

Abstract
Costs and toxicity concerns are at the center of a heated debate regarding the implementation of perovskite solar cells (PSCs) into commercial products. The first bottleneck could be overcome by eliminating the top metal electrode (generally gold) and the underlying hole transporting material and substituting both with one single thick layer of conductive carbon, as in the so‐called carbon‐based PSCs (C‐PSCs). The second issue, related to the presence of lead, can be tackled by resorting to other perovskite structures based on less toxic metallic components. An interesting case is that of the double perovskite Cs2AgBiBr6, which at present still lacks the outstanding optoelectronic performances of the lead‐based counterparts but is very stable to environmental factors. In this work, the processing of carbon electrodes onto Cs2AgBiBr6‐based C‐PSCs was reported, starting from an additive‐free isopropanol ink of a carbon material obtained from the hydrothermal recycling of waste tires and employing a high‐throughput ultrasonic spray coating method in normal environmental conditions. Through this highly sustainable approach that ensures a valuable step from an end‐of‐life to an end‐of‐waste status for used tires, devices were obtained delivering a record open circuit voltage of 1.293 V, which might in the future represent ultra‐cheap solutions to power the indoor Internet of Things ecosystem.
Keywords: carbon electrodes, lead-free, perovskite solar cells, photovoltaics, sustainable chemistry
End‐of‐waste: Waste tires are transformed into conductive carbon black for use as hole‐extracting contact in lead‐free perovskite solar cells. This carbon electrode is processed via spray‐coating from pure isopropanol with no additives, making the whole process highly sustainable. The resulting photovoltaic devices deliver a very high open‐circuit voltage due to the built‐in potential established at the perovskite/carbon interface.
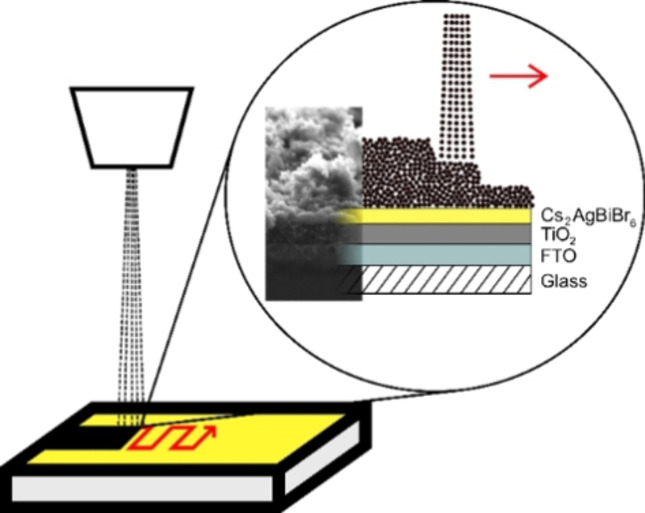
Introduction
Lead‐based perovskite solar cells (PSCs) experienced a decade of tremendous progress, with power conversion efficiency (PCE) starting at 3.8 % in 2009 [1] and reaching >25 %[ 2 , 3 ] nowadays, due to favorable properties such as high absorption coefficients, [4] defects tolerance, [5] and charge carrier mobility. [6] Additionally, the solution‐based preparation of perovskite thin films is rather cost‐effective as it requires low temperatures, small amounts of material, and several high‐throughput processes, such as roll‐to‐roll[ 7 , 8 ] and spray‐coating[ 9 , 10 ] deposition, can be implemented for their production. However, to complete a direct architecture PSC, a hole transporting material (HTM) as well as a back electrode are required, to achieve efficient devices. HTMs such as the commonly employed 2,2’,7,7’‐tetrakis‐(N,N‐di‐4‐methoxyphenylamino)‐9,9’‐spirobifluorene (spiro‐OMeTAD) as well as metal electrodes were identified to play crucial roles in the degradation process of PSCs.[ 11 , 12 , 13 , 14 ] Furthermore, established HTMs and metallic (mostly gold or silver) electrodes are not only expensive, [15] but the latter are additionally non‐abundant materials and have to be deposited through non‐scalable vacuum‐assisted processes. An approach that is emerging in the last few years to overcome these drawbacks is to utilize carbon materials, [16] for example carbon black, [17] graphene, [18] or carbon nanotubes,[ 18 , 19 ] as combined HTM and electrode in one single layer. These layers are generally defined as carbon electrodes (CEs) and the resulting photovoltaic devices are named carbon‐based PSCs (C‐PSCs). CEs are not known to play any role in perovskite degradation and were shown to have an encapsulating effect on PSCs due to their hydrophobicity that can increase device lifetimes.[ 20 , 21 ] Notably, the utilization of CEs as substitution for other HTMs or metal electrodes enables drastic cost reduction for PSC production, due to the high abundancy of carbon (which can even be extracted and processed from waste) [22] and the applicability in solution‐based high‐throughput processes such as spray‐coating.[ 18 , 23 , 24 ]
