

Abstract
On‐surface chemistry holds the potential for ultimate miniaturization of functional devices. Porphyrins are promising building‐blocks in exploring advanced nanoarchitecture concepts. More stable molecular materials of practical interest with improved charge transfer properties can be achieved by covalently interconnecting molecular units. On‐surface synthesis allows to construct extended covalent nanostructures at interfaces not conventionally available. Here, we address the synthesis and properties of covalent molecular network composed of interconnected constituents derived from halogenated nickel tetraphenylporphyrin on Au(111). We report that the π‐extended two‐dimensional material exhibits dispersive electronic features. Concomitantly, the functional Ni cores retain the same single‐active site character of their single‐molecule counterparts. This opens new pathways when exploiting the high robustness of transition metal cores provided by bottom‐up constructed covalent nanomeshes.
Keywords: Nanostructures, Photoelectron Spectroscopy, Porphyrinoids, Scanning Probe Microscopy, X-Ray Absorption Spectroscopy
The on‐surface synthesis of covalent nickel tetraphenylporphyrin network on Au(111) is studied by a multitechnique approach combining local microscopic insights with space‐averaging synchrotron radiation‐based methods. Even though the porphyrin nanomesh is not perfectly ordered extending the π‐system results in the appearance of energy‐dispersive electronic features while the Ni cores remain in the characteristic single‐molecule configuration.
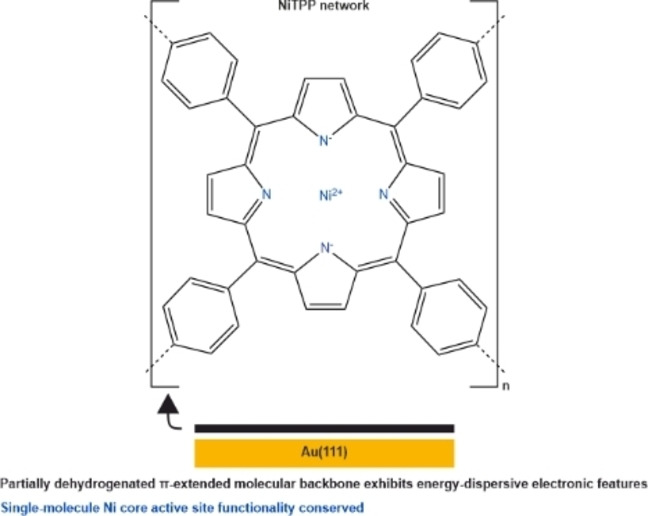
Introduction
Metalloporphyrins portray a class of outstanding heteroaromatic compounds with their on‐surface chemistry relevant for building functional devices precise at the molecular scale. [1] The possibility to vary both the chelated metal ion and peripheral substitution offers the advantage of controlling the molecular arrangement, as well as tuning the molecular substrate interaction.[ 2 , 3 ] This facilitates the realization of self‐assembled nanostructures composed of porphyrin units whose optoelectronic and magnetic properties together with the central metal ion reactivity can be adjusted accurately.[ 4 , 5 ] The tailor‐made performance of single‐active sites that are stabilized within the tetrapyrrolic moiety introduces the unique opportunity to rationally design catalytic materials and chemical sensors.[ 6 , 7 ] Porphyrinic catalysts are relevant for electrochemical CO2, O2 and N2 reduction as well as water splitting and in organic synthesis.[ 8 , 9 ] In photocatalytic applications their importance further lies in enabling oxygen‐mediated radicalic polymerizations and opening pathways in both CO2 reduction and hydrogen evolution without need of noble metal co‐catalyst.[ 10 , 11 , 12 ] Porphyrin units with magnetic cores are also particularly interesting for miniaturized spintronics devices.[ 13 , 14 , 15 ] Within this field of on‐surface magnetochemistry, the possibility to realize spin‐switching of the central metal ion by axial ligands that rely on uniform coordination environments across the interface has become a prominent subject of interest.[ 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ]
The technological exploitation of molecule‐based materials relies on more robust nanostructures composed of covalently linked units.[ 24 , 25 ] Covalent transition metal porphyrin‐based catalytic materials that exhibit improved charge transport and carrier separation properties were proven to outperform their single‐molecule counterparts in electro‐ and photochemical conversion reactions.[ 26 , 27 ] On‐surface synthesis is the method of choice in order to achieve π‐conjugated conductive nanomeshes with red‐shifted absorption characteristics at the interface.[ 28 , 29 , 30 ] Polymerization protocols applied to porphyrin units that yield π‐extended covalent nanostructures are interconnection of activated dehalogenated precursors,[ 24 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 ] Glaser coupling, [39] dehydrogenative[ 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 ] as well as defluorinative [48] coupling schemes, McMurry‐type [49] and Schiff‐base condensation [50] reactions.
Previous studies mainly addressed the properties of these macromolecular compounds by means of local probe methods, namely scanning tunneling microscopy and spectroscopy (STM/STS). Approaches combining local insights with space‐averaging techniques were either limited to porphyrin materials without transition metal chelation or did not include the functional properties of the ligated cores that are provided by covalent metalloporphyrin networks. This work aims at filling this gap by providing joint findings obtained by photoemission and absorption spectroscopy, as well as STM experiments. Thereby, we have chosen a Au(111) substrate as a catalyst to induce Ullmann coupling of a nickel tetra(4‐bromophenyl)porphyrin (NiTBrPP) precursor layer. The weak charge transfer at the interface to the molecular layer allows elucidating the intrinsic properties of the fabricated molecular network, which is composed of in‐plane covalently linked nickel tetraphenylporphyrin (NiTPP) units. Despite deviations from perfect order inherent to the coupling scheme applied, namely by thermal activation, the extended conductive nanomeshes reported here are characterized by exciting functional properties. The creation of a π‐delocalized porphyrin network across the surface is reflected by the appearance of continuous energy‐dispersive electronic valence states that were not observed before for this class of materials. At the same time, the single‐active site character of the Ni cores embedded in the conductive molecular backbone is retained. Thus, our strategy is particularly interesting for advanced devices given the possibility of relying on precisely defined central metal ion functionality, the defined arrangement due to the organic spacer, and the improved stability gained by interconnecting the molecular units.
