008) or varying connectivity with surface aquatic bodies (Velasco Ayuso, Acebes, et al., 2009; Velasco Ayuso, Guerrero, et al., 2009) or coastal seawater (Adyasari et al., 2019; Calvo‐Martin et al., 2022). In our case, all communities contained a high fraction of sequences belonging to unique ASVs (i.e., ASVs that were not detected anywhere else in the aquifer), and ASVs with preference for fresh, brackish or saline groundwater comprised very few reads across the other water types, pointing to a very strong selection of species at each site (Figure 7). Unlike in surface aquatic ecosystems, where the most abundant taxa are often the most ubiquitous even across large spatial scales and broad environmental gradients (Niño‐García et al., 2016a, 2016b; Salazar, Cornejo‐Castillo, Benitez‐Barrios, et al., 2015), we found that most ASVs were rare and were detected only in one or few sampling points across the aquifer. Also, the most abundant ASVs in the data set had very different tolerances and niche breaths, some being dominant across most of the salinity gradient and some appearing at single sites (Figure 6). This points to highly differentiated microbial niches throughout the aquifer and to a reduced dispersal of microbial taxa between aquifer communities. This limited connectivity is most probably related to both the small‐scale hydrogeological heterogeneities found in the Argentona experimental site (e.g., alternances of semiconfining layers, complex interactions between seawater and terrestrial water) and the granular composition of the subterranean estuary (i.e., relatively small pore spaces), which reduces the transport of compounds on the size range of microorganisms (Huettel et al., 2014).
The observed pattern of higher biomass and activity towards saline sites could also be caused by the transport of marine bacteria into the aquifer, which has been suggested to happen in other subterranean estuaries (Calvo‐Martin et al., 2022; Chen et al., 2019; Unno et al., 2015). However, the limited presence of typical marine taxa in the estuary and the highly different communities between the deep saline sites and the surface seawater rejects this possibility. This lack of marine influence in groundwater communities could be linked to the microtidal regime of the Mediterranean Sea, which limits the seawater exchange across the land‐ocean interface, and also to previously mentioned reduced microbial transport due to the granular characteristics and heterogeneities of the system. In turn, very few of the aquifer taxa were also detected in marine waters, suggesting that at least during the sampling period (summer), there is little microbial dispersal between aquifer and marine communities. This transport of microorganisms from the aquifer to the sea might, however, be enhanced in the wet season when groundwater discharge increases, as extreme precipitation episodes typical of spring and autumn in the study site have shown to increase SGD by 1 order of magnitude compared to baseflow conditions (Diego‐Feliu et al., 2022). Regardless of whether dispersal of taxa between the surface, the aquifer and the sea may shape microbial structure and functioning, our data suggest that strong selection by local conditions is the main process structuring the Argentona microbiome, at least during baseflow conditions.
4.5. Different rare Gammaproteobacteria and Alphaproteobacteria ASVs respond fast to changing conditions across aquifer samples
The high degree of endemicity, the very different niche preferences of dominant taxa and the low proportion of active cells implies that it will be difficult to predict how such communities will react to changes in the aquifer environment and how this will affect the aquifer biogeochemical cycles. As an example, when four groundwater communities sampled at different points of the aquifer were exposed to the same experimental manipulation (bottle enclosure in the dark with no carbon or nutrient addition), we observed that the identity of the few individual ASVs growing the most in each incubation differed between the four communities, and that the ASVs that grew the most in one community were rare (i.e., present at low in situ abundances) across all other aquifer sites. These responsive ASVs represented only a small subset of all the aquifer diversity (5–10 responsive ASVs per site) and belonged mostly to Gammaproteobacteria (including some Burkholderiales in the freshwater samples) and Alphaproteobacteria. This supports not only that Proteobacterial groups are viable groups within the aquifer, but also that some rare taxa within them can respond quickly to similar changes in conditions, becoming locally abundant. The associated increases in prokaryote abundance, heterotrophic production and average cell size during incubations further suggests that some of these groups were dormant or miniaturized in the original communities, but that they retain the capacity to respond fast to changes in conditions with significant impacts on the bulk community activity.
5. CONCLUSIONS
Submarine groundwater discharge is considered a major source of terrestrial dissolved solutes to the global ocean, but the nature and magnitude of SGD‐derived solute inputs depend largely on the biogeochemical transformations occurring in the subterranean estuary, many of which are performed by scarcely studied microbial communities. Here, we have shown a large spatial heterogeneity in prokaryotic communities along a Mediterranean subterranean estuary, pointing to clearly differentiated microbial niches across this land‐ocean transition zone and little microbial connectivity between the aquifer and the sea at the time of sampling. The large spatial variations in cell size, abundance, activity and community composition suggest that understanding the role that these coastal bioreactors play in transforming the flowing groundwater requires spatially resolved functional surveys of the complex microbial communities inhabiting these transition zones. Predicting the responses of microbial communities to the dynamic mixing of fresh groundwater and seawater intruded in the subterranean estuary is relevant because climate change scenarios anticipate large changes in groundwater‐seawater interaction in coastal aquifers caused by droughts, storms floods, sea level rise and excessive withdrawal of water for human consumption.
