

Abstract
Introduction/Aims
We hypothesized that early, pretreatment axonal loss would predict long‐term disability, supported by a pilot study of selected patients with chronic inflammatory demyelinating polyneuropathy (CIDP). To further test this hypothesis, we examined a larger consecutive group of CIDP patients.
Methods
Needle electromyography and motor and sensory nerve conduction studies were carried out in 30 CIDP patients at pretreatment and follow‐up 5 to 28 years later. Changes in amplitudes were expressed as axonal Z scores and changes in conduction as demyelination Z scores and correlated with findings of the Inflammatory Rasch‐built Overall Disability Scale (I‐RODS), the Neuropathy Impairment Score (NIS), and isokinetic dynamometry (IKS).
Results
At follow‐up, the median I‐RODS score was 73, the NIS was 23, and the IKS was 56%. The median axonal Z score was unchanged at follow‐up. Conversely, the corresponding demyelination Z scores improved. The initial axonal loss was correlated with the clinical outcome and was an independent predictor of outcome by multivariate regression analysis. Axonal loss at follow‐up was also correlated with the clinical outcome. Only the follow‐up demyelination Z score was correlated with the clinical outcomes. Furthermore, the latency until treatment initiation was predictive of all three clinical outcome scores at follow‐up, and of axonal loss and demyelination at follow‐up.
Discussion
The present study findings indicate that pretreatment axonal loss at diagnosis in CIDP is predictive of long‐term disability, neurological impairment, and strength. A delay in treatment is associated with more pronounced axonal loss and a worse clinical outcome.
Keywords: axonal loss, electrophysiological examination, long‐term follow‐up, prediction of CIDP, prognosis
1. INTRODUCTION
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an immune‐mediated demyelinating polyneuropathy 1 , 2 associated with a variable degree of clinical disability and axonal loss. 3 Evidence of demyelination is mandatory for diagnosis. 4 Recent studies showed that axonal damage occurs early in the disease and it is debated whether the axonal loss occurs secondary to demyelination or is a primary manifestation of the disease due to separate nodal or paranodal disease processes. 5 , 6 , 7 , 8 , 9 Denervation on needle electromyography (EMG), reduced amplitude of the compound muscle action potential (CMAP), and decreased number of motor units on motor unit number estimation (MUNE) have, however, emphasized the frequent occurrence of axonal loss in CIDP. 10 , 11 , 12 , 13 In addition, it is a general assumption that axonal loss is the main pathophysiological mechanism underlying permanent disability. 6 , 9 , 10 , 14 The predictive value of early axonal loss as indicated by a diminished CMAP amplitude is, however, uncertain. In one study of early predictive factors, neither the CMAP amplitude nor other electrophysiological measures were predictive of disability in CIDP. 15 However, in our recent small pilot study of 14 nonconsecutive CIDP patients, we found that the amount of axonal loss before initiation of treatment was predictive of long‐term clinical disability after 10 to 30 years. 16 To address this discrepancy, we conducted a larger study.
2. METHODS
2.1. Study population
All patients fulfilling the revised 2021 European Academy of Neurology/Peripheral Nerve Society criteria for CIDP4 and who were diagnosed at the Departments of Neurology and Clinical Neurophysiology, Rigshospitalet, during the period from 1990 until 2015, with available electrophysiological records and subsequently treated with immune‐modulating therapy (immunoglobulins, plasma exchange, corticosteroids, azathioprine, or cyclophosphamide), were eligible for participation. Exclusion criteria were other neuropathies, other neurological disorders, diabetes, and disabling musculoskeletal disorders.
2.2. Study design
Eligible patients were examined clinically and electrophysiologically at follow‐up and their data were paired with the electrophysiological data at diagnosis. In addition, information regarding disease onset, course, duration, and current treatment were obtained from patients’ records and interviews.
The local Ethics Committee of the Capital Region (H‐17017657) and the Danish Data Protection Agency (2012‐58‐0004) approved the protocol. All study participants gave written informed consent.
2.3. Clinical evaluation
Disability was evaluated using the Rasch‐built Overall Disability Scale (I‐RODS) for immune‐mediated peripheral neuropathies, a self‐report questionnaire evaluating the limitations of physical activity and social participation, with scores ranging from 0 (most severe disability) to 100 (no disability) after translation of the raw sum score to a centile metric value. 17
Neurological impairment was assessed using the neuropathy impairment score (NIS). 18 Grading of muscle strength of 21 pairs of muscle groups was scored as follows: normal strength = 0; 75% of normal strength = 1; 50% = 2; 25% = 3; paralyzed = 4. Five pairs of muscle stretch reflexes and four modalities of sensation at the index finger and hallux at both sides were scored from 0 (normal) to 2 (absent), resulting in a total maximum impairment score of 220.
