

Abstract
Hydrogen‐bonded squaramide (SQ) supramolecular polymers exhibit uncommon thermoreversible polymorph transitions between particle‐ and fiber‐like nanostructures. SQs 1–3, with different steric bulk, self‐assemble in solution into particles (AggI) upon cooling to 298 K, and SQs 1 and 2, with only one dendronic group, show a reversible transformation into fibers (AggII) by further decreasing the temperature to 288 K. Nano‐DSC and UV/Vis studies on SQ 1 reveal a concentration‐dependent transition temperature and ΔH for the AggI‐to‐AggII conversion, while the kinetic studies on SQ 2 indicate the on‐pathway nature of the polymorph transition. Spectroscopic and theoretical studies reveal that these transitions are triggered by the molecular reorganization of the SQ units changing from slipped to head‐to‐tail hydrogen bonding patterns. This work unveils the thermodynamic and kinetic aspects of reversible polymorph transitions that are of interest to develop stimuli‐responsive systems.
Keywords: Hydrogen Bonding, Squaramides, Stimuli-Responsive Systems, Supramolecular Polymers, Supramolecular Polymorphism
Squaramide supramolecular polymers exhibit uncommon thermoreversible polymorph transitions between fiber‐ and particle‐like nanostructures driven by the molecular rearrangement of the squaramide units from slipped to head‐to‐tail hydrogen bonds. Thermodynamic and kinetic studies reveal the concentration dependent nature and the on‐pathway mechanism of the polymorph transitions which are of interest as stimuli‐responsive systems.

Introduction
Understanding the complex self‐assembly pathways of functional molecules is crucial to control the morphology and properties of supramolecular polymers.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 ] In recent years, it has become evident that the occurrence of distinct kinetic and thermodynamic assemblies (pathway complexity) for specific building blocks strongly depends on the molecular structure, the chosen experimental conditions, and the sample processing techniques.[ 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 ] In typical examples of pathway complexity, the kinetically controlled species evolve over time into thermodynamically more stable states, via on‐pathway[ 9 , 12 , 13 , 14 , 15 , 16 ] or off‐pathway[ 10 , 11 , 17 , 18 , 19 , 20 , 21 ] mechanisms. Both processes have been observed for different molecular systems in organic solvents including oligo(p‐phenylenevinylenes), [10] porphyrins,[ 11 , 13 ] perylene bisimides (PBIs), [17] merocyanines, [9] N‐annulated perylenes,[ 15 , 19 ] and naphtalene bisimides, [20] among other monomers. In most literature examples, the kinetic and thermodynamic states present a significant energy difference, which typically prevents the isolation of the kinetic products due to their short lifetimes.[ 2 , 3 , 4 , 5 , 6 , 7 , 8 ]
In some cases, the stabilization energy of the different aggregated states differs only by a few kJ mol−1, and they can be isolated under specific conditions in the same solvent, giving rise to supramolecular polymorphs.[ 22 , 23 , 24 , 25 , 26 , 27 ] For example, the formation of distinct 1D (fibrillar) supramolecular polymorphs with different molecular arrangements has been reported for Pt complexes [24] and PBIs [25] as a function of the sample processing (fast/slow heating‐cooling process and sonication, respectively). Similarly, lamellar and fibrillar polymorphs have also been reported for squaraines, [28] PBIs, [29] and barbiturates. [30] However, the interconversion between supramolecular polymorphs is not straightforward, and commonly requires multistep sample processing methods which, in some cases, result in no direct conversion but via intermediate monomeric or kinetic states.[ 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 ]
Importantly, since supramolecular polymorphs exhibit different molecular packing, morphology, and/or properties, controlled polymorph transformations are of great interest to develop stimuli‐responsive systems. However, direct polymorph transitions are challenging to achieve, particularly in organic solvents where solvophobic effects are attenuated.[ 22 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 ] Among the scarce non‐aqueous examples, a family of ester‐bisurea monomers has been reported that reversibly self‐assemble into three distinct hydrogen‐bonded (H‐bonded) fibrillar nanostructures depending on the temperature, which provide different viscosity to the solution.[ 38 , 39 , 40 , 41 ] These examples demonstrate that supramolecular polymers featuring polymorph transformations are potentially interesting stimuli‐responsive systems, especially if the transitions between the polymorphs are direct, reversible, and accompanied by significant structural/morphological changes.
Herein we report on new H‐bonded squaramide (SQ) supramolecular polymers exhibiting direct thermoreversible polymorph transitions with remarkable morphology change between particle‐ and fiber‐like nanostructures (Figure 1). Our study involves three new SQ monomers (1–3, Figure 1a) with different steric bulk that self‐assemble into AggI (nanoparticles) upon cooling from 365 to 298 K in methylcyclohexane (MCH). Remarkably, the SQ derivatives 1 and 2 with only one bulky 3,4,5‐tridodecyloxybenzyl substituent exhibit uncommon reversible aggregate transitions from AggI to a second polymorph AggII (fibers) upon decreasing the temperature below 288 K. Theoretical studies reveal that this transition is triggered by a molecular reorganization of the SQ groups that switch from slipped (AggI) to head‐to‐tail (AggII) H‐bonded patterns (Figure 1b). The temperature‐driven AggI‐to‐AggII transformation is fast in SQ 1, but proceeds with slower kinetics for SQ 2. Nano‐differential scanning calorimetry (DSC) studies of SQ 1 reveal a concentration/morphology dependence of the thermodynamics of the polymorph transitions, whereas kinetic studies of SQ 2 point to an on‐pathway conversion mechanism.
