

Abstract
Pleiotropy, which consists of a single gene or allelic variant affecting multiple unrelated traits, is common across cancers, with evidence for genome‐wide significant loci shared across cancer and noncancer traits. This feature is particularly relevant in multiple myeloma (MM) because several susceptibility loci that have been identified to date are pleiotropic. Therefore, the aim of this study was to identify novel pleiotropic variants involved in MM risk using 28 684 independent single nucleotide polymorphisms (SNPs) from GWAS Catalog that reached a significant association (P < 5 × 10−8) with their respective trait. The selected SNPs were analyzed in 2434 MM cases and 3446 controls from the International Lymphoma Epidemiology Consortium (InterLymph). The 10 SNPs showing the strongest associations with MM risk in InterLymph were selected for replication in an independent set of 1955 MM cases and 1549 controls from the International Multiple Myeloma rESEarch (IMMEnSE) consortium and 418 MM cases and 147 282 controls from the FinnGen project. The combined analysis of the three studies identified an association between DNAJB4‐rs34517439‐A and an increased risk of developing MM (OR = 1.22, 95%CI 1.13‐1.32, P = 4.81 × 10−7). rs34517439‐A is associated with a modified expression of the FUBP1 gene, which encodes a multifunctional DNA and RNA‐binding protein that it was observed to influence the regulation of various genes involved in cell cycle regulation, among which various oncogenes and oncosuppressors. In conclusion, with a pleiotropic scan approach we identified DNAJB4‐rs34517439 as a potentially novel MM risk locus.
Keywords: genetic susceptibility, multiple myeloma, pleiotropy, pleiotropy scan, polymorphisms
What's new?
Genetic variants can have multiple, seemingly unrelated, effects. Often, these so‐called “pleiotropic” variants play a role in cancer. Here, the authors set out to identify new pleiotropic variants involved in multiple myeloma (MM) risk. They analyzed 28,684 independent single nucleotide polymorphisms (SNPs) that had been identified in genome‐wide association studies as having an effect on a human trait. This analysis revealed an association between increased MM risk and a variant called DNAJB4‐rs34517439‐A. That variant has been associated with changes in expression of a DNA‐ and RNA‐binding protein that helps regulate cell cycle genes.
Abbreviations
- CI:
confidence interval
- eQTL:
expression quantitative trait loci
- GTEx:
genotype‐tissue expression project
- GWAS:
genome‐wide association studies
- HSC:
hematopoietic stem cell
- HWE:
Hardy‐Weinberg equilibrium
- IMMEnSE:
International Multiple Myeloma rESEarch
- IMWG:
International Myeloma Working Group
- InterLymph:
International Lymphoma Epidemiology Consortium
- LD:
linkage disequilibrium
- lncRNA:
long non coding RNA
- MM
multiple myeloma
- OR
odds ratio
- PC
principal component
- SNP
single nucleotide polymorphism
1. INTRODUCTION
Multiple myeloma (MM) is an incurable hematological disease originating from plasma cells in the bone marrow. 1 MM is the third most common hematological tumor with a crude incidence rate of 6.8/100000 new cases per year in Europe 2 (https://gco.iarc.fr/today/home).
Several studies have investigated the genetic susceptibility of MM, using genome‐wide association studies (GWAS) 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 or more targeted approaches. 8 , 11 , 12 , 13 , 14 , 15 Twenty‐four risk loci have been found to be associated with MM risk through GWAS; however, these variants explain approximately only 16% of MM heritability. 3 Due to the large number of tests conducted in a GWAS, a stringent statistical significance threshold, P < 5 × 10−8, is employed. The associated increase in type II error rates means that many true associations may remain undetected and unreported.
One strategy to avoid this loss of information is to investigate pleiotropic variants. Pleiotropy is a genetic phenomenon consisting of a single gene or allelic variant affecting multiple, often unrelated traits. 16 GWASs have identified several loci associated with cancer and noncancer phenotypes. Recently, it was estimated that half of all the SNPs showing an association with a P value threshold of 10−8 are pleiotropic, and 12.34% show associations with 10 or more phenotypes. 17
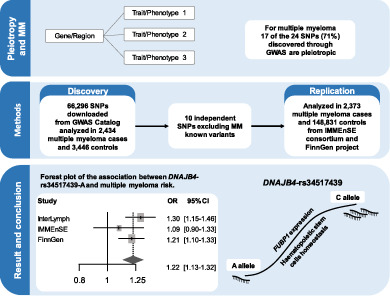
Pleiotropy is frequent across cancers, 18 , 19 , 20 with a portion of genome‐wide associated loci shared with other traits and several regions harboring single nucleotide polymorphisms (SNP) associated with multiple cancer types. For example, the TERT and 8q24 loci are associated with risk of multiple cancer types, including MM. 12 , 21 , 22 , 23 , 24 , 25 This feature is particularly evident in MM since we observed, using GWAS Catalog, that 17 of the 24 associated polymorphisms (~71%) are pleiotropic or in high linkage disequilibrium (LD) (r2 > 0.8) with SNPs associated with other traits (https://www.ebi.ac.uk/gwas/), including telomere length, 26 BMI 27 and risk of various cancers, such as myeloproliferative neoplasms 28 and pancreatic cancer. 25 Therefore, the aim of this study was to test the impact of all the SNPs associated at genome‐wide significant level with any human trait on risk of MM. This strategy has been employed for several cancer sites 18 , 19 , 20 , 29 , 30 , 31 but never for MM.
