

Abstract
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disease among adults. With diagnosis reached relatively late into the disease process, extensive motor cell loss narrows the window for therapeutic opportunities. Clinical heterogeneity in ALS and the lack of disease‐specific biomarkers have so far led to large‐sized clinical trials with long follow‐up needed to define clinical outcomes. In advanced ALS patients, there is presently limited scope to use imaging or invasive cerebrospinal fluid (CSF) collection as a source of disease biomarkers. The development of more patient‐friendly and accessible blood biomarker assays is hampered by analytical hurdles like the matrix effect of blood components. However, blood also provides the opportunity to identify disease‐specific adaptive changes of the stoichiometry and conformation of target proteins and the endogenous immunological response to low‐abundance brain peptides, such as neurofilaments (Nf). Among those biomarkers under investigation in ALS, the change in concentration before or after diagnosis of Nf has been shown to aid prognostication and to allow the a priori stratification of ALS patients into smaller sized and clinically more homogeneous cohorts, supporting more affordable clinical trials. Here, we discuss the technical hurdles affecting reproducible and sensitive biomarker measurement in blood. We also summarize the state of the art of non‐CSF biomarkers in the study of prognosis, disease progression, and treatment response. We will then address the potential as disease‐specific biomarkers of the newly discovered cryptic peptides which are formed down‐stream of TDP‐43 loss of function, the hallmark of ALS pathobiology.
Keywords: amyotrophic lateral sclerosis, blood matrix effect, clinical trials, neurofilaments, pharmacodynamic biomarkers, prognosis
1. INTRODUCTION
1.1. ALS and clinic‐ready disease biomarkers: the challenges
ALS is a relatively rare disorder with an incidence of around 2 in 1,00,000 people and a cumulative lifetime risk of 1 in 400 in Europe. 1 , 2 , 3 Disease onset is commonly seen within a person's mid‐to‐late life. 3 With the pathological process developing to involve lower and upper motor neurons (MN), following different patterns of time and spatial distribution, ALS can present at the outset with predominant involvement of either bulbar or limb‐innervated muscles. As the disease progresses, loss of motor function spreads causing weakness and muscle wasting. 4 Functional decline of the respiratory muscles ultimately leads to the patient's death. Life expectancy from symptom onset is on average 2–5 years, however, the rate of disease progression from symptom onset to death varies greatly, with survival anywhere between a few months and more than 10 years. 5 A subcohort of ALS patients also meet the diagnostic criteria for frontotemporal dementia (FTD), 6 whilst as many as 50% of ALS individuals, according to some reports, suffer from cognitive decline and/or behavioural changes like those seen in FTD. 7 , 8 , 9 ALS poses a significant burden to healthcare systems and to the broader society. The impact of a diagnosis of ALS is deeply felt by families and carers. ALS tops the list of human pathologies for suicide and of request for assisted suicide. 10
ALS appears as a sporadic condition in the large majority of affected individuals, 1 , 3 , 11 whilst in 5%–10% of cases, there is evidence of a family history, with inheritance mostly linked to dominant mutations in an ALS‐causing gene, including the superoxide dismutase (SOD1) and chromosome 9 open‐reading frame 72 (C9orf72) genes. The latter accounts for around 34% of familial ALS cases and about 5% of sporadic cases. 11 Individuals with sporadic ALS may also carry a rare gene variant that increases their susceptibility to develop the disease, acting alone or in concert with other genetic risk factors or still not well‐defined environmental exposures. 3 , 7 , 12 , 13 A common pathological denominator in 97% of ALS cases is nuclear depletion and mislocalisation of the Tar DNA binding protein 43 (TDP‐43), leading to formation of ubiquitinated TDP‐43 positive inclusions in the cytoplasm of neuronal cells. 6 This pathological hallmark is absent only in those individuals with mutations in the SOD1 or fused‐in‐sarcoma (FUS) genes. 14
The clinical diagnosis of ALS is a well‐defined process that leaves little uncertainty in the mind of highly skilled neurologists. In a minority of atypical ALS cases, clinical signs may be more difficult to decipher at the onset. The route to a final diagnosis is based on the recognition – achieved by clinical and neurophysiological testing – of the progressive extension of signs of lower and upper MN involvement to bulbar, cervical, thoracic, and lumbar regions. Recently, emphasis on early recognition of disease has taken centre stage, forming the basis for revised diagnostic criteria. The use of biomarkers is key to earlier diagnosis, overcoming diagnostic uncertainties and thus reducing the number of non‐specialist consultations and the amount of time prior to the patient being referred to a neurologist or motor neuron disease (MND) clinic. Biomarkers may also aid in the identification of a pre‐symptomatic and/or of a prodromal phase of the disease, where novel therapeutic approaches stand more chance to attenuate or revert a pathological process that is confined to a smaller number of motor cells. The latest Gold Coast diagnostic criteria propose that nerve conduction studies and electromyography (EMG); magnetic resonance imaging (MRI); and studies of blood and/or cerebrospinal fluid (CSF) for biomarkers of disease initiation and progression, should be used to exclude other disease processes, improve accuracy and timeliness of diagnosis, and to select more clinically homogeneous ALS patients for clinical trials. 15 , 16 , 17
The discovery and clinical application of easily accessible biomarkers in ALS has been impeded, however, by lack of analytical strategies in more complex matrices like blood. The development of biomarker panels and of multimodal biomarkers, where different modalities of investigations are combined to obtain more sensitive measurements, has also been challenging. CSF is the main reservoir of by‐products of neuro‐axonal loss and of subtle changes in the metabolism of surviving and degenerating neurons and thus a major source of potential biomarkers in ALS. CSF is also a relatively low‐complexity biofluid which simplifies all means of assaying low‐abundance molecules. However, complications arise from the patients' loss of mobility, reduced ability to communicate, and frailty in end‐stage disease. These complications make any invasive procedure in deteriorating ALS patients, such as lumbar puncture for CSF collection, impractical. Additional adverse events that are not primarily linked to ALS, like post‐lumbar puncture headache, can also occur. 18 Positioning individuals living with advanced ALS within a MRI scanner is difficult too due to pooling of secretions and breathing complications. 19 Therefore, the use of CSF or of imaging techniques as sources of biomarkers may suit the initial stage of the disease—to achieve an early diagnosis and for prognostication—but blood or urine‐based biomarkers to monitor disease progression or treatment response may be a more practical option for ALS patients who progress to late‐stage disease.
CSF, blood, and urine are not interchangeable sources of biomarkers. Blood acts as a source of biomarkers of neuroaxonal and muscle degeneration, which may be more representative of ALS pathobiology and of a more comprehensive disease biotype. Skeletal muscle weakness and atrophy define the progression of ALS and as the muscle is the largest contributor to body mass and a highly vascularized tissue, blood is the most important reserve of molecules linked to the neuro‐muscular destruction seen in ALS.
The bioavailability and detectability of blood biomarkers of ALS is a complex and poorly understood phenomenon. While these biomarkers can be more easily accessed, blood remains a more complex matrix than CSF. The analysis of peptides in blood, for example, is hampered by the masking effect and interference of more abundant proteins like albumin and immunoglobulins, as well as by the formation of hetero‐aggregates (see “The blood matrix effect: protein aggregation”) and of immunocomplexes driven by endogenous humoral responses (see “The blood matrix effect: the humoral immune response”). Additionally, blood volume and body mass index may influence the overall concentration of a biomarker, particularly when fluid intake and nutritional state are affected, as observed in the late stage ALS. 20 Thus, using blood as a source of biomarkers comes with its own set of difficulties, which will be explored further throughout this review.
1.2. Protein expression in biofluids, post‐translational modifications, and clearance
The detection and quantification of protein biomarkers in biofluids, and specifically in blood, poses significant challenges. Detection of low‐abundance proteins of brain origin, potential biomarkers of brain disease, is heavily dependent on the molecular processes that condition their bioavailability and modify their structure and sequence, such as post‐translational modifications (PTMs). The release of proteins of brain origin like neurofilaments (Nf), tau, and alpha‐synuclein into peripheral biofluids follows different routes, including intramural peri‐arterial drainage, whereby molecules are drained via basement membranes of smooth muscle cells of cerebral arteries to lymph nodes. 21 , 22 , 23 Upstream to these processes is the leakage of the same proteins from the neuro‐axonal network and the clearance of these molecules in affected tissues. The switch in translation of isoforms of the same protein under a pathological condition may represent an adaptive response for functional preservation of the neuroaxonal structure, which could be relevant as a disease biomarker. 24
The concentration of blood and CSF peptides of brain origin like Nf peptides promising fluid biomarkers of neurodegeneration and informative biomarkers for the definition of survival and prognosis of ALS 25 reflects their release from the neuro‐axonal network. Most Nf isoforms are expressed in neurons and axons in the so‐called stationary Nf network, whilst a small proportion of Nf undergoes slow axonal transport. 26 , 27 In healthy individuals, phosphorylation of the tail regions of the medium‐chain (NfM) and heavy‐chain (NfH) neurofilament isoforms protects them from protease degradation, preserving stability of the cytoskeleton. If the C‐terminal domain is deleted, this results in an increased turnover of the stationary Nf and in an altered Nf content. 28 , 29 When translation of the light‐chain isoform (NfL) is blocked in a transgenic mouse model, Nf content in the axon remains stable and displays a slow turnover with no loss of NfL after 4.5 months, 30 indicating that NfL is a stable and potentially adaptable protein that can support the function and structure in adverse conditions. Previous experiments have shown that a high tissue expression of NfH and NfM, relative to NfL, results in decreased radial growth of axons and reduced formation of dendrites. 31 The stochiometric representation of Nf subunits is, therefore, key to maintaining structural and functional integrity of the axon.
