

Abstract
Objective
To determine the characteristics of participants with amyloid‐related imaging abnormalities (ARIA) in a trial of gantenerumab or solanezumab in dominantly inherited Alzheimer disease (DIAD).
Methods
142 DIAD mutation carriers received either gantenerumab SC (n = 52), solanezumab IV (n = 50), or placebo (n = 40). Participants underwent assessments with the Clinical Dementia Rating® (CDR®), neuropsychological testing, CSF biomarkers, β‐amyloid positron emission tomography (PET), and magnetic resonance imaging (MRI) to monitor ARIA. Cross‐sectional and longitudinal analyses evaluated potential ARIA‐related risk factors.
Results
Eleven participants developed ARIA‐E, including 3 with mild symptoms. No ARIA‐E was reported under solanezumab while gantenerumab was associated with ARIA‐E compared to placebo (odds ratio [OR] = 9.1, confidence interval [CI][1.2, 412.3]; p = 0.021). Under gantenerumab, APOE‐ɛ4 carriers were more likely to develop ARIA‐E (OR = 5.0, CI[1.0, 30.4]; p = 0.055), as were individuals with microhemorrhage at baseline (OR = 13.7, CI[1.2, 163.2]; p = 0.039). No ARIA‐E was observed at the initial 225 mg/month gantenerumab dose, and most cases were observed at doses >675 mg. At first ARIA‐E occurrence, all ARIA‐E participants were amyloid‐PET+, 60% were CDR >0, 60% were past their estimated year to symptom onset, and 60% had also incident ARIA‐H. Most ARIA‐E radiologically resolved after dose adjustment and developing ARIA‐E did not significantly increase odds of trial discontinuation. ARIA‐E was more frequently observed in the occipital lobe (90%). ARIA‐E severity was associated with age at time of ARIA‐E.
Interpretation
In DIAD, solanezumab was not associated with ARIA. Gantenerumab dose over 225 mg increased ARIA‐E risk, with additional risk for individuals APOE‐ɛ4(+) or with microhemorrhage. ARIA‐E was reversible on MRI in most cases, generally asymptomatic, without additional risk for trial discontinuation. ANN NEUROL 2022;92:729–744
Alzheimer disease (AD) is the leading cause of dementia and one of the most important public health concerns worldwide. 1 Effective disease‐modifying treatments that could slow or prevent the disease are an urgent priority. A particular focus of therapeutic strategies has been the use of monoclonal antibodies to prevent or remove β‐amyloid (Aβ) aggregates. 2 , 3 , 4 , 5 , 6 , 7 , 8 In the past two decades, multiple trials using these approaches were successful in decreasing or slowing Aβ burden in mild to moderate sporadic AD (sAD) but have only recently showed some evidence of slowing cognitive decline. 7 , 8 , 9 Potential reasons suggested for this lack of success include treating too late in the disease course, inadequate dosing, and lack of target engagement. 1 Doses may not have been high enough (given the low level of blood–brain barrier penetrance – <1%) due to concern about dose‐related Aβ‐related imaging abnormalities (ARIA) – the most common side effect associated with this drug class. 10 , 11
Recent trials of anti‐amyloid monoclonal antibodies in sAD populations report ARIA episodes at a wide range of incidence rates. 4 , 8 , 12 , 13 Two types of ARIA have been defined on magnetic resonance imaging (MRI): (1) the edema type or ARIA‐E, seen as increased signal intensity on fluid attenuated inversion recovery (FLAIR) sequence in the parenchyma and/or sulcal space, which can be reversible on MRI, and (2) the hemosiderin type or ARIA‐H, seen as signal loss on T2*‐weighted MRI or susceptibility weighted imaging (SWI) sequence in the parenchyma for microhemorrhages (mH) and in the subarachnoid space for superficial siderosis, which corresponds to long‐lasting hemoglobin residual lesions on MRI. ARIA‐E has been associated with higher treatment doses, APOE‐ɛ4(+) status, and the presence of pre‐treatment mH or superficial siderosis. 14 , 15 As shown in previous mild to moderate AD trials, ARIA‐E signs and symptoms are generally reversible and adjusting the anti‐amyloid treatment has often helped their resolution. 4
Although ARIA‐E has been widely described in sporadic late onset AD, the frequency and risk factors for ARIA are less known for younger populations, including those with dominantly inherited Alzheimer disease (DIAD), where a decreased prevalence of comorbid chronic vascular disease may modify the risk of ARIA. Recently, the Dominantly Inherited Alzheimer Network Trials Unit (DIAN‐TU) 16 concluded the first disease‐modifying clinical trials in the DIAD population (DIAN‐TU‐001). 17 Solanezumab (Sola) and gantenerumab (Gant) were evaluated in a Phase II/III randomized, double‐blind, placebo‐controlled study. Although both drugs engaged their respective Aβ targets, neither demonstrated a beneficial effect on cognitive measures compared to the pooled control groups. ARIA‐E was seen in 19.2% participants in the gantenerumab arm, 0% in the solanezumab arm, and 2.5% in the pooled placebo. 17
Further characterization of ARIA may provide useful information to guide the safe use of these anti‐amyloid antibodies. Here we aim to (1) describe clinical and imaging characteristics of ARIA‐E and ARIA‐H in DIAD populations treated with gantenerumab or solanezumab, (2) determine the main risk factors for ARIA, and (3) report the clinical outcomes after ARIA in the DIAN‐TU‐001 trial. This study provides a comprehensive report on ARIA with insights and guidance for future clinical trials. 18
Participants/Material and Methods
Study Participants and Study Design
The study design and the description of participants have been previously reported in the primary DIAN‐TU‐001 trial publication. 17 In brief, the DIAN‐TU‐001 (ClinicalTrials.gov Identifier: NCT01760005), was a phase II/III randomized, double blinded, placebo‐controlled trial conducted between 2012 and 2019 in participants with a family history of DIAD. Eligible participants were at risk members of families with a known DIAD pathogenic mutation on PSEN1, PSEN2, or APP genes, were between −15 to +10 years from the expected age of symptom onset (EYO), 19 and had a Clinical Dementia Rating® 20 (CDR®) score of 0 (no cognitive symptoms) or 0.5–1 (early dementia). One hundred and forty‐four mutation‐carriers (MC) were randomized to gantenerumab (Gant, n = 52), solanezumab (Sola, n = 52), or placebo (n = 40) for a minimum 4‐year course of treatment. The randomization algorithm included age, EYO, and CDR among other factors and balanced study arms for these variables. Cognitive, clinical, imaging, and cerebrospinal fluid (CSF) measures were assessed at randomization/baseline and specific follow‐up time points throughout the duration of the trial as previously described. 17 All participating study sites received Institutional Review Board/Research Ethics Committee approval. All participants gave written informed consent for the study.
Safety Measures and Monitoring of ARIA
To detect and monitor adverse events (AEs) during the trial, participants underwent regular safety assessments including routine laboratory assessments, physical examinations, electrocardiogram (ECG), and MRIs for the detection of ARIA. Safety MRIs were scheduled for each drug arm, according to their respective protocol to allow an appropriate monitoring and dose adjustment. For the gantenerumab arm, MRIs were scheduled before each titration step throughout the study corresponding to an average of every 3 months, while for the solanezumab arm, MRIs were scheduled approximately every 3 months for the first 2 years and then annually (Fig 1A, B). For ARIA management, experienced radiologists at a central reads site (Mayo Clinic) reviewed all MRI scans and provided the type, count, and magnitude of individual lesions over time. Findings were reviewed by the DIAN‐TU medical monitoring team for each MRI evaluation. In the event of new definite ARIA episodes, an intervention algorithm was applied according to study protocol. New ARIA‐E episodes were managed by withholding titration or dosage and resuming at similar or lower doses after resolution of ARIA‐E (Fig 1C). For ARIA‐H, in the presence of 10+ cumulative treatment‐emergent mH, medical review determined management and appropriate action which could include discontinuation of the study drug and continuation of safety MRI monitoring. For each ARIA event a safety report was created by site PIs. Site investigators determined whether symptoms associated with ARIA were present. All clinicians, including radiologists assessing ARIA, were blinded to treatment assignment.
FIGURE 1.