2. MATERIALS AND METHODS
2.1. Study populations
The study employed a two‐step approach, consisting of a discovery phase in which the International Lymphoma Epidemiology Consortium (InterLymph) GWAS data were analyzed, and a validation phase performed using both summary statistics from the FinnGen project MM GWAS (https://www.finngen.fi/en/access_results) and cases and controls of the International Multiple Myeloma rESEarch (IMMEnSE) consortium.
InterLymph has been described elsewhere. 8 Briefly, InterLymph generated GWAS data, using multiple platforms, for 2434 MM cases and 3446 controls of European ancestry from the United States of America, Canada and Australia genotyped using the Affymetrix 6.0 and Illumina (610 Quad, Human660W‐quad Beadchip, Omni5, OmniExpress Beadchip, Oncoarray) platforms. 8 Imputation was performed using the Haplotype Reference Consortium as reference panel, and the Michigan imputation server (https://imputationserver.sph.umich.edu/). 32 After imputation, each site was filtered to include only imputed variants with information score > 0.6 and further quality control checks were implemented (genotype rate > 95%, minor allele frequencies >0.01, and Hardy‐Weinberg equilibrium [HWE] P > 10−5 in controls). Finally, the data were pooled and final quality controls were performed on the pooled GWAS set, including checks for missingness, duplicates, sex mismatch, abnormal heterozygosity, cryptic relatedness, population outliers (through principal component [PC] analysis), and genomic inflation. After applying quality control measures to the imputed data, 5 864 648 SNPs remained for analysis. 8
The FinnGen project 33 is a cohort of 176 899 Finnish individuals genotyped with Illumina and Affymetrix platforms (https://www.finngen.fi). Subjects with sex discrepancies, call rate < 95%, excess heterozygosity (+‐4SD) and non‐Finnish ancestry were removed. SNPs with call rate < 98%, deviation from HWE (P < 10−6) and low minor allele count < 3 were removed. Subsequently, imputation was conducted using the SISu v3 reference panel with Beagle 4.1 (version 08Jun17.d8b). Imputed variants with information score < 0.7 were removed. 33 A GWAS testing association of 16 962 023 SNPs with 2444 endpoints (among which MM risk) was performed. Analyses were adjusted for age, sex, 10 PCs and for genotyping batch. A total of 418 MM cases and 147 282 controls who were cancer‐free at recruitment were evaluated in this study. Summary statistics of genome‐wide associations between SNPs and MM risk were obtained from https://r4.finngen.fi on April 22, 2021.
Information on IMMEnSE is reported in detail elsewhere. 34 Briefly, it consists of a multicentric study involving seven countries (Denmark, France, Hungary, Israel, Italy, Poland, Portugal and Spain). Patients had a confirmed diagnosis of MM in compliance with International Myeloma Working Group (IMWG) criteria, 35 while controls were from the same center or geographic region as the MM patients, including individuals from the general population, blood donors or patients hospitalized for diseases other than cancer. For each participant sex, age (at diagnosis for cases, at recruitment for controls) and country of origin were collected. For this study, 1955 MM cases and 1549 controls were included. Considering all of the studies, the total number of subjects analyzed was 4807 MM cases and 152 277 controls (Table 1).
TABLE 1.
Study populations
| InterLymph | IMMEnSE | FinnGen | Total | |
|---|---|---|---|---|
| Diagnosis | ||||
| MM cases | 2434 | 1955 | 418 | 4807 |
| Controls | 3446 | 1549 | 147 282 | 152 277 |
| Total | 5880 | 3504 | 147 700 | 157 084 |
| Median age (25%‐75%) a | ||||
| MM cases | 61 (26‐90) | 61 (54‐67) | — | — |
| Controls | 51 (43‐61) | — | — | |
| Sex | ||||
| Male | 61% | 52% | — | — |
| Female | 39% | 48% | — | — |
Note: Details on age and sex distribution of FinnGen individuals are not available.
Median age values of MM cases and controls with 25th and 75th percentile.
2.2. SNP identification and selection
We downloaded a list of all the SNPs associated with at least one human phenotype at P < 5 × 10−8 from the GWAS Catalog web site (https://www.ebi.ac.uk/gwas/). The list was downloaded in January 2020 and all SNPs without “rs” identifier (n = 1667) were excluded, leaving 66 296 unique SNPs for analysis.
2.3. Sample preparation, genotyping and quality control in IMMEnSE
DNA samples from IMMEnSE consortium participants were extracted from whole blood and genotyped using TaqMan (Thermo Fisher Applied Biosystems, Waltham MA, USA) assay technology, according to the manufacturer's recommendations. Genotyping was conducted in 384 well plates, including n = 203 duplicate samples (6%) for quality control purpose (concordance rate was higher than 98%). The distribution of cases and controls was unknown to the operator performing the genotyping. The fluorescent emission of the genotyping assay was detected by a QuantStudio 5 Real‐Time PCR system (Thermofisher) and the genotyping calls were made with QuantStudio software (Thermofisher). The average call rate of the SNPs was 86.30%. Subjects with a call rate < 70% (331 cases and 216 controls) were excluded from further analysis. Pearson's chi‐square test (χ2) was performed to assess if genotype frequencies were in HWE. The analysis, restricted to controls, was performed overall and separately for each country. All SNPs were in HWE except for rs10187103 (P = 4.99 × 10−8), rs1063348 (P = 9.27 × 10−4) and rs465530 (P = 8.91 × 10−6) in the controls from Denmark and therefore we excluded the genotypes of Danish subjects for these three SNPs from further analysis.