AUTHOR CONTRIBUTIONS
JGO, JMG, AF and CRG participated in the design of the sampling trip and/or designed the sampling scheme. JGO, VR, JMG, AAK, MDF, CRG participated in the collection of samples and in situ measurements of variables. CRG processed and analyzed all the bacterial production, flow cytometry, CARD‐FISH data and DNA data, with the help of LRP for DNA extractions and PCR and OM for CARD‐FISH processing and image analysis. VR helped compile the nutrient and physicochemical data. CRG conceived the idea of the manuscript and wrote the article with valuable contributions from all coauthors.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Appendix S1
ACKNOWLEDGEMENTS
This work was funded by the Spanish Ministry of Science, Innovation and Universities (MICINN) through the GRAMMI project (RTI2018‐099740‐J‐I00), the Ramon y Cajal contract (RYC2019‐026758‐I) and partly by the project MEDISTRAES III (PID2019‐110311RB‐C22 and PID2019‐110212RB‐C21). Additional financial support was provided by grants SPIP2020‐02595 (Ministry for the Ecological Transition and the Demographic Challenge [MITECO]) with funding from the Spanish government through the ‘Severo Ochoa Centre of Excellence’ accreditation (CEX2019‐000928‐S). JGO acknowledges the financial support of the Spanish Ministry of Science, Innovation and Universities, through the “Maria de Maeztu” programme for Units of Excellence (CEX2019‐000940‐M). VR acknowledges financial support from the Beatriu de Pinós postdoctoral programme of the Catalan Government (2019‐BP‐00241). AAK acknowledges financial support from ICTA “Unit of Excellence” (MinECo, MDM2015‐0552‐17‐1) and PhD fellowship, BES‐2017‐080740. The coauthor AF is a Serra Húnter Fellow. MDF acknowledges the economic support from the FI‐2017 fellowships of the Generalitat de Catalunya autonomous government (2017FI_B_00365). The authors would like to thank the support of the Generalitat de Catalunya to MERS (2017 SGR‐1588), GHS (2017 SGR 1485) and 2017SGR/156, for additional funding. We would like to thank SIMMAR (Serveis Integrals de Manteniment del Maresme) and the Consell Comarcal del Maresme in the construction of the research site. A special word for Jordi Garcia‐Orellana, great mentor and friend, who left us recently and unexpectedly, and without whom this and much of the ongoing work would not exist. May he rest in peace.
Ruiz‐González, C. , Rodríguez‐Pie, L. , Maister, O. , Rodellas, V. , Alorda‐Keinglass, A. , Diego‐Feliu, M. , Folch, A. , Garcia‐Orellana, J. , & Gasol, J. M. (2022). High spatial heterogeneity and low connectivity of bacterial communities along a Mediterranean subterranean estuary. Molecular Ecology, 31, 5745–5764. 10.1111/mec.16695
Handling Editor: Kayla King
DATA AVAILABILITY STATEMENT
DNA sequences and associated metadata: European Nucleotide Archive (ENA) under accession numbers PRJEB52186. The environmental metadata and the nonrarefied ASV and taxonomic tables are provided as Tables S2–S4.
