

Abstract
Objective
To assess the efficacy and safety of subcutaneous administration of 30 mg or 80 mg of ozoralizumab plus methotrexate (MTX) in patients with rheumatoid arthritis (RA) whose disease remained active despite MTX therapy.
Methods
In this multicenter, double‐blind, parallel‐group, placebo‐controlled phase II/III trial, 381 patients were randomized to receive placebo, ozoralizumab 30 mg, or ozoralizumab 80 mg, plus MTX subcutaneously injected every 4 weeks for 24 weeks. The primary end points were the response rates based on the American College of Rheumatology 20% improvement criteria (ACR20) at week 16 and change in the Sharp/van der Heijde score (ΔSHS) from baseline to week 24.
Results
The proportion of patients with an ACR20 response at week 16 was significantly higher (P < 0.001) in both ozoralizumab groups (79.6% for 30 mg, 75.3% for 80 mg), compared with placebo (37.3%); these improvements were observed from the first week of treatment. The proportion of the patients with structural nonprogression (ΔSHS ≤0) was significantly higher in both ozoralizumab groups than in the placebo group. For some secondary end points, significantly greater improvements were observed starting from as early as day 3. Serious adverse events occurred in 4 patients in the ozoralizumab 30‐mg group and 5 patients in the ozoralizumab 80‐mg group.
Conclusion
In patients with active RA who received ozoralizumab in combination with MTX, the signs and symptoms of RA were significantly reduced as compared with the outcomes in those receiving placebo. Ozoralizumab demonstrated acceptable tolerability with no new safety signals when compared with other antibodies against tumor necrosis factor.
INTRODUCTION
Rheumatoid arthritis (RA) treatment has greatly improved the management of RA through a standardized treatment algorithm, treat‐to‐target strategy, and drugs such as biologic disease‐modifying antirheumatic drugs (bDMARDs) and targeted synthetic DMARDs. Despite advances in disease management, there is still an unmet therapeutic need in RA, as current therapeutic agents sometimes only achieve partial response, and only 20–25% of patients achieve complete remission (1, 2).
Tumor necrosis factor (TNF) is deeply implicated in the pathogenesis of RA (3). Ozoralizumab is a next‐generation anti‐TNF antibody. It is a 38‐kd trivalent NANOBODY compound (Ablynx originally discovered and performed initial development of the NANOBODY compound ozoralizumab, and NANOBODY is a registered trademark of Ablynx NV, an affiliate of Sanofi) consisting of the 2 humanized anti‐human TNF VHH antibodies and 1 humanized anti‐human serum albumin (HSA) VHH antibody. VHH antibodies are derived from a special type of heavy‐chain antibody naturally produced by llamas and other camelids (4, 5). Ozoralizumab demonstrates inhibitory activity against human TNF and demonstrates a specific binding ability to HSA, which leads to potent neutralization of the action of TNF and prolonged serum half‐life by interacting with serum albumin (6, 7, 8, 9, 10). Murine surrogate NANOBODY molecules have been shown to accumulate in inflamed tissue in a mouse collagen–induced arthritis model (6). This accumulation in inflamed tissue is expected to occur with the equivalent compound in humans.
Here, we report the first results of a phase II/III trial (JapicCTI identifier: 184029) to evaluate the efficacy and safety of 2 dosage regimens of ozoralizumab concomitant with methotrexate (MTX) therapy, compared with placebo, in patients with active RA who had an inadequate response to MTX treatment alone.
PATIENTS AND METHODS
Trial design
The phase II/III results of the anti‐TNF multivalent NANOBODY compound ozoralizumab in patients with RA (OHZORA) trial (JapicCTI identifier: 184029) was a multicenter, randomized, placebo‐controlled, parallel‐group confirmatory trial with a 24‐week double‐blind treatment period (period A) followed by a 28‐week open‐label treatment period (period B). Period A was conducted between September 2018 and March 2020 at 78 sites in Japan.
In period A, RA patients were randomly allocated in a 2:2:1 ratio to receive ozoralizumab 30 mg, ozoralizumab 80 mg, or placebo under double‐blind conditions, administered subcutaneously every 4 weeks concomitant with MTX therapy (6–16 mg/week) for 24 weeks. The dosage and administration method were determined based on the previous phase I/II study (ClinicalTrials.gov identifier: NCT01007175). At week 16, patients who met the early escape criteria (<20% improvement from baseline in tender joint count in 68 joints [TJC68] and swollen joint count in 66 joints [SJC66]) were moved to the ozoralizumab 30‐mg group from the placebo group and from the ozoralizumab 30‐mg group to the 80‐mg group under double‐blind conditions, starting from week 20. The double‐blind treatment period was followed by a 28‐week open‐label period, with the patients receiving placebo rerandomized (at a 1:1 ratio) to receive treatment with 30 mg or 80 mg of ozoralizumab.
Ethics approval
This clinical trial was conducted in accordance with the ethics principles according to the Declaration of Helsinki Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices; and Japan's Ministerial Ordinance on Good Clinical Practice for Drugs.
Patients
This trial sample included Japanese RA patients ages 20–75 years who had an inadequate response to MTX and met the American College of Rheumatology (ACR)/EULAR 2010 classification criteria for RA (11). The inclusion criteria included active RA (TJC68 score ≥6, SJC66 score ≥6, and high‐sensitivity C‐reactive protein [hsCRP] level of ≥0.6 mg/dl or erythrocyte sedimentation rate [ESR] ≥28 mm/hour), MTX treatment at least 12 weeks prior to the baseline visit, and no change in MTX dose (6–16 mg/week) for at least 6 weeks prior to the baseline visit. Patients with abnormal findings on chest radiography suggestive of a malignant tumor, infection, or interstitial pneumonia were excluded. Patients with active tuberculosis were excluded, and patients with latent tuberculosis were excluded except when antituberculosis pharmacotherapy with isoniazid had been started in advance.