In patients living with ALS, it has been proposed that Nf subunit stoichiometry can adapt itself to limit energy consumption and prevent neuronal death (the adaptive protein stoichiometry hypothesis). This is accomplished by the over‐representation of the less energy‐expensive NfL at the expense of more energy‐demanding NfH and NfM. 24 The shift in Nf isoform composition could explain the observed negative correlation between high NfL levels and survival (see “Prognostic biomarkers: accurate prediction of survival”). Adaptive stoichiometry of Nf and of other proteins may, therefore, be a disease‐specific phenomenon that could be used to develop novel disease biomarkers, informing on prognosis of the disease. Adaptive changes may introduce novel NfL homopolymers, which form far more rigid structures than polymers comprised of the physiological ratio of the three Nf subtypes. 32
Below, this review will cover in more detail the challenges in biomarker discovery specific to blood (the blood matrix effect). We will then proceed to elaborate on the potential clinical applications of Nf peptides and other emerging blood biomarkers for ALS, along with detailing their use as outcome measures in clinical trials. Novel markers of disease, including cryptic peptides, that may prove to be more disease‐specific, will also be discussed.
2. THE BLOOD MATRIX EFFECT
2.1. The blood matrix effect: protein aggregation and clearance
Different matrix components in blood can affect the signal response in bioanalytical processes, such as those used to measure protein or peptide biomarkers. When analysis is undertaken by immunodetection, the conformational and post‐translational variants of serum proteins often bind non‐specifically to analytes or to a reaction surface, resulting in reduced sensitivity. The reduction of the matrix (or interference) effect, resulting in assay improvement, is technically challenging but a required step towards the development of blood assays that are viable for clinical use. 33
The progress towards viable assays to detect Nf isoforms in body fluids is illustrative of the aforementioned complexities, but they apply also to the blood measurement of other heavily post‐translationally modified proteins, including TDP‐43. Immunoassays to detect NfL have been successfully developed, and measurements in CSF and blood are highly correlated. 34 In contrast, accurate measurements in blood of NfM are yet to be accomplished, and tentative progress for NfH has been made only recently. The differing interactions between components of the blood matrix and different Nf isoforms explains the inconsistent detectability across protein isoforms. The linearity of NfL measurements seen in serial sample dilution contrasts with the absence of linearity for NfH in the same experiments, a phenomenon known as the “hook‐effect”. 35 , 36 This reflects the level of sequestration of NfH into immunocomplexes or protein aggregates, a dilution‐dependent phenomenon which interferes with immunodetection (see Figure 1). However, pre‐incubation of plasma samples with urea to dissolve aggregates and overcome the hook effect did not improve the performance of immunoassays for NfH in ALS, 35 , 36 suggesting other components of the matrix effect may also be at play.
FIGURE 1.
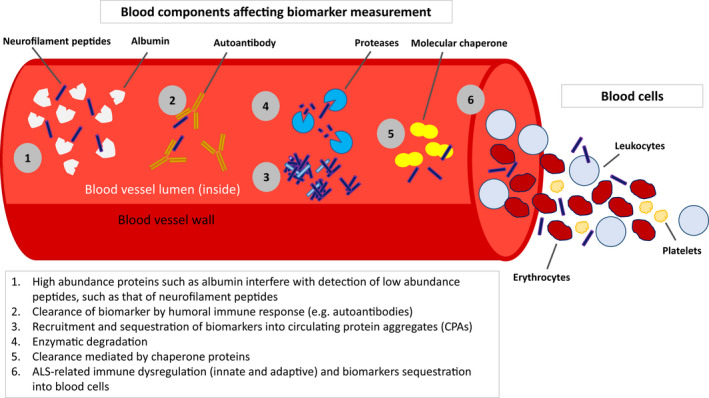
Components of the blood matrix affect the measurement of ALS biomarkers, such as neurofilament peptides
Circulating brain‐derived proteins that are sequestered within aggregates may escape detection using standard immunodetection methods. 37 The observation of sequestration of NfH, and not of NfL, into aggregates has led to further investigations into the brain protein content of circulating protein aggregates (CPAs) in blood 37 and to the identification of up to 5000 peptides of brain origin in these formations, including 30 proteins linked to ALS risk genes. 38 It has also been shown that within Nf‐containing aggregates from healthy individuals, NfL is found in a fragmented form at 30 kDa, while NfM and NfH are found at both their full‐lengths and as fragments. How the neurodegenerative and immunopathological processes underpinning ALS, including proteolysis of Nf isoforms, alters aggregate formation and composition has scarcely been investigated, but early data suggest a differential recruitment of these proteins into aggregates. 38 Thus, disease‐related changes in the proteome of CPAs could be developed as an alternative biomarker for ALS.
Additional mechanisms involved in the disposal of misfolded proteins and of aggregates through molecular chaperones (MC) (see Figure 1) may be targeted by the pathological process in ALS. 39 , 40 In the central nervous system (CNS), heat shock proteins (HSPs) are known to regulate TDP‐43 clearance, and this chaperone‐mediated process has been found to be dysfunctional in ALS. 41 The MC activity of HSP70, HSP27, calreticulin, ERp57, PDI, and CypA is also altered in peripheral blood mononuclear cells (PBMCs) from ALS patients. 42 , 43 The malfunctioning of this important route of clearance of pathological proteins is likely to have a largely underestimated effect on the detection of blood biomarkers in ALS such as Nf and TDP‐43, that have been singled out for being informative of the disease process.
2.2. The blood matrix effect: the humoral immune response
Another important and underestimated confounder of the measurement of proteins in blood is endogenous humoral response. The presence of autoantibodies to self‐antigens is another factor in the physiological clearance of proteins from the circulation (see Figure 1). The affinity of autoantibodies to target proteins changes as a function of immune tolerance, as seen in autoimmune diseases. 44 ALS, particularly faster‐progressing ALS, has been linked to substantial downregulation of T‐regulatory cells (Tregs) – lymphocytes which suppress immune responses and maintain homeostasis and self‐tolerance. 45 , 46 , 47 , 48
Assuming an increased expression of autoantibodies and a state of latent autoimmunity in ALS, natural antibodies (Nabs) against Nf and Nabs affinity to TDP‐43 in blood have been investigated as potential disease biomarkers. 49 , 50 Levels of Nabs to Nf are increased, whilst the affinity/avidity of Nabs to TDP‐43 is decreased in blood from ALS patients, compared to non‐neurological controls. 49 , 50 The overall reduction in Nabs binding capacity to TDP‐43 may reflect loss of tolerance and a reduced clearance of TDP‐43 proteins. More recently, using a blood‐derived phage display antibody library, a number of distinct antibody fragments to TDP‐43 were generated from ALS blood samples but not from non‐neurological control samples, indicating that increased autoantibody formations are possible under pathological conditions. 51 Whatever the role of autoantibodies in ALS progression, the presence of a heightened immunological response to self‐antigens is also likely to act as a confounder by reducing the sensitivity of immunodetection methods, interfering with the binding of primary and secondary antibodies to target proteins or altering the signal‐to‐noise ratio.
