

Abstract
Premise
Understanding evolutionary history and classifying discrete units of organisms remain overwhelming tasks, and lags in this workload concomitantly impede an accurate documentation of biodiversity and conservation management. Rapid advances and improved accessibility of sensitive high‐throughput sequencing tools are fortunately quickening the resolution of morphological complexes and thereby improving the estimation of species diversity. The recently described and critically endangered Banksia vincentia is morphologically similar to the hairpin banksia complex (B. spinulosa s.l.), a group of eastern Australian flowering shrubs whose continuum of morphological diversity has been responsible for taxonomic controversy and possibly questionable conservation initiatives.
Methods
To assist conservation while testing the current taxonomy of this group, we used high‐throughput sequencing to infer a population‐scale evolutionary scenario for a sample set that is comprehensive in its representation of morphological diversity and a 2500‐km distribution.
Results
Banksia spinulosa s.l. represents two clades, each with an internal genetic structure shaped through historical separation by biogeographic barriers. This structure conflicts with the existing taxonomy for the group. Corroboration between phylogeny and population statistics aligns with the hypothesis that B. collina, B. neoanglica, and B. vincentia should not be classified as species.
Conclusions
The pattern here supports how morphological diversity can be indicative of a locally expressed suite of traits rather than relationship. Oversplitting in the hairpin banksias is atypical since genomic analyses often reveal that species diversity is underestimated. However, we show that erring on overestimation can yield negative consequences, such as the disproportionate prioritization of a geographically anomalous population.
Keywords: Banksia, cpDNA, DArTseq, genome‐wide analysis, last glacial maximum, Proteaceae, single nucleotide polymorphism, species boundaries, taxonomy, threatened species
Populations isolated through geographic separation are subject to genetic drift and natural selection and can eventually be rendered reproductively incompatible (i.e., allopatric speciation: Dobzhansky, 1951; Mayr, 1963). This change is gradual, and if a barrier between populations is removed, or only sporadically imposed (e.g., through periodic glaciation) before the tipping point of reproductive isolation is achieved, then gene flow will continue to restrict progress toward lineage divergence and diversification (e.g., Maddison and Knowles, 2006; Mallet et al., 2016). Somewhere throughout this trajectory, however, populations might acquire a dissimilar phenotype, perhaps through drift, environmentally activated plasticity, or localized natural selection, thereby providing a gradient of morphological variability across a given geographic range (Stuglik and Babik, 2016; Hundsdoerfer et al., 2018; Reyes‐Velasco et al., 2018; Georges et al., 2019). In such a situation, distinguishing between what are morphologically variable populations of a species and what are irrevocably separate lineages can be problematic and is the crux of a persistent challenge in systematics and taxonomy (Martin et al., 2013; Dumas et al., 2015; Mallet et al., 2016).
With respect to the unified species concept (de Queiroz, 2005, 2007), multiple lines of evidence are best considered when postulating the boundary between species or between separately evolving metapopulations (Zachos, 2018). Analysis of morphological characters presented in the process described above might support the hypothesis that some populations are distinct species according to a phenetic species concept. However, expanding our examination to consider other lines of evidence might introduce conflict in this conclusion. For instance, phylogenetic relationship or reproductive compatibility would support the hypothesis that these populations are the same species according to a monophyletic or a biological species concept, respectively. In the end, what really matters is whether these are an accurate demonstration of distinct lineages that have embarked on independent evolutionary trajectories, and we would expect them to be less open to erroneous interpretation given a greater amount of evidence. Phenotypic plasticity (Prada et al., 2008), drift and isolation by distance (e.g., Seeholzer, Brumfield, 2018; Sjölund et al., 2019; Vakkari et al., 2020), introgression by neighboring species (Mallet et al., 2016) are processes, to name a few, that will introduce complexity into observed patterns and increase the likelihood of a confounded interpretation.
Reduced representational sampling of the genome, with single nucleotide polymorphisms (SNPs) sampled by techniques such as DArTSeq, ddRAD, and RADSeq, is the next wave revolutionizing our capacity to detect genetic distinctiveness. The techniques can recover a high quantity of highly informative loci over whole genomes (rather than selected regions), and by revealing variation at the individual level, they are proving to be more sensitive than earlier techniques of SNP capture such as RAPD and microsatellites (Narum et al., 2013; Soltis et al., 2013; Fernández‐Mazuecos et al., 2017; Rossetto et al., 2019). They have so far been used for the robust detection of population‐scale admixture, lineage recombination, and other mechanisms responsible for lineage diversification (Eaton et al., 2015; Zhang et al., 2016; Pentinsaari et al., 2017; Edet et al., 2018; Georges et al., 2018; Ortego et al., 2018; Rutherford et al., 2018; Clugston et al., 2019; Hundsdoerfer et al., 2019). They also permit phylogenetic analysis, which has revealed both underestimation and overestimation of specific diversity in previous taxonomic investigations (Nieto‐Montes de Oca et al., 2017; Bryson et al., 2018; Reyes‐Velasco et al., 2018; Rutherford et al., 2018, 2019, 2021; Georges et al., 2018; Hundsdoerfer et al., 2019; Hope and Frey, 2022). The rapid reduction in cost and time of their production has facilitated their pairing with chloroplast and mitochondrial sequence alignments, enabling more robust evaluations of evolutionary rates and mechanisms of inheritance (e.g., Georges et al., 2018). These advances have made genetic analyses more appealing to conservationists eager to maintain robust populations of nonmodel organisms (Baird et al., 2008; Davey and Blaxter, 2011; Devitt et al., 2019; Rossetto et al., 2021; Rutherford et al., 2018, 2019, 2021) because when the path toward extinction is imminent, having a rigorously tested, well‐corroborated phylogenetic model can assist with prioritization.
The critically endangered, iconic species Banksia vincentia Stimpson & P.H. Weston (Proteaceae: Auld and Weston, 2020) consisted of a single population of 14 individuals when it was described (Stimpson et al., 2014). It belongs to the morphologically complex group known as the hairpin banksias (B. spinulosa s.l.), which spans a latitudinal distribution of nearly 3000 km between the southern coast of mainland Australia to the base of the Cape York Peninsula (Figures 1, 2). Following the description of B. spinulosa Sm. (Smith 1793), B. collina R.Br. (Brown 1810) and B. cunninghamii Sieber ex Rchb. (Reichenbach 1827) were described but later synonymized under the former species as varieties (George, 1981, 1988, 1999; Harden, 2002) before B. vincentia and the morphologically similar B. neoanglica (A.S. George) Stimpson & J.J. Bruhl (Stimpson et al., 2012, 2016) were described. However, the condition of monophyly (Mishler and Brandon, 1987; Mishler and Theriot, 2000), where members of a group exclusively share a common ancestor (i.e., a clade; see review by Lee, 2003), has not yet been integrated with an examination of species boundaries for any taxonomic model proposed for the hairpin banksias. And so, while there is consensus across the taxonomic community for the distinctiveness of B. spinulosa Sm., B. neoanglica, and B. vincentia, the status of whether B. cunninghamii Sieber ex Rchb. and B. collina R.Br. are species or varieties of B. spinulosa remains undecided (Australian National Botanical Garden, 2022; Royal Botanic Gardens and Domain Trust, Sydney, 2002 onward) despite evidence supporting the former (Stimpson et al., 2016).
Figure 1.

Morphological diversity of the hairpin banksia species complex. (A) B. vincentia from Vincentia, NSW. (B) B. cunninghamii from Blackheath, NSW. (C) B. neoanglica from Girraween National Park, Qld. (D) Banksia spinulosa from Glenbrook National Park, NSW. (E) B. collina from Kungala, NSW. (F) B. spinulosa from Isla Gorge National Park, NSW. Images not to scale. Image credits: A, K. Coutts‐McClelland; B, J. Allen; C and D, T.C. Wilson; E and F, P. H. Weston. Scale = 25 mm.
Figure 2.
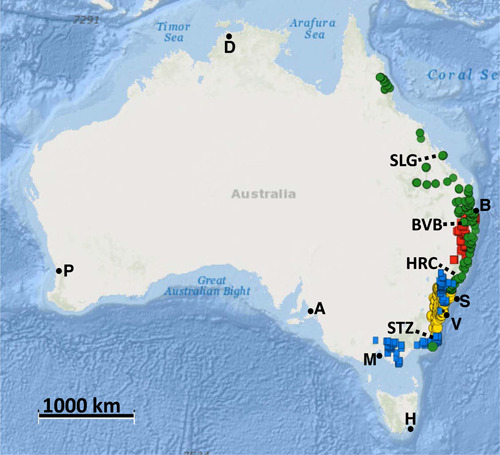
Hairpin banksia complex distribution based on specimen records acquired from the Atlas of Living Australia website (http://www.ala.org.au [accessed 25 April 2020]). Five species are distinguished according to Stimpson et al. (2016): B. collina (green circles), B. cunninghamii (blue squares), B. spinulosa (yellow circles), B. neoanglica (red squares), B. vincentia (V). Dotted lines indicate approximate area of major biogeographic barrier sensu Bryant and Krosch (2016): Brisbane Valley Barrier (BVB), Hunter River Corridor (HRC), Saint Lawrence Gap (SLG), Southern Transition Zone (STZ). State capitals are Adelaide (A), Brisbane (B), Darwin (D), Hobart (H), Melbourne (M), Perth (P), Sydney (S).
The risk of extinction for B. vincentia is extreme since it is found within less than 100 m2 of coastal heathland and is situated next to expanding urban development (Office of Environment and Heritage, 2019). Although an intensive conservation program is currently underway, lack of knowledge of the evolutionary history of the hairpin banksias compromises several aspects of this effort and the future conservation of the entire group. First of all, the relative value to biodiversity of B. vincentia relative to other hairpin banksias is unclear, and thus the efficacy of its conservation in a global context is unknown. Second, without an understanding of species boundaries, genetic purity of B. vincentia cannot be assessed, which is essential for mitigating cryptic hybridization (e.g., Rutherford et al., 2019). It is assumed that there are firm prezygotic factors in the hairpin banksias due to a lack of observed hybrids between sympatric species, such as at the Vincentia site where B. spinulosa grows near B. vincentia (Stimpson et al., 2016). However, this process might not be visible at the Vincentia site since in some research on Banksia it has only been detected using molecular techniques (Carthew et al., 1988; Henderson, 1991; Maguire et al., 1997; Myerscough et al., 2001; Usher et al., 2010). Finally, despite there being a large ex situ collection from seed and cuttings already established, without an understanding of genetic diversity, the maximum potential of this asset remains unharnessed with regards to the maintenance or improvement of species fitness (Ralls et al., 2020), or building and positioning genetically healthy reserve populations.
In this study, we show that an underlying knowledge of evolutionary history is a fundamental aid for appropriate conservation planning as much as it is for the institution of a robust, unequivocal taxonomy (Rossetto et al., 2021). We sought to understand the historic and contemporary evolutionary drivers of speciation and geneflow, rigorously test previous taxonomic conclusions made by phenetic analysis, and detect threatening processes operating at the molecular level (e.g., genetic swamping) and environmental level (e.g., range shift according to climate change) by evaluating results of genome‐wide analysis and modelling past and future climatic conditions. Specifically, we asked what is the evolutionary history of the hairpin banksias and how has it been influenced by landscape and environment? If B. vincentia is a species, what relative value of biodiversity does it represent? This is one of the largest population‐scale examinations we are aware of to date, and the resulting demonstration of a geographically partitioned continuum of populations that displays a geographically localized morphology shows how activities that conserve our global biodiversity heritage can be misinformed.
MATERIALS AND METHODS
Sampling strategy
To provide a comprehensive representation of the variation in morphology and distribution of the hairpin banksias (Banksia spinulosa s.l.) at the population scale, we used the sample set previously collected and analyzed by Stimpson et al. (2016) and augmented the set by additional field collections and herbarium specimens to obtain a total of 597 samples (Appendix S1). For the present study to be relatable to previous research, we continue to treat the hairpin banksias as five species: B. collina, B. cunninghamii, B. neoanglica, B. spinulosa, and B. vincentia (other putative entities are treated as members of B. collina). In selected analyses, we included outgroups of the closest species (B. ericifolia s.l.) and more distantly related species (B. paludosa) to the hairpin banksias (Cardillo and Pratt, 2013).