Isokinetic strength (IKS) at the wrist and ankle on the weakest side was obtained using dynamometry (Biodex Medical Systems, Shirley, New York), with the side selection in patients with symmetrical strength being made at random. To weigh all muscle groups equally, normalized strength was expressed as a ratio between the measured and predicted value, the latter being obtained from data of 178 healthy subjects previously reported. 19 , 20
2.4. Nodal‐paranodal antibodies
Prominent axonal loss has been found in some patients with nodal and paranodal involvement in Guillain‐Barré syndrome (GBS) and CIDP. 21 , 22 To further investigate whether axonal loss in our patients was due to nodo‐paranodopathy, blood samples were taken from 16 patients for determination of paranodal antibodies to neurofascin‐155, neurofascin‐186, caspr1, and contactin‐1. These antibodies induce changes in the nodal architecture, exposing K+ channels at the juxtaparanodal region and ultimately leading to conduction block and slowing. 23 In addition to axonal loss, patients with these antibodies tend to be treatment resistant to immunoglobulin 24 and are considered to represent a specific disease entity. 4
2.5. Electrophysiological studies
2.5.1. EMG
A reduced CMAP amplitude may be due to motor axon loss or distal conduction block. To ascertain the presence of motor fiber loss and denervation, EMG using concentric needle electrodes with a recording area of 0.07 mm2 was performed at follow‐up in the abductor pollicis brevis (APB) and the anterior tibial (AT) muscles in 27 patients. Fibrillation potentials, positive sharp waves, and fasciculation potentials were assessed at 10 sites in the muscle. At these sites, 20 to 71 motor unit potentials (MUPs) were recorded at weak effort and analyzed offline using EMGTools 25 to measure durations, amplitudes, and shapes. In each muscle, the average duration and amplitude of MUPs were calculated and compared with our laboratory age‐matched controls. 26 In each muscle, the Z scores for durations and amplitudes were averaged for the individual patient, and values >2 indicated enlarged MUPs as evidence of chronic partial denervation with collateral reinnervation.
2.5.2. Motor unit number estimation
In chronic neuropathy, the amplitude of the compound muscle action potential (CMAP) may be misleading as to the extent of motor fiber loss due to compensatory collateral reinnervation. To quantify axonal loss, motor unit number estimation (MUNE) was carried out in the APB at follow‐up. Stimulating electrodes were placed over the median nerve at the wrist and the CMAP was recorded from the APB through surface electrodes in a belly‐tendon montage during decreasing stimulus strength from maximal amplitude to zero response, and the number of motor units was calculated using MScanFit. 27 The number of motor units was compared with findings in 41 control subjects, 20 to 74 years or age, who had 89 (interquartile range [IQR], 74 to 121) motor units, similar to other reports. 28 , 29
2.5.3. Nerve conduction studies
Nerve conduction studies (NCS) at follow‐up were intended to be performed in the same nerves as in the initial study at diagnosis using near‐nerve needle or surface electrodes. In patients with both sides examined at diagnosis, the most affected nerves were studied at follow‐up. In most patients, the median (motor and sensory fibers), fibular (motor and sensory fibers), and sural (sensory fibers) nerves were examined. In five patients the tibial nerve rather than the fibular motor nerve was examined during the initial study.
Motor fibers were activated by supramaximal stimulation at the wrist and elbow in the median nerve and at the ankle and fibular head in the fibular nerve. CMAPs were recorded using surface electrodes in a belly‐tendon montage over the APB and the extensor digitorum brevis (EDB) muscles, respectively. In the patients with studies of the tibial nerve, stimulation was carried out at the medial malleolus and the popliteal fossa, and the CMAP was recorded from the abductor hallucis muscle.
In the median and fibular (or tibial) motor nerves, the variables included: amplitudes of the CMAPs evoked at distal and proximal stimulus sites; ratio of CMAP at proximal/distal stimulation sites (P/D ratio); distal motor latency (DML); motor nerve conduction velocities (MNCV) between stimulation sites; and the shortest F‐wave latency in 20 responses.
In the median nerve, ortodromic sensory nerve action potentials (SNAP) evoked by digital ring electrodes were recorded at the wrist using surface or near‐nerve needle electrodes, and, when recorded by needle electrodes, at the elbow as well. In the fibular nerve, SNAPs were recorded through needle electrodes at the fibular head after stimulation at the superior retinaculum, and, in the sural nerve, SNAPs were recorded through needle electrodes at midcalf after stimulation at the lateral malleolus. When recorded with surface electrodes from the sural nerve, the antidromic SNAP was recorded at the lateral malleolus after stimulation at midcalf. Peak‐to‐peak amplitudes were measured and sensory nerve conduction velocities (SNCVs) were calculated to the first positive phase.