Figure 1.

a) Molecular structures of SQ derivatives 1–3. b) Schematic representation of the self‐assembly behavior of SQs 1–3 into AggI and the thermoreversible AggI‐to‐AggII transition for 1 and 2.
Results and Discussion
Self‐Assembly UV/Vis Studies
The aggregation behavior of 1–3 (Supporting Information) was initially assessed through variable‐concentration and variable‐temperature (VT) UV/Vis measurements in MCH between 365 and 298 K (Figure 2 and S5–S14). SQs 1–3 exhibit the characteristic SQ broad monomeric band peaking at 285 nm at high temperatures (365 K)[ 16 , 43 , 44 ] (Figure 2a, black line), ascribed to two bright electronic transitions mainly described by HOMO−1, HOMO→LUMO, LUMO+1 and HOMO−1, HOMO→LUMO+2, LUMO+3 monoexcitations of π‐π* character. [16] The monomeric absorption profile of SQ 1 undergoes a gradual blueshift concomitantly with a substantial broadening covering the region from ca. 255 up to 340 nm by cooling down to 290–298 K (Figure 2a, red line). The broad profile of this absorption band suggests the formation of an ill‐defined aggregate (hereafter AggI). SQs 2 and 3 showed similar VT‐UV behavior (Figures S8 and S9). The cooling curves of SQ 1 show a non‐sigmoidal transition between the monomeric species and AggI, indicating a cooperative self‐assembly process (Figure 2b).[ 45 , 46 ] The experimental data could be fitted to the cooperative nucleation‐elongation model, [47] revealing a cooperativity factor of σ=0.04–0.05 (Table S1). The self‐assembly of SQ 2 followed a far less cooperative mechanism (σ=0.8, Table S2). In contrast, the self‐assembly of SQ 3 was fitted to the isodesmic mechanism, but the observed slight deviations at intermediate temperatures suggest that a weakly anti‐cooperative mechanism might be operative (Table S3).[ 48 , 49 , 50 , 51 ]
Figure 2.

a) VT‐UV absorption spectra of 1 (3×10−5 M, MCH). Cooling rate: 1 K min−1. b) Aggregation degree (αagg) vs. temperature extracted from VT‐UV at λ=319 nm at different concentrations along with the corresponding fits to the nucleation‐elongation model using the global fitting approach. c) ϵ vs. temperature (325–278 K) extracted from VT‐UV at λ=319 nm at different concentrations. d) Polymorph diagram for SQ 1 obtained from cooling cycles of the VT‐UV (dots) and nano‐DSC (squares) experiments.
Unexpectedly, by further cooling down SQ 1 (3×10−5 M, MCH) to 281 K, the UV absorption pattern of AggI evolved into two defined bands at 273 and 319 nm (Figure 2a, blue line), which was not observed for SQs 2 and 3. These spectral changes are attributed to the conversion of AggI into a second aggregate, from now on termed AggII. This two‐band UV pattern of AggII is characteristic for a hydrogen‐bond‐driven head‐to‐tail assembly of SQs (Figure 1b).[ 16 , 43 ] Additional VT‐UV/Vis experiments revealed that this AggI‐to‐AggII transition is reversible (Figures S10 and S11), slightly concentration‐dependent (Figure 2c), and occurs at ca. 288 K (3×10−5 M). Figure 2d shows the polymorph phase diagram of SQ 1 as a function of concentration and temperature extracted from the UV/Vis and nano‐DSC (see below) results. This AggI‐to‐AggII conversion is not directly observed in the VT‐UV/Vis experiments for compounds 2 and 3. However, we found that by aging a solution of SQ 2 (4×10−5 M) for 2–5 hours at 278 K, 2‐AggI also evolved to 2‐AggII (Figure S12), revealing that the process is governed by kinetic effects for this compound. No evolution of 3‐AggI to 3‐AggII was observed by aging the samples at 278 K, even after 1 month.
Morphology Studies
The morphology of AggI and AggII was initially assessed by atomic force microscopy (AFM). Spin‐coating small volumes of AggI (V=10 μL, 5×10−5 M, T=298 K, MCH) onto highly‐oriented pyrolytic graphite (HOPG) revealed the formation of small nanoparticles (height=14.7±3.6 nm) (Figures 3a and S15–S17). Identical results were obtained for SQs 1–3, which agrees with the similar spectroscopic properties of AggI observed for all compounds (Figure S13). In contrast, cooling the solutions of SQ 1 and 2 at 278 K for 3 hours led to the transformation into fibrillar arrangements (≈10 nm) (AggII) that exhibit close lateral contacts into closely packed lamellar arrangements on HOPG (Figures 3b, S15, and S16). Note that a partial fracturing of individual fibers could be observed upon close inspection, which can be attributed to coating and drying effects, commonly observed in AFM studies.[ 52 , 53 ]
Figure 3.

