

Abstract
Over the past two decades, electro‐organic synthesis has gained significant interest, both in technical and academic research as well as in terms of applications. The omission of stoichiometric oxidizers or reducing agents enables a more sustainable route for redox reactions in organic chemistry. Even if it is well‐known that every electrochemical oxidation is only viable with an associated reduction reaction and vice versa, the relevance of the counter reaction is often less addressed. In this Review, the importance of the corresponding counter reaction in electro‐organic synthesis is highlighted and how it can affect the performance and selectivity of the electrolytic conversion. A selection of common strategies and unique concepts to tackle this issue are surveyed to provide a guide to select appropriate counter reactions for electro‐organic synthesis.
Keywords: Electrochemistry, Hydrogen, Oxygen, Paired Electrolyses, Sacrificial Anodes
Every electrochemical synthesis is paired with conversion at the counter electrode. In electro‐organic synthesis, often only the desired half reaction is described well, although good control over the counter reaction is essential. In this Review, the impact of the counter electrode reaction in electro‐organic synthesis is highlighted through a collection of common and unusual counter reactions.

1. Introduction
As a result of climate change, future chemical processes will not only be validated by yield and costs, but also with regard to safety issues and environmental impact. As part of the development of more sustainable routes for the preparation of organic compounds, electro‐organic synthesis has experienced a renaissance in academia and technical synthesis. [1] The use of toxic and/or hazardous oxidants or reducing agents can often be omitted by using an electric current as a reagent. This lowers dramatically the amount of reagent waste generated. Since the electric power can be obtained by renewable sources, this opens the door to sustainable chemistry. [2]
1.1. Basic Concepts
Within an electro‐organic synthesis, electric power is converted into chemical and thermal energy. Organic redox reactions which would not occur spontaneously can be enforced. However, for a good working process, electrolytic conversion at both the cathode (reduction) and anode (oxidation) should happen smoothly (Scheme 1).
Scheme 1.
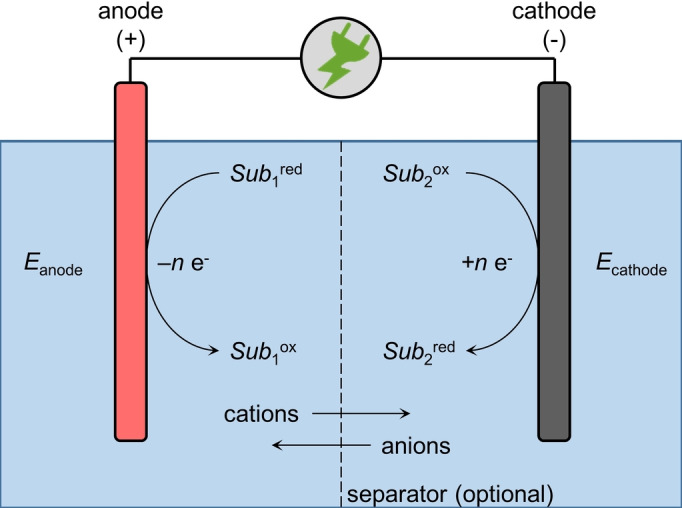
Basic concepts of an electrochemical redox reaction.
Thereby, the reaction with the lowest Gibbs free energy ΔG between the oxidation and reduction is favored to proceed. The Gibbs free energy can, from an electrochemists viewpoint, often be expressed by the difference in the electrode potentials [Eq. (1), where n is the number of electrons transferred and F is the Faraday constant]. The potential of an electron‐transfer reaction is defined by the standard potential, but is affected by a voltage drop that results from many parameters, for example, various over‐potentials (gas evolution, interaction of the electrode surface and substrate, etc.) as well as resistivity of the electrolyte and separator.[ 3 , 4 , 5 ]
| (1) |
The transformation can be initiated by a single‐electron transfer (SET) directly at the electrode, via an electrochemical redox mediator, or with an electrochemically activated electrode surface. The reaction can be controlled by constant potential (CPE) or constant current (CCE) conditions, for which either direct current (DC) or alternating current (AC) can be applied. [6] Those techniques to ensure a selective reaction are well‐known and are not discussed within this Review. The selection of a suitable system and a set of conditions for electrolysis to allow a selective chemical reaction is the main objective of synthetic electrochemists.
The oxidation and the reduction can be carried out in an undivided cell or they can be divided in two half‐cells by a separator (semipermeable ceramic, glass frits, ionic polymer matrices). An undivided setup is simpler in operation, but the oxidation and reduction have to be chosen to be selective for each other to ensure product formation and prevent side reactions. In a divided set‐up, the oxidation and reduction are performed spatially separated and no competing reactions decrease the selectivity. This also ensures that starting materials, products, or intermediates do not migrate to the counter electrode and undergo undesired reactions. In particular, electrochemically reversible transformations can be conducted in this way. Since separators cause substantial ohmic resistivity, more electric power for such electro‐conversions is required.
1.2. Working and Counter Electrode Potential
In general, the operation mode of an electrochemical reaction differs if the reaction is performed under constant current conditions in a two‐electrode set‐up or at a constant potential within a three‐electrode set‐up. The two‐electrode cell is most frequently found in the literature, as the operation of the electrolysis is quite simple (Figure 1, left). By using a power supply, a constant current is applied through electrodes into the electrolyte. The electrode where the synthetically valuable reaction occurs is called the working electrode (WE) and the other one the counter electrode (CE). The operating potential of the electrolysis cell is a result of the current passing through the electrolyte. As the amount of substrate converted at the electrodes decreases, the cell voltage increases over time, whereas undesired side reactions, at both the WE and CE, might become dominant. Within a three‐electrode cell, an additional electrode, the reference electrode (RE), is added (Figure 1, right). Within these kinds of cells, the power supply determines the voltage between the WE and RE and regulates the current between the WE and CE in such a manner that the potential at the WE is kept constant. Therefore, when the concentration of the reactants decreases in the course of the electrolysis, the current flow is regulated down. This allows a selective electron transfer and represses side reactions, such as overoxidations. Even if the three‐electrode cell often has higher selectivity for product formation, the scale‐up is often highly problematic. The decreased current at the end of the electrolysis also leads to prolonged reaction times and the full conversion of starting materials is difficult.[ 3 , 4 ]
Figure 1.

Schematic presentation of a two‐ and three‐electrode cell used in electro‐organic synthesis. The electrode potential is also shown.
If overall an oxidation or reduction of an organic substrate is of interest, the selection of a suitable counter reaction is relevant to ensure a selective and productive electrolysis. Without any reaction at the counter electrode, no faradaic conversion is possible. Extreme potentials should be avoided as high energy can lead to increased and unnecessary reaction temperatures, uncontrolled side reactions happening, and the loss of many benefits. To ensure a selective reaction, the selection of an appropriate counter reaction for an electro‐organic conversion is necessary. In this Review, we will give an overview of common and not‐so‐common counter electrode reactions. Their (dis‐)advantages and importance are also discussed.
2. Cathodic Counter Reactions
2.1. Hydrogen Evolution Reaction (HER)
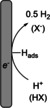
When electro‐organic oxidation processes are described, hydrogen evolution is the most frequently employed counter reaction. Hydrogen can be evolved on a variety of different cathode materials at low energetic costs. Hydrogen is considered to be an easy to control by‐product, which does not interfere with most anodic processes and separates out of the electrolyte by degassing. [7] The latter factor represents a huge benefit in downstream processing. In small amounts, hydrogen can either be liberated into the atmosphere or further utilized as a chemical reagent. [8] In several studies, the HER together with an electro‐organic conversion is described as paired electrolysis. It is noteworthy that this specification can only be used if the hydrogen is produced in a controlled manner and quantified. The standard reduction potential of hydrogen is defined as 0 V under standard conditions (1 atm, pH 0).
In the HER, a proton from the solution or abstracted from an acidic covalent bond (H+) is adsorbed onto the electrode surface (Hads) and released as molecular hydrogen (H2). However, the effective electrode potential to generate hydrogen also depends on the availability of protons in the electrolyte, as well as the over‐potential of the respective cathode for hydrogen evolution (Table 1). [11] Pt is the most abundant example, as it has a very low over‐potential, but it is quite expensive. Notably, nickel can serve as an inexpensive substitute for platinum in many cases. [12] However, readily available steel or stainless steel represent even more attractive alternatives, since dehalogenations are avoided. [13] Electrodes with a high over‐potential (lead or BDD) are not suitable cathode materials if the HER is desired. The use of an acid as a solvent or acidic additives (either mineral or organic acids) ensure an easy formation of hydrogen. To force hydrogen evolution, protic (co‐)solvents such as water, [14] alcohols, [15] fluorinated alcohols, [16] or the addition of protic additives such as TfOH, [17] H2SO4, [18] or acidic ammonium electrolytes [19] play an important role in the electro‐organic oxidations.
Table 1.
Over‐potential for hydrogen evolution at different cathode materials in an acidic aqueous solution.
|
|
Entry |
Electrodes |
Over‐potential[a] η [V] |
Ref. |
|---|---|---|---|---|
|
|
1 |
Pt (plated) |
0,25 (0,01) |
|
|
2 |
Ni |
0,33 |
||
|
3 |
Fe |
0,40 |
||
|
4 |
graphite |
0,47 |
||
|
5 |
lead[b] |
0,91 |
||
|
6 |
BDD[c] |
1,50–2,00 |
[a] Potential vs. SHE, determined in 1 m aq HCl at 1 mA cm−2. [b] Electrodeposition on Cu wire. [c] In 10 % aq H2SO4 at 1 mA cm−2; the over‐potential varies with the doping content.
2.1.1. Impact of the Cathode
Xu and co‐workers described in 2018 a TEMPO (2,2,6,6‐tetramethylpiperidinyloxyl) mediated oxidative C,N coupling of biaryloximes to phenanthridine‐N‐oxides (Scheme 2). When a Pt cathode is used as the counter electrode, the desired N‐oxide is formed with the HER as the counter reaction. However, if a lead cathode with a high over‐potential is used, the HER is suppressed. The N‐oxide is still formed in the course of the electrolysis, but the potential for the HER is more negative than for the reduction of the N‐oxide. Therefore, the product undergoes a subsequent reduction into the corresponding phenanthridine, as it is energetically favored. At graphite electrodes with a moderate over‐potential for H2 evolution, a mixture of both compounds is observed. [20]
Scheme 2.
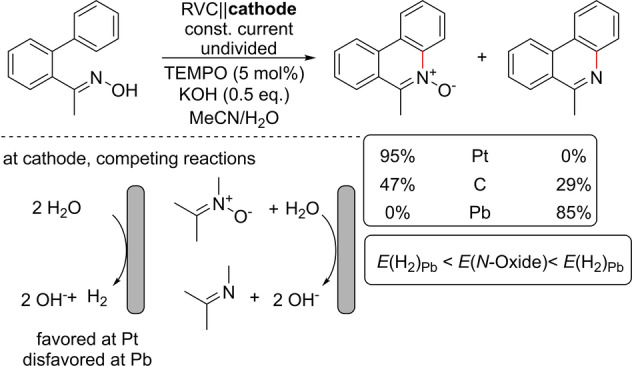
Oxidative cyclization of oximes with different cathode materials. RVC: reticulated vitreous carbon.
2.1.2. Proton Source
Yoshida and Suga developed the “cation pool” as an effective means for electro‐organic chemistry, whereby reactive cations are enriched in a reservoir at a low temperature in a divided electrolysis, either in batch or flow cells, [21] followed by chemical reaction with a nucleophile subsequent to electrolysis (Scheme 3). The success of the reaction depends not only on the stability of the cations generated, but also on them not undergoing reactions with nucleophiles present in the solution. [22]
Scheme 3.
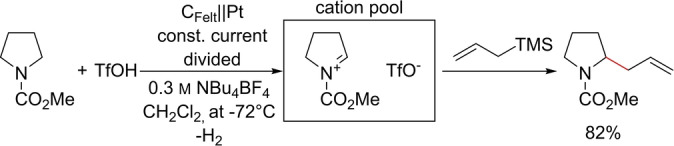
Synthesis of 2‐allylpyrrrolidines by the cation pool method.
Therefore, it is necessary to add a less nucleophilic acid such as TfOH [23] or AcOH [24] into the catholyte. In this way, hydrogen is evolved at a platinum cathode, with the corresponding non‐nucleophilic anions of the acid stabilizing the cations. If the acid is omitted and, thus, the cathodic reaction is suppressed, then the conversion fails. [23] This is caused by an increase in the counter electrode potential, whereas undesired reactions, such as the reduction of solvent or supporting electrolyte, will lead to highly reactive intermediates that react with the stabilized cations.
Lou and co‐workers developed a method for the chiral coupling of tertiary amines and ketones (Scheme 4). Even though some acidic protons were present in the mixture, an efficient conversion could only be achieved by adding protic additives to balance the cathodic hydrogen evolution. [25]
Scheme 4.