Despite their outstanding optoelectronic properties, lead‐based PSCs lack environmental stability [25] and present environmental and human risks due to the high toxicity of the lead component. [26] While the substitution of lead(II) with tin(II) or germanium(II) tackles the material toxicity, these elements’ tendency to oxidize still significantly hinders device stability. [27] A material that combines both low toxicity and high environmental stability is the double perovskite (DP) Cs2AgBiBr6, in which the divalent Pb2+ is substituted with equimolar amounts of monovalent Ag+ and trivalent Bi3+. However, due to many sub‐optimal properties, such as its large indirect bandgap,[ 28 , 29 , 30 ] fast surface charge carrier recombination, [31] and strong electron‐phonon coupling and exciton binding energy, [32] the maximum PCEs achieved from PSCs with Cs2AgBiBr6 as a single absorber rarely exceed 2.8 %. Still, a calculated maximum PCE of 7.92 % for PSCs based on pristine Cs2AgBiBr6 underlines the material's large unexploited potential. [33] On the other hand, it is also true that the silver component in this DP is somewhat suffering from scarcity issues. [34] While there might be alternatives to this DP based on more abundant elements, current research on “lead‐free” perovskites or perovskite‐inspired compounds is very much focusing on it, given its excellent environmental stability and current good level of knowledge about how to produce high‐quality thin films from solution processing, making it a sort of benchmark to carry out fundamental and applied studies on this category of materials.[ 35 , 36 , 37 ]
Although the indirect bandgap of Cs2AgBiBr6 is reported as 2–2.2 eV,[ 28 , 29 , 30 ] PSCs based on this material have been characterized by open‐circuit voltages (V OC) that, to the best of our knowledge, are close to or, in one single case, equal to the 1.2 V threshold. [38] Furthermore, those high values could only be achieved when the DP itself or some interfaces within the device were modified by, for example, substituting the organic HTM with an inorganic CuO2 layer (1.198 V), [39] doping the perovskite with Li+ (1.177 V), [40] making use of polymethyl methacrylate (PMMA) as a passivating capping layer between perovskite and a CE (1.180 V), [41] or by capping the perovskite layer with a 2D/3D mixed perovskite phase (1.18 V). [42] All these approaches aimed at improving the energy level alignment at the perovskite/HTM interface, which emphasizes its crucial role for boosting the V OC. The V OC of PSCs that are based on unmodified Cs2AgBiBr6 has been mostly lower than 1.1 V[ 9 , 42 , 43 ] and has only surpassed this value when Cu2O was applied as an HTM [39] or when CEs were used,[ 40 , 41 ] as it could also be observed for lead‐based PSCs. [44]
C‐PSCs based on the DP have yet been reported only three times: originally, Li et al. achieved a PCE of 1.77 % and a 1.119 V V OC for pristine Cs2AgBiBr6, which could be further improved to 2.57 % and 1.177 V by doping the perovskite with small amounts of Li+. [40] Later, the same authors found that the addition of a thin passivating layer of PMMA enhances the PSC performances of pristine Cs2AgBiBr6 (2.25 %, 1.180 V). [41] Finally, Shao and co‐workers have improved the PCE and V OC of Cs2AgBiBr6‐based C‐PSCs from 1.73 % and 1.13 V to 2.22 % and 1.20 V, respectively, by introducing 1‐butyl‐1‐methylpyrrolidinium chloride (BMPyrCl) into the perovskite structure to pin bromide ions, thus inhibiting their migration towards the interface with the CE. [45] In all cases, for the CEs, commercial carbon paste was employed, deposited on the DP photoactive layer from concentrated chlorobenzene (CB) dispersions through screen printing or blade coating.
Herein, we employ ultrasonic (US) spray‐coating for the deposition on top of Cs2AgBiBr6 thin films of additive‐free CEs processed from isopropanol (iPA), a green and low‐boiling‐point solvent, which provides stable to re‐aggregation colloidal suspensions of a carbon black like material obtained from the recycling of waste tires.
Considering the big environmental concerns that end‐of‐life tires abandoned in open fields all over the world are raising, the production of carbon black from them is highly pursued already by many companies worldwide.[ 46 , 47 ] This process, as of today, can only be carried out employing a pyrolysis step at 1500 °C on pre‐treated tires; [48] therefore, it is of the utmost importance to find applications for the resulting raw materials that ensure a proper payback of the energy consumed for their production. The demonstration of valuable uses from this waste tire‐derived conductive materials is, however, still ongoing at a low technology readiness research level, with examples mostly in the field of energy storage. [48] In optoelectronics, there are, however, no cases reported up to now, to the best of our knowledge.
From the C‐PSCs based on the DP absorber and waste tires‐derived CE, we obtain a record V OC of 1.293 V, which is the highest reported until now for similar device architectures. Although PCEs remain overall lower than 1 %, due to poor fill factor (FF) and short‐circuit current density (J SC), the highly sustainable approach that we report for both the CE component preparation and processing will constitute an interesting proof‐of‐concept from which starts the future realization of better‐performing, environmentally friendly lead‐free C‐PSCs. In particular, we demonstrate a valuable end‐of‐waste (EoW) for used tires, which will serve as a further model of the possibility to effectively implement a circular economy.