Results and Discussion
A saturated layer of NiTBrPP was prepared on Au(111) with the substrate held at room temperature during deposition. All experimental details are given in Chapter 1 of the Supporting Information. The corresponding momentum distribution of the photoelectrons was recorded using an electrostatic photoelectron emission microscope operated in the momentum mode (k‐PEEM). The parallel momentum k // versus binding energy (BE) dependence along the substrate ‐ ‐ direction is presented in Figure 1. This cut represents a waterfall plot of intensity line profiles obtained from the stack of momentum images that were acquired at different BEs. The corresponding momentum‐integrated valence band (VB) spectrum (Figure S2) is included in the Supporting Information and, for better comparison, a normalized valence band spectrum of the bare Au(111) substrate is also depicted. Two additional peaks (highlighted by stars in Figure 1) in the VB region at BEs of 1.1 eV and 1.4 eV appear after NiTBrPP deposition on Au(111) with respect to the clean reference, which shows only contribution from the Au sp‐bands. These peaks arise from the highest occupied molecular orbitals (HOMO and HOMO‐1, respectively) of the anchored NiTBrPP. Essentially, the molecular signals arising in the valence band spectrum produce momentum‐localized maps evidencing the presence of a preferential orientation of the molecular layer with respect to the supporting substrate. [51] The absence of a signal from the molecular levels around the Fermi energy, which are unoccupied in the gas phase and could be partially filled by a strong charge transfer at the interface,[ 4 , 52 ] is a strong indication of weak molecule‐substrate interaction. This is a desired property for this prototype system, whose intrinsic properties upon polymerization can then be further elucidated without being drastically influenced by the supporting substrate.
Figure 1.
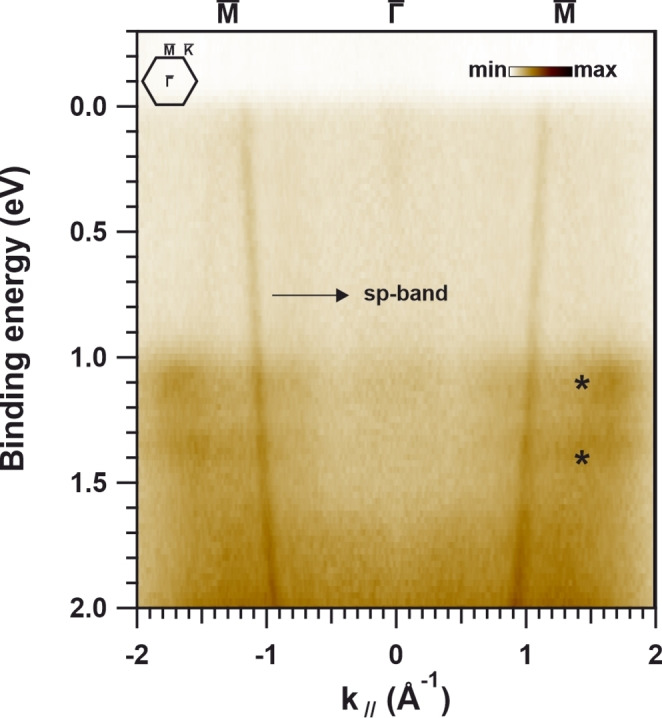
Overview of the momentum‐resolved photoelectron distribution along the substrate ‐ ‐ direction (Au(111) Brillouin zone indicated at the top left) of pristine NiTBrPP/Au(111) that was collected at a photon energy of 30 eV using p‐polarized light.
Intact NiTBrPP units were then annealed stepwise to induce C−Br bond cleavage that results in Ullmann coupling of the activated precursor molecules.[ 24 , 33 ] X‐ray photoelectron spectroscopy (XPS) was performed to identify a suitable temperature range for this polymerization reaction as further elaborated in Chapter 2 of the Supporting Information. We choose direct annealing to 673 K to produce the desired covalent nanomesh making it worth mentioning the observed extremely high thermal stability of the obtained NiTPP‐based covalent network. STM studies were conducted to characterize the chemical changes in the adsorbed molecular film. The topographic images in Figure 2 (top panel), cropped from large‐scale images presented in Figure S3, include a sketch of the molecular geometry. When it comes to pristine NiTBrPP on Au(111), the present contrast allows concluding that the porphyrin macrocycle is characterized by two symmetric bright lobes oriented orthogonally to the surface normal. [53] The phenyl substituents of neighboring molecules, which are rotated off‐plane with respect to the macrocycle, can be identified to be aligned parallel with respect to each other, which, again, confirms the weak molecule‐substrate interaction. [54] In Figure 2 we also present a region of a covalent NiTPP‐based nanomesh created by annealing, which allows concluding that the phenyl substituents are flattened as a consequence of the formation of a π‐extended macromolecule due to intermolecular coupling. [33] From the corresponding large‐scale topographic image presented in Figure S3 it is evident that the covalently interconnected constituents are no longer characterized by any sort of preferential azimuthal orientation. Moreover, randomly distributed defects with various shapes are evident when phenyl groups failed to react with neighboring ones. The nanomesh is highly interconnected since these imperfections almost exclusively appear when one of four phenyl substituents remain unsatured, which approximately applies to one out of ten molecules. When analyzing monomeric units in the network, deviations from a perfect four‐fold symmetry that make the porphyrin nanomesh amorphous become evident. The appearance of the molecular constituents allows us to conclude that these irregularities originate from cyclodehydrogenation side reactions,[ 55 , 56 ] which additionally increases the degree of conjugation of the molecular network. Indeed, four tetraphenylporphyrin derivative products have been reported upon annealing pristine molecules on Au(111) and these species are incorporated into the covalent porphyrin network reported here as confirmed by the STM images. The supporting Au(111) surface appears partially uncovered in comparison to the pristine NiTBrPP layer, which can be attributed to the desorption of some molecular precursors upon thermal activation and in‐plane shrinking of the molecular network. This is in perfect agreement with the corresponding XPS spectra as further elaborated in Chapter 4 of the Supporting Information. The partial dehydrogenation of the monomeric units restricts the growth direction when the covalent nanostructure is formed and causes the monomeric units to exhibit a random azimuthal orientation. Consequently, defects appear when reaction partners are missing due to desorption.