Outcomes
The primary efficacy end points were the ACR criteria for 20% improvement in disease activity (ACR20) response rate at week 16 and a change from baseline in the Sharp/van der Heijde score (SHS) at week 24 (ΔSHS). Radiography was performed at the baseline visit and at week 24 (or treatment discontinuation or week 20, for early escape). Bone erosion and joint space narrowing (JSN) were centrally and independently scored by 2 blinded radiologists (third‐party assessors, as needed). To evaluate structural progression, we used ΔSHS and the proportion of patients with structural nonprogression (ΔSHS ≤0), structural remission (ΔSHS ≤0.5), and ΔSHS less than or equal to smallest detectable change (SDC) (12, 13). In this trial, the SDC in SHS at week 24 was 1.0.
Secondary efficacy end points included ACR50/70 response, Disease Activity Score in 28 joints using the CRP level (DAS28‐CRP), patient global assessment of disease activity (PtGA) score using a visual analogue scale [VAS], patient's assessment of pain using a VAS, Simplified Disease Activity Index (SDAI), Boolean remission, Health Assessment Questionnaire disability index (HAQ DI), erosion score, and JSN score, In addition, pharmacodynamic end points such as hsCRP and ESR were evaluated (for all secondary end points and pharmacodynamics, see Supplementary Information, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.42273).
Safety assessments
For the assessment of safety, adverse events (AEs) (including injection site reactions, severe AEs [SAEs], and AEs of special interest), body weight, vital signs, and laboratory test results were evaluated. For the assessment of immunogenicity, we measured plasma anti‐ozoralizumab antibodies and plasma ozoralizumab‐neutralizing antibodies during administration of the first dose, at week 8, and at week 24 (at week 20 for patients who met the early escape criteria).
Statistical analysis
Sample size (n = 148 for 30 mg and 80 mg ozoralizumab and n = 74 for placebo) was calculated according to the results of phase II studies. The ACR20 response rates (at week 16, i.e., the primary end point) between the ozoralizumab 30 mg, ozoralizumab 80 mg, and placebo groups were assumed to be 66%, 66%, and 40%, respectively. An allocation ratio of 2:2:1 was planned for the ozoralizumab 30‐mg group, ozoralizumab 80‐mg group, and placebo group with 2‐sided significance levels of 2.5% and statistical power of 90%, allowing for a dropout rate and exclusion rate of 10%.
In efficacy and pharmacodynamics evaluations, the full analysis set was used as the primary analysis set (for full analysis set definition, see Supplementary Information, http://onlinelibrary.wiley.com/doi/10.1002/art.42273). Comparisons between the placebo group and ozoralizumab group were conducted using a Cochran‐Mantel‐Haenszel test with history of TNF inhibitor (TNFi) usage as a stratification factor for the ACR20 response rate (week 16), and analysis of covariance, with baseline values and history of TNFi usage as covariates for ΔSHS (week 24). For the primary analysis, the 2‐sided significance level was set at 2.5% and the 2‐sided confidence coefficient was set at 95%. To analyze the primary end point, multiplicity adjustment was conducted using a closed testing procedure (see Supplementary Information, http://onlinelibrary.wiley.com/doi/10.1002/art.42273). For the second analysis, the 2‐sided significance level was set at 5%.
The last observation carried forward method was used for missing ACR20 data and missing data for other secondary end points at weeks 16 and 24 (including early escape at week 24). A linear extrapolation method was used for SHS, erosion score, and JSN score at week 24.
The safety analysis set was used as the analysis set in the safety and immunogenicity assessments (for safety analysis set definition, see Supplementary Information, http://onlinelibrary.wiley.com/doi/10.1002/art.42273). AEs were defined according to the Japanese version of the Medical Dictionary for Regulatory Activities, version 22.1. For details regarding the evaluation of immunogenicity, see Supplementary Information. Statistical analyses were performed using SAS 9.4 software.
RESULTS
Patient demographic and baseline clinical characteristics
In this trial, 587 patients were screened, and 395 patients were randomized to the placebo group, ozoralizumab 30 mg‐group, and ozoralizumab 80‐mg group. Drugs were administered according to the allocation of 381 patients (placebo [n = 75], ozoralizumab 30 mg [n = 152], and ozoralizumab 80 mg [n = 154]). Early escape criteria applied to 21 patients in the placebo group, 9 patients in the ozoralizumab 30‐mg group, and 11 patients in the ozoralizumab 80‐mg group. A total of 29 patients discontinued for reasons other than early escape. The overall rate of trial continuation up to week 24 (including patients who met the early escape criteria at week 20) was 92.4% (Figure 1). Among patients included in the full analysis set used in this trial, the mean disease duration was 7.4 years, the mean DAS28‐CRP at baseline was 5.13, and the mean SHS score was 27.46. Among the included patients, 89.2% were seropositive for rheumatoid factor and/or anti–citrullinated protein antibodies. When other indices were included, there was no major difference in the mean values (Table 1).
Figure 1.
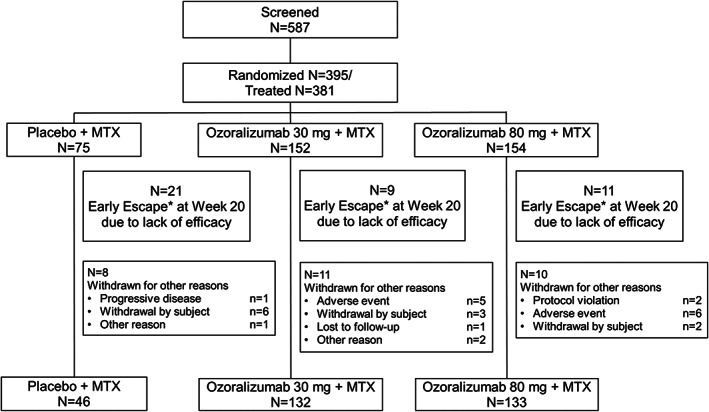
Flow chart showing the disposition of patients into groups to receive either placebo and methotrexate (MTX), ozoralizumab 30 mg and MTX, or ozoralizumab 80 mg and MTX. Early escape (*) indicates that when improvements from baseline in tender joint count and swollen joint count at week 16 were <20%, the patient was transferred to the open‐label follow‐up trial (after administration of the investigational drug) at week 20.