REFERENCES
- Adyasari, D. , Hassenrück, C. , Oehler, T. , Sabdaningsih, A. , & Moosdorf, N. (2019). Microbial community structure associated with submarine groundwater discharge in northern Java (Indonesia). Science of The Total Environment, 689, 590–601. [DOI] [PubMed] [Google Scholar]
- Alonso‐Sáez, L. , Balagué, V. , Sà, E. L. , Sánchez, O. , González, J. M. , Pinhassi, J. , Massana, R. , Pernthaler, J. , Pedrós‐Alió, C. , & Gasol, J. M. (2007). Seasonality in bacterial diversity in north‐west Mediterranean coastal waters: Assessment through clone libraries, fingerprinting and FISH. FEMS Microbiology Ecology, 60, 98–112. [DOI] [PubMed] [Google Scholar]
- Alorda‐Kleinglass, A. , Ruiz‐Mallén, I. , Diego‐Feliu, M. , Rodellas, V. , Bruach‐Menchén, J. M. , & Garcia‐Orellana, J. (2021). The social implications of Submarine Groundwater Discharge from an Ecosystem Services perspective: A systematic review. Earth‐Science Reviews, 221, 103742. [Google Scholar]
- Anantharaman, K. , Brown, C. T. , Hug, L. A. , Sharon, I. , Castelle, C. J. , Probst, A. J. , Thomas, B. C. , Singh, A. , Wilkins, M. J. , Karaoz, U. , Brodie, E. L. , Williams, K. H. , Hubbard, S. S. , & Banfield, J. F. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nature Communications, 7, 13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archana, A. , Francis, C. A. , & Boehm, A. B. (2021). The beach aquifer microbiome: Research gaps and data needs. Frontiers in Environmental Science, 9, 1–13. [Google Scholar]
- Beam, J. P. , Becraft, E. D. , Brown, J. M. , Schulz, F. , Jarett, J. K. , Bezuidt, O. , Poulton, N. J. , Clark, K. , Dunfield, P. F. , Ravin, N. V. , Spear, J. R. , Hedlund, B. P. , Kormas, K. A. , Sievert, S. M. , Elshahed, M. S. , Barton, H. A. , Stott, M. B. , Eisen, J. A. , Moser, D. P. , … Stepanauskas, R. (2020). Ancestral absence of electron transport chains in Patescibacteria and DPANN. Frontiers in Microbiology, 11, 1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier, T. , & del Giorgio, P. A. (2002). Compositional changes in free‐living bacterial communities along a salinity gradient in two temperate estuaries. Limnology and Oceanography, 47, 453–470. [Google Scholar]
- Brown, C. T. , Hug, L. A. , Thomas, B. C. , Sharon, I. , Castelle, C. J. , Singh, A. , Wilkins, M. J. , Wrighton, K. C. , Williams, K. H. , & Banfield, J. F. (2015). Unusual biology across a group comprising more than 15% of domain bacteria. Nature, 523, 208–211. [DOI] [PubMed] [Google Scholar]
- Burnett, W. C. , Bokuniewicz, H. , Huettel, M. , Moore, W. S. , & Taniguchi, M. (2003). Groundwater and pore water inputs to the coastal zone. Biogeochemistry, 66, 3–33. [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from illumina amplicon data. Nature Methods, 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo‐Martin, E. , Teira, E. , Álvarez‐Salgado, X. A. , Rocha, C. , Jiang, S. , Justel‐Díez, M. , & Ibánhez, J. S. P. (2022). On the hidden diversity and niche specialization of the microbial realm of subterranean estuaries. Environmental Microbiology, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Hu, B. X. , Dai, H. , Zhang, X. , Xia, C.‐A. , & Zhang, J. (2019). Characterizing microbial diversity and community composition of groundwater in a salt‐freshwater transition zone. Science of the Total Environment, 678, 574–584. [DOI] [PubMed] [Google Scholar]
- Chen, L. , Zhang, J. , Dai, H. , Hu, B. X. , Tong, J. , Gui, D. , Zhang, X. , & Xia, C. (2020). Comparison of the groundwater microbial community in a salt‐freshwater mixing zone during the dry and wet seasons. Journal of Environmental Management, 271, 110969. [DOI] [PubMed] [Google Scholar]
- Cho, H. M. , Kim, G. , Kwon, E. , Moosdorf, N. , Garcia‐Orellana, J. , & Santos, I. R. (2018). Radium tracing nutrient inputs through submarine groundwater discharge in the global ocean. Scientific Reports, 8, 2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daims, H. , Bruhl, A. , Amann, R. , Schleifer, K. H. , & Wagner, M. (1999). The domain‐specific probe EUB338 is insufficient for the detection of all bacteria: Development and evaluation of a more comprehensive probe set. Systematic and Applied Microbiology, 22, 434–444. [DOI] [PubMed] [Google Scholar]