Figure 2.
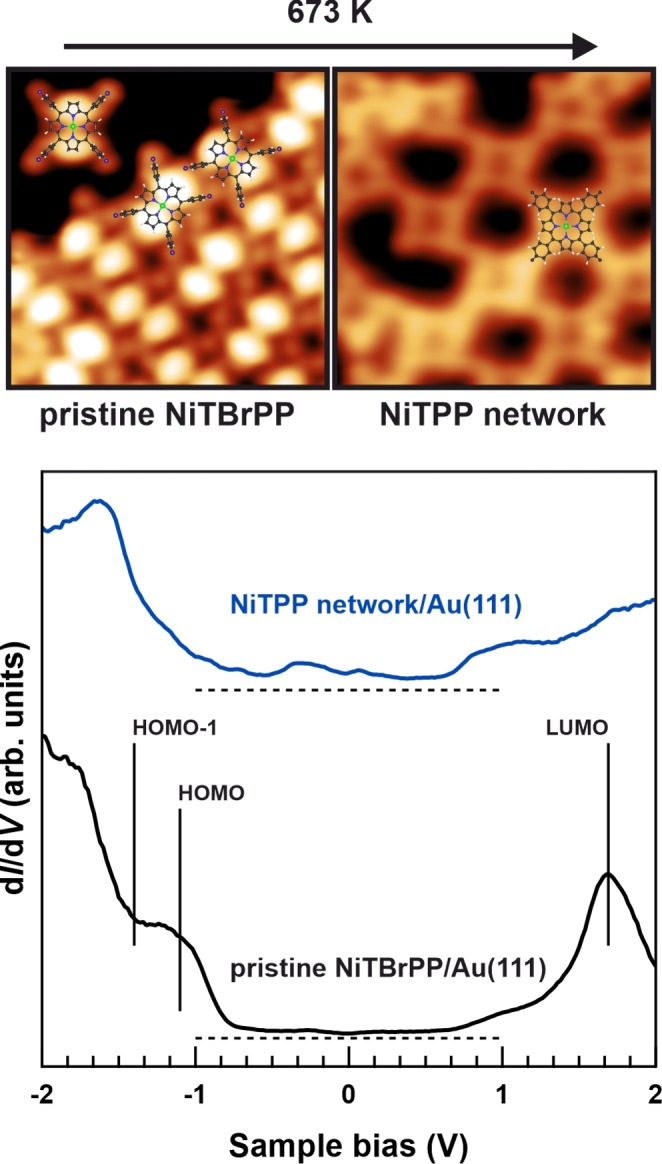
STM topographic viewgraph (top panel) of two 5.2×5.2 nm2 insets highlighting the changes when annealing the pristine NiTBrPP layer on Au(111) (left, V=−1.8 V, I=0.5 nA) to 673 K that induces the formation of NiTPP‐based covalent network (right, V=−0.4 V, I=0.5 nA). The corresponding STS dI/dV textures (bottom panel) obtained for pristine NiTBrPP on Au(111) and after formation of NiTPP‐based network are also included. Thereby, the zero‐levels are indicated by the dashed lines.
Scanning tunneling spectroscopy (STS) was then applied in order to locally characterize the differences between the molecular units embedded in the covalent nanomesh and their single‐molecule counterparts. As evident from the STS dI/dV texture (Figure 2, bottom panel) of pristine NiTBrPP on Au(111), the molecular precursor is characterized by a molecular gap confirming the energy position of the occupied levels observed in the VB spectrum presented in Figure S2. Conversely, the spectrum of the final NiTPP network is not anymore characterized by molecular levels of defined energy but rather by an electronic density of states distributed over the entire bias range. The precursor units being debrominated and interconnected after thermal activation are chemically related to NiTPP and dehydrogenated NiTPP. However, the absence of a clear molecular signature in STS cannot be explained by the distinct dehydrogenation pathways that occurred at this sample position. Indeed, the frontier states of various cyclized porphyrins shift in energy with respect to their initial values but remain defined. [56] It appears that the STS texture of the covalent network probed here is reflecting the chemical changes related to the formation of intermolecular bonds.
Differential conductance (dI/dV) mapping across the occupied states of the sample after the Ullmann coupling allowed us to image the spatial profile of the covalent NiTPP network to further confirm this conclusion. An overview of the dI/dV maps obtained at different voltages is given in Figure S4. There is no more evidence of the occupied molecular level, that is the bright symmetric lobes localized at the macrocycle that allowed us to identify the configuration of the pristine NiTBrPP layer in Figure 2. Instead, we notice that the spatially‐defined states, mainly probed here in the bias range between −0.6 eV and −1.3 eV, are localized at the Ni cores. Both the shape and the energy range strongly resemble the energy position of the central ion d‐states of NiTPP units anchored to the weakly interacting oxygen‐passivated Cu(100) substrate. [5] It turns out that after polymerization no spatially‐confined molecular states can be discriminated within the extended π‐conjugated nanomesh and only the functional Ni cores display localized states.
