

Abstract
Objective
Complete genetic deficiency of the complement component C2 is a strong risk factor for monogenic systemic lupus erythematosus (SLE), but whether heterozygous C2 deficiency adds to the risk of SLE or primary Sjögren's syndrome (SS) has not been studied systematically. This study was undertaken to investigate potential associations of heterozygous C2 deficiency and C4 copy number variation with clinical manifestations in patients with SLE and patients with primary SS.
Methods
The presence of the common 28‐bp C2 deletion rs9332736 and C4 copy number variation was examined in Scandinavian patients who had received a diagnosis of SLE (n = 958) or primary SS (n = 911) and in 2,262 healthy controls through the use of DNA sequencing. The concentration of complement proteins in plasma and classical complement function were analyzed in a subgroup of SLE patients.
Results
Heterozygous C2 deficiency—when present in combination with a low C4A copy number—substantially increased the risk of SLE (odds ratio [OR] 10.2 [95% confidence interval (95% CI) 3.5–37.0]) and the risk of primary SS (OR 13.0 [95% CI 4.5–48.4]) when compared to individuals with 2 C4A copies and normal C2. For patients heterozygous for rs9332736 with 1 C4A copy, the median age at diagnosis was 7 years earlier in patients with SLE and 12 years earlier in patients with primary SS when compared to patients with normal C2. Reduced C2 levels in plasma (P = 2 × 10−9) and impaired function of the classical complement pathway (P = 0.03) were detected in SLE patients with heterozygous C2 deficiency. Finally, in a primary SS patient homozygous for C2 deficiency, we observed low levels of anti–Scl‐70, which suggests a risk of developing systemic sclerosis or potential overlap between primary SS and other systemic autoimmune diseases.
Conclusion
We demonstrate that a genetic pattern involving partial deficiencies of C2 and C4A in the classical complement pathway is a strong risk factor for SLE and for primary SS. Our results emphasize the central role of the complement system in the pathogenesis of both SLE and primary SS.
INTRODUCTION
Deficiencies in genes of the early classical complement pathway (i.e., C1Q, C1R, C1S, C2, and C4) are among the strongest risk factors for monogenic systemic lupus erythematosus (SLE) and lupus‐like disease (1, 2). While rare, C1Q deficiency translates into SLE in ~90% of the described cases, whereas deficiencies of C1R, C1S, and C4 have a penetrance in the range of 65–80% (3). In contrast, complete C2 deficiency is one of the most common complement deficiencies, with an estimated prevalence of 1:20,000 in the Swedish population and a penetrance for SLE of ~25% (4, 5, 6).
The predominant cause of complete C2 deficiency is a 28‐bp deletion (rs9332736) in the exon/intron boundary of exon 6. The deletion introduces an early stop codon in the C2 transcript, thereby leading to the absence of the C2 protein in plasma (7, 8). The minor allele frequency of the 28‐bp C2 deletion is 0.01 in populations of European descent, which means that 1 of 50 individuals are heterozygous carriers of the deleterious variant. Although a few case reports and small studies exist (9, 10, 11, 12), heterozygous C2 deficiency due to rs9332736 and the association with rheumatic disease have not previously been evaluated systematically.
In addition to heterozygous C2 deficiency, a high level of copy number variation is seen for the paralogous C4 genes C4A and C4B. We and others have previously shown that a low copy number of C4A is strongly associated with both SLE and primary Sjögren's syndrome (SS) (13, 14, 15, 16, 17, 18). In the general population, the copy number of C4A ranges between 0 and 5 copies, with ~50% of individuals having 2 copies of C4A. For C4B, the copy number generally ranges between 0 and 4 copies.
The purpose of the current study was to evaluate the interplay between heterozygous C2 deficiency and the common copy number variation of C4 in relation to the risk of SLE and primary SS. Further, we aimed to evaluate the clinical consequences of the partial complement deficiencies in SLE and primary SS.
PATIENTS AND METHODS
Study participants
In the current study, we included patients diagnosed as having SLE or primary SS at Scandinavian rheumatology clinics and healthy blood donors and population controls as previously described (19, 20). SLE patients met ≥4 of the American College of Rheumatology (ACR) 1982 revised criteria for the classification of SLE (21), while primary SS patients fulfilled the American–European Consensus Group criteria for primary SS (22). Patients and controls were analyzed by targeted DNA sequencing as part of the dissecting disease mechanisms in three systemic inflammatory autoimmune diseases with an interferon signature (DISSECT) project, and basic characteristics of the study participants are presented in Supplementary Table 1 (available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.42270). The capturing array (23), targeted sequencing, genotyping of single‐nucleotide variants, and quality control have been described previously (19, 20), and a brief summary of the workflow, including variant‐ and individual‐based quality control, can be found in the Supplementary Information (available at http://onlinelibrary.wiley.com/doi/10.1002/art.42270). In addition, we included 1,000 Swedish population controls analyzed by whole‐genome sequencing as part of the SweGen project (24), and the workflow is presented in the Supplementary Information.
