

Abstract
Pancreatic ductal adenocarcinoma (PDAC) develops via dysplastic changes in the epithelia graded as low‐ and high‐grade with accumulation of molecular alterations. Constitutive activation of mitogen‐activated protein kinase (MAPK) contributed by attenuation of DUSP6 plays a key role in sustaining PDAC. Active MAPK induces various molecules that function as effectors to sustain PDAC. AURKA and SON are downstream effectors that contribute substantially to the proliferation and survival of PDAC cells and are potentially useful as therapeutic targets. Active MAPK also promote microRNAs that modulate the proliferation of PDAC cells and are useful as diagnostic markers. Familial pancreatic cancer kindreds in Japan show various germline mutations supposed to increase a pancreatic cancer risk. Intraductal papillary mucinous neoplasms (IPMNs) consist of dilated ducts lined by papillary neoplastic epithelia of various shapes and varying grades of atypia. Various papillae of IPMNs are classified into four subtypes that are associated with clinicopathological features, including patient prognosis. GNAS is a specific driver gene for the development of IPMN through gain‐of‐function mutations. Tracing of molecular alterations has elucidated the mechanism of progression of IPMN from dysplasia to carcinoma, as well as one type of papillae. Intraductal tubulopapillary neoplasms belong to a distinct class of pancreatic neoplasms.
Keywords: AURKA, DUSP6, GNAS, IPMN, ITPN, MAPK, miRNA, pancreatic cancer, RNF43, SON
Abbreviations
- AURKA
aurora kinase A
- DUSP6
dual specificity phosphatase 6
- Gsα
G‐protein stimulatory α subunit
- IPMN
intraductal papillary mucinous neoplasms
- ITPN
intraductal tubulopapillary neoplasm
- LOH
loss of heterozygosity
- LSL
lox‐STOP‐lox
- MAPK
mitogen‐activated protein kinase
- miRNA
microRNA
- NGS
next generation sequencing
- pAKT
phosphorylated AKT
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- PI3K
phosphatidylinositol 3‐kinase
- PS‐PKA
phosphorylated substrates of protein kinase A
- RNF43
ring finger protein 43
- SON
SON DNA‐and RNA‐binding protein
INTRODUCTION
Pancreatic cancer has the poorest prognosis among all types of cancers. In Japan, approximately 44 000 new cases and 37 000 deaths are estimated in 2021. The average life expectancy after the diagnosis of pancreatic cancer is 1.5 years, and the overall 5‐year survival rate is merely 7% (Projected Cancer Statistics, https://ganjoho.jp). Why is pancreatic cancer so devastating? What can we do to improve the prognosis? These are the main questions for researchers studying pancreatic cancer. In this review, mechanistic insights of development and progression of pancreatic neoplasms and their implications in clinical medicine based on my research results are discussed.
HOW DOES THE PANCREATIC CANCER DEVELOP?
When we review microscopic slides of pancreatic cancer cases, we become aware that dysplastic cells in the pancreatic ducts can be observed around the foci of invasive carcinoma (Figure 1). This phenomenon has been observed and investigated for more than 70 years, and the key findings are that the frequency and degrees of atypia of these dysplastic cells are increased in pancreata with carcinoma compared to those without carcinoma. 1 , 2 , 3 , 4 This indicates not only a close association between these dysplastic cells and carcinoma but also the progression of these dysplastic cells toward carcinoma. During these investigations, the degree of atypia was arbitrarily divided into several classes, such as mild, moderate, and severe. 5 However, it is unknown whether such classification is appropriate, that is, whether it reflects the pathobiological nature. To determine the appropriateness of the statistical meaning, elements for atypia can be translated into several measurable values including nuclear area, nuclear length, nucleocytoplasmic ratio, nuclear polarity (H = H n/H c, where H, H n, and H c indicate the nuclear polarity, height of apical cell membrane from basement membrane, and height of the center of nucleus from basement membrane, respectively), and nuclear form coefficient (Q = 4πS/L 2, where Q, S, and L indicate the nuclear form coefficient, nuclear area, and nuclear perimeter length, respectively); these values can be analyzed statistically to determine appropriate classifications. 6 Such a statistical analysis provides unbiased objective classifications without subjective arbitrariness. In a previous study, degrees of atypia of various dysplastic cells in the pancreatic ducts were measured as noted above and compared with those of normal and carcinoma cells, and these values were analyzed using multivariate cluster analysis. The analysis provided a detailed dendrogram showing closed lesions in morphological values each other and clusters interpretable to neoplasm classes. The dendrogram obtained from the values of 78 lesions with various degrees of atypia in the pancreatic ducts clearly showed that these lesions could be classified into three clusters. 6 Cluster 1 mainly consisted of normal cells, cluster 2 consisted of mild or moderately atypical cells, and cluster 3 consisted of severely dysplastic and invasive carcinoma cells. The separation of clusters was validated using canonical discriminant analysis, which showed the appropriate statistical power in this classification. This result strongly suggests that dysplastic cells in the pancreatic ducts can be appropriately classified into two classes, that is, low‐grade and high‐grade, instead of the arbitrary three tiers of mild, moderate, and severe. This has become a firm basis for the idea of a two‐tiered classification system of pancreatic intraepithelial lesions, which has indeed been accepted for the current classification of pancreatic intraepithelial neoplasia (PanIN) 7 into low‐grade and high‐grade 8 (Figure 1).
Figure 1.
Intraepithelial neoplasias in the pancreas. (a) Normal duct. (b) Pancreatic intraepithelial neoplasia, low‐grade. (c) and (d) Pancreatic intraepithelial neoplasia, high‐grade. All panels are H & E stained, ×200 original magnification.
The two‐tiered grading of pancreatic intraepithelial lesions implies that dysplastic cells would progress from low‐grade to high‐grade and eventually become carcinoma. This corresponds to the multistep carcinogenesis theory described in other organs such as the colon. 9 Multistep progression can be proved by investigating genetic changes as tracers of evolution. Pancreatic adenocarcinoma is known to harbor KRAS mutations in more than 90% of cases. 10 , 11 Investigation of genetic alterations in dysplastic cells in the pancreas also showed frequent mutations in KRAS, even in low‐grade dysplastic cells, 12 , 13 which indicates that low‐grade dysplastic cells would indeed progress to carcinoma. Moreover, aberrant expression of tumor suppressor molecules, including p16, p53, and SMAD4, is associated with the grade of dysplasia, that is, loss of p16 is associated with low‐grade dysplasia, whereas aberrations of p53 and SMAD4 are associated with high‐grade dysplasia. 14 This indicates that the accumulation of molecular aberrations may drive the progression of dysplastic cells to carcinomas.