Experimental Section
All solvents were purchased from Sigma‐Aldrich and used without further purification. Cs2AgBiBr6 crystalline powder was hydrothermally synthesized as described in our earlier publication. [49] Powder X‐ray diffraction (P‐XRD) analysis was carried out through a Panalytical X'Pert MRD Pro diffractometer, equipped with a Cu Kα X‐ray source and a curved graphite secondary monochromator. Thermogravimetric analysis (TGA) was performed with a TG 209 F3 Tarsus instrument by Netzsch. X‐ray photoelectron spectroscopy (XPS) measurements were conducted with a PHI 5000 VersaProbe II Scanning ESCA Microprobe (Physical Electronics) with monochromatized Al Kα X‐ray source in high power mode (beam size 1300 μm×100 μm, X‐ray power: 100 W). Time steps of 50 ms, a step size of 0.2 eV, and an analyzer pass energy of 46.95 eV were used for measuring the detail spectra. The sample surface was charge neutralized with slow electrons and argon ions, and the pressure was in the range from 10−7 to 10−6 Pa during the measurement. Data analysis was performed using the CasaXPS software. Raman spectroscopy was performed on a Bruker Senterra instrument using a 514 nm laser excitation source. The powder sample was displaced over a silicon slide and then analyzed. Dynamic light scattering (DLS) and Zeta potential (ZP) measurements were carried on a Malvern Zetasizer Nano‐ZS device. Grazing incidence X‐ray diffraction (GIXRD) measurements were performed in a range of 3–70° (0.05° step size, 0.014° s−1 scan speed) with a PANalytical B.V. X'Pert Pro diffractometer using CuKα1 radiation. The diffraction patterns were measured along the 2θ axis with a grazing incidence of ω=0.5°. The sheet resistance of the pure CEs was measured with a four‐point probe system (Ossila) controlled by a dedicated software (Ossila Sheet Resistance v2.0.3.3). Scanning electron microscopy (SEM) images were acquired on a Zeiss Merlin instrument operating at a working potential of 20 kV. To calculate the specific surface area, argon physisorption measurements were conducted with an automated gas adsorption station Autosorb iQ2 by Quantachrome Instruments at 87 K by using a cryostat (CryoSync). Prior to the measurements, the samples were degassed in vacuum at 120 °C for 12 h. The surface area was determined using the Brunauer–Emmet–Teller (BET) method as implemented in ASiQwin Version 4.0 from Quantachrome Corporation.
Preparation of the carbon powder from waste tires
A 5‐year used car tire (model Powergy, Pirelli) was washed with water, mechanically reduced into crumbs (diameter: 2–2.5 cm) with a high‐speed grinder, and subsequently soaked in concentrated H2SO4. Temperature was raised up to 125 °C, and the batch was kept warm overnight to yield sulfonated tire rubber. After 15 h, the heating plate was switched off, and the mixture was allowed to reach room temperature. The carbon‐based product was filtered and washed twice with deionized water. Of note, the H2SO4 solution was re‐used seven more times for the same process on other samples before being discarded. The sulfonated tire rubber was dried in the fume hood and then transferred into a tube furnace for the pyrolysis process, which took place under flowing N2 with a ramp rate of 1 °C min−1 up to 450 °C, followed by a 2 °C min−1 ramp up to 1500 °C.
C‐PSCs fabrication and characterization
Cs2AgBiBr6 crystalline powder was dissolved in dimethyl sulfoxide (DMSO) in a glovebox under an argon inert atmosphere to obtain a 0.5 m precursor solution left stirring for several hours. Commercial pre‐patterned substrates from Solaronix, composed of glass covered by fluorine‐doped tin oxide (FTO) and a TiO2 compact layer, were used for the preparation of planar solar cells. Before PSC preparation, the substrates were heated at 450 °C for 45 min. Substrates were consecutively treated in the UV ozone cleaner for 15 min before being transferred into the glove box. Next, 50 μL precursor solution were spread across the substrate before spin‐coating at 4000 rpm for 40 s and finally annealing at 285 °C for 5 min, to prepare Cs2AgBiBr6 thin films as depicted in Figure 2a. The substrates were then transferred into a ND‐SP Mini ultrasonic US spray‐coater (Nadetech, Spain) to deposit the CEs under ambient atmosphere. A 0.1 mg mL−1 colloidal ink for the CE deposition was prepared by dispersing the carbon powder in iPA by ultrasonication for 15 min (3 s on, 3 s off). The substrates were masked to optimize the electrode shapes (active area 0.14 cm2), put onto a hotplate, and heated to 100 °C. US spray‐coating was conducted with a flow rate of 8.00 mL h−1 at a speed of 100.0 mm min−1 for 50 and 100 cycles to deposit CEs with a thickness of circa 4 and 8 μm, respectively. The exact pattern followed for the spray deposition is depicted for the sake of clarity in Figure S2 of the Supporting Information. To screen‐print CEs, both an iPA‐ and a CB‐based precursor paste were used. The former was prepared by first dissolving 400 mg polyvinylpyrrolidone (PVP, M=58000 g mol−1) in 2 mL iPA, while for the latter 240 mg PMMA (M=996000 g mol−1) was dissolved in 2 mL CB, as a first step. Then, within each mixture further 100 mg of the carbon black powder was dispersed through ball milling for 3 h.