Table 1.
Demographic characteristics and disease status of the patients at baseline assigned to receive MTX and either placebo, ozoralizumab 30 mg, or ozoralizumab 80 mg (full analysis set)*
| Ozoralizumab | ||||
|---|---|---|---|---|
| Placebo (n = 75) | 30 mg (n = 152) | 80 mg (n = 154) | Total (n = 381) | |
| Sex, female, no. (%) | 57 (76.0) | 105 (69.1) | 123 (79.9) | 285 (74.8) |
| Age, years | 54.3 ± 12.1 | 54.8 ± 11.2 | 55.5 ± 10.9 | 55.0 ± 11.2 |
| Age <65 years, no. (%) | 56 (74.7) | 119 (78.3) | 116 (75.3) | 291 (76.4) |
| Weight, kg | 58.4 ± 13.5 | 60.0 ± 12.8 | 57.6 ± 11.6 | 58.7 ± 12.5 |
| Disease duration, years | 7.6 ± 7.4 | 6.8 ± 6.4 | 7.8 ± 7.5 | 7.4 ± 7.1 |
| MTX dosage, mg/week | 10.2 ± 3.0 | 10.0 ± 2.9 | 10.1 ± 2.7 | 10.1 ± 2.8 |
| Glucocorticoid use, no. (%) | 37 (49.3) | 62 (40.8) | 64 (41.6) | 163 (42.8) |
| DAS28‐CRP | 5.1 ± 1.0 | 5.2 ± 1.1 | 5.1 ± 0.9 | 5.1 ± 1.0 |
| DAS28‐ESR | 5.8 ± 1.0 | 5.9 ± 1.0 | 5.8 ± 0.9 | 5.8 ± 1.0 |
| TJC68 | 15.5 ± 9.6 | 16.6 ± 8.8 | 15.6 ± 8.9 | 16.0 ± 9.0 |
| SJC66 | 13.2 ± 8.5 | 13.8 ± 7.2 | 12.8 ± 6.4 | 13.3 ± 7.2 |
| SHS | 32.2 ± 50.2 | 25.0 ± 30.9 | 27.6 ± 37.4 | 27.5 ± 38.0 |
| Erosion score | 17.8 ± 28.0 | 14.2 ± 16.4 | 15.2 ± 18.8 | 15.3 ± 20.1 |
| JSN score | 14.5 ± 23.1 | 10.7 ± 15.5 | 12.4 ± 19.4 | 12.1 ± 18.8 |
| hs‐CRP level, mg/dl | 1.3 ± 1.7 | 1.6 ± 2.0 | 1.3 ± 1.8 | 1.4 ± 1.9 |
| ESR, mm/hour | 36.4 ± 17.3 | 40.3 ± 22.3 | 38.6 ± 20.6 | 38.8 ± 20.7 |
| Seropositive RA, no. (%)† | 64 (85.3) | 140 (92.1) | 136 (88.3) | 340 (89.2) |
Except where indicated otherwise, values are the mean ± SD. MTX = methotrexate; DAS28‐CRP = Disease Activity Score in 28 joints using the C‐reactive protein level; ESR = erythrocyte sedimentation rate; TJC68 = tender joint count in 68 joints; SJC68 = swollen joint count in 68 joints; SHS = Sharp/van der Heijde score; JSN = joint space narrowing; hsCRP = high‐sensitivity CRP.
Seropositive rheumatoid arthritis (RA) indicates an anti–citrullinated protein antibody level ≥4.5 units/ml and/or rheumatoid factor >15 IU/ml.
Efficacy
ACR20 and ACR50/70 response
The ACR20 response rate at week 16 was 37.3% in the placebo group, 79.6% in the ozoralizumab 30‐mg group, and 75.3% in the ozoralizumab 80‐mg group. The intergroup difference compared with the placebo group was mean 42.1 (95% confidence interval [95% CI] 28.7–53.7; P < 0.001) in the ozoralizumab 30‐mg group and mean 37.9 (95% CI 24.4–49.7; P < 0.001) in the ozoralizumab 80‐mg group, indicating a significantly higher improvement rate in the ozoralizumab groups compared with the placebo group (Figure 2A). In the post hoc analysis using nonresponder imputation, the ACR20 response rate was also significantly higher in the ozoralizumab groups than in the placebo group (Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.42273). Similarly, at week 24, a significantly higher ACR20 response rate was found in the ozoralizumab groups than in the placebo group (Figure 2B). Furthermore, the onset of ACR20 response with ozoralizumab was rapid (Figure 2C). A similar result compared with the ACR20 was found for ACR50/70, with a rapid onset of response in the ozoralizumab groups, and significantly higher ACR50/70 response rates than in the placebo group at both weeks 16 and 24 (Figures 2A–E).
Figure 2.