The amplitudes of the logarithmically transformed CMAPs and SNAPs, DMLs, F‐wave latencies, MNCVs, and SNCVs were compared with age‐matched normal values from our laboratory. 26 , 30 , 31 The P/D ratio as a function of the conduction distance between elbow and wrist was compared with the findings from 18 control nerves. 31
For each patient, we calculated combined Z scores for axonal loss and demyelination (see Supplementary Material). 16 Due to the more marked motor than sensory deficits in CIDP and the importance of motor conduction properties in the electrodiagnostic criteria of CIDP, 4 we calculated combined Z scores, giving greater weight to motor than sensory recordings (ratio 2:1; Supplementary Material).
2.6. Statistical analysis
The primary parameters used for hypothesis testing were the correlations between the initial and follow‐up axonal Z scores and the three clinical scores of disability at follow‐up: I‐RODS, neurological impairment (NIS), and isokinetic strength. In addition, the correlation between initial and follow‐up axonal Z score was a primary parameter. After Bonferroni correction, the level of significance was P = .007. All other comparisons were considered secondary study parameters, with P < .05 considered statistically significant.
Descriptive data were calculated using nonparametric statistics. Comparisons were performed using the Wilcoxon signed rank test and the Mann‐Whitney U test for paired and nonpaired comparisons, respectively. To assess correlations, the Pearson correlation coefficient (rho) was obtained. Statistics were performed using SAS software (SAS Institute, Cary, North Carolina) and GraphPad Prism (GraphPad Software, San Diego, California).
3. RESULTS
3.1. Characteristics of the CIDP Patients
A total of 38 eligible CIDP patients were identified. Eight patients did not give consent to follow‐up. Thirty patients (22 males), including the 14 from the pilot study fulfilling the revised criteria for CIDP, of whom 1 had possible relapsing multifocal CIDP with a definite response to corticosteroids, participated in the follow‐up study. At diagnosis, electrophysiological motor and sensory nerve abnormalities, according to the current criteria, were present in 29 patients in two to six nerves, whereas 1 patient showed abnormalities in only one nerve. The interval from symptom onset to diagnostic electrophysiological examination was 0.7 year (IQR, 0.01 to 1.5 years), and the interval between the diagnostic and follow‐up electrophysiological studies was 13.4 (IQR, 10.9 to 15.8) years. The age at symptom onset was 47 (IQR, 34 to 58) years and 66 (IQR, 48 to 73) years at follow‐up. At follow‐up, the majority of patients walked independently and half of the patients were in stable inactive condition (Table 1). Fifteen of the 16 patients in whom paranodal antibodies were determined had no antibodies, but 1 patient with a long‐term stable inactive condition had antibodies to neurofascin‐155 at a very low titer (1:32) as compared with previous reports (1:1000 to 1:70 000), 24 , 32 and this was not considered clinically significant.
TABLE 1.
Demographics and clinical data from 30 patients with CIDP
| Variable | Median (IQR) |
|---|---|
| Age (years) | 66.0 (48.0‐73.0) |
| Sex, M:F (n) | 22:8 |
| Age at onset (years) | 47.5 (34.0‐58.0) |
| Time since onset of CIDP (years) | 14.6 (11.5‐19.0) |
| Time since first contact to neurologist (years) | 14.0 (11.0‐16.0) |
| Duration of CIDP until treatment initiation (years) | 0.75 (0.25‐1.5) |
| Interval between initial and follow‐up assessments (years) | 13.4 (10.9‐15.8) |
| Acute, GBS‐like onset (n) | 5 |
| Walking status at initial visit (n) | |
| Walking independently | 22 |
| Walking with aids | 3 |
| No ambulation | 5 |
| Walking status at follow‐up (n) | |
| Walking independently | 28 |
| Walking with aids | 1 |
| No ambulation | 1 |
| I‐RODS score at follow‐up (a.u.) | 73.0 (67.0‐88.0) |
| NIS score at follow‐up (a.u.) | 23.0 (15.0‐30.5) |
| Isokinetic strength, normalized (%) | 56.1 (44.4‐68.7) |
| Current treatment (n) | |
| IgG | 14 |
| Azathioprine | 1 |
| Rituximab | 1 |
| Prednisolone | 1 |
| No treatment | 15 |
| Combined axonal Z score | |
| Initial | −3.6 (−6.3 to −2.3) |
| Follow‐up a | −3.2 (−4.8 to −1.4) |
| Combined demyelination Z score | |
| Initial | −4.1 (−4.8 to −2.5) |
| Follow‐up b | −3.0 (−4.0 to −1.1) |
Abbreviations: CIDP, chronic inflammatory demyelinating polyneuropathy; F, female; GBS, Guillain‐Barré syndrome; IgG, immunoglobulin G; I‐RODS, Inflammatory Rasch‐built Overall Disability Scale; IQR, interquartile range; M, male; NIS, neuropathy impairment score.