Figure 2.

(a) Schematic representation of the preparation process for Cs2AgBiBr6 based C‐PSCs via spin‐coating (perovskite layer) and US spray‐coating (CE). (b) Picture of the resulting device and (c, d) sketch of the overall architecture from top and cross‐section view, respectively. SEM cross‐section images of (e) 4 μm and (f) 8 μm‐thick CE deposited on top of FTO/TiO2/Cs2AgBiBr6 architectures.
C‐PSCs performance was assessed performing current density–voltage (J–V) scans both under dark and under illumination. As a light source, we used a high‐power chip‐on‐board white LED array with an emitting area of 4 cm2 to guarantee a stable and uniform light spot and a fine tuning of the light intensity for prolonged times. In agreement with the International Electrochemical Commission standard IEC 60904‐7‐2019, the LED intensity was calibrated at 1 sun for each sample by means of external‐quantum‐efficiency (EQE) measurements to account for the spectral mismatch between the LED spectrum and the reference spectrum AM1.5G.
Results and Discussion
The carbon‐based material used in this work for the preparation of C‐PSCs CEs was obtained from a 5‐year used car tire. As described in detail in the Experimental Section, tire crumbs were attacked by a re‐usable H2SO4 solution, washed with water, and pyrolyzed at 1500 °C (Figure 1a). XPS analysis carried out on the synthesized carbon material (Figure 1b) shows the presence of a dominant component from π‐conjugated carbon atoms (C=C, graphitic carbon), with minor contributions associated to carbon‐based defects and carbon‐oxygen species like hydroxyls or epoxides and carbonyls.[ 50 , 51 ] However, these last ones are almost negligible as the oxygen in the sample is only of a few at %. The material appears to be rather defects‐rich from Raman analysis (Figure S1a), since the I D/I G ratio is slightly higher than 1 (1.09) and allows for an estimation of the graphitic crystallite sizes (L a) in the material through the Ferrari/Robertson relationship for disordered and amorphous carbon (I D/I G ∝ L a 2) to be around 1.04 nm.[ 52 , 53 ] Thermal and structural analysis of the carbon powder were carried out and are also shown in Figure S1 in the Supporting Information. From TGA, an overall thermal stability in air of up to 400 °C can be assessed (Figure S1b), thus making this carbon powder a suitable component for thermally stable CEs in C‐PSCs. From P‐XRD (Figure S1c), a broad peak around 2θ=27° and a weaker peak at 2θ=44° are detectable, indicating the disordered nature and, overall, the low graphitization of the carbon sheets in the powder sample, respectively. We also calculated the R value, that is, the peak height divided by the background at the position of the peak. An R value equal to 3.18 was obtained, indicating that the pyrolytic process led to a carbon material bearing a large fraction of parallel graphene layers. [54]
Figure 1.
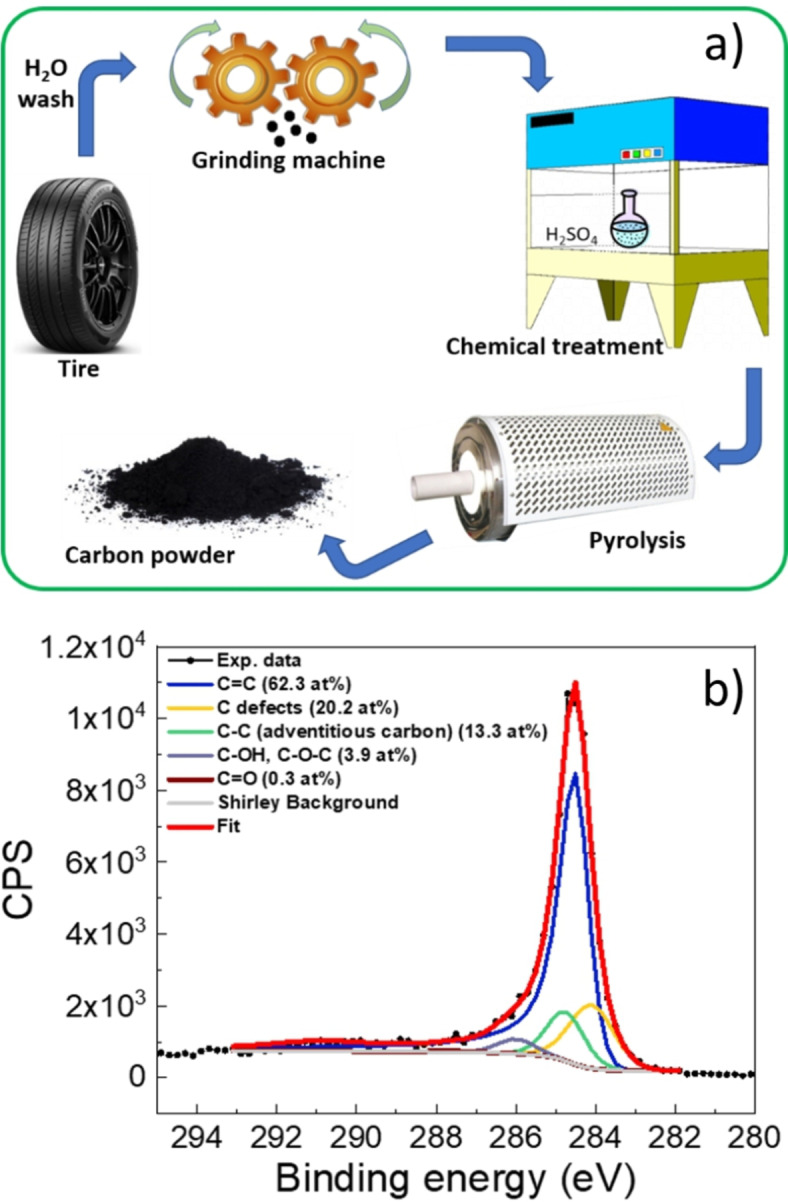
(a) Schematic representation of the process applied for the preparation of the carbon‐based material employed as CE component for C‐PSCs in the present work, starting from waste tires. (b) XPS spectrum of the C 1 s region of the obtained carbon material.