FINDING A TUMOR SUPPRESSOR GENE IN PANCREATIC CANCER
Identifying driver genes in cancer is the key to understanding their pathobiology and determining targets for clinical diagnosis and therapy. The prevailing gain‐of‐function mutation of KRAS in invasive cancer as well as in dysplastic cells clearly indicate that mutated KRAS plays a key oncogenic role in pancreatic cancer. Dysfunction of tumor suppressor genes seems to play critical roles in the progression of dysplastic cells to cancer, and CDKN2A, TP53, and SMAD4 have become well known as such players. Inactivation of a tumor suppressor gene is frequently coupled with loss of heterozygosity (LOH), a major mechanism for two hits that results in complete loss of its function. We conducted a genome‐wide search for LOH and found that multiple loci, including 1p36, 6q21/23‐24, 9p21, 12q21, 17p13, and 18q21, showed frequent LOH. 15 , 16 , 17 Among these loci, it had already been known that 9p21, 17p13, and 18q21 indeed harbored tumor suppressor genes, including CDKN2A, TP53, and SMAD4, respectively. Then, we focused on other loci, particularly on 12q21, and because it was before the human genome project was completed, genomic cloning using a bacterial artificial chromosome library was conducted. As a result of exhaustive investigations, we found that DUSP6/MKP‐3/Pyst1 resided at that locus. 18 DUSP6/MKP‐3/Pyst1 has been cloned by Groom et al. as a gene encoding dual specificity phosphatase 6 (DUSP6) that specifically inactivates mitogen‐activated protein kinase (MAPK). 19 This provoked particular interest among us because MAPK is known to be a major downstream effector molecule of RAS that is activated by the prevailing gain‐of‐function mutation in KRAS in pancreatic cancer. We immediately analyzed whether there is a mutation in this gene in pancreatic cancer cell lines and tissues; however, no mutations were found in the analyzed samples. Instead, we found that the expression of DUSP6 in pancreatic cancer cell lines and tissues was frequently reduced or lost 20 (Figure 2). A subsequent detailed analysis of DUSP6 expression in pancreatic cancer tissues revealed that DUSP6 was expressed evidently in dysplastic cells even in those of high‐grade; while its expression was reduced in invasive cancer cells. 14 DUSP6 forms a negative feedback loop with MAPK, that is, the expression of DUSP6 is induced by active MAPK, which means that active MAPK is inactivated by the induced DUSP6; therefore, sustained activation of MAPK is prevented physiologically. 21 However, in pancreatic cancer cells, the reduced expression of DUSP6 was associated with sustained expression of phosphorylated MAPK, which was observed not only in pancreatic cancer cells with KRAS mutations but also in pancreatic cancer cells with wild‐type KRAS. A detailed analysis of the mechanism of DUSP6 expression revealed that methylation of CpG islands in intron 1 of DUSP6 caused downregulation of its expression. 22 Moreover, extrinsic overexpression of DUSP6 in these DUSP6‐abrogated pancreatic cancer cells by an adenoviral vector resulted in inactivation of MAPK and subsequent apoptosis. 20 These results indicate that DUSP6 plays a tumor‐suppressive role like a gait‐keeper in the progression from high‐grade dysplastic cells to invasive cancer, and that pancreatic cancer cells with abrogation of DUSP6 would require active MAPK for their survival, and inhibition of MAPK activation could be a therapeutic procedure for pancreatic cancer. Recently, it has been reported by Kidger et al. that a genetically engineered mouse model with activated mutation of Kras and deletion of Dusp6 displays accelerated development of pancreatic ductal adenocarcinoma (PDAC) with metastasis, 23 which further supports the tumor‐suppressive role of DUSP6 in pancreatic cancer.
Figure 2.
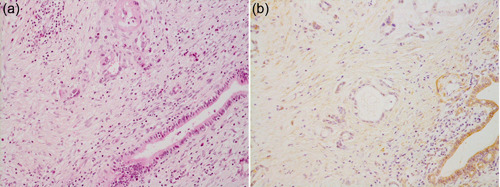
Reduced expression of DUSP6 in invasive ductal adenocarcinoma in the pancreas. (a) H & E staining. (b) Immunohistochemistry using a polyclonal anti‐DUSP6 antibody (Santa Cruz Biotechnology Inc.). Both panels are ×200 original magnification.
ACTIVATION OF MAPK IN PANCREATIC CANCER
Active MAPK appears to play a critical role in the development and progression of pancreatic cancer. A key question is how MAPK plays this role. Active MAPK translocates into the nucleus and activates various transcription factors, which results in the modulation of the expression of many downstream genes. The genes downstream of MAPK are hypothesized to be key molecules for the sustainability of pancreatic cancer. We carried out transcriptome analysis to identify differentially expressed genes between pancreatic cancer cells with active MAPK and those without. The results indicated that 79 and 84 genes were upregulated and downregulated by MAPK, respectively. The upregulated genes include those associated with the cell cycle and mitosis (AURKA, AURKB, HCAP‐G, CNAP1, CCNA2, CCNB1, CCNB2, CCNE2, KIF11, MCM5, MPHOSPH1, CENPA, CENPF, KIF2C, PRC1, SKB1, and TPX2), DNA replication (BLM, MCM4, ORC6L, POLQ, PRIM1, RFC4, and RRM1), receptor signaling pathway (ADORA2B, EPHA2, GCGR, WDR5, and TMEPAI), transcription (ETS1 and MAFF), kinase activity (PBK and RIOK1), and GTPase activity (ARHGAP11A and SIPA1L3). The downregulated genes include those associated with transportation (SAA1, FOLR1, KCNMB1, AQP3, FXYD3, CLDN16, SLC15A3, and SLC2A10), receptor activity (LYPD5, OLFML2A, BTN1A1, KLRC2, KLRC3, TAPBPL, and HLA‐F), cell proliferation (MDK, TGFB2, IFITM1, AKR1C3, RARRES3, and TNFSF13B), signal transduction (DUSP4, MAPK11, CNTNAP1, and CLEC2D), transcriptional regulation (ID3, SOX4, GCM2, and PHF11), immune response (SECTM1, IL32, IFI30, and CD24), metabolism (PMM2, ALDH1B1, ALPP, and DHRS6), and ubiquitin pathway (BARD1, PSMB9, HERC5, and HERC6) 24 (Figure 3). These genes are thought to play essential roles in the maintenance and survival of pancreatic cancer cells.