Catalytic asymmetric coupling of tertiary amines and ketones. TFE: 1,1,1‐trifluoroethanol.
2.1.3. The Relevance of the Counter Ion
The addition of acids or acidic alcohols also plays an important role in the synthesis of bench‐stable hypervalent iodine(III) reagents. These are often prepared in undivided cells by the anodic oxidation of iodoarenes, with the HER as a counter reaction. AcOH, [26] TFE, [27] HFIP, [27] NEt3*nHF, [28] or H2SO4 [29] have been reported to act as proton sources for hydrogen evolution at the cathode. The choice of proton source plays a central role here, as the generated Lewis base often represents a ligand for the iodine reagent (Scheme 5). Different ligands result in different chemical performances of the reagents. When a non‐nucleophilic proton source (H2SO4) is used, the formation of diaryliodonium species is observed.
Scheme 5.

Examples of iodine(III) reagents obtained by the electrolysis of iodoarenes. HFIP: 1,1,1,3,3,3‐hexafluoro‐2‐propanol.
2.2. Electrogenerated Bases (EGB)
2.2.1. Protic Electrolytes
Many examples of dehydrogenative coupling reactions [30] or halide‐mediated oxidation reactions [31] are based on the use of alcohols as a (co‐)solvent in neutral media. The proton can be abstracted very easily, generating the corresponding alkoxide at the cathode. The alkoxide plays a crucial role as base and ensures that the necessary deprotonation occurs in solution and not at the cathode surface. This prevents reactive intermediates from migrating to the cathode. Another advantage of this approach is that the amount of alkoxide corresponds to the amount of oxidized species, which allows selective control over the base added. This effect is often seen to be significant when the electrolysis is carried out in an undivided cell. [32]
One example is the dehydrogenative cyanimination of sulfides (Scheme 6). The electrogenerated base plays a central role in the outcome of the reaction. When MeOH is used with the addition of one equivalent of H2SO4, sulfoxides are formed predominantly, as no crucial deprotonation of the cyanamide is possible. In anhydrous acetonitrile, proton abstraction is possible, but as quite a negative potential is required for this, the formation of sulfilimine is not selective. The combination of MeCN with low amounts of MeOH to generate a base leads to desirable yields. [33]
Scheme 6.

Synthesis of N‐cyanosulfilimines by dehydrogenative coupling.
Another promising tool for a controlled proton concentration in the dehydrogenative coupling is the combination of HFIP and an amine base. This combination has been shown to be successful, since HFIP acts as a proton source, but its acidity is tamed by the amine base to readily generate a conductive solution. [34] Nevertheless, besides having basic properties, the alkoxide can also act as a nucleophile. An example of this is the electrochemical Hofmann rearrangement described by Xu, Zhang, and co‐workers (Scheme 7). The formation of the carbamate from the amide in alcohol is only viable in the presence of the alkoxide formed at the cathode, as a strong base is required for the bromination of the amide as well as for the nitrene formation step.
Scheme 7.
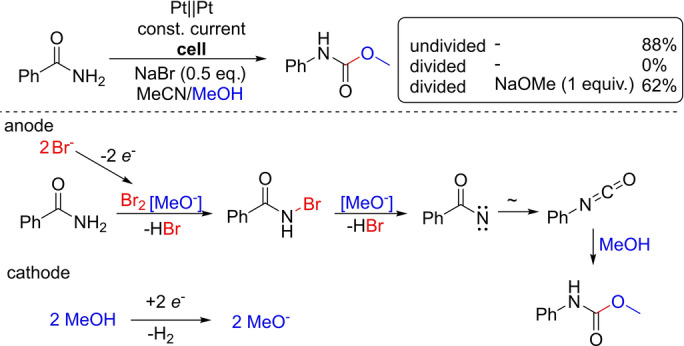
Electrochemical Hofmann rearrangement.
If a divided cell is used, the product formation is completely suppressed by the absence of the EGB. This was proven by the addition of NaOMe into the anolyte leading to product formation in a divided cell. [35]
2.2.2. Aprotic Electrolytes
If aprotic solvents are used without protic additives, the HER is strongly suppressed and reduction can usually only occur in a more negative potential range. However, if cathodes with low over‐potentials are used for hydrogen evolution, the HER can still occur. Proton abstraction can then occur either directly from less acidic substrates, such as benzotriazoles, [36] or proton transport can also occur via hydrogen atom transfer mediators. These carry the abstracted proton to the cathode upon oxidation of a substrate. [37] If no functional group is present that can be easily reduced or deprotonated, even weakly acidic solvents such as nitromethane, [38] DMSO, [39] acetonitrile, [40] or imidazolium‐based ionic liquids [41] can be deprotonated to generate very strong EGBs (Scheme 8). The stability of those anionic bases is determined by the choice of a suitable counter ion.
Scheme 8.

Strong electrogenerated bases from weak acidic solvents. The pKa data refer to the protonated form in DMSO at 25 °C [42] and for [dmim]. [43] [dmim]: N,N‐dimethylimidazolium.
A specific role played by those bases generated at the cathode is the deprotonation of acetonitrile. Although this cathodic process has a high energetic potential barrier, the cyanomethyl anion (pK a=31.3) has unique chemical features as a base. [42]
The formation of cyanomethyl anions can be achieved by ex‐cell electrolysis [44] or directly in situ as a cathodic reaction.[ 40 , 45 ] When no compound for deprotonation or any electrophile is available, it will condensate with itself. [46] Despite the possible use of this strong base or nucleophile as a co‐reagent in electrochemical synthesis, only a few examples have been reported in which this direct synthesis serves as a counter reaction to an anodic process. It is used in the dehydogenative coupling of indoles and ethers by Du and Huang. [45] The Onomura group used MeCN as a solvent in the electrochemical diastereoselective synthesis of β‐lactams by iodide‐mediated oxidation (Scheme 9). The formation of cyanomethyl anions at the cathode was described as a counter reaction. This was crucial for the deprotonation of the reaction intermediate. They compared the electrochemical method with a conventional approach, where iodine was used as the oxidizer with sodium ethoxide as a base. It was shown that the electrochemical route with the stronger base gave significantly better yields of lactam. If the electrochemical method was performed in EtOH instead, the cathodic formation of ethoxide also provided poor yields, similar to the conventional method. [40]
Scheme 9.

The electrochemical synthesis of β‐lactams, promoted by the deprotonation of MeCN at the cathode.
If electrodes with moderate or high over‐potential for hydrogen evolution are used as cathodes, quaternary ammonium ions, which are often used as supporting electrolytes, might be reduced at the cathode.[ 47 , 48 ] This leads to tertiary amines and C‐centered radicals, which either recombine (benzylic substrates) [49] or undergo a second reduction into carbanions (Scheme 10). Aliphatic carbanions can then deprotonate weakly acidic solvents such as acetonitrile, [50] or in less acidic solvents, such as DMF, the amine undergoes a Hofmann elimination. [48] It is noteworthy that the reduction products of ammonium salts are problematic as they contribute to the corrosion of metallic cathodes. [51]
Scheme 10.

Pathways for the electrochemical degradation of quaternary ammonium salts or halogenated solvents.
If halogenated organic solvents or halogenated additives are used without protic additives, the cathodic dehalogenation becomes the dominant reaction at the counter electrode. The cathodic cleavage follows a similar mechanism as the degradation of ammonium salts. The carbanions formed in this way then react as the EGB. [52]
An example where the dehalogenation of halogenated solvents is reported as the counter reaction is the synthesis of α,α‐dihaloketones from alkynes (Scheme 11). The oxidation process at the anode is initiated by chloride oxidation and addition to an alkyne. The chloride was generated by the reduction of chloroform at the cathode, which also captured released protons from water during oxygenation. As both reactions lead in combination to the desired product, this synthesis can be regarded as paired electrolysis. [53]
Scheme 11.