P = .1.
P = .02.
3.1.1. Demographic characteristics of patients in treatment and not in treatment
There were no differences between ages at initial and follow‐up studies, axonal loss or demyelination at baseline, or the latencies until treatment initiation in patients in treatment compared with those with inactive disease (P = .1 to .9).
3.2. Electrophysiological findings at follow‐up
3.2.1. Axonal and demyelinating Z scores
The axonal Z score remained unchanged during the follow‐up period (Table 1). By contrast, the demyelination Z score improved significantly by 0.8 (IQR, −0.6 to 2.2) from diagnosis to follow‐up (P = .02). Axonal Z scores and demyelination Z scores were correlated both initially (rho = 0.52, P = .003) and at follow‐up (rho = 0.82, P < .0001).
3.2.2. EMG and MUNE
At EMG of the APB at follow‐up, fibrillation potentials and positive sharp waves were present in 19 patients and fasciculation potentials in 21 of the 27 patients. At quantitative analysis, the average Z score of amplitudes and durations of motor unit potentials was 3.1 (IQR, 1.8 to 5.8). In the anterior tibial muscle, fibrillation potentials and positive sharp waves occurred in 4 patients and fasciculation potentials in 11 of 27 patients. The Z score at MUP analysis was 2.1 (IQR, 0.9 to 3.7). The changes in MUP parameters were consistent with chronic partial denervation with collateral reinnervation. At MScanFit MUNE, the mean number of motor units in the APB was 57% of control values (IQR, 39% to 72%; P < .0001), consistent with a reduced number of median nerve motor fibers.
3.3. Electrophysiological markers of long‐term outcome
3.3.1. Effect of axonal loss
The initial axonal Z score correlated with disability, I‐RODS score, NIS, and IKS obtained 5 to 28 years after disease onset (Table 2 and Figure 1) as was the follow‐up axonal Z score (rho = 0.47, −0.68, and 0.64, respectively; Figure 1). In addition, the amount of axonal loss at diagnosis correlated significantly with amount of axonal loss at follow‐up (Table 2).
TABLE 2.
Univariate regression analysis of initial combined axonal Z score vs follow‐up combined axonal Z score and clinical measures
| Variable | Effect of a 1‐a.u. increase in initial Z score (95% CI) | rho | P value |
|---|---|---|---|
| Follow‐up combined Z score | 0.6 (0.3‐0.9) | 0.65 | .0001 |
| I‐RODS | 3.2 (1.3‐5.1) | 0.54 | .002 |
| NIS | −3.6 (−5.6 to −1.7) | −0.59 | .0006 |
| Isokinetic strength | 2.8 (1.0‐4.6) | 0.52 | .004 |
Abbreviations: CI, confidence interval; I‐RODS, Inflammatory Rasch‐built Overall Disability Scale; NIS, neuropathy impairment score.
FIGURE 1.
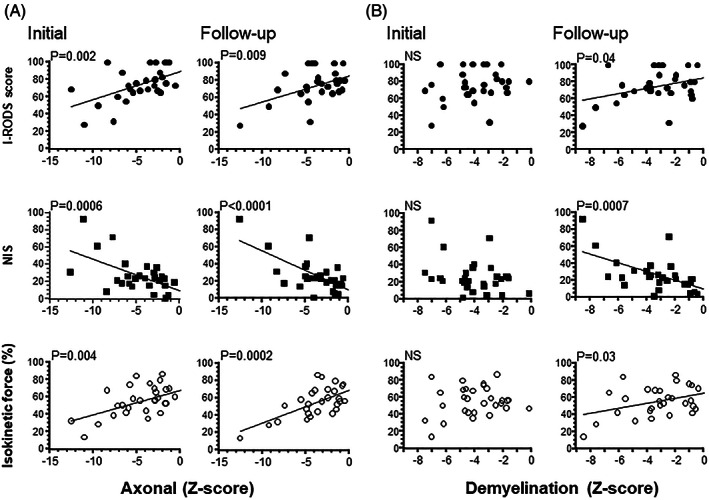
Univariate regression analysis of the follow‐up clinical scores vs the axonal and demyelination Z scores at the initial and follow‐up examinations. Univariate regression analysis of the initial axonal and follow‐up axonal Z scores (A) and initial and follow‐up demyelination Z scores (B) and follow‐up Inflammatory Rasch‐built Overall Disability Scale (I‐RODS), neuropathy impairment score (NIS), and isokinetic strength (%). At the initial axonal Z scores, the correlations were highly significant (P = .0006 to .004) as they were at the follow‐up axonal Z scores (P < .0001 to .009). By contrast, the relationships between clinical scores and demyelination Z scores were only significant at follow‐up (P = .0007 to .04).