- Diego‐Feliu, M. , Rodellas, V. , Alorda‐Kleinglass, A. , Saaltink, M. , Folch, A. , & Garcia‐Orellana, J. (2022). Extreme precipitation events induce high fluxes of groundwater and associated nutrients to the coastal ocean. Hydrology and Earth System Sciences Discussions, 2022, 1–2 [Google Scholar]
- Duque, C. , Michael, H. A. , & Wilson, A. M. (2020). The subterranean estuary: Technical term, simple analogy, or source of confusion? Water Resources Research, 56, e2019WR026554. [Google Scholar]
- Filippidou, S. , Wunderlin, T. , Junier, T. , Jeanneret, N. , Dorador, C. , Molina, V. , Johnson, D. R. , & Junier, P. (2016). A combination of extreme environmental conditions favor the prevalence of endospore‐forming firmicutes. Frontiers in Microbiology, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch, A. , del Val, L. , Luquot, L. , Martinez‐Perez, L. , Bellmunt, F. , Le Lay, H. , et al. (2020). Combining fiber optic (FO‐DTS), cross‐hole ERT and time‐lapse formation electrical conductivity to characterize and monitor a coastal aquifer. Journal of Hydrology, 588, 125050. [Google Scholar]
- Gasol, J. M. , & Del Giorgio, P. A. (2000). Using flow cytometry for counting natural planktonic bacteria and understanding the structure of planktonic bacterial communities. Scientia Marina, 64, 197–224. [Google Scholar]
- Giblin, A. E. , Tobias, C. R. , Song, B. , Weston, N. , Banta, G. T. , & Rivera‐Monroy, V. H. (2013). !e importance of dissimilatory nitrate reduction to ammonium (DNRA) in the nitrogen cycle of coastal ecosystems. Oceanography, 26(3), 124–131. 10.5670/oceanog.2013.54 [DOI] [Google Scholar]
- Gong, J. , Qing, Y. , Guo, X. , & Warren, A. (2014). “Candidatus Sonnebornia yantaiensis”, a member of candidate division OD1, as intracellular bacteria of the ciliated protist Paramecium bursaria (Ciliophora, Oligohymenophorea). Systematic and Applied Microbiology, 37, 35–41. [DOI] [PubMed] [Google Scholar]
- Goyetche, T. , Luquot, L. , Carrera, J. , Martínez‐Pérez, L. , & Folch, A. (2022). Identification and quantification of chemical reactions in a coastal aquifer to assess submarine groundwater discharge composition. Science of the Total Environment, 838, 155978. [DOI] [PubMed] [Google Scholar]
- Griebler, C. , & Lueders, T. (2009). Microbial biodiversity in groundwater ecosystems. Freshwater Biology, 54, 649–677. [Google Scholar]
- Hanson, T. E. , Campbell, B. J. , Kalis, K. M. , Campbell, M. A. , & Klotz, M. G. (2013). Nitrate ammonification by Nautilia profundicola AmH: experimental evidence consistent with a free hyroxylamine intermediate. Frontiers in microbiology, 4, 180. 10.3389/fmicb.2013.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, C. , Keren, R. , Whittaker, M. L. , Farag, I. F. , Doudna, J. A. , Cate, J. H. D. , & Banfield, J. F. (2021). Genome‐resolved metagenomics reveals site‐specific diversity of episymbiotic CPR bacteria and DPANN archaea in groundwater ecosystems. Nature Microbiology, 6, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlemann, D. P. R. , Labrenz, M. , Jürgens, K. , Bertilsson, S. , Waniek, J. J. , & Andersson, A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. The ISME Journal, 5, 1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann, M. , Wegner, C.‐E. , Taubert, M. , Geesink, P. , Lehmann, K. , Yan, L. , Lehmann, R. , Totsche, K. U. , & Küsel, K. (2019). Predominance of Cand. Patescibacteria in groundwater is caused by their preferential mobilization from soils and flourishing under oligotrophic conditions. Frontiers in Microbiology, 10, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herut, B. , Krom, M. D. , Pan, G. , & Mortimer, R. (1999). Atmospheric input of nitrogen and phosphorus to the Southeast Mediterranean: Sources, fluxes, and possible impact. Limnology and Oceanography, 44, 1683–1692. [Google Scholar]
- Héry, M. , Volant, A. , Garing, C. , Luquot, L. , Elbaz Poulichet, F. , & Gouze, P. (2014). Diversity and geochemical structuring of bacterial communities along a salinity gradient in a carbonate aquifer subject to seawater intrusion. FEMS Microbiology Ecology, 90, 922–934. [DOI] [PubMed] [Google Scholar]