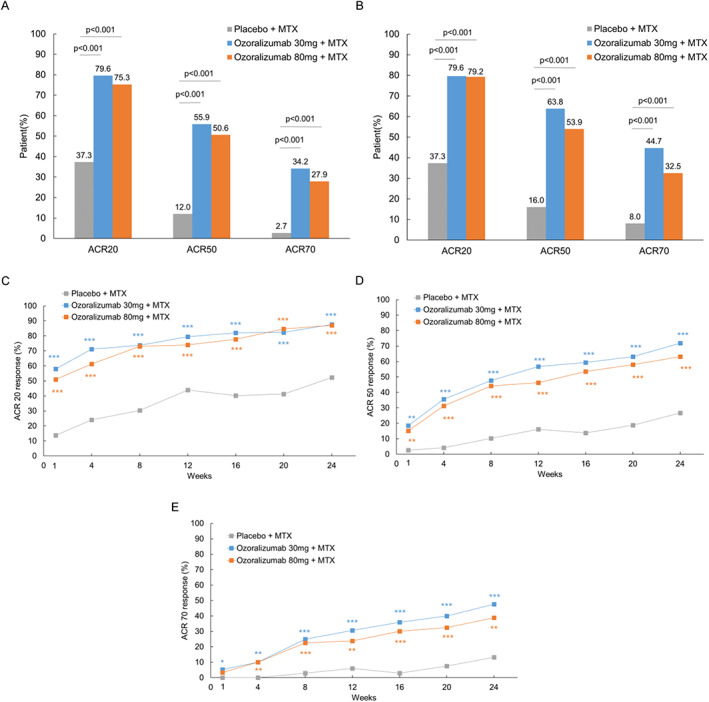
Treatment response rates based on the American College of Rheumatology 20% (ACR20), ACR50, and ACR70 improvement criteria up to week 24. A and B, ACR20, ACR50, and ACR70 improvement response rates at week 16 (A) and week 24 (B). C–E, Changes in ACR20 (C), ACR50 (D), and ACR70 (E) response rates over time. Cochran‐Mantel‐Haenszel test results were used as a stratification factor based on tumor necrosis factor inhibitor usage history. * = P < 0.05; ** = P < 0.01; *** = P < 0.001 versus placebo + methotrexate (MTX).
Other clinical measures and physical function
The ozoralizumab groups showed rapid improvement in DAS28‐CRP compared with the placebo group at day 3 (Figure 3A). Furthermore, the ozoralizumab groups showed a significantly better response to PtGA and patient's assessment of pain than the placebo group (Figures 3B and C). Similarly, rapid reduction was observed for both hsCRP level and ESR (Figures 3D and E). A significant improvement in the other secondary efficacy end points and pharmacodynamic end points were observed in the ozoralizumab groups compared with the placebo group (Supplementary Figure 1 and Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.42273). At week 24, the proportions of patients who achieved an SDAI of ≤3.3 was 5.3% in the placebo group, 25.0% in the ozoralizumab 30‐mg group, and 23.4% in the ozoralizumab 80‐mg group, indicating a significantly higher remission rate in the ozoralizumab groups compared with the placebo group. Boolean remission rates and other indices of clinical remission rate were also significantly higher than that in the placebo group (Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.42273). Improvement in HAQ DI scores in the ozoralizumab groups was observed from week 1 (Figure 3F), and the rates of achieving an HAQ DI score of ≤0.5 at week 24 were significantly higher in both ozoralizumab groups than in the placebo group (Supplementary Table 2).
Figure 3.
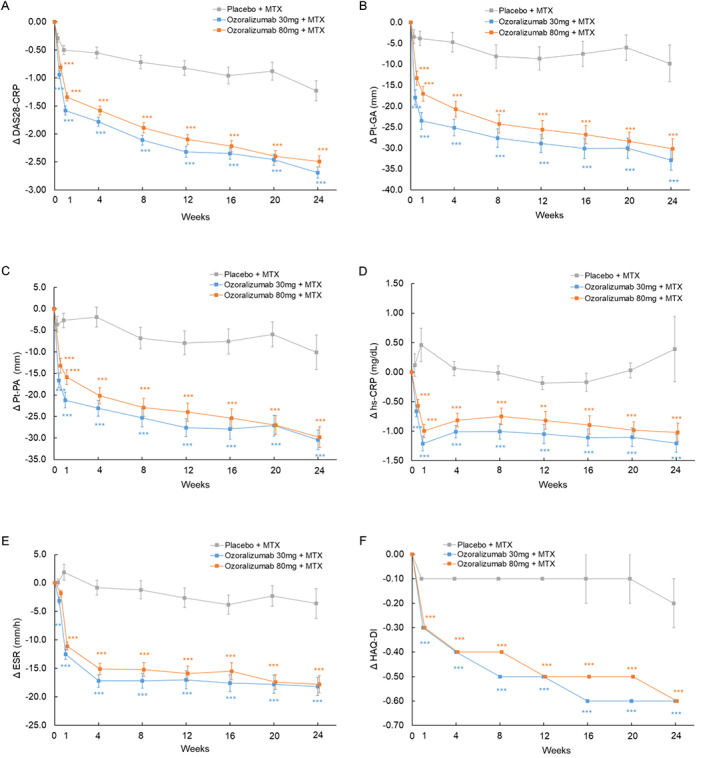
Changes from baseline in various parameters over time, including change in Disease Activity Score in 28 joints using the C‐reactive protein level (DAS28‐CRP) (A), patient global assessment of disease activity (PtGA) (B), patient's assessment of pain (Pt‐PA) (C), high‐sensitivity CRP (hsCRP) level (D), erythrocyte sedimentation rate (ESR) (E), and Health Assessment Questionnaire disability index (HAQ DI) (F), from weeks 0 to 24. Statistical comparisons were determined with an analysis of covariance using the status of prior tumor necrosis factor inhibitor usage and baseline values as covariates. Symbols with error bars show the mean ± SEM. ** = P < 0.01; *** = P < 0.001 versus placebo + methotrexate (MTX).
For all parameters, responses in the ozoralizumab groups were comparable, irrespective of the dose. In the post hoc analysis, there were no clear relationship between serum albumin level and efficacy.
Structural progression
The change from the baseline in SHS score (least squares mean) at week 24 was 0.8 in the placebo group, 0.6 in the ozoralizumab 30‐mg group, and 0.4 in the ozoralizumab 80‐mg group. While the amount of change tended to be lower in the ozoralizumab groups than in the placebo group, no statistically significant inhibition of progression was observed (Figure 4A). On the other hand, the change in SHS score was lower in the ozoralizumab groups than in the placebo group (Figure 4B), and the proportion of patients with structural nonprogression up to week 24 was 56.0% in the placebo group, compared to 73.0% in the ozoralizumab 30‐mg group and 73.4% in the ozoralizumab 80‐mg group. A significant difference was observed in both ozoralizumab groups compared with the placebo group. Also, a significant difference was observed in those with structural remission at week 24. The proportion of patients in whom the change in SHS score from baseline was lower than the SDC was significantly reduced in ozoralizumab 80‐mg group compared with the placebo group (Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.42273).