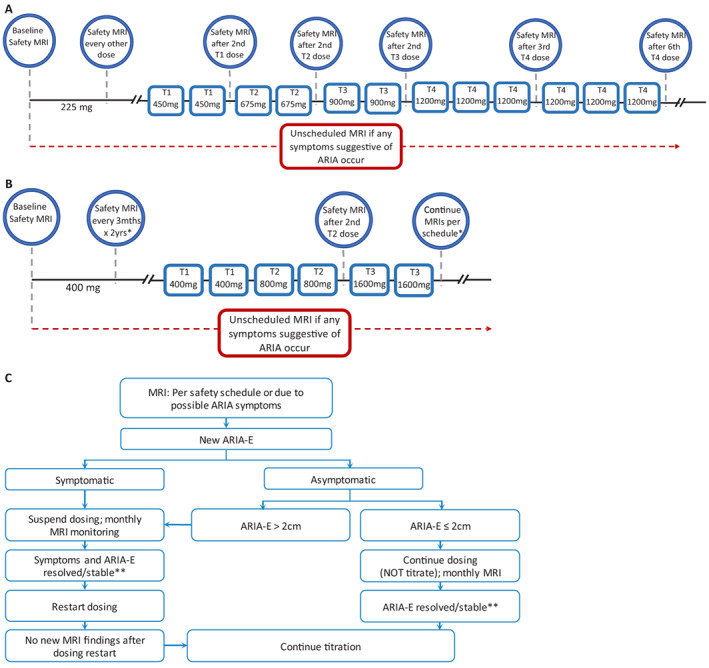
Drug‐specific ARIA monitoring schedules and related intervention procedures. (A) Gantenerumab dosing and ARIA MRI monitoring schedule: Prior to titration, safety MRIs were scheduled before the first dose (baseline scan for safety reads done at V2) and then about 1 week (±4 days) after dose 2, dose 4, dose 6, dose 9, dose 17, and dose 22. After titration started safety MRIs were scheduled 1 week (±4 days) after the second dose of each titration step (225, 450, 675, 900 mg), unless otherwise indicated by the ARIA‐E and ARIA‐H management algorithms. The final titration step (T4) included safety MRIs 1 week (±4 days) after every third dose (or approximately every 3 months) for the next 6 doses. Once participants reached their stable dose (defined as the maximum dose the subject will remain at for the duration of the trial) safety MRIs were scheduled every 6 doses. (B) Solanezumab dosing and ARIA MRI monitoring schedule: Prior to titration, safety MRIs were scheduled before the first dose (baseline scan for safety reads done at V2). *Per schedule, MRIs were done approximately every 3 months for the first 2 years and then annually. During the dose‐escalation period, a safety MRI was conducted after 2 doses of 800 mg (T2) and continued as per protocol (annually). All MRIs were done at least 5 days before escalating dose. (C) Example of diagram of intervention procedures for ARIA‐E in gantenerumab arm. **Appropriate actions to restart dosing after ARIA‐E were discussed among the site PI, DIAN‐TU Medical Director, and PAL (Project Arm Leader). [Color figure can be viewed at www.annalsofneurology.org]
For analyses, Mayo Clinic reported details about ARIA‐E imaging findings, including location, focality (unifocal or multifocal), region (parenchymal, sulcal), and size defined as the longest cross‐sectional axis, and details about ARIA‐H, including type (mH, superficial siderosis) and count. The Barkhof Grand Total Score (BGTS) which ranges from 0 (none) to 60 (severe) with 3 subcategories (parenchymal hyperintensity, sulcal hyperintensity, and generalized swelling) accounting for ARIA‐E size, focality, location and region of the finding, was used for analyses. 21
Clinical and Cognitive Assessments
The primary outcome for the study was a measure of cognition, the DIAN Multivariate Cognitive End Point (DIAN‐MCE), which consisted of joint modelling of four cognitive components, including the Mini‐Mental State Examination (MMSE), as previously described. 22 In this article, we combined each element of the DIAN‐MCE to form a conventional z‐score cognitive composite. Other outcomes included the CDR sum of boxes (CDR‐SB) and the functional assessment scale (FAS). The cognitive composite, MMSE, and CDR‐SB were used in subsequent analyses in the evaluation of ARIA in this study. The participant's EYO was defined as the participant's age at baseline minus their expected age at symptom onset (AAO). 19 , 23 , 24 The AAO was calculated based on either the family mutation‐specific expected age at dementia onset or parental age at first progressive cognitive decline if the expected age at symptom onset for the mutation was unknown. Negative values indicate that an individual is younger than their expected age at onset.
Image Acquisition and Processing for Biomarker Assessment
Standardized procedures were used at all DIAN‐TU sites to ensure consistency in data collection. All study site scanners, protocols, and parameters for MRI and positron emission tomography (PET) were quality controlled by Mayo Clinic Rochester and the University of Michigan. All MRI sessions were conducted on 3 T scanners and included a T1‐weighted MPRAGE image (1 × 1 × 1 mm3 resolution, 2.95 s TE, 2300.00s TR) for volumetric assessment, a T2‐FLAIR sequence (1 × 1 × 5 mm3 resolution, 91 s TE, 9000 s TR) for ARIA‐E detection, and a T2*‐GRE sequence (1 × 1 × 4 mm3 resolution, 20 s TE, 650 s TR) for ARIA‐H detection. 25
In addition to the MRI acquisitions, each participant underwent a 11C‐Pittsburgh Compound B (PiB) PET scan to assess amyloid burden at baseline visit and at the week 52, 104, and 208 visits. The PiB PET scan consisted of 70 minutes of dynamic scanning. Data acquired during the 40–70 min post‐injection window were converted to regional standardized uptake value ratios (SUVRs) using the cerebellar cortex as reference region. Data were partial volume corrected using a geometric transfer matrix approach. 26 A composite mean cortical SUVR to assess overall change in Aβ burden was used as a secondary outcome in the trial. 27 A threshold of SUVR>1.42 defined PiB‐PET positivity (PiB‐PET+).
CSF Measures
Standardized protocols for CSF collection, storage and measures were previously described. 17 CSF samples were performed at baseline visit and at the week 52, 104, and 208 visits. CSF Aβ42, tau, ptau181, and neurofilament light chain (NfL) were quantified using validated and adapted methods as previously described. 17 The CSF NfL measures were log‐transformed for analyses.
Statistical Analyses
The demographics of MC participants were evaluated with the following grouping: all (pooled) placebo, all Sola, Gant non‐ARIA‐E, and Gant ARIA‐E (Table 1A). One ARIA‐E event was observed in the placebo and none in the solanezumab treatment arm. Pairwise comparisons were performed between Gant ARIA‐E and Gant non‐ARIA‐E, and between Gant ARIA‐E and all placebo groups using Mann–Whitney U tests for continuous variables, and chi‐squared test or Fisher's exact tests (as appropriate) for categorical variables. p‐Values were corrected for multiple comparisons using the Benjamini‐Hochberg procedure. Summary statistics based on placebo excluding the ARIA‐E participant were also provided.
TABLE 1A.