Figure 3.
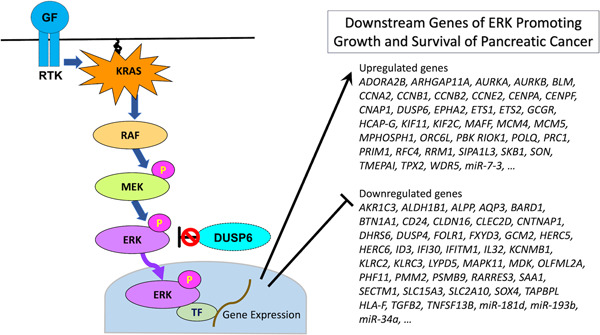
Abrogation of DUSP6 contributes to constitutive activation of ERK, which results in modulation of wide variety of effector genes that promote growth and survival of pancreatic cancer.
The next question that emerged was what would be the most essential key molecule among these downstream genes of MAPK for the proliferation and survival of pancreatic cancer cells. Such molecules may be significant therapeutic targets for pancreatic cancer. One such molecule that was uncovered is AURKA. AURKA encodes aurora kinase A (AURKA), which is known to function in the assembly of centrosomes, spindles, and kinetochores and that play important roles in mitosis. We showed that AURKA was frequently overexpressed in pancreatic cancer due to upregulation of its promoter by active MAPK via Ets‐2. 24 Downregulation of AURKA by RNA interference significantly suppressed the proliferation and survival of pancreatic cancer cells, and the downregulation of AURKA enhanced the effect of taxane, an antimicrotubule drug widely used for pancreatic cancer. 25
We expanded the search for significant molecules downstream of MAPK by assaying the effect of knockdown of each gene on the proliferation and survival of pancreatic cancer cells. These experiments revealed SON/NREBP/BASS1 to be the most significant effector gene that functions in the proliferation and survival of pancreatic cancer cells. 26 SON/NREBP/BASS1 is a gene encoding the SON DNA‐and RNA‐binding protein (SON). SON was found to be overexpressed in pancreatic cancer tissues, specifically in invasive carcinomas (Figure 4). Knockdown of SON most effectively attenuated the proliferation and survival of pancreatic cancer cells among the downstream molecules of MAPK. The exogenous expression of GFP‐labeled SON revealed its localization in a nuclear speckle that is known to be enriched in pre‐messenger RNA splicing factors. These results indicate that SON is required for sustainable proliferation and is a candidate therapeutic target for pancreatic cancer. 26
Figure 4.
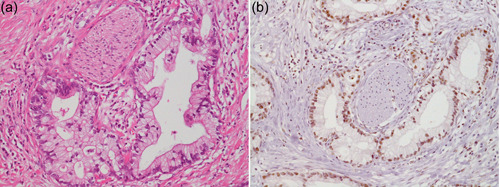
Overexpression of SON in invasive ductal adenocarcinoma of the pancreas. (a) H & E staining. (b) Immunohistochemistry using a polyclonal anti‐SON antibody (Sigma). Both panels are ×200 original magnification.
MicroRNAs (miRNAs) are key molecules in the modulation of gene expression under physiological and pathological conditions. We hypothesized that some miRNAs are regulated by MAPK. We analyzed the altered expression of miRNAs between pancreatic cancer cells with active and inactive MAPK using quantitative real‐time PCR assay. We found that miR‐7‐3, miR‐34a, miR‐181d, and miR‐193b are preferentially associated with MAPK in pancreatic cancer cells. 27 Among these miRNAs, miR‐7‐3 was upregulated, whereas miR‐34a, miR‐181d, and miR‐193b were downregulated by active MAPK. Exogenous miR‐193b expression markedly suppresses the proliferation of pancreatic cancer cells by modulating several genes, including CCND1, NT5E, PLAU, STARD7, STMN1, and YWHAZ, which indicates that miR‐193b is a tumor‐suppressive miRNA in pancreatic cancer. 27 Moreover, these MAPK‐associated miRNAs have proven to be useful serum markers to differentiate patients with pancreatic cancer and pancreatitis. 28 These results indicate that MAPK regulates the expression of miRNAs associated with the proliferation of pancreatic cancer cells and that MAPK‐associated miRNAs could be used as biomarkers for pancreatic cancer.
FAMILIAL PANCREATIC CANCER
Pancreatic cancer often develops in particular kindreds, which is known to be familial pancreatic cancer cases. The familial pancreatic cancer is defined as existence of two or more pancreatic cancer patients in first‐degree relatives. 29 According to an epidemiological study conducted in the United States, the risk of development of pancreatic cancer is significantly elevated as 9.0 fold compared to general population. Moreover, the risk is increased depending on number of patients in first‐degree relatives, that is, 32.0‐fold for three‐, 6.4‐fold for two‐, 4.8‐fold at one‐first degree relative(s) with pancreatic cancer. 29 Therefore, individuals in familial pancreatic cancer kindreds could be a good candidate for pancreatic cancer surveillance for early intervention. To know incidence of familial pancreatic cancer in Japan, we conducted a case‐control study employing 1197 pancreatic cancer cases, and found that 88 cases (7.3%) corresponded to cases of familial pancreatic cancer. 30 Comparisons of onset age, sex, tumor localization, stage, family history except pancreatic cancer, smoking, diabetes mellitus, and history of chronic pancreatitis between the familial pancreatic cancer cases and sporadic cases showed no significant differences. Exome sequencing of 81 cases in this cohort revealed possibly deleterious germline mutations in APC, ATM, BRCA1, BRCA2, CHEK2, ERCC2, FANCA, FANCC, FANCE, FAT4, KMT2C, MLH1, MSH2, NF1, PALB2, POLE, RNF43, SMAD4, TP53, and TSC2. BRCA2, PALB2, and ATM are particularly interest because these genes are reported to be mutated in familial pancreatic cancer kindreds in the United States. 31 Protein‐truncating mutations were indeed found in these genes in this Japanese familial pancreatic cancer cohort including 2 nonsense mutations in BRCA2, one frameshift mutation in PLB2, and 3 nonsense mutations in ATM. 32 These results provide evidences for designing surveillance strategy for Japanese familial pancreatic cancer kindreds.