Figure 4.
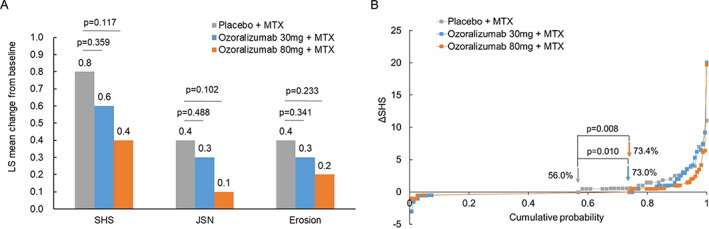
Progression of structural damage (EXTRAP). A, Least squares (LS) mean change from baseline to week 24 in Sharp/van der Heijde (SHS) score, joint space narrowing (JSN) scores, and erosion scores. Statistical comparisons were determined by analysis of covariance using the status of prior tumor necrosis factor inhibitor usage and baseline values as covariates. B, Cumulative probability of change in SHS score at week 24 compared with baseline values. Percentages indicate rates of nonprogression (ΔSHS ≤0) in each treatment group. P values were calculated by chi‐square test without continuity correction or multiplicity adjustment. MTX = methotrexate.
Safety
The occurrence of AEs up to 24 weeks (up to 20 weeks in early escape) is shown in Table 2. The incidence of AEs was 62.7% in the placebo group, 76.3% in the ozoralizumab 30‐mg group, and 72.1% in the ozoralizumab 80‐mg group. There was no clinical difference depending on the dose of ozoralizumab in the incidence of either AEs or adverse reactions. Infection had the highest incidence among AEs. The majority of AEs were mild to moderate; the onset of an SAE was rare. An AE that resulted in death was observed in 1 patient in the ozoralizumab 80‐mg group, in whom disseminated tuberculosis had developed. It was determined to have had a causal relationship with the investigational drug. In the placebo group, 2 patients (2.7%) had serious AEs other than death (perforated appendicitis and pneumonia), 4 patients (2.6%) in the ozoralizumab 30‐mg group (pelvic fracture, diabetes mellitus, lung adenocarcinoma, and interstitial lung disease [ILD]), and 4 patients (2.6%) in the ozoralizumab 80‐mg group (ovarian carcinoma/uterine carcinoma, renal abscess, cerebellar hemorrhage, and ILD). The incidence of injection site reaction was low, and the severity was mild.
Table 2.
Summary of safety through week 24 in patients receiving methotrexate and either placebo, ozoralizumab 30 mg, or ozoralizumab 80 mg*
| Ozoralizumab | |||
|---|---|---|---|
| Placebo (n = 75) | 30 mg (n = 152) | 80 mg (n = 154) | |
| AE | 47 (62.7) | 116 (76.3) | 111 (72.1) |
| Adverse drug reactions | 14 (18.7) | 42 (27.6) | 39 (25.3) |
| AE leading to death | 0 (0) | 0 (0) | 1 (0.6) |
| Other serious AE except death | 2 (2.7) | 4 (2.6) | 4 (2.6) |
| AE leading to discontinuation | 1 (1.3) | 5 (3.3) | 6 (3.9) |
| AE leading to suspension or a dose reduction of the study drug | 2 (2.7) | 8 (5.3) | 11 (7.1) |
| Intensity | |||
| Mild | 42 (56.0) | 101 (66.4) | 101 (65.6) |
| Moderate | 8 (10.7) | 35 (23.0) | 29 (18.8) |
| Severe | 2 (2.7) | 3 (2.0) | 5 (3.2) |
| AEs of special interest | |||
| Infections and infestations | 28 (37.3) | 64 (42.1) | 62 (40.3) |
| Serious infection other than tuberculosis | 2 (2.7) | 7 (4.6) | 3 (1.9) |
| Tuberculosis | 0 (0) | 0 (0) | 1 (0.6) |
| HZ | 0 (0) | 2 (1.3) | 3 (1.9) |
| Malignant tumors | 0 (0) | 1 (0.7) | 1 (0.6) |
| ILD | 0 (0) | 2 (1.3) | 1 (0.6) |
| Injection site reaction | 1 (1.3) | 3 (2.0) | 2 (1.3) |
Values are the number (%) of patients. AE = adverse event; HZ = herpes zoster; ILD = interstitial lung disease.
During the 24‐week treatment period, the generation of a new anti‐ozoralizumab antibody response or an increase in existing anti‐ozoralizumab antibody response was observed in 43 patients in the ozoralizumab 30‐mg group (28.3%), and 41 patients in the ozoralizumab 80‐mg group (26.6%). Among them, 2 patients in the 30‐mg group (1.3%), and 4 patients in the 80 mg group (2.6%) were positive for neutralizing antibodies. The presence of anti‐ozoralizumab antibodies showed no consistent trend in the efficacy or safety of the investigational drug, regardless of dose. Among neutralizing antibody–positive patients, there was no incidence of trial discontinuation due to exacerbation of the underlying illness, and no specific AEs occurred with this drug.
DISCUSSION
This phase II/III trial in RA patients who had an inadequate response to MTX demonstrated that both 30‐mg and 80‐mg ozoralizumab groups achieved statistically significant improvements in ACR20 response, which was one of the primary end points, compared with the placebo group. Furthermore, significant improvements were observed in the ozoralizumab groups compared with the placebo group in terms of other clinical symptoms, such as DAS28‐CRP, PtGA, and patient's assessment of pain. Moreover, a significant HAQ DI response was observed in the ozoralizumab groups compared with the placebo group. Most evaluation indices relating to clinical symptoms and physical function showed improvement starting as early as 3 days or 1 week after ozoralizumab administration. The efficacy of ozoralizumab 30 mg was comparable to 80 mg of ozoralizumab. Several other clinical trials of antirheumatic biologic treatments have demonstrated no dose‐dependency in efficacy above a certain dose (14, 15). In the present trial, there was no major difference between dosages in terms of efficacy, and the 30‐mg dose was considered sufficient for achieving the maximal clinical efficacy of ozoralizumab with concomitant MTX.
