

Abstract
Protein O‐fucosyltransferase 2 (PoFUT2) is an inverting glycosyltransferase (GT) that fucosylates thrombospondin repeats (TSRs) from group 1 and 2. PoFUT2 recognizes a large and diverse number of TSRs through a dynamic network of water‐mediated interactions. By X‐ray structural studies of C. elegans PoFUT2 complexed to a TSR of group 2, we demonstrate that this GT recognizes similarly the 3D structure of TSRs from both groups 1 and 2. Its active site is highly exposed to the solvent, suggesting that water molecules might also play an essential role in the fucosylation mechanism. We applied QM/MM methods using human PoFUT2 as a model, and found that HsPoFUT2 follows a classical SN2 reaction mechanism in which water molecules contribute to a great extent in facilitating the release of the leaving pyrophosphate unit, causing the H transfer from the acceptor nucleophile (Thr/Ser) to the catalytic base, which is the last event in the reaction. This demonstrates the importance of water molecules not only in recognition of the ligands but also in catalysis.
Keywords: Enzyme Catalysis, Fucosylation, Glycosyltransferases, Protein Glycosylation, QM/MM
PoFUT2 is an inverting glycosyltransferase that fucosylates TSR repeats of group 1 and 2. By applying computational methodologies, it has been found that PoFUT2 follows a pure SN2 mechanism in which water molecules play an essential role in the catalysis. Recognition of the TSR repeats by direct and water‐mediated interactions, together with the role of water molecules around the catalytic center, constitute the driving force of the process.
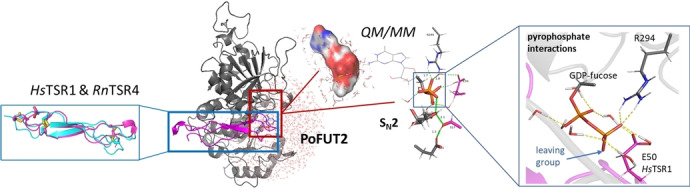
Introduction
Protein O‐fucosyltransferase 2 (PoFUT2), an inverting and GT‐B fold fucosyltransferase located in the endoplasmic reticulum (ER), fucosylates thrombospondin type 1 repeats (TSRs), and is engaged in numerous biological processes such as development, cell proliferation, migration, differentiation, and angiogenesis.[ 1 , 2 ] TSRs are small domains containing three disulfide bridges, and two major groups of TSRs, groups 1 and 2, have been found, which share a similar 3D‐fold and a common core structure. [3] Although TSRs show very variable peptide sequences [4] and group 1 and 2 differ partly in the disulfide bonding patterns, [3] PoFUT2 is capable of glycosylating TSRs containing the minimal consensus sequence (MCS) that is found in both groups.[ 4 , 5 ] This MCS is formed by C1‐X−X‐S/T‐C2 and C2‐X−X‐S/T‐C3 (C1 and C2 represent the second and third cysteine residues, respectively; underlining indicates the residues that are potentially fucosylated; and X denotes variable residues) in TSRs of group 1 and 2, respectively, [5] which might imply some differences in recognition of these groups of TSRs by PoFUT2. This glycosyltransferase (GT) has been suggested to follow the typical SN2‐like mechanism found for most inverting GTs, [6] in which, for this particular GT, a Glu residue acts as the catalytic base. [5] In addition, PoFUT2 possesses an active site exposed to the solvent, and the interface formed between PoFUT2 and the TSRs is also occupied by water molecules that stabilize the PoFUT2‐TSR complex and are key for recognition of dissimilar TSRs. [5]
Although the mechanism for inverting GTs is well accepted, this has been barely studied at computational level for inverting GTs glycosylating proteins. Particularly, only the catalytic mechanisms of O‐GlcNAc transferase (OGT), [7] and PoFUT1 [8] have been explored. The mechanism of OGT is still controversial because in addition to QM/MM and experimental studies suggesting a favorable pathway in which the α‐phosphate acted as the catalytic base,[ 9 , 10 ] structural studies proposed a relay of water molecules together with an Asp residue acting as the catalytic base. [11] For PoFUT1, a distinct mechanism was found in which this enzyme still followed an SN2 mechanism, but an Asn residue rather than a basic residue deprotonated the hydroxyl group of the acceptor nucleophile. The Asn residue bridged the nucleophile residue with the negatively charged β‐phosphate, shuttling a proton between the two groups. [8]
In this study, we conducted a multidisciplinary approach to dissect the catalytic mechanism of PoFUT2 and how it recognizes the TSRs of group 2. In particular, our study suggests that PoFUT2 recognizes the 3D structures of both TSRs of groups 1 and 2 in a similar way, and QM/MM studies suggest that PoFUT2 follows a pure SN2 mechanism in which water molecules around the catalytic center play an essential role. The direct participation of water molecules in enzymatic reaction mechanisms has been observed for other families of enzymes, such as protein kinases, [13] hydrolases, [14] or, particularly, copper nitrite reductase, [15] but it is unprecedented in the case of glycosyltransferases. Our study points out that the enzyme harnesses the reaction medium for its benefit, lowering the energy barrier.