C‐PSCs were fabricated by first processing the Cs2AgBiBr6 DP on the top of compact TiO2 electron transporting layer (ETL) grown onto transparent FTO glass substrates (as schematized in Figure 2a). The procedure was carried out under an inert atmosphere starting from the pre‐synthesized perovskite crystalline powder, as described in the Experimental Section. The obtainment of phase‐pure Cs2AgBiBr6 thin films on the TiO2 substrates is proved by the GIXRD patterns, reported in Figure S3 (no reflexes of the Cs3Bi2Br9 side phase are detected). The films show a compact morphology from top‐view SEM images (Figure S4a), with average grain sizes of 340 nm. The perovskite layer thickness was further examined by looking at the cross‐section SEM images, reported in Figure S4b. The perovskite film growth on compact TiO2 shows an average thickness of 220 nm.
To deposit CEs via common methods such as blade coating or screen‐printing, the precursor paste must be composed not only of the carbon material itself, but also of polymer additives that assure a certain viscosity to the paste for effective deposition. These additives do not only limit the adjustment of the CE thickness but can also negatively affect the conductivity due to the insulating character of commonly utilized polymer binders. In contrast, the precursor for US spray deposition can consist of the sole carbon material, if these can be dispersed efficiently in a solvent medium, and the thickness of deposited CEs can be finely tuned by a variation of the number of spraying cycles (Figure 2e, f). To proceed with US spray coating, a dispersion of the carbon material in iPA was prepared using tip sonication. A concentration high enough to deposit as much material as possible during the process was chosen to reduce the duration of this step (0.1 mg mL−1). By analyzing the 0.1 mg mL−1 carbon dispersion in iPA through DLS (Figure S5), average sizes of 150 nm were found for the dispersed particles and ZP resulted to be around values of −30 mV, indicating negatively charged particles and a good colloidal stability in the additive‐free ink. This is surely an advantage for the use in the US spray deposition process, as this lasts for relatively long times and it is necessary to have in the instrument liquid reservoir a suspension that does not undergo precipitation, which would be noxious for the proper functioning of the instrument (clogging of the needle by agglomerated particles could block the process).
The thickness of spray‐coated CEs is likely to linearly depend on the number of cycles repeated by the US spray‐coater along the defined pattern (see Figure S2 for the exact pattern used). While 50 cycles provided a thickness of around 4 μm for the deposited CE, additional 50 cycles created an 8 μm thick one, as it can be inferred from SEM cross‐section images depicted in Figure 2e, f, respectively. Furthermore, the 4 μm‐thick CE shows a pronounced aerogel‐like morphology, with high porosity, in which the carbon black particles have only small contact to each other. The fact that the CEs could easily be removed from the perovskite by scratching (which is why the CE thicknesses were not measurable by profilometry) supports this observation. While the upper part of the 8 μm‐thick CE resembles the same low‐density agglomeration as the 4 μm one, the bottom part in direct contact with the perovskite layer has a significantly increased density, which we assume is the result of a collapse of the porous structure due to the electrode own weight and ensures good perovskite/CE contact. In order to obtain an estimation of the specific surface area in the US spray‐coated CE, we performed argon physisorption measurements on the carbon black powder (being technically impossible to measure it directly on the deposited electrodes, due to the too low masses). From the application of the BET method, we obtain a surface area for this material of 40 m2 g−1 (see Figure S6). Using this value, we can infer a maximum CE surface area for the 8 μm‐thick layer of around 500 cm2, when considering only the pure powder with no aggregation due to structure collapse. On the other hand, if we had an ideally compact electrode, the overall surface area would be only the top one (i. e., in this case 0.5 cm2). With these two extreme cases in mind, we can presume that the 4 μm layer's surface area is close to the calculated maximum value for it (250 cm2) due to its aerogel‐like morphology, while the 8 μm‐thick layer must have a lower surface area than the thinner one due to its dense bottom part (thus between 500 and 0.5 cm2).