All extant individuals of B. vincentia were sampled, whereas six plant replicates were sampled where possible for each of 102 other hairpin banksia populations (498 samples in total). Outgroup specimens totalled 41 plants, and an additional 58 ex situ samples of B. vincentia were sourced from translocated plants and the nursery. Among the ex situ plants, 31 were propagated from cuttings (Australian National Botanic Gardens, Booderee Botanic Gardens, Wollongong Botanic Gardens) and another 27 were germinated from seed and grown at the Australian Botanic Gardens, Mt Annan, Australia.
DArT extraction and sequencing and quality screening and control of SNPs
Diversity array technology sequencing (DArTseq) analysis is a genotype‐by‐sequencing approach whereby genomic complexity reduction methods are applied to efficiently select low‐copy fractions of genomes and is capable of providing thousands of informative loci or single nucleotide polymorphisms (SNPs). These data were used to infer an evolutionary history and estimate admixture at the individual and population levels for the purposes of assessing genetic diversity, introgression, and hybridization.
DNA was extracted from each sample using the Plant DNA Extraction Protocol for DArT available from the Diversity Arrays Technology Pty Ltd (DArT PL) website (Rossetto et al. 2019). Samples were sent to DArT PL (Canberra, Australia) for the DArT PL genotype by sequencing analysis, according to the documented in‐house procedure. All specimens were co‐analyzed first to create a total data set. Two additional data sets were created by separate reanalysis of the raw data for each subclade identified by the SplitsTree network analysis described below.
For each of the three data sets, the SNP output was checked for quality and samples using the filtering scripts implemented by the in‐house‐designed package RRtools v. 1.0 (Rossetto et al., 2019) in the R environment (R Core Team, 2017). Loci that did not pass standardized quality thresholds were removed from the data and not used in downstream analysis. To ensure that only higher quality DArTseq markers were used for analyses, all SNPs with a reproducibility (proportion of replicate assay pairs for which the marker score is consistent) of less than 96 % and which had more than 30% missing data were excluded from the data set.
Network analysis
In addition to efficiently making a preliminary analysis of major groups across the entire data set of hairpin banksias, we wanted to test for evidence of genetic introgression at several sites where species occur in sympatry, including Vincentia (between B. vincentia and B. spinulosa) and Lamington National Park (between B. neoanglica and B. collina), Australia. A network was therefore constructed for the total filtered SNP matrix using default settings in the SplitsTree program ver. 4.14.6 (Huson and Bryant, 2006). In addition to estimating relationship by proxy of genetic similarity, this analysis was used to identify areas where there might be incomplete lineage sorting and hybridization through a depiction of extensive “cycling” or “webbing” between branches.
Phylogenetic analysis
Coalescent‐based phylogenetic trees were constructed to test species hypotheses and infer evolutionary relationships. Since species trees might not equal diversification of lineages for Banksia, we chose to conduct a coalescent analysis of our sample set with SVDquartets package ver. 1 (Chifman and Kubatko, 2014) implemented in PAUP* version 4.0a (Swofford, 2002). This program is designed to accept SNP data and produces relatively robust phylogenetic results (see Chou et al., 2015 for a critical review of this program). All analyses used at least three runs with the multispecies coalescent tree model selected, evaluating 100,000 quartets and 1000 bootstrap replicates.
Genetic structure
We used the R package adegenet 2.1.1 (version 3.3.0, Jombart, 2008; Jombart and Ahmed, 2011) to perform a principal component analysis (PCA) so that phenetic relationships between individuals and populations collected in situ could be visualized and better compared with ordinations of morphological data produced by Stimpson et al. (2016). This method of PCA derives an ordination based on Euclidean transformed dissimilarity matrix of the data.
F‐statistics were estimated with the SNPrelate package (Zheng et al., 2012) for the same sample set used in the phylogenetic analysis. Isolation by distance was calculated using F ST (i.e., F ST/1 − F ST) and a matrix of log‐transformed geographical distances between populations. Significance of correlation was tested with a Mantel test (10,000 permutations) using vegan version 2.5‐7 (Mantel, 1967; Oksanen, 2011). Comparative, population‐level measures of expected (H E) and observed heterozygosity (H O) and inbreeding coefficient (F IS) were calculated using the diveRsity package (Keenan et al., 2013). Population structure was analyzed using LEA version 2.4.0 (Frichot and François, 2015) to estimate ancestry coefficients from large genotypic matrices and evaluate the number of ancestral populations. LEA implements the snmf function (sparse non‐negative matrix factorization algorithms) and estimates individual admixture coefficients from the genotype matrix. A measure of fit (i.e., the entropy criterion) is evaluated between the statistical model and data, which is used to choose the best number of ancestral populations that explain the data. We examined the minimum cross entropy for a range of ancestral populations (K = 1–11), selecting the optimal number of ancestral populations based on the post‐stabilization of the steepest decline in cross‐entropy values.
TreeMix v. 1.13 (Pickrell and Pritchard, 2012) was used to test for historical admixture between populations of hairpin banksias and B. ericifolia at the Vincentia site where B. ericifolia, B. spinulosa, and B. vincentia are sympatric and at the closely situated Lamington National Park populations of B. collina and B. neoanglica. We analyzed a reduced data set of 23 populations (Appendix S1) representing the basic phylogenetic structure to make runtimes achievable and removed all individuals that the network indicated as likely recent representatives of admixture. TreeMix was run 10 times for each value of m (migration events from 1 to 10) with the settings ‐noss and ‐k 500, and the resulting optimal i value was estimated through the optM R package (Fitak, 2021). A further 100 runs were then produced for the optimal number of migrations using the initial TreeMix settings and were used to compile a consensus tree with bootstrap values using PHYLIP version 3.695 (https://evolution.genetics.washington.edu/phylip.html) and the BITE R package (Milanesi et al., 2017).
Network analysis of chloroplast genome
A separate network analysis of the whole chloroplast genome for 91 specimens representing 57 populations of the nuclear genomic data set was included to test if both genomes evolved independently. Leaf material was sent to the Deakin Genomics Research and Discovery Facility at Deakin University (Geelong, Australia) for DNA extraction, preparation of genomic libraries consisting of paired‐end reads (2 × 150 bp) and paired‐end sequencing using the Illumina NextSeq 500 platform (Illumina, San Diego, CA, USA).
Consistent in silico assembly of chloroplast genomic DNA SNP detection across all Illumina NextSeq libraries were performed to generate a comparable data set from paired‐end libraries according to methods by Yap et al. (2022). Organelle Assembler (http://metabarcoding.org/org.asm; Coissac et al., 2016) was used to assemble library‐specific chloroplast genome sequences de novo from the NGS libraries and then a consensus sequence was created from each of the relevant paired‐end libraries using CLC Bio Genomics Workbench 8.0 (CLC; http://www.clcbio.com). This process involved trimming the raw paired‐end reads, mapping them to library‐specific chloroplast genomic sequence using default settings and then remapping the same quality trimmed reads with more stringent parameters (similarity = 0.8, length fraction = 0.9) to maintain a high‐quality library‐specific consensus sequence with read coverage greater than 5×. GeSeq (Tillich et al., 2017) was used to provide annotations for each sequence that was afterward checked for proper start and stop codon placement using Geneious Pro 2022.9.1.8 (https://www.geneious.com).
To study the distribution of chlorotypes (chloroplast‐derived genotypes), we generated an alignment of relevant library‐specific consensus sequences using MAUVE (Darling et al., 2004) in Geneious Pro. The alignment was checked by removing areas of low coverage and where dubious SNPs exist (i.e., SNPs in the first or second codons and repetitive regions). The resulting chloroplast genome alignment was subdivided into smaller taxonomic groups for median‐joining network analysis with PopArt (Leigh and Bryant, 2015) using default settings.
Niche modelling and overlap
Environmental niche models (ENMs) fit an overall environmental envelope for a select group of populations and permit an examination of spatial distribution of environmental suitability. Originally purposed to provide candidate localities for reserve populations of B. vincentia, we modified ENMs to observe relative environmental niche overlap to test the ecological concept of species and test the hypothesis for dispersal/vicariance of the B. vincentia population since the Last Glacial Maximum (LGM = 22,000 years before present). Our ENM analysis included a comparison of suitability between the present and LGM, using a range of environmental characters pertinent to plant growth (see Appendices S2 and S3 for explanation). We also examined the level of the climatic envelope predicted at the Vincentia site for the group of populations most closely related to B. vincentia (as corroborated by snmf and phylogenetic analysis). An environmental PCA was also generated to examine key differences in environmental characters between groups. MaxEnt niche models (Phillips and Dudik, 2008; Elith et al., 2011) were fitted using the R package maxnet (https://cran.r-project.org/package=maxnet) with a data set of geographic coordinates from our sequenced specimens and records from the Atlas of Living Australia website (http://www.ala.org.au; accessed 25 April 2020). Records were removed if they were earlier than 1990, had spurious locations or unspecific geocoordinates, and lacked herbarium vouchers. After filtering, the B. cunninghamii clade included 459 occurrence records, and the B. spinulosa clade included 423 occurrence records (see Appendices S4 and S5 for relevant details).
RESULTS
Structure and relationships according to nDNA data
The number of SNPs for the raw total data set was 3813. After filtering, 1384 SNPs remained for 642 samples. Network analysis recovered the hairpin banksias as two main clusters (Figure 3), one consisting of all samples of B. cunninghamii, B. neoanglica, and B. vincentia (herein referred to as the B. cunninghamii clade), and the second with B. collina and B. spinulosa s.s. (herein referred to as the B. spinulosa clade). Other clusters and isolated taxa were associated with large cycling and were distantly isolated from their respective populations and hence suspected of recent genetic admixture between the two main clades and outgroup (see admixture section below). Following removal of putative hybrids from the data set, a maximum likelihood tree inferred moderate support for these two separate clades (Figure 4). After excluding outgroup Banksia and ex situ material from the sample set, separate reanalyses of the B. cunninghamii and B. spinulosa clades retrieved 8519 and 8096 SNPs for the raw data set, respectively. Postfiltering, the data were reduced, respectively, to 2404 SNPs for 193 samples and 800 SNPs for 385 samples.
Figure 3.
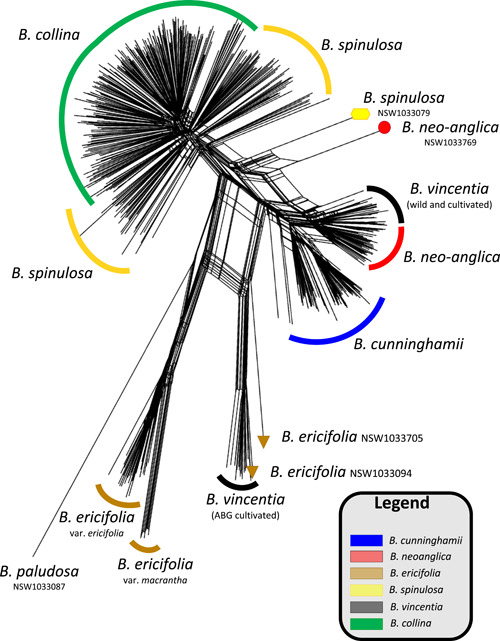
SplitsTree network analysis of 1384 SNP loci for 597 specimens sampled from across the Banksia spinulosa complex using B. ericifolia and B. paludosa as outgroups. The network clusters members of the hairpin banksia complex into two distinct groups/clades: B. cunninghamii clade (B. cunninghamii, B. neoanglica and B. vincentia) and B. spinulosa clade (B. collina and B. spinulosa). The network indicates (1) relative amounts of reticulation through the width of the branches between clusters of samples: extensive reticulation is evident between B. cunninghamii, B. neoanglica, and B. vincentia subclusters, and (2) wild‐sourced specimens of B. neoanglica, B. spinulosa, and B. ericifolia and cultivated seedlings of B. vincentia (Australian Botanic Gardens, ABG) are placed between major species clusters, which is indicative of a hybrid genome
Figure 4.