Participants' Demographics and Vascular‐Related Variables and Radiological Features per Arm and ARIA‐E Status
| Mutation/Arm Group | All Placebo a (ARIA‐E excluded) | Sola | Gant | Unadjusted p‐Value | Adjusted p‐Value | |||
|---|---|---|---|---|---|---|---|---|
| ARIA‐E Status | No ARIA‐E | No ARIA‐E | ARIA‐E | Gant No‐ARIA‐E versus Gant ARIA‐E | Placebo versus Gant ARIA‐E | Gant No‐ARIA‐E versus Gant ARIA‐E | Placebo versus Gant ARIA‐E | |
| N | 40 (39) | 50 | 42 | 10 | – | – | ||
| Age, mean ± SD years |
44.2 ± 9.6 (43.9 ± 9.5) |
42.7 ± 9.6 | 45.5 ± 11.2 | 48.2 ± 8.9 | 0.4 | 0.3 (0.2) | 0.4 | 0.4 (0.4) |
| EYO, mean ± SD years |
−3.5 ± 7.6 (−3.6 ± 7.7) |
−2.4 ± 7.1 | −4.0 ± 7.3 | −1.6 ± 6.2 | 0.4 | 0.5 (0.5) | 0.5 | 0.5 (0.5) |
| Familial AO, mean ± SD years |
47.5 ± 10.1 (47.1 ± 10.0) |
45.9 ± 8.0 | 48.2 ± 8.9 | 49.3 ± 6.4 | 0.5 | 0.4 (0.3) | 0.5 | 0.5 (0.6) |
| Female, n (%) |
22 (55.0) [21 (53.9)] |
29 (58.0) | 17 (40.5) | 4 (40) | 1 | 1 (0.5) | 1.0 | 1.0 (1.0) |
| APOE‐ɛ4 carriers, n (%) |
13 (32.5) [12 (30.8)] |
14 (28.0) | 11 (26.2) | 5 (50) | 0.3 | 0.5 (0.3) | 0.5 | 0.5 (0.3) |
| Mean arterial blood pressure, mean ± SD mmHg |
92.3 ± 9.3 (92.4 ± 9.4) |
92.3 ± 13.7 | 93.4 ± 8.3 | 90.5 ± 9.4 | 0.6 | 0.8 (0.8) | 0.8 | 0.8 (0.8) |
| Hypertension or history of hypertension, n (%) |
2 (5) [2 (5.1)] |
7 (14) | 5 (11.9) | 3 (30) | 0.2 | 0.05 (0.05) | 0.2 | 0.1 (0.1) |
| Baseline mH or SS, n (%) |
1 (2.5) [1 (2.6)] |
3 (6.0) | 1 (2.4) | 3 (30.0) | 0.02 | 0.02 (0.02) | 0.02 | 0.02 (0.02) |
| % with 0/1/2–4/5 mH at baseline |
97.5/2.5/0/0 (97.4/2.6/0/0) |
94/2/4/0 | 98/2/0/0 | 70/30/0/0 | 0.02 | 0.02 (0.02) | 0.02 | 0.02 (0.02) |
| Incident ARIA‐H, n (%) |
4 (10.0) [3 (7.7)] |
6 (12.0) | 6 (14.3) | 6 (60.0) | 0.006 | 0.002 (0.001) | 0.006 | 0.004 (0.002) |
| % with 0/1/2–4/5+ incident mH |
90/2.5/2.5/5 (92.3/2.6/2.6/2.6) |
88/4/4/4 | 86/10/2/2 | 40/0/0/60 | 0.0002 | 0.0004 (0.0001) | 0.0004 | 0.0004 (0.0002) |
| Overall ARIA‐H change over time, mean ± SD count/year |
0.5 ± 2.5 (0.2 ± 0.9) |
0.4 ± 1.9 | 0.2 ± 1.0 | 2.9 ± 3.7 | 0.0006 | 0.0004 (0.0001) | 0.0006 | 0.0006 (0.0002) |
Italics and parentheses denote analyses excluding ARIA‐E case in pooled placebo.
Pooled placebo including case with ARIA‐E.
Factors that could affect the risk of developing ARIA‐E were evaluated separately within the gantenerumab treatment arm using logistic regressions. Factors that were close to significance (p < 0.1) or significantly associated with the risk of ARIA‐E were further evaluated using logistic regression with baseline age and EYO as covariates.
Within the Gant ARIA‐E group, Spearman correlations were used to investigate association of ARIA‐E severity score with variables at ARIA‐E onset including age, EYO, total incident microhemorrhages, and dosage, and with baseline variables such as Aβ burden as measured by PiB‐PET. The annual increase of microhemorrhages was calculated using linear regression separately for each participant.
The difference in change from baseline in clinical, cognitive, and biomarker outcomes between Gant ARIA‐E and Gant non‐ARIA‐E were compared using mixed effects models for repeated measures (MMRM) with unstructured covariance matrix. The fixed effects in the MMRM included the baseline variable of interest, baseline EYO, time and their two‐way interaction with group (Gant non‐ARIA‐E, Gant ARIA‐E, and placebo excluding the ARIA‐E participant).
Statistical analyses were performed using SAS® software version 9.4 (SAS Institute Inc., Cary, NC) and R version 4.0.5 (2021‐03‐31, “Shake and Throw”, Copyright© 2021 The R Foundation for Statistical Computing). All statistical tests were two sided, and a p‐value < 0.05 was considered significant.
Results
Participant Characteristics and ARIA‐E Clinical Presentation
In the DIAN‐TU‐001 trial, 11 participants developed ARIA‐E (8%) and, among those, 3 developed recurrent ARIA‐E (i.e., 2 ARIA‐E events) for a total of 14 ARIA‐E episodes. All ARIA‐E findings were detected on scheduled safety MRIs and 13 of 14 events resolved on MRI following management. Upon retrospective investigation, 3 out of the 11 participants with ARIA‐E (27%) reported mild symptoms that the clinician confirmed were, in view of the MRI findings, probably associated with the ARIA‐E episode. The three reported symptoms were headache, inner ear pain, and dizziness. In summary, ARIA‐E participants corresponded to 2.5% (1/40) of the pooled placebo group, 19% (10/52) of the gantenerumab arm, and 0% (0/50) of the solanezumab arm. Note that the ARIA‐E participant in the placebo group was asymptomatic. Tables 1A and 1B present baseline characteristics for all groups and detailed comparisons for the Gant ARIA‐E group with Gant non‐ARIA‐E and with placebo. Since no incident ARIA‐E episode was observed in participants who received solanezumab, results presented in following sections focus only on the gantenerumab treatment group.
TABLE 1B.
Participants' Baseline Clinical and Biomarker Outcome Measures per Arm and ARIA‐E Status
| Mutation/Arm Group | All Placebo a (ARIA‐E excluded) | Sola | Gant | Unadjusted p‐Value | Adjusted p‐Value | |||
|---|---|---|---|---|---|---|---|---|
| ARIA‐E status | No ARIA‐E | No ARIA‐E | ARIA‐E | Gant No‐ARIA‐E versus Gant ARIA‐E | Placebo versus Gant ARIA‐E | Gant No‐ARIA‐E versus Gant ARIA‐E | Placebo versus Gant ARIA‐E | |
| N | 40 (39) | 50 | 42 | 10 | – | – | ||
| CDR > 0, n (%) |
18 (45.0) [18 (46.2)] |
20 (40.0) | 16 (38.1) | 5 (50.0) | 0.5 | 1 (1) | 1.0 | 1.0 (1) |
| CDR‐SB, mean ± SD |
1.4 ± 1.9 (1.5 ± 1.8) |
1.4 ± 2.0 | 1.2 ± 1.9 | 2.1 ± 2.6 | 0.2 | 0.4 (0.5) | 0.3 | 0.4 (0.5) |
| MMSE, mean ± SD |
26.7 ± 4.0 (26.6 ± 4.0) |
26.7 ± 4.1 | 27.4 ± 3.2 | 25.9 ± 4.5 | 0.4 | 0.7 (0.7) | 0.7 | 0.7 (0.7) |
| Cognitive composite, mean ± SD |
−1.1 ± 1.4 (−1.1 ± 1.4) |
−1.0 ± 1.4 | −0.9 ± 1.4 | −1.5 ± 1.2 | 0.2 | 0.3 (0.3) | 0.3 | 0.3 (0.3) |
| PiB‐PET composite, mean ± SD |
2.6 ± 1.2 (2.6 ± 1.2) |
2.7 ± 1.3 | 2.5 ± 1.2 | 3.0 ± 1.2 | 0.3 | 0.3 (0.6) | 0.3 | 0.3 (0.6) |
| PiB+ cases at baseline, n (%) |
32 (86.5) [31 (86.1)] |
39 (88.6) | 32 (78.1) | 10 (100) | 0.2 | 0.6 (0.3) | 0.4 | 0.6 (0.3) |
| CSF Aβ42, b mean ± SD pg ml−1 |
472.0 ± 228.5 (472.0 ± 228.5) |
‐ | 525.3 ± 205.0 | 418.6 ± 159.3 | 0.1 | 0.5 (0.5) | 0.3 | 0.5 (0.5) |
| CSF total tau, mean ± SD pg ml−1 |
559.5 ± 366.3 (560.5 ± 371.3) |
572.6 ± 277.2 | 572.1 ± 400.9 | 578.6 ± 222.1 | 1.0 | 0.9 (0.9) | 1.0 | 1.0 (1.0) |
| CSF p‐tau181, mean ± SD pg ml−1 |
95.2 ± 71.1 (95.4 ± 72.0) |
95.8 ± 58.4 | 94.6 ± 74.2 | 109.6 ± 50.1 | 0.6 | 0.6 (0.5) | 0.6 | 0.6 (0.6) |
| Log CSF NfL, mean ± SD pg ml−1 |
6.5 ± 0.6 (6.5 ± 0.6) |
6.5 ± 0.7 | 6.6 ± 0.7 | 6.8 ± 0.5 | 0.4 | 0.2 (0.2) | 0.4 | 0.3 (0.3) |
Italics and parentheses denote analyses excluding ARIA‐E case in pooled placebo.
Pooled placebo including case with ARIA‐E.
CSF Aβ42 variable based on Gant‐specific outcome measurement. Placebo values based on 21 participants receiving Gant‐specific placebo. Sola group with different type of CSF Aβ42 measure not reported here.