Oxydihalogenation of alkynes using halogenated solvents as the halide source.
2.3. Sacrificial Additives
If the generation of a base is undesirable and/or the reaction still requires a quite negative cathodic potential, sacrificial additives have been added into the electrolyte or at least into the catholyte. A sacrificial additive, sometimes called an indirect oxidizer, is an additive to enforce counter reactions. It is added in at least equimolar amounts with respect to electron uptake. This additive should be easily reduced at the cathode, but not oxidized at the anode. It is important to prove that the additive does not interfere with the intermediates/products or act as a direct oxidizer of the starting material. It is beneficial to choose inexpensive, stable, and nontoxic compounds as additives, as otherwise the benefits of electro‐organic synthesis get lost. The most prominent example of those is the addition of strong mineral or organic acids, as described previously. Another additive is, for example, sulfur dioxide: In the multicomponent reactions for the synthesis of sulfonates, sulfamides, and sulfonamides, the smooth reduction of SO2 at the cathode is observed as a counter reaction. SO2 has a lower reduction potential than the HER in HFIP. To prevent the loss of reactant, the reaction must be carried out in divided cells. [54]
2.4. CO2 Reduction
The reduction of carbon dioxide is an important field of electrochemistry. The reduction of CO2 takes place in a CO2 atmosphere, [55] in scCO2 (supercritical), [56] dissolved, [57] or by using gas diffusion electrodes (GDEs). [58] The electrochemical reduction of CO2 leads to various synthetically valuable C1 and C2 building blocks such as formic acid, [59] carbon monoxide, [60] methanol, [61] and ethylene. [62] As a consequence of the wide application of these components as synthetic gases or biofuels, the reduction of CO2 has gained huge interest in recent years. [63] Various processes have been developed to obtain specific and selective products. In large‐scale processes, the oxygen evolution reaction (OER) is a frequently listed process at the anode. [64] However, as result of the high over‐potential in the OER, organic oxidation reactions can be used as an energetically favorable counter reaction.[ 65 , 66 ] In this regard, various organic reactions involving CO2 reduction as a counter reaction have been investigated and developed in recent years.
Kenis and co‐workers have performed techno‐economic studies on CO2 reduction as a counter reaction for the oxidation of glycerol. They found that CO2 reduction has a lower energy consumption when coupled with glycerol oxidation than with oxygen evolution. The required cell voltage drops from −2.1 V to −1.2 V, when formate is the desired reduction product. [65] In addition to the energy studies, there is also growing interest in CO2 reduction as a counter reaction in preparative synthesis. Moeller and co‐workers described the reduction of CO2 to carbon monoxide in a divided cell as a counter reaction in the synthesis of benzimidazoles (Scheme 12). The CO2 reduction was performed at a rhenium electrocatalyst with excellent faradaic efficiency. [67] Berlinguette and co‐workers described the reduction of CO2 to carbon monoxide as a counter reaction to the TEMPO‐mediated oxidation of alcohols to aldehydes and ketones in an aqueous electrolyte. [68]
Scheme 12.

Oxidative formation of benzimidazole paired with the reduction of CO2. Ar: 4‐hydroxy‐3,5‐dimethoxybenzene. gC: glassy carbon. CAN: ceric ammonium nitrate.
3. Anodic Counter Reactions
In the development of cathodic processes, the correct choice of a suitable oxidative counter reaction is more challenging, since compatibility with the desired products is often lacking.
3.1. Sacrificial Anodes
The most prominent example of a counter reaction, which is mostly used during initial screening, is sacrificial anodes. Non‐noble and readily available metals such as Mg, Zn, Fe, or Al are used as the anode. [69] During operation, the anode material dissolves into the corresponding stable cations, which might aggregate and, therefore, stabilize the anodically formed anions. This oxidation usually takes place at a quite low potential, which efficiently suppresses other side reactions (Table 2).
Table 2.
Standard oxidation potential for common sacrificial anodes.
|
|
Entry |
Metal oxidation |
Potential[a] Ε 0 [V] |
Ref. |
|
|---|---|---|---|---|---|
|
|
1 |
|
−2.37 |
||
|
2 |
|
−1.66 |
|||
|
3 |
|
−0.76 |
|||
|
4 |
|
−0.48 |
|||
|
5 |
|
−0.26 |
|||
|
6 |
|
0.52 |

[a] Potential vs. SHE at 25 °C and 1 atm.
However, the anode material is consumed in this reaction process, which creates additional reagent waste. In addition, the loss of electrode material alters the inter‐electrode gap, which also influences the electrolysis outcome. [71] Despite this, sacrificial anodes find actual use in the cathodic coupling of prefunctionalized arenes, [72] electroreductive carboxylations,[ 73 , 74 , 75 ] and in cross‐coupling reactions of activated carbon radicals. [76] The cationic polymerization of ϵ‐caprolactam, initiated by a sacrificial anode, has also been reported. [77] It is noteworthy, from the point of view of energy efficiency, that sacrificial anodes are not green at all, since their production consumes large amounts of energy. [71]
3.1.1. The Relevance of the Anode Material
When selecting the appropriate anode material, in addition to the oxidation potential, it is important to note that the released cation goes into solution and can participate in the chemical reaction. Baran and co‐workers described a scalable electrochemical Birch reduction under mild conditions (Scheme 13). Although the process occurs exclusively at the cathode, they were able to show in their reaction that the choice of anode material has a significant effect on the yield and reaction conditions. By using Mg anodes, the reaction can occur at room temperature. By using an aluminum anode under the same conditions, the electrolysis temperature had to be decreased to −78 °C to ensure selective product formation. However, the use of a copper anode or a graphite anode in combination with indirect reductants such as pyrrole, hydrazine, or formate did not give satisfactory yields. [78]
Scheme 13.

Electrochemical Birch reduction at room temperature.
The electrochemical incorporation of carbon dioxide is a promising tool for the reductive synthesis of carboxylic acids. Mg and Al are often reported as sacrificial anodes.[ 75 , 79 ] However, the unreactive Al and Mg carboxylates are formed in situ, which prevents the Kolbe reaction of the formed product as a possible side reaction at the anode. [80] The significant impact of the dissolved cation was demonstrated in the electrochemical Horner–Wadsworth–Emmons reaction for the synthesis of (E)‐unsaturated esters by Frontana‐Uribe and co‐workers (Scheme 14). The cathodic deprotonation of phosphonate promoted the reaction, and no additional base was required. The oxidation of the Mg sacrificial anode was described as being the anodic counter reaction. Cyclic voltammographic measurements showed that the reduction of phosphonate was 0.5 V less negative when Mg2+ ions were present in solution, as they coordinate with the phosphor ylide. [81]
Scheme 14.

Electrochemical Horner–Wadsworth–Emmons reaction. [a] Potential vs. Ag/AgCl.
3.1.2. Utilizing the Released Ions
The ions released by the sacrificial anode can, however, be used not only to stabilize intermediates, but also to actively participate in the reaction. An alternative to the pure sacrifice of the anodes is the synthesis of metal reagents from the anode material. Both the synthesis of inorganic metal reagents, such as FeO4 2−, [82] and the synthesis of organometallic compounds as CuI‐carbenes, [83] or metallasalens have been reported. [84]
Dunach and co‐workers described a SmII system for catalytic coupling reactions that used a magnesium sacrificial anode. [85] Even though several coupling reactions were reported, the presence of a corresponding second metal may favor ligand redistribution and it was not highlighted whether the SmII species is formed cathodically or is reduced by Mg. [86] Thus, Mellah and co‐workers focused on enhancing the efficiency of the SmII electrocatalyst by using inert electrodes (Scheme 15). A RVC anode with a Sm cathode showed similar results and had halide oxidation as a counter reaction. [87] They combined this approach with the use of an Sm sacrificial anode to generate SmII species in situ by pre‐electrolysis. [88]
Scheme 15.

Electrochemical SmII‐enabled coupling of aldehydes by in situ preparation of the catalyst from an Sm sacrificial anode.
Navarro and co‐workers reported an efficient process for the electrocatalytic hydrogenation of alkenes using an iron cathode and a nickel sacrificial anode. The Ni2+ ions of the anode are reduced at the cathode, where they form the catalytically active nickel surface (Scheme 16). This enables steady renewal of the active surface. Various alkenes could be hydrogenated in slightly acidic aqueous media with high faradaic efficiencies. When neutral conditions were used (NaCl instead of NH4Cl), precipitation of Ni(OH)2 was observed, as the pH value increased over the course of the electrolysis. [89] However, if the pH value is basic before or during the course of the electrolysis, the Ni surface is passivated as Ni(OH)2.
Scheme 16.

Application of Ni anodes in either alkaline or acidic conditions. Ni2+ can be applied for electroplating and the following electrocatalytic hydrogenation.
This passivation layer can prevent Ni anodes from corrosion, as the hydroxide is oxidized to NiO(OH).[ 90 , 91 ] With this active surface, the anode can act as an oxygen‐transfer electrode during the oxidation of alcohols,[ 90 , 91 ] the valorization of renewable feedstock chemicals, [92] or effective oxygen evolution in aqueous alkaline media. [93]
3.2. Oxygen Evolution Reaction (OER)
An alternative anodic counter reaction to sacrificial anodes is the oxidation of (co‐)solvent or any additive. For this, water is often used as a harmless and readily available electron source. The oxidation of water leads to the formation of oxygen. The over‐potential depends on the anode material and pH value. [94]
Compared to hydrogen evolution, oxygen evolution usually requires a higher over‐potential. [95] Anode materials having a high over‐potential, such as boron‐doped diamond (BDD) or Pt, are prone to the formation of hydroxyl radials and hydrogen peroxide. [96] So, BDD anodes in the presence of water are used for the mineralization of organic pollutants, but are not a good choice if the OER is a desired counter reaction. [97] To enforce the OER in water splitting processes or as a counter reaction in electro‐organic synthesis, anodes with a moderate over‐potential such as metal oxides or graphite are used. [98] Electrocatalytic electrodes with a low over‐potential have been developed. Anodes consisting of a mixed metal oxide (MMO) such as Ru x Ti1−x O2 or Ir x Ti1−x O2 supported on Ti or Ta fulfill this criterium. [99]
A process for the electrocatalytic hydrogenation of cyanamide in aqueous acetic acid has been described (Scheme 17). Replacing the graphite anode by a MMO anode in a flow cell led to the yield of formamidine acetate being increased, while the cell voltage decreased at same time. This offered a significant increase in energy efficiency. [100] The over‐potential for the OER at graphite is only slightly higher than that of IrO2 at 10 mA cm−2. [101] However, the potential for the C‐oxidation of a graphite surface into CO or CO2 is very low. Therefore, decomposition of the anode surface and modification at graphite proceeds during electrolysis, while the efficiency of the OER decreases during the course of electrolysis. [102]
Scheme 17.