Results and Discussion
To understand how PoFUT2 recognizes TSRs from group 2, we envisaged the same strategy used to determine the structure of CePoFUT2 complexed to GDP and HsTSR1, which is a TSR from group 1. [5] The first fusion protein construct contained CePoFUT2, a flexible linker and the fourth TSR of Rattus norvegicus F‐spondin 1 (RnTSR4) (Figure 1 and Supporting Information). Despite numerous crystallization attempts, crystals were not obtained. Due to that, we rationally designed a double mutant (A418C in CePoFUT2 and E10C in RnTSR4) in the previous fusion protein to form a disulfide bridge between CePoFUT2 and RnTSR4.
Figure 1.

a) Multiple sequence alignment of HsTSR1, HsTSR2 and HsTSR3 of thrombospondin 1 and RnTSR1 and RnTSR4 of F‐spondin. Red arrowhead indicates fucosylated serine and threonine residues, and conserved Cys residues are highlighted in yellow. The numbering for each TSR does not correspond to its location in thrombospondin 1 or F‐spondin.
This double mutant was based on the previous crystal structure of CePoFUT2‐GDP‐HsTSR1 complex, in which the Glu10 in HsTSR1 (Figure 1; conserved in RnTSR4) was in close contact to Ala418. The rationale behind of forming this disulfide bridge was to ensure trapping a productive conformation of RnTSR4 complexed to CePoFUT2 since as we showed previously a limited number of direct interactions maintained the complex between CePoFUT2 and HsTSR1. [4] As expected, we obtained crystals of the complex between CePoFUT2A418C and RnTSR4E10C that diffracted at 2.13 Å (See Supporting Information and Table S1), enabling the structure to be solved and the electron density map to be unambiguously interpreted (Figures 2a and S1; PDB ID: 8AY1). The asymmetric unit (AU) contained two independent binary complexes with the 1 : 1 stoichiometry (one molecule each of CePoFUT2A418C and RnTSR4E10C, Figure 2a).
Figure 2.

a) Cartoon representation of the complex. CePoFUT2A418C and RnTSR4E10C are shown in green and cyan, respectively. Disulfide bridges are indicated in yellow. Carbon atoms of the GlcNAc moiety covalently bound to Asn205 are shown in pink. A sulfate molecule and the acceptor Thr17 of RnTSR4E10C are also shown in the active site. b) Close‐up view of the few direct interactions in the complex. H‐bond interactions are shown as dotted black lines.
The CePoFUT2A418C structure as shown before adopts the typical GT‐B fold formed by two Rossmann‐like domains that face each other. The RnTSR4E10C shows the two typical antiparallel β‐strands found in all TSRs indistinctly of the group, which superpose very well with those of HsTSR1. This was also exemplified by the overall small root‐mean‐square deviation (RMSD of 0.78 Å on 39 residues aligned; see Figure S2a). Note that while the structure of HsTSR1 was complete, the structure of RnTSR4E10C displayed several unstructured regions with no electron density (residues 1–5 and 31–40) that were solvent exposed and not engaged in interactions with CePoFUT2, implying that RnTSR4 is likely more dynamic than HsTSR1. In addition, the superposition between these two types of TSRs revealed that two disulfide bridges occupied the same positions (C18‐C53 RnTSR4 with C18‐C51 HsTSR1, and C14‐C48 RnTSR4 with C14‐C46 HsTSR1). The third disulfide bridge (C3‐C38 RnTSR4) could not be visualized in RnTSR4 (Figure S2a) because it was located in the unstructured regions with no electron density (see below the computational simulations to compare the location of this disulfide bridge with C29‐C36 HsTSR1). Overall, the data show that CePoFUT2 recognizes very similarly the overall structures of TSRs indistinctly from each group.