AFM images of a) 2‐AggI and b) 2‐AggII (5×10−5 M) drop‐casted onto HOPG at 298 and 278 K, respectively. c) SAXS curves of SQ 2 in MCH at 5×10−3 M at 286 (blue triangles) and 298 K (orange squares), and at 1×10−3 M at 298 K (red circles). The samples at 5×10−3 M were best fitted to the flexible cylinder model (orange and blue lines) and the sample at 1×10−3 M was fitted to the sphere customized model (red line). d) SLS counts of SQ 2 at 298 K (AggI) as function of the concentration in MCH.
Small‐angle X‐ray scattering (SAXS) experiments for SQs 1–3 were recorded in MCH at different concentrations and temperatures (Figures 3c, S18, and S19, and Tables S4–S6). These studies revealed SAXS curves with practically no q−1 slope for the AggI of SQs 1 and 2 at 298 K and 1×10−3 M (Figures 3c, red circles, and S18), which are characteristic of discrete aggregates.[ 50 , 54 , 55 , 56 ] The fitting of the experimental curves indicated that SQs 1 and 2 at 298 K form small spherical aggregates (Tables S4 and S5), in line with the AFM observations. However, no suitable SAXS curves (low intensity) were obtained for SQ 3 and for 1 and 2 at lower temperatures. To overcome these experimental issues, additional SAXS experiments were carried out at higher concentrations. Figure 3c shows the SAXS profiles for SQ 2 at 5×10−3 M at 298 (orange) and 288 K (blue). The experimental curves obtained at both temperatures fit best to the flexible cylinder customized model with a radius of r ≈8.2 Å for AggI (298 K) and 8.8 Å for AggII (288 K) (Table S5).[ 22 , 43 , 54 ] Similar results were obtained for SQs 1 and 3 (Tables S4 and S5). These results agree with the AFM results for AggII, but point to a change in the morphology of AggI (298 K) at higher concentrations, from particles to fibers. Nevertheless, the UV patterns of SQs 1–3 at 298 K (AggI) at 1×10−3 and 5×10−3 M are very similar (Figures S13 and S14), suggesting that, despite the distinct morphologies, the molecular packing remains unaltered.
The size/morphology effects of the AggI of 1–3 (MCH, 298 K) were further analyzed by concentration‐dependent static light scattering (SLS) (Figures 3d and S20). At low concentrations (<2×10−3 M), the SLS experiment of SQ 2 (Figure 3d) shows a slight gradual increase in the counts (intensity/kcps) with the concentration, which was linked to slight increments in the size of AggI. In contrast, above a concentration threshold of 2×10−3 M, a more pronounced increase in the counts upon increasing concentration was observed. The same trend was observed for SQs 1 and 3 (Figure S20). In line with the SAXS and AFM findings, these two concentration‐dependent size variation regimes of the AggI of SQs 1–3 were correlated with the formation of two distinct morphologies, particles at lower, and fibers at higher concentrations. Considering that both species have similar UV/Vis profiles and molecular packing, the morphological change of AggI can be attributed to hierarchical effects, where the nanoparticles arrange into fibrillar structures by increasing the concentration (Figure S21). [57]
Self‐Assembly Mode of AggI and AggII
The FT‐IR experiments of SQs 1–3 at 298 and 278 K in MCH‐d14 (Figures 4 and S22) revealed the presence of H‐bonds in AggI and AggII, but the two aggregates present differences in the H‐bonding pattern. For example, the FT‐IR spectrum of 2 (5×10−3 M) at 298 K (AggI) (Figure 4) shows a broad N−H stretching band at 3300 cm−1, while the C=O band appears at 1654 cm−1 with a shoulder at 1670 cm−1. This shoulder of the C=O signal is probably caused by an unsymmetric or ill‐defined interaction pattern between SQs.[ 16 , 43 ] By decreasing the temperature, the FT‐IR spectrum of compound 3 (AggI) remained unaltered (Figure S22), but those registered for 1 and 2 (AggII) display a symmetrization and a sharpening of the N−H and C=O stretching bands together with a displacement towards lower wavenumbers (Figure 4). This behavior is consistent with the formation of symmetric head‐to‐tail H‐bonding interactions in AggII, where the SQs units would display a Z,Z conformation (Figure 1b and Scheme S2),[ 16 , 43 , 58 ] as also supported by NMR (Figure S23).
Figure 4.
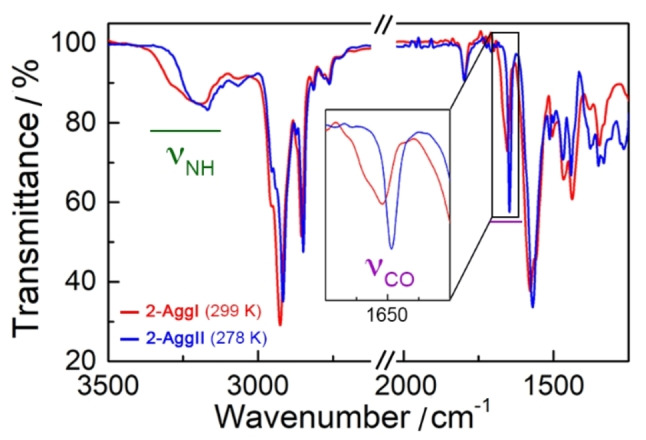
Infrared spectra of 2 (5×10−3 M, MCH‐d14 ) at 299 (AggI, red) and 278 K (AggII, blue). Inset shows the magnification of the C=O signals.
