

Abstract
Myostatin, a negative regulator of skeletal muscle growth, is a therapeutic target in muscle‐wasting diseases. Domagrozumab, a humanized recombinant monoclonal antibody, binds myostatin and inhibits activity. Domagrozumab was investigated in a phase II trial (NCT02310763) as a potential treatment for boys with Duchenne muscular dystrophy (DMD). Pharmacokinetic/pharmacodynamic (PK/PD) modeling is vital in clinical trial design, particularly for determining dosing regimens in pediatric populations. This analysis sought to establish the PK/PD relationship between free domagrozumab and total myostatin concentrations in pediatric patients with DMD using a prior semimechanistic model developed from a phase I study in healthy adult volunteers (NCT01616277) and following inclusion of phase II data. The refined model was developed using a multiple‐step approach comprising structural, random effects, and covariate model development; assessment of model adequacy (goodness‐of‐fit); and predictive performance. Differences in PKs/PDs between healthy adult volunteers and pediatric patients with DMD were quantitatively accounted for and evaluated by predicting myostatin coverage (the percentage of myostatin bound by domagrozumab). The final model parameter estimates and semimechanistic target‐mediated drug disposition structure sufficiently described both domagrozumab and myostatin concentrations in pediatric patients with DMD, and most population parameters were comparable with the prior model (in healthy adult volunteers). Predicted myostatin coverage for phase II patients with DMD was consistently > 90%. Baseline serum myostatin was ~ 65% lower than in healthy adult volunteers. This study provides insights into the regulation of myostatin in healthy adults and pediatric patients with DMD. Clinicaltrials.gov identifiers: NCT01616277 and NCT02310763.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Myostatin, a negative regulator of skeletal muscle growth, is a therapeutic target in dystrophic muscle diseases. Domagrozumab binds myostatin with inhibitory activity and was investigated as a potential treatment for Duchene muscular dystrophy (DMD).
WHAT QUESTION DID THIS STUDY ADDRESS?
The pharmacokinetics and pharmacodynamics (PKs/PDs) of domagrozumab in pediatric patients with DMD are described and compared with a prior model developed from healthy adult volunteers.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
In individuals with DMD, baseline serum myostatin was ~ 65% lower than in healthy adult volunteers. Myostatin coverage was consistently > 90% in individuals with DMD after domagrozumab treatment. The final PK/PD model was generally consistent with the prior model, demonstrating translation and evolution from early PK/PD models.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Sequestering systemic myostatin did not translate to clinical efficacy in DMD, and modulating myostatin a site of action (muscle) or other therapeutic targets should be investigated.
Duchenne muscular dystrophy (DMD) is a recessive X‐linked disease that affects approximately one in 5,000 newborn boys. DMD is caused by mutations in the DMD gene, resulting in the absence, or substantial reduction, of the protein dystrophin. Dystrophin is a rod‐shaped cytoskeletal protein that forms part of the dystrophin‐associated protein complex, which is critical for the plasma membrane stability of muscle cells. 1 DMD is characterized by skeletal and cardiac muscle degeneration. 2 Onset of symptoms usually occurs in early childhood followed by the loss of ambulation by 10–12 years of age and premature death at 20–40 years due to progressive respiratory muscle and cardiac failure. 3 , 4
Myostatin (growth differentiation factor‐8 (GDF‐8) is a negative regulator of skeletal muscle growth. 5 Mutations in the GDF‐8 gene have been associated with increased muscle mass in both animals and humans. 6 , 7 In mdx mouse DMD models, loss or inhibition of myostatin leads to increased muscle mass and strength, and decreased fibrosis and fat substitution. 8 Inhibition of myostatin has therefore been investigated as a therapeutic target in muscle‐wasting diseases, such as DMD.
Domagrozumab, a humanized recombinant immunoglobulin G1 monoclonal antibody that binds myostatin with inhibitory activity, has been investigated in a phase II trial as a potential treatment for ambulatory boys with DMD (Study B5161002; ClinicalTrials.gov: NCT02310763). 9 In clinical development, pharmacokinetic/pharmacodynamic (PK/PD) modeling has a critical role in the identification of relevant doses for early phase trials, bridging dosing between adult and pediatric patient populations, and uncovering sources of variability in drug exposure or observed outcomes. 10
A previous population PK/PD model semimechanistically quantified the exposure‐response relationship between domagrozumab and myostatin in healthy adult volunteers (described by Bhattacharya et al. and Tiwari et al.). 11 , 12 This model was used to predict an appropriate domagrozumab dose in a target population of pediatric patients with DMD, thereby providing maximal myostatin coverage. Coverage was defined as domagrozumab bound to myostatin relative to free myostatin in the peripheral circulation, and is hypothesized to represent the ability of domagrozumab to sequester myostatin from its site of action in the muscles. Domagrozumab administered in the following sequence gave a predicted myostatin coverage in the pediatric patients with DMD population > 90% within the first 4 weeks of dosing: 5 mg/kg every 4 weeks for 16 weeks; followed by 20 mg/kg every 4 weeks for 16 weeks, and 40 mg/kg every 4 weeks for 16 weeks. 12
The prior population PK/PD model served as a platform for learning about the relationship between domagrozumab and myostatin in healthy adult volunteers and was used to confirm that target domagrozumab exposures, and the extent of myostatin coverage, was achieved upon completion of the phase II study in pediatric patients with DMD. The present analysis aimed to establish the relationship between free domagrozumab and total myostatin concentrations in pediatric patients with DMD using the prior semimechanistic model (developed from a domagrozumab phase I study in healthy adult volunteers; Study B5161001; ClinicalTrials.gov: NCT01616277) 13 following the inclusion of data from the double‐blind, randomized phase II study in patients with DMD (Study B5161002). Differences in PKs/PDs between healthy adult volunteers and pediatric patients with DMD were quantitatively accounted for in the present iteration of the semimechanistic model, and the impact of these differences was evaluated by predicting myostatin coverage in the respective populations. 11 , 13
METHODS
Study design
This analysis included data from both the phase I and phase II domagrozumab studies. The complete details of the phase I study in healthy adult volunteers have been previously described. 9 , 13 The phase I study was a randomized, dose‐escalating, parallel group, double‐blind, placebo‐controlled study to evaluate the safety, tolerability, and PKs/PDs of domagrozumab in healthy adult volunteers. Participants received single ascending intravenous (i.v.) doses (n = 8 per cohort, 6 active and 2 placebo) of 1 mg/kg, 3 mg/kg, 10 mg/kg, 20 mg/kg, or 40 mg/kg infused over 2 hours. There was also a single subcutaneous (s.c.) dose cohort of 3 mg/kg (n = 8) and a repeat dose cohort where subjects (n = 17, 11 active and 6 placebo) received 10 mg/kg over 2 hours every 2 weeks for a total of 3 doses.