A close‐up view of the CePoFUT2A418C active site revealed the presence of a sulfate molecule, occupying the same position to that found for GDP alpha phosphate (Figure 2a). The presence of the sulfate molecule was due to the fact that the crystals grew in the presence of ammonium sulfate, which likely excluded the presence of GDP in the active site despite being part of the crystal condition too (see Supporting Information). Regarding the interface between CePoFUT2A418C and RnTSR4E10C, a very limited number of direct interactions was visualized (Figure 2b), resembling in part those found for the CePoFUT2‐GDP‐HsTSR1 complex. The complex is stabilized by hydrogen bonds (H‐bonds) and hydrophobic interactions that are complemented by water‐mediated contacts. Note that the latter interactions will not be discussed further since the importance of water molecules in CePoFUT2 recognition on TSRs was described previously. [5] However, the role of water molecules in catalysis will be discussed below. Of the five residues of RnTSR4E10C engaged in directs interactions with CePoFUT2A418C, only Ser15 and Thr17 are conserved (Figure 2b; see also Figure S1 in ref. [5] that shows a more detailed multiple alignment between TSRs). The hydrophobic interaction between Trp420 and the disulfide bridge formed by E10C and A418C was not considered above because this was an artificial disulfide bridge to trap the complex. The side chain of the acceptor Thr17 establishes a H‐bond with Glu52 (Figure 2b), and the Ser15 backbone is engaged in H‐bond interactions with Arg63. In addition, the Thr17 methyl group is surrounded by a hydrophobic environment formed by Thr383 methyl group and Leu56 side chain, implying as suggested before that these two CePoFUT2 amino acids are important for a better recognition of Thr acceptor sites. This likely explains why PoFUT2 slightly prefers Thr over Ser acceptor sites. In contrast, the interaction of nonconserved RnTSR4E10C residues with CePoFUT2A418C is exclusively through RnTSR4E10C side chains (Figure 2b). In particular, Asp13, Met22 and Ala45 side chains interact with Tyr225, Leu221/Tyr145/Trp141, and Tyr145, respectively (Figure 2b). A comparison between this complex and the previous one with HsTSR1 showed that there are more direct interactions between CePoFUT2 and HsTSR1 (10 residues of HsTSR1 engaged in direct interactions with CePoFUT2 [4] ) than those between CePoFUT2A418C and RnTSR4E10C. This could likely lead to a weaker binding of RnTSR4 to CePoFUT2, which could be behind of our difficulties in getting crystals of CePoFUT2‐RnTSR4 complex. Overall, our results also imply as suggested before that each TSR might adopt a small number of unique direct interactions through their non‐conserved residues.
To assess the molecular basis of the PoFUT2 catalytic mechanism, we carried out computational studies with assembled ternary complexes between human PoFUT2 (HsPoFUT2), GDP‐fucose and TSRs (RnTSR4 and the HsTSR1). The rationale used for constructing suitable ternary complexes was based on previous structures (see Supporting Information). Previous findings [5] as well as the above discussed results show the importance of several residues in binding and catalysis that were confirmed by previous mutagenesis experiments[ 4 , 5 ] Accordingly, a minimum model of the active site (considering HsPoFUT2) should include GDP‐fucose, the acceptor site (either Thr17 in RnTSR4 or Ser17 in HsTSR1), Glu54, acting as the catalytic base and Arg294, which plays an essential role in catalysis. These residues in HsPoFUT2 and HsTSR1 were mutated before confirming their importance in catalysis and binding.[ 4 , 5 ] In addition, the model did not contain a metal in the active site because GT‐B fold GTs, such as PoFUT2, do not require a metal for catalysis. [16]
In the case of PoFUT2, the metal slightly increased the activity of HsPoFUT2 [4] but not that of CePoFUT2, [5] implying, as expected, that metals do not play any role in catalysis. In addition, metals were not visualized in any of the crystal structures of these enzymes. Therefore, we discarded the presence of metals around the pyrophosphate though the pyrophosphate was engaged in interactions with water molecules due to its high exposure to the solvent (see below). In fact, in the case of CePoFUT2‐GDP‐HsTSR1 complex (PDB ID: 5FOE), it can be observed the presence of water molecules coordinating the phosphate oxygen atoms (see Figure S11). The solvent was modelled by a water sphere of 24 Å radius centered at the anomeric carbon of the fucose unit.
Examinations of MD simulations of the starting ternary complexes for RnTSR4 and HsTSR1 models revealed, as the X‐ray structure had pointed out, a great exposure of the active site to the solvent. As expected, both RnTSR4 and HsTSR1 models were superimposable, implying a similar mechanism (Figure 3a). In both types of TSRs, as discussed above, two disulfide bridges occupied the same positions, while the third one was not superimposable (Figure S2b). Interestingly, the pyrophosphate unit of GDP‐fucose was particularly exposed to the solvent (as indicated by Connolly surfaces), showing mostly interactions with water molecules that prevented the presence of metals. (Figure 3b,c). This suggested that water molecules might have a key role in catalysis and contribute to facilitate the leaving of the product (GDP) by interactions with the pyrophosphate, adopting a similar role as that of Arg294.