Less studied in ALS are changes in the innate immune response and the potential impact on protein clearance. The implication of a reported gene dysregulation in monocytes, key players in innate immunological processes, is yet to be elucidated. 52 , 53 In Alzheimer's disease and with ageing, it has recently been shown that clearance of amyloid‐beta (Aβ) by monocytes is impaired. 54
3. UTILISATION OF BLOOD BIOMARKERS IN ALS
3.1. Early diagnosis
Establishing an early diagnosis of ALS using blood biomarkers would allow more effective interventions to attenuate or revert the pathological process, when a larger proportion of motor neurons (MNs) can still be rescued. As such, any effort to improve accuracy and timeliness of diagnosis in ALS is increasingly seen as a priority. Worldwide, diagnostic latency, defined as the time from symptom onset to diagnosis, varies between centres, but the average time to diagnosis for a patient suspected to have ALS is still 12 months (range: 2–27). Different factors have been identified that impact on the speed to reach a diagnosis, including an average of four consultations with healthcare professionals before visiting an ALS multidisciplinary clinic. 55 , 56 , 57
Only those asymptomatic individuals carrying a highly penetrant genetic mutation, known to be associated to familial forms of ALS, can be defined and identified as at risk to develop the disease. In the vast majority of cases, ALS is a sporadic and unpredictable event. As such, the fluid kinetics of molecular biomarkers, particularly Nf isoforms, between the time of symptom onset and of diagnosis has been extensively investigated in the hope that sporadic ALS cases may be identified earlier. One study retrospectively used serum samples that had been collected for routine tests, before and after symptom onset, along with samples taken following diagnosis, to study changes in concentrations of serum phosphorylated NfH (pNfH) – spanning the progression from pre‐symptomatic state to manifest disease. They found a significant increase of pNfH levels up to 18 months before the diagnosis, with a steadier increase towards diagnosis. 58 Similarly, a more recent case–control study, which also used blood samples collected pre‐diagnosis, demonstrated that plasma NfL levels start to increase in the last 12–24 months before a firm diagnosis of ALS is reached. 59
Studies of pre‐symptomatic ALS mutation carriers showed CSF and serum NfL concentrations and CSF pNfH concentrations to be elevated above control levels only in the symptomatic phase of the disease. In the pre‐symptomatic phase, NfL concentrations in blood were not significantly different from those measured in controls, including first‐degree relatives not carrying the mutation. 60 , 61 , 62 , 63 However, Benatar, Malaspina and colleagues demonstrated that serum NfL levels of patients with a SOD1 and FUS mutation were increased up to 1 year before symptom onset. 62 In another study by the same authors, it was shown that serum NfL levels outperformed serum pNfH levels as a biomarker of disease initiation before onset of symptoms. Additionally, in both FUS and C9orf72 repeat expansion carriers, increased serum NfL levels have been reported up to 2 and 3.5 years before symptom onset, respectively. 63 Similarly, CSF pNfH and NfL levels were also reported to be increased up to 2 and 3.5 years before symptom onset in a number of “phenoconverters”. 58 Taken together, these results suggest that the time of the initial increase of CSF and blood Nf, in the pre‐symptomatic and or early stages of disease, may vary from patient to patient, likely reflecting the heterogeneity of the disease process in ALS patients as well as the impact on the disease pathobiology that a specific genetic mutation may have. The study of other blood biomarkers has thus far failed to demonstrate any pattern of pre‐symptomatic increase, including recent investigations into fluid levels of chitinase proteins which are linked to microglia and macrophage homeostasis. 64
3.2. Differentiating ALS from ALS mimics
The role of blood biomarkers in supporting clinical examination to confirm or exclude diagnosis of ALS has been the focus of several investigations, where specificity and sensitivity of a blood or CSF marker were used to separate ALS from disease mimics and other subtypes of MND. Most studies have looked at pNfH and NfL levels in CSF, and all have found these markers to be significantly elevated in ALS compared to ALS mimics. Sensitivity and specificity of CSF NfL and of CSF NfH were found to be above 80% in most instances. 61 , 65 , 66 , 67 , 68 , 69 , 70 , 71 Importantly, given the current delay in patients with suspected ALS being referred to MND clinics, Feneberg and colleagues also showed that these biomarkers were able to differentiate ALS from mimics and most forms of MND, regardless of whether time from symptom onset was below or above 6 months. The AUC, sensitivities, and specificities recorded in this study for ALS patients with symptoms present over 6 months are shown in Table 1 . 72 In most measures, CSF levels of pNfH performed slightly better than CSF NfL in differentiating patients with ALS from ALS mimics (see Table 1). 61 , 68 , 72 However, levels of both CSF NfL and NfH have been reported to be elevated to a similar degree seen in ALS and in some patients with primary lateral sclerosis, another MND variant. 72 Of the 85 MND mimics in their 2015 study, Steinacker and colleagues observed 11 patients with comparable levels of CSF NfL and five patients with comparable levels of CSF pNfH to those levels seen in ALS patients. 70
TABLE 1.
Levels of several different CSF and blood biomarkers are able to differentiate ALS from disease mimics
| Biomarker | Differentiates ALS from mimics in | Study | Higher levels in ALS patients than in ALS mimics in | Area under curve | Sensitivity in blood (%) | Specificity in blood (%) | Sensitivity in CSF (%) | Specificity in CSF (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| blood | CSF | blood | CSF | Blood | CSF | ||||||
| NfL | ✔ | ✔ | Feneberg et al. 2018 72 | Yes, p < .0001 | Yes, p < .0001 | 0.97 (0.94–1) | 0.93 (0.9–0.96) | 100 (95% CI: 84–100) | 84 (95% CI: 76 –90) | 89 (95% CI: 71–98) | 89 (95% CI: 81–93) |
| Poesen et al., 2017 61 | Yes, p < .001 | 0.86 (0.81–0.91) | 85.4 (95% CI: 78.8–90.6) | 78.0 (95% CI: 64.0–88.5) | |||||||
| Behzadi et al., 2021 68 | Yes, p < .05 | Not significant | 0.83 (0.76–0.91) | 0.80 (0.72–0.89) | 76.4 | 83.3 | 80.3 | 81.8 | |||
| Vacchiano et al., 2021 69 | Yes, p < .001 | Yes, p < .001 | 0.87 (0.84–0.91) | 0.92 (0.90–0.95) | 84.7 | 83.3 | 86.8 | 92.4 | |||
| Verde et al., 2019 66 | Yes, p < .0001 | 0.87 (0.81–0.94) | 85.5 (95% CI: 78.0–91.2) | 77.3 (95% CI: 62.2–88.5) | |||||||
| pNfH / NfH | ✔ | Feneberg et al. 2018 72 | Yes, p < .0001 | 0.93 (0.88–0.98) | 78 (95% CI: 58–91) | 94 (95% CI: 88–98) | |||||
| Poesen et al., 2017 61 | Yes, p < .001 | 0.91 (0.86–0.95) | 90.7 (95% CI: 84.9–94.8) | 88.0 (95% CI: 75.7–95.5) | |||||||
| Behzadi et al., 2021 68 | Yes, p < .01 | 0.87 (0.80–0.94) | 82.7 | 83.3 | |||||||
| pTau: tTau | ✔ | Agnello et al., 2021 73 | Lower, p < .001 | 0.78 (0.72–0.83) | 80.2 (95% CI: 73.9 to 85.5) | 76.7 (95% CI: 57.7–90.1) | |||||
| CHIT1 | ✔ | Gille et al. 2019 74 | Yes, p < .001 | 0.79 (0.70–0.86) | 66.7 (95% CI: 56.8–75.6) | 81.3 (95% CI: 54.4–96.0) | |||||
| Thompson et al., 2019 75 | Yes, p < .001 | 0.84 (0.72–0.95) | Not reported | Not reported | |||||||
| CHIT1 + pNfH | ✔ | Thompson et al., 2019 75 | Yes | 0.94 (0.88–1.00) | Not reported | Not reported | |||||
| YKL‐40 | ✔ | Gille et al. 2019 74 | Yes, p < .05 | 0.72 (0.63–0.80) | 70.5 (95% CI: 60.8–79.0) | 68.8 (95% CI: 41.3–89.0) | |||||
| Thompson et al., 2019 75 | Yes, p < .05 | 0.73 (0.58–0.88) | Not reported | Not reported | |||||||
| YKL‐39 | ✔ | Thompson et al., 2019 75 | Yes, p < .001 | 0.88 (0.81–0.95) | Not reported | Not reported | |||||
| hs‐cTnT | ✔ | Kläppe et al., 2022 76 | Yes, p < .001 | 0.70 (0.61–0.79) | Not reported | Not reported | |||||
Note: Combined use of biomarkers, such as hs‐cTnT with NfL, appears to increase discriminatory power. Data shown limited to what was reported by authors in original paper and supplemental material; due to high number of studies in NfL and NfH, where appropriate, for each research group only one paper is shown.
Abbreviations: CHIT1, chitotriosidase; hs‐cTnT, high‐sensitivity cardiac troponin T; NfL, neurofilament light chain; pNfH/NfH, (phosphorylated) neurofilament heavy chain; pTau:tTau, ratio of phosphorylated to total tau; YKL‐39, chitinase‐3‐like protein 2; YKL‐40, chitinase‐3‐like protein 1.
Along with CSF NfL, serum NfL can differentiate ALS from disease mimics (see Tables 1 and 2). 66 , 68 , 69 , 72 To date, only patients with Guillain‐Barré syndrome (GBS) 34 and Creutzfeldt‐Jakob disease 66 have been shown to have serum NfL levels comparable to those seen in ALS, but these conditions are clinically distinct from ALS, thus unlikely to constitute a risk of misdiagnosis.
TABLE 2.
Utility of blood biomarkers in ALS and utilisation in ongoing clinical trials
| Blood biomarker | Diagnostic utility | Prognostic utility | Commercial immunoassays | Clinical trial(s) | Status of trial | Key references |
|---|---|---|---|---|---|---|
| NfL | ✔ | ✔ | ✔ | AP‐101 | Recruiting | Thompson et al., 2022 75 ; Behzadi et al., 2021 68 ; Bjornevik et al., 2021 59 ; Benatar et al., 2019 25 ; Feneberg et al., 2018 72 |
| BIIB067 (Tofersen) | Failed at Phase 3 | |||||
| Rapamycin | Ongoing | |||||
| Aldesleukin | Ongoing | |||||
| pNfH/NfH | ✔ | ✔ | AP‐101 | Recruiting | Adiutori et al., 2021 37 ; Lu, Petzold et al., 2015 36 ; Lu et al., 2011 35 | |
| BIIB067 (Tofersen) | Failed at Phase 3 | |||||
| Rapamycin | Ongoing | |||||
| AMX0035 | Ongoing | |||||
| miRNAs | ✔ | REALS‐1 | Recruiting | Magen et al., 2021 77 | ||
| Tregs | ✔ | ✔ | Rapamycin | Ongoing | Camu et al., 2020 78 ; Rajabinejad et al., 2020 48 ; Beers, 2017 79 | |
| Aldesleukin | Ongoing | |||||
| hs‐cTnT | ✔ | ✔ | Kläppe et al., 2022 76 | |||
| CK | ✔ | ✔ | Chen et al. 2021 41 | |||
| anti‐NFs | ✔ | Puentes et al., 2021 49 ; Puentes et al., 2014 80 |
Abbreviations: anti‐NFs, antibodies against neurofilament peptides; CK, creatine kinase; hs‐cTnT, high‐sensitivity cardiac troponin T; miRNAs, micro‐RNAs; NfL, neurofilament light chain; pNfH/NfH, (phosphorylated) neurofilament heavy chain; Tregs, T‐regulatory cells.
