

Abstract
Modifying cyclic cell‐penetrating deca‐arginine (cR10) peptides with 4‐(4‐dimethylaminophenylazo)benzoic acid (DABCYL) improves the uptake efficiency of synthetic ubiquitin (Ub) cargoes into living cells. To probe the role of the DABCYL moiety, we performed time‐lapse microscopy and fluorescence lifetime imaging microscopy (FLIM) of fluorescent DABCYL‐R10 to evaluate the impact on cell entry by the formation of nucleation zones. Furthermore, we performed a structure–uptake relationship study with 13 DABCYL derivatives coupled to CPP to examine their effect on the cell‐uptake efficiency when conjugated to mono‐Ub through disulfide linkages. Our results show that through structure variations of the DABCYL moiety alone we could reach, at nanomolar concentration, an additional threefold increase in the cytosolic delivery of Ub, which will enable studies on various intracellular processes related to Ub signaling.
Keywords: Cell-Penetrating Peptides, Chemical Protein Synthesis, Fluorescence, Intracellular Protein Delivery, Nucleation Zones
Modifying cyclic cell‐penetrating peptides with DABCYL derivatives can enhance the formation of nucleation zones for small fluorescent cargoes. Structure variations of the DABCYL moiety significantly increased the cellular uptake efficiency of ubiquitin, resulting in efficient cytosolic transport at nanomolar concentrations.
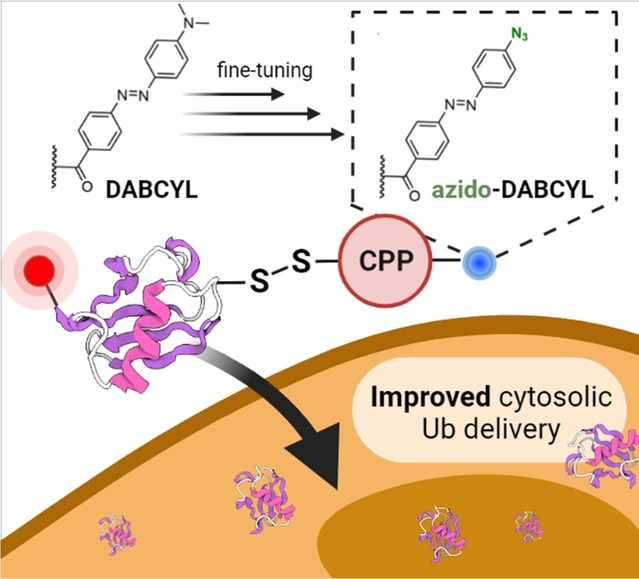
Introduction
Cellular delivery of functional proteins offers great opportunities in therapeutic applications and for a basic understanding of cellular function. [1] Recent reports show that live‐cell delivery of synthetic mono‐ubiquitin (Ub) and/or poly‐Ub‐based probes are important tools to study the deubiquitinating enzymes (DUBs) and to have an insight into their functions in a native environment.[ 2 , 3 , 4 ] In this regard, protein chemistry allows for the synthesis of uniquely modified proteins such as post‐translationally modified proteins and activity‐based probes in which their delivery could be a powerful strategy to probe their cellular functions in a unique way.[ 5 , 6 , 7 ] As of today, the effective delivery of proteins to the cytosol remains a challenge due to their large size, polar nature, and endosomal entrapment. Therefore, to cross the plasma membrane, one must utilize various delivery systems such as liposomes, nanoparticles, cell‐penetrating peptides (CPPs), zinc fingers, supercharged proteins, etc. [8] Among these methods, CPPs have gained a great deal of attention due to their capability to deliver various macromolecules into live cells. [9] This concept was used by the groups of Hackenberger and Cardoso by covalently attaching the CPP to a protein cargo through a cleavable or a stable linker to ensure non‐endosomal intracellular protein delivery in living cells. [10] The CPP‐cargo attachment through a cleavable disulfide bond is a preferred strategy due to the high abundance of thiol‐reducing systems (predominantly glutathione) in the cytosol, which assists in retaining the native protein structure after delivery and avoiding their nucleolar localization. [11]
The development of polyarginine‐based cyclic CPPs has gained particular attention due to their proteolytic stability and better performance of intracellular protein delivery.[ 12 , 13 , 14 ] Several studies showed that the cell‐uptake efficacy of cyclic CPPs conjugated to small molecule cargoes not only depends on the primary structure of the peptide but also on the number of cationic and hydrophobic residues, the presence of chiral amino acid residues, and the extent of hydrogen bonding with the membrane.[ 15 , 16 ] In this context, Marsault and co‐workers reported that modification of the ring size, site of cyclization, and stereochemistry in macrocyclic arginine‐rich cell‐penetrating peptides impact cellular internalization. [17] In addition, the groups of Burlina [18] and Pei [19] showed independently that fluorescent cyclic Arg‐rich CPPs containing either a hydrophobic amino acid or hydrophilic groups attached to amino acid side chains can vary in their cellular uptake mechanism, either by endocytosis or direct transduction. During these investigations, the most active CPP described by the Pei group displayed a direct translocation mechanism for the transport of naphthofluorescein as a fluorescent small model cargo, [19] which led directly after addition to the cells to the formation of bright fluorescent spots at the cellular membrane, so‐called nucleation zones.