Supporting‐electrolyte‐free electrocatalytic hydrogenation of cyanamide at Ni‐foam cathodes with the OER as the counter reaction. [a] 0.5 m H2SO4 at j=10 mA cm−2 obtained from Ref. [101].
In addition to oxygen evolution, dimensionally stable anodes (DSAs) also play an important role in the evolution of chlorine gas from chloride, as in chlorine‐alkali electrolysis. [103] However, this plays only a minor role as a counter reaction in electro‐organic synthesis. The oxidation of water is also described in the Baizer process for the electrochemical hydrodimerization of acrylonitrile at a platinum anode in a divided cell. [104]
As water is often not suitable as a solvent for organic reactions, because of the poor solubility of most organic substrates in water, the addition of only equimolar amounts of water or its use as a co‐solvent in the electrolyte is often used to ensure oxygen formation in a predominantly organic solvent. In such a case, water can also act as a proton source as many reductions occur with the uptake of stoichiometric amounts of protons. An often‐described method is the combination of MeCN or alcohols with water at a glassy carbon anode. [105] With this method, even nitrones and amines can be synthesized in an undivided cell without re‐oxidation, as the OER is favored. This counter reaction was also described in the reduction of prochiral ketones by Yadav and Singh, who used DMF/water 9 : 1 as the anolyte. [106]
3.3. The Oxidation of Alcohols
In addition to the oxidation of water, the oxidation of short alcohols also plays an important role as a counter reaction. Of particular interest here is the oxidation of methanol. In the case of complete oxidation, MeOH is converted via formaldehyde, formic acid, or formate ultimately into carbon dioxide. The oxidation potential of alcohols depends on the anode material as well as the pH value of the solution. [107]
Our group described the electrochemical synthesis of benzo[d]triazoles (Scheme 18). The initial reduction was carried out in an acetonitrile/water mixture, thereby enforcing oxygen evolution. However, the yield was indeed significantly increased when switching the solvent to methanol. It was assumed that the benefit arose from the enhanced solubility of starting materials as well as the preferred counter reaction. The oxidation of MeOH was confirmed by the detection of sodium formate in the crude NMR spectrum. The use of NaOH as a supporting electrolyte had a beneficial influence on the formation of formate, as it stabilizes the formate as well as decreasing the oxidation potential of MeOH. [108]
Scheme 18.

Cathodic synthesis of benzotriazoles, with the oxidation of MeOH as the counter reaction.
A process for the reductive coupling of styrene derivatives and aliphatic carbonyl compounds in an undivided cell was developed at BASF (Scheme 19). The oxidation of methanol as a solvent is described as the counter reaction. The selectivity and the yield of the reductive coupling depend strongly on the efficiency of the counter reaction. Thus, adding catalytic amounts of 4‐hydoxy‐TEMPO led to the necessary potential of the counter reaction being lowered and the product yield increasing. The lowering of the anode potential also has the benefit that the anodic oxidation of aromatic vinyl compound is suppressed. [109]
Scheme 19.

Reductive addition of styrenes and aliphatic carbonyl compounds. GDL: gas diffusion layer. MTBS: methyltributylammonium methyl sulfate.
3.4. Anodic Additives
If the addition of water, alcohols, or the use of a sacrificial anode is undesirable or leads to unwanted by‐products in the reaction setup, additives are inevitably incorporated to support the oxidation process. The most prominent examples are formates or oxalates. These are both subject to oxidation to CO2 at a less positive potential. In this process, different equivalents of protons are released. In both cases, the carboxylates serve as indirect reducing agents. In such cases, it is always necessary to check whether the actual reduction occurs via the cathode or the additive itself. Wang et al. reported the electrochemical formation of pinacols from ketones (Scheme 20). Formic acid was added as the sacrificial additive for the anode. When it was omitted, the yield of pinacol drastically decreased. Sodium azide, used as a supporting electrolyte, was shown to have a positive effect on the oxidation of formic acid, as the electron transfer was enforced. When acetic acid was used instead of formic acid, diphenylmethanol was found as the main product from the hydrogenation of benzophenone. [110]
Scheme 20.

Electrochemical reduction of ketones to pinacols and alcohols.
Another non‐green but practical laboratory solution is the oxidation of sterically hindered amines. Xiang and co‐workers reported the reduction of cyclic imides to hydroxylactams (Scheme 21). If the sterically hindered amine is not used, a significant loss in yield is observed. The oxidation of the amine results in the formation of an aminyl radical. This species is very acidic and will form neutral radicals after proton exchange with the solvent. After further oxidation and H‐abstraction, those radicals undergo imine formation with the intermediate formed at the cathode. [111]
Scheme 21.

Electrochemical reduction of phthalimides using a sterically demanding amine as the electron source.
However, especially sterically demanding sec‐amines or tertiary with α‐protons such as piperidine, [112] diisopropylethylamine, [113] trimethylamine, [114] or triethanolamine [115] are often used. Less‐hindered amines also undergo a slight anodic oxidation, but the resulting radical can covalently bind to several anodes and chemically modify them. [116] Amines without α‐H atoms undergo either decomposition [117] or form stable radical cations (triarylamine mediators). [118]
Baran and co‐workers described a similar deoxygenation procedure for phthalimides by using rapidly alternating polarity (rAP, Scheme 22). By applying rAP, better yields and unusually high selectivity were obtained. To achieve a desired counter reaction, pivaloic acid was used as an additive. This undergoes non‐Kolbe electrolysis and is converted into MTBE (methyl tert‐butyl ether) after reaction with MeOH. In various mechanistic studies, it could be shown that the Shono oxidation, which occurs as a by‐product at the counter electrode, could be suppressed and, thus, the desired anodic counter reaction could be driven more selectively. [119]
Scheme 22.

Electrochemical deoxygenation of phtalimides by rAP. Boc: tert‐butoxycarbonyl. Piv: Pivalyol.
3.5. The Oxidation of Halides
If halides are released during a reaction or if halides are used as supporting electrolytes, oxidation to form an oxidized halogen species (X+, X⋅, or X2) can occur. The oxidation usually takes place at a low potential, which allows an easy reduction on the other hand. However, the released active halogen species is a strong oxidant, which can either have a positive effect on the desired reaction performance if oxidation/halogenation is desired or must be removed from the reaction solution as an undesired by‐product. The group of Manthiram developed a procedure for the reductive electrocarboxylation of alkyl halides (Scheme 23). In contrast to other synthetic approaches, which are often limited by the use of a sacrificial anode, they were able to avoid this. The addition of MgBr2 achieved a stabilization of the carboxylate, coordinating the Mg2+ ion which is otherwise released during sacrificial anode methods. The reaction can be performed in an undivided flow cell. The oxidation of bromide to bromine is described as the counter reaction. When chlorides were present at electrolysis, it was necessary to add an increased amount of bromide in the form of NBu4Br to prevent Cl2 formation, as otherwise undesired by‐products would be formed by chlorination. [80]
Scheme 23.

Carboxylation of halides with CO2 as well as bromide oxidation.
The electroreductive carboxylation of imines at an Al anode has been described by Li et al. (Scheme 24). However, the anode only undergoes partial sacrifice. Bromide from the supporting electrolyte is also oxidized at the anode into bromine. This acts, after the reductive carboxylation, as a brominating agent to form an N‐bromoamino acid. [74]
Scheme 24.

Electrochemical synthesis of N‐bromoamino acids.
4. Paired Electrolysis
4.1. Organic Synthesis as a Counter Reaction
In recent years, the term “paired electrolysis” has become widely established in the design of sustainable and efficient processes. [120] Paired electrolysis is when both the cathode and the anode reactions merge to generate synthetically valuable product(s). Such electrolysis is energetically useful and a sustainable approach in electrochemistry, since even the otherwise harmless side streams of oxidation or reductions can be further avoided. However, paired electrolysis is often not easy to perform, since both the anode and cathode reactions must be coordinated precisely, and the resulting products must be able to be separated from each other. There are four types of paired electrolysis: parallel, consecutive, convergent, and divergent (Scheme 25).
Scheme 25.

Common types of paired electrolyses. Parallel (A), consecutive (B), convergent (C), and divergent (D).
When choosing the reaction, it must be considered that both reactions take place in the same base electrolyte. The required voltage must be matched in both cases and care must be taken above all to ensure that the typical counter reactions mentioned above do not occur as competing reactions.
The simplest example is parallel paired electrolysis. Here, two independent organic reactions are run in the same electrolyte. The synthesis can be divided, which has the advantage that the half cells can be processed independently and side reactions can be suppressed, but ion exchange has to be allowed. However, the use of a separator leads to an ohmic drop. Of particular interest is, therefore, parallel paired electrolysis in an undivided cell. Such a process is used at BASF, among others. The anodic methoxylation of tert‐butyltoluene into a protected aldehyde is coupled with the reduction of phthalic acid into phthalide (Scheme 26). Both electrochemical reactions can be carried out in MeOH and the protons are balanced. [121]
Scheme 26.