The diagnostic performance of other biomarkers has been the focus of intense investigation, with significance mostly recorded in CSF studies. The ratio of phosphorylated tau to total tau protein (pTau:tTau) in CSF could differentiate patients with ALS from ALS mimics, with an area under the curve (AUC) of 0.78. The sensitivity and specificity of this ratio were equal to 80% and 77%, respectively (see Table 1). 73 In CSF, a number of chitinase and chitinase‐like proteins have been shown to be significantly elevated in ALS patients compared to mimics, including chitotriosidase (CHIT1), chitinase‐3‐like protein 1 (YKL‐40; or CHI3L1), and chitinase‐3‐like protein 2 (YKL‐39; or CHI3L2) (see Table 1). 74 , 75 In serum, a significant difference was not reported when the same analyses were conducted. 74 , 75 Monocyte chemoattractant protein‐1 (MCP‐1) was also tested but whilst sensitivity was high (93.3%), specificity was low (50%). 74 The best discriminatory performance given in these two studies was accomplished through combined use of CHIT1 with pNfH (AUC: 0.94 [0.88–1.00]), which was a slight improvement on that given by pNfH alone (AUC: 0.91 [0.83–0.99]). 75
In blood, concentrations of high‐sensitivity cardiac troponin T (hs‐cTnT), a protein of muscle origin, have been recently shown to differentiate ALS from disease mimics (AUC 0.70; 0.61–0.79) (see Tables 1 and 2). The combined use of this marker and CSF NfL concentrations in the same samples did not improve the power of discrimination past that of CSF NfL alone, and the combined power of plasma hs‐cTnT and plasma NfL was not tested. 76 Several cytokines and immune mediators, including those linked to TDP‐43 aggregation, which causes initiation of the nuclear factor and kappa light chain enhancer of activated B cells (NF‐κB) in microglia and secretion of the proinflammatory cytokines IL‐1β and IL‐18, have been tested in blood for biomarker potential in ALS. The blood expression of these markers has not been found to have any utility in the separation of ALS from disease mimics. 79 , 81 , 82
3.3. Prognostic blood biomarkers: accurate prediction of survival
In a recent metanalysis of multivariable survival studies looking at protective or risk effects of pre‐defined non‐genetic factors in ALS, NfL emerged as the most significant predictor. 83 Nf blood concentrations have been found to strongly associate to the rate of disease progression and survival in ALS. Faster‐progressing ALS (ALS‐F) patients identified by a more rapid decline of their revised ALS Functional Rating Scale (ALSFRS‐r) score from disease onset display significantly higher plasma NfL levels than slower‐progressing ALS (ALS‐S) patients. This is also true of patients with a shorter disease duration. 84 Furthermore, a recent multi‐centre study has shown that first‐visit plasma NfL concentration is strongly associated with survival and rate of disability progression, independent from other accepted prognostic factors such as age, ALSFRS‐r at baseline, and bulbar site of symptoms onset (see Table 2). 85 The severity and speed of progression of ALS is dictated by the spread of the pathological process and by the relative proportion of lower (LMN) vs upper motor neuron (UMN) loss in the regions involved, the bulbar being the most impactful in terms of survival. It is to date unclear whether loss of UMNs or of LMNs contributes more to the pool of detectable Nf in CSF and blood. CSF concentrations of NfL have been linked to degeneration of UMNs in a multimodal biomarker study looking at this neurochemical marker in combination with corticospinal involvement, measured by MRI, 86 , 87 but findings have not been confirmed by larger investigations. As such, the rate of increase of Nf in the early stage of disease may also serve as a predictor of speed of disease progression in a later stage of the disease and of survival. 88
A recent study of non‐coding micro‐RNA (miRNA) in blood using a hypothesis‐free approach identified miR‐181 as a strong prognostic marker (see Table 2). ALS patients with high plasma levels of miR‐181 were almost five times more likely to die during the study period in a multivariate model inclusive of previously established prognostic markers. 77 When blood measurement of miR‐181 was combined with that of NfL, the prognostic performance in ALS was even stronger. 77 , 89
Blood TDP‐43 has also shown promise as a prognostic biomarker. In a study of sporadic ALS patients, plasma TDP‐43 levels were found to positively correlate with the patients' time to generalisation, defined as the interval between reported disease onset and the appearance of signs of bulbar and spinal involvement. 90 A negative correlation between plasma TDP‐43 and split hand index, a measure of localised muscle wasting commonly seen in ALS, and a positive correlation with slow vital capacity have also been reported. 91 These data suggest that higher levels of plasma TDP‐43 may be seen in slower disease progression, possibly because of a higher blood clearance of this pathological protein, reflecting a reduced TDP‐43 pathological burden in the brain.
Immune dysregulation is increasingly being seen as a driver of disease progression in ALS. As mentioned, Tregs maintain immunological homeostasis and self‐tolerance; an inverse correlation between Tregs levels in blood and disease progression rate in ALS has been reported (see Table 2). 45 , 46 , 47 As Tregs impact upon levels of proinflammatory markers such as cytokines, these too may also provide a viable source of blood biomarkers in the prediction of survival. 82
The regulation of factors involved in lipid and glucose metabolism have also been shown to have a prognostic significance in ALS. In a large prospective population (the UK Biobank), a recent study looking at the effect of a range of metabolic markers on the risk of a subsequent ALS diagnosis found an association between HDL, apoA1, and LDL levels and ALS. This is in line with the body of evidence that premorbid metabolic dysfunction may play a role in the pathogenesis of ALS. 92 A similar approach in a large cohort of ALS patients has shown that lower than median levels of serum creatinine (hazard ratio [HR]: 1.67; 95% CI: 1.31–2.12) or albumin (HR: 1.49; 95% CI: 1.13–1.96) and a higher than median level of log‐transformed CRP (HR: 1.33; 95% CI: 1.04–1.71) or glucose (HR: 1.34; 95% CI: 1.01–1.78), at baseline, are associated with higher mortality risk in ALS. 93
Among other non‐neurocentric biomarkers, factors released from the perivascular space and muscle have been investigated for their prognostic relevance in ALS. Creatine kinase (CK) has shown potential as a biomarker of prognosis and survival (see Table 2). Higher blood creatine kinase (CK) concentrations were found in ALS‐S than ALS‐F and correlated with lower ALSFRS‐r scores. 94 Similarly, a recent study on a larger ALS cohort linked higher CK blood concentration to longer survival, adjusting for known prognostic covariates and using a Cox regression analysis. 95 In contrast, the prognostic role for serum CK was not confirmed in a more recent investigation, whilst the lower serum creatinine levels emerged as a strong predictor of shorter survival in ALS. 96 Emphasis on perivascular changes in early ALS has led to investigations into fibroblast‐derived markers as disease biomarkers. Levels of the perivascular fibroblasts' phosphoprotein 1 (SPP1) have been shown to be increased in blood from ALS patients at diagnosis and to be predictive of a shorter survival (HR = 1.82, p < .0001). 97 Whilst a good diagnostic candidate, high plasma hs‐cTnT concentrations were not associated with shorter survival in a multivariable survival model, where known clinical predictors of survival and NfL levels in CSF were incorporated as covariates. 76
3.4. Monitoring of disease progression in clinic
In multidisciplinary ALS clinics, foreseeing the timing of appearance of clinical milestones, like the need for enteric feeding and assisted ventilation, is of utmost importance. Adding to clinical observation, the use of blood biomarkers for monitoring purposes would be the strategy of choice, as longitudinal sampling of CSF for this purpose may be impractical. However, while it is accepted that blood samples are easier to obtain than CSF, venepuncture in advanced ALS patients can also be distressing for the patient and time‐consuming. The development of remote collection of micro‐samples and of assay miniaturization so that only small blood quantities of biofluids are required for biomarkers measurement is becoming an increasingly popular strategy in biomarker development. 98
Several studies have shown a stable expression of Nf biomarkers in blood in the progression of ALS. 25 , 36 , 84 , 85 In a recent multi‐centre study, raising NfL blood concentrations were reported only within the first 12 months after reported symptom onset, followed by a flat trajectory of expression with disease progression. 85 As reported, NfH immunoassays have not, in the past, delivered the expected accuracy in blood measurements. Therefore, the seemingly steady NfH concentrations thus far reported in longitudinal studies have to be interpreted with caution (see Table 2). 36
Unlike neurofilaments, the concentration in blood of mediators of the systemic immune response, including IL‐6, appear to follow an upwards trend with ALS progression. However, confirmation by larger multi‐centre studies for most putative biomarkers is lacking. 82 A recent study reported a steep increase of monocyte cells expressing the inflammation suppressing active CD11b integrin in blood from patients with ALS, with a slow disease progression. 99 The observation of a peripheral rise of myeloid cells is in line with the reported longitudinal increase of neurotrophin receptor p75 extracellular domain (p75ECD) in urine, from patients with ALS compared to healthy controls. Baseline levels of p75ECD levels also predict disease progression and survival (HR = 1.3, p < .001). 100 A similar pattern of steady increase across time points in urine from patients with ALS has been recently described for neopterin, that like p75 is a marker of microglia and macrophage activation. 101
From the pre‐symptomatic to the manifesting stage of the disease, a stable increase in concentration of the products of translation of the C9orf72 intronic expansion, a poly‐GP dipeptide repeat, has also been recently observed in CSF and PBMCs from ALS patients. 102 In blood, higher levels of immunocomplexes containing this expanded C9orf72 peptide have been reported in ALS patients than healthy controls; higher antibody levels to the poly‐GP dipeptide repeat were also seen in C9orf72‐positive patients than ALS patients without the mutation. 49 Upregulation of autoantibodies and immunocomplexes against NfL and NfH in longitudinal blood samples from faster progressing ALS patients is another example of a mounting immune response with the progression of ALS. 49 The highest levels of NfL antibodies have been detected in those patients with the lowest ALSFRS‐r scores (the most advanced disease). 80 It has been proposed that the humoral response to Nf and other proteins derived from ALS risk genes may exert a toxic effect on neurons, by inducing blood–CSF barrier impairment and by hastening neuronal loss, which is at first instigated by these dysfunctional proteins. 103 , 104 The rise of autoantibodies in ALS ties very well also with the reported loss of immunological tolerance linked to the reduced immunoregulatory functions of Tregs. 45 , 46 , 47 , 82 Antibodies against Nf or other pathogenic proteins may represent an alternative biomarker to the detection of the same low‐abundance antigens, given their greater stability and their relative abundance in blood (see Table 2). 80
Recently, it has also been shown that biomarkers may be able to differentiate different patient phenotypes. Glial fibrillary acidic protein (GFAP), a marker of astrogliosis, was shown to be higher in ALS patients with cognitive and/or behavioural impairment compared to those without, with the highest levels present in patients with confirmed ALS‐FTD. 105 Further investigations into this preliminary observation are needed. The ability to separate patients by phenotype, earlier in the disease progression, may be particularly relevant for treatment decisions and allow for development of treatment strategies targeted to the individual patient. Given the heterogeneity of the disease, such strategies may have an impact in slowing the progression of the disease and in the absence of any preventative strategy, they may improve the patient's quality of life.