The phase II study was a randomized, double‐blind, placebo‐controlled, two‐period (48 weeks each), multiple ascending dose study to evaluate the safety, efficacy, PKs, and PDs of domagrozumab in ambulatory boys with DMD. In period 1, patients with DMD were administered domagrozumab or placebo in a sequential dose escalation (5 mg/kg, 20 mg/kg, and 40 mg/kg), with each dose level administered every 4 weeks for a total duration of 16 weeks. In period 2, patients who received domagrozumab in period 1 continued to receive the highest tolerated domagrozumab dose or placebo. Patients who received placebo in period 1 received the same domagrozumab treatment schedule as the active patients in period 1 (3 consecutive dose levels, each level every 4 weeks for 16 weeks, for a total of 12 doses). Serum samples to evaluate domagrozumab and myostatin concentrations were obtained predose and 2 and 6 hours postdose. In both studies, serum concentrations of free domagrozumab were assayed using a validated enzyme‐linked immunosorbent assay (lower limit of quantification (LLOQ) was 0.2 and 2.67 nM for phase I and phase II studies, respectively). Total myostatin serum concentrations were assayed using a validated immunoprecipitation liquid chromatography tandem mass spectroscopy assay (LLOQ was 0.04 and 0.008 nM for phase I and phase II studies, respectively).
Both studies were conducted in accordance with legal and regulatory requirements, as well as the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Subjects, guidelines for Good Clinical Practice, and the Declaration of Helsinki. The protocols, amendments, and informed consent/assent documents were approved by the institutional review board or ethics committee at each study center. Each adult subject and parent/legal guardian in the case of pediatric patients with DMD provided written, informed consent.
Development of a population PK/PD model in pediatric patients with DMD
The prior population PK/PD analysis of adult healthy volunteers modeled free domagrozumab and total myostatin concentrations simultaneously using a quasi‐approximation of the steady‐state target‐mediated drug disposition (TMDD) model. 11 The domagrozumab PKs was best described by a two‐compartment model with elimination described by both linear (first‐order) and nonlinear (Michaelis–Menten) mechanisms. 12 In the phase I study, domagrozumab concentrations were in excess compared with myostatin concentrations; therefore, it was considered that binding of domagrozumab to myostatin would not impact free domagrozumab concentrations. Free domagrozumab concentrations and total myostatin concentrations were modeled in natural log or linear domains, respectively. Domagrozumab dose amounts and rates were input as nM/kg and nM/kg/hour, respectively, inherently linearly scaling PK parameters by total body weight. Allometric scaling approaches were also previously examined with four‐species data for scaling clearance (CL) and volume of distribution (V). 11 Baseline body mass index (BMI) was a significant covariate with a quantitative effect on the volume of the central compartment. Final parameter estimates of the prior TMDD model and a description of the approximations examined to determine final parameters are described in the studies by Bhattacharya et al. 11 and Tiwari et al. 12
The objective of the population PK/PD modeling analysis described herein was to confirm the structural parameterization of the prior TMDD model's ability to describe newly available data from pediatric patients with DMD, and re‐evaluate the random effects and covariate models (where appropriate). NONMEM version VII Level 3.0 (ICON Development Solutions, Ellicott City, MD) with the ADVAN13 subroutine (TOL = 9) was used for model development.
Population parameter estimation used the first‐order conditional estimation method with interaction and individual parameters obtained from empirical Bayes estimates (EBE).
Structural equations for the prior TMDD model are shown in Eqs. (1), (2), (3), (4), (5), (6):
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
DEPOT is the concentration of domagrozumab at the injection site when administered s.c. (nM). CENT is the concentration of domagrozumab in the central compartment when administered i.v. (nM). PERI is the concentration of domagrozumab in the peripheral compartment (nM). Myo represents total myostatin (free and drug‐myostatin complex, nM). CL is the clearance of domagrozumab (L/hour/kg). V1 is the volume of distribution of the central compartment (L/kg), V2 is the volume of distribution of the peripheral compartment (L/kg). Q is the intercompartmental clearance between the central and peripheral compartments (L/hour/kg). InputSC is the amount of domagrozumab administered to the injection site for s.c. administration (nM/kg). InputIV is the rate of drug administered to the central compartment when given as a 2‐hour infusion (nM/kg/hour), ka is the first‐order absorption rate constant for the movement of domagrozumab from DEPOT to CENT following s.c. administration (hour−1).
Vmax is the maximum elimination rate of domagrozumab via nonlinear pathways (nM/kg/hour), km is the Michaelis–Menten constant (concentration of domagrozumab when 50% of Vmax is achieved, nM), BASE is baseline concentration of myostatin (nM), kdeg is the first‐order rate constant for degradation (elimination) of myostatin (hour−1), ksyn is the zero‐order rate constant for synthesis of myostatin (nM/hour), kint is the first‐order internalization rate constant of the drug‐myostatin complex (hour−1), kSS is the half‐saturation/steady‐state constant for the binding of drug to myostatin (nM) and is assumed to be at equilibrium (where kint is considered to be slower than the dissociation rate for the binding of domagrozumab to myostatin).
Interindividual variability (IIV) was assumed to be log‐normally distributed for both PK and PD parameters. The suitability of additional random effect parameters, and models with and without covariance for random effects using an off‐diagonal variance–covariance matrix, were also re‐investigated. Random unexplained variability for domagrozumab concentrations was described using an additive error model in the natural log domain, and a proportional error model in the linear domain was used for total myostatin concentrations.
Covariate analyses quantified the effects of key characteristics to improve the predictive performance of the model. Baseline patient characteristics investigated for covariate relationships included age, study population, albumin, and race. Weight‐based metrics, such as total body weight and lean body weight, were not considered for covariate testing on PK parameters as these parameters were inherently scaled by weight in the model. However, the effect of baseline BMI on the volume of the central compartment, included in the prior PK/PD model, was re‐evaluated following incorporation of the phase II population. Allometric scaling was only revisited as a comparison with the final model with domagrozumab dose input in nM, and exponents of 0.75 for CL, Q, and Vmax, and 1 for V1 and V2, with reference to a 70 kg individual. Sex and presence of anti‐drug antibodies were not tested for covariate relationships as the entire phase II population was male gender and all anti‐drug antibody samples were negative or below the limit of quantification.
Potential covariate relationships were initially inspected graphically by plotting EBEs against the range of covariate values for the population. Covariates with clear trends were carried forward into covariate model development. The effect of a categorical covariate on a parameter was represented as a discrete relationship. For example, the effect of study population (SPOP) on a parameter P, was described as:
| (7) |
| (8) |
where θSPOPP is the estimable parameter for the effect of pediatric patients with DMD on P.
The effect of a continuous covariate on a parameter was represented as a power model referenced to the median of the observed data. For example, the effect of age on P was described as:
| (9) |
where AGE i is the age (years) in the ith subject, AGEref is the median age in the observed population, and AGE is the parameter for the effect of age on P.