PATHOLOGY OF INTRADUCTAL PAPILLARY MUCINOUS NEOPLASMS (IPMNs) OF THE PANCREAS
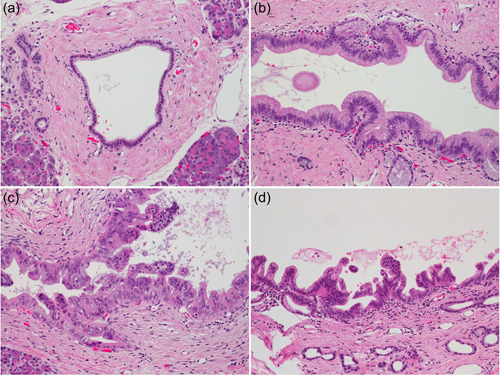
Intraductal papillary mucinous neoplasms are grossly visible pancreatic cystic neoplasms. Cysts are composed of dilated ducts lined with neoplastic epithelial cells with various grades of atypia that grow in various shapes of papillae. 33 Atypia is classified as low‐ and high‐grade. 8 Papillae of various shapes are classified into four subtypes based on their histologic characteristics, namely, gastric, intestinal, pancreatobiliary, and oncocytic (Figure 5). These subtypes are associated with the expression of specific mucin proteins, that is, MUC5AC and MUC6 in the gastric subtype; MUC2 and MUC5AC in the intestinal subtype; MUC1, MUC5AC, and MUC6 in the pancreatobiliary subtype; MUC5AC and MUC6 in the oncocytic subtype. 34 To determine the clinicopathological significance of this subtyping of IPMNs, 283 IPMNs were collected in collaboration with multiple institutions in the United States and Japan, and the associations between the subtypes and clinicopathological features, including prognosis, were analyzed. The analysis indicated that the subtypes were significantly associated with grade, type of invasive carcinoma, and more importantly, patient prognosis, in which gastric‐type IPMNs revealed favorable prognosis, intestinal‐type and oncocytic‐type IPMNs revealed modest prognoses, and pancreatobiliary‐type IPMNs revealed the poorest prognosis. 35 The oncocytic subtype is reclassified into intraductal oncocytic papillary neoplasms in the current fifth edition of the WHO Classification of Tumours in the Digestive System. 36
Figure 5.
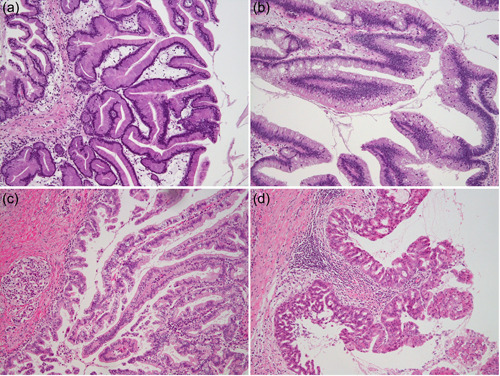
Variations in intraductal papillary mucinous neoplasms of the pancreas including intraductal oncocytic papillary neoplasm. IPMNs of (a) gastric type, (b) intestinal type, and (c) pancreatobiliary type. (d) Intraductal oncocytic papillary neoplasm. All panels are H & E stained, ×100 original magnification. IPMNs, intraductal papillary mucinous neoplasms.
The various grades of neoplastic cells in IPMN suggest multistep tumor development. We conducted a study to map neoplastic cells according to ductal contours in three dimensions by reconstruction from serial sections, which showed that low‐grade neoplastic cells were distributed in a multifocal fashion, while high‐grade neoplastic cells were distributed in a unified continuous fashion. This result suggests that low‐grade neoplastic cells may develop polyclonally, whereas high‐grade neoplastic cells develop from a low‐grade neoplasm and spread as a monoclone. 37 This assumption has been proven by recent molecular studies, which indicate that low‐grade dysplastic lesions harbor various distinct mutations; however, high‐grade dysplastic lesions harbor less heterogeneous mutations than multiple low‐grade and high‐grade dysplastic lesions in the same pancreas. 38
MOLECULAR INSIGHTS OF IPMN
In 2009, molecular alterations specifically associated with IPMNs were unknown. In that year, the next‐generation sequencer became available in Japan, and we immediately decided to perform a study using the newly developed next‐generation sequencing (NGS) technology and exome methodology to identify mutated genes specific for IPMN. A sample of a typical IPMN with high‐grade dysplasia of the intestinal type was subjected to microdissection of neoplastic cells for DNA preparation. Elaborated exome sequencing revealed various mutations in previously unappreciated genes, including GNAS and RNF43. 39 GNAS was particularly interesting because it functions as an oncogene and has a hotspot for activating mutations, such as R201H and R201C. 40 Indeed, the mutation detected by the exome sequencing was R201H, and the hotspot GNAS mutations involving R201 were examined in 118 additional samples of IPMN using conventional Sanger sequencing. We found that 40% of the IPMNs harbored the GNAS mutations R201H or R201C. We also examined KRAS mutations, which are known to be common in IPMN, and found that 48% of the same samples harbored KRAS mutations. We also found that 30% of the samples harbored mutations in both KRAS and GNAS, suggesting an independent oncogenic function of these genes. Mutations in GNAS were not associated with the grade or prognosis of IPMNs but with subtype differentiation, where they were more common in intestinal‐type IPMNs than in other subtypes. We then analyzed the expression of G‐protein stimulatory α subunit (Gsα) encoded by GNAS and phosphorylated substrates of protein kinase A (PS‐PKA) in IPMNs. We found that Gsα and PS‐PKA were strongly expressed in neoplastic cells of IPMN, which indicates that Gsα has higher activity and upregulates G‐protein signaling in IPMNs. On the other hand, an examination of 32 PDACs revealed no GNAS mutation and low expression of Gsα. 39 In the following expanded study analyzing 172 IPMNs, 48% and 56% of them harbored GNAS mutations and KRAS mutations, respectively, whereas no mutation was found in related genes of GNAS, namely, GNAO1, GNAQ, or GNAI2. 41 These results clearly indicate that mutations of GNAS are common and specific for IPMNs.
Next, we conducted an in vitro study to determine the functions of the active Gsα in pancreatic cancer cells. Conventional pancreatic cancer cell lines and an immortalized normal pancreatic ductal epithelial cell line, HPDE6, 42 were transfected with wild‐type and R201H mutant GNAS cDNAs, and their phenotypes were examined. The results showed that the mutant GNAS induced marked elevation of intracellular cyclic AMP, and subsequently the expression of various genes including mucin genes. 43 These results indicate that the mutated GNAS indeed causes upregulation of PKA signaling and secretion of excess mucin.