Figure 3.
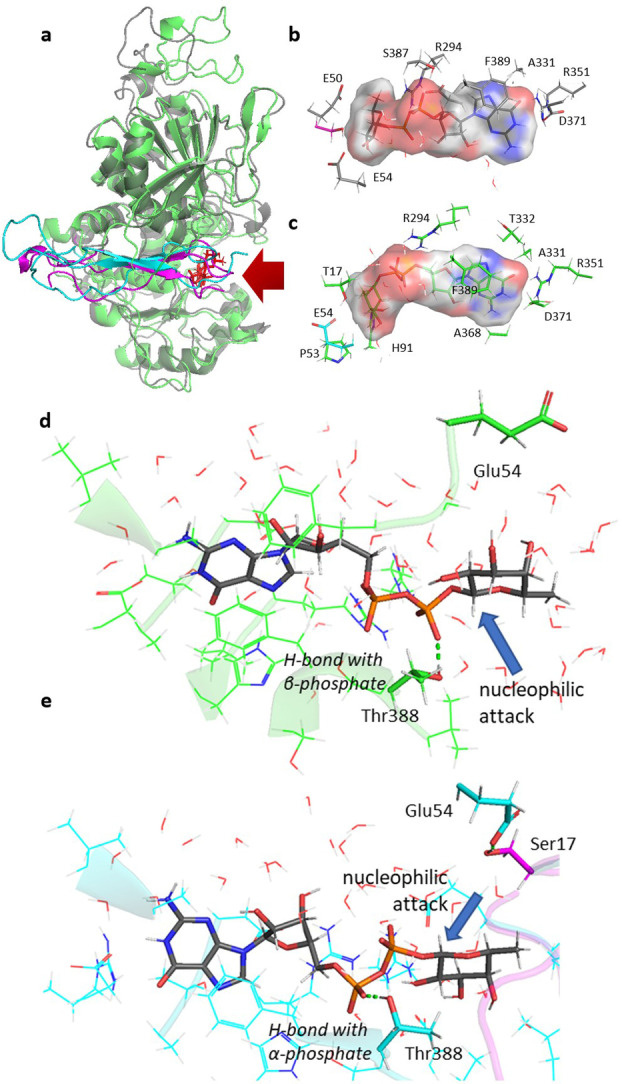
a) Superimposed and aligned structures of the initial points (MD) corresponding to PoFUT2 (green) in complex with GDP‐fucose and RnTSR4 (cyan) and PoFUT2 (gray) in complex with GDP‐fucose and HsTSR1 (magenta). GDP‐fucose is showed in red at the active site exposed to solvent (red arrow). b), c) Detail of residues of PoFUT2 interacting with GDP‐fucose in the complex with HsTSR1 (b) and RnTSR4 (c). The Connolly surface for GDP‐fucose reflects the high accessibility (red) of the solvent to the pyrophosphate unit in both cases. d) Snapshot of MD simulations corresponding to the binary complex between HsPoFUT2 (green) and GDP‐fucose (gray). The reactive face of fucose anomeric carbon is oriented towards the opposite face of the catalytic base. e) Snapshot of MD simulations corresponding to the ternary complex between HsPoFUT2 (cyan), GDP‐fucose (gray) and HsTSR1 (magenta) The reactive face of fucose anomeric carbon has rotated to be oriented towards the same side in which Ser17 (the nucleophile) and Glu54 (the catalytic base) are placed. Both snapshots (d) and (e) have the same orientation view.
A common feature of GTs that use sugar‐nucleotide donors as substrates is their tendency to hydrolyze them. Initially, we inspected the binary complex formed by HsPoFUT2 and GDP‐fucose by MD simulations (starting from PDB ID: 4AP6), [4] showing that GDP‐fucose is well exposed to the solvent as it has been reported for the complex with PoFUT1. [12] However the mechanism in which these enzymes circumvent the undesired hydrolysis of the sugar nucleotide is different. Whereas in the complex with PoFUT1, the lack of a proper amino acid acting as a catalytic base might impede the hydrolysis, in the complex with HsPoFUT2, this is prevented by orienting the reactive face of the fucose anomeric carbon on the opposite side of the location of Glu54 (Figure 3d). Such orientation is stabilized by a H‐bond between the hydroxyl group of Thr388 and the α‐phosphate. MD simulations of our ternary complex showed that the recognition of the HsTSR1 results in the switching of the H‐bond between the Thr388 and α‐phosphate to that with the β‐phosphate. This minimal change allows the fucose to rotate 180 degrees orienting the reactive face of the anomeric carbon towards Glu54 and Ser17 of HsTSR1 (Figure 3e). Therefore, in a similar manner as described before for PoFUT1, PoFUT2 avoids the hydrolysis due to the acceptor TSR substrate, which protects the area around the anomeric carbon from bulk water.