The self‐assembly mode of AggI and AggII was further studied at atomistic scale by quantum‐chemical calculations (GFN2‐xTB) [59] for 20‐mers of SQs 1–3. To build these oligomers, all the SQ units were disposed in Z,Z conformation according to the experimental results. For the supramolecular growth, we considered slipped and head‐to‐tail interacting H‐bonding patterns between vicinal SQs according to the literature.[ 16 , 43 , 54 , 58 ] Figure 5 displays two optimized 20‐mers for SQ 1 associated with AggI and AggII whereas Figures S30 and S31 show the optimized models for SQ 2 and 3, respectively. For SQ 1, the self‐assembly process giving rise to AggI can be seen as a supramolecular growth of a dimer in which the SQ units are slightly slipped and the 3,4,5‐tridodecyloxybenzyl units are in alternating positions (Figure 5a). This agrees with the information inferred from 1H NMR‐NOE experiments of AggI for SQs 1 and 2 (Figures S24–S29). Considering the centroids of the SQ units, the next interacting dimer in the aggregate is rotated by ca. 27° (θ) and displaced by around 10.70 Å (d c). In the resulting aggregate, only a carbonyl group of each SQ forms H‐bonds with the two N−H groups of the vicinal SQ with similar distances around 1.95 Å (d HB). Importantly, this supramolecular arrangement implies no significant internal reorganization of the interacting SQ monomers and avoids hindrance effects between the bulky trisdodecyloxybenzyl groups. Additionally, it can easily give rise to oligomers of different size that may interact between them without any long‐range hierarchical order, in line with the nanoparticle‐like morphology observed for AggI (Figure 3a).
Figure 5.

Minimum‐energy structures calculated at the GFN2‐xTB level, including solvent effects (n‐hexane), for the AggI (a) and AggII (b) models of SQ 1. Relevant intermolecular distances (d c and d HB) are indicated in Å and the rotational angle between vicinal SQ dimers θ in degrees (°). Color coding: C in green, N in blue, O in red and H in white.
In contrast to AggI, a well‐organized head‐to‐tail H‐bonding pattern is predicted for AggII of 1 (Figure 5b). In this aggregate, the two C=O groups of each SQ unit form H‐bonds with the respective N−H groups of the vicinal SQ with distances of around 1.82 Å in a very symmetric way. Similar to AggI, the interacting SQs are disposed in an alternating manner to mitigate the repulsion between the bulky chains. This linear head‐to‐tail arrangement generates a permanent dipole moment that is likely to be compensated by an antiparallel π‐π stacking between SQs of a similar linear assembly.[ 16 , 43 ] Note that similar structural models were obtained for AggI and AggII of SQs 2 and 3 (Figures S30 and S31). For the latter, however, the head‐to‐tail arrangement implies an important hindrance effect between the side dendrons of the symmetric structure of SQ 3 (Figure S31b).
To analyze the relative stability of AggI and AggII for SQs 1–3, three SQ units were removed from each end of the optimized 20‐mer (i.e. a 14‐mer) to mitigate terminal effects due to the lack of periodic boundary conditions and environmental effects. Single‐point calculations were then performed at the GFN2‐xTB level for 14‐mers of 1–3. For SQ 1, similar energies per monomer unit were obtained for both aggregates AggI and AggII with a difference of 2.97 kJ mol−1, AggII being the most stable due to the slightly more favorable H‐bonding network (4 linear H‐bonds per SQ unit). This trend is also maintained for SQ 2 with an energy difference per monomeric unit between both aggregates of 5.58 kJ mol−1. In contrast, AggII for SQ 3 is less stable than AggI by 0.86 kJ mol−1 per monomeric unit due to steric repulsion. These theoretical findings support the experimental detection of two different supramolecular polymorphs (AggI and AggII) for SQ 1 and 2 under different temperature conditions, AggII being the most stable structure, whereas only one aggregate (AggI) is characterized for SQ 3. According to this, it is apparent that the AggI‐AggII transformation in SQs 1 and 2 is strongly influenced by steric effects, while changes in solvation with temperature may also play a role. [57]
Kinetic Effects in the AggI‐to‐AggII Conversion
Kinetic effects of the polymorph transitions were studied by UV/Vis time‐dependent experiments utilizing SQ 2, which exhibits a slow AggI‐to‐AggII conversion upon cooling. We prepared different solutions of SQ 2 (3–6×10−5 M) in MCH (Figure S32) and the samples were quickly cooled down from 298 (AggI) to 278 K (AggII), and subsequently monitored over time (Figure 6a top). Figure 6b depicts the evolution of the UV pattern from 0 to 120 min, which shows the transformation of the broad AggI UV pattern into the two‐band pattern characteristic of AggII. The time‐dependent curves (followed at 324 nm) display a lag time of several minutes before the conversion starts (Figure 6c). The polymorph transition was faster at higher concentrations characteristic of an on‐pathway mechanism, which proceeds via a molecular rearrangement without previous disassembly.[ 9 , 12 , 13 , 14 , 15 , 16 ]
Figure 6.