Recently, our group has reported a new class of modified cyclic Arg‐rich peptides, where conjugation of DABCYL to a lysine side chain in the exocyclic position of a cyclic deca‐arginine peptide (cR10) improves the cellular uptake of synthetic Ub and small Ub‐like modifier‐2 (SUMO‐2) up to three folds compared to the unmodified cR10.[ 20 , 21 ] The impact of the DABCYL moiety to improve cellular internalization for various types of cargo was also observed by other research groups. Bánóczi et al. reported that the attachment of DABCYL to oligoarginine‐based cell‐penetrating peptides improves cellular uptake efficiency. [22] Other than CPP‐based cell delivery, the effect of DABCYL was also reported in the cellular internalization of a surface‐modified micellar nanoparticle. [23]
Motivated by these recent observations, we now aimed to investigate the improved cellular uptake of DABCYL‐modified CPPs further. In this, we started by probing whether a DABCYL‐modified cR10 conjugated to a small fluorescent cargo shows—similar to previously reported hydrophobic Arg‐rich CPPs[ 18 , 19 ]—the formation of nucleation zones and whether direct transduction or endosomal uptake mechanisms are preferred. Second, we planned to screen the impact of various DABCYL derivatives linked to cyclic CPPs to improve the cellular delivery of ubiquitin cargoes systematically, which would be important to design even better protein delivery systems in the future.
In the course of our study, we have prepared 13 CPP‐Ub analogues with different DABCYL derivatives including the parent DABCYL analogue and studied their cell uptake efficiency. Our results show that through structure variations of the DABCYL alone, we could reach up to threefold increase of Ub delivery over the parent DABCYL‐conjugate, up to 5‐fold increase compared to unmodified cyclic CPP, and up to 10‐fold increase compared to natural TAT CPP.
Results and Discussion
Cellular Uptake of Fluorescent DABCYL‐cR10 Peptides
In order to investigate the potential role of DABCYL, we synthesized C‐terminally Cyanine5‐labeled cR10 (cR10‐Cy5) and its DABCYL conjugate (DABCYL‐cR10‐Cy5) (Scheme S1 and Figure S1–S2) and used these for time‐lapse microscopy in human Bone Osteosarcoma Epithelial Cells (U2OS) cells. We observed nucleation zones after 30 s of incubation for both peptide treatments (Figure 1A, white arrows). Interestingly, U2OS cells treated with 500 nM of DABCYL‐cR10‐Cy5 at 37 °C showed more nucleation zones compared to cR10‐Cy5 treated cells. Nucleation zones are areas on the cell membrane that are considered the points of non‐endocytic entry for CPPs.[ 24 , 25 ] Parallel to previous studies, [13] we observed the disappearance of initially formed nucleation zones (white arrows in Figure 1A), followed by gradual diffusion of fluorescent signal from these nucleation zones to cover the cytoplasm. CPPs are also known to accumulate on the surface of cells, which could trigger endocytosis and macropinocytosis.[ 26 , 27 , 28 ] However, we did not observe the formation of endosomes (commonly seen as highly fluorescent dot‐like signals) originating from the nucleation zones (Supporting Information Movie 1 and 2). Previous studies have shown that the parts of the cell membrane, in which these nucleation zones are observed, have loose membrane lipid packing, thereby allowing CPPs to enter more readily into the cytosol.[ 29 , 30 , 31 ] Consequently, we were curious whether DABCYL conjugation to cR10 has a similar impact on the membrane. To study this, we used the Flipper‐TR probe to monitor changes in membrane tension by measuring the change in its fluorescence lifetime (τ). [32] A lower fluorescence lifetime (τ) value suggests a lower membrane tension, thus loose membrane lipid packing, and vice versa. We observed a significant reduction of fluorescence lifetime (τ) around the areas of nucleation zones in both cR10 and DABCYL‐cR10 treatments indicated by a yellow‐to‐green shift of pixels within nucleation zone areas (Figure 1B, i and C, i). Quantifying the fluorescence lifetime (τ) of 10 nucleation zones across three biological replicates showed a statistically significant decrease in fluorescence lifetime (τ) after peptide treatment (Figure 1B, ii and C, ii). This was not observed in the control setup, in which the fluorescence lifetime remained unchanged after the addition of media (Figure S4). The emission maxima of Flipper‐TR probe is at 600 nm, [33] which is close to the excitation of Cy5 (λ ex=633 nm). Fluorescence resonance energy transfer may occur and could result in decreased fluorescence lifetime. We addressed this by performing the FLIM measurement on cells treated with 1 : 5 DABCYL‐cR10‐Cy5/DABCYL‐cR10—having an excess of unlabeled DABCYL‐cR10 will dampen the energy transfer from excited Flipper‐TR probes while keeping the same treatment concentration. Figure S5 (A) and (B) shows a similar decrease in τ, parallel to Figure 1C. This result suggests that energy transfer should not contribute greatly to the observed decrease in τ.