Once that we reasonably explained why PoFUT2 might ameliorate hydrolysis of GDP‐fucose in the presence of water molecules, we studied the mechanism of the glycosylation of HsTSR1 and RnTSR4. [4]
We selected a minimal QM region (QM1) consisting of the fucose ring, the pyrophosphate unit, and sidechains of Glu54, Arg294 of the enzyme, and Ser17 and Glu50 of HsTSR1 (82 atoms). Then we defined additional QM regions (QM2‐QM6) that contained an increasing number of water molecules showing H‐bonds with the pyrophosphate unit (Figure 4a).
Figure 4.
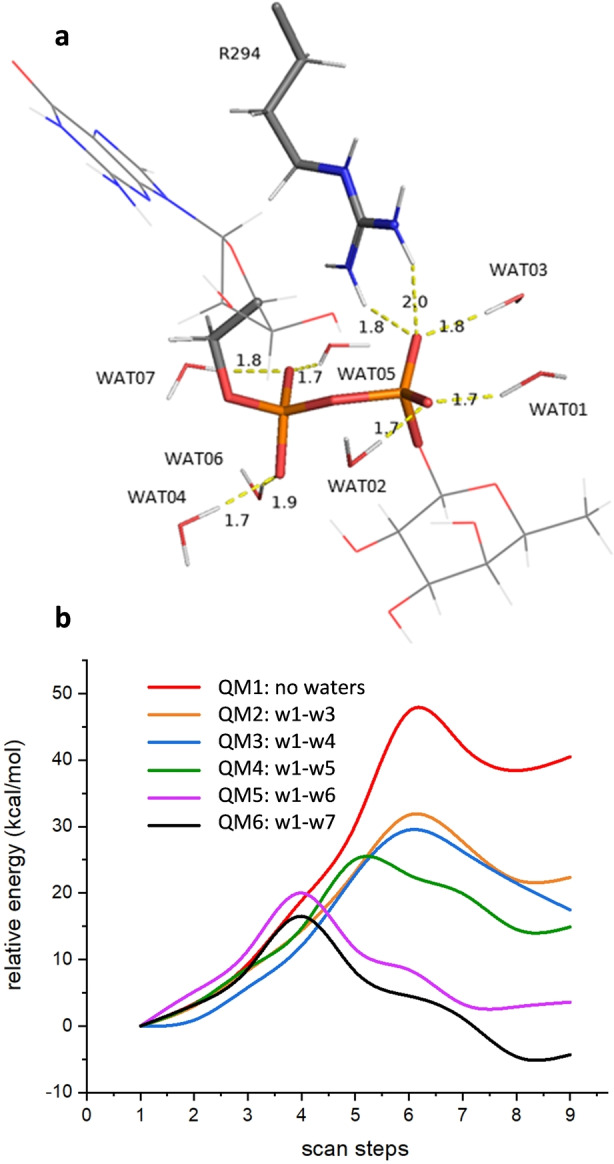
a) Detail of water molecules considered in the QM region, illustrated for the reagent corresponding to the glycosylation of HsTSR1. b) Potential energy surfaces for the different QM models tested with HsTSR1. QM1 corresponds to the model without waters (82 atoms). Successive models, QM2‐QM6, correspond to further incorporation of water molecules to the QM system as indicated. The model in QM6 (103 atoms), containing seven water molecules, showed the best result.
QM6 contained the highest number of water molecules reaching up to 103 atoms in the system. Additional water molecules were not considered of catalytic importance since they do not present direct interactions with GDP‐fucose. We also discard the presence of an additional divalent cation on the basis of previous experiments that had identified PoFUT2 as a GT‐B fold glycosyltrasferase. [4]
The study of the potential energy surfaces was made at BP86/SVP level of theory, [17] after testing several reaction coordinates (see Figure S3), by varying the distance between the anomeric carbon C1 of the fucose and the nucleophile oxygen of the acceptor hydroxyl group of Ser17. This distance provides information of the nucleophilic attack of the serine acceptor on the anomeric carbon of the donor GDP‐fucose, and the formation of the new glycosidic linkage. The rest of distances evolved smoothly towards the product. Any attempt of locating a stepwise mechanism considering a previous deprotonation of Ser17 failed. On the other hand, the concerted reaction was successful for all QM systems studied. We initiated the studies with HsTSR1 and found a barrier of ca. 47 kcal mol−1 for QM1 without consideration of water molecules. As the water molecules were incorporated into the QM system, the barrier was lowering till 16.5 kcal mol−1 when all the seven water molecules showing H‐bonds with the pyrophosphate unit were considered; notably, only in this case the reaction showed to be exothermic by 4.3 kcal mol−1 (Figure 4b).