3.3.2. Effect of demyelination
In contrast to the effect of axonal loss, no correlation was found between the initial demyelination Z score and clinical measures at follow‐up, whereas the demyelination Z score at follow‐up correlated with I‐RODS score, NIS, and IKS (rho = 0.37, −0.58, and 0.39, respectively; Figure 1). Furthermore, the amount of initial demyelination was predictive of level of axonal loss at follow‐up (rho = 0.42, P = .02).
There was no significant correlation between the percentage decay of CMAP between wrist and elbow and IKS at the wrist (P = .4), whereas there was a strong correlation between wrist force and amplitude of the APB CMAP evoked by stimulation at the wrist (P = .0003).
3.4. Effect of treatment
The latency until treatment initiation was predictive of all three clinical outcome scores at follow‐up (│rho│= 0.38 to 0.56, P = .001 to .04), of axonal loss (rho = −0.47, P = .009), and of demyelination (rho = −0.42, P = .02) at follow‐up.
3.5. Predictors of outcome
After multivariate regression analysis, including the initial axonal Z score, the initial demyelination Z score, the latency until treatment initiation, age at onset, duration of treatment, and type of onset (ie, acute or chronic), only the initial axonal Z score was predictive of follow‐up I‐RODS score (P = .02) as well as follow‐up IKS (P = .04), whereas both initial axonal Z score and latency until treatment initiation were predictive of follow‐up NIS, with P = .01 and P = .04, respectively.
4. DISCUSSION
It is well established that the severity of weakness is determined by the amount of axonal loss in CIDP. 6 , 9 , 14 The novel finding in our study is that the degree of axonal loss at the time of diagnosis is predictive of long‐term disability, impairment, and muscle strength in CIDP patients, confirming findings from our previous pilot study. 16 The strong predictive value of early axonal loss is further corroborated by the fact that, after a multivariate regression analysis, including various presumed predictors of outcome, only early axonal loss had an independent predictive value. This observation points to a potential role for early axonal loss determined by electrophysiology as a biomarker for long‐term disability in patients treated for CIDP and for prompt initiation of treatment.
To allow comparison of electrophysiological findings years apart, patients were diagnosed and re‐examined at follow‐up in the same laboratory to allow calculation of Z scores using our validated, age‐controlled normal material. In this consecutive cohort, the correlation was confirmed, but was diminished from absolute values of 0.75 to 0.87 down to 0.52 to 0.59, which may be due to a larger proportion of patients with stable inactive disease not requiring continued immune‐modulating treatment. The age at onset, age at follow‐up, male:female ratio, and proportion of patients with acute GBS‐like onset were similar in the two studies. In the present study, the follow‐up period in the two treatment groups was similar, but the duration of treatment differed significantly, being 12.0 (IQR, 8.5 to 14.0) years in the treatment group compared with 4.0 (IQR, 1.2 to 8.0) years in patients with a stable inactive condition not receiving treatment at the time of follow‐up (P = .0003).
To distinguish axonal degeneration from demyelination we calculated axonal scores from Z scores of the amplitudes of motor and sensory responses and demyelination scores based on motor and sensory conduction properties. Nevertheless, these parameters may not be distinct aspects of the pathophysiological changes in CIDP as the CMAP amplitudes may be diminished by conduction failure in demyelinated nerve fibers. Fibrillation potentials and positive sharp waves or signs of chronic denervation at EMG examination support that reduced CMAP amplitudes in the APB were associated with axonal degeneration in 82%, whereas 18% had reduced CMAP but no evidence of denervation. By contrast, in chronic disorders the amplitudes of CMAPs may underestimate the extent of axonal loss due to collateral sprouting and reinnervation of muscle fibers, as indicated by the increases in motor unit potential durations and amplitudes during EMG. To quantify the extent of motor axonal loss, we used MUNE, which showed a loss of about 50%, in agreement with other studies. 13 , 28 Focal conduction block due to demyelination of motor fibers is conventionally considered a factor causing weakness. Nevertheless, using quantitative force measurements in the wrist in our patients, the degree of CMAP amplitude decay between elbow and wrist did not correlate with the amount of weakness. Other studies also showed that conduction block did not influence clinical scores 33 and confirmed axonal loss as being the decisive factor. We cannot exclude that block of fibers at more proximal sites not tested in this study can contribute to weakness together with the axonal loss. The observed strong correlation between the CMAP amplitude reduction at the APB and the isokinetic strength at the wrist in the present study is of limited significance as the two assessments were performed on different sites of the median nerve.