Anodic methoxylation of tert‐butyltoluene paired with the reductive synthesis of phthalide.
A recently formulated example of how counter reactions can be paired was given by Waldvogel, Morandi, and co‐workers. In electron‐shuttle electrolysis, the cathodic dehalogenation of vicinal dihalides is combined with the dihalogenation of alkenes (Scheme 27). In this method, the reversible transport of vicinal dihalides is used for selective synthesis. Both the generation of dihalides as products without the use of hazardous oxidants (replacing chlorine by ClCH2CH2Cl) and the preparative degradation of persistent pollutants were shown. [122]
Scheme 27.
Consecutive paired synthesis for the electron‐shuttle transfer of halo substituents.
Consecutive electrolysis is, as a principle, also known for the synthesis of nitriles from the oxidation of aldoximes into nitrile oxides and their reduction, [123] the preparation of nitrosobenzenes by the oxidation of hydroxylamines from the reduction of nitrobenzene during the preparation of heterocycles, [124] and the arylation of heteroarenes with aryldiazonium salts. [125]
In a convergent synthesis, the anodic and cathodic reactions form reactive intermediates. Those migrate into solution and undergo a merging reaction. Lei and co‐workers describe a method for the S,N coupling of thiols and (sulfon)amides to sulfenamides (Scheme 28). In an upstream reaction, the thiol is oxidized to the disulfide. In this process, the HER occurs as a counter reaction. In the second phase of the electrolysis, the disulfide is reduced to the S‐centered radical. This occurs with N oxidation of the (sulfone)amidyl radical in the anodic phase. The success of the reaction is coupled to the non‐necessity for mass transfer. To suppress secondary or side reactions resulting from slow mass transfer, both the anode and cathode reactions can be performed by applying an alternating current (AC) to the same electrode. The basis for this is that both the anode and cathode materials are the same. If the reaction is carried out under direct current (DC) electrolysis conditions, the S,N product is not detected. [126]
Scheme 28.

Convergent synthesis of sulfenamides using alternating current.
Another method for a convergent synthesis has been described by the Hilt group. They reported a method for the dibromination of alkenes by anodic oxidation of bromide paired with oxygen reduction as a counter reaction. The hydrogen peroxide formed from the reduction also acted as an oxidizer for the oxidation of bromide into bromine. This method allows the formation of bromine at both the cathode and anode. [127]
4.2. Electromediated Ni Catalysis
The combination of transition‐metal catalysis and electro‐organic synthesis has gained importance in recent years. [128] Through this combined approach, electrochemical reactions can be driven mildly and stereoselectively. In the case of oxidative or reductive coupling reactions, either the catalyst is (re)activated or the organic substrate is (re)activated at the corresponding electrode. In this case, easy‐to‐perform counter reactions such as the HER, sacrificial anodes, or halide oxidation are usually carried out. [129] Special attention is paid to paired transition‐metal‐catalyzed coupling reactions. Baran and co‐workers described a method for the amination of aryl halides (Scheme 29). Herein, the electroreductive activation of the NiII catalyst to NiI is the initial step. The active nickel species inserts into the aryl halide and the arene complex is reduced at the cathode. After ligand exchange with the amine, the complex is oxidized at the anode. After oxidation, reductive elimination of the C,N coupled product occurs. When the RVC anode was replaced by a Zn sacrificial anode, the yield drastically decreased and large amounts of biaryl were generated as the product. [130] A similar mechanism is found in the arylation of NH‐sulfoximines, [131] the thiolation of aryl halides, [132] and the oxygenation of sulfides. [133]
Scheme 29.

Ni‐catalyzed amination of aryl halides. Ligand: 4,4′‐di‐tert‐butyl‐2,2′‐dipyridyl.
5. Conclusions
In the execution and development of new electrochemical routes, the choice of the counter reaction is an important means of enabling targeted reaction control. The proper operation of the counter reaction not only serves to enable a faradic conversion, but often also actively contributes to the outcome of the reaction. Therefore, when carrying out an electrosynthesis, it is important to think about possible counter reactions and to investigate their influence on the reaction. The development of paired electrolysis is of particular interest for further investigations, since organic reactions are run as counter reactions. Here, reagent wastes and side‐stream products are largely omitted. Nevertheless, the classical side streams of oxidation are one of the special features of electrosynthesis. Hydrogen and biomethanol from the reduction of CO2 are already being considered as sustainable fuels of the future and can, thus, be used efficiently as storage forms of excess energy from chemical processes.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Martin Klein studied biomedical chemistry in Mainz. He received his BSc at Johannes Gutenberg University in 2017 (Prof. S. R. Waldvogel) and in 2019 he received his MSc at Mainz (Prof. S. R. Waldvogel). He is currently conducting PhD research, which focuses on electro‐organic synthesis.

Biographical Information
Siegfried R. Waldvogel studied chemistry in Konstanz and received his PhD in 1996 from the University of Bochum/Max Planck Institute for Coal Research (Prof. M. T. Reetz). After postdoctoral research at La Jolla, CA (Prof. J. Rebek, Jr.), he started his own research at the Universities of Münster and Bonn. He became full professor at JGU Mainz in 2010. His research interests are novel electro‐organic transformations including bio‐based feedstocks. In 2018, he cofounded ESy‐Labs GmbH, which provides custom electrosynthesis and contract R&D.