- Hofmann, R. , & Griebler, C. (2018). DOM and bacterial growth efficiency in oligotrophic groundwater: absence of priming and co‐limitation by organic carbon and phosphorus. Aquatic Microbial Ecology, 81, 55–71. [Google Scholar]
- Huettel, M. , Berg, P. , & Kostka, J. E. (2014). Benthic exchange and biogeochemical cycling in permeable sediments. Annual Review of Marine Science, 6, 23–51. [DOI] [PubMed] [Google Scholar]
- Jewell, T. N. M. , Karaoz, U. , Brodie, E. L. , Williams, K. H. , & Beller, H. R. (2016). Metatranscriptomic evidence of pervasive and diverse chemolithoautotrophy relevant to C, S, N, and Fe cycling in a shallow alluvial aquifer. The ISME Journal, 10, 2106–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, L. , Wu, J. , He, X. , Wen, X. , Li, Y. , & Hong, Y. (2018). Significant microbial nitrogen loss from denitrification and anammox in the land‐sea interface of low permeable sediments. International Biodeterioration & Biodegradation, 135, 80–89. [Google Scholar]
- Jones, S. E. , & Lennon, J. T. (2010). Dormancy contributes to the maintenance of microbial diversity. Proceedings of the National Academy of Sciences of the United States of America, 107, 5881–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman, D. , Knees, E. , & Hodson, R. (1985). Leucine incorporation and its potential as a measure of protein‐synthesis by bacteria in natural aquatic systems. Applied and Environmental Microbiology, 49, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Herrmann, M. , Blohm, A. , Hilke, I. , Frosch, T. , Trumbore, S. , & Küsel, K. (2018). Thiosulfate‐ and hydrogen‐driven autotrophic denitrification by a microbial consortium enrichedfrom groundwater of an oligotrophic limestone aquifer. FEMS Microbiology Ecology, 94, y141. [DOI] [PubMed] [Google Scholar]
- Kwon, E. Y. , Kim, G. , Primeau, F. , Moore, W. S. , Cho, H.‐M. , DeVries, T. , Sarmiento, J. L. , Charette, M. A. , & Cho, Y. K. (2014). Global estimate of submarine groundwater discharge based on an observationally constrained radium isotope model. Geophysical Research Letters, 41, 8438–8444. [Google Scholar]
- Lecher, A. L. , & Mackey, K. R. M. (2018). Synthesizing the effects of submarine groundwater discharge on marine biota. Hydrology, 5, 60. [Google Scholar]
- Loy, A. , Lehner, A. , Lee, N. , Adamczyk, J. , Meier, H. , Ernst, J. , Schleifer, K. H. , & Wagner, M. (2002). Oligonucleotide microarray for 16S rRNA gene‐based detection of all recognized lineages of sulfate‐reducing prokaryotes in the environment. Applied and Environmental Microbiology, 68, 5064–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. A. , & Knight, R. (2007). Global patterns in bacterial diversity. Proceedings of the National Academy of Sciences, 104, 11436–11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, W. , Dumont, E. , Meybeck, M. , & Heussner, S. (2009). River discharges of water and nutrients to the Mediterranean and Black Sea: Major drivers for ecosystem changes during past and future decades? Progress in Oceanography, 80, 199–217. [Google Scholar]
- Luef, B. , Frischkorn, K. R. , Wrighton, K. C. , Holman, H. Y. N. , Birarda, G. , Thomas, B. C. , Singh, A. , Williams, K. H. , Siegerist, C. E. , Tringe, S. G. , Downing, K. H. , Comolli, L. R. , & Banfield, J. F. (2015). Diverse uncultivated ultra‐small bacterial cells in groundwater. Nature Communications, 6, 6372. [DOI] [PubMed] [Google Scholar]
- Manz, W. , Amann, R. , Ludwig, W. , Vancanneyt, M. , & Schleifer, H. (1996). Application of a suite of 16S rRNA‐specific oligonucleotide probes designed to investigate bacteria of the phylum Cytophaga–Flavobacter–Bacteroides in the natural environment. Microbiology, 142, 1097–1106. [DOI] [PubMed] [Google Scholar]
- Manz, W. , Amann, R. , Ludwig, W. , Vancanneyt, M. , & Schleifer, K. H. (1992). Phylogenetic oligodeoxynucleotide probes for the major subclasses of Proteobacteria: Problems and solutions. Systematic and Applied Microbiology, 15, 593–600. [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal, 17(1), 10–12. [Google Scholar]
- Martínez‐Pérez, L. , Luquot, L. , Carrera, J. , Angel Marazuela, M. , Goyetche, T. , Pool, M. , et al. (2022). A multidisciplinary approach to characterizing coastal alluvial aquifers to improve understanding of seawater intrusion and submarine groundwater discharge. Journal of Hydrology, 607, 127510. [Google Scholar]