3.5. Clinical trials and the use of pharmacodynamic and target engagement biomarkers
There is currently no cure for ALS, and treatment options are limited. Therapeutic management of ALS in the UK includes the use of riluzole, non‐invasive or invasive ventilation, and endoscopic gastrostomy. 5 , 106 , 107 , 108 The glutamate‐uptake enhancer riluzole is linked to a modest increase in life expectancy in ALS. 5 , 106 , 108 Edaravone, available in the US and Japan, is thought to dampen oxidative stress and has been reported to modestly reduce functional decline when administered to patients at an earlier stage of disease. 108
The design of clinical trials for ALS hinges on survival and functional decline as primary outcomes. Rate of disease progression and survival vary significantly across affected individuals. 108 Large size cohorts and long follow‐up periods are therefore needed to power clinical trials a challenging and expensive option for a relatively rare condition. Furthermore, enrolment of ALS patients into clinical trials is on average 1 or 2 years from disease onset, narrowing the window for early disease modification. Easily accessible biomarkers that allow for earlier diagnosis and recruitment, complementing clinical outcome measures, are thus the obligate route towards more affordable, and hopefully successful studies. As the most studied biomarker for ALS, it is unsurprising that Nf peptides are already used as exploratory outcome measures in most clinical trials (see Table 2). Statistical modelling recently suggested that plasma NfL has the potential to reduce the duration (and costs) of clinical trials. 85 However, this assumes that disease modification preserving neuroaxonal structures is accompanied by a reduced Nf release and concentration in biofluids. This modelling of Nf behaviour may not consider mechanisms of Nf‐clearance, which may alter the biomarker concentration independently from the pathological process that is the target of the therapeutic intervention.
Clinical trials in ALS using antisense oligonucleotides (ASO) directed against known genetic causative factors are currently ongoing and/or in a final stage in some cases. Plasma and CSF levels of both NfL and pNfH have been included as exploratory measures, along with more specific biomarkers of target engagement, in an ASO trial designed to degrade SOD1 mRNA and reduce pathological protein levels in ALS patients carrying a SOD1 gene mutation (BIIB067, Tofersen) (see Table 2). Published data from phase 1 and 2, and reports on a phase 3 study, pointed to a signifcant decrease a signifcant NfL and CSF and SOD1 protein in ALS patients treated with the ASO intrathecally as opposed to placebo, particularly in the group who received the highest dose of the drug. 109 The phase 3 study did not meet the primary end point represented by the reduction of the participants' ALSFRS‐r scores at week 28. However positive trends across multiple clinical outcome measures and exploratory endpoints were, nevertheless, reported in these studies and observed specifically. 110 Another phase 1 clinical trial looking at reducing SOD1 protein involves intravenous administration of a human IgG1 against misfolded SOD1 (AP‐101). Plasma and CSF levels of both NfL and pNfH have been included as secondary outcome measures (see Table 2). 111 The ASO therapeutic approach has also been extended to the treatment of ALS induced by the C9orf72 gene mutation (BIIB078), 112 with a novel immunoassay to test CSF levels of GP dipeptide repeat used as a disease biomarker. 113 It was recently announced, however, that this trial is being stopped due to a lack of efficacy. 114
The administration of sodium phenylbutyrate–taurursodiol (AMX0035), a drug thought to reduce endoplasmic reticulum stress and restore mitochondrial energy deficits, has given promising results. 115 Preliminary data from a phase 2 trial, however, showed that plasma pNfH levels included as a secondary outcome measure (see Table 2) were not different between placebo and the treated group. In contrast, the change in ALSFRS‐r score was significantly smaller in the group who received the drug. 115 These data point towards the lack of a full understanding of how changes in known measures of clinical efficacy associate with the change in concentration of putative biomarkers of treatment response, making the interpretation of clinical trial results difficult.
Manipulation of the immune system, including using intra‐spinal allogenic stem cell injection and immunosuppression by repurposed drugs, has not yet shown any therapeutic benefit or an effect in restoring Treg levels to normal levels. 9 , 116 , 117 However, Treg modification and enhancement in ALS patients is currently being pursued as a viable therapeutic strategy, 48 , 116 , 118 and a clinical trial using a single repurposed Tregs‐enhancing drug, rapamycin, is ongoing. 117 Additionally, low‐dose interleukin‐2 (IL‐2) (aldesleukin), a booster of Treg levels, has thus far shown a successful increase of Tregs with no severe side effects. Any effect on disease progression is yet to be reported (see Table 2). 78
4. CONCLUSIONS: CRYPTIC PEPTIDES AND THE NEW FRONTIER OF BIOMARKERS DISCOVERY IN ALS
TDP‐43 proteinopathy is the defining neuropathological event in most ALS cases and in a percentage of FTD individuals. TDP‐43 mis‐localization from the nucleus to the cytosol, resulting in nuclear loss of function, affects RNA translation of a wide range of proteins, including the protein unc‐13 homolog A (UNC13A). In the case of UNC13A, this leads to random inclusion of so‐called cryptic exons, derived from non‐conserved intronic sequences, and formation of cryptic peptides upon translation into the final protein. 14 , 119 UNC13A acts as regulator of synaptic function and neurotransmitter release. The cryptic exon inclusion in UNC13A, and in the translation of other proteins subjected to a dysfunctional TDP‐43 control, is likely to generate an aberrant and possibly toxic proteome in ALS. The risk of cryptic exon inclusion in UNC13A is increased in the presence of two single‐nucleotide polymorphisms (SNP), rs12973192 and rs12608932, which lie within or near to cryptic exon insertion site. 14 , 119 Both have previously been shown to contribute to the risk to develop ALS in genome‐wide association studies. In sporadic ALS patients, for example, the rs12608932 SNP is linked to an earlier onset of the disease in European and American ALS populations, 120 , 121 to a shorter survival time 122 and to frontotemporal degeneration. 123 The loss of UNC13A function causes reduction of miR‐3911 expression, normally detected in neuron‐derived extracellular plasma vesicles. 124 The detection of miR‐3911 downregulation is, therefore, a surrogate of UNC13A‐related pathology, which could be detected without resorting to expensive whole‐genome sequencing.
The premature incorporation of cryptic exons and the formation of aberrant proteins as shown for UNC13A and for other proteins downstream of TDP‐43 loss of function, may be the defining pathological event in ALS. It could constitute a paradigm shift in the search for biomarkers in this neurodegenerative disorder, particularly as TDP‐43 pathology is largely ALS/FTD‐specific. As such, cryptic peptide formation is a potential source of biomarkers to be used for early diagnosis and for stratification of ALS patients within specific endophenotypes. Loss of UNC13A normal function and cryptic exon inclusion could also represent the pharmacodynamic biomarker of future therapeutic intervention aimed at arresting cryptic peptide formation in ALS. Whilst Nfs are beyond doubt shaping the landscape of disease monitoring in neurodegenerative disorders and helping with the prognostic characterization of ALS, the discovery of cryptic proteins in ALS may provide the ground of next‐generation biomarkers for this incurable condition.
FUNDING INFORMATION
Ellie Rebecca Sturmey is a PhD student funded by Barts and the London Charities and has no conflicts of interest to declare. Andrea Malaspina has acted as a consultant and received personal fees from Roche and Pfizer; he reports grant funding from the National Institutes of Health, the ALS Association, MND Association UK, Wellcome Trust UK, NIHR UK, Barts Charity UK, and MRC.
CONFLICT OF INTEREST
The Authors have no conflict of interest that would affect the publication of this article
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/ane.13698.
ACKNOWLEDGMENTS
We thank all individuals living with ALS, their families, and carers for the extraordinary support they provide to the longitudinal biobanking projects that have supported research into biomarkers, including the ALS biomarker study and A Multicentre Biomarker Resource Strategy In ALS (AMBRoSIA). We are grateful for the advice and guidance we have received from investigators based at the Queen Square MND Centre, Institute of Neurology, on topics discussed in the paper.
Sturmey, E. & Malaspina, A. (2022). Blood biomarkers in ALS: challenges, applications and novel frontiers. Acta Neurologica Scandinavica, 146, 375–388. 10.1111/ane.13698
Contributor Information
Ellie Sturmey, Email: e.r.sturmey@qmul.ac.uk.