Acknowledgements
We acknowledge the Deutsche Forschungsgemeinschaft (Wa1276/17‐2) for financial support. Support by SusInnoScience (Forschungsinitiative des Landes Rheinland‐Pfalz) and by ECHELON (Carl Zeiss Foundation) is highly appreciated. Open Access funding enabled and organized by Projekt DEAL.
M. Klein, S. R. Waldvogel, Angew. Chem. Int. Ed. 2022, 61, e202204140; Angew. Chem. 2022, 134, e202204140.
References
- 1.
- 1a. Waldvogel S. R., Janza B., Angew. Chem. Int. Ed. 2014, 53, 7122–7123; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7248–7249; [Google Scholar]
- 1b. Wiebe A., Gieshoff T., Möhle S., Rodrigo E., Zirbes M., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 5594–5619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5694–5721; [Google Scholar]
- 1c. Möhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149; [Google Scholar]
- 1d. Pollok D., Waldvogel S. R., Chem. Sci. 2020, 11, 12386–12400; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Kärkäs M. D., Chem. Soc. Rev. 2018, 47, 5786–5865; [DOI] [PubMed] [Google Scholar]
- 1g. Yoshida J.-I., Kataoka K., Horcajada R., Nagaki A., Chem. Rev. 2008, 108, 2265–2299; [DOI] [PubMed] [Google Scholar]
- 1h. Little R. D., Moeller K. D., Chem. Rev. 2018, 118, 4483–4484; [DOI] [PubMed] [Google Scholar]
- 1i. Zhu C., Ang N. W. J., Meyer T. H., Qiu Y., Ackermann L., ACS Cent. Sci. 2021, 7, 415–431; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1j. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Frontana-Uribe B. A., Little R. D., Ibanez J. G., Palma A., Vasquez-Medrano R., Green Chem. 2010, 12, 2099; [Google Scholar]
- 2b. Yuan Y., Lei A., Nat. Commun. 2020, 11, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hammerich O., Speiser B., Organic Electrochemistry, CRC Press, Boca Raton, 2015. [Google Scholar]
- 4. Moeller K. D., Chem. Rev. 2018, 118, 4817–4833. [DOI] [PubMed] [Google Scholar]
- 5. Nutting J. E., Gerken J. B., Stamoulis A. G., Bruns D. L., Stahl S. S., J. Org. Chem. 2021, 86, 15875–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Meyer T. H., Choi I., Tian C., Ackermann L., Chem 2020, 6, 2484–2496; [Google Scholar]
- 6b. Hilt G., ChemElectroChem 2020, 7, 395–405; [Google Scholar]
- 6c. Kingston C., Palkowitz M. D., Takahira Y., Vantourout J. C., Peters B. K., Kawamata Y., Baran P. S., Acc. Chem. Res. 2020, 53, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Heard D. M., Lennox A. J. J., Angew. Chem. Int. Ed. 2020, 59, 18866–18884; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 19026–19044; [Google Scholar]
- 7b. Seidler J., Strugatchi J., Gärtner T., Waldvogel S. R., MRS Energy Sustain. 2020, 7, e42; [Google Scholar]
- 7c. Waldvogel S. R., Lips S., Selt M., Riehl B., Kampf C. J., Chem. Rev. 2018, 118, 6706–6765; [DOI] [PubMed] [Google Scholar]
- 7d. Röckl J. L., Pollok D., Franke R., Waldvogel S. R., Acc. Chem. Res. 2020, 53, 45–61. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Wu T., Nguyen B. H., Daugherty M. C., Moeller K. D., Angew. Chem. Int. Ed. 2019, 58, 3562–3565; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3600–3603; [Google Scholar]
- 8b. Nguyen B. H., Perkins R. J., Smith J. A., Moeller K. D., J. Org. Chem. 2015, 80, 11953–11962. [DOI] [PubMed] [Google Scholar]
- 9. Hickling A., Salt F. W., Trans. Faraday Soc. 1940, 36, 1226–1235. [Google Scholar]
- 10.
- 10a. Katsuki N., Takahashi E., Toyoda M., Kurosu T., Iida M., Wakita S., Nishiki Y., Shimamune T., J. Electrochem. Soc. 1998, 145, 2358–2362; [Google Scholar]
- 10b. Waldvogel S. R., Mentizi S., Kirste A., Boron-Doped Diamond Electrodes for Electroorganic Chemistry. In Radicals in Synthesis III. Topics in Current Chemistry, Vol. 320 (Eds.: Heinrich M., Gansäuer A.), Springer, Berlin, Heidelberg, 2011. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Dubouis N., Grimaud A., Chem. Sci. 2019, 10, 9165–9181; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Couper A. M., Pletcher D., Walsh F. C., Chem. Rev. 1990, 90, 837–865. [Google Scholar]
- 12. Wang D., Wang P., Wang S., Chen Y.-H., Zhang H., Lei A., Nat. Commun. 2019, 10, 2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Park H., Vecitis C. D., Hoffmann M. R., J. Phys. Chem. A 2008, 112, 7616–7626; [DOI] [PubMed] [Google Scholar]
- 13b. Elinson M. N., Feducovich S. K., Lizunova T. L., Nikishin G. I., Tetrahedron 2000, 56, 3063–3069; [Google Scholar]
- 13c. Lugiņina J., Linden M., Bazulis M., Kumpiņš V., Mishnev A., Popov S. A., Golubeva T. S., Waldvogel S. R., Shults E. E., Turks M., Eur. J. Org. Chem. 2021, 2557–2577. [Google Scholar]
- 14. Kashiwagi Y., Anzai J., Chem. Pharm. Bull. 2001, 49, 324–326. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Shirai K., Hamamoto T., Maki T., Onomura O., Kise N., Aoyama Y., Matsumara Y., J. Electroanal. Chem. 2001, 507, 191–197; [Google Scholar]
- 15b. Nikishin G. I., Elinson M. N., Lizunova T. L., Ugrak B. I., Tetrahedron Lett. 1991, 32, 2655–2656; [Google Scholar]
- 15c. Kong X., Tian Y., Chen X., Chen Y., Wang W., J. Org. Chem. 2021, 86, 13610–13617. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Hielscher M. M., Gleede B., Waldvogel S. R., Electrochim. Acta 2021, 368, 137420; [Google Scholar]
- 16b. Hielscher M., Oehl E. K., Gleede B., Buchholz J., Waldvogel S. R., ChemElectroChem 2021, 8, 3904–3910; [Google Scholar]
- 16c. Selt M., Gleede B., Franke R., Stenglein A., Waldvogel S. R., J. Flow Chem. 2021, 11, 143–162; [Google Scholar]
- 16d. Schulz L., Waldvogel S., Synlett 2019, 30, 275–286; [Google Scholar]
- 16e. Lipp A., Ferenc D., Gütz C., Geffe M., Vierengel N., Schollmeyer D., Schäfer H. J., Waldvogel S. R., Opatz T., Angew. Chem. Int. Ed. 2018, 57, 11055–11059; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11221–11225; [Google Scholar]
- 16f. Kirste A., Hayashi S., Schnakenburg G., Malkowsky I. M., Stecker F., Fischer A., Fuchigami T., Waldvogel S. R., Chem. Eur. J. 2011, 17, 14164–14169. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Morofuji T., Shimizu A., Yoshida J.-I., J. Am. Chem. Soc. 2013, 135, 5000–5003; [DOI] [PubMed] [Google Scholar]
- 17b. Herold S., Möhle S., Zirbes M., Richter F., Nefzger H., Waldvogel S. R., Eur. J. Org. Chem. 2016, 1274–1278; [Google Scholar]
- 17c. Möhle S., Herold S., Hillerson N. D., Waldvogel S. R., ChemElectroChem 2019, 6, 121–125; [Google Scholar]
- 17d. Möhle S., Herold S., Richter F., Nefzger H., Waldvogel S. R., ChemElectroChem 2017, 4, 2196–2210; [Google Scholar]
- 17e. Wesenberg L. J., Diehl E., Zähringer T. J. B., Dörr C., Schollmeyer D., Shimizu A., Yoshida J.-I., Hellmich U. A., Waldvogel S. R., Chem. Eur. J. 2020, 26, 17574–17580; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17f. Wesenberg L. J., Herold S., Shimizu A., Yoshida J.-I., Waldvogel S. R., Chem. Eur. J. 2017, 23, 12096–12099. [DOI] [PubMed] [Google Scholar]
- 18. Zollinger D., Griesbach U., Pütter H., Comninellis C., Electrochem. Commun. 2004, 6, 600–604. [Google Scholar]
- 19.
- 19a. Siu T., Yudin A. K., Org. Lett. 2002, 4, 1839–1842; [DOI] [PubMed] [Google Scholar]
- 19b. Siu T., Yudin A. K., J. Am. Chem. Soc. 2002, 124, 530–531. [DOI] [PubMed] [Google Scholar]
- 20. Zhao H.-B., Xu P., Song J., Xu H.-C., Angew. Chem. Int. Ed. 2018, 57, 15153–15156; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15373–15376. [Google Scholar]
- 21. Yoshida J.-I., Suga S., Chem. Eur. J. 2002, 8, 2650. [Google Scholar]
- 22. Yoshida J.-I., Murata T., Isoe S., J. Organomet. Chem. 1988, 345, C23–C27. [Google Scholar]
- 23. Yoshida J.-I., Suga S., Suzuki S., Kinomura N., Yamamoto A., Fujiwara K., J. Am. Chem. Soc. 1999, 121, 9546–9549. [Google Scholar]
- 24. Kim S., Hayashi K., Kitano Y., Tada M., Chiba K., Org. Lett. 2002, 4, 3735–3737. [DOI] [PubMed] [Google Scholar]
- 25. Fu N., Li L., Yang Q., Luo S., Org. Lett. 2017, 19, 2122–2125. [DOI] [PubMed] [Google Scholar]
- 26. Zu B., Ke J., Guo Y., He C., Chin. J. Chem. 2021, 39, 627–632. [Google Scholar]
- 27. Elsherbini M., Winterson B., Alharbi H., Folgueiras-Amador A. A., Génot C., Wirth T., Angew. Chem. Int. Ed. 2019, 58, 9811–9815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9916–9920. [Google Scholar]
- 28. Herszman J. D., Berger M., Waldvogel S. R., Org. Lett. 2019, 21, 7893–7896. [DOI] [PubMed] [Google Scholar]
- 29. Watts K., Gattrell W., Wirth T., Beilstein J. Org. Chem. 2011, 7, 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang S., Liu Y., Lei A., Chem 2018, 4, 27–45. [Google Scholar]
- 31. Tang H.-T., Jia J.-S., Pan Y.-M., Org. Biomol. Chem. 2020, 18, 5315–5333. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Gieshoff T., Kehl A., Schollmeyer D., Moeller K. D., Waldvogel S. R., J. Am. Chem. Soc. 2017, 139, 12317–12324; [DOI] [PubMed] [Google Scholar]
- 32b. Gieshoff T., Schollmeyer D., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 9437–9440; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9587–9590; [Google Scholar]
- 32c. Gieshoff T., Trieu V., Heijl J., Waldvogel S. R., Beilstein J. Org. Chem. 2018, 14, 1578–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klein M., Waldvogel S. R., Angew. Chem. Int. Ed. 2021, 60, 23197–23201; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 23382–23387. [Google Scholar]