For the genetic analysis, we combined all population controls from the SweGen project and the 2 targeted sequencing studies previously conducted by our group (19, 20), excluding related individuals and genetic population outliers (Supplementary Information). Findings on the prevalence of heterozygous carriers of the C2 variant rs9332736 in the individual study cohorts of SLE and primary SS patients and healthy controls, as well as the association between the rs9332736 variant and C4A copy number, can be found in Supplementary Figure 1 (available at http://onlinelibrary.wiley.com/doi/10.1002/art.42270). In order to increase the power of the functional and clinical analysis of SLE and primary SS patients, we included all patients (including population outliers) who passed the quality filters for genotype calls of the 28‐bp C2 deletion and C4 copy number (Supplementary Information, http://onlinelibrary.wiley.com/doi/10.1002/art.42270). The individual studies were approved by local ethics committees, and all study participants gave informed consent.
Genotyping of the 28‐bp C2 deletion
As the previous analyses of genetic variation in the targeted sequencing data by our group comprised single‐nucleotide variants only (19, 20), a focused reanalysis of the sequencing data was performed in order to genotype the 28‐bp deletion rs9332736 in C2 as described in the Supplementary Information, including quality control of the genotype calls. We excluded 1 SLE patient with a heterozygous call for rs9332736 that clustered with homozygous carriers of the C2 deletion (Supplementary Information, http://onlinelibrary.wiley.com/doi/10.1002/art.42270).
Analysis of C4 copy number and HLA alleles from DNA sequencing data
The complement C4 copy numbers for both targeted sequencing data and whole‐genome sequencing data were estimated based on read depth using the GermlineCNVCaller (Genome Analysis Toolkit) as described previously (18), and a brief description can be found in the Supplementary Information. The integer copy number of C4A and C4B was estimated using the relative read depths of 5 single‐nucleotide variants in exon 26 specific for C4A and C4B while accounting for the total copy number of C4. Alleles of the 6 HLA genes HLA–A, B, C, DPB1, DQB1, and DRB1 were called from sequencing reads at 2‐field (i.e., 4‐digit) resolution using xHLA (25) as described previously (18).
Clinical data, analysis of complement proteins in plasma, and autoantibody status
Plasma levels of C2 had been analyzed in a subset of SLE patients by electroimmunoassay as previously described (26), with the C2 level being reported relative to the concentration of a reference serum. Measurement of plasma C3 levels and plasma C4 levels in SLE patients (27) and function of the classical complement pathway (28, 29) have been described previously. Clinical information including autoantibody status was extracted from medical records.
Statistical analysis
R version 4.0.4 (30) was used for statistical analyses that included logistic regression, analysis of variance, and Cox proportional hazards regression. Two‐tailed P values less than 0.05 were considered significant, and the models and covariates that were included are described in the text or in the figure legends.
Data availability
Raw data for individual figures are available in the Supplementary Data (http://onlinelibrary.wiley.com/doi/10.1002/art.42270). Genotype data at the individual level are not publicly available since some of the information could compromise research participant privacy and consent. Scripts for calling C4 copy number in GATK GermlineCNVCaller are available upon request.
RESULTS
Heterozygous C2 deficiency in combination with low C4A copy number associated with SLE and primary SS
Initially, we analyzed the occurrence of a 28‐bp deletion in complement C2 (i.e., rs9332736) in our cohort and found that 3.3% of both SLE patients (n = 31) and primary SS patients (n = 30) were heterozygous for the variant compared to 1.9% of healthy controls (n = 43). Further, we identified 3 patients who were homozygous for the deletion. The heterozygous presence of the 28‐bp C2 deletion rs9332736 was associated with an increased risk for both SLE and primary SS (odds ratio [OR] 1.75 [95% confidence interval (95% CI) 1.08–2.81] and OR 1.72 [95% CI 1.05–2.81], respectively) (Figure 1A).
Figure 1.