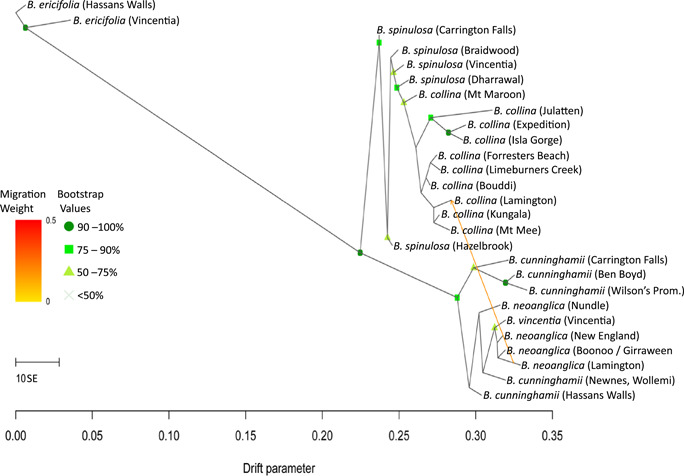
TreeMix maximum likelihood tree of the relationships among representative populations of the hairpin banksia complex indicating one migration event and its direction between sympatric species at the Lamington site. Migration events are modelled for populations that do not fit well into the bifurcating tree model, because they have ancestry from multiple parental populations, and they are indicated as edges. The number of migration events presented is based on an examination of the progressive improvement in fit (Appendix S18). The color of the edges reflects the relative weight of migration, i.e., the fraction of alleles in the descendant population that originated in each parental population (m = 0 → 1, yellow: small fraction of alleles, red: large fraction). Bootstrap support values below 50% are not reported. Refer to Appendix S1 for detailed locality data.
The B. cunninghamii clade consisted of two subclusters in the network, the first consisting of all B. cunninghamii populations and the second consisting of B. vincentia and B. neoanglica (Figure 3). The PCA ordination demonstrated this separation but also articulated B. cunninghamii as two clusters, with populations south of the Towamba River (southern New South Wales) as the first and populations found farther north as the second cluster (Appendix S6). These three groups were also identifiable in the results of the snmf analysis, and with the exception of B. vincentia, occupied three separate regions across southeastern Australia (Figure 5A; see Appendices S7–S10 for corresponding Q values). Although the southern B. cunninghamii and the B. neoanglica plus B. vincentia clusters were recovered as clades in the phylogeny, the northern B. cunninghamii populations formed a paraphyletic grade that was more closely related to B. neoanglica plus B. vincentia than to the southern B. cunninghamii clade, albeit without bootstrap support (Figure 5C). Banksia vincentia was deeply nested within a strongly supported B. neoanglica clade, sharing the closest relationship to the Girraween NP and Boonoo Boonoo populations, and similarly, was shown to have the closest genetic similarity with the most northern populations of B. neoanglica by snmf and PCA (Figure 5; Appendices S6, S7). A low (20%) incomplete quartets value indicated a low incidence of incomplete lineage sorting. Calculations of genetic diversity between populations generally retrieved low F ST values (<0.5), with the highest values (0.5–0.73) being produced between the populations of the southern B. cunninghamii clade and those of B. vincentia and B. neoanglica (Appendix S11). A curve representing isolation by distance (IBD) was produced when transformed F ST values were graphed with log‐transformed distance (Appendix S12a), and levels of heterozygosity (FIS) were generally low (63% below 0.2), including for the B. vincentia population (Appendix S13).
Figure 5.
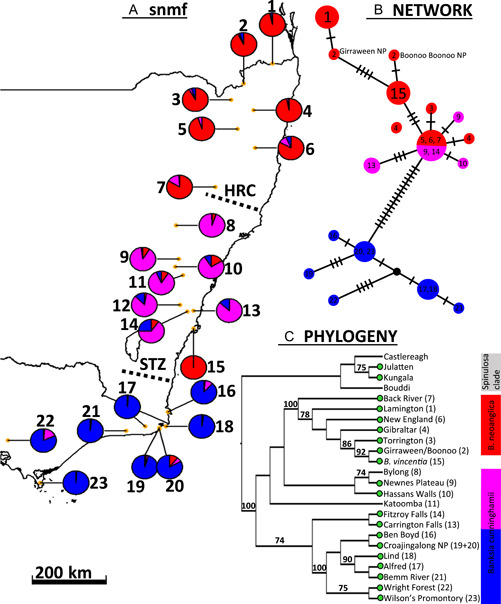
Sampling and genomic analysis of the Banksia cunninghamii clade for B. cunninghamii (8–14, 16–23), B. neoanglica (1–7), and B. vincentia (15) across southeastern Australia. (A) Pie charts represent averaged snmf Q values according to three ancestral populations (K = 3) for each population. (B) Network analysis of the chloroplast genome where each hash on a branch represents a mutational change and color corresponds to results from snmf analysis. (C) SVDQ quartets coalescent tree, with total incompatible quartets = 20475 (20.475%), branch tips representing a population of up to six individuals, bootstrap support values >70% placed above branches and green circles indicating inclusion in the chloroplast genomic dataset; Mount Mitchel (5) and Medway (12) were not included; proximally located specimens were amalgamated as one population in two places: Secret River (19) and Shipwreck Creek (20) as “Croajingalong NP”, and Girraween NP (2) and Boonoo Boonoo NP (2) as “Girraween/Boonoo”. Refer to Appendices S7–S10 for cross entropy vs. number of ancestral populations and corresponding averaged Q values, Appendix S1 for detailed locality and sampling information for all analyses, and Bryant and Kosch (2016) for description of the Hunter Valley Corridor (HRC) and Southern Transition Zone (STZ) biogeographical barriers.
The network of the B. spinulosa cluster did not recover B. spinulosa and B. collina samples as separate subclusters (Figure 3). The corresponding B. spinulosa OTUs in the PCA ordination were more or less situated at one end of a large cluster of B. collina found south of Tewantin Queensland (Appendix S14). Remaining populations farther north from Tewantin Queensland clustered in an adjacently placed and widely spaced cluster. Similar to PCA, snmf graphs demonstrated that these northern and northwestern populations provided the most distinct genetic signature; however, a large portion of this genetic signature was found in neighboring populations farther south, nearly resembling the gradual proportional shift of other genetic signatures throughout the southern populations (Figure 6A; Appendix S15). Banksia spinulosa and B. collina were not recovered as clades, and many populations of B. spinulosa were recovered as separate early‐divergent branches of the phylogeny (Figure 6C) and network (Figure 3). Similar to the PCA, phylogenetic analysis generally organized populations according to latitude, and similar to all results, the most northern and western populations were markedly distinct, each being recovered as a clade with strong branch support (Figure 6C). The same analysis reported high genetic admixture between populations, coinciding with the general trend of low F ST values (Appendix S16), the highest of which were observed between either of the most northerly distributed clades and populations farther south. A distinct curve demonstrating IBD was produced when transformed F ST values were graphed with log‐transformed distance (Appendix S12b). Heterozygosity (F IS) was generally low across the B. spinulosa clade (63% were below 0.2), with the maximum score found at 0.454 for B. collina at Calga (Appendix S13).
Figure 6.
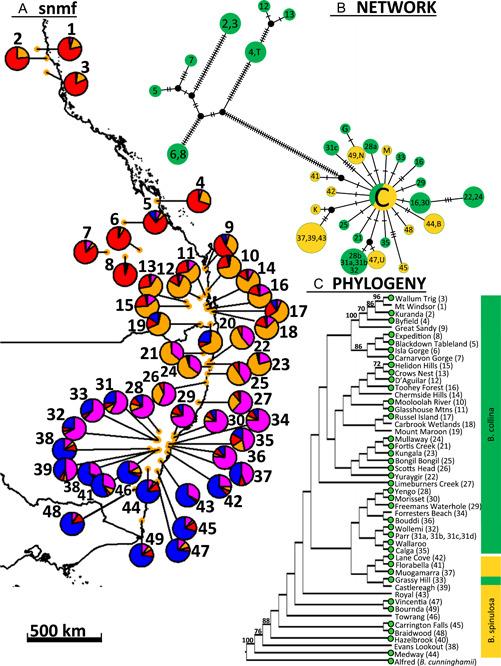
Sampling and nuclear and plastid genomic analysis of the Banksia spinulosa clade for B. collina (1–36) and B. spinulosa (37–49) across eastern Australia. (A) Pie charts represent averaged snmf Q values according to four ancestral populations (K = 4) for each population. (B) Network analysis of the chloroplast genome. Hashes on a branch represent number of mutational changes, and color indicates species (B. collina = green; B. spinulosa = yellow). Populations are organized as two discrete haplogroups based on 59 mutational changes, the smallest haplogroup consisting of seven haplotypes (more northern populations) and the largest consisting of southern populations with a large central haplotype joined to numerous satellite haplotypes. (C) SVDQ quartets coalescent tree, with total incompatible quartets = 42,964 (45.923%), branch tips representing a population of up to six individuals, bootstrap support values >70% placed above branches, and green circles indicating inclusion in the chloroplast genomic data set; Banksia spinulosa is rendered paraphyletic with respect to B. collina. Lamington (20) was not included in phylogeny or chloroplast; Wallaroo and Barren Grounds (B) were included with Limeburners (27) and Carrington Falls (45) for snmf, respectively; Parr (31a = NSW1033917, 31b = NSW1033272, 31c = NSW1033274, 31d = NSW1033541) and Yengo (28a = NSW1033276, 28b = NSW1033944, 28c = NSW1033269) samples were not recovered together in the network; Coondella (N), Mogo (G), Ulladulla (U), Katoomba (K), McMahons (M), Tewantin (T) samples were only included in network analysis; the central chloroplast network haplotype (C) consisted of Wallaroo, Mooloolah River (10), Glasshouse Mountains (11), Helidon Hills (15), Russel Island (17), Kungala (23), Scotts Head (26), Limeburners Creek (27), Parr (31d), Forrester's Beach (34), Bouddi (36), Evan's Lookout (38) and Bournda (49). Refer to Appendices S7–S10 for cross entropy vs. number of ancestral populations and corresponding averaged Q values and Appendix S1 for all locality and sampling information.
Relationships according to chloroplast genome
Chloroplast genomic sequencing yielded 32 paired‐end libraries with an average of 5,398,937 quality reads for the B. cunninghamii clade and 59 paired‐end libraries with an average of 5,903,501 quality reads for the B. spinulosa clade. The final chloroplast sequence length used in the analyses for both data sets was 129,427 bp with 41 polymorphic sites detected within the B. cunninghamii clade and 170 polymorphic sites detected within the B. spinulosa clade (Appendix S17).
Separate analysis of the two largest hairpin banksia clades (B. cunninghamii or B. spinulosa clusters from Figure 3) did not produce discrete haplogroups with respect to the current species hypothesis (Figures 5, 6). Distinctively unlike the nDNA phylogeny, the B. cunninghamii clade was structured as two large haplogroups consisting of populations distributed north and south of the Towamba River of southern New South Wales. As in the nDNA phylogeny, the haplotype of B. vincentia clustered with the most northern haplotypes of B. neoanglica and was most similar to that of the Boonoo Boonoo population (Figure 5B). However, unlike the nDNA phylogeny, B. neoanglica populations were not recovered as a distinct haplogroup since they shared a most recent ancestor with the northern B. cunninghamii populations. Analysis of the B. spinulosa clade also was dissimilar to the nDNA phylogeny since it produced two discrete haplogroups separated by the relatively highest number (59) of mutational changes (Figure 6B). The constituents of one of the haplogroups consisted of all populations north from Tewantin, and although a clade in the phylogeny similarly recovered most of these populations, in addition to having low branch support, it did not include D'Aguilar National Park, Byfield, Crows Nest and Tewantin populations (Figure 6C). The structure of the southern haplogroup was also in conflict with the phylogeny, since few (if any) mutational differences contributed to the substructure between haplotypes within the southern haplogroup, rather than a grade structured according to latitude.
Evidence of contemporary and historical admixture
Network analysis recovered ex situ B. vincentia (Australian Botanic Garden cultivated), B. neoanglica (NSW1033769), B. spinulosa s.l. (NSW1033079) and B. ericifolia (NSW1033705) as positioned away from other samples collected at their respective populations, all suggesting contemporary hybridization (Figure 3). Seed‐grown ex situ plants either demonstrated typical B. vincentia leaf morphology, or leaf morphology similar to B. ericifolia (Appendix S18). Although no quantitative analysis for morphology here is provided, some of the plants identified as ex situ hybrids produced leaves that are intermediate between B. ericifolia and individuals described as B. vincentia (Appendix S18); these do not fit the description of B. vincentia because their leaves are half as long and with fewer teeth along the leaf margin (PlantNet 2022). We also found that some individuals that were initially identified as B. ericifolia and B. spinulosa in the immediate vicinity of B. vincentia also are likely to have hybrid origins (Figure 3).