Assessment of model fit was performed using standard goodness‐of‐fit diagnostic plots and the Akaike information criterion (AIC) to compare non‐nested models and candidate models with lower AIC values. The square root of the ratio of the highest and lowest eigenvalues for a model (condition number) was also used to compare models, where models with condition numbers < 100 were considered. 14 Covariate effect relationships were retained if the addition of an estimable parameter reduced the objective function value (OFV) by > 10.8 units (P < 0.001 according to the likelihood ratio test) and reduced IIV of the targeted parameter, and in conjunction with a relative standard error (i.e., 95% confidence interval of parameter estimates does not include 0).
The predictive performance of the final model was evaluated by a visual predictive check (VPC) based on 1000 simulations of the analysis dataset.
Simulation of myostatin coverage
The final parameter estimates of the final model were used to simulate free domagrozumab concentrations, total myostatin concentrations, and expected myostatin coverage (Eq. 10) for 500 pediatric patients with DMD following the administration of (1) 5 mg/kg every 4 weeks for 16 weeks, followed by 20 mg/kg every 4 weeks for 16 weeks, followed by 40 mg/kg every 4 weeks for 16 weeks, and (2) 0, 1, 2, 3, 5, 7, 10, 20, 30, 40, 60, and 80 mg/kg every 4 weeks for 48 weeks (to construct a dose‐myostatin coverage curve).
Domagrozumab, total myostatin concentrations, and myostatin coverage were calculated predose, and 2 and 6 hours postdose. The median and 95% prediction intervals of myostatin coverage were calculated for the population according to Eq. 10 and were summarized at each sampling time.
| (10) |
The proportion of total myostatin at time (t) inhibited by domagrozumab as a function of the model predicted free myostatin at time (t) and baseline myostatin (assumed to be 100% unbound prior to first administration of domagrozumab), where BMyo is the baseline myostatin (nM), and all other parameters are as previously described in Eqs. (1), (2), (3), (4), (5), (6).
RESULTS
Demographics of healthy adult volunteers and pediatric patients with DMD
The analysis dataset consisted of 193 individuals who contributed 5181 evaluable free domagrozumab and 8001 evaluable total myostatin concentrations. There were 5.79% of free domagrozumab and 0.075% total myostatin concentrations below the LLOQ. Demographic and baseline characteristics for adult healthy volunteers and pediatric patients with DMD are summarized in Table 1 . There was an expected difference in median age (range); 37.0 (19.0–61.0) years in healthy adult volunteers and 9.0 (6.0–15.0) years in patients with DMD. Baseline BMI also differed, with a median baseline BMI (range) of 25.1 kg/m2 (19.0–30.1) and 19.2 kg/m2 (11.7–39.7) in adult healthy volunteers and pediatric patients with DMD, respectively.
Table 1.
Analysis population baseline characteristics by patient type
| Phase I healthy adult volunteers | Phase II pediatric patients with DMD | Total | |
|---|---|---|---|
| Age, years | |||
| Median | 37 | 9 | 10 |
| Minimum | 19 | 6 | 6 |
| Maximum | 61 | 15 | 61 |
| Mean | 37.7 | 8.72 | 19.7 |
| SD | 11.2 | 1.96 | 15.7 |
| n Available | 73 | 120 | 193 |
| n Missing | 0 | 0 | 0 |
| BWT, kg | |||
| Median | 76.9 | 28.9 | 39.2 |
| Minimum | 56.8 | 14.8 | 14.8 |
| Maximum | 99.8 | 86.4 | 99.8 |
| Mean | 76.9 | 31.8 | 48.9 |
| SD | 11.9 | 11.1 | 24.7 |
| n Available | 73 | 120 | 193 |
| n Missing | 0 | 0 | 0 |
| LBW, kga | |||
| Median | 58.5 | 24.9 | 31.7 |
| Minimum | 42 | 14.1 | 14.1 |
| Maximum | 73 | 55.5 | 73 |
| Mean | 59.1 | 26.3 | 38.7 |
| SD | 7.5 | 6.77 | 17.4 |
| n Available | 73 | 120 | 193 |
| n Missing | 0 | 0 | 0 |
| Body mass index, kg/m2 | |||
| Median | 25.1 | 19.2 | 21.5 |
| Minimum | 19 | 11.7 | 11.7 |
| Maximum | 30.1 | 39.7 | 39.7 |
| Mean | 25 | 19.9 | 21.8 |
| SD | 2.96 | 4.6 | 4.75 |
| n Available | 73 | 120 | 193 |
| n Missing | 0 | 0 | 0 |
| Albumin, g/dL | |||
| Median | 4.7 | 4.4 | 4.5 |
| Minimum | 4.2 | 3.7 | 3.7 |
| Maximum | 5.4 | 5 | 5.4 |
| Mean | 4.68 | 4.36 | 4.48 |
| SD | 0.252 | 0.279 | 0.311 |
| n Available | 73 | 120 | 193 |
| n Missing | 0 | 0 | 0 |
| Total myostatin, nM | |||
| Median | 0.134 | 0.0496 | 0.072 |
| Minimum | 0.06 | 0.025 | 0.025 |
| Maximum | 0.296 | 0.151 | 0.296 |
| Mean | 0.14 | 0.0548 | 0.09 |
| SD | 0.0468 | 0.0216 | 0.0543 |
| n Available | 73 | 104 | 177 |
| n Missingb | 0 | 16 | 16 |
| Treatment Group, n (%) | |||
| 1 mg/kg Single i.v. | 6 (8.2) | 0 | 6 (3.1) |
| 10 mg/kg Multiple i.v. | 11 (15.1) | 0 | 11 (5.7) |
| 10 mg/kg Single i.v. | 12 (16.4) | 0 | 12 (6.2) |
| 20 mg/kg Single i.v. | 6 (8.2) | 0 | 6 (3.1) |
| 3 mg/kg Single i.v. | 6 (8.2) | 0 | 6 (3.1) |
| 3 mg/kg Single s.c. | 6 (8.2) | 0 | 6 (3.1) |
| 40 mg/kg Single i.v. | 6 (8.2) | 0 | 6 (3.1) |
| Placebo | 20 (27.4) | 0 | 20 (10.4) |
| Sequence 1c | 0 | 41 (34.2) | 41 (21.2) |
| Sequence 2d | 0 | 39 (32.5) | 39 (20.2) |
| Sequence 3e | 0 | 40 (33.3) | 40 (20.7) |
| Sex, n (%) | |||
| Female | 7 (9.6) | 0 | 7 (3.6) |
| Male | 66 (90.4) | 120 (100) | 186 (96.4) |
| Race, n (%) | |||
| Asian | 3 (4.1) | 15 (12.5) | 18 (9.3) |
| Black | 22 (30.1) | 2 (1.7) | 24 (12.4) |
| White | 31 (42.5) | 101 (84.2) | 132 (68.4) |
| Other | 17 (23.3) | 2 (1.7) | 19 (9.8) |
BWT, total body weight; BHT, height; DMD, Duchenne muscular dystrophy; LBW, lean body weight.