Recipients of gantenerumab were more likely to develop ARIA‐E compared to placebo (odds ratio [OR] = 9.1, 95% confidence interval [CI] [1.2, 412.3], p‐value< 0.05). At baseline, all Gant participants who developed ARIA‐E were PiB‐PET+, 30% had at least one microhemorrhage at baseline, and 50% were CDR > 0. Baseline characteristics of the Gant ARIA‐E group were not significantly different from the Gant non‐ARIA‐E group or the pooled placebo group, except for the number of participants with baseline microhemorrhage (30% vs 2.5%). Incident ARIA‐H, including mH, were also significantly higher in the Gant ARIA‐E group compared to Gant non‐ARIA‐E or to placebo. Moreover, 60% of Gant ARIA‐E participants developed more than five new mHs as opposed to 2% in Gant non‐ARIA‐E and 5% in all placebo (3% in ARIA‐E‐excluded placebo) groups, respectively (Table 1A). Note that the ARIA‐E group tended to have more individuals with history of hypertension than the non‐ARIA‐E group (30% vs 12%).
The first ARIA‐E episode was observed in the gantenerumab arm during the first titration step of 450 mg (after the second dose at this level) and no ARIA‐E events were observed at initial 225 mg dose. Overall, 70% of participants with ARIA‐E developed their first episode during higher doses (900 and 1,200 mg, titration step 3 and 4, respectively). Participants with ARIA‐E at lower titrations (450 or 675 mg, n = 3) were APOE‐ɛ4(+) and appeared to be at advanced disease stage at the start of the trial (mean CDR‐SB of 3.7 ± 4.0 and mean MMSE of 23.0 ± 7.0, Table 2). Although no statistical comparisons could be done (small sample size), they also appeared to experience larger ARIA‐E size, more incident ARIA‐H, and longer ARIA‐E resolution time (16.4 ± 8.3 weeks vs 7.8 ± 2.8 weeks for the higher titration doses, Table 2). On average, the Gant ARIA‐E participants at time of first ARIA‐E, were 50.1 ± 9.0 years old, with an EYO of 3.3 ± 5.8 years, CDR‐SB of 3.0 ± 3.2, MMSE of 24.2 ± 6.0, and cognitive composite of −1.7 ± 1.6. Moreover, 60% were CDR >0 and 60% were EYO > 0 at time of ARIA‐E. All Gant ARIA‐E participants paused dosing with 7 out of 10 resuming at same dose or continuing titration to the maximum dose per protocol, and three discontinued treatments. Overall, developing ARIA‐E did not significantly increase odds of trial discontinuation (OR = 1.6, 95% CI [0.2, 8.8], p‐value = 0.7).
TABLE 2.
Baseline and Clinical Characteristics at First ARIA‐E of Gant ARIA‐E Participants per Lower versus Higher Titrations and per Baseline CDR Status
| Gant ARIA‐E Participants per | Titration Step at First ARIA | CDR Status at Baseline | ||
|---|---|---|---|---|
| <900 mg (n = 3, 30%) | ≥900 mg (n = 7, 70%) | CDR = 0 (n = 5, 50%) | CDR > 0 (n = 5, 50%) | |
| % of APOE‐e4 carriers, n (%) | 100 | <30 | 40 | 60 |
| Baseline characteristics | ||||
| Age at baseline, mean ± SD years | 46.7 ± 6.7 | 48.9 ± 10.1 | 45.8 ± 6.5 | 50.6 ± 11.0 |
| EYO at baseline, mean ± SD years | −1.7 ± 4.0 | −1.6 ± 7.2 | −4.0 ± 7.6 | 0.8 ± 3.7 |
| Case EYO >0 at baseline, n (%) | 2 (66.7%) | 4 (57.1%) | 2 (40.0%) | 4 (80.0%) |
| Familial AO, mean ± SD years | 48.3 ± 9.3 | 49.8 ± 5.4 | 52.0 ± 4.7 | 47.2 ± 7.2 |
| Baseline PiB mean cortical, mean ± SD | 2.9 ± 1.8 | 3.1 ± 1.1 | 2.5 ± 1.1 | 3.6 ± 1.2 |
| CSF Aβ42 at baseline, mean ± SD pg ml−1 | 416.3 ± 224.9 | 419.6 ± 145.6 | 520.2 ± 165.8 | 317.0 ± 61.7 |
| CSF total tau at baseline, mean ± SD pg ml−1 | 475.3 ± 232.2 | 622.9 ± 219.9 | 582.0 ± 280.9 | 575.2 ± 179.0 |
| CSF p‐tau181 at baseline, mean ± SD pg ml−1 | 90.3 ± 51.7 | 117.8 ± 51.1 | 108.3 ± 70.2 | 110.9 ± 26.9 |
| Log CSF NfL at baseline, mean ± SD pg ml−1 | 6.7 ± 0.8 | 6.9 ± 0.4 | 6.5 ± 0.5 | 7.1 ± 0.4 |
| mH count baseline, mean ± SD | 0.7 ± 0.6 | 0.1 ± 0.4 | 0 | 0.6 ± 0.6 |
| Mean arterial blood pressure, mean ± SD mmHg | 89.1 ± 6.9 | 91.1 ± 10.7 | 89.1 ± 12.0 | 92.0 ± 6.9 |
| CDR‐SB at baseline, mean ± SD | 3.7 ± 4.0 | 1.4 ± 1.7 | 0.2 ± 0.3 | 3.9 ± 2.6 |
| MMSE at baseline, mean ± SD | 23.0 ± 7.0 | 27.1 ± 2.9 | 29.2 ± 1.3 | 22.6 ± 4.2 |
| Cognitive composite at baseline, mean ± SD | −2.1 ± 1.5 | −1.3 ± 1.1 | −0.5 ± 0.5 | −2.5 ± 0.6 |
| Clinical characteristics at first incident ARIA‐E | ||||
| Age at ARIA‐E, mean ± SD years | 48.0 ± 7.0 | 51.0 ± 10.1 | 47.8 ± 6.6 | 52.4 ± 11.2 |
| EYO at ARIA‐E, mean ± SD years | 3.0 ± 4.0 | 3.5 ± 6.7 | 1.4 ± 6.3 | 5.3 ± 5.0 |
| Case EYO > 0 at ARIA‐E, n (%) | 2 (66.7%) | 4 (57.1%) | 2 (40%) | 4 (80%) |
| CDR‐SB at ARIA‐E, mean ± SD | 4.5 ± 5.1 | 2.4 ± 2.3 | 0.6 ± 1.3 | 5.4 ± 2.7 |
| Case CDR > 0 at ARIA‐E, n (%) | 2 (66.7%) | 4 (57.1%) | 1 (20%) | 5 (100%) |
| MMSE at ARIA‐E, mean ± SD | 21.0 ± 8.2 | 25.6 ± 5.0 | 28.6 ± 1.3 | 19.8 ± 5.6 |
| Cognitive composite at ARIA‐E, mean ± SD | −2.4 ± 1.7 | −1.4 ± 1.6 | −0.4 ± 0.9 | −3.0 ± 0.7 |
| ARIA‐E severity | ||||
| Mean BGTS, mean ± SD | 3.3 ± 0.6 | 2.7 ± 1.3 | 2.2 ± 0.8 | 3.6 ± 0.9 |
| Averaged count of ARIA‐E, mean ± SD | 2.7 ± 1.2 | 2.3 ± 1.1 | 2.4 ± 1.1 | 2.4 ± 1.1 |
| Cases with worsening ARIA‐E size, n (%) | 3 (100%) | 2 (28.57%) | 3 (60%) | 2 (40%) |
| ARIA‐E averaged length, mean ± SD mm | 30.5 ± 8.3 | 22.8 ± 10.8 | 19.5 ± 9.6 | 30.7 ± 8.3 |
| Resolution time, mean ± SD weeks | 16.4 ± 8.3 | 7.8 ± 2.8 | 8.9 ± 2.6 | 11.8 ± 8.5 |
| ARIA‐H at first incident ARIA‐E | ||||
| Total incident ARIA‐H count, mean ± SD | 6.3 ± 7.1 | 3.1 ± 3.4 | 1.8 ± 2.5 | 6.4 ± 5.3 |
| ARIA‐E related incident ARIA‐H count, mean ± SD | 4.7 ± 6.4 | 2 ± 2.5 | 1.4 ± 2.2 | 4.2 ± 4.9 |
| Total incident mH count, mean ± SD | 5.3 ± 6.1 | 2.9 ± 3.4 | 1.8 ± 2.5 | 5.4 ± 5.0 |
| ARIA‐E related incident mH count, mean ± SD | 4.3 ± 5.9 | 1.9 ± 2.6 | 1.4 ± 2.2 | 3.8 ± 4.7 |
| Timeline and management | ||||
| Time post‐titration step, mean ± SD wks/dose count | 4.8 ± 0.5/1 | 10.5 ± 7.3/3 ± 1.7 | 7.7 ± 5.6/2.4 ± 1.5 | 9.9 ± 8.0/2.4 ± 2.1 |
| Had dosing escalation paused or dosing reduction after ARIA, n (%) | 2 (66.7%) | 3 (42.9%) | 2 (40%) | 3 (60%) |
| Discontinued treatment, n (%) | 2 (66.7%) | 1 (14.3%) | 0 (0%) | 3 (60%) |
Risk Factors Associated with Developing ARIA‐E
Figure 2 and Supplemental table summarize results of all evaluated potential risk factors within the DIAD mutation carriers who received gantenerumab. Overall, 24% of individuals who were CDR > 0 at start of the trial (5/21) developed at least one ARIA‐E episode during the trial, compared to 16% for individuals asymptomatic at the start of the trial (5/31). Moreover, 27% of individuals past their EYO at start of the trial (6/22) developed at least one ARIA‐E episode during the trial, compared to 13% for individuals who were at EYO <0 (4/30). Note that no individuals who started treatment at 10 years ahead of their expected onset (EYO < −10) developed an ARIA‐E event. Despite this trend, being CDR > 0 or at EYO > 0 at start of the trial was not significantly associated with higher risk for an ARIA‐E episode. CDR‐SB, MMSE, and cognitive composite scores were not significantly associated with higher odds for developing ARIA‐E. Global Aβ burden at baseline as estimated with PiB‐PET composite SUVR was not significantly associated with higher odds of developing ARIA‐E during the trial (Fig 2).