Covalently linking NiTPP units leads to drastic changes in the VB spectrum that are evident in Figure S2. The discrete molecular levels disappear upon annealing the molecular precursor and evolve into a continuum of states that is largely spread in energy and arising from few tenths of eV below the Fermi level. Broadening of molecular levels was observed for oligomerized metal‐free porphyrin units, suggesting that the continuum observed here is related to the formation of an extended covalent nanostructure. [43] Looking at the photoelectron k // distribution along the Au(111) substrate ‐ ‐ direction after NiTPP network creation that is shown in Figure 3, the formation of a dispersive band of parabolic shape is evident. The corresponding photoelectron distribution characteristic for the clean Au(111) substrate proves the parabola of states to be related to the covalent nanostructure and is displayed in Figure S5. The molecule‐related states are fully absent when using s‐polarized light instead of p‐polarized light indicating the π‐delocalization in the covalent nanomesh. All directions in reciprocal space yield the same dispersion relation for the molecular network when using p‐polarized light. The band structure is equally broadened along any direction and within the full energy range. This is demonstrated by the constant binding energy momentum images (Figure S6) obtained by k‐PEEM for both Au(111) and after formation of the NiTPP‐based network. The covalent nanomesh produces a ring structure in reciprocal space that is closing with the binding energy increasing. Similar findings were observed for graphene when more than one rotational domain was present. [57] We note that a broadening of the bands of graphene was further observed when introducing nitrogen heteroatoms into graphene and, simultaneously, creating topological defects. [58] It is very peculiar, that the present graphene‐related NiTPP‐based polymer structure, though amorphous and defect‐rich, is also characterized by broad, and yet, continuous spectrum of states. This supports the picture of a uniform and efficient charge transport across the metal‐like molecular layer, the stability of which is increased as a consequence of creating an extended aromatic system. This further allows concluding that the previously discussed STS dI/dV texture of NiTPP network reflects the local density of states of the conjugated NiTPP network. We also emphasize that so far dispersive electronic features that are exhibited by bottom‐up constructed nanostructures were rarely observed, limited to smaller monomeric units without any transition metal ions embedded.[ 59 , 60 , 61 , 62 , 63 , 64 , 65 ]
Figure 3.
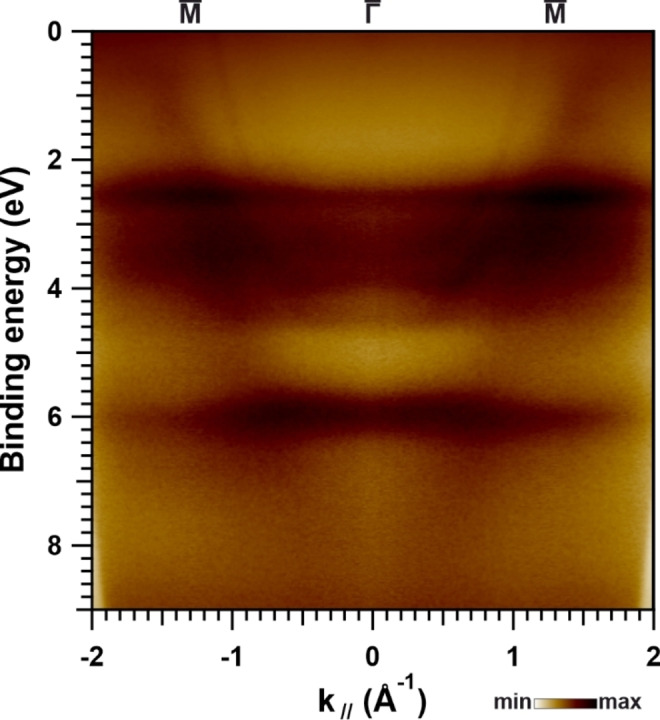
Photoelectron k // distribution obtained for covalent NiTPP‐based network on Au(111) along the substrate ‐ ‐ direction recorded at a photon energy of 30 eV using p‐polarized light.
This finding raises the question of how the individual porphyrin components are influenced by the creation of the porphyrin network. For this reason, near‐edge x‐ray absorption fine structure (NEXAFS) measurements were conducted for both the pristine NiTBrPP layer on Au(111) and after the formation of the NiTPP‐based network. Molecular layers with a preferential on‐surface orientation give rise to distinct linear dichroism in NEXAFS, which is particularly helpful in the case of porphyrin units for determining the geometric arrangement. [66] Their absorption spectra turned out to be a superposition of the different molecular moieties. [67] This is already demonstrated by NEXAFS performed at the C 1s threshold depicted in Figure 4 for the as‐deposited NiTBrPP layer on Au(111) substrate. Specific transitions into unoccupied π*‐symmetric molecular states are fully distinguishable as they are mostly localized on either the macrocycle (index m, 283.9 eV and 284.2 eV) or the phenyl substituents (index p, 285.2 eV) and, thus, can be used to evaluate the molecular geometric configuration.[ 33 , 68 , 69 , 70 , 71 ] The aforementioned characteristic π*‐macrocycle resonances are almost absent for s‐polarized light, which confirms a nearly flat absorption of the macrocycle on the Au(111) substrate. On the contrary, the π*‐phenyl transitions are observed in both polarizations. These findings correspond directly to those results obtained by STM, where a significant tilt of the phenyl groups with respect to the macrocycle plane was evidenced. C K‐edge spectra recorded after annealing the parent NiTBrPP layer on Au(111) are also included in Figure 4, where all π*‐resonances nearly vanish in s‐polarization. This leads us to conclude that all aromatic rings are forced into a flat configuration as already observed by STM. At the same time, the shape and relative intensity of the π*‐resonances in the C K‐edge in p‐polarization significantly differ from those measured for the pristine NiTBrPP/Au(111) layer. Instead of sharp molecular absorption peaks corresponding to NiTBrPP molecular levels, a continuum of states is probed when performing absorption spectroscopy on the covalent NiTPP network. As concluded by STM, annealing of NiTBrPP leads to cyclodehydrogenation side reactions accompanied by the formation of new carbon‐carbon intermolecular bonds. Interestingly, Di Santo and co‐workers, who performed NEXAFS studies on the cyclodehydrogenation reaction of 2H‐TPP on Ag(111) substrate, [72] found no significant changes in the C 1s NEXAFS spectrum of planar cyclodehydrogenated 2H‐TPP compared to parent 2H‐TPP species, since both give rise to clear molecular absorption features. Indeed, changes similar to the ones obtained here were observed when oligomerizing porphyrin units. [43] Thus, we attribute the findings observed for NiTPP‐based network to the formation of a π‐extended macromolecular covalent structure. As a matter of fact, the NEXAFS spectra of graphene on various surfaces appear similar to that obtained for NiTPP network, which highlights the formation of a well‐extended conjugated nanomesh exhibiting dispersive electronic features in full agreement with k‐PEEM findings. [73] This is strongly supported by the chemical changes observed for the C 1s XPS spectra discussed in Chapter 4 of the Supporting Information.