Lean body weight was calculated using the James formula; Men, LBW = (1.10∙BWT)‐(128∙BWT2/BHT2); Women, LBW = (1.07∙BWT)‐(148∙BWT2/BHT2).
Missing baseline myostatin values were below the lower limit of quantification of 0.008 nM.
Sequence 1 = 5 mg/kg every 4 weeks for 16 weeks, then 20 mg/kg every 4 weeks for 16 weeks, then 40 mg/kg every 4 weeks for 16 weeks, then 40 mg/kg every 4 weeks.
Sequence 2 = 5 mg/kg every 4 weeks for 16 weeks, then 20 mg/kg every 4 weeks for 16 weeks, then 40 mg/kg every 4 weeks for 16 weeks, then placebo.
Sequence 3 = Placebo for 48 weeks then 5 mg/kg every 4 weeks for 16 weeks, then 20 mg/kg every 4 weeks for 16 weeks, then 40 mg/kg every 4 weeks for 16 weeks.
Final TMDD model
The structural model of the prior TMDD model with updated parameter estimates with the addition of pediatric patients with DMD adequately predicted free domagrozumab and total myostatin concentrations as depicted by goodness‐of‐fit diagnostics (Figures S1 , S2 ).
Model diagnostics of the final structural model demonstrated a distinct bimodal distribution for the EBEs of the BASE parameter (Figure 1 a ), because of differences between healthy adult volunteers and pediatric patients with DMD. The addition of a study population effect on the BASE parameter demonstrated statistical significance (P value < 0.001, 242 unit decrease in OFV) and normalized the distribution of individual random effect parameters (Figure 1 b ). The addition of study population to BASE, resulted in the emergence of bimodal distributions for the EBEs of the random effect parameter for kdeg and kint. Subsequently, the effect of study population was tested on both kdeg and kint as a single fixed effect parameter (Figure 1 ), and demonstrated statistical significance (P value < 0.001, 508 unit decrease in OFV). Based on graphical inspection of goodness‐of‐fit diagnostics, other covariate effects, such as age and body weight, were not tested on population PD parameters to quantify differences between healthy adult volunteers and pediatric patients with DMD (Figure S3 ).
Figure 1.
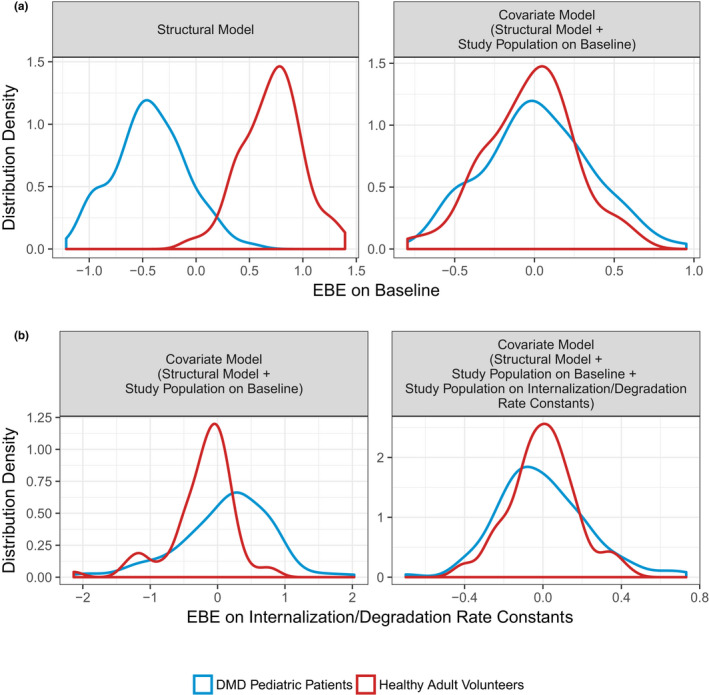
Distribution density of empirical Bayes estimates (EBE) for baseline and internalization/degradation rate constants parameters. (a) Distribution density for EBEs on baseline. Red and blue lines are the distribution densities of individual random effects for baseline for healthy adult volunteers and pediatric patients with DMD, respectively. (b) Distribution density EBE on internalization/degradation rate constants. Red and blue lines are the distribution densities of individual random effects on internalization/degradation rate constants (kdeg − kint) for healthy adult volunteers and pediatric patients with DMD, respectively. DMD, Duchenne muscular dystrophy.
The effect of baseline BMI on V1 was present in the prior TMDD model and was re‐evaluated in the model after the incorporation of the study population covariate effects. The addition of BMI to V1 in the model was not statistically significant and did not improve the model fit from a graphical perspective. Therefore, the effect of baseline BMI on V1 was not retained in the model.
The final parameter estimates for the final TMDD model and a model with allometric scaling are presented in Table 2 . An evaluation of EBEs on CL vs. age and total body weight for the final model and a model with allometric scaling is presented in Figure 2 . Both models adequately account for differences in CL owing to body weight between healthy adult volunteers and pediatric patients with DMD.
Table 2.