FIGURE 2.
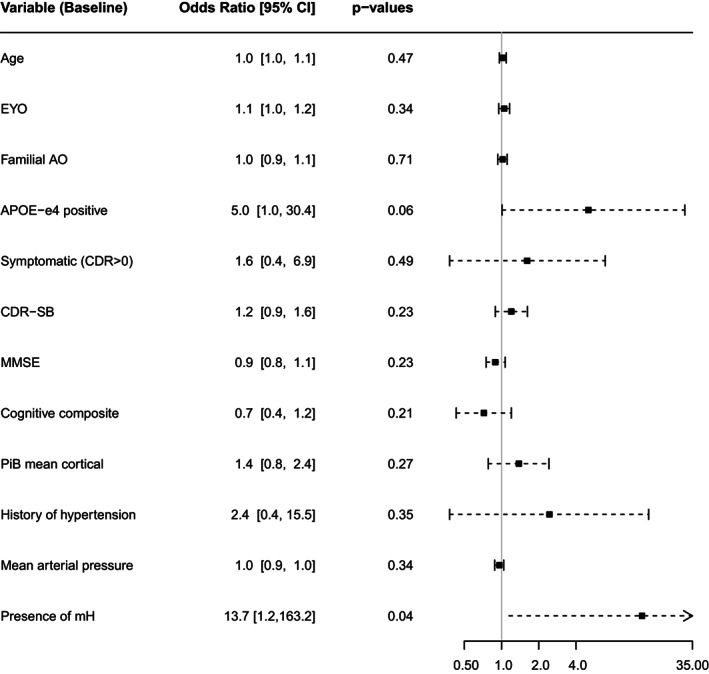
Forest plot of risk factors for developing ARIA‐E under gantenerumab. Baseline variables and associated odds for having ARIA‐E during the trial. APOE‐e4 status tends to be associated with odds for developing ARIA‐E during the trial. Presence of microhemorrhages (mH) at baseline increased odds for ARIA‐E. Annotation: AO = age of onset; APOE = apolipoprotein‐E; CDR = clinical dementia rating; CI = confidence interval; EYO = estimated year to symptom onset; MMSE = Mini‐Mental State Examination; OR = odds ratio; PiB = Pittsburgh compound B.
As for genetic factors, APOE‐ɛ4(+) were more likely to develop ARIA‐E (OR = 5.0, 95% CI [1.0, 30.4], adjusted p‐value = 0.055, Fig 2). Overall, 31% of APOE‐ɛ4(+) (5/16) developed at least one ARIA‐E episode during the trial, compared to only 14% for APOE‐ɛ4(−) (5/36). The potential effects of the number of APOE‐ɛ4 alleles on ARIA‐E outcome was not evaluated due to the very small number of cases. Similarly, the effect of DIAD mutation type PSEN1, PSEN2, or APP could not be evaluated.
Concerning vascular factors or comorbidities, 38% of individuals with a history of hypertension (3/8) developed at least one ARIA‐E episode, compared to 16% of individuals without any history of hypertension (7/44), although this association was not significant (Fig 2). The presence of microhemorrhages at baseline was significantly associated with greater risk of developing ARIA‐E during the trial.
Imaging Characteristics and Evolution of ARIA Findings
Under gantenerumab, ARIA‐E episodes were mostly multifocal (62%), found in the occipital (85%), and occurring in both sulcal and parenchyma (54%). At first episode, 60% of the ARIA‐E participants presented with associated ARIA‐H (mH and/or superficial siderosis). The longest cross‐sectional axis of ARIA‐E at initial findings ranged from 0.9 to 4.8 cm. The severity of ARIA‐E, as assessed by the BGTS, ranged from 1 to 8. As an example, the first case presented with edema in three different locations and a BGTS score of 4. All ARIA‐E participants had titration or dosing withheld when incident ARIA‐E was detected, and 60% of them had an increase in ARIA‐E size. Figure 3 shows an example of a participant who developed ARIA‐E with associated ARIA‐H and a focal reduction of PiB‐uptake in the region containing ARIA‐E.
FIGURE 3.
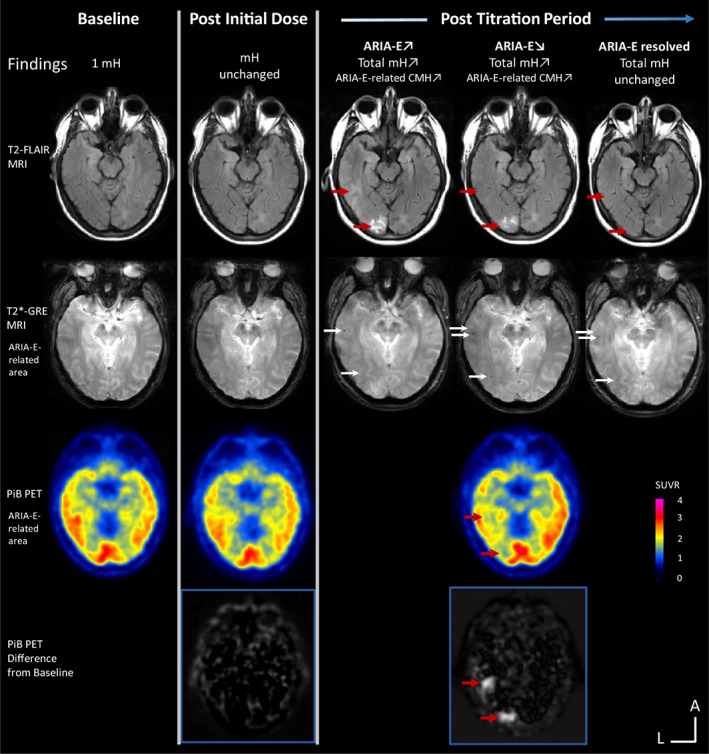
MRI and PET follow‐up of an individual treated with gantenerumab who had an ARIA‐E episode. The participant had one microhemorrhage (mH) at baseline and no additional MRI or PET findings post initial dose. During the participant's post‐titration period, a safety MRI showed an incident parenchymal ARIA‐E in the left occipital region with the longest cross‐sectional axis <2 cm (first row, red arrows). This episode was associated with incident mHs both spatially and temporally (second row, white arrows). Thirty days after the first ARIA‐E episode, a safety MRI showed increase size to ~2 cm of the left occipital ARIA‐E and a new definite parenchymal and sulcal ARIA‐E in the left occipital/temporal region with the longest cross‐sectional axis >5 cm. PiB PET showed substantial removal of Aβ burden co‐localized with the ARIA‐E lesions (third and fourth rows, red arrows). The dose of the study drug was withheld. ARIA‐E resolved after ~6 months. No neurological symptoms were described associated with the ARIA‐E episode. Annotations: FLAIR = fluid attenuated inversion recovery; GRE = gradient echo; PiB = Pittsburgh compound B. L = left and A = anterior for MR and PET image orientations. Color bar scale: PET SUVR with highest values in red. Hyperintense voxels in the subtracted PET images represent highest SUVR decrease from baseline. [Color figure can be viewed at www.annalsofneurology.org]
ARIA‐E severity was not significantly associated with the total count of microhemorrhages at time of ARIA‐E (Spearman's rho = 0.5, 95% CI [−0.2, 0.9], p‐value = 0.12) and, although not significant, was moderately associated with incident microhemorrhages at time of ARIA‐E (rho = 0.6, 95% CI [−0.1, 0.9], p‐value = 0.08). ARIA‐E severity was not significantly associated with disease stage as measured by EYO, CDR‐SB, and MMSE (rho = 0.5, 95% CI [−0.2, 0.9], p‐value = 0.12, rho = 0.3, 95% CI [−0.4, 0.8], p‐value = 0.47, and rho = −0.21, 95% CI [−0.7, 0.5], p‐value = 0.56, respectively) but was associated with age at time of ARIA‐E (rho = 0.9, 95% CI [0.6, 1.0], p‐value < 0.001). The dosage at time of ARIA‐E was not significantly associated with the severity of the episode (rho = −0.05, 95% CI [−0.66, 0.60], p‐value = 0.90). There was low to moderate association between the number of incident ARIA‐H at time of ARIA‐E and age at time of ARIA‐E (rho = 0.4) and disease stage (rho = 0.6), however it was not statistically significant.