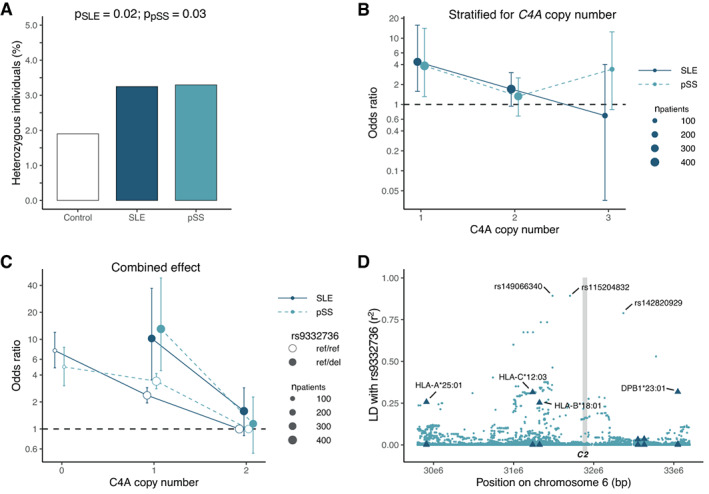
Heterozygosity of the 28‐bp C2 deletion rs9332736 in systemic lupus erythematosus (SLE) patients and primary Sjögren's syndrome (SS) patients. A, Prevalence of heterozygous carriers of the C2 loss‐of‐function variant rs9332736 in SLE patients (n = 955), primary SS patients (n = 910), and healthy controls (n = 2,262). Individuals homozygous for the rs9332736 variant (2 SLE patients and 1 primary SS patient) were excluded. B, Risk of association with SLE or primary SS according to number of C4A copies in rs9332736, with odds calculated relative to healthy controls. C, Risk of association with SLE or primary SS according to the combined effect of C4A copy number and rs9332736 heterozygosity (ref/del), with odds calculated relative to a C4A copy number of 2 and normal C2 (ref/ref). Due to rs9332736 segregating with C4A, no individuals heterozygous for rs9332736 have 0 C4A copies. Data were analyzed using logistic regression and adjusted for sex (A, B, and C) and C4B copy number (B and C). In B and C, bars show the 95% confidence intervals. D, Linkage disequilibrium (LD; r2) between the 28‐bp C2 deletion rs9332736 and HLA alleles/biallelic single‐nucleotide polymorphisms in the HLA region. LD with HLA alleles for 6 HLA genes are indicated by triangles, including HLA alleles A, C, B, DRB1, DQB1, and DPB1. The vertical gray‐shaded line indicates the genomic position of C2. LD was estimated using SweGen whole‐genome sequencing samples (n = 1,000). Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42270/abstract.
A low copy number of C4A is a strong risk factor for both SLE and primary SS (17, 18), and since both C4 and C2 are part of the early classical complement pathway, we investigated whether heterozygous C2 deficiency in combination with a low copy number of C4A would impose an even greater disease risk. The copy number of C4A ranged between 0 and 5 copies for patients and healthy controls. However, all individuals heterozygous for rs9332736 carried 1–3 copies of C4A due to linkage disequilibrium between C4 and C2 (both genes are located in the HLA region on chromosome 6). Interestingly, we detected an increased risk of both SLE and primary SS for patients heterozygous for rs9332736 when the deletion was present in combination with a C4A copy number of 1 (Figure 1B). In contrast, heterozygous C2 deficiency was not associated with SLE or primary SS when present with a C4A copy number of 2 or 3, and no association was seen with a low copy number of C4B.
We next evaluated the combined effect of low C4A copy number and heterozygous C2 deficiency in comparison to individuals with 2 C4A copies and normal C2. A C4A copy number of 1 in combination with heterozygous C2 deficiency was associated to an even greater extent with a substantially increased risk of both SLE and primary SS (OR 10.2 and OR 13.0, respectively) (Figure 1C) than was a C4A copy number of 0 (OR 7.5 for SLE and OR 4.9 for primary SS). Further, we noted a tendency toward a significant interaction between heterozygous C2 deficiency and C4A copy number (for SLE and primary SS combined, P = 0.06 by logistic regression adjusted for C4B copy number and sex).
Due to C2 being located in the HLA region, we assessed the linkage between the 28‐bp C2 deletion rs9332736 and disease‐associated HLA alleles. We noted a strong linkage disequilibrium between rs9332736 and multiple single‐nucleotide polymorphisms, but the linkage to HLA alleles was limited (Figure 1D), indicating that the effect of 28‐bp C2 deletion was not through an indirect link to a disease‐associated HLA allele such as DRB1*03:01 or DRB1*15:01.
In summary, we identified heterozygous C2 deficiency as a risk factor for SLE and primary SS when it occurs in combination with low C4A copy number, highlighting the role of the early classical complement pathway in the pathogenesis of SLE and primary SS.
Decreased classical complement function for rs9332736 carriers
Having demonstrated a genetic association between the 28‐bp C2 deletion and SLE and primary SS, we continued evaluating the functional consequences of heterozygous C2 deficiency. Levels of complement proteins were analyzed in a subgroup of SLE patients. As expected, patients heterozygous for the deleterious C2 variant rs9332736 had lower plasma C2 levels when compared to patients without the genetic variant (P for C2 = 2 × 10−9) (Figure 2A). Further, we detected lower function of the classical complement pathway in patients heterozygous for rs9332736 (P = 0.03) (Figure 2B).
Figure 2.