a) Procedure to carry out the time‐dependent AggI‐to‐AggII transformation without (top) and with (bottom) addition of seeds. b) Time‐dependent UV/Vis absorption spectra at 278 K of 2 (4×10−5 M, MCH) after rapid cooling of the solution from 298 to 278 K. c) Normalized absorbance of SQ 2 vs. time plots extracted from the time‐dependent UV/Vis experiments at λ=324 nm at different concentrations (5×10−5 M (orange), 4×10−5 M (purple), and 3×10−5 M (grey)). d) Normalized absorbance vs. time plots (2, 4×10−5 M, MCH) extracted from the time‐dependent UV/Vis experiments at λ=324 nm, without seeding (violet dots), adding 10 μL of MCH (green triangles), and seeding with a 10 μL solution of 2‐AggII (4×10−5 M, kept for 2 h at 278 K) (blue squares). The aliquots were added at t=10 min as indicated in the plot with a blue arrow.
The lag time observed in the kinetic AggI‐to‐AggII conversion allowed us to carry out seeding experiments, which are widely performed in off‐pathway systems,[ 10 , 11 , 17 , 18 , 19 , 20 , 21 ] but little explored in on‐pathway conversions.[ 9 , 12 , 13 , 14 , 15 , 16 ] Thus, a solution of SQ 2 (4×10−5 M, MCH) was rapidly cooled down from 298 to 278 K, and after waiting for 10 min an aliquot of seeds was added. It is noteworthy that we used preformed 2‐AggII for the seeding experiments, since the typical procedure to prepare small seeds by sonication was unsuccessful, yielding 2‐AggI (Figure S33). Figure 6d shows the time‐dependent evolution of AggI into AggII (followed by UV/Vis at 324 nm at 278 K) with and without the addition of seeds (Figure S34). Remarkably, the transformation started much faster (after 5 min) in the seeded solution (seed: 10 μL of 2 (4×10−5 M) in MCH, kept for 2 h at 278 K) than in the reference sample where we added pure MCH (10 μL) (Figure 6d). This demonstrates that the addition of preformed‐AggII influences the AggI‐to‐AggII on‐pathway kinetic conversion, like previously described for off‐pathway processes.[ 10 , 11 , 17 , 18 , 19 , 20 , 21 ] It is noteworthy, that the kinetic profile of the seeded supramolecular polymerization follows a sigmoidal transition, [61] rather than the exponential transition commonly observed for systems following a primary seeded nucleation mechanism.[ 11 , 13 , 17 ] Then, a surface‐catalyzed transformation of the aggregates induced by the addition of seeds is inferred. [62] This type of transformation has been previously observed in the seeded self‐assembly of porphyrins, [13] barbiturates, [25] and naphthalene diimides, [63] for instance.
Thermodynamics of the AggI‐to‐AggII Conversion
The thermodynamic aspects of the temperature‐driven AggI‐to‐AggII conversion were studied using SQ 1, which showed relatively fast and reversible transitions and no hysteresis in the VT‐UV/Vis experiments (Figures S10 and S11). Additional nano‐DSC studies between 2×10−4 and 2.5×10−2 M (Figure S35 and Table S7) were carried out in MCH. The DSC heating and cooling traces (Figures 7a and S35) show no transitions between 363 and 298 K during the assembly/disassembly of AggI. Interestingly, clear transition peaks were observed around 290–295 K in the heating and cooling processes, which were attributed to the AggI‐to‐AggII transformation. These peaks are consistent with first‐order transitions and are analogous to those observed in liquid crystal phase transitions.[ 64 , 65 ] Integration of the DSC peaks revealed that the temperature‐driven AggII‐to‐AggI transition (heating) is endothermic, while the reverse process is exothermic (cooling). DSC analysis also showed that AggII is around 4 kJ mol−1 more stable than AggI (5×10−4 M) (Figure 7a) which is in good agreement with the theoretical value discussed above and being a consistent energy difference between polymorphs.[ 38 , 39 , 40 , 41 ]
Figure 7.

a) Nano‐DSC heating (red) and cooling (blue) traces for SQ 1 (5×10−4 M, MCH). Heating/cooling rate 1 K min−1. MHC: Molar heat capacity. B) Stacked plots of T I‐II (top), ΔH, and ΔS (bottom) of the AggI‐to‐AggII conversion (obtained from nano‐DSC cooling experiments in MCH) as function of the concentration of SQ 1. Green lines indicate the variation regimes of the parameters with concentration.
The thermodynamic parameters obtained from the nano‐DSC experiments revealed that not only the T I‐II (as well T II‐I) varies with the concentration, but also the ΔH and the ΔS of the AggI‐to‐AggII conversion (Figure 7b). The ΔH decreased from −3.6 to −7.6 kJ mol−1 between 2×10−4 and 5×10−3 M, but it remained relatively constant by further increasing the concentration. The ΔS followed a similar trend varying from −12.2 to −25.7 J K−1 mol−1 in the same concentration range. Remarkably, the concentration‐dependent behavior of the thermodynamic parameters of the AggI‐to‐AggII transition (nano‐DSC) was found to match well with the morphology behavior of AggI observed by AFM, SAXS, and SLS experiments (Figure 3d, 7b, and S36). According to this, at lower concentrations, small changes of the size of the AggI particles strongly affect the thermodynamics (T I‐II, ΔH, and ΔS) of the polymorph transition. However, at higher concentrations, AggI and AggII exist as fibers and the thermodynamic parameters are constant with the concentration.