With SplitsTree‐identified plants of putative hybrid ancestry removed from the network, the snmf results did not reveal introgression from neighboring populations of B. cunninghamii into the B. vincentia population (Figure 5A). The maximum likelihood tree generated by TreeMix (Figure 4) produced a substructure topology within each of the two main clades with few support values much like the svdq phylogenies (Figures 5, 6), likewise recovering the Vincentia population in a clade with the most northern populations of the B. cunninghamii clade. One migration event was the most optimal for describing the demographic history of the hairpin banksia complex populations (Figure 4; Appendix S19). This migration was consistently found to originate and travel from the B. cunninghamii clade to the B. spinulosa clade, specifically between the functionally sympatric populations at Lamington National Park, although the migration only consisted of a relatively small fraction of alleles.
Environmental niche modelling and overlap
Model performance was uniformly good (Appendix S20) with Akaike information criterion (AIC) values well above 0.7 for all four groupings of occurrence records. Diagnostics indicated that niche modelling results were of very good quality for the B. cunninghamii and B. spinulosa clades (Boyce's continuous index = 0.91 and 0.895, respectively). The first three principal components for the environmental PCA of each clade accounted for 72.7% and 75.0% of variation, respectively (Appendices S21, S22). The operational taxonomic units (OTUs) for both analyses were distinctively sorted according to latitudinal cline but did not form discrete clusters. The LGM projection of environmental suitability above zero for each clade covered the same general area as the present‐day area, although B. cunninghamii included additional area either to the north or south (Figure 7; Appendix S23). However, suitability values for the LGM were always lower compared to the present‐day values. The highest suitability for B. cunninghamii was concentrated inland at higher elevation along the eastern portion of the Australian continent, whereas for B. spinulosa it was highest along the coast. Separate analyses for the northern, central, and southern groups of the B. cunninghamii populations (as recovered by snmf at K = 3) each showed a more restricted area of suitability above 0 compared to the results from the total data. The suitability did not exceed 0.2 in the LGM projections, and although this suitability covered a greater area than the present, it did not include the coastal areas of Australia in the analysis of the northern and central groups (Figure 7).
Figure 7.
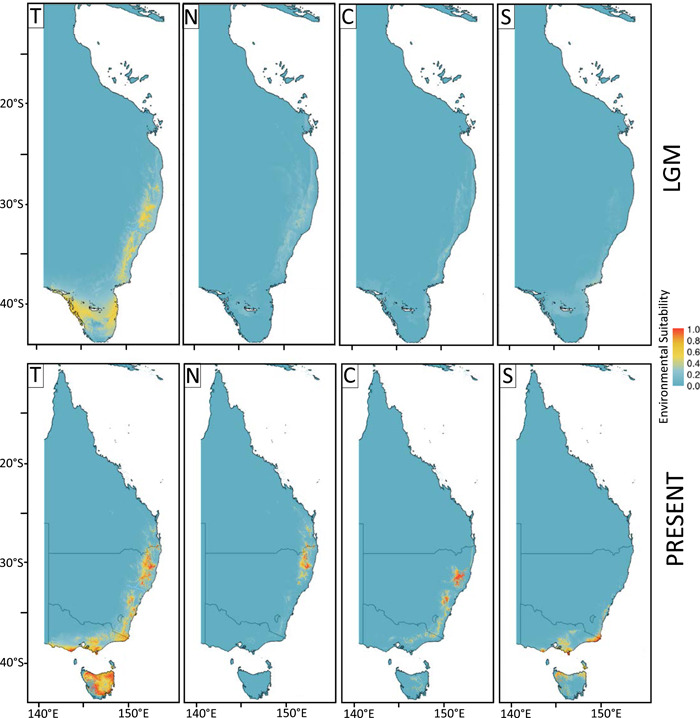
Last glacial maximum (LGM) and present environmental niche modeling projections provided for the samples of the hairpin banksia B. cunninghamii clade nDNA data set, with B. vincentia removed and augmented with specimen records from the Atlas of Living Australia website (http://www.ala.org.au [accessed 25 April 2020]). Modelling was provided for the (T) total data set and for the three groups of populations identified from analysis of the nDNA data set: (N) northern group found north of the Hunter river in New South Wales; (S) southern group found south of the Towamba River in southern New South Wales; (C) central group found between the Hunter river and Towamba River. The total data set showed a present‐day optimal climatic envelope similar to its southeastern Australia range but extending much farther south. The highest optimality occurred away from the coast along most of the eastern side of the continent, along the southern coast of the mainland, and extensively throughout the island of Tasmania. When the data set was restricted to southern, central, or northern groups, the projected optimality envelope was generally similar to the current distribution, although it extended north of the Hunter River corridor for the central group, and south to the north coast of the island of Tasmania for the southern group. Projections of each data set for the LGM were all provided a lower suitability, although the range of suitability was more extensive by latitude.
DISCUSSION
Hairpin banksias: a case of oversplitting
Geographically correlated morphological variation amongst populations of hairpin banksias seemingly presents a typical case of allopatric differentiation of numerous morphologically similar, but narrowly distinct taxa. However, in testing this model by analyzing nuclear and chloroplast genomic data sets at a population level, we have shown that the hairpin banksias form two main clades of populations, within each of which genetic variation is more complex than expected, only partly correlated with morphological variation and taxonomic diversification is incomplete. The first, the B. cunninghamii clade, consists of all populations currently recognized as B. cunninghamii, B. neoanglica, and B. vincentia, and the second, the B. spinulosa clade, consists of all populations currently recognized as B. collina and B. spinulosa. Within these clades, genetic variation is often morphologically cryptic, is clinal in some parts of the distribution, forms discrete metapopulations in other parts, and only some morphological gaps correspond to genetic discontinuities. Because of these complexities, our results do not support the recognition of more than two taxa.
Satisfying a phenetic concept of species, the clustering of these two groups is broadly similar, although not strictly congruent. Only two major incongruencies between them are evident: (1) the morphometric analysis (Stimpson et al., 2016) does not resolve the basal split between the two main clades, clustering B. spinulosa with the B. cunninghamii clade, not with B. collina; (2) the Shipwreck Creek population (at Croajingalong National Park) is clustered with B. cunninghamii populations in our results, rather than the expected B. spinulosa clade. Both of these discrepancies are probably the result of overweighting in the phenetic morphometric analysis of style color, in which it was represented by two binary characters, color before and after anthesis (Stimpson et al., 2016). The yellow style color found in Croajingalong is also widespread in the B. spinulosa clade north of the Hawkesbury River but had never previously been observed in the B. cunninghamii clade (Stimpson et al., 2016). Satisfying a phylogenetic concept of species, there is unequivocal evidence that these two groups are reciprocally monophyletic (Figures 4, 5, 6). Finally, satisfying an ecological concept of species, each clade shows a distinct area of environmental optimum despite some overlap: where latitude is standardized, higher altitudes away from the coast are mostly suitable for the B. cunninghamii clade and lower altitudes adjacent the coastline are mostly suitable for B. spinulosa (Figure 7; Appendix S23). Unequivocal evidence of recurrent hybridization between B. cunninghamii and B. spinulosa (Figures 3, 4) might for these two clades be used to argue they are not distinct species. However, B. spinulosa and B. cunninghamii are sympatric over a large area in the Central Tablelands of New South Wales, and despite hybridization reported here for the first time, there remains strong genetic and morphological signal separating these two lineages.
Extracting further taxonomic resolution from within the two hairpin banksia clades becomes challenging based on the much higher amount of genetic admixture within each of the clades relative to that between them. Relationships based on nDNA and cpDNA do not correspond with each other, nor do they correspond with those based on phenetic morphometric analysis (Stimpson et al., 2016). Recognizing previously described taxa even as early divergent lineages within either clade would necessitate the recognition of paraphyletic subspecies (Crisp and Chandler, 1996; de Queiroz, 2020). However, although taxonomic recognition of such metapopulations may be acceptable in some cases, here each main clade shows little restriction of gene flow across its range, according to low F ST values and a characteristic isolation by distance (IBD) trend, which is what would be expected among populations of a single species rather than between several species of a genus.
Within each of the two main clades, patterns of nuclear and chloroplast genomic variation differ from morphometric variation as well as taxonomic hypotheses, but there are also differences between the two genomes as well. For both clades, the chloroplast network generally shows less structure, but in some specific areas it also shows incongruence with the nDNA phylogeny. For example, the reciprocally monophyletic clades of northern B. collina populations shown in our analysis of the nuclear genomic data set (Figure 6C) did not form separate haplogroups, and two populations of B. cunninghamii were clustered with either northern populations of the B. cunninghamii clade (cpDNA) or southern populations (nDNA). Although not the only cause of cytonuclear incongruence (Funk and Omland, 2003; Ballard and Whitlock, 2004; Barrett and Schluter, 2008; Sloan et al., 2017; Nge et al., 2021), incomplete lineage sorting is likely to be involved. The nDNA phylogeny retrieved a high number of total incompatible quartets, and despite relatively close proximity to each other, populations of B. cunninghamii across the Central Tablelands of New South Wales were clustered with either northern or southern clades rather than forming a clade of their own (Figure 5C).
Although the hairpin banksias can be organized into several groups according to morphological characters, based on the evidence of genetic variation, the group would most logically be interpreted as two distinct species. The current taxonomy therefore requires change, as it is not compatible with the findings here, or even the findings from previous morphological analysis. Taxonomic reorganization is a constant accompaniment to discovery through technological and theoretical advances, and the need of such a change is unsurprising given that other profound reinterpretations of morphological diversity in Proteaceae have recently been made or are currently underway (George, 2008, 2012, 2014; Weston, 2014; Mast et al., 2005, 2008, 2015; Thiele et al., 2015).
A biogeographic anomaly: long‐distance dispersal or a relictual population?
Our results show that such a high morphological similarity between B. neoanglica and B. vincentia (Stimpson et al., 2016) is simply explained by the fact that they are the same species. This situation would not be a surprising if B. vincentia were distributed at the northern end of the distribution of B. cunninghamii. However, it is not; it occurs 600 km south of its genetically closest relatives, and the gap between them is scattered with many populations of the B. cunninghamii clade that are less genetically similar to it. An explanation for such a peculiar anomaly in genetic distribution could be that the individuals at Vincentia are a relictual population of a once wider distribution of the northern populations currently described as B. neoanglica that otherwise contracted northward. However, it seems unlikely that the Vincentia population is merely genetic residue from a once wider distribution since traces of admixture between the Vincentia population and populations situated proximally north or south of it (i.e., populations of B. cunninghamii) are effectively nil. If the Vincentia population was relictual, it would be expected to have acquired at least a trace of genetic signature from neighboring populations of B. cunninghamii. Geographic barriers have not been identified in the area that might have created such a distinct contrast in genetic structure between the Vincentia site and adjacent populations of B. cunninghamii. Furthermore, it is unlikely that there are many (if any) prezygotic barriers between these neighboring populations that could prevent gene flow since we show that historical and contemporary gene flow has occurred between B. vincentia and other cooccurring species that are more genetically different (e.g., B. ericifolia and B. spinulosa). Modelling also shows that the Vincentia site does not have an ideal climatic envelope for northern populations that are most closely related to B. vincentia, both now and at the height of the LGM (Figure 7).
With results not in favor of a relictual hypothesis, such marked separation in the closely related Vincentia and Girraween populations might also be explained by a recent, highly unusual long‐distance dispersal, especially given that Banksia seeds can be wind‐dispersed (He et al., 2004, 2009). Long‐distance dispersal might seem unlikely based on what evidence has currently been gathered for vectors, seed dormancy, and dispersal distances for Banksia (Abbott, 1985; Carthew, 1993; Myerscough et al., 2001; Whelan and Ayre, 2020); however, as seen in other Proteaceae with low vagility, there is good evidence to suggest that long‐distance dispersal events have happened, albeit extremely rarely (Mast et al. 2008). On the other hand, successful establishment after long‐distance dispersal would appear unlikely given that the Vincentia site does not have an optimal climatic envelope. Furthermore, a strong isolation‐by‐distance signal across the whole group suggests that long distance dispersal is not a prevalent feature in this group. While more examination of the reproductive biology of other Banksia species is warranted to study this anomaly further, another hypothesis in need of testing is that of recent anthropogenic dispersal, since none of our results can be used to reject this possibility. That there is evidence for human‐mediated dispersal of plants including Banksia (e.g., Rossetto et al., 2017; Silcock, 2018) should inspire further exploration into the movement of this group of plants. The Vincentia population might not be ecologically important if dispersed anthropogenically; however, it might still be valuable for understanding human history.