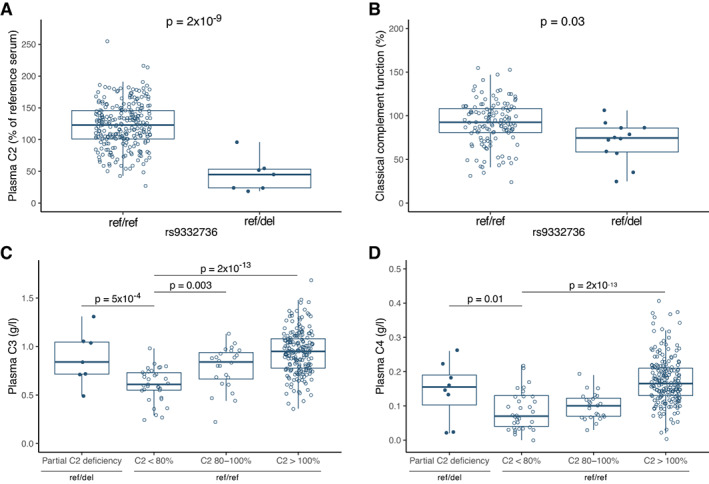
Concentration of plasma complement components in systemic lupus erythematosus (SLE) patients with the 28‐bp C2 deletion rs9332736 (ref/del) and in SLE patients with normal C2 (ref/ref), including plasma C2 concentration relative to a reference serum (n = 261) (A), classical complement function (n = 140) (B), plasma C3 (C) and plasma C4 (D) concentration stratified by presence of the 28‐bp C2 deletion rs9332736 and plasma C2 concentration (n = 258 for C and D). Data were analyzed by analysis of variance and adjusted for sex and age at sampling (A, C, and D), and for copy number of C4A and C4B (B). Plasma concentration of C4 was square root transformed. Data are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median, and whiskers extend to 1.5 times the interquartile range. Solid circles represent individual SLE patients heterozygous for rs9332736; open circles represent individual SLE patients with normal C2. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42270/abstract.
Nevertheless, a substantial number of SLE patients without the deleterious rs9332736 variant presented with low levels of plasma C2 (defined here as <80%) (Figure 2A), and we investigated whether the low concentration could be explained by other, nongenetic effects. Analysis of C3 and C4 levels in plasma revealed lower concentration of both proteins for this subgroup of SLE patients, whereas patients heterozygous for rs9332736 had normal levels of C3 and C4 (Figures 2C and D). These data suggest that the lower plasma concentration of C2 in SLE patients without a genetic cause for low C2 may be due to complement activation and consumption of proteins in the complement pathway.
Lower age at diagnosis in patients with heterozygous C2 deficiency
Next, we evaluated the association between heterozygous C2 deficiency and clinical manifestations in SLE. However, we did not detect any associations between the 28‐bp C2 deletion and the ACR criteria used for clinical classification of SLE patients (Supplementary Figure 2, http://onlinelibrary.wiley.com/doi/10.1002/art.42270). The relatively low number of patients carrying the rs9332736 variant likely limited the power in cross‐sectional analyses, and we reasoned that time‐dependent analyses would be more suitable.
Therefore, we evaluated how the deleterious C2 variant rs9332736 affected the progression of SLE and primary SS. Intriguingly, SLE patients heterozygous for rs9332736 who had a C4A copy number of 1 were diagnosed earlier when compared to patients with normal C2 (P for rs9332736 = 0.002) (Figure 3A), with the difference in median age at diagnosis being 7 years. In contrast, rs9332736 was not found to have an effect on disease progression in SLE patients with 2 copies of C4A (P for rs9332736 = 0.70) (Figure 3A). A similar pattern was seen for primary SS patients, where patients with heterozygous C2 deficiency and a C4A copy number of 1 tended to have lower age at diagnosis (P for rs9332736 = 0.05) (Figure 3B), the difference in median age at diagnosis being 12 years. Again, rs9332736 was not found to have an effect on disease progression in patients with primary SS with 2 C4A copies (P for rs9332736 = 0.48) (Figure 3B).
Figure 3.
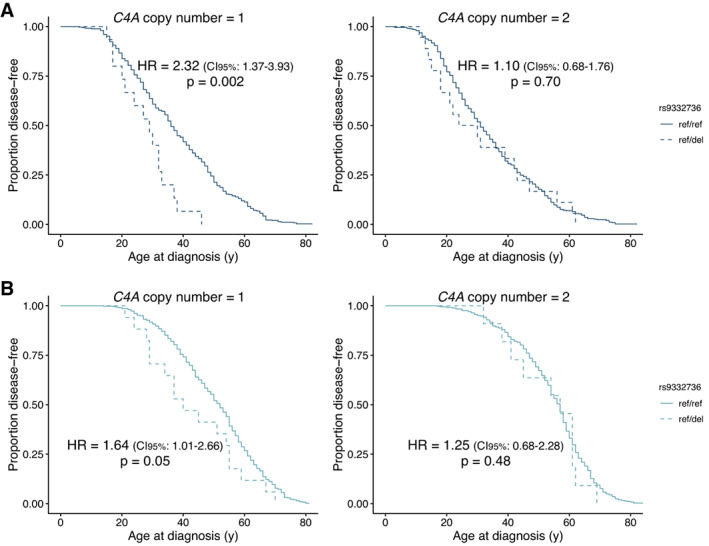
Age at diagnosis of systemic lupus erythematosus (SLE) and primary Sjögren's syndrome (SS) among patients with the 28‐bp C2 deletion rs9332736 (ref/del) relative to SLE patients and primary SS patients with normal C2 (ref/ref). Kaplan‐Meier plots depict the age at diagnosis of SLE in those with the 28‐bp C2 deletion rs9332736 and 1 C4A copy (n = 15) or 2 C4A copies (n = 18), relative to SLE patients with normal C2 and 1 C4A copy (n = 352) or 2 C4A copies (n = 446) (A), and age at diagnosis of primary SS in those with the 28‐bp C2 deletion rs9332736 and 1 C4A copy (n = 17) or 2 C4A copies (n = 11), relative to primary SS patients with normal C2 and 1 C4A copy (n = 412) or 2 C4A copies (n = 363) (B). Data were analyzed using a Cox proportional hazards regression model adjusted for sex and C4B copy number. HR = hazard ratio; 95% CI = 95% confidence interval. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42270/abstract.