Figure 1.

A) Time‐lapse microscopy images of U2OS cells treated with 500 nM cR10‐Cy5 or DABCYL‐cR10‐Cy5. Images were taken at 15 s intervals. White arrows indicate nucleation zones. B), C) Fluorescence lifetime imaging microscopy (FLIM) images of U2OS cells before and after the addition of B) cR10‐Cy5 and C) DABCYL‐cR10‐Cy5. (i) upper panel represents Cy5 photon count and lower panels shows FastFLIM images. Scale bar=10 μm. (ii) Graphs showing the fluorescence lifetime (τ in ns) of 10 nucleation zones across three biological replicates. Red line indicates the mean τ. Statistical significance was determined by Student's t‐test. (***, P<0.001; ****, P<0.0001).
Overall, these findings suggest that DABCYL‐cR10 creates nucleation zones on the cell membrane more easily than cR10. Additionally, these nucleation zones create loose membrane packing areas, which further facilitate the CPP entry into cells. Moreover, extending FLIM measurements revealed that the initial formation of nucleation zones results in the highest decrease in membrane tension [Figure S5 (C)]. In addition to these data, our previous report shows that modification of CPP with Black Hole Quencher 2 (BHQ2, with structural similarities to DABCYL), has poor cell delivery efficacy compared to CPP modified with DABCYL. [20] Inspired by these results, we sought to evaluate the effect of different synthetic DABCYL derivatives on the cellular uptake of ubiquitin cargoes in live cells.
Design and Synthesis
Knowing DABCYL as an influential ligand for the cellular internalization of synthetic protein substrates whenever linked to CPP, we envisioned to explore the structural features of the DABCYL ligand which could 1) push the limit of its efficacy and 2) give insight into the structural features that give its properties in the cellular environment. To do so, we intended to generate a library of new CPPs linked to synthetic proteins. To compare the efficiency of the new CPPs, we chose Ub as our model system due to our long‐standing interest in Ub related chemical biology studies.[ 34 , 35 ] The intracellular delivery of Ub conjugates and probes is a promising strategy to shed light on Ub‐regulated biological processes such as protein trafficking and degradation, signal transduction, DNA repair, and cell division, which are related to health and diseases.[ 36 , 37 ]
The DABCYL unit has two aromatic rings connected via a diazo bond where one of the aromatic rings is substituted with N,N‐dimethylamine (named as “ring A”), and the second (named as “ring B”) is connected to a carboxylic acid group. The latter is used to couple with the model CPP. The designed DABCYL derivatives should assist in shedding light on the contribution of aromatic rings, diazo unit, and dimethylamine substitution on ring A (Figure 2A).
Figure 2.

A) Structural features of the DABCYL unit. B)–E) Synthesis of DABCYL derivatives D4–D13.
To investigate the involvement of rings A and B linked via the diazo bond, we added to our list of compounds, the commercially available 4‐(dimethylamino)benzoic acid (D2) and 4‐(phenylazo)benzoic acid (D3) (Figure 4). On the other hand, to better understand the effect of hydrophilic, hydrophobic, and electron donating or withdrawing group on ring A, we synthesized compounds D4–D9 (Figure 2B). To comprehend the participation of the diazo bond in DABCYL, we synthesized compound D10, where the diazo bond is replaced by the ethylene mimic (Figure 2C). A neutral but polar azido group that also has a unique electronic property, may favor interactions with the polar surface of the cell membrane. Hence, the azido substituted DABCYL, D11 (azido‐DABCYL) was also synthesized (Figure 2D). Moreover, aromatic azides could be easily functionalized by the well‐known azide–alkyne click chemistry to install a triazole unit. [38] Therefore, we synthesized D12 and D13 where the triazole unit is present on ring A of DABCYL (Figure 2E). All the compounds were prepared by the known literature procedures and characterized by NMR and HRMS, discussed in Supporting Information section 11 and 24.
Figure 4.
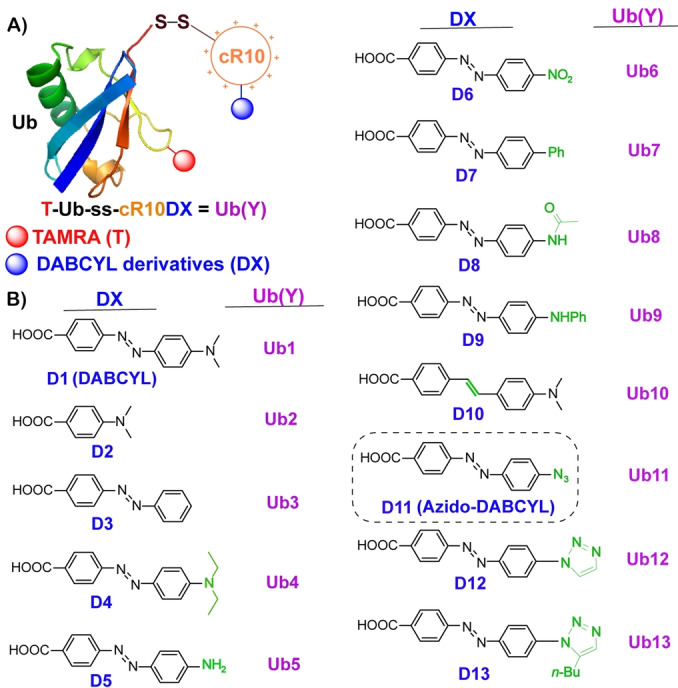
A) General structure of DABCYL derivatives linked to cR10 and conjugated with fluorescently labeled TAMRA Ub. B) DABCYL and its derivatives (D1–D13) were used to modify CPP to generate the corresponding Ub conjugates (Ub1–Ub13).