In a previous study of early predictive factors of disability in long‐term CIDP, neither amplitude nor conduction velocity was associated with long‐term disability. 15 However, in that study, the composite score of axonal loss was based on mean raw CMAP values of various nerves of arms and legs differing between patients and irrespective of patient age. In addition, the outcome was dichotomized into a modified Rankin scale score of above or below 4. Hence, a potential association between axonal loss and disability may have been obscured. 15 In other reports in the literature the correlation between axonal loss and clinical disability was found in studies of patients with a disease duration of only 4 to 6 years, whereas the time of assessment in relation to disease start and treatment initiation was less well‐defined. 6 , 11 One study identified CMAP amplitude as a correlate of the short‐term intravenous immunoglobulin response, supporting the role of axonal loss. 34 These findings cannot be directly compared with ours because we only assessed long‐term outcome. Terminally damaged nerves may, however, lead to a short‐term lack of treatment response as well as to long‐term severe muscle impairment. 16 , 34 Our findings are in accordance with previous observations in 22 CIDP patients indicating a correlation between axonal loss at follow‐up and the clinical condition. 10
CIDP is a heterogeneous condition composed of diverse rare subtypes aside from the typical subtype, which may have different treatment responses and outcomes. In our study, two patients had multifocal acquired demyelinating sensory and motor neuropathy and one had pure motor CIDP, the remainder with typical CIDP. In addition, five patients with typical CIDP had an acute GBS‐like onset. One study demonstrated similar outcomes in acute, GBS‐like CIDP and typical CIDP with chronic onset. 35 Our study did not include any patients with pure sensory CIDP, which may follow a different clinical course, and none of our 16 patients in whom assessment for paranodal antibodies was performed had a significant titer.
Unlike the effect of early axonal loss, initial demyelination did not correlate with long‐term clinical outcome, in agreement with our previous study. 16 This raises the question of the relationship between demyelination and axonal loss early in CIDP, at which time there was a clear correlation between demyelination and axonal loss. The lesion in early demyelination is localized at or near the nodes of Ranvier with macrophage stripping and inflammatory infiltrates followed by axonal degeneration. 1 , 8 In our study, there was a strong correlation between follow‐up demyelination, axonal loss, and poorer clinical outcome, consistent with progressive disease activity and secondary axonal loss. 6 , 9 , 36 We found a clear relationship between delay in onset of treatment and long‐term axonal, demyelinating, and clinical features, indicating that prompt initiation of treatment is needed to reduce the risk of axonal loss and for long‐term prognosis in CIDP. 35 It is of interest that long‐term demyelination and axonal loss had improved in patients not requiring continued treatment. Our data do not allow for prediction at onset of which patients will require long‐term treatment and those who will become stable and not require continuous therapy.
AUTHOR CONTRIBUTIONS
A.A. contributed to design of the study, writing of the protocol, coordination, clinical assessment, statistical analyses, and writing and revision of the manuscript. J.J. contributed to design of the study and writing and revision of the manuscript. C.K. contributed to design of the study, electrophysiological examination, statistical analyses, and writing and revision of the manuscript.
FUNDING INFORMATION
Takeda Pharma A/S, Grant Number: IISR‐2019‐104354
CONFLICT OF INTEREST
J.J. received a research grant from Takeda Pharmaceutical Co. The remaining authors declare no potential conflicts of interest.
ETHICAL PUBLICATION STATEMENT
I confirm that I have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Abbreviations
- ABP
abductor pollicis brevis
- AT
anterior tibial
- CIDP
chronic inflammatory demyelinating polyneuropathy
- CMAP
compound muscle action potential
- DML
distal motor latency
- EDB
extensor digitorum brevis
- EMG
electromyography
- GBS
Guillain‐Barré syndrome
- I‐RODS
Inflammatory Rasch‐built Overall Disability Scale
- IKS
isokinetic strength
- MNCV
motor nerve conduction velocity
- MUNE
motor unit number estimation
- NCS
nerve conduction studies
- NIS
neuropathy impairment score
- SNAP
sensory nerve action potential
- SNCV
sensory nerve conduction velocity
Supporting information
APPENDIX S1 Supporting information
ACKNOWLEDGMENT
This study was partially funded as Investigator‐Initiated Research Grant (No. IISR‐2019‐104354) from Takeda Pharma A/S, a company incorporated in Denmark.
Al‐Zuhairy A, Jakobsen J, Moldovan M, Krarup C. Axonal loss at time of diagnosis as biomarker for long‐term disability in chronic inflammatory demyelinating polyneuropathy. Muscle & Nerve. 2022;66(6):715‐722. doi: 10.1002/mus.27722
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Funding information Takeda Pharmaceutical Company, Grant/Award Number: IISR‐2019‐104354
DATA AVAILABILITY STATEMENT
Data will be available upon contact to the corresponding author.