We moved on to generate a genetically engineered mouse model to determine whether mutant GNAS would indeed cause the development of IPMN. Mouse lines with a transgene of the CAG promoter‐lox‐STOP‐lox (LSL)‐GNAS R201H were established and bred with Cre‐ptf1a mice. These mice expressed mutant GNAS in a pancreas‐specific manner and showed microscopic dilatation of the pancreatic ducts with focal atrophy of the surrounding parenchyma; however, there was no gross tumor. Then, the mouse line was crossed with the LSL‐Kras G12D mouse line to observe the cooperative role of GNAS and KRAS in pancreatic tumorigenesis, which showed a large multicystic tumor in the pancreas within 5 weeks. The tumor consisted of multiple dilated ducts with mucinous contents lined with papillary neoplastic cells, which closely mimicked human IPMNs (Figure 6). The neoplastic tissues of this tumor showed elevated cAMP levels and PS‐PKA expression. Mice with Kras G12D but no GNAS R201H developed PanINs, but no cystic tumor. 44 These results indicate that mutant GNAS plays a causative role in the development of IPMN in vivo. These mouse models die within 6 weeks after birth because of acute pancreatitis due to multiple clogging of the pancreatic ducts with thick mucus; therefore, no long‐term effect of the mutated GNAS in the pancreas can be observed. Patra et al. and Ideno et al. generated mouse models of IPMNs using Cre‐and doxycycline‐inducible Gnas R201C. 45 , 46 They showed that induction of Gnas R201C at 4 or 8 weeks after the birth of mice expressing Kras G12D from gestation resulted in the development of IPMN‐like tumors, which is consistent with our study. Patra et al. showed that an additional Trp53 loss in Gnas R201C; Kras G12D pancreas resulted in the development of an invasive carcinoma associated with IPMN. Ideno et al. showed that Gnas R201C; Kras G12D mice developed invasive carcinoma 30 weeks after the initiation of Gnas R201C expression; however, mice with only Kras G12D expression also developed invasive carcinoma in the pancreas. These results indicate that the development of invasive carcinoma associated with IPMN may be accelerated by p53 inactivation. These genetically engineered mouse models of IPMN with mutated GNAS, including ours, do not show development of IPMNs with intestinal type differentiation although GNAS mutations are common in intestinal‐type IPMNs in human, which is an important remaining question.
Figure 6.
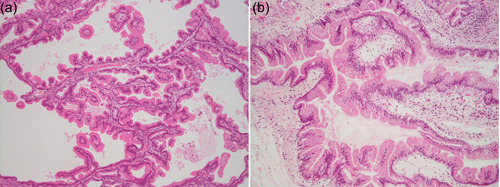
Comparison of IPMN in a model mouse and that of a human. (a) IPMN developed in a genetically engineered mouse conditionally expressing GNAS R201H and Kras G12D in the pancreas. (b) IPMN of a human. Both panels are H & E stained, ×100 original magnification. IPMN, intraductal papillary mucinous neoplasm.
RNF43 encodes the ring finger protein 43 (RNF43). RNF43 is a ubiquitin E3 ligase whose specific substrate is Frizzled, which is a Wnt receptor. Therefore, RNF43 functions as a negative regulator of the Wnt signaling pathway. Because we found an RNF43 mutation in IPMN using exome sequencing, we searched for mutations in RNF43 and examined the expression of its product in 176 IPMNs. Among them, 57 frozen IPMN tissues were subjected to targeted NGS analysis and it was found that 14% of them harbored mutations, mostly protein truncating ones. The expression of RNF43 was downregulated in 30% of the 176 IPMNs. RNF43 mutations were significantly associated with GNAS mutations and mural nodules in the imaging studies. 47 These results indicate that RNF43 is frequently inactivated in IPMNs; hence, dysregulated Wnt signaling may play a role in the development and/or progression of IPMN.
INVASIVE CARCINOMA ASSOCIATED WITH IPMN
The development of invasive carcinoma associated with IPMN is critical for patient survival. Invasive carcinomas that develop in the pancreas with IPMN are classified as derived carcinomas and concomitant carcinomas. 48 Derived carcinoma is defined as a carcinoma that develops in association with IPMN, such as carcinoma that develops from neoplastic cells of IPMN. On the other hand, the concomitant carcinoma is defined as a carcinoma that develops independently from IPMN existing in the same pancreas. Therefore, the derived carcinoma usually shows histological continuity with IPMN, whereas concomitant carcinoma usually exists independently of IPMN. However, there is a carcinoma close to the IPMN without obvious direct continuity, which is called adjacent carcinoma. Omori et al. investigated the molecular mechanism of invasive carcinoma development in IPMN by careful microdissection of IPMN and carcinoma and performed targeted sequencing of 18 genes to compare mutations in each lesion. 49 They showed that invasive carcinoma may develop in three different ways: sequential, branch‐off, and de novo. Sequential development indicates that IPMN progresses to invasive carcinoma, which is proved by mutations shared between IPMN and carcinoma, with some additional mutations in the latter. The branch‐off development indicates that IPMN and carcinoma derive from a common ancestry clone but develop as branched, which is proven by the common and distinct mutations detected in IPMN and carcinoma. De novo development indicates that IPMN and carcinoma develop independently, as proven by independent mutations in IPMN and carcinoma. Sequential, branch‐off, and de novo carcinomas are likely to correspond to derived, adjacent, and concomitant carcinomas, respectively. Intriguingly, patients with branch‐off carcinoma showed better survival than those with sequential and de novo carcinomas. Although the exact cause of this difference in prognosis has not been clarified, this finding indicates that these pathways can have a clinical impact.