Synthesis of CPP–Ub Conjugates
For the preparation of the primary sequence of deca‐arginine, all the amino acid residues were coupled in presence of HATU and DIEA on a pre‐swollen Rink amide resin using Fmoc‐SPPS. Subsequent cyclization and then conjugation with various ligands (D1–D13) to the exocyclic lysine residue gave the ligand conjugated cyclic deca‐arginine in an isolated yield of ≈10–20 % (cR10DX, X=1–13, Figure 3A, Scheme S7, and Figure S6–S18). We also synthesized the ligand unmodified CPP, cR10 (Scheme S8 and Figure S19). Finally, the Cys residue was activated using 2,2′‐dithiobis(5‐nitropyridine) (DTNP) [20] to enable conjugation with the model protein (Figure 3B).
Figure 3.

A) General scheme of synthesis of DABCYL derivatives linked to cyclic deca‐arginine (cR10DX, X=1–13). B) General scheme of activated CPP (cR10DX‐TNP) synthesis. C) Synthesis of TAMRA labeled Ub in CPP‐Ub conjugates (TAMRA‐Ub‐ss‐cR10DX). D) Analytical HPLC and mass analysis of purified Azido‐DABCYL conjugated cyclic deca‐arginine (cR10D11) with the observed mass 2645.4±0.6 Da, calcd 2646 Da (average isotopes). E) Analytical HPLC and mass analysis of purified TAMRA‐Ub‐ss‐cR10D11 (Ub11). HPLC chromatogram detected at 214 nm.
The synthesis of the Ub(1–76) unit was carried out on pre‐swollen Rink amide resin using Fmoc‐SPPS, as previously reported.[ 20 , 39 ] Upon completion of synthesis, a PEG linker was coupled followed by coupling of a Cys residue. Consequently, this was coupled with the fluorophore 5‐carboxytetramethylrhodamine (TAMRA) to afford TAMRA‐Cys‐PEG‐Ub(1–76)‐CONH2 in 25 % isolated yield (Figure 3C, Scheme S9, and Figure S22). Finally, the DTNP activated CPP was conjugated with the free Cys residue present in the TAMRA labeled Ub (TAMRA‐Ub). This step was completed within 5 min in 6 M Gn⋅HCl, pH 7.3 at 37 °C for each analogue to give Ub1–Ub13 with an isolated yield of 50–60 % (Figure 3C, Scheme S10, and Figure S23–S35). The unmodified cR10 and Ub conjugate were also obtained using a similar procedure to achieve TAMRA‐Ub‐ss‐cR10 (Ub14) with an isolated yield of 45 % (Figure S36). All the final CPP‐Ub conjugates with various DABCYL derivatives (Ub1–Ub13) are shown in Figure 4.
It is important to mention that we have prepared all these CPPs and their protein conjugates under dark conditions, as we observed that sunlight could influence cis–trans isomerization of DABCYL derivatives. [40] To examine the isomerization effect under light exposure, we have prepared D3 and D11 conjugated LAKAG (as a model peptide, Supporting Information section 15). Under sunlight, a considerable portion of the purified trans conjugates was converted to the cis isomer, which is more polar than the trans isomer (Figure S20, S21). Importantly, we did not detect presence of the cis conjugates when the solutions were kept in dark under our experimental conditions (Figure S20, S21). Isomerization studies of DABCYL derivatives conjugated to CPP‐Ub can be quite challenging, we decided to analyze the HPLC traces for all the conjugated CPPs before Ub conjugation, which did not show any cis/trans isomerization. Therefore, we believe that in the CPP‐Ub conjugates, which were used for cellular delivery studies, the trans isomer of the DABCYL derivatives is predominant.
Live‐Cell Delivery of CPP‐Ub
Having all the fluorescently labeled CPP‐Ub conjugates in hand (Ub1–Ub14), we screened the efficacy of intracellular delivery of the protein conjugates in live U2OS cells. The cells were treated with the synthesized CPP‐Ub analogues at a fixed concentration of 2 μM in serum‐free culture media for 1 h. To exclude the signal of endosomal entrapped proteins from our calculations, we utilized a nuclear masking strategy to measure the effective cytoplasmic delivery of protein conjugates.[ 20 , 41 ] The absolute nuclear TAMRA intensity inside the nucleus of U2OS cells (>150 cells) was quantified by analyzing the high‐resolution CLSM images using Fiji software and comparing the cell delivery efficiency of our listed CPP‐Ub analogues (Figure 4, Supporting Information Section 7).