Evolutionary context highlights a classification in need of reinterpretation
From the discussion above, it is evident that the hairpin banksias do not correspond to more than two species. Instead, with exception to the unique situation of the Vincentia population as described above, we assert that the pattern across the landscape in each of the hairpin banksia species is an example of a morphologically variable continuum structured by pronounced interruptions to gene flow at specific areas or biogeographic barriers.
Across the landscape, genomic diversity is partitioned along with the morphology‐based taxonomic boundaries by the Hunter River Corridor and Southern Transition Zone in the B. cunninghamii clade, and Brisbane Valley Barrier and St. Lawrence Gap in the B. spinulosa clade (Figure 2). The most outstanding pattern in this data set, which corresponds closely with morphological differentiation, is the disruption of gene flow at the Hunter River Corridor in the B. cunninghamii clade, or slightly south of this location at the Hawkesbury River for the B. spinulosa clade. The Hunter River Corridor is a dry, open woodland and savannah that bisects upland closed forest and appears to have had a similar marked disruption of gene flow between or within several other taxa (Chapple et al., 2011; Milner et al., 2012; 2015; Rix and Harvey, 2012; Bryant and Krosch, 2016 and references therein). However, chloroplast results instead show a major difference elsewhere in each clade: across the Southern Transition Zone for the B. cunninghamii clade (Figure 5B) and around the St. Lawrence Gap for the B. spinulosa clade (Figure 6B). Differing evolutionary histories are not rare (e.g., Acosta and Premoli, 2010; Barrett et al., 2015; Folk et al., 2017; Vargas et al., 2017) and cytonuclear incongruence (i.e., between chloroplast and nuclear genome) is frequently indicative of incomplete lineage sorting (Birky, 1995; Soltis and Kuzoff, 1995; Yoo et al., 2002; but see Lee‐Yaw et al., 2018). This explanation seems most likely given that, in at least the B. cunninghamii clade, the svdq analysis returned a high number of incompatible quartets, and despite their high genetic similarity, populations of the central group were not recovered as a single clade in the phylogeny.
A growing number of studies link incomplete lineage sorting to historical range expansions and contractions, whereby isolated populations reach preliminary levels of vicariance, although not reproductive isolation, before they expand and overlap again (e.g., Georges et al., 2018; Simpson et al., 2018; Liu et al., 2019; Nistelberger et al., 2020). Indeed, our models suggest that during the LGM there was some level of environmental suitability for each hairpin banksia clade within their present‐day distributions (Figure 7; Appendix S23). Although this suitability was relatively lower, it suggests that several populations had the opportunity to persist in refugia along this latitudinal distribution throughout the LGM, thereby reinforcing an IBD pattern. Cyclical patterns of glaciation over the last 10 million years (Ehlers and Gibbard, 2007; Colhoun and Barrows, 2011) in combination with the effects of geographic barriers could have repetitively separated populations, permitting some degree of drift and adaptation to localized environments, while not extensive enough to allow for the evolution of reproductive incompatibility. Porous reproductive barriers between B. cunninghamii and B. spinulosa clades might equally be explained by this process, given that historically their total range distributions lay adjacent to each other at the LGM (Figure 7; Appendix S23). If groups of populations with particularly distinct genetic signatures reconnected in postglacial encounters, then introgression could have occurred, as supported by the numerous hybridizations we have detected (Figures 3, 4).
The apparently complicated evolutionary history described here emphasizes the desirability of critically testing morphology‐based taxonomic models using genomic data sets, especially around biogeographic barriers. It is evident in this example that, although a previous analysis of morphology was based on a suitably comprehensive sampling across both distributional range and morphological variation, incongruent signal between genomic and morphological variation within the two main clades did not support finer taxonomic division within them. That chloroplast results showed a bigger change than nuclear genomic data across the Hunter River Corridor furthermore underscores that no single line of evidence adequately represents evolution in this group. As such, our results make it clear that species hypotheses (especially with reference to widely distributed species complexes) associated with complex geomorphological partitioning require rigorous testing.
Evolutionary perspective and implications for conservation
At a time when species loss is increasing and climate is changing, oversplitting might seemingly be the safest plan in a vacuum of knowledge when the known consequences (i.e., extinction) are irreversible. The case of the hairpin banksias is not a singular occurrence where recognizably distinct and attractive taxa have been extracted from morphological grades with a complicated taxonomic history (Georges et al., 2018; Hundsdoerfer et al., 2019). Seemingly innocuous, recognizing too much diversity (in this case based on morphology of banksias) has the capacity to draw resources away from effective conservation of species diversity. Thus, interpreting morphological variability as a proxy for genetic variation (and thus resilience of a population) should be tested directly, not assumed.
Banksia vincentia should not be accepted as a distinct taxon at the species level, and intuitively, the endangered species list should be reduced to liberate resources for other conservation priorities. There is no question that species recognition is critical for conservation, and there is a long list of undescribed species and taxonomic complexes awaiting treatment by an underfunded taxonomic community. Banksia has several other critically endangered species in need of conservation (Galea and Lamont, 1993; Lamont, 1996; Day et al., 1997; Millar et al., 2010; Coates et al., 2013; Bradbury et al., 2019; Millar and Byrne, 2020) that represent a greater proportion of the genetic biodiversity of the genus than the population at Vincentia (Cardillo and Pratt, 2013). Therefore, maximizing conservation efforts for this population would draw from our capacity to save other endangered flora that represent a higher value of biodiversity. Furthermore, the anticipated intensive repropagation and translocation of populations at Vincentia would not only hamper conservation, it would also be capable of producing a human‐induced bottleneck through amplification of a relatively (i.e., with respect to the B. cunninghamii s.l.) genetically poor and highly inbred population. Reciprocally, such an introduction of individuals that apparently rely on hybridization (i.e., that which we observed between it and B. ericifolia) to balance the lack of natural diversity would also increase the occurrence of unwanted admixture among other locally occurring Banksia populations.
Our results also show that, in addition to an inaccurate estimation of species, over‐splitting has the capability of cryptically eroding species resilience through the means of obscuring genetically diverse populations. In the case of B. cunninghamii s.l., there is disproportionately more importance placed on northern populations through the recognition of B. neoanglica and B. vincentia, despite that southern populations have an equal amount of genetic diversity. Similarly, a recognition of B. spinulosa and B. collina conceals the more genetically diverse northern populations of B. spinulosa s.l.
It had been earlier concluded that there were no hybrids at the Vincentia site, as exhaustive searches recovered no morphologically intermediate forms between cooccurring Banksias (Stimpson et al., 2012). However, we show that genetic swamping and cryptic hybridization have occurred at the Vincentia site, bringing into picture a major pitfall for conservation work that relies solely on morphology to detect genetic admixture. A demonstration of such admixture with other sympatric species of Banksia (Usher, 2011; Figure 3) also suggests that such genetic introgression is neither rare nor a recent occurrence and can be problematic for taxonomic assumptions in general. For instance, a similar lack of visual evidence for intermediate forms at a contact zone between two species, such as at Lamington National Park where B. neoanglica and a morphologically distinct population of B. collina occur in close proximity, suggests reproductive isolation and hence support for distinct species (Stimpson et al., 2016). Yet, this idea does not take into account the possibility that at contact zones a certain portion of genetic material has already been dispersed throughout a recipient population, and hence corresponding morphological characters are already stabilized such that no hybridization grade amongst individuals visibly exists. Based on our results that suggest historical migration between the B. cunninghamii and B. spinulosa clade at Lamington National Park, it appears that there has been time for stabilization to have occurred across proximally located and morphologically similar (Stimpson et al., 2016) populations at Mt Maroon (Mt Barney National Park) and Lamington National Park.
CONCLUSIONS
Inextricably linked to the efficacy of conservation management is the way a taxon is defined, a process which is now assisted by an understanding of evolutionary processes. In the case of the hairpin banksias, genomewide scans (i.e., nuclear SNP data and chloroplast genome sequence) have shown that the number of species has been overestimated. Although such an outcome is typically the exception of the result of investigation into these problems, it is nonetheless as important as the identification of cryptic species. An incorrectly applied taxonomy such as the case of the hairpin banksias has the capacity to put into effect the gradual erosion of species resilience via human‐mediated measures by overemphasizing the conservation of genetically poor populations and masking others that are more genetically diverse. Therefore, like other studies (e.g., Ahrens et al., 2020a, 2020b; Rossetto et al., 2021; Rutherford et al., 2019; Nielsen et al., 2020; Hope and Frey, 2022), our study here has shown that a genomic analysis complemented by a good sampling strategy is a well‐justified step at the beginning of a conservation campaign. This step has enabled a dramatic reduction in time and cost, and although these factors still impose some limitations, where protection measures are urgently needed for putative entities with conservation value, the step should at least be used as soon as possible.
AUTHOR CONTRIBUTIONS
Conceptualization (T.C.W., M.R., M.L.S., P.H.W., and D.B.); data curation (T.C.W., M.L.S., and J.‐Y.S.Y.); formal analysis (T.C.W., J.‐Y.S.Y., and P.D.W.); funding acquisition (M.R. and D.B.); investigation and supervision, project administration, visualization, writing: original draft (T.C.W. and M.R.); methodology (T.C.W., M.R., and L.C.), resources (T.C.W., M.R., M.L.S., P.H.W., L.C., and D.B.); writing: review, editing (T.C.W., M.R., J.‐Y.S.Y., P.H.W., D.B., and P.D.W).
CONFLICT OF INTEREST
All authors declare no conflict of interest.
Supporting information
Appendix S1. Wild‐collected and cultivated hairpin banksia specimens sampled for genomic sequencing and chloroplast genome sequencing.
Appendix S2. Environmental niche modelling for the Banksia cunninghamii clade: methods.
Appendix S3. The 19 standard bioclimatic variables used to estimate an overall environmental envelope to project spatial distribution of environmental suitability for the hairpin banksias using a select group of populations.
Appendix S4. Cleaned data set of spatial records used to project spatial distribution of environmental suitability for the Banksia cunninghamii clade of the hairpin banksias.
Appendix S5. Cleaned data set of spatial records used to project spatial distribution of environmental suitability for the Banksia spinulosa clade of the hairpin banksias.
Appendix S6. Principal component analysis of nDNA data for the hairpin banksia B. cunninghamii clade identified in the network analysis.
Appendix S7. LEA snmf analysis for the nDNA data set of the hairpin banksia B. cunninghamii clade, as recovered by network analysis of genomic data.
Appendix S8. Averaged Q values (at K = 2 ancestral populations) for each population of hairpin banksia from separate LEA snmf analyses for the B. cunninghamii clade and the B. spinulosa clade.
Appendix S9. Averaged Q values (at K = 3 ancestral populations) for each population of hairpin banksia from separate LEA snmf analyses for the B. cunninghamii clade and the B. spinulosa clade.
Appendix S10. Averaged Q values (at K = 4 ancestral populations) for each population of hairpin banksia from separate LEA snmf analyses for the B. cunninghamii clade and the B. spinulosa clade.
Appendix S11. F ST pairwise matrix for the hairpin banksia B. cunninghamii clade identified from network analysis of nDNA.
Appendix S12. Isolation by distance graphs derived from nDNA data for the (a) Banksia cunninghamia clade and (b) B. spinulosa clade, as identified by network analysis.
Appendix S13. Heterozygosity values retrieved from two separate analyses of the genomic data for the hairpin banksia complex.
Appendix S14. Principal component analysis of genetic diversity from nDNA data for the hairpin banksia Banksia spinulosa clade identified in the network analysis.
Appendix S15. LEA snmf analysis for nDNA data set of the hairpin banksia B. spinulosa clade, as recovered by network analysis of genomic data.