2.4. Statistical analysis
Unconditional logistic regression analysis was performed using InterLymph data to assess the association between the pleiotropic SNPs and risk of developing MM, reporting odds ratios (ORs) with 95% confidence intervals (CIs). Analyses were adjusted for age at diagnosis/recruitment, sex, study site and for the first five PCs. LD pruning (r 2 = 0.8) was applied to eliminate SNPs representing the same locus or variants in LD with known MM susceptibility regions. All independent variants that showed an association at P < 10−4 (arbitrary threshold) were then analyzed in the validation populations. Summary statistics were used for FinnGen, whereas for IMMEnSE, additive and co‐dominant unconditional logistic regression analysis was performed, adjusted for age at diagnosis/recruitment, sex and country of origin.
Finally, random‐effects meta‐analysis was performed with the PLINK software to combine results of the two phases. Heterogeneity was quantified with the I2 metric and evaluated with the Cochran's Q statistic test for each SNP. To account for multiple testing, we considered LD (r 2 > 0.8) among the SNPs used in the discovery phase to obtain a list of independent variants (n = 28 684). The threshold for statistical significance was therefore set to P = 0.05/28684 = 1.74 × 10−6 using Bonferroni's correction.
2.4.1. Annotation of functional effect of the SNPs
Several bioinformatic tools were used to evaluate possible functional features of the SNPs associated with MM. The occurrence of regulatory motif alterations was investigated through HaploReg (https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php) 36 and RegulomeDB (https://regulomedb.org/regulome-search). 37 The Genotype‐Tissue Expression project (GTEx) portal 38 (https://www.gtexportal.org/home/) was used to assess if selected SNPs are expression quantitative trait loci (eQTLs), that is, their alleles are associated with differential expression of genes in cis.
3. RESULTS
3.1. Discovery
Among the 66 296 variants selected for testing, 6438 were not genotyped or imputed in the InterLymph MM GWAS and therefore could not be analyzed. Of the 59 858 remaining SNPs, 2997 SNPs were associated with risk of developing MM at P < 0.05. Within that group, 56 SNPs with P < 10−4 were selected for the validation studies (Table S1). Forty SNPs already known to be associated with the risk of developing MM or in LD (r 2 ≥ 0.8) with these known SNPs were further excluded. Residual LD (r 2 > 0.4) between the remaining 16 SNPs was evaluated, resulting in the exclusion of an additional six SNPs. The remaining 10 SNPs were selected for evaluation in FinnGen and IMMEnSE. Figure S1 shows a flowchart of the process.
3.2. Replication and combined analysis
We performed the combined analysis using exclusively the allelic model, since this was available for the FinnGen dataset. We found a statistically significant association (using P < 1.74 × 10−6) between the A allele of DNAJB4‐rs34517439 and an increased risk of developing MM (ORcombined‐analysis = 1.22, 95%CI 1.13‐1.32, P = 4.81 × 10−7) with very low study heterogeneity (I 2 = 7.30%, Q = 2.25) (Table 2 and Figure 1). However, we observed some degree of heterogeneity in the combined analysis for the SNPs that did not show any statistically significant results, that could be due to the different direction of the association (Table 2) or to differences of the allelic frequencies between Finnish and non‐Finnish Europeans (Table S2).
TABLE 2.
Combined analysis using InterLymph, IMMEnSE and FinnGen results
| SNP | Region | (Alleles) M/m a | InterLymph | IMMEnSE | FinnGen | Combined analysis | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95%CI) | P value | OR (95%CI) | P value | OR (95%CI) | P value | OR (95%CI) | P value | I 2 b | Q b | Q‐P value | |||
| rs34517439 | 1p31.1 | C/A | 1.30 (1.15‐1.48) | 4.70 × 10−5 | 1.09 (0.89‐1.34) | .402 | 1.21 (1.09‐1.34) | .071 | 1.22 (1.13–1.32) | 4.81 × 10 −7 | 7.30% | 2.25 | .325 |
| rs6674512 | 1p22.3 | G/A | 1.40 (1.21‐1.62) | 8.71 × 10−6 | 1.10 (0.88‐1.36) | .402 | 1.04 (0.91‐1.20) | .765 | 1.17 (0.96‐1.43) | .115 | 77.50% | 9.74 | .008 |