Andrea Malaspina, Email: a.malaspina@ucl.ac.uk.
DATA AVAILABILITY STATEMENT
Data sharing not applicable as no datasets were generated or analysed for the writing of this article
REFERENCES
- 1. Cook C, Petrucelli L. Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron. 2019;101(6):1057‐1069. doi: 10.1016/j.neuron.2019.02.032 [DOI] [PubMed] [Google Scholar]
- 2. Chiò A, Logroscino G, Traynor BJ, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118‐130. doi: 10.1159/000351153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162‐172. doi: 10.1056/NEJMra1603471 [DOI] [PubMed] [Google Scholar]
- 4. Tortelli R, Copetti M, Ruggieri M, et al. Cerebrospinal fluid neurofilament light chain levels: marker of progression to generalized amyotrophic lateral sclerosis. Eur J Neurol. 2014;22(1):215‐218. doi: 10.1111/ene.12421 [DOI] [PubMed] [Google Scholar]
- 5. Knibb JA, Keren N, Kulka A, et al. A clinical tool for predicting survival in ALS. J Neurol Neurosurg Psychiatry. 2016;87(12):1361‐1367. doi: 10.1136/jnnp-2015-312908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in als and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416‐438. doi: 10.1016/j.neuron.2013.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morgan S, Shatunov A, Sproviero W, et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain. 2017;140(6):1611‐1618. doi: 10.1093/brain/awx082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Therrien M, Dion PA, Rouleau GA. ALS: recent developments from genetics studies. Curr Neurol Neurosci Rep. 2016;16(6):59. doi: 10.1007/s11910-016-0658-1 [DOI] [PubMed] [Google Scholar]
- 9. Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197‐206. doi: 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maessen M, Veldink JH, Van Den Berg LH, Schouten HJ, Van Der Wal G, Onwuteaka‐Philipsen BD. Requests for euthanasia: origin of suffering in ALS, heart failure, and cancer patients. J Neurol. 2010;257(7):1192‐1198. doi: 10.1007/s00415-010-5474-y [DOI] [PubMed] [Google Scholar]
- 11. Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci. 2019;13:1‐27. doi: 10.3389/fnins.2019.01310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corcia P, Couratier P, Blasco H, et al. Genetics of amyotrophic lateral sclerosis. Rev Neurol (Paris). 2017;173:254‐262. doi: 10.1007/s00115-013-3898-1 [DOI] [PubMed] [Google Scholar]
- 13. Volk AE, Weishaupt JH, Andersen PM, Ludolph AC, Kubisch C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet. 2018;30(2):252‐258. doi: 10.1007/s11825-018-0185-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown A‐L, Wilkins OG, Keuss MJ, et al. TDP‐43 loss and ALS‐risk SNPs drive mis‐splicing and depletion of UNC13A. Nature. 2022;603:131‐137. doi: 10.1038/s41586-022-04436-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shefner JM, Al‐Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020;131:1975‐1978. doi: 10.1016/j.clinph.2020.04.005 [DOI] [PubMed] [Google Scholar]
- 16. Turner MR. Diagnosing ALS: the Gold Coast criteria and the role of EMG. Pract Neurol. 2022;22:176‐178. doi: 10.1136/practneurol-2021-003256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen D, Yang X, Wang Y, et al. The Gold Coast criteria increases the diagnostic sensitivity for amyotrophic lateral sclerosis in a Chinese population. Transl Neurodegener. 2021;10(1):2‐9. doi: 10.1186/s40035-021-00253-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duits FH, Martinez‐Lage P, Paquet C, et al. Performance and complications of lumbar puncture in memory clinics: results of the multicenter lumbar puncture feasibility study. Alzheimers Dement. 2016;12(2):154‐163. doi: 10.1016/j.jalz.2015.08.003 [DOI] [PubMed] [Google Scholar]
- 19. Ferraro PM, Agosta F, Riva N, et al. Multimodal structural MRI in the diagnosis of motor neuron diseases. NeuroImage Clin. 2017;16:240‐247. doi: 10.1016/j.nicl.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manouchehrinia A, Piehl F, Hillert J, et al. Confounding effect of blood volume and body mass index on blood neurofilament light chain levels. Ann Clin Transl Neurol. 2020;7(1):139‐143. doi: 10.1002/acn3.50972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nimmo J, Johnston DA, Dodart JC, et al. Peri‐arterial pathways for clearance of α‐synuclein and tau from the brain: implications for the pathogenesis of dementias and for immunotherapy. Alzheimer's Dement (Amst). 2020;12(1):1‐10. doi: 10.1002/dad2.12070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Preston SD, Steart PV, Wilkinson A, Nicoll JAR, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid β from the human brain. Neuropathol Appl Neurobiol. 2003;29(2):106‐117. doi: 10.1046/j.1365-2990.2003.00424.x [DOI] [PubMed] [Google Scholar]
- 23. Hawkes CA, Gatherer M, Sharp MM, et al. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid‐β from the mouse brain. Aging Cell. 2013;12(2):224‐236. doi: 10.1111/acel.12045 [DOI] [PubMed] [Google Scholar]
- 24. Zucchi E, Lu CH, Cho Y, et al. A motor neuron strategy to save time and energy in neurodegeneration: adaptive protein stoichiometry. J Neurochem. 2018;146(5):631‐641. doi: 10.1111/jnc.14542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benatar M, Zhang L, Wang L, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):59‐69. doi: 10.1212/WNL.0000000000009559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yuan A, Rao MV, Veeranna NRA. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb Perspect Biol. 2017;9(4):a018309. doi: 10.1101/cshperspect.a018309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nixon RA, Logvinenko KB. Multiple fates of newly synthesized neurofilament proteins: evidence for a stationary neurofilament network distributed nonuniformly along axons of retinal ganglion cell neurons. J Cell Biol. 1986;102(2):647‐659. doi: 10.1083/jcb.102.2.647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pant HC. Dephosphorylation of neurofilament proteins enhances their susceptibility to degradation by calpain. Biochem J. 1988;256(2):665‐668. doi: 10.1042/bj2560665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rao MV, Yuan A, Campbell J, Kumar A, Nixon RA. The C‐Terminal Domains of NF‐H and NF‐M Subunits Maintain Axonal Neurofilament Content by Blocking Turnover of the Stationary Neurofilament Network. PLoS One. 2012;7(9):1‐10. doi: 10.1371/journal.pone.0044320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Millecamps S, Gowing G, Corti O, Mallet J, Julien JP. Conditional NF‐L transgene expression in mice for in vivo analysis of turnover and transport rate of neurofilaments. J Neurosci. 2007;27(18):4947‐4956. doi: 10.1523/JNEUROSCI.5299-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kong J, Tung VWY, Aghajanian J, Xu Z. Antagonistic roles of neurofilament subunits NF‐H and NF‐M against NF‐L in shaping dendritic arborization in spinal motor neurons. J Cell Biol. 1998;140(5):1167‐1176. doi: 10.1083/jcb.140.5.1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beck R, Deek J, Safinya CR. Structures and interactions in “bottlebrush” neurofilaments: the role of charged disordered proteins in forming hydrogel networks. Biochem Soc Trans. 2012;40(5):1027‐1031. doi: 10.1042/BST20120101 [DOI] [PubMed] [Google Scholar]
- 33. Wood G. “Matrix effects” in immunoassays. Scand J Clin Lab Invest. 1991;51:105‐112. doi: 10.3109/00365519109104608 [DOI] [PubMed] [Google Scholar]
- 34. Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One. 2013;8(9):1‐9. doi: 10.1371/journal.pone.0075091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu CH, Kalmar B, Malaspina A, Greensmith L, Petzold A. A method to solubilise protein aggregates for immunoassay quantification which overcomes the neurofilament “hook” effect. J Neurosci Methods. 2011;195(2):143‐150. doi: 10.1016/j.jneumeth.2010.11.026 [DOI] [PubMed] [Google Scholar]
- 36. Lu CH, Petzold A, Topping J, et al. Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry. 2015;86(5):565‐573. doi: 10.1136/jnnp-2014-307672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adiutori R, Aarum J, Zubiri I, et al. The proteome of neurofilament‐containing protein aggregates in blood. Biochem Biophys Reports. 2018;14:168‐177. doi: 10.1016/j.bbrep.2018.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adiutori R, Puentes F, Bremang M, et al. Analysis of circulating protein aggregates as a route of investigation into neurodegenerative disorders. Brain Commun. 2021;3(3):1‐16. doi: 10.1093/braincomms/fcab148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wyatt AR, Yerbury JJ, Berghofer P, et al. Clusterin facilitates in vivo clearance of extracellular misfolded proteins. Cell Mol Life Sci. 2011;68(23):3919‐3931. doi: 10.1007/s00018-011-0684-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chaudhury S, Keegan BM, Blagg BSJ. The role and therapeutic potential of Hsp90, Hsp70, and smaller heat shock proteins in peripheral and central neuropathies. Med Res Rev. 2021;41(1):202‐222. doi: 10.1002/med.21729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen HJ, Mitchell JC, Novoselov S, et al. The heat shock response plays an important role in TDP‐43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain. 2016;139(5):1417‐1432. doi: 10.1093/brain/aww028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nardo G, Pozzi S, Pignataro M, et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One. 2011;6(10):e25545. doi: 10.1371/journal.pone.0025545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vats A, Gourie‐Devi M, Ahuja K, et al. Expression analysis of protein homeostasis pathways in the peripheral blood mononuclear cells of sporadic amyotrophic lateral sclerosis patients. J Neurol Sci. 2018;387:85‐91. doi: 10.1016/j.jns.2018.01.035 [DOI] [PubMed] [Google Scholar]