Of note, the MTX dosage used in this study, which was within the dosage range approved in Japan, is lower (10 mg on average and a maximum approved dosage of 16 mg/week) than typical dosages in North America and Europe. More robust MTX treatment regimens may leave less room for improvement from baseline in ACR20 response. However, even using more stringent criteria, such as ACR50/70, ozoralizumab showed significant improvement. Also, it is remarkable that the difference in ACR responses between the placebo group and ozoralizumab groups was comparable or greater than responses in previous trials of TNFi in Japanese patients conducted at even lower MTX doses (6–8 mg) (14, 16). Finally, it has been shown that MTX–polyglutamate concentrations between Japanese RA patients receiving a lower dose of MTX and those in North America receiving a higher dose were comparable, in part, because of the lower body mass index and body weight of Japanese RA patients (17).
Ozoralizumab is an antibody with a structure that greatly differs from that of conventional IgG antibodies. The rate of absorption of subcutaneously injected drugs is highly dependent on molecular weight (18, 19, 20). In addition, VHH antibodies have demonstrated good tissue penetration (21, 22, 23, 24). Furthermore, since serum albumin accumulates in inflammatory tissue in RA and other diseases, serum albumin–mediated drug transport has also been reported (25, 26, 27). Due to its structural characteristics (e.g., low molecular weight, HSA binding ability), ozoralizumab is predicted to quickly accumulate in the inflamed tissue. In this trial, an improvement was observed in terms of biochemical indices and clinical symptoms and physical function from day 3. Thus, it was found that ozoralizumab causes a change in biochemical indices soon after administration. As a result, early improvement was observed in indices of disease activity and in subjective indices, such as PtGA and patient's assessment of pain.
The change in SHS score from baseline, which was another primary end point (to evaluate prevention of structural damage in the joints), was not significantly different between the ozoralizumab groups and placebo group. We attribute this result to less change in SHS in the placebo group, compared with the change in SHS observed in phase III trials of other anti‐TNFi (14, 16). In recent years, the treatment of RA has improved; there has been an overall declining trend in the progression of structural damage in patients (28, 29). There are examples of similar trends in trials conducted in Japanese patients (30). However, regarding the proportion of patients without progression of structural damage and those who achieved structural remission, a significant response was observed in both ozoralizumab groups compared with the placebo group. Significant changes in SHS scores from baseline were not demonstrated at a group level. Thus, prevention of joint destruction by ozoralizumab was not demonstrated. However, a significant difference was demonstrated in terms of the proportion of patients with structural nonprogression, suggesting that ozoralizumab prevents or reduces progression of structural damage to joints in the majority of patients with active RA.
Our trial also included patients who had previously received antirheumatic biologic treatments. The subpopulation analysis showed no difference in terms of ACR20 response rates between patients with and those without previous use of antirheumatic biologic treatments (Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.42273). Moreover, this trial also included a small number of patients who had discontinued the use of therapeutic drugs (anti‐TNF antibody) due to secondary ineffectiveness. All of those who received treatment with ozoralizumab achieved an ACR20 response, which suggests that ozoralizumab might improve the condition of patients with secondary ineffectiveness to other anti‐TNF antibodies. It is thought that this could be attributed to the characteristic structure of ozoralizumab. It is necessary to understand the actual effect on patients who previously received bDMARDs with further evaluation in the future.
New expression or induction of anti‐ozoralizumab antibodies was observed in 28.3% of patients in the ozoralizumab 30‐mg group and in 26.6% of patients in the ozoralizumab 80‐mg group. At week 16, 81.4% (30‐mg group) and 67.5% (80‐mg group) of these patients achieved an ACR20 response. This finding indicates that the effect of antidrug antibodies on the investigational drug is limited; it is expected that efficacy and safety would be maintained in many subjects, but longer‐term observation is required to determine whether the development of antidrug antibodies affects the efficacy (including the prevention of structural damage) and safety of ozoralizumab.
Ozoralizumab had good tolerability up to 24 weeks. Most AEs were mild or moderate. The incidences of AE in the ozoralizumab 30‐mg and 80‐mg groups were comparable. AEs observed during administration of ozoralizumab were comparable to that of other TNFi approved by regulators in terms of frequency and type (14, 16, 31).
Treatment with bDMARDs has the potential to increase the risk of infections, including tuberculosis. In this trial, 11 cases of serious infection (including 1 case of tuberculosis in the ozoralizumab 80‐mg group) were reported in ozoralizumab groups, compared with 2 cases reported in the placebo group. Overall, the frequency of serious infections reported in this trial, including tuberculosis, was consistent with other TNFi, such as infliximab, etanercept, adalimumab, and golimumab in the Japanese population (32, 33, 34, 35).
This trial has limitations, including the fact that it is a clinical trial that included only Japanese patients who had an inadequate response to prior therapy with MTX. The sample size was small, with inclusion/exclusion criteria that included history of MTX use and disease activity. The unique genetic, environmental, and medical background of the Japanese population may affect the efficacy and safety of biologic agents in RA patients (36). In addition, since this trial was not designed to be active‐controlled, it is impossible to compare the results with other biologic agents. In this trial, eligibility criteria for active RA included TJC levels, SJC levels, and CRP levels, but not the number of bone erosions. This may have been a reason for the difficulty in assessing radiographic progression. Here we present an interim analysis of the results up to week 24. The safety and efficacy of ozoralizumab requires further assessment in the open‐label period, up to week 52 of the trial.