The resulting photovoltaic devices were characterized as described in the Experimental Section. First, EQE measurements were performed in order to precisely determine J SC complying with the AM1.5G standard reference. As reported in Figure 3a, the EQE spectrum of a prototypical cell is comprised between 300 and 560 nm, compatible with a bandgap of around 2.2 eV, and two dominant peaks at 366 nm (3.39 eV) and 438 nm (2.83 eV) are visible. Although the contribution at lower energies can be safely ascribed to the photo‐generation of charge carriers directly in the Cs2AgBiBr6 film (maximum absorption of the excitonic feature of Cs2AgBiBr6 onto compact TiO2 is located at 438 nm, see Figure S7) the high energy contribution can be tentatively associated to charge photo‐generation and separation at the perovskite/TiO2 interface (TiO2 energy gap is 3.2 eV). [55] Figure 3b shows the J–V characteristics of the best cell performed under dark (red solid line) and under 1 sun illumination (solid blue line) from which a record V OC of 1.293 V is detected. Despite the high V OC, a low PCE (0.19 %) results, due to a poor FF (0.3). However, it is important to point out that the here presented C‐PSCs feature an active area (0.14 cm2) that is roughly four times bigger than that used for the champion Cs2AgBiBr6‐based C‐PSC present in literature [45] (PCE=2.2 %, active are=0.04 cm2). From J–V curves recorded under dark, it is possible to notice that the cells do not present shunts and short‐circuit paths between FTO and the CE, indicating that this is not the reason for such a low FF.
Figure 3.

(a) EQE measurement, performed under 1 sun light intensity, from which the calibration value of J SC is extracted. (b) J–V characteristics of the cell performed under dark (solid red line), light (solid blue line), and light with another FTO electrode gently displaced on top of CE (dashed black line) conditions.
To understand whether the carbon layer itself is responsible for such low FF, we attempted further engineering of the top contact by gently displacing a clean FTO substrate on top of the CE (the relative EQE spectra are shown in Figure S8 for the sake of completeness). In this way, we obtained the J–V curve in Figure 3b (dashed black line), from which an improved efficiency results with respect to the sample with the sole CE as top contact (0.37 %). The relevant figures of merit under investigation are the V OC and the FF, because the J SC is hugely dependent on the sample. The outcomes (V OC=1.28 V, FF=0.39) show that the compression of the carbon layer given by the FTO electrode strongly affects the conductivity of the top contact CE (i. e., by importantly reducing the contact resistance), thus boosting the FF value, whereas the V OC remains almost unchanged. Therefore, we can safely assume that the carbon layer allows to obtain high V OC, but the top contact architecture needs to be further optimized in order to provide a state‐of‐the‐art photocurrent. In addition, it is clear that the main reason for the low FF is to be identified in the sub‐optimal conductivity across the CE and not in the DP absorber itself.
To further verify that the strong PCE limitations that we have encountered are not deriving from the specific US spray coating processing method utilized for the CE, we prepared analogous C‐PSCs by depositing the CE via screen‐printing, thus resorting to a more conventional approach for the preparation of similar devices. [14] To do this, we could not resort to the relatively diluted ink used for US spray coating but had to prepare a viscous paste of the carbon material in iPA, for which the addition of a polymer binder was strictly necessary (see details in the Experimental Section). We also prepared a second reference CE employing CB as the dispersing medium, which is a better dispersant for the carbon material than iPA, as well as being the solvent normally used to process CEs on top of perovskites, but at the same time is also more toxic and thus less sustainable for industrial processing. Also in this case, we were forced to add a polymer to the mixture to have the suitable viscosity for screen‐printing. In contrast to the US spray coated CEs, the SEM cross‐sections of the screen‐printed CE (see Figure S9) are characterized by a homogeneous and dense morphology in which several single carbon black nanoparticles are observable as well as a thickness of around 2 μm for the iPA‐based and 3 μm for the CB‐based paste. The increased density is due to the addition of the binders, which work as a glue between carbon black particles, while the film thickness is almost only influenced by the precursor viscosity. Representative J–V characteristics obtained for these two types of C‐PSCs based on screen‐printed CEs are reported in Figure S10. While for the device whose top contact was deposited from CB, PCE is comparable (0.18 %) to that obtained from the US spray coating processed one, but with a better FF (0.51), most likely due to higher conductivity in the carbon layer, and slightly lower V OC (1.18 V), for the one screen‐printed from the iPA‐based paste efficiency drops considerably (0.03 %), due to very low J SC and FF. Overall, we can conclude that the US spray coating is a fully comparable method to state‐of‐the‐art ones for the processing of CEs, while at the same time allowing for the use of additive‐free inks in non‐toxic solvents. Furthermore, when trying to resort to a commercial carbon paste (used as received) to prepare reference devices, we could never obtain suitable J–V curves (V OC always below 1 V and S‐shapes, which do not allow proper FF evaluation), and for this reason we do not show them here as valuable comparisons for our US spray‐coated CEs.