- McAllister, S. M. , Barnett, J. M. , Heiss, J. W. , Findlay, A. J. , MacDonald, D. J. , Dow, C. L. , Luther, G. W., III , Michael, H. A. , & Chan, C. S. (2015). Dynamic hydrologic and biogeochemical processes drive microbially enhanced iron and sulfur cycling within the intertidal mixing zone of a beach aquifer. Limnology and Oceanography, 60, 329–345. [Google Scholar]
- Menning, D. M. , Carraher‐Stross, W. A. , Graham, E. D. , Thomas, D. N. , Phillips, A. R. , Scharping, R. J. , & Garey, J. R. (2018). Aquifer discharge drives microbial community change in karst estuaries. Estuaries and Coasts, 41, 430–443. [Google Scholar]
- Mohan, S. B. , Schmid, M. , Jetten, M. , & Cole, J. (2004). Detection and widespread distribution of the nrfA gene encoding nitrite reduction to ammonia: A short circuit in the biological nitrogen cycle that competes with denitri!cation. FEMS Microbiology Ecology, 49, 433–443. 10.1016/j.femsec.2004.04.012 [DOI] [PubMed] [Google Scholar]
- Moore, W. S. (1999). The subterranean estuary: A reaction zone of ground water and sea water. Marine Chemistry, 65, 111–125. [Google Scholar]
- Moore, W. S. (2010). The effect of submarine groundwater discharge on the ocean. Annual Review of Marine Science, 2, 59–88. [DOI] [PubMed] [Google Scholar]
- Neef, A. (1997). Anwendung der in situ‐Einzelzell‐Identifizierung von bakterin zur populationsanalyse in komplexen mikrobiellen Biozo ¨nosen. Ph.D. thesis. Technische Universität München, Munich, Germany. [Google Scholar]
- Nelson, W. C. , & Stegen, J. C. (2015). The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Frontiers in Microbiology, 6, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niño‐García, J. P. , Ruiz‐González, C. , & del Giorgio, P. A. (2016a). Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. The ISME Journal, 10, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niño‐García, J. P. , Ruiz‐González, C. , & del Giorgio, P. A. (2016b). Landscape‐scale spatial abundance distributions discriminate core from random components of boreal lake bacterioplankton. Ecology Letters, 19, 1506–1515. [DOI] [PubMed] [Google Scholar]
- Oksanen, F. J. , Blanchet, G. , Kindt, R. , Legendre, P. , Minchin, P. R. , O'Hara, R. B. , et al. (2015). Vegan: Community ecology package. R package version 2.3‐0. [Google Scholar]
- Overholt, W. A. , Trumbore, S. , Xu, X. , Bornemann, T. L. V. , Probst, A. J. , Krüger, M. , Herrmann, M. , Thamdrup, B. , Bristow, L. A. , Taubert, M. , Schwab, V. F. , Hölzer, M. , Marz, M. , & Küsel, K. (2022). Carbon fixation rates in groundwater similar to those in oligotrophic marine systems. Nature Geoscience, 15, 561–567. [Google Scholar]
- Palacios, A. , Ledo, J. J. , Linde, N. , Luquot, L. , Bellmunt, F. , Folch, A. , Marcuello, A. , Queralt, P. , Pezard, P. A. , Martínez, L. , del Val, L. , Bosch, D. , & Carrera, J. (2020). Time‐lapse cross‐hole electrical resistivity tomography (CHERT) for monitoring seawater intrusion dynamics in a Mediterranean aquifer. Hydrology and Earth System Sciences, 24, 2121–2139. [Google Scholar]
- Pernthaler, A. , Pernthaler, J. , & Amann, R. (2002). Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Applied and Environmental Microbiology, 68, 3094–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peura, S. , Eiler, A. , Bertilsson, S. , Nykänen, H. , Tiirola, M. , & Jones, R. I. (2012). Distinct and diverse anaerobic bacterial communities in boreal lakes dominated by candidate division OD1. The ISME Journal, 6, 1640–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing. http://www.R‐project.org/ [Google Scholar]
- Robinson, C. E. , Xin, P. , Santos, I. R. , Charette, M. A. , Li, L. , & Barry, D. A. (2018). Groundwater dynamics in subterranean estuaries of coastal unconfined aquifers: Controls on submarine groundwater discharge and chemical inputs to the ocean. Advances in Water Resources, 115, 315–331. [Google Scholar]