Ozoralizumab, at doses of 30 mg and 80 mg once every 4 weeks, demonstrated significant therapeutic effects on clinical symptoms and physical functions, as well as the prevention of structural damage to the joints, in RA patients who had inadequate response to MTX. The onset of therapeutic effects was rapid after administration. Ozoralizumab was well tolerated in the trial period. The efficacy and tolerability of ozoralizumab were comparable between 30‐mg and 80‐mg doses.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Tanaka had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Takeuchi, Nakanishi, Yamasaki, Tanaka
Acquisition of data
Nakanishi.
Analysis and interpretation of data
Takeuchi, Kawanishi, Nakanishi, Yamasaki, Tanaka.
ROLE OF THE STUDY SPONSOR
Taisho Pharmaceutical Co., Ltd. was involved in the study design and in the collection, analysis, and interpretation of the data, the writing of the manuscript, and the decision to submit the manuscript for publication. Publication of this article was contingent upon approval by Taisho Pharmaceutical Co., Ltd.
Supporting information
Disclosure Form
Appendix S1 Supplementary Information
Supplementary Table 1 ACR20 response rates at Week 16
Supplementary Table 2. Summary of clinical efficacy at Week 24
Supplementary Table 3. Subpopulation analysis of ACR20 response rate at Week 16, based on the previous use of antirheumatic biologics
Supplementary Figure 1 Changes from baseline over time
Change from baseline over time in A) DAS28‐ESR, B) ACR‐N, C) TJC68, D) SJC66, E) CDAI, F) SDAI, G) Ph‐GA, H) morning stiffness, I) MMP‐3, J) IL‐6, K) ACPA and L) RF. Observed data for weeks 0–24. Statistical comparisons were conducted according to analysis of covariance using the status of prior TNF inhibitor usage and baseline values as covariates. The data are presented as the mean ± standard error. *p < 0.05, **p < 0.01, ***p < 0.001
Abbreviations: ESR, erythrocyte sedimentation rate; DAS28‐ESR, disease activity score in 28 joints based on ESR; ACR‐N, American College of Rheumatology N; TJC68, tender joint count of 68 joints; SJC66, swollen joint count of 66 joints; CDAI, Clinical Disease Activity Index; SDAI, Simplified Disease Activity Index; Ph‐GA, physician's global assessment of disease activity; MMP‐3, matrix metalloproteinase 3; IL‐6, interleukin 6; ACPA, anti‐cyclic citrullinated peptide antibody; RF, rheumatoid factor
ACKNOWLEDGMENTS
We would like to thank the patients who were involved in this trial, as well as the investigators and the trial team. Ablynx originally discovered and performed initial development of the NANOBODY compound ozoralizumab. (NANOBODY is a registered trademark of Ablynx NV. Ablynx is an affiliate of Sanofi.)
Supported by Taisho Pharmaceutical Co., Ltd.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42273&file=art42273‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Zampeli E, Vlachoyiannopoulos PG, Tzioufas AG. Treatment of rheumatoid arthritis: unraveling the conundrum. J Autoimmun 2015;65:1–18. [DOI] [PubMed] [Google Scholar]
- 2. Smolen JS, Aletaha D, Barton A, et al. Rheumatoid arthritis. Nat Rev Dis Primers 2018;4:18001. [DOI] [PubMed] [Google Scholar]
- 3. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis ‐ shaping the immunological landscape [review]. Nat Rev Rheumatol 2016;12:63–8. [DOI] [PubMed] [Google Scholar]
- 4. Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single‐domain antibody fragments. Appl Microbiol Biotechnol 2007;77:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khodabakhsh F, Behdani M, Rami A, et al. Single‐domain antibodies or nanobodies: a class of next‐generation antibodies. Int Rev Immunol 2018;37:316–22. [DOI] [PubMed] [Google Scholar]
- 6. Coppieters K, Dreier T, Silence K, et al. Formatted anti–tumor necrosis factor α VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen‐induced arthritis. Arthritis Rheum 2006;54:1856–66. [DOI] [PubMed] [Google Scholar]
- 7. Beirnaert E, Desmyter A, Spinelli S, et al. Bivalent llama single‐domain antibody fragments against tumor necrosis factor have picomolar potencies due to intramolecular interactions. Front Immunol 2017;8:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Faassen H, Ryan S, Henry KA, et al. Serum albumin‐binding VHHs with variable pH sensitivities enable tailored half‐life extension of biologics. FASEB J 2020;34:8155–71. [DOI] [PubMed] [Google Scholar]
- 9. Dennis MS, Zhang M, Meng YG, et al. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem 2002;277:35035–43. [DOI] [PubMed] [Google Scholar]
- 10. Ishiwatari‐Ogata C, Kyuuma M, Ogata H, et al. Ozoralizumab, a humanized anti‐TNFα NANOBODY compound, exhibits efficacy not only at the onset of arthritis in a human TNF transgenic mouse but also during secondary failure of administration of an anti‐TNFα IgG. Front Immunol 2022;13:853008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 12. Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1986;1:307–10. [PubMed] [Google Scholar]
- 13. Bruynesteyn K, Boers M, Kostense P, et al. Deciding on progression of joint damage in paired films of individual patients: smallest detectable difference or change. Ann Rheum Dis 2005;64:179–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamamoto K, Takeuchi T, Yamanaka H, et al. Efficacy and safety of certolizumab pegol plus methotrexate in Japanese rheumatoid arthritis patients with an inadequate response to methotrexate: the J‐RAPID randomized, placebo‐controlled trial. Mod Rheumatol 2014;24:715–24. [DOI] [PubMed] [Google Scholar]
- 15. Aletaha D, Bingham CO III, Tanaka Y, et al. Efficacy and safety of sirukumab in patients with active rheumatoid arthritis refractory to anti‐TNF therapy (SIRROUND‐T): a randomised, double‐blind, placebo‐controlled, parallel‐group, multinational, phase 3 study. Lancet 2017;389:1206–17. [DOI] [PubMed] [Google Scholar]