Conclusion
In conclusion, we have reported three new squaramides (1–3) presenting distinct supramolecular polymorphism. While the three SQ derivatives 1–3 self‐assemble into nanoparticle‐like aggregates (AggI), compounds 1 and 2 can also evolve into a fiber‐like polymorph (AggII) at lower temperatures. Squaramide 1 presents fast and reversible temperature‐induced transitions between AggI and AggII, which permits the elucidation of the thermodynamic parameters of these uncommon polymorph transitions and rationalize its dependence on the concentration and morphology/size of the aggregates. On the other hand, the AggI‐AggII transition in squaramide 2 is governed by a slow kinetic regime, which permits unveiling the on‐pathway mechanism of the transition and the catalyzing effects of the seeded process. Theoretical calculations revealed that the different aggregates are formed via distinct hydrogen bonding patterns, which, together with steric effects, produce substantial changes in the morphology. This study sheds light into unusual thermoreversible supramolecular polymorph transitions and open new avenues towards the development of new stimuli‐responsive supramolecular polymers.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank J. Cifre and B. Martorell for the technical support. We acknowledge the MICINN/AEI of Spain (projects PID2019‐107779GA‐I00/AEI/10.13039/501100011033, PID2021‐128569NB‐I00, PID2020‐115637GB‐I00, EQC2018‐004206‐P, RED2018‐102331‐T, Unidad de Excelencia María de Maeztu CEX2019‐000919‐M and AEI/FEDER funds), the GOIB (AAEE 116/2017) and the Generalitat Valenciana (PROMETEO/2020/077) for financial support. B.S. and J.A. are thankful to the MICINN/AEI for their “Ramón‐y‐Cajal” fellowships (RYC‐2017‐21789 and RyC‐2017‐23500, respectively). A.D.‐G. thanks the Generalitat Valenciana for her predoctoral scholarship. N.B. gratefully acknowledges the Deutsche Forschungsgemeinschaft (GRK 2678—437785492) for funding.
S. Bujosa, A. Doncel-Giménez, N. Bäumer, G. Fernández, E. Ortí, A. Costa, C. Rotger, J. Aragó, B. Soberats, Angew. Chem. Int. Ed. 2022, 61, e202213345; Angew. Chem. 2022, 134, e202213345.
Contributor Information
Dr. Juan Aragó, Email: juan.arago@uv.es.
Dr. Bartolome Soberats, Email: b.soberats@uib.es.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Aida T., Meijer E. W., Stupp S. I., Science 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matern J., Dorca Y., Sánchez L., Fernández G., Angew. Chem. Int. Ed. 2019, 58, 16730–16740; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16884–16895. [Google Scholar]
- 3. Lutz J.-F., Lehn J.-M., Meijer E. W., Matyjaszewski K., Nat. Rev. Mater. 2016, 1, 16024. [Google Scholar]
- 4. Ghosh G., Dey P., Ghosh S., Chem. Commun. 2020, 56, 6757–6769. [DOI] [PubMed] [Google Scholar]
- 5. Hashim P. K., Bergueiro J., Meijer E. W., Aida T., Prog. Polym. Sci. 2020, 105, 101250. [Google Scholar]
- 6. Wehner M., Würthner F., Nat. Chem. Rev. 2020, 4, 38–53. [Google Scholar]
- 7. Korevaar P. A., de Greef T. F. A., Meijer E. W., Chem. Mater. 2014, 26, 576–586. [Google Scholar]
- 8. MacFarlane L., Zhao C., Cai J., Qiu H., Manners I., Chem. Sci. 2021, 12, 4661–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lohr A., Lysetska M., Würthner F., Angew. Chem. Int. Ed. 2005, 44, 5071–5074; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5199–5202. [Google Scholar]
- 10. Korevaar P. A., George S. J., Markvoort A. J., Smulders M. M. J., Hilbers P. A. J., Schenning A. P. H. J., De Greef T. F. A., Meijer E. W., Nature 2012, 481, 492–496. [DOI] [PubMed] [Google Scholar]
- 11. Ogi S., Sugiyasu K., Manna S., Samitsu S., Takeuchi M., Nat. Chem. 2014, 6, 188–195. [DOI] [PubMed] [Google Scholar]
- 12. Korevaar P. A., Newcomb C. J., Meijer E. W., Stupp S. I., J. Am. Chem. Soc. 2014, 136, 8540–8543. [DOI] [PubMed] [Google Scholar]
- 13. Fukui T., Kawai S., Fujinuma S., Matsushita Y., Yasuda T., Sakurai T., Seki S., Takeuchi M., Sugiyasu K., Nat. Chem. 2017, 9, 493–499. [DOI] [PubMed] [Google Scholar]
- 14. Kemper B., Zengerling L., Spitzer D., Otter R., Bauer T., Besenius P., J. Am. Chem. Soc. 2018, 140, 534–537. [DOI] [PubMed] [Google Scholar]
- 15. Greciano E. E., Matarranz B., Sánchez L., Angew. Chem. Int. Ed. 2018, 57, 4697–4701; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4787–4791. [Google Scholar]