We continued by analyzing the association between rs9332736 and age at first event of nephritis in SLE patients and identified a similar pattern. It was found that SLE patients heterozygous for 28‐bp C2 deletion who had 1 copy of C4A tended to have earlier occurrences of nephritis when compared to SLE patients with normal C2, although the difference was not significant (P for rs9332736 = 0.17) (Figure 4). No difference was seen in the age at first occurrence of nephritis between SLE patients with normal C2 and SLE patients heterozygous for 28‐bp C2 deletion who had 2 copies of C4A (P for rs9332736 = 0.73) (Figure 4).
Figure 4.
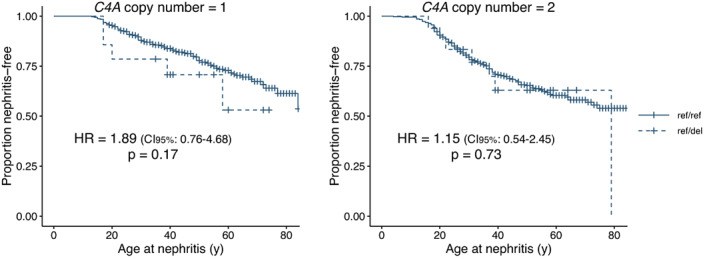
Age at first occurrence of nephritis in SLE patients. The Kaplan‐Meier plot depicts age at first nephritis event in SLE patients with the 28‐bp C2 deletion rs9332736 and 1 C4A copy (n = 14) or 2 C4A copies (n = 18), and in SLE patients with normal C2 and 1 C4A copy (n = 322) or 2 C4A copies (n = 412). Data were analyzed using a Cox proportional hazards regression model adjusted for sex and C4B copy number. See Figure 3 for definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42270/abstract.
In a previous study by our group, we found a strong association between C4A copy number and the presence of autoantibodies against SSA/Ro and SSB/La in systemic inflammatory autoimmune diseases (18), and therefore, we evaluated whether rs9332736 affected the presence of autoantibodies in SLE and primary SS. However, analysis of SSA/Ro, SSB/La, anti–U1 RNP, anti‐Sm, and antiphospholipid antibodies in SLE and primary SS patients only showed minor differences when evaluating the role of the 28‐bp C2 deletion (Supplementary Figures 3A–E, http://onlinelibrary.wiley.com/doi/10.1002/art.42270). Further, the sex distribution of patients with heterozygous C2 deficiency did not differ from that of patients with normal C2 (Supplementary Figure 3F).
Overall, we detected earlier onsets of disease for both SLE and primary SS patients heterozygous for the 28‐bp C2 deletion rs9332736 when present in combination with a C4A copy number of 1. A similar but not significant association was seen between heterozygous 28‐bp C2 deletion and age at the first occurrence of nephritis in SLE patients, although this needs to be verified in a larger cohort.
Monogenic SLE and primary SS due to complete C2 deficiency
Complete C2 deficiency constitutes a major risk factor for monogenic SLE (6), and we identified 2 SLE patients homozygous for the 28‐bp C2 deletion rs9332736. Interestingly, we also identified 1 primary SS patient with complete C2 deficiency due to the rs9332736 variant, and a brief clinical summary of the 3 C2‐deficient patients is presented in Supplementary Figure 4 (http://onlinelibrary.wiley.com/doi/10.1002/art.42270). None of the population controls were homozygous for the rs9332736 deletion.
DISCUSSION
In the current study, we observed that heterozygous C2 deficiency in combination with a low copy number of C4A substantially increases the risk of both SLE and primary SS (summarized in Figure 5). Further, this genetic combination was also found to be associated with lower age at diagnosis. Although only 1.5% of the patient cohort was observed to have heterozygous C2 deficiency in combination with a C4A copy number of 1, these characteristics still explain a likely genetic cause of disease at a much larger proportion of patients than can be explained by monogenic disease. In comparison, 0.18% of population controls carried the combination of rs9332736 heterozygosity and a C4A copy number of 1. As we had no clinical information for the population controls, we were not able to describe whether these individuals were diagnosed as having SLE or primary SS. Further, a previous study on combined heterozygous deficiencies of C2 and C4 involving 6 families did not find that all individuals with the C2/C4A risk combination had clinical symptoms (10), suggesting an incomplete penetrance.
Figure 5.