Figure 4.

Near‐edge x‐ray absorption fine structure spectra recorded across the C K‐edge, N K‐edge and Ni L3‐edge for both the pristine NiTBrPP layer on Au(111) and after the formation of the NiTPP‐based network.
Information on the Ni cores stabilized within the porphyrin N‐moiety is of major interest in order to evaluate to what extent their main single‐molecule functionalities are retained when embedded in more stable π‐delocalized covalent nanomeshes. The N 1s and Ni 2p3/2 core‐level photoelectron lines before and after polymerization discussed in Chapter 4 of the Supporting Information indicate no chemical changes. NEXAFS spectra recorded across the N K and Ni L3 thresholds allow us to get a better insight into the details of the electronic structure of the functional porphyrin center provided by the NiTPP network and the single‐molecule layer counterpart. The low energy resonances apparent in the N K‐edge spectra at ≈399 eV shown in Figure 4 result from the mixing of Ni 3d with ligand 2p levels, as emphasized by the chosen index Ni. A σ*‐resonance is associated with the covalent N‐bonding (ligand 2px,y and Ni 3dx2‐y2), while the Ni 3dxz,yz to ligand 2pz backbonding corresponds to the first π*‐resonance.[ 4 , 68 , 69 ] Although these features are similar in energy, the flat macrocycle configuration already discussed enables us to easily recognize them in the NEXAFS spectra acquired with different polarizations. The σ*‐resonance, well‐evident in the spectrum recorded using s‐polarized light, is known to only show up in the case of a weak molecule to substrate interaction. [4] Indeed, no change in the energy position of this σ*‐resonance is observed upon creation of the covalent porphyrin macromolecule, while the π*‐resonance corresponding to the Ni‐to‐ligand‐backbonding is only slightly shifted by ≈0.2 eV towards lower photon energies. This indicates a very similar coordination environment of the Ni cores embedded in the covalent nanomesh with respect to the NiTBrPP precursor. The higher energy features in the N K‐edge spectra that correspond to higher‐lying unoccupied π*‐symmetric pure macrocyclic molecular levels rather turned out to be more affected by the polymerization reaction.[ 68 , 69 ] These resonances display an overall broadening upon creation of the covalent NiTPP network, which suggests an increase of the orbital delocalization (faster decay of the excited state). The formation of a polymeric π‐conjugated nanostructure, thus, involves also the Ni‐coordinating porphyrin N‐moiety without major impacts on the coordination characteristics. This is strongly confirmed by the corresponding Ni L3‐edge spectra presented in Figure 4. Upon NiTPP‐based network formation, the Ni main absorption line is shifted towards higher photon energies, which also reflects the increased Ni‐to‐N‐backbonding. Since no further changes characteristic for the NiII low‐spin state in both the spectral shape and satellite structure are observed, we can assume the Ni core single‐active site character to be preserved.[ 5 , 74 ] The Ni ion cores embedded in the polymeric molecular backbone are barely involved in the extended π‐bonding as desired for a multifunctional device with active centers, whose performance benefits from the larger stability of the polymerized backbone. It should be emphasized that the imperfections in the covalent nanomesh reported here do not lead to any visible broadening of the Ni‐related states indicating a well‐defined functionality that is uniform across the interface. Furthermore, the multifunctional properties of the NiTPP network are not altered when changing the interface to the more reactive Ag(111) substrate. Indeed, the parabola of states exhibited by the molecular backbone is shifted towards higher binding energies due to the more pronounced charge transfer on Ag(111). The spectroscopic fingerprint of π‐extended covalent nanomesh observed by NEXAFS on the Au(111) surface is reproduced on Ag(111) and further indicates that the electronic structure of the Ni cores can be tuned by charge transfer without changing their single‐active site character. This is further elaborated in Chapter 5 of the Supporting Information.
Conclusion
Following the on‐surface chemistry approach, this work addressed the functional properties of a conjugated NiTPP‐based polymer. Covalently linking individual molecular units facilitates the formation of a π‐extended aromatic nanomesh that exhibits dispersive electronic states and, at the same time, is stable up to very high temperatures. Insights gained from a multitechnique approach revealed that the single‐active site functionality of Ni incorporated in the porphyrin coordination pocket is preserved and remains unaltered upon polymerization. The increased structural robustness, consequence of the creation of a conjugated system, introduces a certain rigidity to the molecular network since the molecular units are interconnected. Upon exposing the functional transition metal cores to external stimuli like axial ligands, an interaction sensitive to the transition metal and axial ligand combination can thus be expected when paving the road to advanced applications based on tailored reactivity.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge access to Elettra, the Italian synchrotron facility in Trieste. Open Access funding enabled and organized by Projekt DEAL.