- 44. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, et al. Mechanisms of autoantibody‐induced pathology. Front Immunol. 2017;8:603. doi: 10.3389/fimmu.2017.00603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheean RK, McKay FC, Cretney E, et al. Association of regulatory T‐cell expansion with progression of amyotrophic lateral sclerosis a study of humans and a transgenic mouse model. JAMA Neurol. 2018;75(6):681‐689. doi: 10.1001/jamaneurol.2018.0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beers DR, Henkel JS, Zhao W, et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134(5):1293‐1314. doi: 10.1093/brain/awr074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henkel JS, Beers DR, Wen S, et al. Regulatory T‐lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO ssol Med. 2013;5(1):64‐79. doi: 10.1002/emmm.201201544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rajabinejad M, Ranjbar S, Afshar Hezarkhani L, Salari F, Gorgin Karaji A, Rezaiemanesh A. Regulatory T cells for amyotrophic lateral sclerosis/motor neuron disease: a clinical and preclinical systematic review. J Cell Physiol. 2020;235(6):5030‐5040. doi: 10.1002/jcp.29401 [DOI] [PubMed] [Google Scholar]
- 49. Puentes F, Lombardi V, Lu CH, et al. Humoral response to neurofilaments and dipeptide repeats in ALS progression. Ann Clin Transl Neurol. 2021;8:1831‐1844. doi: 10.1002/acn3.51428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nielsen AK, Folke J, Owczarek S, et al. TDP‐43‐specific autoantibody decline in patients with amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. 2021;8(2):1‐12. doi: 10.1212/NXI.0000000000000937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Williams SM, Khan G, Harris BT, Ravits J, Sierks MR. TDP‐43 protein variants as biomarkers in amyotrophic lateral sclerosis. BMC Neurosci. 2017;18(1):1‐12. doi: 10.1186/s12868-017-0334-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gustafson MP, Staff NP, Bornschlegl S, et al. Comprehensive immune profiling reveals substantial immune system alterations in a subset of patients with amyotrophic lateral sclerosis. PLoS One. 2017;12(7):1‐21. doi: 10.1371/journal.pone.0182002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao W, Beers DR, Hooten KG, et al. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol. 2017;74(6):677‐685. doi: 10.1001/jamaneurol.2017.0357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen SH, Tian DY, Shen YY, et al. Amyloid‐beta uptake by blood monocytes is reduced with ageing and Alzheimer's disease. Transl Psychiatry. 2020;10(1):423. doi: 10.1038/s41398-020-01113-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matharan M, Mathis S, Bonabaud S, Carla L, Soulages A, Le Masson G. Minimizing the diagnostic delay in amyotrophic lateral sclerosis: the role of nonneurologist practitioners. Neurol Res Int. 2020;2020:1‐8. doi: 10.1155/2020/1473981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Galvin M, Ryan P, Maguire S, et al. The path to specialist multidisciplinary care in amyotrophic lateral sclerosis: a population‐based study of consultations, interventions and costs. PLoS One. 2017;12(6):1‐12. doi: 10.1371/journal.pone.0179796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Househam E, Swash M. Diagnostic delay in amyotrophic lateral sclerosis: what scope for improvement? J Neurol Sci. 2000;180(1–2):76‐81. doi: 10.1016/S0022-510X(00)00418-4 [DOI] [PubMed] [Google Scholar]
- 58. De Schaepdryver M, Goossens J, De Meyer S, et al. Serum neurofilament heavy chains as early marker of motor neuron degeneration. Ann Clin Transl Neurol. 2019;6(10):1971‐1979. doi: 10.1002/acn3.50890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bjornevik K, O'Reilly EJ, Molsberry S, et al. Prediagnostic neurofilament light chain levels in amyotrophic lateral sclerosis. Neurology. 2021;97(15):e1466‐e1474. doi: 10.1212/wnl.0000000000012632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weydt P, Oeckl P, Huss A, et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol. 2016;79(1):152‐158. doi: 10.1002/ana.24552 [DOI] [PubMed] [Google Scholar]
- 61. Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology. 2017;88(24):2302‐2309. doi: 10.1212/WNL.0000000000004029 [DOI] [PubMed] [Google Scholar]
- 62. Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. 2018;84(1):130‐139. doi: 10.1002/ana.25276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Benatar M, Wuu J, Lombardi V, et al. Neurofilaments in pre‐symptomatic ALS and the impact of genotype. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(7–8):538‐548. doi: 10.1080/21678421.2019.1646769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gray E, Thompson AG, Wuu J, et al. CSF chitinases before and after symptom onset in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2020;7(8):1296‐1306. doi: 10.1002/acn3.51114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reijn TS, Abdo WF, Schelhaas HJ, Verbeek MM. CSF neurofilament protein analysis in the differential diagnosis of ALS. J Neurol. 2009;256(4):615‐619. doi: 10.1007/s00415-009-0131-z [DOI] [PubMed] [Google Scholar]
- 66. Verde F, Steinacker P, Weishaupt JH, et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(2):157‐164. doi: 10.1136/jnnp-2018-318704 [DOI] [PubMed] [Google Scholar]
- 67. Halbgebauer S, Steinacker P, Verde F, et al. Comparison of CSF and serum neurofilament light and heavy chain as differential diagnostic biomarkers for ALS. J Neurol Neurosurg Psychiatry. 2022;93(1):68‐74. doi: 10.1136/jnnp-2021-327129 [DOI] [PubMed] [Google Scholar]
- 68. Behzadi A, Pujol‐Calderón F, Tjust AE, et al. Neurofilaments can differentiate ALS subgroups and ALS from common diagnostic mimics. Sci Rep. 2021;11(1):22128. doi: 10.1038/s41598-021-01499-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vacchiano V, Mastrangelo A, Zenesini C, et al. Plasma and CSF neurofilament light chain in amyotrophic lateral sclerosis: a cross‐sectional and longitudinal study. Front Aging Neurosci. 2021;13:1‐11. doi: 10.3389/fnagi.2021.753242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Steinacker P, Feneberg E, Weishaupt J, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry. 2016;87(1):12‐20. doi: 10.1136/jnnp-2015-311387 [DOI] [PubMed] [Google Scholar]
- 71. Ganesalingam J, An J, Bowser R, Andersen PM, Shaw CE. PNfH is a promising biomarker for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(2):146‐149. doi: 10.3109/21678421.2012.729596 [DOI] [PubMed] [Google Scholar]
- 72. Feneberg E, Oeckl P, Steinacker P, et al. Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology. 2018;90(1):e22‐e30. doi: 10.1212/WNL.0000000000004761 [DOI] [PubMed] [Google Scholar]
- 73. Agnello L, Colletti T, Lo Sasso B, et al. Tau protein as a diagnostic and prognostic biomarker in amyotrophic lateral sclerosis. Eur J Neurol. 2021;28(6):1868‐1875. doi: 10.1111/ene.14789 [DOI] [PubMed] [Google Scholar]
- 74. Gille B, De Schaepdryver M, Dedeene L, et al. Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2019;90(12):1338‐1346. doi: 10.1136/jnnp-2018-319586 [DOI] [PubMed] [Google Scholar]
- 75. Thompson AG, Gray E, Bampton A, Raciborska D, Talbot K, Turner MR. CSF chitinase proteins in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(11):1215‐1220. doi: 10.1136/jnnp-2019-320442 [DOI] [PubMed] [Google Scholar]
- 76. Kläppe U, Chamoun S, Shen Q, et al. Cardiac troponin T is elevated and increases longitudinally in ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. 2022;23(1–2):58‐65. doi: 10.1080/21678421.2021.1939384 [DOI] [PubMed] [Google Scholar]
- 77. Magen I, Coenen‐Stass A, Yacovzada NS, et al. Circulating miR‐181a‐5p is a prognostic biomarker for amyotrophic lateral sclerosis. BioRxiv. 2019;373426. https://www.nature.com/articles/s41593‐021‐00936‐z [DOI] [PubMed] [Google Scholar]
- 78. Camu W, Mickunas M, Veyrune JL, et al. Repeated 5‐day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): a phase 2a randomised, double‐blind, placebo‐controlled trial. EBioMedicine. 2020;59:102844. doi: 10.1016/j.ebiom.2020.102844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019;18:211‐220. doi: 10.1016/S1474-4422(18)30394-6 [DOI] [PubMed] [Google Scholar]
- 80. Puentes F, Topping J, Kuhle J, et al. Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2014;85(3):274‐278. doi: 10.1136/jnnp-2013-305494 [DOI] [PubMed] [Google Scholar]
- 81. Moreno‐Martinez L, Calvo AC, Muñoz MJ, Osta R. Are circulating cytokines reliable biomarkers for amyotrophic lateral sclerosis? Int J Mol Sci. 2019;20(11):2759. doi: 10.3390/ijms20112759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lu C‐H, Allen K, Oei F, et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. 2016;3(4):e244. doi: 10.1212/nxi.0000000000000244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Su WM, Cheng YF, Jiang Z, et al. Predictors of survival in patients with amyotrophic lateral sclerosis: a large meta‐analysis. EBioMedicine. 2021;74:103732. doi: 10.1016/j.ebiom.2021.103732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lu CH, Macdonald‐Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84(22):2247‐2257. doi: 10.1212/WNL.0000000000001642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thompson AG, Gray E, Verber N, et al. Multicentre appraisal of amyotrophic lateral sclerosis biofluid biomarkers shows primacy of blood neurofilament light chain. Brain Commun. 2022;4(1):1‐11. doi: 10.1093/braincomms/fcac029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Menke RAL, Gray E, Lu CH, et al. CSF neurofilament light chain reflects corticospinal tract degeneration in ALS. Ann Clin Transl Neurol. 2015;2(7):748‐755. doi: 10.1002/acn3.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gille B, De Schaepdryver M, Goossens J, et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2019;45(3):291‐304. doi: 10.1111/nan.12511 [DOI] [PubMed] [Google Scholar]