ARIA‐E and Change in Primary and Secondary Outcomes
No significant changes in the primary outcome (cognitive composite score, Fig 4A) were observed between Gant ARIA‐E and non‐ARIA‐E groups (Table 3). Although changes from baseline in MMSE or CDR‐SB measures of the ARIA‐E group were consistently smaller (indicating slow decline) than in the non‐ARIA‐E group, the differences between the groups were not statistically significant (Fig 4B, C, Table 3). Similarly, changes in mean PiB composite from baseline were also consistently larger across visits (indicating more Aβ removal) but were not significant (Fig 4D). None of the CSF biomarker changes showed significant difference between the two groups, and only the change in CSF‐ptau181 from baseline to year 4 tended to be larger (indicating greater decrease) in the ARIA‐E group in the adjusted model (p‐value = 0.05, Fig 4E–H).
FIGURE 4.
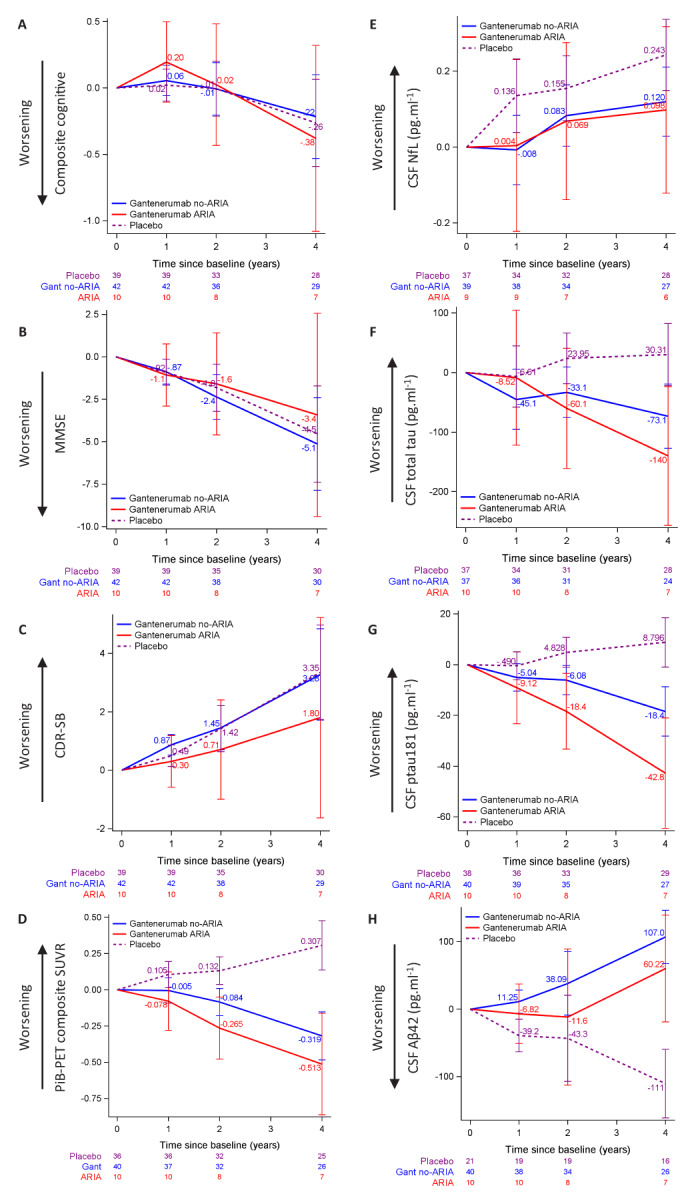
Longitudinal changes from baseline in outcome variables of placebo without ARIA‐E, Gant ARIA‐E, and Gant non‐ARIA‐E. Plots of mixed effects models for repeated measures, showing the placebo group without ARIA‐E in dashed purple, Gant ARIA‐E in solid red and Gant non‐ARIA‐E in solid blue. The placebo group is represented for reference. (A–C) Evaluation of cognitive measures. No significant difference between the Gant groups for cognitive composite (A), MMSE (B), and CDR‐SB (C). (D) Longitudinal changes in PiB‐PET composite measure was not different between the Gant groups. (E–H) Evaluation of CSF biomarkers. No significant difference between the Gant groups for changes in log‐transformed CSF NfL (E), CSF total tau (F), CSF ptau181 (G), or CSF Aβ42 (H) measures between groups. [Color figure can be viewed at www.annalsofneurology.org]
TABLE 3.
Results of Mixed Model for Repeated Measures to Assess ARIA and Change in Clinical and Biomarker Outcomes
| Biomarker | Time (years) | Difference in Change from Baseline between Gant ARIA‐E and Gant Non‐ARIA‐E | SE | p‐Value |
|---|---|---|---|---|
| Cognitive composite | 1 | 0.14 | 0.16 | 0.39 |
| 2 | 0.03 | 0.25 | 0.90 | |
| 3 | −0.005 | 0.30 | 0.99 | |
| 4 | −0.16 | 0.39 | 0.67 | |
| CDR‐sum of boxes | 1 | −0.57 | 0.48 | 0.24 |
| 2 | −0.75 | 0.93 | 0.43 | |
| 3 | −1.34 | 1.30 | 0.31 | |
| 4 | −1.48 | 1.87 | 0.43 | |
| MMSE | 1 | −0.19 | 0.99 | 0.85 |
| 2 | 0.77 | 1.66 | 0.64 | |
| 3 | 1.58 | 2.44 | 0.52 | |
| 4 | 1.71 | 3.29 | 0.61 | |
| PiB‐PET composite | 1 | −0.07 | 0.11 | 0.51 |
| 2 | −0.18 | 0.12 | 0.13 | |
| 4 | −0.19 | 0.19 | 0.32 | |
| CSF‐Aβ42 | 1 | −18.07 | 23.61 | 0.45 |
| 2 | −49.72 | 55.77 | 0.38 | |
| 4 | −46.80 | 44.13 | 0.29 | |
| CSF‐ptau181 | 1 | −4.08 | 7.61 | 0.59 |
| 2 | −12.28 | 8.01 | 0.13 | |
| 4 | −24.33 | 11.94 | 0.05 | |
| CSF‐tau | 1 | 36.57 | 62.33 | 0.56 |
| 2 | −27.01 | 54.18 | 0.62 | |
| 4 | −66.85 | 63.47 | 0.30 | |
| CSF‐NfL | 1 | 0.01 | 0.12 | 0.92 |
| 2 | −0.01 | 0.11 | 0.90 | |
| 4 | −0.02 | 0.12 | 0.86 |
Discussion
In the double‐blind phase II/III DIAN‐TU‐001 trial, gantenerumab and solanezumab, two anti‐Aβ monoclonal antibodies, were investigated in participants with dominantly inherited Alzheimer disease. In the current study, we give a comprehensive description of ARIA in the context of the DIAN‐TU‐001 trial. Gantenerumab, but not solanezumab, was associated with ARIA‐E events compared to placebo. This is consistent with prior published data on these two antibodies. 3 , 9 We found that APOE‐ɛ4(+) tended to be associated with higher risk for developing ARIA‐E, and the presence of microhemorrhages at baseline showed significantly higher risk for ARIA‐E. Most episodes were asymptomatic and mild, when symptomatic. Most episodes were transient and reversible on MRI after intervention to manage ARIA, consisting of withheld titration or withheld dosage.