D. Baranowski, I. Cojocariu, A. Sala, C. Africh, G. Comelli, L. Schio, M. Tormen, L. Floreano, V. Feyer, C. M. Schneider, Angew. Chem. Int. Ed. 2022, 61, e202210326; Angew. Chem. 2022, 134, e202210326.
Contributor Information
Daniel Baranowski, Email: d.baranowski@fz-juelich.de.
Dr. Vitaliy Feyer, Email: v.feyer@fz-juelich.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Liu Z., Yasseri A. A., Lindsey J. S., Bocian D. F., Science 2003, 302, 1543–1545. [DOI] [PubMed] [Google Scholar]
- 2. Auwärter W., Écija D., Klappenberger F., Barth J. V., Nat. Chem. 2015, 7, 105–120. [DOI] [PubMed] [Google Scholar]
- 3. Gottfried J. M., Surf. Sci. Rep. 2015, 70, 259–379. [Google Scholar]
- 4. Cojocariu I., Sturmeit H. M., Zamborlini G., Cossaro A., Verdini A., Floreano L., D'Incecco E., Stredansky M., Vesselli E., Jugovac M., Cinchetti M., Feyer V., Schneider C. M., Appl. Surf. Sci. 2020, 504, 144343. [Google Scholar]
- 5. Sturmeit H. M., Cojocariu I., Windischbacher A., Puschnig P., Piamonteze C., Jugovac M., Sala A., Africh C., Comelli G., Cossaro A., Verdini A., Floreano L., Stredansky M., Vesselli E., Hohner C., Kettner M., Libuda J., Schneider C. M., Zamborlini G., Cinchetti M., Feyer V., Small 2021, 17, 2104779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paolesse R., Nardis S., Monti D., Stefanelli M., di Natale C., Chem. Rev. 2017, 117, 2517–2583. [DOI] [PubMed] [Google Scholar]
- 7. Wang A., Li J., Zhang T., Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar]
- 8. Yang H., Shi R., Shang L., Zhang T., Small Struct. 2021, 2, 2100007. [Google Scholar]
- 9. Cong M., Chen X., Xia K., Ding X., Zhang L., Jin Y., Gao Y., Zhang L., J. Mater. Chem. A 2021, 9, 4673–4678. [Google Scholar]
- 10. Lu Z., Yang H., Fu X., Zhao Y., Lin Q., Xiao L., Hou L., Mater. Today Chem. 2022, 25, 100959. [Google Scholar]
- 11. Zhao X., Zhang X., Liang Y., Hu Z., Huang F., Macromolecules 2021, 54, 4902–4909. [Google Scholar]
- 12. Lu M., Liu J., Li Q., Zhang M., Liu M., Wang J., Yuan D., Lan Y., Angew. Chem. Int. Ed. 2019, 58, 12392–12397; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12522–12527. [Google Scholar]
- 13. Wende H., Bernien M., Luo J., Sorg C., Ponpandian N., Kurde J., Miguel J., Piantek M., Xu X., Eckhold P., Kuch W., Baberschke K., Panchmatia P. M., Sanyal B., Oppeneer P. M., Eriksson O., Nat. Mater. 2007, 6, 516–520. [DOI] [PubMed] [Google Scholar]
- 14. Natterer F. D., Yang K., Paul W., Willke P., Choi T., Greber T., Heinrich A. J., Lutz C. P., Nature 2017, 543, 226–228. [DOI] [PubMed] [Google Scholar]
- 15. Khajetoorians A. A., Wiebe J., Chilian B., Wiesendanger R., Science 2011, 332, 1062–1064. [DOI] [PubMed] [Google Scholar]
- 16. Chang M. H., Chang Y. H., Kim N.-Y., Kim H., Lee S.-H., Choi M.-S., Kim Y.-H., Kahng S.-J., Phys. Rev. B 2019, 100, 245406. [Google Scholar]
- 17. Flechtner K., Kretschmann A., Steinrück H.-P., Gottfried J. M., J. Am. Chem. Soc. 2007, 129, 12110–12111. [DOI] [PubMed] [Google Scholar]
- 18. Chang M. H., Kim N.-Y., Chang Y. H., Lee Y., Jeon U. S., Kim H., Kim Y.-H., Kahng S.-J., Nanoscale 2019, 11, 8510–8517. [DOI] [PubMed] [Google Scholar]
- 19. Gottfried M., Marbach H., Z. Phys. Chem. (Muenchen Ger.) 2009, 223, 53–74. [Google Scholar]
- 20. Wäckerlin C., Tarafder K., Girovsky J., Nowakowski J., Hählen T., Shchyrba A., Siewert D., Kleibert A., Nolting F., Oppeneer P. M., Jung T. A., Ballav N., Angew. Chem. Int. Ed. 2013, 52, 4568–4571; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4666–4669. [Google Scholar]
- 21. Wäckerlin C., Chylarecka D., Kleibert A., Müller K., Iacovita C., Nolting F., Jung T. A., Ballav N., Nat. Commun. 2010, 1, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ballav N., Wäckerlin C., Siewert D., Oppeneer P. M., Jung T. A., J. Phys. Chem. Lett. 2013, 4, 2303–2311. [Google Scholar]
- 23. Cojocariu I., Carlotto S., Zamborlini G., Jugovac M., Schio L., Floreano L., Casarin M., Feyer V., Schneider C. M., J. Mater. Chem. C 2021, 9, 12559–12565. [Google Scholar]
- 24. Grill L., Dyer M., Lafferentz L., Persson M., Peters M. V., Hecht S., Nat. Nanotechnol. 2007, 2, 687–691. [DOI] [PubMed] [Google Scholar]
- 25. Côté A. P., Benin A. I., Ockwig N. W., O'Keeffe M., Matzger A. J., Yaghi O. M., Science 2005, 310, 1166–1170. [DOI] [PubMed] [Google Scholar]