- 88. McCombe PA, Pfluger C, Singh P, Lim CYH, Airey C, Henderson RD. Serial measurements of phosphorylated neurofilament‐heavy in the serum of subjects with amyotrophic lateral sclerosis. J Neurol Sci. 2015;353(1–2):122‐129. doi: 10.1016/j.jns.2015.04.032 [DOI] [PubMed] [Google Scholar]
- 89. Westeneng HJ, Debray TPA, Visser AE, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17(5):423‐433. doi: 10.1016/S1474-4422(18)30089-9 [DOI] [PubMed] [Google Scholar]
- 90. Ren Y, Li S, Chen S, et al. TDP‐43 and phosphorylated TDP‐43 levels in paired plasma and CSF samples in amyotrophic lateral sclerosis. Front Neurol. 2021;12:1‐8. doi: 10.3389/fneur.2021.663637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kojima Y, Kasai T, Noto YI, et al. Amyotrophic lateral sclerosis: correlations between fluid biomarkers of NfL, TDP‐43, and tau, and clinical characteristics. PLoS One. 2021;16(11):1‐13. doi: 10.1371/journal.pone.0260323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Thompson AG, Talbot K, Turner MR. Higher blood high density lipoprotein and apolipoprotein A1 levels are associated with reduced risk of developing amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2022;93(1):75‐81. doi: 10.1136/jnnp-2021-327133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sun J, Carrero JJ, Zagai U, et al. Blood biomarkers and prognosis of amyotrophic lateral sclerosis. Eur J Neurol. 2020;27(11):2125‐2133. doi: 10.1111/ene.14409 [DOI] [PubMed] [Google Scholar]
- 94. Ceccanti M, Pozzilli V, Cambieri C, et al. Creatine kinase and progression rate in amyotrophic lateral sclerosis. Cell. 2020;9(5):1174. doi: 10.3390/cells9051174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chen XP, Wei QQ, Ou RW, et al. Creatine kinase in the diagnosis and prognostic prediction of amyotrophic lateral sclerosis: a retrospective case‐control study. Neural Regen Res. 2021;16(3):591‐595. doi: 10.4103/1673-5374.293159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Guo QF, Hu W, Xu LQ, Luo H, Wang N, Zhang QJ. Decreased serum creatinine levels predict short survival in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2021;8(2):448‐455. doi: 10.1002/acn3.51299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Månberg A, Skene N, Sanders F, et al. Altered perivascular fibroblast activity precedes ALS disease onset. Nat Med. 2021;27(4):640‐646. doi: 10.1038/s41591-021-01295-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lombardi V, Carassiti D, Giovannoni G, Lu CH, Adiutori R, Malaspina A. Theme 7 pre‐clinical therapeutic strategies. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:217‐245. doi: 10.1080/21678421.2019.1646995 [DOI] [PubMed] [Google Scholar]
- 99. Yildiz O, Schroth J, Lombardi V, et al. The expression of active CD11b monocytes in blood and disease progression in amyotrophic lateral sclerosis. Int J Mol Sci. 2022;23(6):3370. doi: 10.3390/ijms23063370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shepheard SR, Wuu J, Cardoso M, et al. Urinary p75(ECD): a prognostic, disease progression, and pharmacodynamic biomarker in ALS. Am Acad Neurol. 2017;12(88):1137‐1143. doi: 10.1212/WNL.0000000000003741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shepheard SR, Karnaros V, Benyamin B, et al. Urinary neopterin: a novel biomarker of disease progression in amyotrophic lateral sclerosis. Eur J Neurol. 2022;29(4):990‐999. doi: 10.1111/ene.15237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gendron TF, Chew J, Stankowski JN, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72‐associated amyotrophic lateral sclerosis. Sci Transl Med. 2017;9(383):aai7866. doi: 10.1126/scitranslmed.aai7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Fialová L, Švarcová J, Bartos A, et al. Cerebrospinal fluid and serum antibodies against neurofilaments in patients with amyotrophic lateral sclerosis. Eur J Neurol. 2010;17(4):562‐566. doi: 10.1111/j.1468-1331.2009.02853.x [DOI] [PubMed] [Google Scholar]
- 104. Puentes F, van der Star BJ, Boomkamp SD, et al. Neurofilament light as an immune target for pathogenic antibodies. Immunology. 2017;152(4):580‐588. doi: 10.1111/imm.12797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Falzone YM, Domi T, Mandelli A, et al. Integrated evaluation of a panel of neurochemical biomarkers to optimize diagnosis and prognosis in amyotrophic lateral sclerosis. Eur J Neurol. 2022;29(7):1930‐1939. doi: 10.1111/ene.15321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942‐955. doi: 10.1016/S0140-6736(10)61156-7 [DOI] [PubMed] [Google Scholar]
- 107. Verber NS, Shepheard SR, Sassani M, et al. Biomarkers in motor neuron disease: a state of the art review. Front Neurol. 2019;10:1‐28. doi: 10.3389/fneur.2019.00291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39(2):733‐748. doi: 10.1002/med.21528 [DOI] [PubMed] [Google Scholar]
- 109. Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1–2 trial of antisense oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109‐119. doi: 10.1056/nejmoa2003715 [DOI] [PubMed] [Google Scholar]
- 110. Biogen . Biogen Announces Topline Results from the Tofersen Phase 3 Study and its Open‐Label Extension in SOD1‐ALS. 2021. Accessed 20 March 2022. http://media.biogen.com/news‐releases/news‐release‐details/biogen‐announces‐topline‐results‐tofersen‐phase‐3‐study‐and‐its
- 111. U.S. National Libary of Medicine . A study to evaluate, safety, tolerability, pharmacodynamic (PD) markers and pharmacokinetics (PK) of AP‐101 in participants with amyotrophic lateral sclerosis (ALS). Clinical Trials .Gov. 2022. Accessed 20 March 2022. https://clinicaltrials.gov/ct2/show/NCT05039099 [Google Scholar]
- 112. Tran H, Moazami MP, Yang H, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28(1):117‐124. doi: 10.1038/s41591-021-01557-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wilson KM, Katona E, Glaria I, et al. Development of a sensitive trial‐ready poly(GP) CSF biomarker assay for C9orf72 ‐associated frontotemporal dementia and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2022. 0, 1‐11. doi: 10.1136/jnnp-2021-328710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Biogen . Biogen and Ionis Announce Topline Phase 1 Study Results of Investigational Drug in C9orf72 Amyotrophic Lateral Sclerosis. 2022. Accessed 22 April 2022. https://investors.biogen.com/news‐releases/news‐release‐details/biogen‐and‐ionis‐announce‐topline‐phase‐1‐study‐results
- 115. Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate‐Taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919‐930. doi: 10.1056/NEJMoa1916945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Beers DR. Tregs in ALS are dysfunctional and predict progression rate and severity. JCI Insight. 2017;2(5):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mandrioli J, D'Amico R, Zucchi E, et al. Rapamycin treatment for amyotrophic lateral sclerosis protocol for a phase II randomized, double‐blind, placebo‐controlled, multicenter, clinical trial (RAP‐ALS trial). Medicine (Baltimore). 2018;97(24):e11119. doi: 10.1097/MD.0000000000011119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Fournier CN, Schoenfeld D, Berry JD, et al. An open label study of a novel immunosuppression intervention for the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(3–4):242‐249. doi: 10.1080/21678421.2017.1421666 [DOI] [PubMed] [Google Scholar]
- 119. Ma XR, Prudencio M, Koike Y, et al. TDP‐43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature. 2022;603:124‐130. doi: 10.1038/s41586-022-04424-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. The ALSGEN Consortium . Age of onset of amyotrophic lateral sclerosis is modulated by a locus on 1p34.1. Neurobiol Aging. 2013;34(1):357.e7‐357.e19. doi: 10.1016/j.neurobiolaging.2012.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yang B, Jiang H, Wang F, et al. UNC13A variant rs12608932 is associated with increased risk of amyotrophic lateral sclerosis and reduced patient survival: a meta‐analysis. Neurol Sci. 2019;40(11):2293‐2302. doi: 10.1007/s10072-019-03951-y [DOI] [PubMed] [Google Scholar]
- 122. Diekstra FP, van Vught PWJ, van Rheenen W, et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(3):630.e3‐630.e8. doi: 10.1016/j.neurobiolaging.2011.10.029 [DOI] [PubMed] [Google Scholar]
- 123. Placek K, Baer GM, Elman L, et al. UNC13A polymorphism contributes to frontotemporal disease in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2019;73:190‐199. doi: 10.1016/j.neurobiolaging.2018.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Katsu M, Hama Y, Utsumi J, et al. MicroRNA expression profiles of neuron‐derived extracellular vesicles in plasma from patients with amyotrophic lateral sclerosis. Neurosci Lett. 2019;708(March):134176. doi: 10.1016/j.neulet.2019.03.048 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable as no datasets were generated or analysed for the writing of this article