The cell delivery efficacy for each of Ub1–Ub13 was screened to explore the effect of the structural variations in the DABCYL unit. To investigate the role of the substituents on ring A, we compared the cell delivery efficacy of Ub3 and Ub5 with the parent DABCYL in Ub1 (Figure 5B and Figure S37). We found that the absence of any substitution in ring A (D3) in Ub3 and the presence of primary amine substitution on ring A (D5) in Ub5 did not show a substantial difference in cell delivery compared to the parent Ub1.
Figure 5.

A) Schematic presentation of cellular uptake of CPP‐Ub conjugates; nuclear TAMRA intensity is the measure of active cytosolic delivery of the protein cargo. B)–G) Quantified live‐cell delivery of synthetic CPP‐Ub analogues, Ub1–Ub13 at a fixed concentration of 2 μM using CLSM by nuclear masking method. Data were plotted as mean ± SEM of four independent biological experiments and statistical significance was determined by unpaired t‐test (*, P<0.1; **, P<0.01; ***, P<0.001; ****, P<0.0001; and ns, nonsignificant).
We then continued in further examining the role of the substituents on ring A and the contribution of ring A or B. For this, we included Ub4 and Ub2 and compared their cell delivery efficacy with Ub1 (Figure 5C and Figure S38). We found that increasing the chain length only by the addition of another carbon atom to the N,N‐dimethyl substitution on ring A (D1) to generate D4, present in Ub4, improved the cell delivery up to ≈1.5 fold. This indicates that the nature of the substituent on ring A of DABCYL could play a role in the cell delivery efficiency. Such observation motivated us to investigate the site of modification on ring A more extensively. Moreover, the presence of D2 reduced the cell delivery of Ub2 up to ≈1.4‐fold.
With this initial screen, we concluded that the presence of rings A and B is an essential part of DABCYL's assistance in cell delivery. This initial screen showed an increase in the order of cell delivery, Ub2<Ub1<Ub4, which correlates with increased hydrophobicity of the DABCYL derivatives and aligned well with their calculated logP (Figure 5C and Supporting Information Section 12).
To further check the effect of substitution on ring A, we tested the cell delivery of Ub6 and Ub7 (Figure 5D and Figure S39). Compound Ub6 showed almost similar cell delivery efficacy compared to the parent Ub1, whereas compound Ub7 showed up to ≈2.1‐fold improvement. Clearly, this shows that the presence of phenyl group on ring A (D7) in Ub7, which provides further hydrophobicity (logP D7 =5.68), improved the cell delivery efficacy.
To examine the role of various amine functionalities on ring A, considering the activity of Ub4, we screened Ub8, Ub9, and Ub11 (Figure 5E and Figure S40). The screening results showed improvement in cell delivery efficacy for Ub9 and Ub11 up to 1.7‐fold and 2.7‐fold, respectively, while Ub8 showed a decrease up to 2.3‐fold compared to the parent compound Ub1. Apparently, the presence of hydrophobic phenylamine functionality in ring A (D9, logP D9 =5.48) in Ub9 improves hydrophobic interaction with the cell membrane and the subsequent cell delivery.[ 19 , 42 ] The presence of the partial polar acetamide functionality on ring A of D8 in Ub8 decreased the cell delivery. Importantly, the neutral but polar azido functionality instead of dimethylamine on the DABCYL moiety (Azido‐DABCYL, D11) in Ub11 favored the cell permeability. Furthermore, we have compared the cell‐uptake efficiency of Ub11 with CPP‐Ub analogues linked to DABCYL derivative, containing triazole small unit, Ub12, and Ub13 (Figure 5F and Figure S41). This screening showed a decrease in the cell delivery efficacy of triazole‐containing compounds. The slight improvement in cell delivery of Ub13 over Ub12 could be due to the hydrophobicity provided by the butyl moiety attached to the triazole unit.
To comprehend the participation of the diazo bond in DABCYL, we have screened Ub10. However, the compound showed high cytotoxicity in U2OS cells at 2 μM. Hence, no further alterations to the diazo bond were carried out.
Finally, to find the most effective DABCYL derivative, we picked the most active compounds (Ub4, Ub7, Ub9, and Ub11) over various screening steps, Figure 5(B–F), and examined their cell delivery efficiency in a single frame (Figure 5G and Figure S42). The increased order of cell delivery efficacy, Ub1<Ub4<Ub9<Ub7<Ub11 revealed that the azido‐DABCYL derivatives in compound Ub11 have the highest cell delivery efficacy among all the potent compounds. The quantified result showed up to 3‐fold improved cell delivery efficiency of Ub11 than Ub1.