- Rodellas, V. , Garcia‐Orellana, J. , Masqué, P. , Feldmane, M. , & Weinsteine, Y. (2015). Submarine groundwater discharge as a major source of nutrients to the Mediterranean Sea. Proceedings of the National Academy of Sciences of the United States of America, 112, 3926–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodellas, V. , Stieglitz, T. C. , Andrisoa, A. , Cook, P. G. , Raimbault, P. , Tamborski, J. J. , van Beek, P. , & Radakovitch, O. (2018). Groundwater‐driven nutrient inputs to coastal lagoons: The relevance of lagoon water recirculation as a conveyor of dissolved nutrients. Science of the Total Environment, 642, 764–780. [DOI] [PubMed] [Google Scholar]
- Roller, C. , Wagner, M. , Amann, R. , Ludwig, W. , & Schleifer, K. H. (1994). In situ probing of gram‐positive bacteria with hight DNA G+C content using 23S ribosomal RNA‐targeted oligonucleotides. Microbiology (Reading), 140, 2849–2858. [DOI] [PubMed] [Google Scholar]
- Rufí‐Salís, M. , Garcia‐Orellana, J. , Cantero, G. , Castillo, J. , Hierro, A. , Rieradevall, J. , & Bach, J. (2019). Influence of land use changes on submarine groundwater discharge. Environmental Research Communications, 1, 031005. [Google Scholar]
- Ruiz‐González, C. , Proia, L. , Ferrera, I. , Gasol, J. M. , & Sabater, S. (2013). Effects of large river dam regulation on bacterioplankton community structure. FEMS Microbiology Ecology, 84, 316–331. [DOI] [PubMed] [Google Scholar]
- Ruiz‐González, C. , Rodellas, V. , & Garcia‐Orellana, J. (2021). The microbial dimension of submarine groundwater discharge: Current challenges and future directions. FEMS Microbiology Reviews, 45, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar, G. , Cornejo‐Castillo, F. M. , Benitez‐Barrios, V. , Fraile‐Nuez, E. , Álvarez‐Salgado, X. A. , Duarte, C. M. , Gasol, J. M. , & Acinas, S. G. (2015). Global diversity and biogeography of deep‐sea pelagic prokaryotes. The ISME Journal, 10, 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang, S. , Zhang, X. , Dai, H. , Hu, B. X. , Ou, H. , & Sun, L. (2018). Diversity and predictive metabolic pathways of the prokaryotic microbial community along a groundwater salinity gradient of the Pearl River Delta, China. Scientific Reports, 8, 17317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, A. E. (2010). Microbial nitrogen cycling at the saltwater–freshwater interface. Hydrogeology Journal, 18, 187–202. [Google Scholar]
- Santoro, A. E. , Francis, C. A. , de Sieyes, N. R. , & Boehm, A. B. (2008). Shifts in the relative abundance of ammonia‐oxidizing bacteria and archaea across physicochemical gradients in a subterranean estuary. Environmental Microbiology, 10, 1068–1079. [DOI] [PubMed] [Google Scholar]
- Santos, I. R. , Chen, X. , Lecher, A. L. , Sawyer, A. H. , Moosdorf, N. , Rodellas, V. , Tamborski, J. , Cho, H. M. , Dimova, N. , Sugimoto, R. , Bonaglia, S. , Li, H. , Hajati, M. C. , & Li, L. (2021). Submarine groundwater discharge impacts on coastal nutrient biogeochemistry. Nature Reviews Earth & Environment, 2, 307–323. [Google Scholar]
- Sebastián, M. , Auguet, J.‐C. , Restrepo‐Ortiz, C. X. , Sala, M. M. , Marrasé, C. , & Gasol, J. M. (2018). Deep ocean prokaryotic communities are remarkably malleable when facing long‐term starvation. Environmental Microbiology, 20, 713–723. [DOI] [PubMed] [Google Scholar]
- Sebastián, M. , Estrany, M. , Ruiz‐González, C. , Forn, I. , Sala, M. M. , Gasol, J. M. , & Marrasé, C. (2019). High growth potential of long‐term starved deep ocean opportunistic heterotrophic bacteria. Frontiers in Microbiology, 10, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert, S. L. , Degenhardt, J. , Ahrens, J. , Reckhardt, A. , Schwalfenberg, K. , & Waska, H. (2020). Investigating the land‐sea transition zone. In Jungblut S., Liebich V., & Bode‐Dalby M. (Eds.), YOUMARES 9 – The oceans: Our research, our future (pp. 225–242). Springer Nature. [Google Scholar]
- Smith, D. , & Azam, F. (1992). A simple, economical method for measuring bacteria protein synthesis rates in seawater using 3H‐leucine. Marine Microbial Food Webs, 6, 107–114. [Google Scholar]
- Sola, F. , Vargas‐García, M. d. C. , & Vallejos, A. (2020). Interrelation prokaryotic community‐aquifer in a carbonate coastal environment. Aquatic Sciences, 82, 13. [Google Scholar]
- Taniguchi, M. , Dulai, H. , Burnett, K. M. , Santos, I. R. , Sugimoto, R. , Stieglitz, T. , Kim, G. , Moosdorf, N. , & Burnett, W. C. (2019). Submarine groundwater discharge: Updates on its measurement techniques, geophysical drivers, magnitudes, and effects. Frontiers in Environmental Science, 7, 141. [Google Scholar]