| rs10187103 | 2q24.3 | C/T | 0.84 (0.77‐0.91) | 6.06 × 10−5 | 0.99 (0.88–1.12) | .915 | 0.99 (0.90‐1.09) | .924 | 0.95 (0.83–1.08) | .397 | 81.10% | 10.38 | .006 |
| rs1022206 | 3q13.13 | C/T | 1.18 (1.09‐1.28) | 8.61 × 10−5 | 1.11 (1.00–1.24) | .061 | 0.99 (0.92‐1.07) | .923 | 1.09 (0.97‐1.22) | .136 | 80.00% | 10.25 | .006 |
| rs4143832 | 5q31.1 | G/T | 1.24 (1.12‐1.37) | 4.66 × 10−5 | 0.95 (0.83–1.08) | .439 | 0.91 (0.84‐0.99) | .275 | 1.02 (0.84‐1.25) | .819 | 91.30% | 22.96 | 1.03 × 10−5 |
| rs537930 | 5q31.1 | G/T | 0.83 (0.76‐0.91) | 5.09 × 10−5 | 0.97 (0.86–1.09) | .571 | 0.96 (0.88‐1.04) | .596 | 0.92 (0.83‐1.01) | .088 | 70.10% | 6.51 | 3.80 × 10−2 |
| rs1063348 | 6p21.32 | G/A | 1.19 (1.09‐1.30) | 7.44 × 10−5 | 0.95 (0.85–1.07) | .398 | NA c | NA | 1.07 (0.87‐1.32) | .534 | 89.6% | 14.49 | .001 |
| rs13252276 | 8p23.1 | C/T | 0.77 (0.68‐0.87) | 3.10 × 10−5 | 1.01 (0.91–1.12) | .870 | 1.03 (0.96‐1.11) | .711 | 0.93 (0.79–1.10) | .415 | 88.00% | 17.50 | 2.00 × 10−4 |
| rs465530 | 12q14.3 | G/T | 1.18 (1.09–1.28) | 4.94 × 10−5 | 1.01 (0.89–1.13) | .926 | 0.95 (0.88‐1.02) | .458 | 1.03 (0.90‐1.19) | .657 | 87.80% | 17.06 | 2.00 × 10−4 |
| rs8132680 | 21q22.12 | T/C | 1.21 (1.11‐1.33) | 2.96 × 10−5 | 0.99 (0.88–1.12) | .924 | 1.04 (0.96‐1.13) | .646 | 1.08 (0.96‐1.21) | .197 | 77.40% | 8.06 | .018 |
M = the most common allele in controls; m = the less common allele in controls. The more common allele found in the control population has been used as reference and the other allele was defined as the exposure factor.
Measures of heterogeneity between the combined analysis components.
This polymorphism is not present in FinnGen, and therefore the combined analysis refers only to InterLymph and IMMEnSE populations.
FIGURE 1.
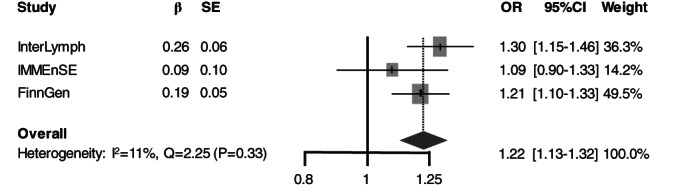
Forest plot of the association between DNAJB4‐rs34517439‐A and MM risk
Though not statistically significant, we observed an association between LOC105374037‐rs1022206 and risk of developing MM in the IMMEnSE population when comparing homozygotes for the rare allele (T/T) to homozygotes for the common allele (C/C) (ORhomozygptes = 1.29, 95%CI 1.03‐1.62, P = .028) and in the recessive model (ORrecessive = 1.28, 95%CI 1.04‐1.57, P = .019) in the IMMEnSE population (Table 3).
TABLE 3.
Associations between MM risk and the top 10 SNPs in IMMEnSE population
| SNP | Region | (Alleles) M/m a | Allelic model b | Codominant model c | Dominant model d | Recessive model e | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR f | 95% CI | P value | OR het f | 95% CI | P value | OR hom f | 95% CI | P value | OR f | 95% CI | P value | OR f | 95% CI | P value | |||
| rs34517439 | 1p31.1 | C/A | 1.09 | 0.89‐1.34 | .402 | 1.10 | 0.88‐1.38 | .395 | 1.09 | 0.44‐2.66 | .853 | 1.10 | 0.88‐1.37 | .387 | 1.07 | 0.44‐2.62 | .878 |
| rs6674512 | 1p22.3 | G/A | 1.10 | 0.88‐1.36 | .402 | 1.14 | 0.90‐1.44 | .267 | 0.74 | 0.24‐2.28 | .600 | 1.12 | 0.89‐1.41 | .320 | 0.73 | 0.24‐2.24 | .580 |
| rs10187103 | 2q24.3 | C/T | 0.99 | 0.88‐1.12 | .915 | 0.90 | 0.75‐1.07 | .215 | 1.09 | 0.83‐1.43 | .540 | 0.93 | 0.79‐1.10 | .407 | 1.15 | 0.89‐1.49 | .287 |
| rs1022206 | 3q13.13 | C/T | 1.11 | 1.00‐1.24 | .061 | 1.01 | 0.86‐1.20 | .889 | 1.29 | 1.03–1.62 | .028 | 1.08 | 0.92‐1.26 | .348 | 1.28 | 1.04–1.57 | .019 |
| rs4143832 | 5q31.1 | G/T | 0.95 | 0.83‐1.08 | .439 | 0.94 | 0.79–1.10 | .428 | 0.94 | 0.66‐1.32 | .709 | 0.94 | 0.80‐1.10 | .406 | 0.96 | 0.68‐1.35 | .807 |
| rs537930 | 5q31.1 | G/T | 0.97 | 0.86‐1.09 | .571 | 0.86 | 0.73‐1.00 | .061 | 1.14 | 0.84‐1.55 | .385 | 0.90 | 0.77‐1.05 | .168 | 1.22 | 0.90‐1.64 | .198 |
| rs1063348 | 6p21.32 | G/A | 0.95 | 0.85‐1.07 | .398 | 0.92 | 0.77‐1.10 | .368 | 0.92 | 0.72‐1.17 | .487 | 0.92 | 0.77‐1.09 | .336 | 0.96 | 0.77‐1.20 | .731 |
| rs13252276 | 8p23.1 | C/T | 1.01 | 0.91‐1.12 | .870 | 1.02 | 0.86‐1.21 | .825 | 1.01 | 0.82‐1.25 | .895 | 1.02 | 0.87‐1.19 | .826 | 1.00 | 0.83‐1.21 | .973 |
| rs465530 | 12q14.3 | G/T | 1.01 | 0.89‐1.13 | .926 | 0.83 | 0.67‐1.01 | .064 | 1.03 | 0.81‐1.31 | .820 | 0.89 | 0.73‐1.07 | .220 | 1.16 | 0.95‐1.42 | .147 |
| rs8132680 | 21q22.12 | T/C | 0.99 | 0.88–1.12 | .924 | 1.01 | 0.86‐1.19 | .869 | 0.96 | 0.72‐1.28 | .785 | 1.00 | 0.86‐1.17 | .960 | 0.96 | 0.72‐1.27 | .751 |
Note: In bold it is reported the statistically significant association.