- 26. Marianov A. N., Kochubei A. S., Gu S., Jiang Y., ACS Catal. 2022, 12, 8610–8622. [Google Scholar]
- 27. Lin S., Diercks C. S., Zhang Y.-B., Kornienko N., Nichols E. M., Zhao Y., Paris A. R., Kim D., Yang P., Yaghi O. M., Chang C. J., Science 2015, 349, 1208–1213. [DOI] [PubMed] [Google Scholar]
- 28. Tsuda A., Osuka A., Science 2001, 293, 79–82. [DOI] [PubMed] [Google Scholar]
- 29. Nitzan A., Ratner M. A., Science 2003, 300, 1384–1389. [DOI] [PubMed] [Google Scholar]
- 30. Grill L., Hecht S., Nat. Chem. 2020, 12, 115–130. [DOI] [PubMed] [Google Scholar]
- 31. Mallada B., Błoński P., Langer R., Jelínek P., Otyepka M., de la Torre B., ACS Appl. Mater. Interfaces 2021, 13, 32393–32401. [DOI] [PubMed] [Google Scholar]
- 32. Lafferentz L., Eberhardt V., Dri C., Africh C., Comelli G., Esch F., Hecht S., Grill L., Nat. Chem. 2012, 4, 215–220. [DOI] [PubMed] [Google Scholar]
- 33. Krasnikov S. A., Doyle C. M., Sergeeva N. N., Preobrajenski A. B., Vinogradov N. A., Sergeeva Y. N., Zakharov A. A., Senge M. O., Cafolla A. A., Nano Res. 2011, 4, 376–384. [Google Scholar]
- 34. Beggan J. P., Boyle N. M., Pryce M. T., Cafolla A. A., Nanotechnology 2015, 26, 365602. [DOI] [PubMed] [Google Scholar]
- 35. Smykalla L., Shukrynau P., Korb M., Lang H., Hietschold M., Nanoscale 2015, 7, 4234–4241. [DOI] [PubMed] [Google Scholar]
- 36. Mateo L. M., Sun Q., Liu S., Bergkamp J. J., Eimre K., Pignedoli C. A., Ruffieux P., Decurtins S., Bottari G., Fasel R., Torres T., Angew. Chem. Int. Ed. 2020, 59, 1334–1339; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1350–1355. [Google Scholar]
- 37. Mateo L. M., Sun Q., Eimre K., Pignedoli C. A., Torres T., Fasel R., Bottari G., Chem. Sci. 2021, 12, 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin T., Shang X. S., Adisoejoso J., Liu P. N., Lin N., J. Am. Chem. Soc. 2013, 135, 3576–3582. [DOI] [PubMed] [Google Scholar]
- 39. Saywell A., Browning A. S., Rahe P., Anderson H. L., Beton P. H., Chem. Commun. 2016, 52, 10342–10345. [DOI] [PubMed] [Google Scholar]
- 40. Bischoff F., He Y., Riss A., Seufert K., Auwärter W., Barth J. V., Angew. Chem. Int. Ed. 2018, 57, 16030–16035; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16262–16267. [Google Scholar]
- 41. Floris A., Haq S., In't Veld M., Amabilino D. B., Raval R., Kantorovich L., J. Am. Chem. Soc. 2016, 138, 5837–5847. [DOI] [PubMed] [Google Scholar]
- 42. In't Veld M., Iavicoli P., Haq S., Amabilino D. B., Raval R., Chem. Commun. 2008, 13, 1536. [DOI] [PubMed] [Google Scholar]
- 43. Wiengarten A., Seufert K., Auwärter W., Ecija D., Diller K., Allegretti F., Bischoff F., Fischer S., Duncan D. A., Papageorgiou A. C., Klappenberger F., Acres R. G., Ngo T. H., Barth J. V., J. Am. Chem. Soc. 2014, 136, 9346–9354. [DOI] [PubMed] [Google Scholar]
- 44. Seufert K., McBride F., Jaekel S., Wit B., Haq S., Steiner A., Poli P., Persson M., Raval R., Grill L., J. Phys. Chem. C 2019, 123, 16690–16698. [Google Scholar]
- 45. Haq S., Hanke F., Sharp J., Persson M., Amabilino D. B., Raval R., ACS Nano 2014, 8, 8856–8870. [DOI] [PubMed] [Google Scholar]
- 46. Xiang F., Lu Y., Wang Z., Ju H., di Filippo G., Li C., Liu X., Leng X., Zhu J., Wang L., Schneider M. A., J. Phys. Chem. C 2019, 123, 23007–23013. [Google Scholar]
- 47. Sun Q., Mateo L. M., Robles R., Lorente N., Ruffieux P., Bottari G., Torres T., Fasel R., Angew. Chem. Int. Ed. 2021, 60, 16208–16214; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 16344–16350. [Google Scholar]
- 48. Cirera B., de la Torre B., Moreno D., Ondráček M., Zbořil R., Miranda R., Jelínek P., Écija D., Chem. Mater. 2019, 31, 3248–3256. [Google Scholar]
- 49. Cao N., Riss A., Corral-Rascon E., Meindl A., Auwärter W., Senge M. O., Ebrahimi M., Barth J. V., Nanoscale 2021, 13, 19884–19889. [DOI] [PubMed] [Google Scholar]
- 50. Joshi T., Chen C., Li H., Diercks C. S., Wang G., Waller P. J., Li H., Bredas J. L., Yaghi O. M., Crommie M. F., Adv. Mater. 2019, 31, 1805941. [DOI] [PubMed] [Google Scholar]
- 51. Puschnig P., Berkebile S., Fleming A. J., Koller G., Emtsev K., Seyller T., Riley J. D., Ambrosch-Draxl C., Netzer F. P., Ramsey M. G., Science 2009, 326, 702–706. [DOI] [PubMed] [Google Scholar]