Final parameter estimates of TMDD models (with and without allometric scaling)
| Parameter | Final model estimate (95% CIa) | Model with allometric scalingb estimate (95% CI) | |
|---|---|---|---|
| Objective function value | −55597.41 | −55939.59 | |
| Condition numberc | 14.8 | 17.4 | |
| Population parameter | |||
| Clearance (CL) | 0.0000982 (0.0000935, 0.000103) L/hour/kg | 0.00644 (0.00618, 0.00670) L/hour | |
| Volume of the central compartment (V1) | 0.0415 (0.0395, 0.0436) L/kg | 3.10 (2.95, 3.24) L | |
| Inter‐compartmental clearance (Q) | 0.000306 (0.000283, 0.000329) L/hour/kg | 0.0219 (0.0202, 0.0237) L/hour | |
| Volume of the peripheral compartment (V2) | 0.0416 (0.0405, 0.0426) L/kg | 2.85 (2.77, 2.93) L | |
| First‐order absorption rate constant (ka) | 0.00769 (0.00544, 0.00994) hour−1 | 0.00820 (0.00521, 0.0112) hour−1 | |
| Relative bioavailability for s.c. administration (F) | 0.858 (0.363, 1.35) | 0.822 (0.267, 1.38) | |
| Michaelis–Menten constant (km) | 12.2 (10.9, 13.4) nM | 12.3 (11.1, 13.4) nM | |
| Maximum nonlinear elimination rate (Vmax) | 0.00251 (0.00191, 0.00311) nM/hour/kg | 0.111 (0.0769, 0.145) nM/hour | |
| First‐order degradation rate constant (kdeg) | 0.0381 (0.0357, 0.0404) hour−1 | 0.0385 (0.0360, 0.041) hour−1 | |
| Baseline total myostatin (BASE) | 0.156 (0.141, 0.171) nM | 0.156 (0.141, 0.171) nM | |
| Steady‐state binding constant (kSS) | 7.76 (7.37, 8.14) nM | 7.19 (6.82, 7.55) nM | |
| First‐order internalization rate constant (kint) | 0.00716 (0.0068, 0.00751) hour−1 | 0.00730 (0.00693, 0.00767) hour−1 | |
| Ratio of SD for ηkint relative to ηkdeg | −0.295 (−0.383, −0.208) | −0.182 (−0.254, −0.110) | |
| Effect of DMD pediatric patients on BASE | −0.641 (−0.685, −0.598) | −0.640 (−0.680, −0.600) | |
| Effect of DMD pediatric patients on kdeg and kint | −0.900 (−0.905, −0.895) | −0.904 (−0.909, −0.899) | |
| Population parameter variability | |||
| ωCL (% CV) | 24.3 (20.8, 27.4); 7.77% shrinkage | 21.0 (18.0, 23.6); 8.24% shrinkage | |
| ωV1 (% CV) | 23.4 (20.3, 26.0); 7.55% shrinkage | 22.1 (19.0, 24.8); 7.73% shrinkage | |
| (% CV) | 104 (78.6, 124); 20.7% shrinkage | 119 (84.9, 145); 24.3% shrinkage | |
| ωBASE (% CV) | 31.8 (27.5, 35.7); 2.23% shrinkage | 32.2 (28.0, 35.9); 2.28% shrinkage | |
| (% CV) | 23.3 (19.8, 26.4); 10.5% shrinkage | 25.7 (21.8, 29.1); 10.3% shrinkage | |
| Covariance | |||
| ρCL‐V1 | 0.0363 (0.0243, 0.0482) | 0.0247 (0.0162, 0.0332) | |
|
|
−0.0122 (−0.0726, 0.0481) | 0.00383 (−0.0604, 0.0680) | |
|
|
0.00352 (−0.0643, 0.0713) | −0.00215 (−0.0715, 0.0672) | |
| ρCL‐BASE | 0.0048 (−0.0105, 0.0201) | 0.000591 (−0.0118, 0.0129) | |
| ρV1‐BASE | −0.00284 (−0.0187, 0.0131) | −0.00392 (−0.0175, 0.00963) | |
|
|
−0.0265 (−0.0924, 0.0394) | −0.0515 (−0.135, 0.0316) | |
|
|
−0.024 (−0.035, −0.013) | −0.0178 (−0.0277, −0.00787) | |
|
|
−0.018 (−0.0302, −0.00577) | −0.0172 (−0.0302, −0.00415) | |
|
|
−0.00303 (−0.0642, 0.0581) | 0.000371 (−0.0758, 0.0832) | |
|
|
−0.0216 (−0.0368, −0.00646) | −0.0256 (−0.0423, −0.00889) | |
| Random unexplained variability | |||
| σadd (SD) | 0.142 (0.141, 0.143) | 0.137 (0.136, 0.138) | |
| σpro (% CV) | 20.6 (20.5, 20.8) | 20.7 (20.5, 20.9) | |
CI, confidence interval; CV, coefficient of variation; DMD, Duchenne muscular dystrophy; TMDD, target‐mediated drug disposition.
95% CI are calculated based on the standard error obtained from the variance–covariance matrix of parameter estimates assuming a normal distribution.
Allometric scaling for body weight was applied to the following pharmacokinetic parameters referenced to a 70 kg individual: CL (exponent 0.75), V1 (exponent 1), Q (exponent 0.75), V2 (exponent = 1), Vmax (exponent = 0.75).
Condition number is calculated as the square root of ratio of largest to smallest eigenvalues of the correlation matrix.
Figure 2.
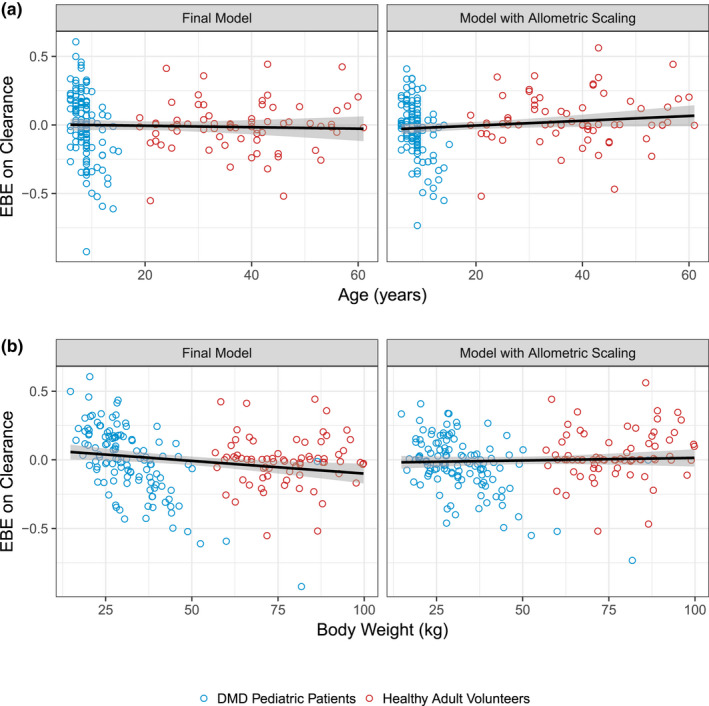
Evaluation of empirical Bayes estimates (EBEs) on domagrozumab clearance vs (a) age (years) and (b) total body weight (kg) for the final model and model with allometric scaling. Circles are the EBEs for clearance vs. age (a) and body weight (b) for the final model (left) and a model with allometric scaling (right). Blue circles represent pediatric patients with DMD from the phase II study and red circles represent the healthy adult volunteers from the phase I study. Black solid lines and shaded areas are the linear regression and 95% confidence interval, respectively, for the relationship of EBEs vs. covariate values. DMD, Duchenne muscular dystrophy.
Final model predictive performance
VPCs of the pediatric patients with DMD for free domagrozumab and total myostatin concentrations are presented in Figures 3 and 4 , respectively. The final model's VPC supports that the final model parameter estimates and the semimechanistic TMDD structure sufficiently describe both domagrozumab and myostatin concentrations in pediatric patients with DMD.
Figure 3.