M = the more common allele in controls; m = the less common allele in controls. The more common allele found in the control population has been used as the reference and the other allele was defined as the exposure factor.
Also termed quantitative additive (m vs M).
Mm vs MM (het) and mm vs MM (hom).
Mm + mm vs MM.
mm vs Mm + MM.
All the analyses were adjusted for age at diagnosis/recruitment, sex and country of origin. Results in bold show associations with P < .05.
3.3. Functional effect of the SNPs
RegulomeDB assigned a rank of 5 to both SNPs, implying a possible influence on a transcription factor binding site. In addition, GTEx and HaploReg indicated that DNAJB4‐rs34517439 is an eQTL affecting far upstream element binding protein 1 gene (FUBP1) expression in several human tissues. This included a statistically significant correlation between the A allele of this SNP and a reduced expression of FUBP1 in cultured fibroblasts (P = 1.10 × 10−4). GTEx also indicated an association between the T allele of LOC105374037‐rs1022206 and increased expression of the nectin cell adhesion molecule 3 (NECTIN3) gene in cultured fibroblasts (P = 2.10 × 10−20).
4. DISCUSSION
Pleiotropic variants are commonly found associated with complex diseases, like cancer. To investigate the possible association between common SNPs and the risk of developing MM, we analyzed all the genetic variants associated with any human trait in 4807 patients and 152 277 controls. We observed a statistically significant association (P < 1.74 × 10−6) between DNAJB4‐rs34517439‐A and risk of developing MM. This SNP is pleiotropic since it is associated with numerous traits such as cutaneous melanoma, diastolic blood pressure, fat‐free mass, hair color, hand grip strength, height, lung cancer, noncognitive aspects of educational attainment, psoriasis, serum alkaline phosphatase levels, smoking initiation and BMI, with a wide range of effect sizes and P values as reported in Table S3. The association between this SNP and BMI is of particular interest since BMI is also associated with MM risk. 39 The A allele that was associated with increased MM risk in our study has previously been associated with increased risk of lung cancer. 40 This pleiotropic effect, especially in cancers, may be attributed to the LD of rs34517439 with the variants inside or in the proximity of DnaJ heat shock protein family (Hsp40) member B4 (DNAJB4), situated on chromosome 1. This protein is a molecular chaperone specifically recognizing wild‐type from mutant E‐cadherin protein. Since E‐cadherin is an important element in the suppression of tumor invasion, the function of DNAJB4 represents a significant pleiotropic mechanism of tumoral invasion inhibition. 41 , 42 DNAJB4 also acts as a co‐chaperone, forming a complex with Hsp70 which participates in protein folding. 43 The expression and activity of heat shock proteins increases during cellular stress, 44 and DNAJB4 has been observed to reduce tumor metastasis and progression in several cancer types, including lung carcinoma and melanoma. 45 , 46 This mechanism could also be relevant to MM. In bioinformatic analysis, GTEx showed an association between DNAJB4‐rs34517439‐A and a reduced expression of the FUBP1 gene in various tissues, including lung, skin (sun exposed and not sun exposed), adipose tissue, the arteries and the heart. The altered expression of the gene in these tissues mirrors the associations found in the GWASs (eg, altered expression in lung for lung cancer, or skin for melanoma). FUBP1 encodes a multifunctional DNA and RNA‐binding protein involved in cell cycle regulation and self‐renewal of hematopoietic stem cells (HSCs). 47 FUBP1 acts both as oncogene and as tumor suppressor, and its activity is tissue‐dependent. 48 In various tumor types FUBP1 promotes the overexpression of the MYC oncogene, 48 whereas FUBP1 silencing does not influence the expression of MYC in normal fibroblasts, prostate and bladder cancer cells. 48 Moreover, FUBP1 is involved in the upregulation and downregulation of the cell cycle inhibitor p21. 49 , 50 The pleiotropic role of rs34517439 could be explained by its involvement in the altered expression of FUBP1 in different tumors, that determines their development, by interacting with specific factors in each tissue. Specifically for hematopoietic lineages, the cooperation of FUBP1 with RUNX1 in promoting the expression of the c‐KIT oncogene was reported. 51 In summary, a possible explanation for the DNAJB4‐rs34517439 association with increased risk of developing MM could be that the A allele modifies the expression of DNAJB4 leading to an incorrect folding of FUBP1. In turn, this mechanism could be responsible for the disruption of the HSCs homeostasis equilibrium, thus increasing the risk of developing MM. Although we lack an experimental validation of the proposed mechanism in MM cell lines, our hypothesis relies on the experimental and genomic data collected from GTEx, which are broadly used in the scientific community.