The observations of ARIA in the gantenerumab but not in the solanezumab treatment arm corroborate previous findings in sporadic AD populations. Gantenerumab targets fibrillar Aβ in plaques, and the increased Aβ clearance appears to disrupt the blood–brain barrier and exacerbate inflammation, potentially facilitating ARIA. 28 Solanezumab, by contrast, targets soluble Aβ and has been shown to be associated with ARIA to a lesser extent. 29
The overall prevalence of ARIA‐E observed in the DIAN study for gantenerumab was lower than that observed in some clinical trials in sAD populations. For instance, in the DIAN‐TU‐001, 19% of participants receiving gantenerumab developed ARIA‐E, while in the SCarlet RoAD and Marguerite RoAD open label extension (OLE) studies, 28.6 and 29.2% of participants receiving gantenerumab developed at least one ARIA‐E episode, respectively. 9 Several reasons may explain the differences in ARIA‐E frequency between DIAD and sAD trials. Sporadic AD trials have typically included mild to moderate AD participants and more recently targeted prodromal to mild AD populations; for example, the SCarlet RoAD and Marguerite RoAD OLE studies included participants from prodromal/asymptomatic to moderate AD at baseline while DIAN included asymptomatic or mild AD participants, which may account for the observed differences in the overall ARIA‐E frequency. In fact, in the DIAN‐TU‐001, 24% of symptomatic carriers receiving gantenerumab developed ARIA‐E. Most ARIA‐E individuals were symptomatic or past their EYO at the time their first ARIA‐E was detected. On average they appeared at advanced disease stage based on the CDR‐SB and MMSE, but we did not confirm that disease stage was clearly associated with developing ARIA‐E, likely due to our limited sample size. Another possible confound is the prevalence of APOE‐ɛ4(+) in the DIAD population relative to the sporadic AD trial population. For instance, sporadic AD gantenerumab trials had almost twice the proportion of APOE‐ɛ4(+) than the DIAN‐TU (~60% vs 31%). 4 , 9 Possible differences in the number of baseline microhemorrhages between DIAN‐TU and sporadic AD trials may also explain the observed differences. Nevertheless, we did not observe any ARIA‐E event at doses lower than 450 mg in the DIAN‐TU gantenerumab arm, whereas ARIA‐E was observed after the initial doses of 105 mg in the sAD trial, suggesting that the younger DIAD population may tolerate higher doses. Additionally, higher doses may be needed in DIAD to counteract higher Aβ aggregation rates and total load in this population relative to sAD. 30 Some of these questions will be explored during the ongoing open‐label extension of the DIAN‐TU‐001 study with gantenerumab.
Concerning solanezumab, in previous phase III trial studies in participants with mild to moderate AD such as the EXPEDITION trials, ARIA‐E was observed in 0.9% for combined EXPEDITION 1 and 2 trials and in 0.09% in the EXPEDITION 3 trial and no statistically significant difference was observed compared to their placebo groups which displayed 0.4 and 0.19%, respectively. 3 , 31 In the DIAN‐TU trial, ARIA‐E was not observed in the DIAD participants receiving solanezumab and was observed in one case in the corresponding placebo group. Note that, overall, ARIA‐E events have been observed in 1 to 3% in placebo arms from other trials of anti‐amyloid antibodies such as gantenerumab, 4 lecanemab, 5 or aducanumab 15 in early AD. During clinical trials, individuals who develop ARIA‐E while receiving a placebo may in fact be experiencing disease‐related spontaneous events such as cerebral amyloid angiopathy‐related inflammation (CAA‐ri). These findings, radiographically identical to drug‐related ARIA‐E, can be observed without treatment in DIAD and sporadic AD populations although they are relatively rare. 32 , 33
We observed that most of the first incident ARIA‐E occurred at gantenerumab doses higher than 675 mg. This is consistent with dose‐dependency observed in previous clinical trials of gantenerumab and several other anti‐amyloid monoclonal antibody drugs. 4 , 14 , 15 However, ARIA‐E appeared more severe at lower doses with longer resolution times and larger sizes, which may be related in part to confounding effect due to APOE‐ɛ4(+) status. In the DIAN‐TU‐001 trial, the individuals who developed ARIA‐E at lower titration steps and who may have been more sensitive to developing ARIA were all carriers of at least one APOE‐ɛ4 allele and at advanced disease stage. Although we did not find that disease stage was significantly associated with higher risk, APOE‐ɛ4(+) were more likely to have doubled risk for developing ARIA‐E. This suggests that the risk to develop ARIA‐E, specially at lower doses, is higher in APOE‐ɛ4 carriers. This is consistent with previous trials in sporadic AD reporting the increased risk for developing ARIA. 4 , 9 , 14 , 15
It has also been reported that the presence of microhemorrhages or other ARIA‐H type are associated with risk for developing ARIA‐E. 15 DIAD individuals can spontaneously develop ARIA‐H and we reported in a previous study that spontaneous microhemorrhages and superficial siderosis can be found in 8 and 1% of carriers of a DIAD mutation, respectively. 25 Furthermore, we found that having 2 or more microhemorrhages at baseline was associated with higher risk for incident ARIA‐H (microhemorrhages) as part of the disease progression outside of a clinical trial. Here, we found that individuals with ARIA‐H at baseline were at higher risk for developing ARIA‐E under gantenerumab with high incident associated ARIA‐H, supporting our previous findings and corroborating previous trial reports in sporadic AD populations. 15
Concerning the imaging characteristics of the ARIA‐E findings, most were in the occipital lobe and were associated with concurrent and colocalized ARIA‐H. The severity of the ARIA‐E findings in the DIAN‐TU‐001 was variable, although overall appeared lower than in trials in sporadic AD populations. Such differences can be explained by the younger age of this population compared to late‐onset sporadic AD. This is supported by the strong association we found between severity score of ARIA‐E and age at time of ARIA‐E. The less severe ARIA‐E in younger populations might be related to a lesser extent of vascular amyloid deposition and higher vascular resilience and/or a less detrimental inflammatory response to vascular Aβ removal.
We also observed in an example case that amyloid was removed locally in the same area as ARIA‐E. This observation has also been reported in other trials of anti‐amyloid monoclonal antibodies targeting amyloid plaques. 14 Although this removal was visible locally and longitudinally, no significant difference in global PiB changes was observed between the ARIA‐E group and the non‐ARIA‐E group who received gantenerumab, even after >4 years. In previous trials of gantenerumab in sporadic populations, a trend toward slightly higher reductions in the ARIA‐E group was observed but global amyloid load at baseline and during the trial was not significantly different between the ARIA‐E and non‐ARIA‐E groups. 9 In the DIAN‐TU‐001 trial, these groups did not differ on primary or secondary cognitive measures, nor on secondary biomarker outcome measures. Due to the limited size of the trial population, no statistical significance could reasonably be seen on most measures. It was noted that participants with ARIA‐E tended to experience slower CDR‐SB progression and MMSE decline, and greater amyloid removal and CSF tau and ptau181 decrease. Another trial showing similar biomarker profiles in ARIA‐E participants suggested that ARIA was a marker for better treatment response with reductions towards normalization of the downstream biomarkers. 34 Furthermore, unlike rare reports of serious radiographic or clinical presentations of ARIA‐E in sporadic AD anti‐amyloid antibody trials, no serious ARIA‐E radiographic or clinical presentations were observed in this study at the doses tested.
We recognize the present study has limitations, the largest of which was drawing definitive conclusions given our small size. As evidenced by the wide confidence intervals, our sample size limited some of the power estimations for ARIA‐E risk factors. Second, we did not collect extensive cognitive measures, CSF‐ or blood‐based biomarkers immediately after ARIA‐E episodes, limiting our ability to estimate the impact of ARIA‐E on cognition and biomarkers. Third, potential treatment‐induced ARIA‐H were not evaluated due to limited data. Some of these issues may be addressed by the ongoing exploratory open‐label extension of the DIAN‐TU‐001 study with gantenerumab.
To conclude, this is the first study to report ARIA‐E frequency and risk factors in a DIAD population, helping to address key questions about dose escalation and treatment in this population. As DIAN‐TU continues to expand clinical trials in DIAD populations, including primary and secondary prevention, these results may inform effective and safe doses for therapeutic strategies similar to gantenerumab. Finally, our findings may also apply to future prevention trials in sporadic AD, particularly our observation that risk of ARIA‐E is lower during asymptomatic phases of the disease and in younger populations (e.g., sporadic EOAD).