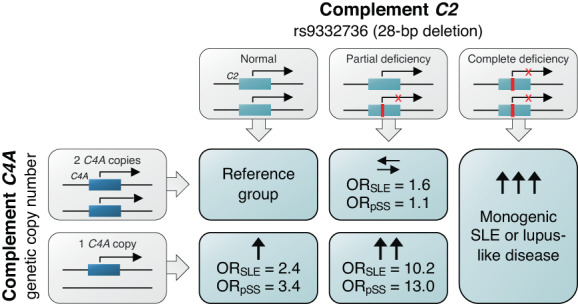
Summary of the conduct of the study. Homozygosity of the 28‐bp deletion rs9332736 in C2 is associated with monogenic lupus. Heterozygous individuals are not at risk of SLE or primary SS if they have 2 or more copies of C4A, whereas individuals heterozygous for rs9332736 who have 1 copy of C4A are at substantial risk of both SLE and primary SS. OR = odds ratio (see Figure 3 for other definitions).
In addition to heterozygous C2 deficiency, we also identified 3 patients with homozygous C2 deficiency. Interestingly, one of the patients with homozygous C2 deficiency was diagnosed as having primary SS. Homozygous C2 deficiency has mainly been associated with the risk of severe infections and SLE, but other rheumatic diseases, such as undifferentiated connective tissue disease and vasculitis, have also been described (5, 6, 11). Further, Sjögren's syndrome has been described as a condition secondary to SLE and vasculitis in patients with complete C2 deficiency, showing that the absence of C2 may cause a range of different manifestations (6, 10). In the present study, low levels of anti–Scl‐70 were observed in the patient with primary SS, suggesting a risk of developing systemic sclerosis or a potential overlap between primary SS and other systemic autoimmune diseases (31).
Both C2 and C4A are located in the HLA region on chromosome 6, and previous studies have shown that the copy number of C4A is inversely correlated with the DRB1*03:01 allele that is a risk factor for both SLE and primary SS in populations of European ancestry (32). Still, the findings of recent studies involving participants of various ancestries suggest that the copy number of C4A is a causative factor for SLE and primary SS as it has consistently been found to be a risk factor across all populations studied (14, 17). In addition, complete deficiency of the genes C1Q, C1R, C1S, C4, and C2 are among the strongest risk factors for monogenic SLE. The apparent interaction between C4A copy number and heterozygous C2 deficiency that was found in this study further strengthens the pivotal role of the classical complement pathway in the pathogenesis of both SLE and primary SS, indicating that impaired function of this pathway may lead to disease.
The increased risk of disease for individuals heterozygous for rs9332736 who also had a C4A copy number of 1 translated directly into lower age at diagnosis for both SLE and primary SS patients, whereas heterozygous C2 deficiency did not affect the age of diagnosis among patients with 2 C4A copies. This is in line with the observation that heterozygous C2 deficiency is only a genetic risk factor for SLE and primary SS when present in combination with a C4A copy number of 1. Also, we noted a weak tendency toward earlier development of nephritis for SLE patients with heterozygous C2 deficiency and a low C4A copy number, although this was not significant and should be verified in a larger cohort. We did not see any associations between rs93322736 and specified autoantibodies and other clinical manifestations, which may have been due to low statistical power in the cross‐sectional analyses.
The relationship between deficiencies in the early classical complement pathway and SLE has generally been ascribed to impaired clearance of apoptotic cells and defective handling of immune complexes, which may lead to the loss of self tolerance, the activation of self‐reactive immune cells, and the production of autoantibodies (1, 33). Alternatively, aberrant activation of the complement system may also lead to disease, and levels of C3 and C4 together with classical complement function (e.g., CH50) are currently used as biomarkers for complement activation in the classification of SLE (34) as well as in the evaluation of disease activity in patients with SLE (35). However, the common genetic causes of low complement, such as variation in C4 copy number and heterozygous C2 deficiency, obscure the diagnostic utility of these measures, and genetic analyses may add useful information in this regard. In patients with primary SS, the complement cascade has been studied to a lesser extent, but the results of the present study further highlight the role of the classical complement pathway in primary SS patients, and previous studies have shown that complement status may have prognostic value with regard to disease activity and adverse outcomes in primary SS (36, 37).
Finally, while the 28‐bp C2 deletion rs9332736 exists in all populations of European descent, it is relatively uncommon in African and Asian populations (Supplementary Tables 2 and 3, http://onlinelibrary.wiley.com/doi/10.1002/art.42270), and therefore, the current results do not apply globally. However, other loss‐of‐function C2 variants have been described (38, 39, 40), and as a low C4A copy number has been found to be common in all populations (14, 17), the combination of heterozygous deficiencies of C2 and C4A may also be a risk factor for patients of other ethnicities.
In conclusion, we report that heterozygous C2 deficiency is a strong risk factor for SLE and primary SS when present together with a low C4A copy number. These results show that partial deficiencies affecting multiple genes of the classical complement pathway may increase the risk of disease substantially when present in combination, thereby emphasizing the role of the complement system in systemic inflammatory autoimmune diseases.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Lundtoft had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Lundtoft, Sjöwall, Rantapää‐Dahlqvist, Bengtsson, Jönsen, Svenungsson, Leonard, Rönnblom.