In addition, the T allele of LOC105374037‐rs1022206 in homozygosity showed an increased risk of developing MM in the IMMEnSE population. LOC105374037 is an uncharacterized long noncoding RNA (lncRNA) for which possible functional roles are unknown. Interestingly, LOC105374037‐rs1022206 is an eQTL for NECTIN3 gene expression in cultured fibroblasts. Fibroblasts are the cells with the highest expression of NECTIN3 in humans. 52 Cancer‐associated fibroblasts (CAFs) are one of the known cellular elements participating in MM tumoral microenvironment in the BM. 53 In fact, they show a bidirectional loop with myeloma cells providing chemotaxis, adhesion, apoptosis resistance and proliferation through cytokines, growth factors, angiogenetic factors and cell‐cell contact. 54 However, considering the lack of data in the literature on the topic it is not clear how the increased expression of NECTIN3 mediated by LOC105374037‐rs1022206 might be involved in MM risk. Furthermore, Nectin‐3 is involved in cell survival through PDGF receptor signal and in inhibition of cell movement. 52 , 55 The SNP LOC105374037‐rs1022206 is in LD with variants associated in with various traits: balding type 1, smoking initiation, self‐reported math ability, feeling hurt and neuroticism. Although these traits apparently do not show a shared mechanism or pathway, the LOC105374037 lncRNA could be the common element explaining the pleiotropic effect of the SNP on different cellular functions.
A possible limitation of this study could be that only the allelic model was available for all the datasets and therefore it was the only one assessed in the final combined analysis using InterLymph, IMMEnSE and FinnGen populations. In fact, we were not able to confirm the promising association detected in the IMMEnSE population for LOC105374037‐rs1022206‐T, that needs to be further investigated. Another possible limitation is the ethnic origin of the populations used in the replication phase, due to the known genetic differences between the Finnish and non‐Finnish Europeans. 56 , 57 Specifically, for the 10 SNPs considered in the replication phase of our study, the average difference in allelic frequency is 6%, and for the two SNPs that show significant association this difference is lower than 5%. The heterogeneity value obtained in the combined analysis for DNAJB4‐rs34517439 is very low (I 2 = 7.30%) and not significant, and therefore, it is highly unlikely that differences in the allelic frequencies might have affected the results. However, we have observed some degrees of heterogeneity in the other SNPs. An additional limitation in our study is that the number of SNPs chosen for replication was limited. We chose a threshold of P < 10−4 as a good compromise allowing us to select a suitably small number of variants to genotype in IMMEnSE and to maximize chances of finding significant results in the overall analysis. It would have not been worth choosing SNPs showing a weaker association in InterLymph, because we would not have sufficient statistical power to find genome‐wide significant associations in the combined results.
Finally, considering that the P value we observed, although statistically significant considering the Bonferroni correction for multiple testing, does not reach genome‐wide significance, further studies are warranted to establish this SNP as a new MM risk locus.
In conclusion, our results suggest the involvement of a pleiotropic region on chromosome 1 in MM development and highlight pleiotropy as an approach to uncover additional risk variants in cancer susceptibility.
AUTHOR CONTRIBUTIONS
Daniele Campa conceived and designed the study. Marco Dicanio and Matteo Giaccherini performed the lab work, Marco Dicanio, Matteo Giaccherini and Alyssa Clay‐Gilmour analyzed the data. All the authors contributed with the interpretation of the data. Marco Dicanio, Matteo Giaccherini and Daniele Campa wrote the first draft of the manuscript and all authors contributed to the writing and approve of the final version of the manuscript. The work reported in the paper has been performed by the authors, unless clearly specified in the text.
FUNDING INFORMATION
This study was partially supported by intramural funds of University of Pisa and DKFZ.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The IMMEnSE study protocol was approved by the Ethics Committee of the Medical Faculty of the University of Heidelberg (reference number: S‐004/2020). Following the guidelines of the Declaration of Helsinki, written informed consent was obtained from each participant For InterLymph, contributing studies were approved by local ethics review committees. Summary statistics were used from the FinnGen study, that was approved by the ethical Review Board of the Hospital District of Helsinki and Uusimaa. FinnGen participants provided written, informed consent.
Supporting information
TABLE S1 SNPs associated with MM risk in InterLymph (P < 10−4)
TABLE S2: Minor allelic frequency (MAF) of the 10 SNPs analyzed in the replication phase.
TABLE S3: effect sizes and P values reported in GWAS catalog for the SNPs selected for replication.
FIGURE S1. Flowchart of the process performed to select the SNPs to be replicated in IMMEnSE and FinnGen
ACKNOWLEDGMENT
Open Access Funding provided by Universita degli Studi di Pisa within the CRUI‐CARE Agreement.
Dicanio M, Giaccherini M, Clay‐Gilmour A, et al. A pleiotropic variant in DNAJB4 is associated with multiple myeloma risk. Int J Cancer. 2023;152(2):239‐248. doi: 10.1002/ijc.34278
Marco Dicanio, Matteo Giaccherini and Alyssa Clay‐Gilmour share the first position and Celine Vachon, Federico Canzian and Daniele Campa share the last position.