- 52. Zamborlini G., Lüftner D., Feng Z., Kollmann B., Puschnig P., Dri C., Panighel M., di Santo G., Goldoni A., Comelli G., Jugovac M., Feyer V., Schneider C. M., Nat. Commun. 2017, 8, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fratesi G., Achilli S., Ugolotti A., Lodesani A., Picone A., Brambilla A., Floreano L., Calloni A., Bussetti G., Appl. Surf. Sci. 2020, 530, 147085. [Google Scholar]
- 54. Buchner F., Kellner I., Hieringer W., Görling A., Steinrück H.-P., Marbach H., Phys. Chem. Chem. Phys. 2010, 12, 13082. [DOI] [PubMed] [Google Scholar]
- 55. Yin C., Peng Z., Liu D., Song H., Zhu H., Chen Q., Wu K., Molecules 2020, 25, 3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu J., Da B., Xiong W., Du R., Hao Z., Ruan Z., Zhang Y., Sun S., Gao L., Cai J., Phys. Chem. Chem. Phys. 2021, 23, 11784–11788. [DOI] [PubMed] [Google Scholar]
- 57. Emtsev K. V., Speck F., Seyller T., Ley L., Riley J. D., Phys. Rev. B 2008, 77, 155303. [Google Scholar]
- 58. Sala A., Zamborlini G., Menteş T. O., Locatelli A., Small 2015, 11, 5927–5931. [DOI] [PubMed] [Google Scholar]
- 59. Galeotti G., de Marchi F., Hamzehpoor E., MacLean O., Rajeswara Rao M., Chen Y., Besteiro L. V., Dettmann D., Ferrari L., Frezza F., Sheverdyaeva P. M., Liu R., Kundu A. K., Moras P., Ebrahimi M., Gallagher M. C., Rosei F., Perepichka D. F., Contini G., Nat. Mater. 2020, 19, 874–880. [DOI] [PubMed] [Google Scholar]
- 60. Stredansky M., Sala A., Fontanot T., Costantini R., Africh C., Comelli G., Floreano L., Morgante A., Cossaro A., Chem. Commun. 2018, 54, 3971–3973. [DOI] [PubMed] [Google Scholar]
- 61. Vasseur G., Fagot-Revurat Y., Sicot M., Kierren B., Moreau L., Malterre D., Cardenas L., Galeotti G., Lipton-Duffin J., Rosei F., di Giovannantonio M., Contini G., le Fèvre P., Bertran F., Liang L., Meunier V., Perepichka D. F., Nat. Commun. 2016, 7, 10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. García-Fernández C., Sierda E., Abadía M., Bugenhagen B., Prosenc M. H., Wiesendanger R., Bazarnik M., Ortega J. E., Brede J., Matito E., Arnau A., J. Phys. Chem. C 2017, 121, 27118–27125. [Google Scholar]
- 63. Klappenberger F., Hellwig R., Du P., Paintner T., Uphoff M., Zhang L., Lin T., Moghanaki B. A., Paszkiewicz M., Vobornik I., Fujii J., Fuhr O., Zhang Y.-Q., Allegretti F., Ruben M., Barth J. V., Small 2018, 14, 1704321. [DOI] [PubMed] [Google Scholar]
- 64. Basagni A., Vasseur G., Pignedoli C. A., Vilas-Varela M., Peña D., Nicolas L., Vitali L., Lobo-Checa J., de Oteyza D. G., Sedona F., Casarin M., Ortega J. E., Sambi M., ACS Nano 2016, 10, 2644–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vasseur G., Abadia M., Miccio L. A., Brede J., Garcia-Lekue A., de Oteyza D. G., Rogero C., Lobo-Checa J., Ortega J. E., J. Am. Chem. Soc. 2016, 138, 5685–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stöhr J., Outka D. A., Phys. Rev. B 1987, 36, 7891–7905. [DOI] [PubMed] [Google Scholar]
- 67. Cudia C. C., Vilmercati P., Larciprete R., Cepek C., Zampieri G., Sangaletti L., Pagliara S., Verdini A., Cossaro A., Floreano L., Morgante A., Petaccia L., Lizzit S., Battocchio C., Polzonetti G., Goldoni A., Surf. Sci. 2006, 600, 4013–4017. [Google Scholar]
- 68. Krasnikov S. A., Preobrajenski A. B., Sergeeva N. N., Brzhezinskaya M. M., Nesterov M. A., Cafolla A. A., Senge M. O., Vinogradov A. S., Chem. Phys. 2007, 332, 318–324. [Google Scholar]
- 69. Krasnikov S. A., Sergeeva N. N., Brzhezinskaya M. M., Preobrajenski A. B., Sergeeva Y. N., Vinogradov N. A., Cafolla A. A., Senge M. O., Vinogradov A. S., J. Phys. Condens. Matter 2008, 20, 235207. [DOI] [PubMed] [Google Scholar]
- 70. Chen M., Feng X., Zhang L., Ju H., Xu Q., Zhu J., Gottfried J. M., Ibrahim K., Qian H., Wang J., J. Phys. Chem. C 2010, 114, 9908–9916. [Google Scholar]
- 71. Schmidt N., Fink R., Hieringer W., J. Chem. Phys. 2010, 133, 054703. [DOI] [PubMed] [Google Scholar]
- 72. Di Santo G., Blankenburg S., Castellarin-Cudia C., Fanetti M., Borghetti P., Sangaletti L., Floreano L., Verdini A., Magnano E., Bondino F., Pignedoli C. A., Nguyen M.-T., Gaspari R., Passerone D., Goldoni A., Chem. Eur. J. 2011, 17, 14354–14359. [DOI] [PubMed] [Google Scholar]
- 73. Voloshina E. N., Generalov A., Weser M., Böttcher S., Horn K., Dedkov Y. S., New J. Phys. 2011, 13, 113028. [Google Scholar]
- 74. Wang H., Butorin S. M., Young A. T., Guo J., J. Phys. Chem. C 2013, 117, 24767–24772. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.