- 16. Tanaka Y, Harigai M, Takeuchi T, et al. Golimumab in combination with methotrexate in Japanese patients with active rheumatoid arthritis: results of the GO‐FORTH study. Ann Rheum Dis 2012;71:817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takahashi C, Kaneko Y, Okano Y, et al. Association of erythrocyte methotrexate‐polyglutamate levels with the efficacy and hepatotoxicity of methotrexate in patients with rheumatoid arthritis: a 76‐week prospective study. RMD Open 2017;3:e000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Porter CJ, Charman SA. Lymphatic transport of proteins after subcutaneous administration. J Pharm Sci 2000;89:297–310. [DOI] [PubMed] [Google Scholar]
- 19. McLennan DN, Porter CJ, Charman SA. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov Today Technol 2005;2:89–96. [DOI] [PubMed] [Google Scholar]
- 20. Tibbitts J, Canter D, Graff R, et al. Key factors influencing ADME properties of therapeutic proteins: a need for ADME characterization in drug discovery and development [review]. MAbs 2016;8:229–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wesolowski J, Alzogaray V, Reyelt J, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol 2009;198:157–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mir MA, Mehraj U, Sheikh BA, et al. Nanobodies: the "magic bullets" in therapeutics, drug delivery and diagnostics. Hum Antibodies 2020;28:29–51. [DOI] [PubMed] [Google Scholar]
- 23. Jovčevska I, Muyldermans S. The therapeutic potential of nanobodies. BioDrugs 2020;34:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bao G, Tang M, Zhao J, et al. Nanobody: a promising toolkit for molecular imaging and disease therapy. EJNMMI Res 2021;11:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levick JR. Permeability of rheumatoid and normal human synovium to specific plasma proteins. Arthritis Rheum 1981;24:1550–60. [DOI] [PubMed] [Google Scholar]
- 26. Wunder A, Muller‐Ladner U, Stelzer EH, et al. Albumin‐based drug delivery as novel therapeutic approach for rheumatoid arthritis. J Immunol 2003;170:4793–801. [DOI] [PubMed] [Google Scholar]
- 27. Larsen MT, Kuhlmann M, Hvam ML, et al. Albumin‐based drug delivery: harnessing nature to cure disease. Mol Cell Ther 2016;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rahman MU, Buchanan J, Doyle MK, et al. Changes in patient characteristics in anti‐tumour necrosis factor clinical trials for rheumatoid arthritis: results of an analysis of the literature over the past 16 years. Ann Rheum Dis 2011;70:1631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van der Heijde D, Landewé R. Should radiographic progression still be used as outcome in RA? Clin Immunol 2018;186:79–81. [DOI] [PubMed] [Google Scholar]
- 30. Matsubara T, Inoue H, Nakajima T, et al. Abatacept in combination with methotrexate in Japanese biologic‐naive patients with active rheumatoid arthritis: a randomised placebo‐controlled phase IV study. RMD Open 2018;4:e000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takeuchi T, Yamanaka H, Ishiguro N, et al. Adalimumab, a human anti‐TNF monoclonal antibody, outcome study for the prevention of joint damage in Japanese patients with early rheumatoid arthritis: the HOPEFUL 1 study. Ann Rheum Dis 2014;73:536–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takeuchi T, Tatsuki Y, Nogami Y, et al. Postmarketing surveillance of the safety profile of infliximab in 5000 Japanese patients with rheumatoid arthritis. Ann Rheum Dis 2008;67:189–94. [DOI] [PubMed] [Google Scholar]
- 33. Koike T, Harigai M, Inokuma S, et al. Postmarketing surveillance of safety and effectiveness of etanercept in Japanese patients with rheumatoid arthritis. Mod Rheumatol 2011;21:343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koike T, Harigai M, Ishiguro N, et al. Safety and effectiveness of adalimumab in Japanese rheumatoid arthritis patients: postmarketing surveillance report of 7740 patients. Mod Rheumatol 2014;24:390–8. [DOI] [PubMed] [Google Scholar]
- 35. Kanbori M, Suzuka H, Yajima T, et al. Postmarketing surveillance evaluating the safety and effectiveness of golimumab in Japanese patients with rheumatoid arthritis. Mod Rheumatol 2018;28:66–75. [DOI] [PubMed] [Google Scholar]
- 36. Takeuchi T, Kameda H. The Japanese experience with biologic therapies for rheumatoid arthritis [review]. Nat Rev Rheumatol 2010;6:644–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1 Supplementary Information
Supplementary Table 1 ACR20 response rates at Week 16
Supplementary Table 2. Summary of clinical efficacy at Week 24
Supplementary Table 3. Subpopulation analysis of ACR20 response rate at Week 16, based on the previous use of antirheumatic biologics
Supplementary Figure 1 Changes from baseline over time
Change from baseline over time in A) DAS28‐ESR, B) ACR‐N, C) TJC68, D) SJC66, E) CDAI, F) SDAI, G) Ph‐GA, H) morning stiffness, I) MMP‐3, J) IL‐6, K) ACPA and L) RF. Observed data for weeks 0–24. Statistical comparisons were conducted according to analysis of covariance using the status of prior TNF inhibitor usage and baseline values as covariates. The data are presented as the mean ± standard error. *p < 0.05, **p < 0.01, ***p < 0.001
Abbreviations: ESR, erythrocyte sedimentation rate; DAS28‐ESR, disease activity score in 28 joints based on ESR; ACR‐N, American College of Rheumatology N; TJC68, tender joint count of 68 joints; SJC66, swollen joint count of 66 joints; CDAI, Clinical Disease Activity Index; SDAI, Simplified Disease Activity Index; Ph‐GA, physician's global assessment of disease activity; MMP‐3, matrix metalloproteinase 3; IL‐6, interleukin 6; ACPA, anti‐cyclic citrullinated peptide antibody; RF, rheumatoid factor