MECHANISM OF DEVELOPMENT OF IPMN SUBTYPE
How does the epithelial subtype of IPMN develop? To address this question, we collected lesions of various subtypes of IPMNs, including 49 gastric‐type lesions, 26 intestinal‐type lesions, 22 pancreatobiliary‐type lesions, and 6 oncocytic‐type lesions by microdissection, and compared the mutations by targeted sequencing of 37 genes. 50 The analysis revealed that gastric‐type lesions harbored a significantly smaller number of mutations than the other lesions. Mutations in GNAS and RNF43 were more frequent in intestinal‐type lesions, whereas mutations in TP53 were more frequent in pancreatobiliary‐type lesions. STK11 mutations were more frequent in pancreatobiliary‐ and oncocytic‐type lesions (Figure 7). To understand the driving forces of genetic mutations in the development of lesions of various subtypes, a comparison of mutations in multiple subtypes of IPMN in the same pancreas would provide clues. This presumption led us to investigate the genetic mutations in each subtype in 36 IPMNs with multiple subtypes in the same pancreas. Most of these IPMNs with multiple subtypes consisted of gastric‐type low‐grade lesions and high‐grade lesions of other types. Comparison of mutations among different subtypes revealed three distinct pathways, namely, progressive, divergent, and independent, that are most likely to play a role in the development of multiple subtypes. 50 The progressive pathway indicates the direct progression of one type of lesion into another type of lesion, which is evidenced by the gradual accumulation of mutations, that is, genetic mutations in one type of lesion are preserved in another type of lesion that harbors additional mutations. An example of this pathway revealed a gastric‐type lesion harboring GNAS and TP53 mutations seemed to progress to an intestinal‐type lesion in the same pancreas, harboring the same GNAS and TP53 mutations and additional RNF43 mutations. 50 The divergent pathway indicates that lesions of different subtypes develop divergently from an ancestral clone that harbors fundamental mutations, while acquiring additional distinct mutations in other lesions. An example of this pathway revealed that a gastric‐type lesion harboring a GNAS mutation, an intestinal‐type lesion harboring GNAS and RNF43 mutations, and a pancreatobiliary‐type lesion harboring GNAS, APC, and SMARCA4 mutations existed in the same pancreas, in which the gastric‐type lesion seemed to be an ancestral clone and the intestinal‐type and pancreatobiliary‐type lesions seemed to be divergent clones. 50 The independent pathway indicates that lesions of different subtypes develop independently, which is revealed by complete distinct mutations among them. An example of this pathway revealed that a gastric‐type lesion harboring GNAS and CTNNB1 mutations, and a pancreatobilary‐type lesion harboring KRAS and RNF43 mutations, existed in the same pancreas. 50 These results indicate that gastric‐type low‐grade lesions can transform into high‐grade lesions of other subtypes in IPMN, which may be the reason why continuous surveillance is recommended for small cystic lesions that are likely to be low‐grade gastric lesions that could develop into high‐grade lesions in the international consensus guidelines for the management of patients with IPMN.
Figure 7.
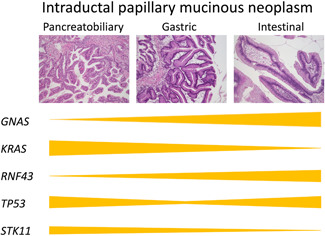
Molecular aberrations pertaining to development of subtypes of intraductal papillary mucinous neoplasms.
IDENTIFICATION OF INTRADUCTAL TUBULOPAPILLARY NEOPLASM (ITPN)
In 2008, we encountered a peculiar case of a pancreatic tumor with an intraductal mass without cystic changes in the pancreas. The mass appeared to clog the main pancreatic duct, without obvious mucin secretion. Microscopically, the mass consisted of complex tubulopapillary glands with minimal mucin. Necrotic foci were also observed. Cells of the glands were cuboidal to columnar, with markedly enlarged atypical nuclei and loss of polarity. Mitoses were frequent. Immunohistochemical analysis of mucin proteins showed no expression of MUC5AC, a hallmark of IPMN. Obviously, it was different from IPMN, and there seemed to be no class of pancreatic neoplasms to belong. Astonishingly, we had a similar case shortly after; then, we decided to collect as many similar cases as possible through multi‐institutional collaboration. As a result, we collected 11 cases and performed a clinicopathological study including molecular analysis. The tumors showed characteristic features, including a solid mass with no visible mucin, tubulopapillary growth, uniform high‐grade atypia with frequent mitoses, frequent necrotic foci, no expression of MUC2 or MUC5AC, and occasional expression of MUC6 (Figure 8). Some cases showed invasion of the parenchyma and vessels. Strikingly, these tumors did not harbor KRAS or BRAF mutations. We reported these cases as a new class of pancreatic neoplasms and coined the term ITPN, 51 which is incorporated into the World Health Organization Classification of Tumours of Digestive System, fourth edition. 52 Subsequently, we conducted a mutation analysis of candidate genes and found that some of these tumors harbored PIK3CA mutations, suggesting the activation of the phosphatidylinositol 3‐kinase (PI3K) pathway in this tumor. We examined the expression of phosphorylated AKT (pAKT) and found that most of these tumors showed strong expression of pAKT, which indeed indicated the activation of PI3K signaling. In contrast, the 50 IPMNs examined simultaneously did not show mutations in PIK3CA. 53 These results indicate that ITPN is distinct from IPMN and that activation of the PI3K pathway plays a decisive role in its development. Furthermore, we carried out an NGS study including whole genome analysis and fusion gene analysis of ITPNs through an international collaboration. This study uncovered mutations in PI3K‐associated genes, including PIK3CA, PIK3CB, INPP4A, and PTEN, mutations in chromatin remodeling genes, including KMT2C and KMT2D, and fusion genes involving FGFR2, in which some are potentially targetable. 54
Figure 8.
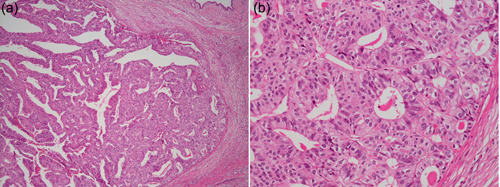
Intraductal tubulopapillary neoplasm (ITPN) of the pancreas. (a) A clogging tumor in the pancreatic duct consisted of tubulopapillary glands without obvious mucin (H & E staining, ×40 original magnification). (b) Neoplastic cells of ITPN show high‐grade atypia (H & E staining, ×200 original magnification).
CLOSING REMARK
The goal of my research on the pathology of pancreatic neoplasms is to find clues for the prevention, diagnosis, and treatment of patients with these neoplasms. When I was a trainee, I was in charge of a patient with pancreatic cancer. He was admitted to the hospital with jaundice and underwent bypass surgery of the biliary tract because curative resection was not possible. After the surgery, jaundice remitted, and the patient was allowed to have a meal with obvious joy. However, 2 weeks after discharge, he was admitted again because of vomiting. The patient had duodenal obstruction due to the growth of pancreatic cancer. He soon passed away. This was a painful experience that brought me to research on pancreatic cancer. Although I am still far away from reaching this goal, I strongly believe that pathological research will bring important results to help cure patients. For that, I would like to stress that daily pathological practice is very important to advance understanding of the nature of diseases such as this intractable neoplasm. I hope that this review will provide some ideas to pathologists, especially young pathologists, to identify and pursue interesting research topics.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The author would like to dedicate this article to the memory of Professor Emeritus Tohru Takahashi and Professor Emeritus Hiroshi Nagura of the Tohoku University. This article summarizes a commemorative lecture on the Japan Pathology Award 2019 given to the author. This work was supported by JSPS KAKENHI Grant Number JP22H02839 to T.F.