These results provide substantial evidence of the crucial role exerted by the solvent in the reaction, contributing to stabilize the GDP‐fucose and facilitate the departure of the leaving group (GDP). Also, elimination of Arg294 in the model resulted in a barrier of 31.9 kcal mol−1 confirming the experimental observation and a synergistic effect of that residue with the solvent. We also considered a similar possibility to that reported for O‐GlcNAc transferase (OGT) in which α‐phosphate could act as the catalytic base. [9] All attempts for orienting the substrates on that route failed and no rational scans could be obtained. Thus, we can conclude that QM6, considering Glu54 as the catalytic base is the proper system to study the catalytic glycosylation of HsPoFUT2.
The obtained results from the scans represent reasonable approaches for an SN2 reaction mechanism. The optimized reagents RE1 (for HsTSR1) and RE2 (for RnTS4)) confirmed the high exposure of the pyrophosphate unit to the solvent. To refine the energy barrier, we used the structure with the maximum energy to explore the conformational variability and locate the transition structure TS1. The final point of the scan was also optimized to find the final product PR1. After optimization of the stationary points, the barrier for the fucose transfer mechanism via the [RE→TS→PR] pathway was calculated to be 17.0 kcal mol−1. More accurate calculations were made at 3ξ level, and the single‐point energy barriers were in the range 17.1–30.2 kcal mol−1 (Table S2). The barriers calculated at the b3lyp and M06‐2X functionals were higher than those obtained with other functionals. The rest of functionals remain in the range 17–19 kcal mol−1. In particular, bp86‐d3 in combination with Pople basis sets like 6–311*G(d,p) (Table S1, entry 1), which has been considered to be a level with very low mean absolute errors, [18] gave 17.2 kcal mol−1. Then, we applied the same protocol to the glycosylation of RnTSR4, which provided very similar results to those shown above. We optimized the corresponding stationary points TS2 and PR2, and after 3ξ single point calculations barriers of ca 12–14 kcal mol−1 were obtained (with exception of b3lyp and m062x functionals that again, showed higher values) predicting a slightly faster reaction. To the best of our knowledge, only the kinetic constant for the glycosylation of RnTSR4 has been reported, with a value of k cat of 2.4 s−1, [4] corresponding to a barrier of ca. ≈17.0 kcal mol−1 (Eyring equation). This experimental value is in good agreement with those calculated (Table S2).
The optimized transition structures TS1 and TS2 (Figure 5a,b) keep the water molecules close to the pyrophosphate unit, which donate H‐bonds to all the pyrophosphate oxygens except that lnked to the carbihydrate, directly involved in the reaction.
Figure 5.
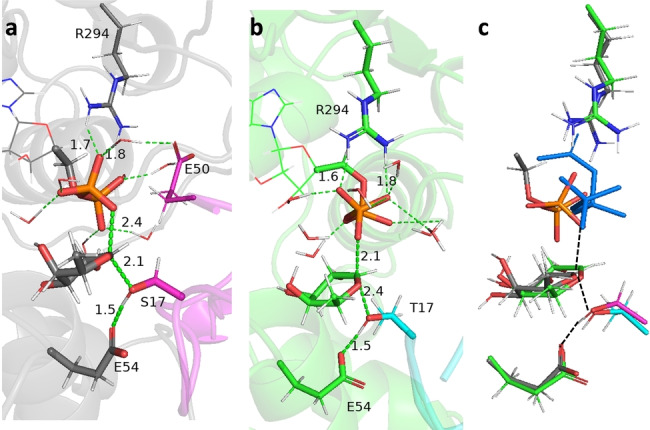
a) Transition structure TS1 corresponding to the glycosylation of HsTSR1 (cyan). b) Transition structure TS2 corresponding to the glycosylation of RnTSR4 (magenta). c) Overlay of the QM regions of the transition structures TS1 and TS2. TS1 and TS2 corresponding to the glycosylation of RnTSR4 (green, nucleophilic Thr17 in cyan) and HsTSR1 (gray, nucleophilic Ser17 in magenta), respectively. Note the alignment of the breaking/forming bonds (dashed black line). Pyrophosphate of TS2 is displayed in blue to distinguish the different orientation allowed by exposure to the solvent (the leaving oxygen is, however, perfectly aligned). QM regions are given in sticks.