To deeper investigate the performance of the carbon layer used in this study, we performed sheet resistance (R sheet) measurements of the CEs spray coated over a glass substrate. From this characterization, we extracted average value of R sheet=2.2 kΩ square−1, from which we can estimate the resistivity of the CE to be ρ=17 mΩ m. This high value of resistivity (compared to 2 ⋅ 10−5 mΩ m of gold) produces the important parasitic series resistance that limits carrier extraction, explaining the shape and FF of the curve in Figure 3b. In addition, by looking at the EQE data on samples with and without the additional FTO (Figure S8), we can observe that the addition of FTO improves only the high‐frequency contribution to the spectrum (the one related to the TiO2/perovskite interface), thus confirming the idea that the perovskite/carbon layer interface is not affected by the extra FTO and that, reasonably, the external compression improves the photoelectrons collection by boosting the adhesion of the perovskite layer to the underlying TiO2 ETL. This result is in agreement with some recent calculations in which a 20 % higher stiffness was found for the fully inorganic Cs2AgBiBr6 perovskite with respect to the classical organic–inorganic methylammonium–lead‐iodide perovskite. [56]
Within this scenario, the main question still to be answered is how to correlate the high V oc value found for the C‐PSCs based on US spray coated CEs with the device architecture. One possible explanation can be obtained by analyzing the performance of the devices under dark conditions: in this way, the eventual presence of electrical dipoles at the perovskite/CE interface can be studied through the application of proper modelling. Therefore, in order to understand the energy level alignment between the CE and Cs2AgBiBr6, we performed dark J–V measurement from −1.5 to +1.5 V. Figure 4a highlights the rectifying behavior of our cells that displays a strong asymmetry between positive and negative voltages, indicating the formation of a strong built‐in potential (V bi) formed within the Cs2AgBiBr6 layer. Accordingly, the current density under dark conditions (J DARK) can be described by the Mott–Gurney model [Eq. 1]: [57]
| (1) |
Figure 4.

(a) J–V characteristics of a C‐PSC based on a US spray coated CE from −1.5 to 1.5 V measured under dark conditions. (b) Proposed energy band diagram for the FTO/TiO2/Cs2AgBiBr6/CE architecture under flat‐band conditions. Valence and conduction bands values for the perovskite are average values according to literature.[ 40 , 43 , 63 , 64 ] (c) Lumped equivalent circuit model of the tested PSCs under illumination. (d) Comparison between under illumination J–V data and our model: the black solid line is the measured current; the blue dotted line is the numerical fitting applying the model; the red solid line is a model simulation with R S=0 Ω.
Where ϵ 0 is the vacuum permittivity, ϵ r=6.1 is the Cs2AgBiBr6 relative permittivity, [58] μ is the carrier mobility, and L is the thickness of the perovskite layer, which is 220 nm in our samples (estimated from SEM cross section images in Figure 2e, f).
Equation (1) can be applied to the data in Figure 4a, leading to the estimation of V bi as well as the carrier mobility (in the order of 10−6–10−5 cm2 V s−1). Neglecting the voltage drop on the TiO2 layer and on the TiO2/Cs2AgBiBr6 interface, V bi is defined as [Eq. 2]:
| (2) |
where is the conduction band of TiO2, and is the effective work function of the CE. From J–V curve under dark, we measured V bi=1 V and, assuming ,[ 55 , 59 ] we can estimate .
For CEs, a work function of is typically reported, [60] indicating the presence of an electric dipole at the Cs2AgBiBr6/CE interface (≈0.5 eV) that contributes to explain the high V OC found in our cells. Figure 4b shows the proposed scenario that justifies the experimental data. However, it is already known from literature that such inorganic double perovskite allows to generally obtain high V OC (1.20 V is the record value as of today to the best of our knowledge, obtained not on the pure double perovskite but after the addition of the ionic liquid BMPyrCl to pin bromide ions), [45] and the reasons are normally ascribed to the very good alignment between the perovskite and the HTM. In addition, recent relevant literature further asserts that V bi itself is the main cause of a high V OC; [61] therefore, even considering the band alignment between the CE and the double perovskite valence/conduction band in our architecture, we can further confirm the validity of our previous discussion.
To further support our findings, we developed a qualitative model to describe the obtained J–V characteristics. As sketched in Figure 4c, the PSC current (J PSC) of an illuminated device is given by two contributions (namely, the photo‐generated current J PH minus the dark current J DARK):
| (3) |
Where V IN is the internal C‐PSC voltage that differs from the applied potential V by the voltage drop across the series resistance R S [i. e., V IN=V−R S I PSC(V)]. In our modelling, R S is a lumped parameter used to describe the distributed resistivity of the extracting contacts, thus avoiding the complexity of finite element computation. [62]
J PH can be describe by a modified Sokel–Hughes model, [65] particularly suited for materials with low conductivity. Furthermore, based on the band diagram in Figure 4b, we can assume that the TiO2 layer acts as a selective contact for electrons, thus J PH can be written as Equation 4:
| (4) |
where q is the electrons charge, k B the Boltzmann constant, T the absolute temperature in K, G is the photo‐generation rate of free charges, and V FB is the quasi‐flat band voltage corresponding to the condition of zero electric‐field in the bulk of the Cs2AgBiBr6 layer, and, ideally, we can assume that V OC=V FB.