Author Contributions
N.J‐M., J.J.L‐G., A.A.M, C.H., J.W., S.P.S., T.L.S.B., and D.B.C. contributed to the conception and design of the study; N.J‐M., J.J.L‐G., Y.L., A.A.M, E.P., G.M.P., N.T.A., S.B.B., T.D.B., S.E.B., B.B., W.S.B., R.C., C.C., A.M.F., M.F., S.G., J.H., L.S.H., G.R.H., C.R.J., I.Z.J., M.M., C.J.M., J.M.R., B.J.S., D.W., E.M., R.J.B., S.P.S., T.L.S.B., D.B.C. and DIAN‐TU Study group (see Supplementary Material) contributed to the acquisition and analysis of data; N.J‐M., J.J.L‐G., Y.L., A.A.M, E.P., G.M.P., C.R.J., S.P.S., D.B.C. contributed to drafting the text or preparing the figures.
Potential Conflicts of Interest
CH, JW, and GK are employees of and have stock in F. Hoffmann‐La Roche, Ltd, which manufactures gantenerumab one of the drugs tested in this study, and CH also has a patent planned for gantenerumab. KCH, MM, and RY are employees of and have stock in Eli Lilly and Company, which manufactures solanezumab the other drug tested in this study. SEB, NCF, JH, CJM received consulting fees and JL speaker fees from F. Hoffman‐La Roche, Ltd. EM reports receiving personal honorarium for Continuing Medical Education activities for Eli Lilly and Company. SPS is the Gantenerumab Project Arm Leader for the DIAN‐TU‐001 study and receives consulting fees from F. Hoffmann‐La Roche, Ltd. and Eli Lilly and Company. TLSB reports receiving radiopharmaceuticals and technology from Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company. DBC is Medical Director for the DIAN‐TU‐001 study and has received consulting fees from F. Hoffmann‐La Roche, Ltd./Genentech. The former Department Head of Neurology at Washington University, Dr. David Holtzman, is an inventor on patents for solanezumab which was tested in this study. If solanezumab is approved as a treatment for Alzheimer's disease or dominantly inherited Alzheimer's disease, Washington University and Dr. Holtzman will receive part of the net sales of solanezumab from Eli Lilly and Company.
Supporting information
Appendix S1. Statistical parameters of logistic regressions investigating the baseline continuous variable shown in Figure 2 and the baseline CSF measures as potential risk factors for developing ARIA‐E.
Acknowledgments
Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Numbers U01AG042791, R01AG046179, R01AG053267. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The research for the DIAN‐TU‐001 trial, solanezumab and gantenerumab drug arms, was also supported by the Alzheimer's Association, Eli Lilly and Company, F. Hoffmann‐La Roche Ltd., Avid Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly and Company), GHR Foundation, an anonymous organization, FNIH and Accelerating Medicines Partnership, Cogstate, and Signant. The DIAN‐TU has received funding from the DIAN‐TU Pharma Consortium. N.J‐M. and J.J.L‐G. gratefully acknowledge supports from the Alzheimer's Association Research Fellowship to promote diversity (AARFD‐20‐681815 and AARFD‐21‐851415, respectively). We acknowledge the altruism of the participants and their families and contributions of the DIAN, DIAN Expanded Registry, and DIAN‐TU research and support staff at each of the participating sites (see DIAN‐TU Study Team) for their contributions to this study.
Nelly Joseph‐Mathurin and Jorge J. Llibre‐Guerra contributed equally to this study.
References
- 1. McDade E, Llibre‐Guerra JJ, Holtzman DM, et al. The informed road map to prevention of Alzheimer disease: a call to arms. Mol Neurodegener 2021;16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cummings J, Lee G, Zhong K, et al. Alzheimer's disease drug development pipeline: 2021. Alzheimers Dement 2021;7:e12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Honig LS, Vellas B, Woodward M, et al. Trial of Solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med 2018;378:321–330. [DOI] [PubMed] [Google Scholar]
- 4. Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alz Res Ther 2017;9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double‐blind, phase 2b proof‐of‐concept clinical trial in early Alzheimer's disease with lecanemab, an anti‐Aβ protofibril antibody. Alzheimers Res Ther 2021;13:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 2016;537:50–56. [DOI] [PubMed] [Google Scholar]
- 7. Guthrie H, Honig LS, Lin H, et al. Safety, tolerability, and pharmacokinetics of Crenezumab in patients with mild‐to‐moderate Alzheimer's disease treated with escalating doses for up to 133 weeks. J Alzheimers Dis 2020;76:967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med 2021;384:1691–1704. [DOI] [PubMed] [Google Scholar]
- 9. Klein G, Delmar P, Voyle N, et al. Gantenerumab reduces amyloid‐β plaques in patients with prodromal to moderate Alzheimer's disease: a PET substudy interim analysis. Alz Res Ther 2019;11:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sperling RA, Jack CR, Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: recommendations from the Alzheimer's association research roundtable workgroup. Alzheimers Dement 2011;7:367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cogswell PM, Barakos JA, Barkhof F, et al. Amyloid‐related imaging abnormalities with emerging Alzheimer disease therapeutics: detection and reporting recommendations for clinical practice. Am J Neuroradiol 2022;43:E19–E35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carlson C, Siemers E, Hake A, et al. Amyloid‐related imaging abnormalities from trials of solanezumab for Alzheimer's disease. Alzheimers Dement 2016;2:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. VandeVrede L, Gibbs DM, Koestler M, et al. Symptomatic amyloid‐related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement 2020;12:e12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sperling R, Salloway S, Brooks DJ, et al. Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 2012;11:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salloway S, Chalkias S, Barkhof F, et al. Amyloid‐related imaging abnormalities in 2 phase 3 studies evaluating Aducanumab in patients with early Alzheimer disease. JAMA Neurol 2022;79:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mills SM, Mallmann J, Santacruz AM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN‐TU trial. Rev Neurol 2013;169:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Nat Med 2021;27:1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bateman RJ, Benzinger TL, Berry S, et al. The DIAN‐TU next generation Alzheimer's prevention trial: adaptive design and disease progression model. Alzheimers Dement 2017;13:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ryman DC, Acosta‐Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta‐analysis. Neurology 2014;83:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 21. Barkhof F, Daams M, Scheltens P, et al. An MRI rating scale for amyloid‐related imaging abnormalities with edema or effusion. Am J Neuroradiol 2013;34:1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang G, Berry S, Xiong C, et al. A novel cognitive disease progression model for clinical trials in autosomal‐dominant Alzheimer's disease. Stat Med 2018;37:3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moulder KL, Snider BJ, Mills SL, et al. Dominantly inherited Alzheimer network: facilitating research and clinical trials. Alzheimers Res Ther 2013;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Joseph‐Mathurin N, Wang G, Kantarci K, et al. Longitudinal accumulation of cerebral microhemorrhages in dominantly inherited Alzheimer disease. Neurology 2021;96:e1632–e1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage 2015;107:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su Y, D'Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB‐PET with FreeSurfer ROIs. PLoS One 2013;8:e73377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bohrmann B, Baumann K, Benz J, et al. Gantenerumab: a novel human anti‐Aβ antibody demonstrates sustained cerebral amyloid‐β binding and elicits cell‐mediated removal of human amyloid‐β. JAD 2012;28:49–69. [DOI] [PubMed] [Google Scholar]
- 29. DeMattos RB, Bales KR, Cummins DJ, et al. Peripheral anti‐A antibody alters CNS and plasma A clearance and decreases brain A burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci 2001;98:8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen CD, Joseph‐Mathurin N, Sinha N, et al. Comparing amyloid‐β plaque burden with antemortem PiB PET in autosomal dominant and late‐onset Alzheimer disease. Acta Neuropathol 2021;142:689–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement 2016;12:110–120. [DOI] [PubMed] [Google Scholar]
- 32. Ryan NS, Lashley T, Revesz T, et al. Spontaneous ARIA (amyloid‐related imaging abnormalities) and cerebral amyloid angiopathy related inflammation in Presenilin 1‐associated familial Alzheimer's disease. JAD 2015;44:1069–1074. [DOI] [PubMed] [Google Scholar]
- 33. Plotzker AS, Henson RL, Fagan AM, et al. Clinical and paraclinical measures associated with outcome in cerebral amyloid angiopathy with related inflammation. J Alzheimers Dis 2021;80:133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu E, Wang D, Sperling R, et al. Biomarker pattern of ARIA‐E participants in phase 3 randomized clinical trials with bapineuzumab. Neurology 2018;90:e877–e886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Statistical parameters of logistic regressions investigating the baseline continuous variable shown in Figure 2 and the baseline CSF measures as potential risk factors for developing ARIA‐E.