These results were further confirmed by measuring the nuclear TAMRA signal using an automated digital fluorescence microscope CytationTM 5 (Figure S43). These results correlate well with the increased order of logP value for compounds and with their cell delivery (Ub1<Ub4<Ub9<Ub7). However, the most active compound Ub11 has the lowest logP compared to Ub4, Ub9, and Ub7, which may indicate that the contribution of the azide functionality is more prominent than the added hydrophobicity by the other substituents to interact with the cell membrane. [43] It is worth mentioning that Ub11 retained its chemical integrity, based on our HPLC‐MS analysis, under our cell delivery assay conditions (Supporting Information section 20 and Figure S44). Moreover, Ub11 (CPP modified by Azido‐DABCYL) shows up to 5‐fold higher cell delivery efficiency compared to unmodified cR10 in Ub14 (Figure 6). Including Ub1 (CPP modified by DABCYL) in the same experimental frame shows, our stepwise improvement in the intracellular protein delivery area in a unique way, simply by exocyclic side‐chain modification in cyclic CPP using small molecule ligands (Figure 6Q). We also compared the delivery efficacy of Ub11 to a natural CPP, HIV1(47–57) TAT peptide [44] conjugated to Ub, Ub15 (Supporting Information section 21 and Figure S45, S46). The AzidoDABCYL‐cR10 in Ub11 shows up to 10‐fold higher delivery efficacy compared to the TAT peptide in Ub15. This supports the usefulness of synthetic AzidoDABCYL‐cR10 CPP over a natural CPP, (Figure S47).
Figure 6.

Representative images of delivery of Ub analogues Ub (A)–(D), Ub14 (E)–(H), Ub1 (I)–(L), and Ub11 (M)–(P) to live U2OS cells at 2 μM. A), E), I), M) Ub, Ub14, Ub1, and Ub11 Hoechst (blue). B), F), J), N) (TAMRA, red). C), G), K), O) TAMRA and Hoechst channels combined. D), H), L), P) Bright field channel. Scale bars 20 μm. Q) Quantification of nuclear TAMRA intensity of the cell images after delivery of Ub, Ub14, Ub1, and Ub11 under Hoechst relative to the untreated cells. Data were plotted as mean ± SEM of three independent biological experiments (over 150 cells per experiment) and statistical significance was determined by an unpaired t‐test (****, P<0.0001).
Having the most effective CPP in hand, we further optimized the minimum concentration for cytosolic delivery of Ub11. The concentration‐dependent uptake results showed that at only 500 nM, Ub11 can be internalized and freely diffused into live cells with up to 3‐fold higher efficiency compared to Ub1 (Figure 7(A–I)). From the confocal images, we also quantified the percentage of cells having nuclear TAMRA intensity relative to the total number of cells which was found ≈25 % for Ub1 and ≈55 % for Ub11, these results correlate well with the improved cell delivery of Ub11 (Figure 7J).
Figure 7.

Representative images of delivery of Ub analogues Ub1 (A)–(D) and Ub11 (E)–(H) to live U2OS cells at 500 nM. A), E) Ub1 and Ub11 Hoechst (blue). B), F) (TAMRA, red). C), G) TAMRA and Hoechst channels combined. D), H) Bright field channel. Scale bars 10 μm. I) Quantification of nuclear TAMRA intensity of the cell images after delivery of Ub1 and Ub11 under Hoechst relative to the untreated cells. J) Quantification of the percentage of cells to have cytosol‐delivered Ub1 and Ub11 which was achieved by calculating the total number of cells and the number of cells having TAMRA signal using FiJi software. Data were plotted as mean ± SEM of three independent biological experiments (over 150 cells per experiment) and statistical significance was determined by unpaired t‐test (****, P<0.0001 and **, P<0.01).
To validate the effect of serum on live‐cell delivery of CPP‐Ub conjugates, we compared cytosolic delivery of Ub1 and Ub11 in presence of 5 % fetal bovine serum (Supporting Information section 22 and Figure S48), and the result showed up to 4‐fold higher cell delivery efficiency in the case of AzidoDABCYL‐cR10 in Ub11 compared to DABCYL‐cR10 in Ub1. This shows that in presence of serum does not negatively affect the relative cytosolic delivery of the Ub11.
CPPs are prone to trap in the cell membranes at high concentrations subsequently damaging cell membranes. [45] To validate the membrane integrity at various concentrations of Ub11, we have used a well‐known SYTOX blue assay (Supporting Information section 23). [3] In our delivery conditions of Ub11, we did not detect any cells with the compromised plasma membrane, with up to 4 μM of our compound (Figure S49), which supports the conjugation of Azido‐DABCYL to cR10 does not compromise membrane integrity in the cell delivery experiments.