- 16. Orvay F., Cerda J., Rotger C., Ortí E., Aragó J., Costa A., Soberats B., Small 2021, 17, 2006133. [DOI] [PubMed] [Google Scholar]
- 17. Ogi S., Stepanenko V., Sugiyasu K., Takeuchi M., Würthner F., J. Am. Chem. Soc. 2015, 137, 3300–3307. [DOI] [PubMed] [Google Scholar]
- 18. Ogi S., Grzeszkiewicz C., Würthner F., Chem. Sci. 2018, 9, 2768–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greciano E. E., Calbo J., Ortí E., Sánchez L., Angew. Chem. Int. Ed. 2020, 59, 17517–17524; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 17670–17677. [Google Scholar]
- 20. Ghosh G., Ghosh S., Chem. Commun. 2018, 54, 5720–5723. [DOI] [PubMed] [Google Scholar]
- 21. Sarkar S., Sarkar A., George S. J., Angew. Chem. Int. Ed. 2020, 59, 19841–19845; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 20013–20017. [Google Scholar]
- 22. Matsumoto N. M., Lafleur R. P. M., Lou X., Shih K.-C., Wijnands S. P. W., Guibert C., van Rosendaal J. W. A. M., Voets I. K., Palmans A. R. A., Lin Y., Meijer E. W., J. Am. Chem. Soc. 2018, 140, 13308–13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langenstroer A., Kartha K. K., Dorca Y., Droste J., Stepanenko V., Albuquerque R. Q., Hansen M. R., Sánchez L., Fernández G., J. Am. Chem. Soc. 2019, 141, 5192–5200. [DOI] [PubMed] [Google Scholar]
- 24. Wehner M., Röhr M. I. S., Bühler M., Stepanenko V., Wagner W., Würthner F., J. Am. Chem. Soc. 2019, 141, 6092–6107. [DOI] [PubMed] [Google Scholar]
- 25. Datta S., Kato Y., Higashiharaguchi S., Aratsu K., Isobe A., Saito T., Prabhu D. D., Kitamoto Y., Hollamby M. J., Smith A. J., Dalgliesh R., Mahmoudi N., Pesce L., Perego C., Pavan G. M., Yagai S., Nature 2020, 583, 400–405. [DOI] [PubMed] [Google Scholar]
- 26. Suda N., Saito T., Arima H., Yagai S., Chem. Sci. 2022, 13, 3249–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grabicki N., Dumele O., Sai H., Powers-Riggs N. E., Phelan B. T., Sangji M. H., Chapman C. T., Passarelli J. V., Dannenhoffer A. J., Wasielewski M. R., Stupp S. I., Chem. Mater. 2021, 33, 706–718. [Google Scholar]
- 28. Shen C.-A., Bialas D., Hecht M., Stepanenko V., Sugiyasu K., Würthner F., Angew. Chem. Int. Ed. 2021, 60, 11949–11958; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 12056–12065. [Google Scholar]
- 29. Hecht M., Leowanawat P., Gerlach T., Stepanenko V., Stolte M., Lehmann M., Würthner F., Angew. Chem. Int. Ed. 2020, 59, 17084–17090; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 17232–17238. [Google Scholar]
- 30. Aizawa T., Aratsu K., Datta S., Mashimo T., Seki T., Kajitani T., Silly F., Yagai S., Chem. Commun. 2020, 56, 4280–4283. [DOI] [PubMed] [Google Scholar]
- 31. Kim D.-Y., Christoff-Tempesta T., Lamour G., Zuo X., Ryu K.-H., Ortony J. H., Nano Lett. 2021, 21, 2912–2918. [DOI] [PubMed] [Google Scholar]
- 32. Pochan D. J., Schneider J. P., Kretsinger J., Ozbas B., Rajagopal K., Haines L., J. Am. Chem. Soc. 2003, 125, 11802–11803. [DOI] [PubMed] [Google Scholar]
- 33. Grande V., Soberats B., Herbst S., Stepanenko V., Würthner F., Chem. Sci. 2018, 9, 6904–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato K., Hendricks M. P., Palmer L. C., Stupp S. I., Chem. Soc. Rev. 2018, 47, 7539–7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim H.-J., Kim T., Lee M., Acc. Chem. Res. 2011, 44, 72–82. [DOI] [PubMed] [Google Scholar]
- 36. Datta S., Chaudhuri D., Angew. Chem. Int. Ed. 2022, 61, e202201956; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202201956. [DOI] [PubMed] [Google Scholar]
- 37. Chen Z., Liu Y., Wagner W., Stepanenko V., Ren X., Ogi S., Würthner F., Angew. Chem. Int. Ed. 2017, 56, 5729–5733; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5823–5827. [Google Scholar]
- 38. Isare B., Pensec S., Raynal M., Bouteiller L., C. R. Chim. 2016, 19, 148–156. [Google Scholar]
- 39. Bouteiller L., Colombani O., Lortie F., Terech P., J. Am. Chem. Soc. 2005, 127, 8893–8898. [DOI] [PubMed] [Google Scholar]
- 40. Ayzac V., Sallembien Q., Raynal M., Isare B., Jestin J., Bouteiller L., Angew. Chem. Int. Ed. 2019, 58, 13849–13853; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13987–13991. [Google Scholar]