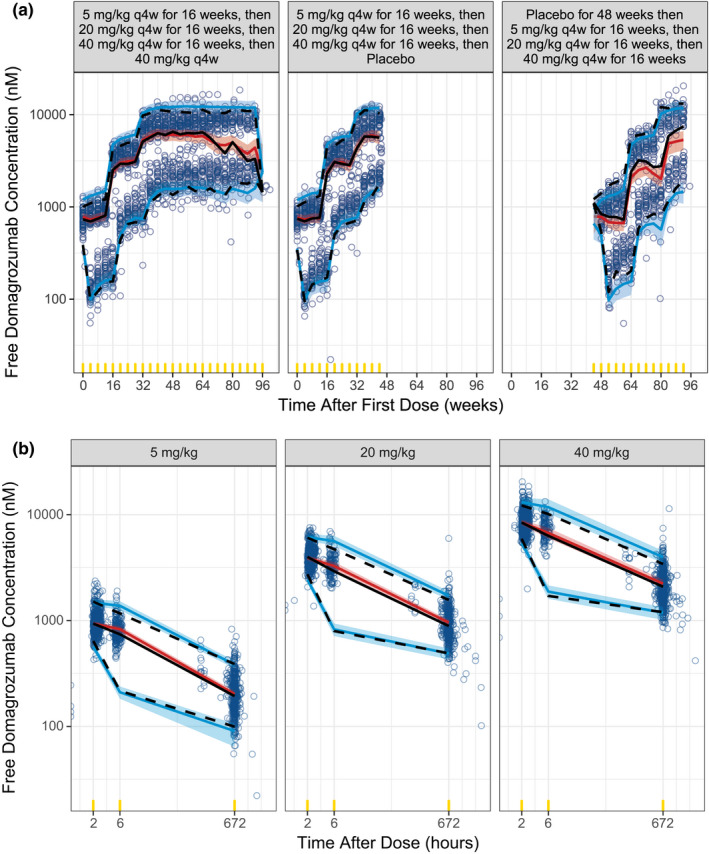
Final TMDD model visual predictive check for free domagrozumab concentrations in pediatric patients with DMD. Visual predictive checks are presented for time after first dose (a) and time after dose (b). Observed data are represented by blue circles and the black lines (median (solid), 5th and 95th percentiles (dashed)). The simulated free domagrozumab concentrations for phase II patients with DMD are represented by the red line and red shaded ribbon (median and 95% prediction intervals (PIs) of the median, respectively), and the blue lines and blue shaded ribbons (median and 95% PIs of the 5th and 95th percentiles, respectively). Yellow indicators are the times where observed and simulated data are binned for summaries. DMD, Duchenne muscular dystrophy; TMDD, target‐mediated drug disposition.
Figure 4.
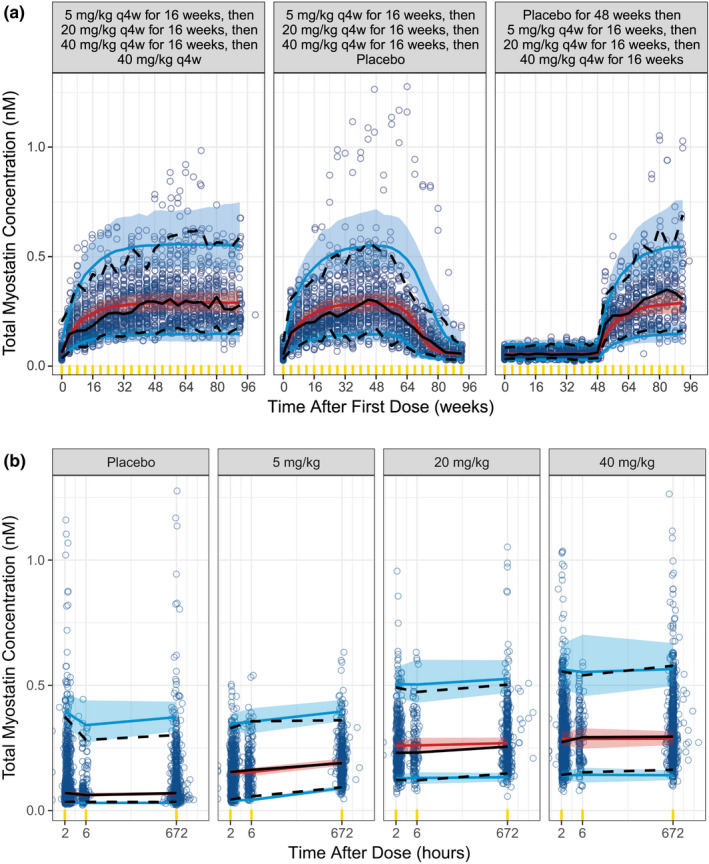
Final TMDD model visual predictive check for total myostatin in pediatric patients with DMD. Visual predictive checks are presented for time after first dose (a) and time after dose (b). Observed data are represented by blue circles and the black lines (median (solid), 5th and 95th percentiles (dashed)). The simulated total myostatin concentrations for phase II patients with DMD are represented by the red line and red shaded ribbon (median and 95% prediction intervals (PIs) of the median, respectively), and the blue lines and blue shaded ribbons (median and 95% PIs of the 5th and 95th percentiles, respectively). Yellow indicators are the times where observed and simulated data are binned for summaries. DMD, Duchenne muscular dystrophy; TMDD, target‐mediated drug disposition.
Simulation of myostatin coverage
Population parameters and IIV estimates for PK/PD parameters were comparable with those of the prior model developed using data from healthy adult volunteers only. 11 Median and 95% prediction intervals (PIs) of simulated myostatin coverage for healthy adult and pediatric patients with DMD, using the final parameter estimates of either the prior or final TMDD models, are depicted in Figure 5 . For the final model, the median (95% PI) for myostatin coverage in pediatric patients with DMD for 5, 20, and 40 mg/kg was 86.9% (69.1, 92.9), 96.6% (93.8, 98.2), and 98.3% (96.8, 99.1), respectively. Additional simulations constructing the dose–response relationship of domagrozumab and myostatin coverage demonstrated that doubling the highest dose of 40 mg/kg to 80 mg/kg resulted in a < 1% increase in myostatin coverage (median coverage for 80 mg/kg was 99.1%).
Figure 5.