Conclusion
In summary, we present further experimental evidence that supports the benefits of DABCYL modifications to improve the cellular uptake of cargoes conjugated to cyclic Arg‐rich peptides. First, we showed that a fluorescent DABCYL‐modified cR10‐peptide yielded increased formation of nucleation zones, i.e., points of entry for CPPs, with reduced membrane tension on the cell membrane without the direct formation of endosomes, which further validated the transduction mechanism of these cyclic CPPs. To further enhance previously introduced DABCYL‐cR10 peptides for the cellular uptake of Ub, we have synthesized a library of CPP‐Ub analogues with different DABCYL derivatives linked to the exocyclic lysine residue of the cyclic CPP. Our results show that the cellular uptake efficiency is influenced by minor structural variations in the DABCYL. This work examined the possible role of aromatic rings, dimethylamine substitution, and diazo unit in DABCYL towards its assistance in intracellular protein delivery perspective. The relative cell‐uptake screening of CPP‐Ub analogues resulted in Azido‐DABCYL as the most effective derivative for intracellular Ub delivery with up to 3‐fold and 5‐fold improvement compared to the DABCYL modified and unmodified cR10, respectively. Moreover, the presence of serum does not negatively affect the relative cytosolic delivery by the cyclic CPPs.
In summary, this study shows the usefulness of synthetic DABCYL‐based cyclic CPPs at nanomolar concentration of the protein conjugates. We expect that our continuous improvement in delivering ubiquitin conjugates into living cells will help our understanding of Ub and Ub‐like modifiers involved in numerous biological processes and ultimately help in the development of pharmacologically relevant applications in near future.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
A.B. holds The Jordan and Irene Tark Academic Chair. This research was supported by a joint grant to A.B. and C.P.R.H. from the GIF, the German‐Israeli Foundation for Scientific Research and Development (I‐1488‐302.5/2019). C.P.R.H. acknowledges support from the DFG (RTG2473 “Bioactive Peptides”, Project‐number 392923329). J.V.V.A. is funded by the Alexander von Humboldt Fellowship for Postdocs and J.G.G. is funded by Margarita Salas Fellowship from Spanish Ministry of Universities. We thank Dr. Gandhesiri Satish, Dr. Ganga B. Vamisetti, and Dr. Guy Mann (Technion‐Haifa) for their valuable suggestions. We also thank Dr. Martin Lehmann and Dr. Agata Witkowska (FMP‐Berlin) for their assistance with the FLIM measurement. We also acknowledge BioRender.com for assistance in schematic figure preparations.
A. Saha, S. Mandal, J. V. V. Arafiles, J. Gómez-González, C. P. R. Hackenberger, A. Brik, Angew. Chem. Int. Ed. 2022, 61, e202207551; Angew. Chem. 2022, 134, e202207551.
Contributor Information
Prof. Christian P. R. Hackenberger, Email: Hackenbe@fmp-berlin.de.
Prof. Ashraf Brik, Email: abrik@technion.ac.il.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Fu A., Tang R., Hardie J., Farkas M. E., Rotello V. M., Bioconjugate Chem. 2014, 25, 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gui W., Ott C. A., Yang K., Chung J. S., Shen S., Zhuang Z., J. Am. Chem. Soc. 2018, 140, 12424–12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mann G., Satish G., Meledin R., Vamisetti G. B., Brik A., Angew. Chem. Int. Ed. 2019, 58, 13540–13549; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13674–13683. [Google Scholar]
- 4. Wang Y., Chen J., Hua X., Meng X., Cai H., Wang R., Shi J., Deng H., Liu L., Li Y., Angew. Chem. Int. Ed. 2022, 61, e202203792; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202203792. [DOI] [PubMed] [Google Scholar]
- 5. David Y., Vila-Perelló M., Verma S., Muir T. W., Nat. Chem. 2015, 7, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rabideau A. E., Pentelute B. L., ACS Cent. Sci. 2015, 1, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mann G., Satish G., Sulkshane P., Mandal S., Glickman M. H., Brik A., Chem. Commun. 2021, 57, 9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deprey K., Becker L., Kritzer J., Plückthun A., Bioconjugate Chem. 2019, 30, 1006–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sawant R. R., Patel N. R., Torchilin V. P., Eur. J. Nanomed. 2013, 5, 141–158. [Google Scholar]
- 10. Nischan N., Herce H. D., Natale F., Bohlke N., Budisa N., Cardoso M. C., Hackenberger C. P. R., Angew. Chem. Int. Ed. 2015, 54, 1950–1953; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1972–1976. [Google Scholar]
- 11. Schneider A. F. L., Wallabregue A. L. D., Franz L., Hackenberger C. P. R., Bioconjugate Chem. 2019, 30, 400–404. [DOI] [PubMed] [Google Scholar]