- Tian, R. , Ning, D. , He, Z. , Zhang, P. , Spencer, S. J. , Gao, S. , Shi, W. , Wu, L. , Zhang, Y. , Yang, Y. , Adams, B. G. , Rocha, A. M. , Detienne, B. L. , Lowe, K. A. , Joyner, D. C. , Klingeman, D. M. , Arkin, A. P. , Fields, M. W. , Hazen, T. C. , … Zhou, J. (2020). Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome, 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar‐Sánchez, A. , Basterretxea, G. , Rodellas, V. , Sánchez‐Quiles, D. , García‐Orellana, J. , Masqué, P. , Jordi, A. , López, J. M. , & Garcia‐Solsona, E. (2014). Contribution of groundwater discharge to the coastal dissolved nutrients and trace metal concentrations in majorca island: Karstic vs detrital Systems. Environmental Science & Technology, 48, 11819–11827. [DOI] [PubMed] [Google Scholar]
- Troussellier, M. , Schafer, H. , Batailler, N. , Bernard, L. , Courties, C. , Lebaron, P. , et al. (2002). Bacterial activity and genetic richness along an estuarine gradient (Rhone River plume, France). Aquatic Microbial Ecology, 28, 13–24. [Google Scholar]
- Unno, T. , Kim, J. , Kim, Y. , Nguyen, S. G. , Guevarra, R. B. , Kim, G. P. , Lee, J. H. , & Sadowsky, M. J. (2015). Influence of seawater intrusion on microbial communities in groundwater. Science of the Total Environment, 532, 337–343. [DOI] [PubMed] [Google Scholar]
- Varela, M. M. , van Aken, H. M. , Sintes, E. , & Herndl, G. J. (2007). Latitudinal trends of Crenarchaeota and bacteria in the meso‐ and bathypelagic water masses of the Eastern North Atlantic. Environmental Microbiology, 10, 110‐124. [DOI] [PubMed] [Google Scholar]
- Velasco Ayuso, S. , Acebes, P. , López‐Archilla, A. I. , Montes, C. , & Guerrero, M. d. C. (2009). Environmental factors controlling the spatiotemporal distribution of microbial communities in a coastal, sandy aquifer system (Doñana, southwest Spain). Hydrogeology Journal, 17, 767–780. [Google Scholar]
- Velasco Ayuso, S. , Guerrero, M. C. , Montes, C. , & López‐Archilla, A. I. (2009). Spatiotemporal distribution of microbial communities in a coastal, sandy aquifer system (Doñana, SW Spain). Geobiology, 7, 66–81. [DOI] [PubMed] [Google Scholar]
- Velimirov, B. (2001). Nanobacteria, ultramicrobacteria and starvation forms: A search for the smallest metabolizing bacterium. Microbes and Environments, 16, 67–77. [Google Scholar]
- Vila‐Costa, M. , Gasol, J. M. , Sharma, S. , & Moran, M. A. (2012). Community analysis of high‐ and low‐nucleic acid‐containing bacteria in NW Mediterranean coastal waters using 16S rDNA pyrosequencing. Environmental Microbiology, 14, 1390–1402. [DOI] [PubMed] [Google Scholar]
- Waite, D. W. , Chuvochina, M. , Pelikan, C. , Parks, D. H. , Yilmaz, P. , Wagner, M. , Loy, A. , Naganuma, T. , Nakai, R. , Whitman, W. B. , Hahn, M. W. , Kuever, J. , & Hugenholtz, P. (2020). Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. International Journal of Systematic and Evolutionary Microbiology, 70, 5972–6016. [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein, Y. , Yechieli, Y. , Shalem, Y. , Burnett, W. C. , Swarzenski, P. W. , & Herut, B. (2011). What is the role of fresh groundwater and recirculated seawater in conveying nutrients to the coastal ocean? Environmental Science & Technology, 45, 5195–5200. [DOI] [PubMed] [Google Scholar]
- West, N. J. , Schonhuber, W. A. , Fuller, N. J. , Amann, R. I. , Rippka, R. , Post, A. F. , & Scanlan, D. J. (2001). Closely related Prochlorococcus genotypes show remarkably different depth distributions in two oceanic regions as revealed by in situ hybridization using 16S rRNA‐targeted oligonucleotides. Microbiology (Reading), 147, 1731–1744. [DOI] [PubMed] [Google Scholar]
- Wisnoski, N. I. , Muscarella, M. E. , Larsen, M. L. , Peralta, A. L. , & Lennon, J. F. (2020). Metabolic insight into bacterial community assembly across ecosystem boundaries. Ecology, 101, e02968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
DNA sequences and associated metadata: European Nucleotide Archive (ENA) under accession numbers PRJEB52186. The environmental metadata and the nonrarefied ASV and taxonomic tables are provided as Tables S2–S4.