Funding information University of Pisa and DKFZ
DATA AVAILABILITY STATEMENT
The primary data for this work will be made available to researchers who submit a reasonable request to the corresponding author, conditional to approval by the centers participating in this study. Data will be stripped from all information allowing identification of study participants.
REFERENCES
- 1. Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Prim. 2017;3:17046. [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Colombet M, Soerjomataram I, et al. Cancer statistics for the year 2020: An overview. Int J Cancer. 2021;149(4):778‐789. [DOI] [PubMed] [Google Scholar]
- 3. Went M, Sud A, Försti A, et al. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat Commun. 2018;9(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swaminathan B, Thorleifsson G, Jöud M, et al. Variants in ELL2 influencing immunoglobulin levels associate with multiple myeloma. Nat Commun. 2015;6:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitchell JS, Li N, Weinhold N, et al. Genome‐wide association study identifies multiple susceptibility loci for multiple myeloma. Nat Commun. 2016;7:12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chubb D, Weinhold N, Broderick P, et al. Common variation at 3q26.2, 6p21.33, 17p11.2 and 22q13.1 influences multiple myeloma risk. Nat Genet. 2013;45(10):1221‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broderick P, Chubb D, Johnson DC, et al. Common variation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nat Genet. 2012;44(1):58‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clay‐Gilmour AI, Hildebrandt MAT, Brown EE, et al. Coinherited genetics of multiple myeloma and its precursor, monoclonal gammopathy of undetermined significance. Blood Adv. 2020;4(12):2789‐2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pertesi M, Went M, Hansson M, Hemminki K, Houlston RS, Nilsson B. Genetic predisposition for multiple myeloma. Leukemia. 2020;34(3):697‐708. [DOI] [PubMed] [Google Scholar]
- 10. Duran‐Lozano L, Thorleifsson G, de Lapuente L, et al. Germline variants at SOHLH2 influence multiple myeloma risk. Blood Cancer J. 2021;11(4):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Giaccherini M, Macauda A, Orciuolo E, et al. Genetically determined telomere length and multiple myeloma risk and outcome. Blood Cancer J. 2021;11(4):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campa D, Martino A, Varkonyi J, et al. Risk of multiple myeloma is associated with polymorphisms within telomerase genes and telomere length. Int J Cancer. 2015;136(5):E351‐E358. [DOI] [PubMed] [Google Scholar]
- 13. Campa D, Martino A, Macauda A, et al. Genetic polymorphisms in genes of class switch recombination and multiple myeloma risk and survival: an IMMEnSE study. Leuk Lymphoma. 2019;60(7):1803‐1811. [DOI] [PubMed] [Google Scholar]
- 14. Melaiu O, Macauda A, Sainz J, et al. Common gene variants within 3′‐untranslated regions as modulators of multiple myeloma risk and survival. Int J Cancer. 2020;148:1887‐1894. [DOI] [PubMed] [Google Scholar]
- 15. Rand KA, Song C, Dean E, et al. A meta‐analysis of multiple myeloma risk regions in African and European ancestry populations identifies putatively functional loci. Cancer Epidemiol Biomarkers Prev. 2016;25(12):1609‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paaby AB, Rockman MV. The many faces of pleiotropy. Trends Genet. 2013;29(2):66‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shikov AE, Skitchenko RK, Predeus AV, Barbitoff YA. Phenome‐wide functional dissection of pleiotropic effects highlights key molecular pathways for human complex traits. Sci Rep. 2020;10(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pierce BL, Ahsan H. Genome‐wide “pleiotropy scan” identifies HNF1A region as a novel pancreatic cancer susceptibility locus. Cancer Res. 2011;71(13):4352‐4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Campa D, Barrdahl M, Tsilidis KK, et al. A genome‐wide “pleiotropy scan” does not identify new susceptibility loci for estrogen receptor negative breast cancer. PLoS One. 2014;9(2):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Panagiotou OA, Travis RC, Campa D, et al. A genome‐wide pleiotropy scan for prostate cancer risk. Eur Urol. 2015;67(4):649‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Campa D, Rizzato C, Stolzenberg‐Solomon R, et al. TERT gene harbors multiple variants associated with pancreatic cancer susceptibility. Int J Cancer. 2015;137(9):2175‐2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Campa D, Martino A, Sainz J, et al. Comprehensive investigation of genetic variation in the 8q24 region and multiple myeloma risk in the IMMEnSE consortium. Br J Haematol. 2012;157(3):331‐338. [DOI] [PubMed] [Google Scholar]
- 23. Giaccherini M, Macauda A, Sgherza N, et al. Genetic polymorphisms associated with telomere length and risk of developing myeloproliferative neoplasms. Blood Cancer J. 2020;10(8):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilson C, Kanhere A. 8q24.21 locus: a paradigm to link non‐coding RNAs, genome polymorphisms and cancer. Int J Mol Sci. 2021;22(3):1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wolpin BM, Rizzato C, Kraft P, et al. Genome‐wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46(9):994‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Codd V, Nelson CP, Albrecht E, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45(4):422‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoffmann TJ, Choquet H, Yin J, et al. A large multiethnic genome‐wide association study of adult body mass index identifies novel loci. Genetics. 2018;210(2):499‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Macauda A, Giaccherini M, Sainz J, et al. Do myeloproliferative neoplasms and multiple myeloma share the same genetic susceptibility loci? Int J Cancer. 2021;148(7):1616‐1624. [DOI] [PubMed] [Google Scholar]