Acquisition of data
Lundtoft, Sjöwall, Rantapää‐Dahlqvist, Bengtsson, Jönsen, Pucholt, Wu, Lundström, Eloranta, Gunnarsson, Baecklund, Jonsson, Hammenfors, Forsblad‐d'Elia, Eriksson, Mandl, Bucher, Norheim, Johnsen, Omdal, Kvarnström, Wahren‐Herlenius, Truedsson, Nilsson, Kozyrev, Bianchi, Lindblad‐Toh, Yu, Nordmark, Sandling, Svenungsson, Leonard, Rönnblom.
Analysis and interpretation of data
Lundtoft, Sjöwall, Rantapää‐Dahlqvist, Bengtsson, Jönsen, Pucholt, Yu, Sandling, Svenungsson, Leonard, Rönnblom.
ADDITIONAL DISCLOSURES
Author Pucholt is an employee of Olink Proteomics. Author Mandl is an employee of Novartis.
Supporting information
Disclosure Form
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
DNA sequencing and genotyping were performed at the SNP&SEQ Technology Platform in Uppsala, part of the National Genomics Infrastructure Sweden and Science for Life Laboratory. Computations were enabled by resources in projects sens2017142 and sens2020577, provided by the Swedish National Infrastructure for Computing at the Uppsala Multidisciplinary Center for Advanced Computational Science. Members of the DISSECT consortium and the ImmunoArray consortium are listed in the Supplementary Information.
Supported by the Swedish Research Council for Medicine and Health, the Swedish Rheumatism Association, King Gustaf V's 80‐Year Foundation, the Swedish Heart‐Lung Foundation, ALF funding from Stockholm County and Region Östergötland, the Swedish Society for Medical Research, the Swedish Society of Medicine, the Ingegerd Johansson donation, and the Gustafsson Family Foundation. Dr. Lindblad‐Toh's work was supported by a Wallenberg Scholarship.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42270&file=art42270‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Sturfelt G, Truedsson L. Complement in the immunopathogenesis of rheumatic disease [review]. Nat Rev Rheumatol 2012;8:458. [DOI] [PubMed] [Google Scholar]
- 2. Macedo ACL, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway [review]. Front Immunol 2016;7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lintner KE, Wu YL, Yang Y, et al. Early components of the complement classical activation pathway in human systemic autoimmune diseases [review]. Front Immunol 2016;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Truedsson L, Sturfelt G, Nived O. Prevalence of the type I complement C2 deficiency gene in Swedish systemic lupus erythematosus patients. Lupus 1993;2:325–7. [DOI] [PubMed] [Google Scholar]
- 5. Jönsson G, Truedsson L, Sturfelt G, et al. Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Medicine (Baltimore) 2005;84:23–34. [DOI] [PubMed] [Google Scholar]
- 6. Jönsson G, Sjöholm AG, Truedsson L, et al. Rheumatological manifestations, organ damage and autoimmunity in hereditary C2 deficiency. Rheumatology (Oxford) 2007;46:1133–9. [DOI] [PubMed] [Google Scholar]
- 7. Johnson CA, Densen P, Hurford RK, et al. Type I human complement C2 deficiency. A 28‐base pair gene deletion causes skipping of exon 6 during RNA splicing. J Biol Chem 1992;267:9347–53. [PubMed] [Google Scholar]
- 8. Belot A, Rice GI, Omarjee SO, et al. Contribution of rare and predicted pathogenic gene variants to childhood‐onset lupus: a large, genetic panel analysis of British and French cohorts. Lancet Rheumatol 2020;2:e99–109. [DOI] [PubMed] [Google Scholar]
- 9. Sullivan KE, Petri MA, Schmeckpeper BJ, et al. Prevalence of a mutation causing C2 deficiency in systemic lupus erythematosus. J Rheumatol 1994;21:1128–33. [PubMed] [Google Scholar]
- 10. Hartmann D, Fremeaux‐Bacchi V, Weiss L, et al. Combined heterozygous deficiency of the classical complement pathway proteins C2 and C4. J Clin Immunol 1997;17:176–84. [DOI] [PubMed] [Google Scholar]
- 11. Lipsker DM, Schreckenberg‐Gilliot C, Uring‐Lambert B, et al. Lupus erythematosus associated with genetically determined deficiency of the second component of the complement. Arch Dermatol 2000;136:1508–14. [DOI] [PubMed] [Google Scholar]
- 12. Yang WJ, Rich E, Saint‐Cyr C, et al. Hereditary heterozygous C2 deficiency: variable clinical and serological manifestations among three sisters [review]. Curr Rheumatol Rev 2017;13:158–60. [DOI] [PubMed] [Google Scholar]
- 13. Yang Y, Chung EK, Wu YL, et al. Gene copy‐number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in european americans. Am J Hum Genet 2007;80:1037–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen JY, Ling Wu Y, Yin Mok M, et al. Effects of complement C4 gene copy number variations, size dichotomy, and C4A deficiency on genetic risk and clinical presentation of systemic lupus erythematosus in East Asian populations. Arthritis Rheumatol 2016;68:1442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsang‐A‐Sjoe MW, Bultink IE, Korswagen LA, et al. Comprehensive approach to study complement C4 in systemic lupus erythematosus: gene polymorphisms, protein levels and functional activity. Mol Immunol 2017;92:125–31. [DOI] [PubMed] [Google Scholar]