Appendix S16. F ST pairwise matrix for the hairpin banksia B. spinulosa clade identified from network analysis of nDNA.
Appendix S17. Chloroplast sequence reads and coverage for members of the B. cunninghamii (cun) and B. spinulosa (spin) clades as defined from network analysis of the nDNA data set of the hairpin banksias.
Appendix S18. Morphological variation across the seed‐grown ex situ Banksia collection at Australian Botanic Garden (ABG) sourced from Vincentia, NSW (living collection number 20160262).
Appendix S19. The output produced by TreeMix and OptM for the nDNA data set for 23 populations of the hairpin banksias with recent hybrids removed.
Appendix S20. Summary of environmental niche model performance data for the total occurrence records of the Banksia cunninghamii clade (Total) or partitioned as three separate groups (North, Central, and South) according to the results from snmf analysis.
Appendix S21. Principal component analysis of environmental data for the hairpin banksia B. cunninghamii clade without B. vincentia.
Appendix S22. Principal component analysis of environmental data for samples of the hairpin banksia B. spinulosa clade nDNA data set.
Appendix S23. Present‐day and last glacial maximum (LGM) environmental niche modeling projections provided for the samples of the hairpin banksia B. spinulosa clade nDNA data set.
ACKNOWLEDGMENTS
We acknowledge the Traditional Custodians of the land on which this study was undertaken and pay respect to Elders past and present. The present study would not have been possible without the assistance of Jeremy J. Bruhl (N.C.W. Beadle Herbarium, University of New England, Australia) in sampling key populations. The authors also thank Stig Peterson (Bouderee Botanic Gardens), James Beattie (Wollongong Botanic Gardens), and Joel Cohen for providing additional research material; Jason Bragg for assistance with analyses; staff at the Australian Botanic Gardens and Australian National Botanic Gardens for assistance with providing ex situ material; Jan Allen and Kylie Coutts‐McClelland for photographs; Hanno Schaefer and an anonymous reviewer for improvements on the manuscript. This work was supported by the Saving Our Species initiative of the Department of Planning and Environment (NSW, Australia) and from the Royal Botanic Gardens and Domain Trust. Open access publishing facilitated by New South Wales Department of Planning Industry and Environment, as part of the Wiley ‐ New South Wales Department of Planning Industry and Environment agreement via the Council of Australian University Librarians.
Wilson, T. C. , Rossetto M., Bain D., Yap J.‐Y.S., Wilson P. D., Stimpson M. L., Weston P. H., and Croft L.. 2022. A turn in species conservation for hairpin banksias: demonstration of oversplitting leads to better management of diversity. American Journal of Botany 109(10): 1652–1671. 10.1002/ajb2.16074
DATA AVAILABILITY STATEMENT
The nuclear genomic data sets and the chloroplast genome sequence alignment are available in the Dryad Digital Repository: https://doi.org/10.5061/dryad.69p8cz94x (Wilson et al., 2022).
REFERENCES
- Abbott, I. 1985. Reproductive ecology of Banksia grandis (Proteaceae). New Phytologist 99: 129–148. [Google Scholar]
- Acosta, M. C. , and Premoli A. C.. 2010. Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Molecular Phylogenetics and Evolution 54: 235–242. [DOI] [PubMed] [Google Scholar]
- Ahrens, C. W. , James E. A., Miller A. D., Scott F., Aitken N. C., Jones A. W., Lu‐Irving P., et al. 2020a. Spatial, climate and ploidy factors drive genomic diversity and resilience in the widespread grass Themeda triandra . Molecular Ecology 29: 3872–3888. [DOI] [PubMed] [Google Scholar]
- Ahrens, C. W. , Supple M. A., Aitken N. C., Cantrill D., Borevitz J. O., and James E. A. 2020b. Genomic diversity guides conservation strategies among rare terrestrial orchid. Annals of Botany 119: 1267–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Australian National Botanical Garden . 2022. Australian Plant Name Index (APNI), Vascular plants, Banksia spinulosa [online; part of IBIS database]. Website: https://biodiversity.org.au/nsl/services/search/names?product=APNIandtree.id=andname=Banksia%2Bspinulosaandinc._scientific=andinc.scientific=onandinc._cultivar=andinc._other=andmax=100anddisplay=apniandsearch=true [accessed 22 February 2022]. Australian National Botanical Garden and Centre for Australian National Biodiversity Research, Canberra, Australia.
- Auld, T. , and Weston P.. 2020. Banksia vincentia Stimpson and P.H.Weston. The IUCN red list of threatened species 2020. Website: 10.2305/IUCN.UK.2020-2.RLTS.T117474000A117474507.en [retrieved 22 February 2022]. [DOI]
- Baird, N. A. , Etter P. D., Atwood T. S., Currey M. C., Shiver A. L., Lewis Z. A., Selker E. U., et al. 2008. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3: e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard, J. W. and Whitlock M. C.. 2004. The incomplete natural history of mitochondria. Molecular Ecology 13: 729–744. [DOI] [PubMed] [Google Scholar]
- Barrett, R. D. and Schluter D.. 2008. Adaptation from standing genetic variation. Trends in Ecology and Evolution 23: 38–44. [DOI] [PubMed] [Google Scholar]
- Barrett, R. A. , Bayly M. J., Duretto M. F., Forster P. I., Ladiges P. Y. and Cantrill D. J.. 2015. A chloroplast phylogeny of Zieria (Rutaceae) in Australia and New Caledonia shows widespread incongruence with species‐level taxonomy. Australian Systematic Botany 27: 427–449. [Google Scholar]
- Birky, C. W 1995. Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proceedings of the National Academy of Sciences, USA 92: 11331–11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, D. , Binks R. M., Coates D. J. and Byrne M.. 2019. Conservation genomics of range disjunction in a global biodiversity hotspot: a case study of Banksia biterax (Proteaceae) in southwestern Australia. Biological Journal of the Linnean Society 127: 390–406. [Google Scholar]
- Brown, R. 1810. On the Proteaceae of Jussieu. Transactions of the Linnean Society of London 10: 215–226. [Google Scholar]
- Bryant, L. M. and Krosch M. N.. 2016. Lines in the land: a review of evidence for eastern Australia's major biogeographical barriers to closed forest taxa. Biological Journal of the Linnean Society 119: 238–264. [Google Scholar]
- Bryson, R. W. , Zarza E., Grummer J. A., Parra‐Olea G., Flores‐Villela O., Klicka J., and McCormack J. E.. 2018. Phylogenomic insights into the diversification of salamanders in the Isthmura bellii group across the Mexican highlands. Molecular Phylogenetics and Evolution 125: 78–84. [DOI] [PubMed] [Google Scholar]
- Cardillo, M. , and Pratt R.. 2013. Evolution of a hotspot genus: geographic variation in speciation and extinction rates in Banksia (Proteaceae). BMC Evolutionary Biology 13: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew, S. M. , Ayre D. J. and Whelan R. J.. 1988. High levels of outcrossing in populations of Banksia spinulosa R. Br. and Banksia paludosa Smith. Australian Journal of Botany 36: 217–223. [Google Scholar]
- Carthew, S. M. 1993. Patterns of flowering and fruit production in a natural population of Banksia spinulosa . Australian Journal of Botany 41: 465–480. [Google Scholar]
- Chapple, D. G. , Hoskin C. J., Chapple S. N., and Thompson M. B.. 2011. Phylogeographic divergence in the widespread delicate skink (Lampropholis delicata) corresponds to dry habitat barriers in eastern Australia. BMC Evolutionary Biology 11: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman, J. and Kubatko L.. 2014. Quartet inference from SNP data under the coalescent model. Bioinformatics 30: 3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, J. , Gupta A., Yaduvanshi S., Davidson R., Nute M., Mirarab S. and Warnow T.. 2015. A comparative study of SVDquartets and other coalescent‐based species tree estimation methods. BMC Genomics 16: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clugston, J. A. R. , Kenicer G. J., Milne R., Overcast I., Wilson T. C. and Nagalingum N. S.. 2019. RADseq as a valuable tool for plants with large genomes—A case study in cycads. Molecular Ecology Resources 19: 1610–1622. [DOI] [PubMed] [Google Scholar]
- Coates, D. J. , Williams M. R. and Madden S.. 2013. Temporal and spatial mating‐system variation in fragmented populations of Banksia cuneata, a rare bird‐pollinated long‐lived plant. Australian Journal of Botany 61: 235–242. [Google Scholar]
- Coissac, E. , Hollingsworth P. M., Lavergne S. and Taberlet P.. 2016. From barcodes to genomes: extending the concept of DNA barcoding. Molecular Ecology 25: 1423–1428. [DOI] [PubMed] [Google Scholar]
- Colhoun, E. A. , and Barrows T. T.. 2011. The glaciation of Australia. Developments in Quaternary Science 15: 1037–1045. [Google Scholar]
- Crisp, M. D. , and Chandler G. T.. 1996. Paraphyletic species. Telopea 6: 813–838. [Google Scholar]
- Darling, A. C. , Mau B., Blattner F. R., and Perna N. T.. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Research 14: 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, D. , Collins B. G., and Rees R. G.. 1997. . Reproductive biology of the rare and endangered Banksia brownii Baxter ex R. Br. (Proteaceae). Australian Journal of Ecology 22: 307–315. [Google Scholar]
- Davey, J. W. , and Blaxter M. L.. 2011. RADSeq: next‐generation population genetics. Briefings in Functional Genomics 9: 416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Queiroz, K. 2005. Ernst Mayr and the modern concept of species. Proceedings of the National Academy of Sciences, USA 102: 6600–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Queiroz, K. 2007. Species concepts and species delimitation. Systematic Biology 6: 879–886. [DOI] [PubMed] [Google Scholar]
- de Queiroz, K. 2020. An updated concept of subspecies resolves a dispute about the taxonomy of incompletely separated lineages. Herpetological Review 51: 459–461. [Google Scholar]
- Devitt, T. J. , Wright A. M., Cannatella D. C., and Hillis D. M.. 2019. Species delimitation in endangered groundwater salamanders: implications for aquifer management and biodiversity conservation. Proceedings of the National Academy of Sciences, USA 116: 2624–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky, T. 1951. Genetics and the origin of species, 3rd ed. Columbia University Press, NY, NY, USA. [Google Scholar]
- Dumas, P. , Barbut J., Le Ru B., Silvain J.‐F., Clamens A.‐L., d'Alençon E., and Kergoat G. J.. 2015. Phylogenetic molecular species delimitations unravel potential new species in the pest genus Spodoptera Guenée. PLoS One 10: e0122407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton, D. A. , Hipp A. L., González‐Rodríguez A., and Cavender‐Bares J.. 2015. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69: 2587–2601. [DOI] [PubMed] [Google Scholar]
- Edet, O. U. , Gorafi Y. S. A., Nasuda S., and Tsujimoto H.. 2018. DArTseq‐based analysis of genomic relationships among species of tribe Triticeae. Scientific Reports 8: e16397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers, J. , and Gibbard P. L.. 2007. The extent and chronology of Cenozoic global glaciation. Quaternary International 164–165: 6–20. [Google Scholar]
- Elith, J. , Phillips S. J., Hastie T., Dudík M., Chee Y. E., and Yates C. J.. 2011. A statistical explanation of MaxEnt for ecologists. Diversity and Distributions 17: 43–57. [Google Scholar]
- Fernández‐Mazuecos, M. , Mellers G., Vigalondo B., Sáez L., Vargas P., and Glover B.J.. 2017. Resolving recent plant radiations: power and robustness of genotyping‐by‐sequencing. Systematic Biology 67: 250–268. [DOI] [PubMed] [Google Scholar]
- Fitak, R. R. 2021. optM: an R package to optimize the number of migration edges using threshold models. Journal of Heredity 6: bpab017. [Google Scholar]
- Folk, R. A. , Mandel J. R., and Freudenstein J. V.. 2017. Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Systematic Biology 66: 320–337. [DOI] [PubMed] [Google Scholar]
- Frichot, E. , and François, O. 2015. LEA: an R package for landscape and ecological association studies. Methods in Ecology, 6: 925–929. [Google Scholar]
- Funk, D. J. , and Omland K. E.. 2003. Species‐level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annual Review of Ecology, Evolution, and Systematics 34: 397‐423. [Google Scholar]
- Galea, H. , and Lamont B.. 1993. Population ecology of the rare and endangered Banksia brownii. Research report to the Department of Conservation and Land Management. Curtin University of Technology, Western Australia, Australia. [Google Scholar]
- George, A. S. 1981. The genus Banksia L.f. (Proteaceae). Nuytsia 3: 239–473. [Google Scholar]
- George, A. S. 1988. New taxa and notes on Banksia L.f. (Proteaceae). Nuytsia 6: 309–317. [Google Scholar]
- George, A. S. 1999. Banksia. Flora of Australia; 17B: 175–251. [Google Scholar]
- George, A. S. 2008. You don't have to call Dryandra Banksia . Wildflower Society of Western Australia Newsletter 46: 7–9. [Google Scholar]
- George, A. S. 2012. A new combination in Dryandra (Proteaceae). Western Australian Naturalist 28: 266–268. [Google Scholar]
- George, A. S. 2014. The case against the transfer of Dryandra to Banksia (Proteaceae) 1. Annals of the Missouri Botanical Garden 100: 32–49. [Google Scholar]
- Georges A., Gruber B., Pauly G. B., White D., Adams M., Young M. J., Kilian A., et al. 2018. Genomewide SNP markers breathe new life into phylogeography and species delimitation for the problematic short‐necked turtles (Chelidae: Emydura) of eastern Australia. Molecular Ecology 27: 5195–5213. [DOI] [PubMed] [Google Scholar]
- Harden, G. J. 2002. Banksia . In Harden G. J. [ed.], Flora of New South Wales, 2nd ed., 82–86. UNSW Press, Sydney, Australia. [Google Scholar]
- He, T. , Krauss S. L., Lamont B. B., Miller B. P., and Enright N. J.. 2004. Long distance dispersal in a metapopulation of Banksia hookeriana inferred by population allocation from AFLP data. Molecular Ecology 13: 1099–1109. [DOI] [PubMed] [Google Scholar]
- He, T. , Lamont B. B., Krauss S. L., Enright N. J., and Miller B. P.. 2009. Long distance dispersal of seeds in the fire‐tolerant shrub Banksia attenuate . Ecography 32: 571–580. [Google Scholar]
- Henderson, M. 1991. Aspects of the mating system of Banksia ericifoliaB. spinulosa and (Proteaceae). B.Sc. Honours dissertation, University of Wollongong, New South Wales, Australia.