- 34. Röckl J. L., Dörr M., Waldvogel S. R., ChemElectroChem 2020, 7, 3686–3694. [Google Scholar]
- 35. Li L., Xue M., Yan X., Liu W., Xu K., Zhang S., Org. Biomol. Chem. 2018, 16, 4615–4618. [DOI] [PubMed] [Google Scholar]
- 36. Wu J., Zhou Y., Zhou Y., Chiang C.-W., Lei A., ACS Catal. 2017, 7, 8320–8323. [Google Scholar]
- 37. Wang F., Stahl S. S., Acc. Chem. Res. 2020, 53, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.
- 38a. Saraswat A., Sharma L. K., Singh S., Siddiqui I. R., Singh R. K. P., Res. Chem. Intermed. 2013, 39, 1393–1399; [Google Scholar]
- 38b. Kim S., Uchiyama R., Kitano Y., Tada M., Chiba K., J. Electroanal. Chem. 2001, 507, 152–156. [Google Scholar]
- 39. Lund H., J. Electroanal. Chem. 2005, 584, 174–181. [Google Scholar]
- 40. Minato D., Mizuta S., Kuriyama M., Matsumura Y., Onomura O., Tetrahedron 2009, 65, 9742–9748. [Google Scholar]
- 41.
- 41a. Baslé O., Borduas N., Dubois P., Chapuzet J. M., Chan T.-H., Lessard J., Li C.-J., Chem. Eur. J. 2010, 16, 8162–8166; [DOI] [PubMed] [Google Scholar]
- 41b. Feroci M., Chiarotto I., Orsini M., Sotgiu G., Inesi A., Adv. Synth. Catal. 2008, 350, 1355–1359. [Google Scholar]
- 42. Bordwell F. G., Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar]
- 43. Magill A. M., Cavell K. J., Yates B. F., J. Am. Chem. Soc. 2004, 126, 8717–8724. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Haouas B., Saied T., Ayari H., Arfaoui Y., Benkhoud M. L., Boujlel K., J. Sulfur Chem. 2016, 37, 391–400; [Google Scholar]
- 44b. Sbei N., Batanero B., Barba F., Haouas B., Benkhoud M. L., Barba I., Tetrahedron 2018, 74, 2068–2072. [Google Scholar]
- 45. Du K.-S., Huang J.-M., Org. Lett. 2018, 20, 2911–2915. [DOI] [PubMed] [Google Scholar]
- 46. Foley J. K., Korzeniewski C., Pons S., Can. J. Chem. 1988, 66, 201–206. [Google Scholar]
- 47.
- 47a. Mayell J. S., Bard A. J., J. Am. Chem. Soc. 1963, 85, 421–425; [Google Scholar]
- 47b. Ross S. D., Finkelstein M., Petersen R. C., J. Am. Chem. Soc. 1960, 82, 1582–1585. [Google Scholar]
- 48. Dahm C. E., Peters D. G., J. Electroanal. Chem. 1996, 402, 91–96. [Google Scholar]
- 49. Brown O. R., Gonzalez E. R., J. Electroanal. Chem. Interfacial Electrochem. 1972, 35, 13–19. [Google Scholar]
- 50. Pons S., Khoo S. B., Electrochim. Acta 1982, 27, 1161–1169. [Google Scholar]
- 51. Wirtanen T., Prenzel T., Tessonnier J.-P., Waldvogel S. R., Chem. Rev. 2021, 121, 10241–10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barhdadi R., Gal J., Heintz M., Troupel M., Périchon J., Tetrahedron 1993, 49, 5091–5098. [Google Scholar]
- 53. Meng X., Zhang Y., Luo J., Wang F., Cao X., Huang S., Org. Lett. 2020, 22, 1169–1174. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Blum S. P., Karakaya T., Schollmeyer D., Klapars A., Waldvogel S. R., Angew. Chem. Int. Ed. 2021, 60, 5056–5062; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 5114–5120; [Google Scholar]
- 54b. Blum S. P., Schollmeyer D., Turks M., Waldvogel S. R., Chem. Eur. J. 2020, 26, 8358–8362; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54c. Blum S. P., Schäffer L., Schollmeyer D., Waldvogel S. R., Chem. Commun. 2021, 57, 4775–4778; [DOI] [PubMed] [Google Scholar]
- 54d. Blum S. P., Hofman K., Manolikakes G., Waldvogel S. R., Chem. Commun. 2021, 57, 8236–8249. [DOI] [PubMed] [Google Scholar]
- 55. Bazzi S., Le Duc G., Schulz E., Gosmini C., Mellah M., Org. Biomol. Chem. 2019, 17, 8546–8550. [DOI] [PubMed] [Google Scholar]
- 56. Melchaeva O., Voyame P., Bassetto V. C., Prokein M., Renner M., Weidner E., Petermann M., Battistel A., ChemSusChem 2017, 10, 3660–3670. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Naragino H., Saitoh Y., Honda K., Electrochem. Commun. 2022, 134, 107164; [Google Scholar]
- 57b. Nakata K., Ozaki T., Terashima C., Fujishima A., Einaga Y., Angew. Chem. Int. Ed. 2014, 53, 871–874; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 890–893. [Google Scholar]
- 58.
- 58a. Bevilacqua M., Filippi J., Lavacchi A., Marchionni A., Miller H. A., Oberhauser W., Vesselli E., Vizza F., Energy Technol. 2014, 2, 522–525; [Google Scholar]
- 58b. Medvedeva X. V., Medvedev J. J., Tatarchuk S. W., Choueiri R. M., Klinkova A., Green Chem. 2020, 22, 4456–4462. [Google Scholar]
- 59. Zhang R., Lv W., Lei L., Appl. Surf. Sci. 2015, 356, 24–29. [Google Scholar]
- 60.
- 60a. Ma M., Djanashvili K., Smith W. A., Phys. Chem. Chem. Phys. 2015, 17, 20861–20867; [DOI] [PubMed] [Google Scholar]
- 60b. Rosen J., Hutchings G. S., Lu Q., Rivera S., Zhou Y., Vlachos D. G., Jiao F., ACS Catal. 2015, 5, 4293–4299. [Google Scholar]
- 61.
- 61a. Irfan Malik M., Malaibari Z. O., Atieh M., Abussaud B., Chem. Eng. Sci. 2016, 152, 468–477; [Google Scholar]
- 61b. Kobayashi T., Takahashi H., Energy Fuels 2004, 18, 285–286. [Google Scholar]
- 62. Kuhl K. P., Cave E. R., Abram D. N., Jaramillo T. F., Energy Environ. Sci. 2012, 5, 7050. [Google Scholar]
- 63.
- 63a. Finn C., Schnittger S., Yellowlees L. J., Love J. B., Chem. Commun. 2012, 48, 1392–1399; [DOI] [PubMed] [Google Scholar]
- 63b. Lu X., Leung D. Y. C., Wang H., Leung M. K. H., Xuan J., ChemElectroChem 2014, 1, 836–849; [Google Scholar]
- 63c. Costentin C., Robert M., Savéant J.-M., Chem. Soc. Rev. 2013, 42, 2423–2436. [DOI] [PubMed] [Google Scholar]
- 64. Park S., Wijaya D. T., Na J., Lee C. W., Catalysts 2021, 11, 253. [Google Scholar]
- 65. Verma S., Lu S., Kenis P. J. A., Nat. Energy 2019, 4, 466–474. [Google Scholar]
- 66. Martínez N. P., Isaacs M., Nanda K. K., New J. Chem. 2020, 44, 5617–5637. [Google Scholar]
- 67. Llorente M. J., Nguyen B. H., Kubiak C. P., Moeller K. D., J. Am. Chem. Soc. 2016, 138, 15110–15113. [DOI] [PubMed] [Google Scholar]
- 68. Li T., Cao Y., He J., Berlinguette C. P., ACS Cent. Sci. 2017, 3, 778–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.
- 69a. Chaussard J., Folest J.-C., Nedelec J.-Y., Perichon J., Sibille S., Troupel M., Synthesis 1990, 1990, 369–381; [Google Scholar]
- 69b. Silvestri G., Gambino S., Filardo G., Acta Chem. Scand. 1991, 45, 987–992. [Google Scholar]
- 70. Vansek P., in CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 8-21–8-23. [Google Scholar]
- 71. Beil S. B., Pollok D., Waldvogel S. R., Angew. Chem. Int. Ed. 2021, 60, 14750–14759; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 14874–14883. [Google Scholar]
- 72.
- 72a. Jiao K.-J., Liu D., Ma H.-X., Qiu H., Fang P., Mei T.-S., Angew. Chem. Int. Ed. 2020, 59, 6520–6524; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 6582–6586; [Google Scholar]
- 72b. Barhdadi R., Courtinard C., Nédélec J. Y., Troupel M., Chem. Commun. 2003, 1434–1435; [DOI] [PubMed] [Google Scholar]
- 72c. Kweon D.-K., Jang Y.-S., Kim H.-B., Bull. Korean Chem. Soc. 2003, 24, 1049–1050; [Google Scholar]
- 72d. Torii S., Tanaka H., Morisaki K., Tetrahedron Lett. 1985, 26, 1655–1658. [Google Scholar]
- 73.
- 73a. Tokuda M., J. Nat. Gas Chem. 2006, 15, 275–281; [Google Scholar]
- 73b. Chowdhury M. A., Senboku H., Tokuda M., Tetrahedron 2004, 60, 475–481. [Google Scholar]
- 74. Li C.-H., Song X.-Z., Tao L.-M., Li Q.-G., Xie J.-Q., Peng M.-N., Pan L., Jiang C., Peng Z.-Y., Xu M.-F., Tetrahedron 2014, 70, 1855–1860. [Google Scholar]
- 75. Qu Y., Tsuneishi C., Tateno H., Matsumura Y., Atobe M., React. Chem. Eng. 2017, 2, 871–875. [Google Scholar]
- 76.
- 76a. Koyanagi T., Herath A., Chong A., Ratnikov M., Valiere A., Chang J., Molteni V., Loren J., Org. Lett. 2019, 21, 816–820; [DOI] [PubMed] [Google Scholar]
- 76b. Liu Y., Tao X., Mao Y., Yuan X., Qiu J., Kong L., Ni S., Guo K., Wang Y., Pan Y., Nat. Commun. 2021, 12, 6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pierre G., Limosin D., Djelali N.-E., Makromol. Chem. 1991, 192, 2767–2775. [Google Scholar]
- 78. Peters B. K., Rodriguez K. X., Reisberg S. H., Beil S. B., Hickey D. P., Kawamata Y., Collins M., Starr J., Chen L., Udyavara et al S., Science 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.
- 79a. Courtois G., Miginiac L., J. Organomet. Chem. 1974, 69, 1–44; [Google Scholar]
- 79b. Kamekawa H., Senboku H., Tokuda M., Tetrahedron Lett. 1998, 39, 1591–1594; [Google Scholar]
- 79c. Tokuda M., Kabuki T., Katoh Y., Suginome H., Tetrahedron Lett. 1995, 36, 3345–3348; [Google Scholar]
- 79d. Isse A. A., Durante C., Gennaro A., Electrochem. Commun. 2011, 13, 810–813. [Google Scholar]
- 80. Corbin N., Yang D.-T., Lazouski N., Steinberg K., Manthiram K., Chem. Sci. 2021, 12, 12365–12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Palma A., Frontana-Uribe B. A., Cárdenas J., Saloma M., Electrochem. Commun. 2003, 5, 455–459. [Google Scholar]
- 82.
- 82a. Sun X., Zu K., Liang H., Sun L., Zhang L., Wang C., Sharma V. K., J. Hazard. Mater. 2018, 344, 1155–1164; [DOI] [PubMed] [Google Scholar]
- 82b. Wang J., Zheng T., Liu H., Wang G., Zhang Y., Cai C., Electrochim. Acta 2020, 356, 136706. [Google Scholar]
- 83.