Glycosylation of HsTSR1 follows a pure SN2 mechanism without apparent formation of an oxocarbenium ion in any extent. In the transition structure, the HSer17⋅⋅⋅OGlu54 and HSer17⋅⋅⋅OSer17 distances are 1.5 Å and 1.0 Å indicating that the nucleophile was not deprotonated yet. The nucleophile OSer17 is positioned at 2.1 Å from the anomeric carbon and 2.4 Å from the leaving oxygen, in agreement with a concomitant deprotonation, nucleophilic attack and simultaneous leaving of the pyrophosphate unit. In fact, according to the reaction path, the formation of the glycosydic bond, the breaking of the phosphate bond and the H‐transfer from Ser17 to Glu54 takes place in a full concerted way after the transition structure (Figure S6).
The sugar moiety adopts a 3H4 conformation, according to Cremer–Pople parameters (ϕ=13° and θ=143°); in this configuration, the C5‐O5‐C1‐C2 group of atoms exhibits an almost coplanar rearrangement (dihedral=−13°), in which the distance C1‐O5 change minimally (1.4 Å and 1.3 Å in RE1 and TS1, respectively; 1.4 Å in PR1) disregarding the development of any appreciable positive charge as expected in a pure SN2 mechanism. The electrostatic contributions to the electronic stabilization/destabilization of the transition structure (typically more than 2 kcal mol−1) were evaluated through a charge deletion analysis, [19] allowing us to determine the contribution of individual residues close to the QM region. There were no residues showing prominent effects with the exception of Asp333 that destabilizes the transition structure by −4.3 and −2.8 kcal mol−1 for HsPoFUT2‐GDP‐fucose‐HsTSR1 and HsPoFUT2‐GDP‐fucose‐RnTSR4 complexes, respectively. This effect is caused by a H‐bond interaction with the guanidine group of Arg294 (Figure S7), which impedes an optimal interaction with the leaving pyrophosphate group. The length of the H‐bond (1.9 Å for the complex with HsTSR1 and 2.9 Å for the complex with RnTS4) is in agreement with the observed destabilization energy. Inclusion of Asp333 in the QM region resulted in negligible differences confirming that the QM/MM partition used satisfactorily describes the system. Overlay of the QM regions of the optimized transition structures revealed a very similar situation (Figure 5c). Noteworthy, only the pyrophosphate unit, highly exposed to solvent, has enough flexibility to change orientation, while the rest of chemical entities involved in the reaction are perfectly aligned. This causes that Arg294 promotes the pyrophosphate departure in two different ways, both equally efficient (see Figure S8). Topological analyses by ELF [20] and NCI [21] of the transition structure for the glycosylation of HsTSR1 corroborated the interactions discussed above (see Figure S9). Finally, replacing the Ser residue in HsTSR1 by a Thr residue yields a very similar reaction to that observed with the original substrate, and the same takes place when replacing the Thr residue in RnTSR4 by a Ser residue (Figure S10). This further demonstrates at computational level that PoFUT2 fucosylates indistinctly both Ser and Thr acceptor sites.
Conclusion
In summary, we demonstrate here that PoFUT2 recognizes very similarly the 3D‐structures of TSRs of group 1 and 2. Yet, depending on the TSR sequence, some changes might be found at the level of the interacting residues. Furthermore, by applying computational methodologies, we thoroughly find that PoFUT2 follows a pure SN2 mechanism in which water molecules around the catalytic center play an essential role in catalysis. Water molecules interacting directly with the pyrophosphate contribute to stabilizing the leaving group GDP, adopting a similar role to that found for Arg294. In addition, we found that both chemical entities might have a synergistic effect, as was demonstrated by calculations that suppressed either waters or Arg294. Overall, we think that the driving force of the reaction is the recognition of the TSR repeats, by direct and water‐mediated interactions, together with the role of water molecules around the catalytic center. This provides a remarkable example of how this particular enzyme uses water molecules not only to account for its specificity and promiscuity on TSRs, as reported, [5] but also to drive the carbohydrate transfer. This dual role of water molecules in GTs has no precedent and open the door to consider alternative mechanisms for other GTs in which water molecules might play a direct role in catalysis.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
We thank the Spanish MICINN (grants PID2019‐104090RB‐100 to P.M. and PID2019‐105451GB‐I00 to R.H.‐G., and predoctoral FPI fellowship to I.S.‐M.) for financial support. We also thank ARAID, Gobierno de Aragón (E34_20R to P.M. and LMP58_18 to R.H.‐G.) with FEDER (2014‐2020) funds for “Building Europe from Aragón” for financial support, and the COST Action CA18103 INNOGLY: Innovation with Glycans new frontiers from synthesis to new biological targets. ALBA (Barcelona, Spain) synchrotron beamline XALOC and resources from the super‐computers “Memento” and “Cierzo” provided by BIFI‐ZCAM (University of Zaragoza, Spain) are gratefully acknowledged.