- 41. Burger N. A., Pembouong G., Bouteiller L., Vlassopoulos D., Loppinet B., Macromolecules 2022, 55, 2609–2614. [Google Scholar]
- 42. Aratsu K., Takeya R., Pauw B. R., Hollamby M. J., Kitamoto Y., Shimizu N., Takagi H., Haruki R., Adachi S.-i., Yagai S., Nat. Commun. 2020, 11, 1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saez Talens V., Englebienne P., Trinh T. T., Noteborn W. E. M., Voets I. K., Kieltyka R. E., Angew. Chem. Int. Ed. 2015, 54, 10502–10506; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10648–10652. [Google Scholar]
- 44. Soberats B., Martinez L., Sanna E., Sampedro A., Rotger C., Costa A., Chem. Eur. J. 2012, 18, 7533–7542. [DOI] [PubMed] [Google Scholar]
- 45. De Greef T. F. A., Smulders M. M. J., Wolffs M., Schenning A. P. H. J., Sijbesma R. P., Meijer E. W., Chem. Rev. 2009, 109, 5687–5754. [DOI] [PubMed] [Google Scholar]
- 46. Kaiser T. E., Stepanenko V., Würthner F., J. Am. Chem. Soc. 2009, 131, 6719–6732. [DOI] [PubMed] [Google Scholar]
- 47. ten Eikelder H. M. M., Markvoort A. J., de Greef T. F. A., Hilbers P. A. J., J. Phys. Chem. B 2012, 116, 5291–5301. [DOI] [PubMed] [Google Scholar]
- 48. Gershberg J., Fennel F., Rehm T. H., Lochbrunner S., Würthner F., Chem. Sci. 2016, 7, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Herkert L., Droste J., Kartha K. K., Korevaar P. A., de Greef T. F. A., Hansen M. R., Fernández G., Angew. Chem. Int. Ed. 2019, 58, 11344–11349; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11466–11471. [Google Scholar]
- 50. Dorca Y., Naranjo C., Ghosh G., Soberats B., Calbo J., Ortí E., Fernández G., Sánchez L., Chem. Sci. 2022, 13, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van der Weegen R., Korevaar P. A., Voudouris P., Voets I. K., de Greef T. F. A., Vekemans J. A. J. M., Meijer E. W., Chem. Commun. 2013, 49, 5532–5534. [DOI] [PubMed] [Google Scholar]
- 52. Tamaki K., Datta S., Tashiro K., Isobe A., Silly F., Yagai S., Asian J. Org. Chem. 2021, 10, 257–261. [Google Scholar]
- 53. Fukushima T., Tamaki K., Isobe A., Hirose T., Shimizu N., Takagi H., Haruki R., Adachi S.-i., Hollamby M. J., Yagai S., J. Am. Chem. Soc. 2021, 143, 5845–5854. [DOI] [PubMed] [Google Scholar]
- 54. Saez Talens V., Davis J., Wu C.-H., Wen Z., Lauria F., Gupta K. B. S. S., Rudge R., Boraghi M., Hagemeijer A., Trinh T. T., Englebienne P., Voets I. K., Wu J. I., Kieltyka R. E., J. Am. Chem. Soc. 2020, 142, 19907–19916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Helmers I., Hossain M. S., Bäumer N., Wesarg P., Soberats B., Shimizu L. S., Fernández G., Angew. Chem. Int. Ed. 2022, 61, e202200390; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202200390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Draper E. R., Dietrich B., McAulay K., Brasnett C., Abdizadeh H., Patmanidis I., Marrink S. J., Su H., Cui H., Schweins R., Seddon A., Adams D. J., Matter 2020, 2, 764–778. [Google Scholar]
- 57. Su L., Mosquera J., Mabesoone M. F. J., Schoenmakers S. M. C., Muller C., Vleugels M. E. J., Dhiman S., Wijker S., Palmans A. R. A., Meijer E. W., Science 2022, 377, 213–218. [DOI] [PubMed] [Google Scholar]
- 58. Rotger M. C., Pina M. N., Frontera A., Martorell G., Ballester P., Deya P. M., Costa A., J. Org. Chem. 2004, 69, 2302–2308. [DOI] [PubMed] [Google Scholar]
- 59. Bannwarth C., Ehlert S., Grimme S., J. Chem. Theory Comput. 2019, 15, 1652–1671. [DOI] [PubMed] [Google Scholar]
- 60. Ghosh G., Chakraborty A., Pal P., Jana B., Ghosh S., Chem. Eur. J. 2022, 28, e202201082. [DOI] [PubMed] [Google Scholar]
- 61. Meisl G., Kirkegaard J. B., Arosio P., Michaels T. C. T., Vendruscolo M., Dobson C. M., Linse S., Knowles T. P. J., Nat. Protoc. 2016, 11, 252–272. [DOI] [PubMed] [Google Scholar]
- 62. Laishram R., Sarkar S., Seth I., Khatun N., Aswal V. K., Maitra U., George S. J., J. Am. Chem. Soc. 2022, 144, 11306–11315. [DOI] [PubMed] [Google Scholar]
- 63. Sarkar S., Sarkar A., Som A., Agasti S. S., George S. J., J. Am. Chem. Soc. 2021, 143, 11777–11787. [DOI] [PubMed] [Google Scholar]
- 64. Bujosa S., Greciano E. E., Martínez M. A., Sánchez L., Soberats B., Chem. Eur. J. 2021, 27, 14282–14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Uchida J., Soberats B., Gupta M., Kato T., Adv. Mater. 2022, 34, 2109063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.