- 83a. Chapman M. R., Shafi Y. M., Kapur N., Nguyen B. N., Willans C. E., Chem. Commun. 2015, 51, 1282–1284; [DOI] [PubMed] [Google Scholar]
- 83b. Lake B. R. M., Bullough E. K., Williams T. J., Whitwood A. C., Little M. A., Willans C. E., Chem. Commun. 2012, 48, 4887–4889. [DOI] [PubMed] [Google Scholar]
- 84. Chapman M. R., Henkelis S. E., Kapur N., Nguyen B. N., Willans C. E., ChemistryOpen 2016, 5, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hébri H., Duñach E., Périchon J., Synlett 1992, 1992, 293–294. [Google Scholar]
- 86. Nomura R., Matsuno T., Endo T., J. Am. Chem. Soc. 1996, 118, 11666–11667. [Google Scholar]
- 87. Sun L., Sahloul K., Mellah M., ACS Catal. 2013, 3, 2568–2573. [Google Scholar]
- 88. Sahloul K., Sun L., Requet A., Chahine Y., Mellah M., Chem. Eur. J. 2012, 18, 11205–11209. [DOI] [PubMed] [Google Scholar]
- 89. Santana D. S., Melo G. O., Lima M. V. F., Daniel J. R. R., Areias M. C. C., Navarro M., J. Electroanal. Chem. 2004, 569, 71–78. [Google Scholar]
- 90. Robertson P. M., J. Electroanal. Chem. Interfacial Electrochem. 1980, 111, 97–104. [Google Scholar]
- 91. Kaulen J., Schäfer H.-J., Tetrahedron 1982, 38, 3299–3308. [Google Scholar]
- 92.
- 92a. Breiner M., Zirbes M., Waldvogel S. R., Green Chem. 2021, 23, 6449–6455; [Google Scholar]
- 92b. Zirbes M., Schmitt D., Beiser N., Pitton D., Hoffmann T., Waldvogel S. R., ChemElectroChem 2019, 6, 155–161; [Google Scholar]
- 92c. Rauen A. L., Weinelt F., Waldvogel S. R., Green Chem. 2020, 22, 5956–5960; [Google Scholar]
- 92d. Schmitt D., Regenbrecht C., Hartmer M., Stecker F., Waldvogel S. R., Beilstein J. Org. Chem. 2015, 11, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shinagawa T., Ng M. T.-K., Takanabe K., Angew. Chem. Int. Ed. 2017, 56, 5061–5065; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5143–5147. [Google Scholar]
- 94. Hickling A., Hill S., Discuss. Faraday Soc. 1947, 1, 236. [Google Scholar]
- 95. McCrory C. C. L., Jung S., Ferrer I. M., Chatman S. M., Peters J. C., Jaramillo T. F., J. Am. Chem. Soc. 2015, 137, 4347–4357. [DOI] [PubMed] [Google Scholar]
- 96. Lips S., Waldvogel S. R., ChemElectroChem 2019, 6, 1649–1660. [Google Scholar]
- 97.
- 97a. Ganiyu S. O., dos Santos E. V., Martínez-Huitle C. A., Waldvogel S. R., Curr. Opin. Electrochem. 2022, 32, 100903; [Google Scholar]
- 97b. Oturan M. A., Curr. Opin. Solid State Mater. Sci. 2021, 25, 100925. [Google Scholar]
- 98.
- 98a. Matsumoto Y., Sato E., Mater. Chem. Phys. 1986, 14, 397–426; [Google Scholar]
- 98b. Devilliers D., Mahé E., Electrochim. Acta 2010, 55, 8207–8214; [Google Scholar]
- 98c. Scialdone O., Electrochim. Acta 2009, 54, 6140–6147. [Google Scholar]
- 99. Krstić V., Pešovski B., Hydrometallurgy 2019, 185, 71–75. [Google Scholar]
- 100. Klein M., Güthner T., Sans J., Thalhammer F., Waldvogel S. R., Green Chem. 2021, 23, 3289–3294. [Google Scholar]
- 101. Zhao X., Su H., Cheng W., Zhang H., Che W., Tang F., Liu Q., ACS Appl. Mater. Interfaces 2019, 11, 34854–34861. [DOI] [PubMed] [Google Scholar]
- 102. Yi Y., Tornow J., Willinger E., Willinger M. G., Ranjan C., Schlögl R., ChemElectroChem 2015, 2, 1929–1937. [Google Scholar]
- 103.
- 103a. Chen R., Trieu V., Schley B., Natter H., Kintrup J., Bulan A., Weber R., Hempelmann R., Z. Phys. Chem. 2013, 227, 651–666; [Google Scholar]
- 103b. Karlsson R. K. B., Cornell A., Chem. Rev. 2016, 116, 2982–3028. [DOI] [PubMed] [Google Scholar]
- 104. Baizer M. M., J. Electrochem. Soc. 1964, 111, 215. [Google Scholar]
- 105.
- 105a. Rodrigo E., Baunis H., Suna E., Waldvogel S. R., Chem. Commun. 2019, 55, 12255–12258; [DOI] [PubMed] [Google Scholar]
- 105b. Rodrigo E., Waldvogel S. R., Green Chem. 2018, 20, 2013–2017; [Google Scholar]
- 105c. Rodrigo E., Waldvogel S. R., Chem. Sci. 2019, 10, 2044–2047; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105d. Wirtanen T., Rodrigo E., Waldvogel S. R., Adv. Synth. Catal. 2020, 362, 2088–2101. [Google Scholar]
- 106. Yadav A. K., Singh A., Bull. Chem. Soc. Jpn. 2002, 75, 587–588. [Google Scholar]
- 107. Kwon Y., Lai S. C. S., Rodriguez P., Koper M. T. M., J. Am. Chem. Soc. 2011, 133, 6914–6917. [DOI] [PubMed] [Google Scholar]
- 108. Wirtanen T., Rodrigo E., Waldvogel S. R., Chem. Eur. J. 2020, 26, 5592–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Aust N. C., Griesbach U., Pelzer R., Haag T., Berens U., Botzem J., EP3180459B1, 2015.
- 110. Wang Y., Zhao J., Qiao T., Zhang J., Chen G., Chin. J. Chem. 2021, 39, 3297–3302. [Google Scholar]
- 111. Bai Y., Shi L., Zheng L., Ning S., Che X., Zhang Z., Xiang J., Org. Lett. 2021, 23, 2298–2302. [DOI] [PubMed] [Google Scholar]
- 112. Wen J., Qin H., Yan K., Yang X., Sun X., Wei W., Yang J., Wang H., Org. Lett. 2020, 22, 8824–8828. [DOI] [PubMed] [Google Scholar]
- 113. Wang H.-B., Huang J.-M., Adv. Synth. Catal. 2016, 358, 1975–1981. [Google Scholar]
- 114. Li H., Breen C. P., Seo H., Jamison T. F., Fang Y.-Q., Bio M. M., Org. Lett. 2018, 20, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Alkayal A., Tabas V., Montanaro S., Wright I. A., Malkov A. V., Buckley B. R., J. Am. Chem. Soc. 2020, 142, 1780–1785. [DOI] [PubMed] [Google Scholar]
- 116.
- 116a. Adenier A., Chehimi M. M., Gallardo I., Pinson J., Vilà N., Langmuir 2004, 20, 8243–8253; [DOI] [PubMed] [Google Scholar]
- 116b. Ghilane J., Martin P., Randriamahazaka H., Lacroix J.-C., Electrochem. Commun. 2010, 12, 246–249. [Google Scholar]
- 117. Gallardo I., Vilà N., J. Org. Chem. 2008, 73, 6647–6656. [DOI] [PubMed] [Google Scholar]
- 118. Seo E. T., Nelson R. F., Fritsch J. M., Marcoux L. S., Leedy D. W., Adams R. N., J. Am. Chem. Soc. 1966, 88, 3498–3503. [Google Scholar]
- 119. Kawamata Y., Hayashi K., Carlson E., Shaji S., Waldmann D., Simmons B. J., Edwards J. T., Zapf C. W., Saito M., Baran P. S., J. Am. Chem. Soc. 2021, 143, 16580–16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.
- 120a. Ibanez J. G., Frontana-Uribe B. A., Vasquez-Medrano R., J. Mex. Chem. Soc. 2016, 60, 247–260; [Google Scholar]
- 120b. Liu H., Li W., Curr. Opin. Electrochem. 2021, 30, 100795; [Google Scholar]
- 120c. Marken F., Cresswell A. J., Bull S. D., Chem. Rec. 2021, 21, 2585–2600; [DOI] [PubMed] [Google Scholar]
- 120d. Sbei N., Hardwick T., Ahmed N., ACS Sustainable Chem. Eng. 2021, 9, 6148–6169. [Google Scholar]
- 121. Pütter H., Hannebaum H., DE19618854 A1, 1997.
- 122. Dong X., Roeckl J. L., Waldvogel S. R., Morandi B., Science 2021, 371, 507–514. [DOI] [PubMed] [Google Scholar]
- 123.
- 123a. Hartmer M. F., Waldvogel S. R., Chem. Commun. 2015, 51, 16346–16348; [DOI] [PubMed] [Google Scholar]
- 123b. Gütz C., Grimaudo V., Holtkamp M., Hartmer M., Werra J., Frensemeier L., Kehl A., Karst U., Broekmann P., Waldvogel S. R., ChemElectroChem 2018, 5, 247–252. [Google Scholar]
- 124.
- 124a. Frontana-Uribe B. A., Moinet C., Tetrahedron 1998, 54, 3197–3206; [Google Scholar]
- 124b. Frontana-Uribe B. A., Moinet C., Toupet L., Eur. J. Org. Chem. 1999, 419–430. [Google Scholar]
- 125. Jiang Y.-Y., Dou G.-Y., Zhang L.-S., Xu K., Little R. D., Zeng C.-C., Adv. Synth. Catal. 2019, 361, 5170–5175. [Google Scholar]
- 126. Yuan Y., Qi J.-C., Wang D.-X., Chen Z., Wan H., Zhu J.-Y., Yi H., Chowdhury A. D., Lei A., CCS Chem. 2021, 3, 3027–3038. [Google Scholar]
- 127. Strehl J., Abraham M. L., Hilt G., Angew. Chem. Int. Ed. 2021, 60, 9996–10000; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 10084–10088. [Google Scholar]
- 128.
- 128a. Chakraborty P., Mandal R., Garg N., Sundararaju B., Coord. Chem. Rev. 2021, 444, 214065; [Google Scholar]
- 128b. Dhawa U., Kaplaneris N., Ackermann L., Org. Chem. Front. 2021, 8, 4886–4913; [Google Scholar]
- 128c. Malapit C. A., Prater M. B., Cabrera-Pardo J. R., Li M., Pham T. D., McFadden T. P., Blank S., Minteer S. D., Chem. Rev. 2022, 122, 3180–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.
- 129a. Ackermann L., Acc. Chem. Res. 2020, 53, 84–104; [DOI] [PubMed] [Google Scholar]
- 129b. Cheng X., Lei A., Mei T.-S., Xu H.-C., Xu K., Zeng C., CCS Chem. 2022, 4, 1–33; [Google Scholar]
- 129c. Ma C., Fang P., Mei T.-S., ACS Catal. 2018, 8, 7179–7189. [Google Scholar]
- 130.
- 130a. Kawamata Y., Vantourout J. C., Hickey D. P., Bai P., Chen L., Hou Q., Qiao W., Barman K., Edwards M. A., Garrido-Castro et al A. F., J. Am. Chem. Soc. 2019, 141, 6392–6402; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130b. Li C., Kawamata Y., Nakamura H., Vantourout J. C., Liu Z., Hou Q., Bao D., Starr J. T., Chen J., Yan et al M., Angew. Chem. Int. Ed. 2017, 56, 13088–13093; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13268–13273. [Google Scholar]
- 131. Liu D., Liu Z.-R., Ma C., Jiao K.-J., Sun B., Wei L., Lefranc J., Herbert S., -S Mei T., Angew. Chem. Int. Ed. 2021, 60, 9444–9449; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 9530–9535. [Google Scholar]
- 132. Liu D., Ma H.-X., Fang P., Mei T.-S., Angew. Chem. Int. Ed. 2019, 58, 5033–5037; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5087–5091. [Google Scholar]
- 133. Liang Y., Shi S.-H., Jin R., Qiu X., Wei J., Tan H., Jiang X., Shi X., Song S., Jiao N., Nat. Catal. 2021, 4, 116–123. [Google Scholar]