- 12. Lättig-Tünnemann G., Prinz M., Hoffmann D., Behlke J., Palm-Apergi C., Morano I., Herce H. D., Cardoso M. C., Nat. Commun. 2011, 2, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duchardt F., Fotin-Mleczek M., Schwarz H., Fischer R., Brock R., Traffic 2007, 8, 848–866. [DOI] [PubMed] [Google Scholar]
- 14. Herce H. D., Schumacher D., Schneider A. F. L., Ludwig A. K., Mann F. A., Fillies M., Kasper M.-A., Reinke S., Krause E., Leonhardt H., Hackenberger C. P. R., Nat. Chem. 2017, 9, 762–771. [DOI] [PubMed] [Google Scholar]
- 15. Bechara C., Sagan S., FEBS Lett. 2013, 587, 1693–1702. [DOI] [PubMed] [Google Scholar]
- 16. Dougherty P. G., Sahni A., Pei D., Chem. Rev. 2019, 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Traboulsi H., Larkin H., Bonin M.-A., Volkov L., Lavoie C. L., Marsault É., Bioconjugate Chem. 2015, 26, 405–411. [DOI] [PubMed] [Google Scholar]
- 18. Amoura M., Illien F., Joliot A., Guitot K., Offer J., Sagan S., Burlina F., Chem. Commun. 2019, 55, 4566–4569. [DOI] [PubMed] [Google Scholar]
- 19. Song J., Qian Z., Sahni A., Chen K., Pei D., ChemBioChem 2019, 20, 2085–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mandal S., Mann G., Satish G., Brik A., Angew. Chem. Int. Ed. 2021, 60, 7333–7343; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 7409–7419. [Google Scholar]
- 21. Arafiles J. V. V., Futaki S., Nat. Chem. 2021, 13, 517–519. [DOI] [PubMed] [Google Scholar]
- 22. Szabó I., Illien F., Dókus L. E., Yousef M., Baranyai Z., Bősze S., Ise S., Kawano K., Sagan S., Futaki S., Amino Acids 2021, 53, 1033–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roloff A., Nelles D. A., Thompson M. P., Yeo G. W., Gianneschi N. C., Bioconjugate Chem. 2018, 29, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karikó K., Muramatsu H., Keller J. M., Weissman D., Mol. Ther. 2012, 20, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Verdurmen W. P. R., Thanos M., Ruttekolk I. R., Gulbins E., Brock R., J. Controlled Release 2010, 147, 171–179. [DOI] [PubMed] [Google Scholar]
- 26. Nakase I., Hirose H., Tanaka G., Tadokoro A., Kobayashi S., Takeuchi T., Futaki S., Mol. Ther. 2009, 17, 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Futaki S., Nakase I., Acc. Chem. Res. 2017, 50, 2449–2456. [DOI] [PubMed] [Google Scholar]
- 28. Kosuge M., Takeuchi T., Nakase I., Jones A. T., Futaki S., Bioconjugate Chem. 2008, 19, 656–664. [DOI] [PubMed] [Google Scholar]
- 29. Sakai T., Kawano K., Iino M., Takeuchi T., Imanishi M., Futaki S., ChemBioChem 2019, 20, 2151–2159. [DOI] [PubMed] [Google Scholar]
- 30. Murayama T., Masuda T., Afonin S., Kawano K., Takatani-Nakase T., Ida H., Takahashi Y., Fukuma T., Ulrich A. S., Futaki S., Angew. Chem. Int. Ed. 2017, 56, 7644–7647; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7752–7755. [Google Scholar]
- 31. Schneider A. F. L., Kithil M., Cardoso M. C., Lehmann M., Hackenberger C. P. R., Nat. Chem. 2021, 13, 530–539. [DOI] [PubMed] [Google Scholar]
- 32. Colom A., Derivery E., Soleimanpour S., Tomba C., Molin M. D., Sakai N., González-Gaitán M., Matile S., Roux A., Nat. Chem. 2018, 10, 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Soleimanpour S., Colom A., Derivery E., Gonzalez-Gaitan M., Roux A., Sakai N., Matile S., Chem. Commun. 2016, 52, 14450–14453. [DOI] [PubMed] [Google Scholar]
- 34. Mali S. M., Singh S. K., Eid E., Brik A., J. Am. Chem. Soc. 2017, 139, 4971–4986. [DOI] [PubMed] [Google Scholar]
- 35. Weller C. E., Pilkerton M. E., Chatterjee C., Biopolymers 2014, 101, 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Glickman M. H., Ciechanover A., Physiol. Rev. 2002, 82, 373–428. [DOI] [PubMed] [Google Scholar]
- 37. van Wijk S. J. L., Fulda S., Dikic I., Heilemann M., EMBO Rep. 2019, 20, e46520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2708–2711. [Google Scholar]
- 39. Erlich L. A., Kumar K. S. A., Haj-Yahya M., Dawson P. E., Brik A., Org. Biomol. Chem. 2010, 8, 2392–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Siampiringue N., Guyot G., Monti S., Bortolus P., J. Photochem. 1987, 37, 185–188. [Google Scholar]
- 41. Luther D. C., Jeon T., Goswami R., Nagaraj H., Kim D., Lee Y.-W., Rotello V. M., Bioconjugate Chem. 2021, 32, 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tripathi S. K., Singh V. P., Gupta K. C., Kumar P., J. Mater. Chem. B 2013, 1, 2515–2524. [DOI] [PubMed] [Google Scholar]
- 43. Zhang R., Qin X., Kong F., Chen P., Pan G., Drug Delivery 2019, 26, 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Torchilin V. P., Adv. Drug Delivery Rev. 2008, 60, 548–558. [DOI] [PubMed] [Google Scholar]
- 45. Iwata T., Hirose H., Sakamoto K., Hirai Y., Arafiles J. V. V., Akishiba M., Imanishi M., Futaki S., Angew. Chem. Int. Ed. 2021, 60, 19804–19812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 19957–19965. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.