REFERENCES
- 1. Prineas JW, McLeod JG. Chronic relapsing polyneuritis. J Neurol Sci. 1976;27:427‐458. doi: 10.1016/0022-510x(76)90213-6 [DOI] [PubMed] [Google Scholar]
- 2. McCombe PA, Pollard JD, McLeod JG. Chronic inflammatory demyelinating polyradiculoneuropathy. A clinical and electrophysiological study of 92 cases. Brain. 1987;110:1617‐1630. [DOI] [PubMed] [Google Scholar]
- 3. Nagamatsu M, Terao S, Misu K, et al. Axonal and perikaryal involvement in chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 1999;66:727‐733. doi: 10.1136/jnnp.66.6.727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society Guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force‐‐‐second revision. Eur J Neurol. 2021;28:3556‐3583. doi: 10.1111/ene.14959 [DOI] [PubMed] [Google Scholar]
- 5. Barnett MH, Mathey E, Kiernan MC, Pollard JD. Axonal damage in central and peripheral nervous system inflammatory demyelinating diseases: common and divergent pathways of tissue damage. Curr Opin Neurol. 2016;29:213‐221. doi: 10.1097/WCO.0000000000000334 [DOI] [PubMed] [Google Scholar]
- 6. Bouchard C, Lacroix C, Planté V, et al. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1999;52:498‐503. doi: 10.1212/wnl.52.3.498 [DOI] [PubMed] [Google Scholar]
- 7. van Doorn PA, Hadden RDM, Van den Bergh PYK. Elucidating autoimmune nodopathies and the CIDP spectrum. Brain. 2021;144:1043‐1045. doi: 10.1093/brain/awab116 [DOI] [PubMed] [Google Scholar]
- 8. Said G. Chronic inflammatory demyelinating polyneuropathy. Neuromuscul Disord. 2006;16:293‐303. doi: 10.1016/j.nmd.2006.02.008 [DOI] [PubMed] [Google Scholar]
- 9. Said G, Saida K, Saida T, Asbury AK. Axonal lesions in acute experimental demyelination: a sequential teased nerve fiber study. Neurology. 1981;31:413‐421. doi: 10.1212/wnl.31.4.413 [DOI] [PubMed] [Google Scholar]
- 10. Harbo T, Andersen H, Jakobsen J. Length‐dependent weakness and electrophysiological signs of secondary axonal loss in chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 2008;38:1036‐1045. doi: 10.1002/mus.21000 [DOI] [PubMed] [Google Scholar]
- 11. Sghirlanzoni A, Solari A, Ciano C, Mariotti C, Fallica E, Pareyson D. Chronic inflammatory demyelinating polyradiculoneuropathy: long‐term course and treatment of 60 patients. Neurol Sci. 2000;21:31‐37. doi: 10.1007/s100720070116 [DOI] [PubMed] [Google Scholar]
- 12. Kuwabara S, Misawa S, Mori M, Tamura N, Kubota M, Hattori T. Long term prognosis of chronic inflammatory demyelinating polyneuropathy: a five year follow up of 38 cases. J Neurol Neurosurg Psychiatry. 2006;77:66‐70. doi: 10.1136/jnnp.2005.065441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paramanathan S, Tankisi H, Andersen H, Fuglsang‐Frederiksen A. Axonal loss in patients with inflammatory demyelinating polyneuropathy as determined by motor unit number estimation and MUNIX. Clin Neurophysiol. 2016;127:898‐904. doi: 10.1016/j.clinph.2015.05.004 [DOI] [PubMed] [Google Scholar]
- 14. Mathey EK, Park SB, Hughes RAC, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. 2015;86:973‐985. doi: 10.1136/jnnp-2014-309697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spina E, Topa A, Iodice R, et al. Early predictive factors of disability in CIDP. J Neurol. 2017;264:1939‐1944. doi: 10.1007/s00415-017-8578-9 [DOI] [PubMed] [Google Scholar]
- 16. Al‐Zuhairy A, Jakobsen J, Krarup C. Early axonal loss predicts long‐term disability in chronic inflammatory demyelinating polyneuropathy. Clin Neurophysiol. 2021;132:1000‐1007. doi: 10.1016/j.clinph.2020.12.017 [DOI] [PubMed] [Google Scholar]
- 17. van Nes SI, Vanhoutte EK, van Doorn PA, et al. Rasch‐built Overall Disability Scale (R‐ODS) for immune‐mediated peripheral neuropathies. Neurology. 2011;76:337‐345. doi: 10.1212/WNL.0b013e318208824b [DOI] [PubMed] [Google Scholar]
- 18. Dyck PJ, Davies JL, Litchy WJ, O'Brien PC. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester Diabetic Neuropathy Study cohort. Neurology. 1997;49:229‐239. [DOI] [PubMed] [Google Scholar]