Furukawa T. Mechanisms of development and progression of pancreatic neoplasms. Pathol. Int. 2022;72:529–540. 10.1111/pin.13272
REFERENCES
- 1. Sommers SC, Murphy SA, Warren S. Pancreatic duct hyperplasia and cancer. Gastroenterology. 1954;27:629–40. [PubMed] [Google Scholar]
- 2. Kozuka S, Sassa R, Taki T, Masamoto K, Nagasawa S, Saga S, et al. Relation of pancreatic duct hyperplasia to carcinoma. Cancer. 1979;43:1418–28. [DOI] [PubMed] [Google Scholar]
- 3. Cubilla AL, Fitzgerald PJ. Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res. 1976;36:2690–8. [PubMed] [Google Scholar]
- 4. Klöppel G, Bommer G, Ruckert K, Seifert G. Intraductal proliferation in the pancreas and its relationship to human and experimental carcinogenesis. Virchows Arch A Pathol Anat Histol. 1980;387:221–33. [DOI] [PubMed] [Google Scholar]
- 5. Klimstra DS, Longnecker DS. K‐ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145:1547–50. [PMC free article] [PubMed] [Google Scholar]
- 6. Furukawa T, Chiba R, Kobari M, Matsuno S, Nagura H, Takahashi T. Varying grades of epithelial atypia in the pancreatic ducts of humans. Classification based on morphometry and multivariate analysis and correlated with positive reactions of carcinoembryonic antigen. Arch Pathol Lab Med. 1994;118:227–34. [PubMed] [Google Scholar]
- 7. Hruban RH, Adsay NV, Albores–Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–86. [DOI] [PubMed] [Google Scholar]
- 8. Basturk O, Hong SM, Wood LD, Adsay NV, Albores‐Saavedra J, Biankin AV, et al. A revised classification system and recommendations from the baltimore consensus meeting for neoplastic precursor lesions in the pancreas. Am J Surg Pathol. 2015;39:1730–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Day DW, Morson BC. The adenoma‐carcinoma sequence. Major Probl Pathol. 1978;10:58–71. [PubMed] [Google Scholar]
- 10. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c‐K‐ras genes. Cell. 1988;53:549–54. [DOI] [PubMed] [Google Scholar]
- 11. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yanagisawa A, Ohtake K, Ohashi K, Hori M, Kitagawa T, Sugano H, et al. Frequent c‐Ki‐ras oncogene activation in mucous cell hyperplasias of pancreas suffering from chronic inflammation. Cancer Res. 1993;53:953–6. [PubMed] [Google Scholar]
- 13. Furukawa T, Chiba R, Kobari M, Matsuno S, Sasaki H, Obinata M, et al. Reproducible categorization of atypia in pancreatic duct epithelia based on morphometry and cluster analysis, correlating with CEA‐positive reactions and Ki‐ras gene alterations. Ann Cancer Res Ther. 1993;2:121–2. [Google Scholar]
- 14. Furukawa T, Fujisaki R, Yoshida Y, Kanai N, Sunamura M, Abe T, et al. Distinct progression pathways involving the dysfunction of DUSP6/MKP‐3 in pancreatic intraepithelial neoplasia and intraductal papillary‐mucinous neoplasms of the pancreas. Mod Pathol. 2005;18:1034–42. [DOI] [PubMed] [Google Scholar]
- 15. Kimura M, Abe T, Sunamura M, Matsuno S, Horii A. Detailed deletion mapping on chromosome arm 12q in human pancreatic adenocarcinoma: identification of a 1‐cM region of common allelic loss. Genes Chromosomes Cancer. 1996;17:88–93. [DOI] [PubMed] [Google Scholar]
- 16. Abe T, Makino N, Furukawa T, Ouyang H, Kimura M, Yatsuoka T, et al. Identification of three commonly deleted regions on chromosome arm 6q in human pancreatic cancer. Genes Chromosomes Cancer. 1999;25:60–4. [PubMed] [Google Scholar]
- 17. Kimura M, Furukawa T, Abe T, Yatsuoka T, Youssef EM, Yokoyama T, et al. Identification of two common regions of allelic loss in chromosome arm 12q in human pancreatic cancer. Cancer Res. 1998;58:2456–60. [PubMed] [Google Scholar]
- 18. Furukawa T, Yatsuoka T, Youssef EM, Abe T, Yokoyama T, Fukushige S, et al. Genomic analysis of DUSP6, a dual specificity MAP kinase phosphatase, in pancreatic cancer. Cytogenet Cell Genet. 1998;82:156–9. [DOI] [PubMed] [Google Scholar]
- 19. Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual‐specificity phosphatase. EMBO J. 1996;15:3621–32. [PMC free article] [PubMed] [Google Scholar]
- 20. Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP‐3 in pancreatic cancer. Am J Pathol. 2003;162:1807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Furukawa T, Tanji E, Xu S, Horii A. Feedback regulation of DUSP6 transcription responding to MAPK1 via ETS2 in human cells. Biochem Biophys Res Commun. 2008;377:317–20. [DOI] [PubMed] [Google Scholar]
- 22. Xu S, Furukawa T, Kanai N, Sunamura M, Horii A. Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J Hum Genet. 2005;50:159–67. [DOI] [PubMed] [Google Scholar]
- 23. Kidger AM, Saville MK, Rushworth LK, Davidson J, Stellzig J, Ono M, et al. Suppression of mutant Kirsten‐RAS (KRAS(G12D))‐driven pancreatic carcinogenesis by dual‐specificity MAP kinase phosphatases 5 and 6. Oncogene. 2022;41:2811–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furukawa T, Kanai N, Shiwaku HO, Soga N, Uehara A, Horii A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene. 2006;25:4831–9. [DOI] [PubMed] [Google Scholar]
- 25. Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, et al. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 2005;65:2899–905. [DOI] [PubMed] [Google Scholar]
- 26. Furukawa T, Tanji E, Kuboki Y, Hatori T, Yamamoto M, Shimizu K, et al. Targeting of MAPK‐associated molecules identifies SON as a prime target to attenuate the proliferation and tumorigenicity of pancreatic cancer cells. Mol Cancer. 2012;11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ikeda Y, Tanji E, Makino N, Kawata S, Furukawa T. MicroRNAs associated with mitogen‐activated protein kinase in human pancreatic cancer. Mol Cancer Res. 2012;10:259–69. [DOI] [PubMed] [Google Scholar]