- Hope, A. G. , and Frey J. K.. 2022. Misinterpretation of genomic data matters for endangered species listing: the sub‐specific status of the Peñasco least chipmunk (Neotamias minimus atristriatus). Frontiers in Conservation Science 2: 793277. [Google Scholar]
- Hundsdoerfer, A. K. , Lee K. M., Kitching I. J., and Mutanen M.. 2019. Genome‐wide SNP data reveal an overestimation of species diversity in a group of hawkmoths. Genome Biology and Evolution 11: 2136–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H. , and Bryant L. M.. 2006. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Jombart, T. 2008. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405. [DOI] [PubMed] [Google Scholar]
- Jombart T., and Ahmed I.. 2011. adegenet 1.3‐1: new tools for the analysis of genome‐wide SNP data. Bioinformatics 27: 3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan, K. , McGinnity P., Cross T. F., Crozier W. W., and Prodöhl P. A.. 2013. diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods in Ecology and Evolution 4: 782–788. [Google Scholar]
- Lamont, B. B. 1996. Conservation biology of banksias in southwestern Australia. In Hopper S. D., Harvey M., Chappill J., and George A. S. [eds.], Gondwanan heritage: past, present and future of the Western Australian biota, 292–298. Surrey Beatty, Chipping Norton, NSW, Australia. [Google Scholar]
- Lee, M. S. Y. 2003. Species concepts and species reality: salvaging a Linnaean rank. Journal of Evolutionary Biology 16: 179–188. [DOI] [PubMed] [Google Scholar]
- Lee‐Yaw, J. A. , Grassa C. J., Andrew R. A., and Rieseberg L. A.. 2018. An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytologist 221: 515–526. [DOI] [PubMed] [Google Scholar]
- Leigh, J. W. , and Bryant D.. 2015. Popart: full‐feature software for haplotype network construction. Methods in Ecology and Evolution 6: 1110–1116. [Google Scholar]
- Liu, T. , Sun K., Csorba G., Zhang K., Zhang L., Zhao H., Jin L., et al. 2019. Species delimitation and evolutionary reconstruction within an integrative taxonomic framework: a case study on Rhinolophus macrotis complex (Chiroptera: Rhinolophidae). Molecular Phylogenetics and Evolution 139: 106544. [DOI] [PubMed] [Google Scholar]
- Maddison, W. , and Knowles L. L.. 2006. Inferring phylogeny despite incomplete lineage sorting. Systematic Biology 55: 21–30. [DOI] [PubMed] [Google Scholar]
- Maguire, T. L. , Conran J. G., Collins G. G., and Sedgley M.. 1997. Molecular analysis of interspecific and intergeneric relationships of Banksia using RAPDS and non‐coding chloroplast DNA sequences. Theoretical and Applied Genetics 95: 253–260. [Google Scholar]
- Mallet, J. , Besansky N., and Hahn M. W.. 2016. How reticulated are species? Bioessays 16: 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel, N. 1967. The detection of disease clustering and a generalized regression approach. Cancer Research 27: 209–220. [PubMed] [Google Scholar]
- Martin, S. H. , Dasmahapatra K. K., Nadeau N. J., Salazar C., Walters J. R., Simpson F., Blaxter M., et al. 2013. Genome‐wide evidence for speciation with gene flow in Heliconius butterflies. Genome Research 23: 1817–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast, A. R. , Jones E. H., and Havery S. P.. 2005. An assessment of old and new DNA sequence evidence for the paraphyly of Banksia with respect to Dryandra (Proteaceae). Australian Systematic Botany 18: 75–88. [Google Scholar]
- Mast, A. R. , Olde P. M., Makinson R. O., Jones E., Kubes A., Miller E. T., and Weston P. H.. 2015. Paraphyly changes understanding of timing and tempo of diversification in subtribe Hakeinae (Proteaceae), a giant Australian plant radiation. American Journal of Botany 102: 1634–1646. [DOI] [PubMed] [Google Scholar]
- Mast, A. R. , Willis C. L., Jones E. H., Downs K. M., and Weston P. H. 2008. A smaller Macadamia from a more vagile tribe: inference of phylogenetic relationships and divergence times in Macadamia and relatives (tribe Macadamieae; Proteaceae). American Journal of Botany 95: 843‐870. [DOI] [PubMed] [Google Scholar]
- Mayr, E. 1963. Animal species and evolution. Harvard University Press, Cambridge, MA, USA. [Google Scholar]
- Milanesi, M. , Capomaccio S., Vajana E., Bomba L., Garcia J. F., Ajmone‐Marsan P. A‐M., and Colli L.. 2017. BITE: an R package for biodiversity analyses. bioRxiv 10.1101/181610 [Preprint]. [DOI]
- Millar, M. A. , and Byrne M.. 2020. Variable clonality and genetic structure among disjunct populations of Banksia mimica . Conservation Genetics 21: 803–818. [Google Scholar]
- Millar, M. A. , Byrne M., and Coates D.. 2010. The maintenance of disparate levels of clonality, genetic diversity and genetic differentiation in disjunct subspecies of the rare Banksia ionthocarpa . Conservation Genetics 19: 4217–4227. [DOI] [PubMed] [Google Scholar]
- Milner, M. L. , Rossetto M., Crisp M. D., and Weston P. H.. 2012. The impact of multiple biogeographic barriers and hybridization on species‐level differentiation. American Journal of Botany 99: 2045–2057. [DOI] [PubMed] [Google Scholar]
- Milner, M. L. , Weston P. H., Rossetto M., and Crisp M. D.. 2015. Biogeography of the Gondwanan genus Lomatia (Proteaceae): vicariance at continental and intercontinental scales. Journal of Biogeography 42: 2440–2451. [Google Scholar]
- Mishler, B. , and Brandon R. N.. 1987. Individuality, pluralism, and the phylogenetic species concept. Biological Philosophy 2: 397–414 [Google Scholar]
- Mishler, B. , and Theriot E. C.. 2000. The phylogenetic species concept (sensu Mishler and Theriot): monophyly, apomorphy and phylogenetic species concepts. In Wheeler Q. D. and Meier R. [eds.], Species concepts and phylogenetic theory: a debate, 44–54. Columbia University Press, NY, NY, USA. [Google Scholar]
- Myerscough, P. J. , Whelan R. J., and Bradstock R. A.. 2001. Ecology of Proteaceae with special reference to the Sydney region. Cunninghamia 6: 951–1015. [Google Scholar]
- Narum, S. R. , Buerkle C. A., Davey J. W., Miller M. R., and Hohenlohe P. A.. 2013. Genotyping‐by‐sequencing in ecological and conservation genomics. Molecular Ecology 22: 2841–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nge, F. J. , Biffin E., Thiele K. R., and Waycott M.. 2021. Reticulate evolution, ancient chloroplast haplotypes, and rapid radiation of the Australian plant genus Adenanthos . Frontiers in Ecology and Evolution 8: 616741. [Google Scholar]
- Nielsen, E. S. , Beger M., Henriques R., and von der Heyden S.. 2020. A comparison of genetic and genomic approaches to represent evolutionary potential in conservation planning Biological Conservation 251: 108770. [Google Scholar]
- Nieto‐Montes de Oca, A. , Barley A. J., Meza‐Lázaro R. N., García‐Vázquez U. O., Zamora‐Abrego J. G., Thomson R. C., and Leaché A. D.. 2017. Phylogenomics and species delimitation in the knob‐scaled lizards of the genus Xenosaurus (Squamata: Xenosauridae) using ddRADseq data reveal a substantial underestimation of diversity. Molecular Phylogenetics and Evolution 106: 241–253. [DOI] [PubMed] [Google Scholar]
- Nistelberger, H. M. , Tapper S.‐L., Coates D. J., McArthur S. L., and Byrne M.. 2020. As old as the hills: Pliocene palaeogeographical processes influence patterns of genetic structure in the widespread common shrub Banksia sessilis . Ecology and Evolution 11: 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Office of Environment and Heritage 2019. Banksia vincentia ‐ profile. Department of Planning and Environment, New South Wales Government, Parramatta, Australia. Website: https://www.environment.nsw.gov.au/threatenedSpeciesApp/profile.aspx?id=20291 [accessed 3 March 2022].
- Oksanen, J. 2011. vegan: community ecology package. R package version 2.5‐7. Available at https://cran.r-project.org/package=vegan [accessed April 2020].