Dedicated to Professor Miguel Yus on occasion of his 75th birthday
I. Sanz-Martínez, A. García-García, T. Tejero, R. Hurtado-Guerrero, P. Merino, Angew. Chem. Int. Ed. 2022, 61, e202213610; Angew. Chem. 2022, 134, e202213610.
Contributor Information
Dr. Ramón Hurtado‐Guerrero, Email: rhurtado@bifi.es.
Prof. Dr. Pedro Merino, Email: pmerino@unizar.es.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Schneider M., Al-Shareffi E., Haltiwanger R. S., Glycobiology 2017, 27, 601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lira-Navarrete E., Hurtado-Guerrero R., Acta Crystallogr. Sect. F 2018, 74, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan K., Duquette M., Liu J. H., Dong Y., Zhang R., Joachimiak A., Lawler J., Wang J. H., J. Cell Biol. 2002, 159, 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen C.-I., Keusch J. J., Klein D., Hess D., Hofsteenge J., Gut H., EMBO J. 2012, 31, 3183–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valero-González J., Leonhard-Melief C., Lira-Navarrete E., Jiménez-Osés G., Hernández-Ruiz C., Pallarés M. C., Yruela I., Vasudevan D., Lostao A., Corzana F., Takeuchi H., Haltiwanger R. S., Hurtado-Guerrero R., Nat. Chem. Biol. 2016, 12, 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moremen K. W., Haltiwanger R. S., Nat. Chem. Biol. 2019, 15, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saha A., Bello D., Fernandez-Tejada A., Chem. Soc. Rev. 2021, 50, 10451–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Piniello B., Lira-Navarrete E., Takeuchi H., Takeuchi M., Haltiwanger R. S., Hurtado-Guerrero R., Rovira C., ACS Catal. 2021, 11, 9926–9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumari M., Kozmon S., Kulhanek P., Stepan J., Tvaroska I., Koca J., J. Phys. Chem. B 2015, 119, 4371–4381. [DOI] [PubMed] [Google Scholar]
- 10. Schimpl M., Zheng X., Borodkin V. S., Blair D. E., Ferenbach A. T., Schuttelkopf A. W., Navratilova I., Aristotelous T., Albarbarawi O., Robinson D. A., Macnaughtan M. A., van Aalten D. M., Nat. Chem. Biol. 2012, 8, 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lazarus M. B., Jiang J., Gloster T. M., Zandberg W. F., Whitworth G. E., Vocadlo D. J., Walker S., Nat. Chem. Biol. 2012, 8, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Z., Han K., Pak J. E., Satkunarajah M., Zhou D., Rini J. M., Nat. Chem. Biol. 2017, 13, 757–763. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Knight J. D., Hamelberg D., McCammon J. A., Kothary R., Proteins Struct. Funct. Bioinf. 2009, 76, 527–535; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Setny P., Proteins Struct. Funct. Bioinf. 2020, 88, 1578–1591. [DOI] [PubMed] [Google Scholar]
- 14. Cui P., Wang Y., Chu W., Guo X., Yang F., Yu M., Zhao H., Dong Y., Xie Y., Gong W., Wu Z., Sci. Rep. 2014, 4, 7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rose S. L., Antonyuk S. V., Sasaki D., Yamashita K., Hirata K., Ueno G., Ago H., Eady R. R., Tosha T., Yamamoto M., Hasnain S. S., Sci. Adv. 2021, 7, eabd8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lairson L. L., Henrissat B., Davies G. J., Whiters S. G., Annu. Rev. Biochem. 2008, 77, 521–555. [DOI] [PubMed] [Google Scholar]
- 17. Schäfer A., Horn H., Ahlrichs R., J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar]
- 18.J. Sirirak, N. Lawan, M. W. van der Kamp, J. N. Harvey, A. J. Mulholland, PeerJ Phys. Chem. 2020, 2, e8.
- 19. Liao R.-Z., Thiel W., J. Comput. Chem. 2013, 34, 2389–2397. [DOI] [PubMed] [Google Scholar]
- 20.Electron localization function (ELF) analysis shows where electron density is concentrated. See. Silvi B., Savin A., Nature 1994, 371, 683, and [Google Scholar]; Savin A., Nesper R., Wengert S., Fässler T. F., Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832; [Google Scholar]; Angew. Chem. 1997, 109, 1892–1918. [Google Scholar]
- 21.Non-covalent interactions (NCI) analysis shows strong and weak interactions not involved in formed bonds. See: Johnson E. R., Keinan S., Mori-Sanchez P., Contreras-Garcia J., Cohen A. J., Yang W., J. Am. Chem. Soc. 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.