- 28. Akamatsu M, Makino N, Ikeda Y, Matsuda A, Ito M, Kakizaki Y, et al. Specific MAPK‐associated microRNAs in serum differentiate pancreatic cancer from autoimmune pancreatitis. PLoS One. 2016;11:e0158669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64:2634–8. [DOI] [PubMed] [Google Scholar]
- 30. Takai E, Yachida S, Shimizu K, Furuse J, Kubo E, Ohmoto A, et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget. 2016;7:74227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nat Rev Cancer. 2013;13:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takai E, Nakamura H, Chiku S, Kubo E, Ohmoto A, Totoki Y, et al. Whole‐exome sequencing reveals new potential susceptibility genes for Japanese familial pancreatic cancer. Ann Surg. 2022;275:e652‐8. [DOI] [PubMed] [Google Scholar]
- 33. Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores‐Saavedra J, Biankin AV, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–87. [DOI] [PubMed] [Google Scholar]
- 34. Furukawa T, Klöppel G, Volkan Adsay N, Albores‐Saavedra J, Fukushima N, Horii A, et al. Classification of types of intraductal papillary‐mucinous neoplasm of the pancreas: a consensus study. Virchows Arch. 2005;447:794–9. [DOI] [PubMed] [Google Scholar]
- 35. Furukawa T, Hatori T, Fujita I, Yamamoto M, Kobayashi M, Ohike N, et al. Prognostic relevance of morphological types of intraductal papillary mucinous neoplasms of the pancreas. Gut. 2011;60:509–16. [DOI] [PubMed] [Google Scholar]
- 36. Basturk O, Esposito I, Fukushima N, Furukawa T, Hong SM, Klöppel G, et al. Pancreatic intraductal oncocytic papillary neoplasm. In: Gill AJ, Klimstra DS, Lam AK, Washington MK editors. WHO classification of digestive system tumours. 1. Lyon: International Agency for Research on Cancer; 2019. p. 315–6. [Google Scholar]
- 37. Furukawa T, Takahashi T, Kobari M, Matsuno S. The mucus‐hypersecreting tumor of the pancreas. Development and extension visualized by three‐dimensional computerized mapping. Cancer. 1992;70:1505–13. [DOI] [PubMed] [Google Scholar]
- 38. Fischer CG, Beleva Guthrie V, Braxton AM, Zheng L, Wang P, Song Q, et al. Intraductal papillary mucinous neoplasms arise from multiple independent clones, each with distinct mutations. Gastroenterology. 2019;157:1123–37.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Furukawa T, Kuboki Y, Tanji E, Yoshida S, Hatori T, Yamamoto M, et al. Whole‐exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–6. [DOI] [PubMed] [Google Scholar]
- 41. Kuboki Y, Shimizu K, Hatori T, Yamamoto M, Shibata N, Shiratori K, et al. Molecular biomarkers for progression of intraductal papillary mucinous neoplasm of the pancreas. Pancreas. 2015;44:227–35. [DOI] [PubMed] [Google Scholar]
- 42. Furukawa T, Duguid WP, Rosenberg L, Viallet J, Galloway DA, Tsao MS. Long‐term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am J Pathol. 1996;148:1763–70. [PMC free article] [PubMed] [Google Scholar]
- 43. Komatsu H, Tanji E, Sakata N, Aoki T, Motoi F, Naitoh T, et al. A GNAS mutation found in pancreatic intraductal papillary mucinous neoplasms induces drastic alterations of gene expression profiles with upregulation of mucin genes. PLoS One. 2014;9:e87875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taki K, Ohmuraya M, Tanji E, Komatsu H, Hashimoto D, Semba K, et al. GNAS(R201H) and Kras(G12D) cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm. Oncogene. 2016;35:2407–12. [DOI] [PubMed] [Google Scholar]
- 45. Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA‐mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. 2018;20:811–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ideno N, Yamaguchi H, Ghosh B, Gupta S, Okumura T, Steffen DJ, et al. GNAS(R201C) induces pancreatic cystic neoplasms in mice that express activated KRAS by inhibiting YAP1 signaling. Gastroenterology. 2018;155:1593–607.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sakamoto H, Kuboki Y, Hatori T, Yamamoto M, Sugiyama M, Shibata N, et al. Clinicopathological significance of somatic RNF43 mutation and aberrant expression of ring finger protein 43 in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol. 2015;28:261–7. [DOI] [PubMed] [Google Scholar]
- 48. Yamaguchi K, Kanemitsu S, Hatori T, Maguchi H, Shimizu Y, Tada M, et al. Pancreatic ductal adenocarcinoma derived from IPMN and pancreatic ductal adenocarcinoma concomitant with IPMN. Pancreas. 2011;40:571–80. [DOI] [PubMed] [Google Scholar]
- 49. Omori Y, Ono Y, Tanino M, Karasaki H, Yamaguchi H, Furukawa T, et al. Pathways of progression from intraductal papillary mucinous neoplasm to pancreatic ductal adenocarcinoma based on molecular features. Gastroenterology. 2019;156:647–61. [DOI] [PubMed] [Google Scholar]
- 50. Kobayashi T, Omori Y, Ono Y, Karasaki H, Mizukami Y, Makino N, et al. Pathways for the development of multiple epithelial types of intraductal papillary mucinous neoplasm of the pancreas. J Gastroenterol. 2021;56:581–92. [DOI] [PubMed] [Google Scholar]
- 51. Yamaguchi H, Shimizu M, Ban S, Koyama I, Hatori T, Fujita I, et al. Intraductal tubulopapillary neoplasms of the pancreas distinct from pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2009;33:1164–72. [DOI] [PubMed] [Google Scholar]
- 52. Kloppel G, Basturk O, Schlitter AM, Konukiewitz B, Esposito I. Intraductal neoplasms of the pancreas. Semin Diagn Pathol. 2014;31:452–66. [DOI] [PubMed] [Google Scholar]
- 53. Yamaguchi H, Kuboki Y, Hatori T, Yamamoto M, Shiratori K, Kawamura S, et al. Somatic mutations in PIK3CA and activation of AKT in intraductal tubulopapillary neoplasms of the pancreas. Am J Surg Pathol. 2011;35:1812–7. [DOI] [PubMed] [Google Scholar]
- 54. Basturk O, Berger MF, Yamaguchi H, Adsay V, Askan G, Bhanot UK, et al. Pancreatic intraductal tubulopapillary neoplasm is genetically distinct from intraductal papillary mucinous neoplasm and ductal adenocarcinoma. Mod Pathol. 2017;30:1760–72. [DOI] [PubMed] [Google Scholar]