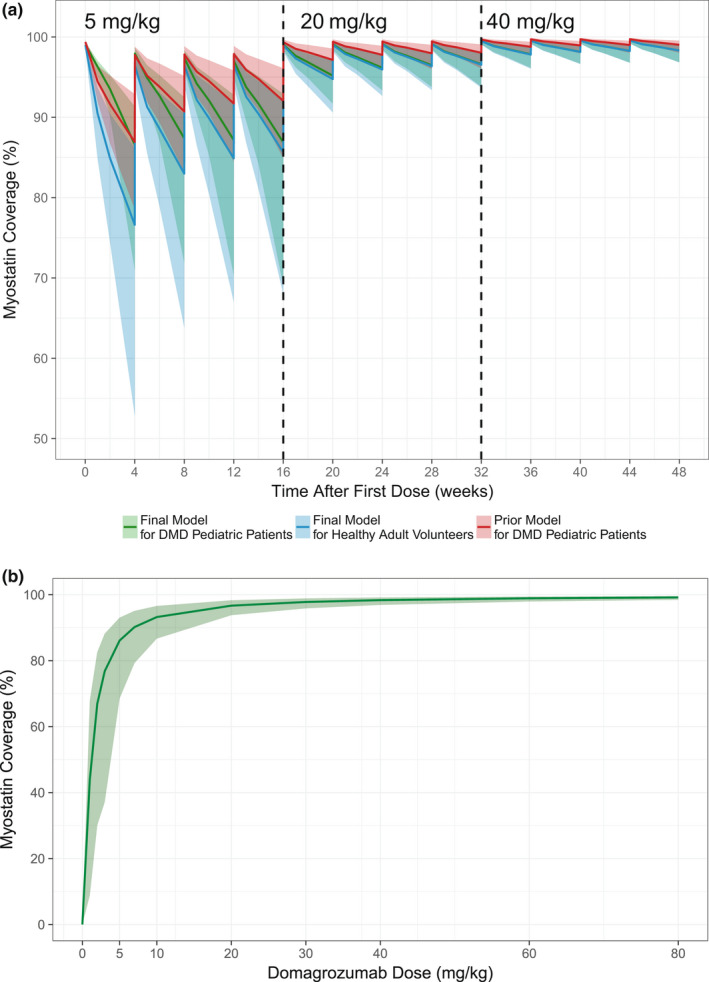
Simulated myostatin coverage. (a) The predicted myostatin coverage following 4 doses of each of 5 mg/kg, 20 mg/kg, and 40 mg/kg every 28 days were simulated using the prior model for pediatric patients with DMD (green), the final model for healthy adult volunteers (blue), and the final model for pediatric patients with DMD (red). Lines represent the median, and ribbons depict the 95% prediction intervals (PIs) for a population of 500 subjects. (b) The predicted myostatin coverage following the administration of 0, 1, 2, 3, 5, 7, 10, 20, 30, 40, 60, and 80 mg/kg of domagrozumab every 4 weeks for 48 weeks using the final model for 500 pediatric patients with DMD. Lines represent the median, and ribbons depict the 95% prediction intervals. DMD, Duchenne muscular dystrophy.
DISCUSSION
The modeling analysis presented is an example of utilizing prior modeling knowledge and the need for informed decision making under an accelerated drug development paradigm for a rare disease such as DMD. The final TMDD model was used to rapidly confirm (1) domagrozumab exposures in the pediatric patients with DMD were as expected based on prior modeling efforts in animals and the same healthy adult volunteers from the phase I study (Figure 3 ), 11 (2) domagrozumab binds to myostatin in pediatric patients with DMD despite downregulation when compared with healthy adult volunteers (Figure 4 ), and (3) expected coverage as per the prior model of the targeted biomarker, myostatin, was achieved (Figure 5 ). The final TMDD model in pediatric patients with DMD is a final chapter in the book of the model‐informed drug development for domagrozumab in targeting myostatin in pediatric patients with DMD.
Despite myostatin coverage being as expected and mostly exceeding 90% in pediatric patients with DMD from the phase II study, domagrozumab failed to meet its primary end point of mean change in 4‐stair climb time at week 49 vs. placebo. 9 The final TMDD model demonstrated a significant role in determining that the observed efficacy results were not associated with less than expected domagrozumab exposure or lack of target engagement in the pediatric patients with DMD.
Several efforts led the timely analysis of domagrozumab and myostatin data obtained from the pediatric patients with DMD upon conclusion of the phase II study, and delivery of interpretations based on its results. These included the extensive structural characterization of free domagrozumab and total myostatin in healthy adult volunteers, 11 , 12 and assessment of the impact of body weight on domagrozumab PK parameters using preclinical data and data from healthy adult volunteers. 11
With the addition of data from pediatric patients with DMD, herein we report that the final TMDD model was generally consistent with the prior developed model. 11 First, the final TMDD model supported the relationship of total body weight and domagrozumab PK parameters assumed by the prior model. For example, Figure 2 depicts EBEs for CL vs. body weight and age (as in a pooled adult and pediatric population, age and body weight are highly correlated) for the final model with linearly scaled PK parameters by body weight and a model with allometric scaling. Both models adequately account for trends in CL with respect to body weight. In context of model parameters for total myostatin, the greatest parameter estimate change was for kss, which increased from 4.32 hour−1 in the prior model 11 to 7.76 hour−1 in the present final model. This change is reflective of the inclusion of pediatric patients with DMD to the overall analysis population. Figure 5 depicts slight differences in the predicted myostatin coverage between the prior model and the final present model as kss is a variable in the calculation of myostatin coverage (as given by Eq. 10).
Total myostatin in pediatric patients with DMD from the phase II study were lower compared with healthy adult volunteers from the phase I study, as might be expected. The lower myostatin concentrations in patients with DMD may be due to altered homeostasis, where the level of circulating myostatin is hypothesized to be a marker of muscle health. 15 Hence, lower levels of myostatin in patients with DMD could be a marker for the loss of functional muscle in these patients. 16 Whereas the DMD population in the phase II study was younger (aged 6–16 years) and with a lower body weight, age‐ and weight‐related differences in the overall population did not explain differences between patients with DMD and healthy adult volunteers in myostatin model parameters. Age and/or body weight could be important explanatory variables for quantifying the differences in myostatin between patients with DMD.
For the purpose of quantifying differences between pediatric patients with DMD and healthy adult volunteers, population status on key myostatin model parameters was used (i.e., parameters as product of synthesis, BASE, and degradation of myostatin, kdeg). Compared with the healthy adult volunteers, patients with DMD were estimated to exhibit a 90% reduction in kdeg, and consequently, ksyn (Table 2 ). Additionally, kint (first‐order internalization rate constant of drug‐myostatin complex) was equally impacted in patients with DMD relative to healthy adult volunteers (Table 2 ). These differences in myostatin homeostasis in the DMD population are most evident at baseline and time to achieve an equilibrium in myostatin coverage (Figure 3 ). Based on the final model, pediatric patients with DMD had, on average, a 64.1% lower baseline myostatin than healthy adult volunteers. This is consistent with previous observations that demonstrated patients with DMD had an ~ 69% lower serum myostatin compared with a < 25‐year‐old healthy control group. 16 Despite differences in myostatin homeostasis, predicted myostatin coverage in pediatric patients with DMD using the final TMDD model confirmed expectations made from the prior model based on healthy adult volunteers.