- 16. Jüptner M, Flachsbart F, Caliebe A, et al. Low copy numbers of complement C4 and homozygous deficiency of C4A may predispose to severe disease and earlier disease onset in patients with systemic lupus erythematosus. Lupus 2018;27:600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kamitaki N, Sekar A, Handsaker RE, et al. Complement genes contribute sex‐biased vulnerability in diverse disorders. Nature 2020;582:577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lundtoft C, Pucholt P, Martin M, et al. Complement C4 copy number variation is linked to SSA/Ro and SSB/La autoantibodies in systemic inflammatory autoimmune diseases. Arthritis Rheumatol 2022;74:1440–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sandling JK, Pucholt P, Rosenberg LH, et al. Molecular pathways in patients with systemic lupus erythematosus revealed by gene‐centred DNA sequencing. Ann Rheum Dis 2021;80:109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thorlacius GE, Hultin‐Rosenberg L, Sandling JK, et al. Genetic and clinical basis for two distinct subtypes of primary Sjögren's syndrome. Rheumatology (Oxford) 2021;60:837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271–7. [DOI] [PubMed] [Google Scholar]
- 22. Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American‐European Consensus Group. Ann Rheum Dis 2002;61:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eriksson D, Bianchi M, Landegren N, et al. Extended exome sequencing identifies BACH2 as a novel major risk locus for Addison's disease. J Intern Med 2016;280:595–608. [DOI] [PubMed] [Google Scholar]
- 24. Ameur A, Dahlberg J, Olason P, et al. SweGen: a whole‐genome data resource of genetic variability in a cross‐section of the Swedish population. Eur J Hum Genet 2017;25:1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xie C, Yeo ZX, Wong M, et al. Fast and accurate HLA typing from short‐read next‐generation sequence data with xHLA. Proc Natl Acad Sci 2017;114:8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grosso G, Sandholm K, Antovic A, et al. The complex relationship between C4b‐binding protein, warfarin, and antiphospholipid antibodies. Thromb Haemost 2021;121:1299–309. [DOI] [PubMed] [Google Scholar]
- 27. Svenungsson E, Gustafsson JT, Grosso G, et al. Complement deposition, C4d, on platelets is associated with vascular events in systemic lupus erythematosus. Rheumatology (Oxford) 2020;59:3264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nilsson UR, Nilsson B. Simplified assays of hemolytic activity of the classical and alternative complement pathways. J Immunol Methods 1984;72:49–59. [DOI] [PubMed] [Google Scholar]
- 29. Angioi A, Fervenza FC, Sethi S, et al. Diagnosis of complement alternative pathway disorders. Kidney Int 2016;89:278–88. [DOI] [PubMed] [Google Scholar]
- 30. R Core Team . R: A language and environment for statistical computing: R Foundation for Statistical Computing; 2018. URL: https://www.R-project.org/.
- 31. Ramos‐Casals M, Nardi N, Brito‐Zerón P, et al. Atypical autoantibodies in patients with primary Sjögren Syndrome: clinical characteristics and follow‐up of 82 cases. Semin Arthritis Rheum 2006;35:312–21. [DOI] [PubMed] [Google Scholar]
- 32. Boteva L, Morris DL, Cortés‐Hernández J, et al. Genetically determined partial complement C4 deficiency states are not independent risk factors for SLE in UK and Spanish populations. Am J Hum Genet 2012;90:445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis 2014;73:1601. [DOI] [PubMed] [Google Scholar]
- 34. Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2002;29:288. [PubMed] [Google Scholar]
- 36. Ramos‐Casals M, Brito‐Zerón P, Yagüe J, et al. Hypocomplementaemia as an immunological marker of morbidity and mortality in patients with primary Sjögren's syndrome. Rheumatology (Oxford) 2004;44:89–94. [DOI] [PubMed] [Google Scholar]
- 37. Jordán‐González P, Gago‐Piñero R, Varela‐Rosario N, et al. Characterization of a subset of patients with primary Sjögren's syndrome initially presenting with C3 or C4 hypocomplementemia. Eur J Rheumatol 2020;7:112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wetsel RA, Kulics J, Lokki ML, et al. Type II human complement C2 deficiency: allele‐specific amino acid substitutions (Ser189 →Phe; Gly444 →Arg) cause impaired C2 secretion. J Biol Chem 1996;271:5824–31. [DOI] [PubMed] [Google Scholar]
- 39. Wang X, Circolo A, Lokki M, et al. Molecular heterogeneity in deficiency of complement protein C2 type I. Immunology 1998;93:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. El Sissy C, Rosain J, Vieira‐Martins P, et al. Clinical and genetic spectrum of a large cohort with total and sub‐total complement deficiencies. Front Immunol 2019;10:1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1 Supporting Information
Data Availability Statement
Raw data for individual figures are available in the Supplementary Data (http://onlinelibrary.wiley.com/doi/10.1002/art.42270). Genotype data at the individual level are not publicly available since some of the information could compromise research participant privacy and consent. Scripts for calling C4 copy number in GATK GermlineCNVCaller are available upon request.