- 29. Cheng I, Kocarnik JM, Dumitrescu L, et al. Pleiotropic effects of genetic risk variants for other cancers on colorectal cancer risk: PAGE, GECCO and CCFR Consortia. Gut. 2014;63(5):800‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang X, Finucane HK, Schumacher FR, et al. Shared heritability and functional enrichment across six solid cancers. Nat Commun. 2019;10(1):4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sampson JN, Wheeler WA, Yeager M, et al. Analysis of heritability and shared heritability based on genome‐wide association studies for thirteen cancer types. J Natl Cancer Inst. 2015;107(12):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McCarthy S, Europe PMC. Funders group Europe PMC funders author manuscripts a reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tabassum R, Rämö JT, Ripatti P, et al. Genetic architecture of human plasma lipidome and its link to cardiovascular disease. Nat Commun. 2019;10(1):4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martino A, Sainz J, Buda G, et al. Genetics and molecular epidemiology of multiple myeloma: the rationale for the IMMEnSE consortium (review). Int J Oncol. 2012;40(3):625‐638. [DOI] [PubMed] [Google Scholar]
- 35. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538‐e548. [DOI] [PubMed] [Google Scholar]
- 36. Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44(D1):D877‐D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22(9):1790‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lonsdale J, Thomas J, Salvatore M, et al. The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45(6):580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wallin A, Larsson SC. Body mass index and risk of multiple myeloma: a meta‐analysis of prospective studies. Eur J Cancer. 2011;47(11):1606‐1615. [DOI] [PubMed] [Google Scholar]
- 40. McKay JD, Hung RJ, Han Y, et al. Large‐scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49(7):1126‐1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simões‐correia J, Silva DI, Melo S, et al. DNAJB4 molecular chaperone distinguishes WT from mutant E‐cadherin, determining their fate in vitro and in vivo. Hum Mol Genet. 2014;23(8):2094‐2105. [DOI] [PubMed] [Google Scholar]
- 42. Mitra A, Shevde LA, Samant RS. Multi‐faceted role of HSP40 in cancer. Clin Exp Metastasis. 2009;26(6):559‐567. [DOI] [PubMed] [Google Scholar]
- 43. Qiu X‐B, Shao Y‐M, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63(22):2560‐2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Feder ME, Hofmann GE. Heat‐shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol. 1999;61:243‐282. [DOI] [PubMed] [Google Scholar]
- 45. Chen CH, Chang WH, Su KY, et al. HLJ1 is an endogenous Src inhibitor suppressing cancer progression through dual mechanisms. Oncogene. 2016;35(43):5674‐5685. [DOI] [PubMed] [Google Scholar]
- 46. Miao W, Li L, Wang Y. A targeted proteomic approach for heat shock proteins reveals DNAJB4 as a suppressor for melanoma metastasis. Anal Chem. 2018;90(11):6835‐6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rabenhorst U, Thalheimer FB, Gerlach K, et al. Single‐stranded DNA‐binding transcriptional regulator FUBP1 is essential for fetal and adult hematopoietic stem cell self‐renewal. Cell Rep. 2015;11(12):1847‐1855. [DOI] [PubMed] [Google Scholar]
- 48. Kang M, Kim HJ, Kim T‐J, et al. Multiple functions of Fubp1 in cell cycle progression and cell survival. Cell. 2020;9(6):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duan J, Bao X, Ma X, et al. Upregulation of far upstream element‐binding protein 1 (FUBP1) promotes tumor proliferation and tumorigenesis of clear cell renal cell carcinoma. PLoS One. 2017;12(1):e0169852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rabenhorst U, Beinoraviciute‐Kellner R, Brezniceanu M‐L, et al. Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology. 2009;50(4):1121‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Debaize L, Jakobczyk H, Avner S, et al. Interplay between transcription regulators RUNX1 and FUBP1 activates an enhancer of the oncogene c‐KIT and amplifies cell proliferation. Nucleic Acids Res. 2018;46(21):11214‐11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mandai K, Rikitake Y, Mori M, Takai Y. Nectins and Nectin‐Like Molecules in Development and Disease. Curr Top Dev Biol. 2015;112:197‐231. [DOI] [PubMed] [Google Scholar]
- 53. Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. 2015;125:3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Frassanito MA, Rao L, Moschetta M, et al. Bone Marrow Fibroblasts Parallel Multiple Myeloma Progression in Patients and Mice: In Vitro and In Vivo Studies. Leukemia. 2014;28(4):904‐16. [DOI] [PubMed] [Google Scholar]
- 55. Fujito T, Ikeda W, Kakunaga S, et al. Inhibition of cell movement and proliferation by cell‐cell contact‐induced interaction of Necl‐5 with nectin‐3. J Cell Biol. 2005;171(1):165‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nelis M, Esko T, Mägi R, et al. Genetic structure of Europeans: a view from the North‐east. PLoS One. 2009;4(5):e5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 SNPs associated with MM risk in InterLymph (P < 10−4)
TABLE S2: Minor allelic frequency (MAF) of the 10 SNPs analyzed in the replication phase.
TABLE S3: effect sizes and P values reported in GWAS catalog for the SNPs selected for replication.
FIGURE S1. Flowchart of the process performed to select the SNPs to be replicated in IMMEnSE and FinnGen
Data Availability Statement
The primary data for this work will be made available to researchers who submit a reasonable request to the corresponding author, conditional to approval by the centers participating in this study. Data will be stripped from all information allowing identification of study participants.