Median myostatin coverage predicted based on healthy adult volunteer data and the prior model at the completion of each dosing level, 5, 20, and 40 mg/kg, was 92%, 98%, and 99%, respectively. 11 For the final model, the median for myostatin coverage in pediatric patients with DMD for 5, 20, and 40 mg/kg was 86.9%, 96.6%, and 98.3%, respectively. Examination of the dose–response curve for the domagrozumab‐myostatin coverage relationship demonstrated that further increases in dose do not result in substantial increases in myostatin coverage (Figure 5 ). Other clinical trials of myostatin inhibitors, including bimagrumab (inclusion body myositis; ClinicalTrials.gov: NCT01925209), 17 ACE‐083 (Charcot–Marie–Tooth Disease; ClinicalTrials.gov: NCT03124459), and RG6206 (DMD; ClinicalTrials.gov: NCT03039686) have also reported insufficient efficacy in muscle‐wasting diseases to warrant further development. It has been suggested that the lack of improvement in muscle strength or physical function following treatment with myostatin inhibitors in patients with muscle‐wasting diseases may be due to downregulation of myostatin and its pathway. 15 The downregulation of myostatin signaling may be most evident in patients affected by the most muscle atrophying diseases, such as DMD. 15 As skeletal muscle is the primary tissue source of myostatin expression, lower circulating myostatin levels in patients with DMD may also reflect the inherent loss of functional muscle mass with disease progression. 16
Limitations of the final model include the appropriateness of estimating myostatin inhibition by plasma levels, such that true inhibition at the target in muscles is not known. The inhibition of, or binding of domagrozumab to, myostatin in peripheral circulation is expected to sequester the myostatin away from its site of action, thereby less myostatin is available in the muscles to exert its homeostatic effect, as evidenced by downregulation at the mRNA and protein level. 15 Additionally, it is not known if sensitivity to myostatin mediates inhibition of myogenesis in patients with DMD. Nevertheless, the TMDD model described herein provides insights into myostatin coverage in pediatric patients with DMD as compared with healthy adult volunteers. Ensuring that the appropriate dosing rationale and regimen are used in pediatric clinical trials, as well as identifying potential patient subgroups who may be more sensitive to treatment (in terms of efficacy and safety), remains a challenge in pediatric drug development. 18 , 19 It is therefore essential to accurately characterize the underlying PK/PD relationships, especially in children as these are likely to change as they age.
The present analysis quantified variation in parameters defining the disposition of domagrozumab and myostatin, and established confidence in the use of PK/PD modeling in translating dosing of myostatin modulators from healthy adult volunteers to pediatric patients with DMD. The final model confirmed the hypothesis of achieving greater than 90% myostatin coverage in pediatric patients with DMD at the doses evaluated in the phase II study. However, due to lack of efficacy, this mechanism of action may not be suitable for altering the progression of disease in patients with DMD.
FUNDING
The studies were sponsored by Pfizer.
CONFLICTS OF INTEREST
B.T., P.D., and L.O.H. were employees of and hold stock in Pfizer; V.S.P., T.N., and J.W. are employees of and hold stock in Pfizer.
AUTHOR CONTRIBUTIONS
J.W., V.S.P., L.O.H., P.D., B.T., and T.N. wrote the manuscript. J.W. designed the research. J.W., V.S.P., L.O.H., P.D., B.T., and T.N. performed the research. J.W., V.S.P., L.O.H., P.D., B.T., and T.N. analyzed the data.
Supporting information
Data S1
ACKNOWLEDGMENTS
Medical writing support was provided by Charles Cheng, MS, and Leon Adams, PhD, of Engage Scientific Solutions, and funded by Pfizer.
DATA AVAILABILITY STATEMENT
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.
References
- 1. Gao, Q.Q. & McNally, E.M. The dystrophin complex: structure, function, and implications for therapy. Compr. Physiol. 5, 1223–1239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendell, J.R. & Lloyd‐Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 48, 21–26 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Werneck, L.C. , Lorenzoni, P.J. , Ducci, R.D. , Fustes, O.H. , Kay, C.S.K. & Scola, R.H. Duchenne muscular dystrophy: an historical treatment review. Arq. Neuropsiquiatr. 77, 579–589 (2019). [DOI] [PubMed] [Google Scholar]
- 4. Koeks, Z. et al. Clinical outcomes in Duchenne muscular dystrophy: a study of 5345 patients from the TREAT‐NMD DMD global database. J. Neuromuscul. Dis. 4, 293–306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McNally, E.M. Powerful genes‐myostatin regulation of human muscle mass. N. Engl. J. Med. 350, 2642–2644 (2004). [DOI] [PubMed] [Google Scholar]
- 6. Mosher, D.S. et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 3, e79 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schuelke, M. et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 350, 2682–2688 (2004). [DOI] [PubMed] [Google Scholar]
- 8. Bogdanovich, S. et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature 420, 418–421 (2002). [DOI] [PubMed] [Google Scholar]
- 9. Wagner, K.R. et al. Randomized phase 2 trial and open‐label extension of domagrozumab in Duchenne muscular dystrophy. Neuromuscul. Disord. 30, 492–502 (2020). [DOI] [PubMed] [Google Scholar]
- 10. Huang, S.M. , Abernethy, D.R. , Wang, Y. , Zhao, P. & Zineh, I. The utility of modeling and simulation in drug development and regulatory review. J. Pharm. Sci. 102, 2912–2923 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Bhattacharya, I. , Manukyan, Z. , Chan, P. , Heatherington, A. & Harnisch, L. Application of quantitative pharmacology approaches in bridging pharmacokinetics and pharmacodynamics of domagrozumab from adult healthy subjects to pediatric patients with Duchenne muscular disease. J. Clin. Pharmacol. 58, 314–326 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Tiwari, A. , Bhattacharya, I. , Chan, P.L.S. & Harnisch, L. Comparing model performance in characterizing the PK/PD of the anti‐myostatin antibody domagrozumab. Clin. Transl. Sci. 13, 125–136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhattacharya, I. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of domagrozumab (PF‐06252616), an antimyostatin monoclonal antibody, in healthy subjects. Clin. Pharmacol. Drug Dev. 7, 484–497 (2018). [DOI] [PubMed] [Google Scholar]
- 14. Mould, D.R. & Upton, R.N. Basic concepts in population modeling, simulation, and model‐based drug development‐part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst. Pharmacol. 2, e38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mariot, V. et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti‐myostatin therapeutic approaches. Nat. Commun. 8, 1859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burch, P.M. et al. Reduced serum myostatin concentrations associated with genetic muscle disease progression. J. Neurol. 264, 541–553 (2017). [DOI] [PubMed] [Google Scholar]
- 17. Hanna, M.G. et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): a randomised, double‐blind, placebo‐controlled phase 2b trial. Lancet Neurol. 18, 834–844 (2019). [DOI] [PubMed] [Google Scholar]
- 18. Bi, Y. et al. Model‐informed drug development in pediatric dose selection. J. Clin. Pharmacol. 61(Suppl 1), S60–S69 (2021). [DOI] [PubMed] [Google Scholar]
- 19. Green, F.G. , Park, K. & Burckart, G.J. Methods used for pediatric dose selection in drug development programs submitted to the US FDA 2012–2020. J. Clin. Pharmacol. 61(Suppl 1), S28–S35 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Data Availability Statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.