- Ortego, J. , Gugger P. F., and Sork V. L.. 2018. Genomic data reveal cryptic lineage diversification and introgression in Californian golden cup oaks (section Protobalanus). New Phytologist 218: 804–818. [DOI] [PubMed] [Google Scholar]
- Pentinsaari, M. , Vos R., and Mutanen M.. 2017. Algorithmic single‐locus species delimitation: effects of sampling effort, variation and nonmonophyly in four methods and 1870 species of beetles. Molecular Ecology Resources 17: 393–404. [DOI] [PubMed] [Google Scholar]
- Phillips, S. J. , and Dudik M.. 2008. Modeling of species distributions with MaxEnt: new extensions and a comprehensive evaluation. Ecography 31: 161–175. [Google Scholar]
- Pickrell, J. K. , and Pritchard J. K.. 2012. Inference of population splits and mixtures from genome‐wide allele frequency data. PLoS Genetics 8: e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada, C. , Schizas N. V., and Yoshioka P. M.. 2008. Phenotypic plasticity or speciation? A case from a clonal marine organism. BMC Evolutionary Biology 8: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Website: https://www.R-project.org/ [Google Scholar]
- Ralls, K. , Sunnucks P., Lacy R. C., and Frankham R.. 2020. Genetic rescue: A critique of the evidence supports maximizing genetic diversity rather than minimizing the introduction of putatively harmful genetic variation. Biological Conservation 251:108784. [Google Scholar]
- Reichenbach, H. G. L. 1827. Iconographia botanica exotica, 1, 58. Self‐published, Leipzig, Germany. [Google Scholar]
- Reyes‐Velasco, J. , Manthey J. D., Bourgeouis Y., Freilich X., and Boissinot S.. 2018. Revisiting the phylogeography, demography and taxonomy of the frog genus Ptychadena in the Ethiopian highlands with the use of genome‐wide SNP data. PLoS One 13: e0190440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix, M. G. , and Harvey M. S.. 2012. Phylogeny and historical biogeography of ancient assassin spiders (Araneae: Archaeidae) in the Australian mesic zone: evidence for Miocene speciation within Tertiary refugia, Molecular Phylogenetics and Evolution 62: 375–396. [DOI] [PubMed] [Google Scholar]
- Rossetto, M. , Bragg J., Kilian A., McPherson H., van der Merwe M., and Wilson P. D.. 2019. Restore and Renew: a genomics‐era framework for species provenance delimitation. Restoration Ecology 27: 538–548. [Google Scholar]
- Rossetto, M. , Ens E. J., Honings T., Wilson P. D., Yap J.‐Y. S., Costello O., Round E. R., and Bowern C.. 2017. From songlines to genomes: prehistoric assisted migration of a rain forest tree by Australian Aboriginal people. PLoS One 12: e0186663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto, M. , Yap J.‐Y. S., Lemmon J., Bain D., Bragg J., Hogbin P., Gallagher R., et al. 2021. A conservation genomics workflow to guide practical management actions. Global Ecology and Conservation 26: e01492. [Google Scholar]
- Royal Botanic Gardens and Domain Trust, Sydney . 2002. onward [continuously updated]. PlantNET (The NSW Plant Information Network System). Website: https://plantnet.rbgsyd.nsw.gov.au/cgi-bin/NSWfl.pl?page=nswflandlvl=gnand%20name=Banksia. [accessed 22 February 2022].
- Rutherford, S. , Rossetto M., Bragg J. G., McPherson H., Benson D., Bonser S. P., and Wilson P. G.. 2018. Speciation in the presence of gene flow: population genomics of closely related and diverging Eucalyptus species. Heredity 121: 126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, S. , van der Merwe M., Wilson P. G., Kooyman R., and Rossetto M.. 2019. Managing the risk of genetic swamping of a rare and restricted tree. Conservation Genetics 20: 1113–1131. [Google Scholar]
- Rutherford, S. , Wan S. H., Cohen J. M., Benson D., and Rossetto M.. 2021. Looks can be deceiving: speciation dynamics of co‐distributed Angophora (Myrtaceae) species in a varying landscape Evolution 75: 310–329. [DOI] [PubMed] [Google Scholar]
- Seeholzer, G. F. , and Brumfield R. T.. 2018. Isolation by distance, not incipient ecological speciation, explains genetic differentiation in an Andean songbird (Aves: Furnariidae: Cranioleuca antisiensis, line‐cheeked spinetail) despite near threefold body size change across an environmental gradient. Molecular Ecology 27: 279–296. [DOI] [PubMed] [Google Scholar]
- Silcock, J. L. 2018. Aboriginal translocations: the intentional propagation and dispersal of plants in Aboriginal Australia. Journal of Ethnobiology 38: 390–405. [Google Scholar]
- Simpson, L. , Clements M. A., Crayn D. M., and Nargar K.. 2018. Evolution in Australia's mesic biome under past and future climates: insights from a phylogenetic study of the Australian Rock Orchids (Dendrobium speciosum complex, Orchidaceae). Molecular Phylogenetics and Evolution 118: 32–46. [DOI] [PubMed] [Google Scholar]
- Sjölund, M. J. , González‐Díaz P., Moreno‐Villena J. J., and Jump A. S.. 2019. Gene flow at the leading range edge: the long‐term consequences of isolation in European beech (Fagus sylvatica L. Kuhn). Journal of Biogeography 46: 2787–2799. [Google Scholar]
- Sloan, D. B. , Havird J. C., and Sharbrough J.. 2017. The on‐again, off‐again relationship between mitochondrial genomes and species boundaries. Molecular Ecology 26: 2212–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. E. 1793. A specimen of the botany of New Holland, vol. 1, 13, t. 4. J. Sowerby, London, Great Britain. [Google Scholar]
- Soltis, D. E. , Gitzendanner M. A., Stull G., Chester M., Chanderbali A., Chamala S., Jordon‐Thaden I., et al. 2013. The potential of genomics in plant systematics. Taxon 62: 886–898. [Google Scholar]
- Soltis, D. E. , and Kuzoff R. K.. 1995. Discordance between nuclear and chloroplast phylogenies in the Heuchera group (Saxifragaceae). Evolution 49: 727–742. [DOI] [PubMed] [Google Scholar]
- Stimpson M. L., Weston P. H., Telford I. R. H., and Bruhl J. J.. 2012. First instalment in resolution of the Banksia spinulosa complex (Proteaceae): B. neoanglica, a new species supported by phenetic analysis, ecology and geography. PhytoKeys 14: 57–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimpson, M. L. , Bruhl J. J., and Weston P. H.. 2014. Could this be Australia's rarest Banksia? Banksia vincentia (Proteaceae), a new species known from fourteen plants from south‐eastern New South Wales, Australia. Phytotaxa 163: 269–286. [Google Scholar]
- Stimpson, M. L. , Weston P. H., Whalley R. W. D. B., and Bruhl J. J.. 2016. A morphometric analysis of the Banksia spinulosa complex (Proteaceae) and its complex taxonomic implications. Australian Systematic Botany 29: 55–86. [Google Scholar]
- Stuglik, M. T. , and Babik W.. 2016. Genomic heterogeneity of historical gene flow between two species of newts inferred from transcriptome data. Ecology and Evolution 6: 4513–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford, D. L. 2002. PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4.0a. Sinauer, Sunderland, MA, USA. [Google Scholar]
- Thiele, K. R. , Weston P. H., and Mast A. R.. 2015. Paraphyly, modern systematics and the transfer of Dryandra into Banksia (Proteaceae): a response to George. Australian Systematic Botany 28: 194–202. [Google Scholar]
- Tillich, M. , Lehwark P., Pellizzer T., Ulbricht‐Jones E. S., Fischer A., Bock R., and Greiner S.. 2017. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Research 45: W6–W11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usher, A. V. 2011. Complex and dynamic hybridization between Banksia robur and Banksia oblongifolia is revealed by genetic and morphological surveys and experimental manipulations. Ph.D. dissertation, University of Wollongong, NSW, Australia.
- Usher, A. V. , Whelan R. J., and Ayre D. J.. 2010. Window of opportunity: an episode of recruitment in a Banksia hybrid zone demonstrates continuing hybridization and phenotypic plasticity. Annals of Botany 105: 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakkari, P. , Rusanen M., Heikkinen J., Huotari T., and Karkkainen K.. 2020. Patterns of genetic variation in leading‐edge populations of Quercus robur: genetic patchiness due to family clusters. Tree Genetics and Genomes 16: 73. [Google Scholar]
- Vargas, O. M. , Ortiz E. M., and Simpson B. B.. 2017. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high‐Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytologist 214: 1736–1750. [DOI] [PubMed] [Google Scholar]
- Weston, P. H. 2014. What has molecular systematics contributed to our knowledge of the Proteaceae? In Besse P. [ed.], Molecular plant taxonomy. Methods in molecular biology, 1115, 365–397. Humana Press, Totowa, NJ, USA. [DOI] [PubMed] [Google Scholar]
- Whelan, R. J. , and Ayre D. J.. 2020. Long inter‐fire intervals do not guarantee a large seed bank in a serotinous shrub (Banksia spinulosa Sm.). Journal of Ecology 108: 1690–1702. [Google Scholar]
- Wilson, T. C. , Rossetto M., Bain D., Yap J.‐Y. S., Wilson P. D., Stimpson M. L., Weston P. H., and Croft L.. 2022. A turn in species conservation for hairpin banksias: Demonstration of oversplitting leads to a better management of diversity. Dryad , Dataset. 10.5061/dryad.69p8cz94x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap, J.‐Y. S. , Rossetto M., Das S., Wilson P. D., Beaumont L. J., and Henry R. J.. 2022. Tracking habitat or testing its suitability? Similar distributional patterns can hide very different histories of persistence versus nonequilibrium dynamics. Evolution 76: 1209–1228. [DOI] [PubMed] [Google Scholar]
- Zachos, F. 2018. Mammals and meaningful taxonomic units: the debate about species concepts and conservation. Mammal Review 43: 153–159. [Google Scholar]
- Zhang, C. , Stadler T., Klopfstein S., Heath T. A., and Ronquist F.. 2016. Total‐evidence dating under the fossilized birth–death process. Systematic Biology 65: 228–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X. , Levine D., Shen J., Gogarten S. M., Laurie C., and Weir B. S.. 2012. A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28: 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Wild‐collected and cultivated hairpin banksia specimens sampled for genomic sequencing and chloroplast genome sequencing.
Appendix S2. Environmental niche modelling for the Banksia cunninghamii clade: methods.
Appendix S3. The 19 standard bioclimatic variables used to estimate an overall environmental envelope to project spatial distribution of environmental suitability for the hairpin banksias using a select group of populations.
Appendix S4. Cleaned data set of spatial records used to project spatial distribution of environmental suitability for the Banksia cunninghamii clade of the hairpin banksias.
Appendix S5. Cleaned data set of spatial records used to project spatial distribution of environmental suitability for the Banksia spinulosa clade of the hairpin banksias.
Appendix S6. Principal component analysis of nDNA data for the hairpin banksia B. cunninghamii clade identified in the network analysis.
Appendix S7. LEA snmf analysis for the nDNA data set of the hairpin banksia B. cunninghamii clade, as recovered by network analysis of genomic data.
Appendix S8. Averaged Q values (at K = 2 ancestral populations) for each population of hairpin banksia from separate LEA snmf analyses for the B. cunninghamii clade and the B. spinulosa clade.
Appendix S9. Averaged Q values (at K = 3 ancestral populations) for each population of hairpin banksia from separate LEA snmf analyses for the B. cunninghamii clade and the B. spinulosa clade.
Appendix S10. Averaged Q values (at K = 4 ancestral populations) for each population of hairpin banksia from separate LEA snmf analyses for the B. cunninghamii clade and the B. spinulosa clade.
Appendix S11. F ST pairwise matrix for the hairpin banksia B. cunninghamii clade identified from network analysis of nDNA.
Appendix S12. Isolation by distance graphs derived from nDNA data for the (a) Banksia cunninghamia clade and (b) B. spinulosa clade, as identified by network analysis.
Appendix S13. Heterozygosity values retrieved from two separate analyses of the genomic data for the hairpin banksia complex.
Appendix S14. Principal component analysis of genetic diversity from nDNA data for the hairpin banksia Banksia spinulosa clade identified in the network analysis.
Appendix S15. LEA snmf analysis for nDNA data set of the hairpin banksia B. spinulosa clade, as recovered by network analysis of genomic data.
Appendix S16. F ST pairwise matrix for the hairpin banksia B. spinulosa clade identified from network analysis of nDNA.
Appendix S17. Chloroplast sequence reads and coverage for members of the B. cunninghamii (cun) and B. spinulosa (spin) clades as defined from network analysis of the nDNA data set of the hairpin banksias.
Appendix S18. Morphological variation across the seed‐grown ex situ Banksia collection at Australian Botanic Garden (ABG) sourced from Vincentia, NSW (living collection number 20160262).
Appendix S19. The output produced by TreeMix and OptM for the nDNA data set for 23 populations of the hairpin banksias with recent hybrids removed.
Appendix S20. Summary of environmental niche model performance data for the total occurrence records of the Banksia cunninghamii clade (Total) or partitioned as three separate groups (North, Central, and South) according to the results from snmf analysis.
Appendix S21. Principal component analysis of environmental data for the hairpin banksia B. cunninghamii clade without B. vincentia.
Appendix S22. Principal component analysis of environmental data for samples of the hairpin banksia B. spinulosa clade nDNA data set.
Appendix S23. Present‐day and last glacial maximum (LGM) environmental niche modeling projections provided for the samples of the hairpin banksia B. spinulosa clade nDNA data set.
Data Availability Statement
The nuclear genomic data sets and the chloroplast genome sequence alignment are available in the Dryad Digital Repository: https://doi.org/10.5061/dryad.69p8cz94x (Wilson et al., 2022).