- 19. Harbo T, Andersen H, Overgaard K, Jakobsen J. Muscle performance relates to physical function and quality of life in long‐term chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2008;13:208‐217. doi: 10.1111/j.1529-8027.2008.00179.x [DOI] [PubMed] [Google Scholar]
- 20. Harbo T, Brincks J, Andersen H. Maximal isokinetic and isometric muscle strength of major muscle groups related to age, body mass, height, and sex in 178 healthy subjects. Eur J Appl Physiol. 2012;112:267‐275. doi: 10.1007/s00421-011-1975-3 [DOI] [PubMed] [Google Scholar]
- 21. Cifuentes‐Diaz C, Dubourg O, Irinopoulou T, et al. Nodes of ranvier and paranodes in chronic acquired neuropathies. PLoS One. 2011;6:e14533. doi: 10.1371/journal.pone.0014533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Uncini A. A common mechanism and a new categorization for anti‐ganglioside antibody‐mediated neuropathies. Exp Neurol. 2012;235:513‐516. doi: 10.1016/j.expneurol.2012.03.023 [DOI] [PubMed] [Google Scholar]
- 23. Lewis RA. Chronic inflammatory demyelinating polyneuropathy. Curr Opin Neurol. 2017;30:508‐512. doi: 10.1097/WCO.0000000000000481 [DOI] [PubMed] [Google Scholar]
- 24. Querol L, Nogales‐Gadea G, Rojas‐Garcia R, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology. 2014;82:879‐886. doi: 10.1212/WNL.0000000000000205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nikolic M, Krarup C. EMGTools, an adaptive and versatile tool for detailed EMG analysis. IEEE Trans Biomed Eng. 2011;58:2707‐2718. doi: 10.1109/TBME.2010.2064773 [DOI] [PubMed] [Google Scholar]
- 26. Rosenfalck P, Rosenfalck A. Electromyography—Sensory and Motor Conduction. Findings in Normal Subjects. Laboratory of Clinical Neurophysiology, Rigshospitalet; 1975. [Google Scholar]
- 27. Bostock H. Estimating motor unit numbers from a CMAP scan. Muscle Nerve. 2016;53:889‐896. doi: 10.1002/mus.24945 [DOI] [PubMed] [Google Scholar]
- 28. Garg N, Howells J, Yiannikas C, et al. Motor unit remodelling in multifocal motor neuropathy: the importance of axonal loss. Clin Neurophysiol. 2017;128:2022‐2028. doi: 10.1016/j.clinph.2017.07.414 [DOI] [PubMed] [Google Scholar]
- 29. Kristensen RS, Bostock H, Tan SV, et al. MScanFit motor unit number estimation (MScan) and muscle velocity recovery cycle recordings in amyotrophic lateral sclerosis patients. Clin Neurophysiol. 2019;130:1280‐1288. doi: 10.1016/j.clinph.2019.04.713 [DOI] [PubMed] [Google Scholar]
- 30. Horowitz SH, Krarup C. Conduction studies of the normal sural nerve. Muscle Nerve. 1992;15:374‐383. doi: 10.1002/mus.880150318 [DOI] [PubMed] [Google Scholar]
- 31. Krarup C, Trojaborg W. Sensory pathophysiology in chronic acquired demyelinating neuropathy. Brain. 1996;119:257‐270. doi: 10.1093/brain/119.1.257 [DOI] [PubMed] [Google Scholar]
- 32. Garg N, Park SB, Yiannikas C, et al. Neurofascin‐155 IGG4 neuropathy: pathophysiological insights, spectrum of clinical severity and response to treatment. Muscle Nerve. 2018;57:848‐851. doi: 10.1002/mus.26010 [DOI] [PubMed] [Google Scholar]
- 33. Garg N, Park SB, Howells J, et al. Conduction block in immune‐mediated neuropathy: paranodopathy versus axonopathy. Eur J Neurol. 2019;26:1121‐1129. doi: 10.1111/ene.13953 [DOI] [PubMed] [Google Scholar]
- 34. Iijima M, Yamamoto M, Hirayama M, et al. Clinical and electrophysiologic correlates of IVIg responsiveness in CIDP. Neurology. 2005;64:1471‐1475. doi: 10.1212/01.WNL.0000158680.89323.F8 [DOI] [PubMed] [Google Scholar]
- 35. Al‐Zuhairy A, Sindrup SH, Andersen H, Jakobsen J. A population‐based study of long‐term outcome in treated chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2020;61:316‐324. doi: 10.1002/mus.26772 [DOI] [PubMed] [Google Scholar]
- 36. Köller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med. 2005;352:1343‐1356. doi: 10.1056/NEJMra041347 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1 Supporting information
Data Availability Statement
Data will be available upon contact to the corresponding author.