Figure 4d shows the resulting fitting of experimental applying the model already described, allowing the estimation of the photocurrent parameters G=1.4×1020 cm−3 s−1 (a value in agreement with the methyl ammonium lead iodide perovskite) [66] and V FB=1.3 V.
Notably, the flat‐band voltage is larger than the V bi estimated by J–V measurements under dark conditions. This is consistent with previous findings showing that, under illumination, the accumulation of mobile ions at the anode and cathode interfaces increases the internal electric field; [67] therefore, higher flat‐band voltages are expected under illumination explaining the high V OC in our cells.
The proposed model also highlights the importance of the resistivity of the extracting contacts described by the in‐series lumped resistance R S, which we calculated to be around 8 kΩ. Such resistance is the main contribution responsible for the low FF of our cells. In fact, we can use our model to simulate an ideal scenario by setting R S=0 Ω (without changing the previously fitted parameters) and the performance of the cell would drastically improve. In particular, from our simulation we can estimate an ideal FF as high as 76 %, pointing out the need of improving the conductivity of selective contacts to increase the performance of Cs2AgBiBr6‐based cells and architectures.
Conclusions
In this work, we describe the ultrasonic (US) spray deposition of carbon electrodes (CEs) on top of Cs2AgBiBr6‐based carbon perovskite solar cells (C‐PSCs) employing a highly sustainable ink, containing a carbon black like material obtained from the recycling of waste tires, thus also implementing a virtuous concept of the circular economy. In this way, we both ensure a suitable end‐of‐waste for a highly environmentally cumbersome end‐of‐life product and, given the proposed use in renewable energy harvesting devices, we also prospect an effective payback of the energy necessary to produce such electrode material.
From the resulting devices, we achieve a very high open‐circuit voltage (the current record for PSCs containing this double perovskite light absorber, to the best of our knowledge), while the other figures of merit still lag behind the state‐of‐the‐art for analogous architectures prepared by others through more classical blade coating or screen‐printing methods. These approaches, however, always resort to toxic solvents and polymeric additives, which are completely avoided in the present work. On the other hand, electrical conductivity in the US spray‐coated CEs will require improvement through the possible addition of conductive additives like carbon nanotubes or graphene, which will be the subject of future investigations aimed at increasing the overall power conversion efficiency. Similar C‐PSC devices, with relatively modest performances but extremely low costs of production and toxicity issues, might have an actual economic impact for powering many of the technological items belonging to the burgeoning Internet of Things ecosystem.[ 35 , 68 ]
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
T. G. would like to acknowledge financial support of the European commission through the H2020 FET‐PROACTIVE‐EIC‐07‐2020 project LIGHT‐CAP (project number 101017821) and of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the project GA 3052/1‐1. N. L. and A. C. thank financial support of the Italian project “Bifacial Efficient Solar cell Technology with 4 terminal architecture for Utility scale” – BEST4U (PONARS01_00519). Open Access funding enabled and organized by Projekt DEAL.
F. Schmitz, N. Lago, L. Fagiolari, J. Burkhart, A. Cester, A. Polo, M. Prato, G. Meneghesso, S. Gross, F. Bella, F. Lamberti, T. Gatti, ChemSusChem 2022, 15, e202201590.
Contributor Information
Prof. Federico Bella, Email: federico.bella@polito.it.
Dr. Francesco Lamberti, Email: francesco.lamberti@unipd.it.
Dr. Teresa Gatti, Email: teresa.gatti@phys.chemie.uni-giessen.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Kojima A., Teshima K., Shirai Y., Miyasaka T., J. Am. Chem. Soc. 2009, 131, 6050–6051. [DOI] [PubMed] [Google Scholar]
- 2. Yoo J. J., Seo G., Chua M. R., Park T. G., Lu Y., Rotermund F., Kim Y. K., Moon C. S., Jeon N. J., Correa-Baena J. P., Bulović V., Shin S. S., Bawendi M. G., Seo J., Nature 2021, 590, 587–593. [DOI] [PubMed] [Google Scholar]
- 3. Jeong J. et al., Nature 2021, 592, 381–385. [DOI] [PubMed] [Google Scholar]
- 4. De Wolf S., Holovsky J., Moon S. J., Löper P., Niesen B., Ledinsky M., Haug F. J., Yum J. H., Ballif C., J. Phys. Chem. Lett. 2014, 5, 1035–1039. [DOI] [PubMed] [Google Scholar]
- 5. Kang J., Wang L. W., J. Phys. Chem. Lett. 2017, 8, 489–493. [DOI] [PubMed] [Google Scholar]
- 6. Herz L. M., ACS Energy Lett. 2017, 2, 1539–1548. [Google Scholar]
- 7. Hwang K., Jung Y. S., Heo Y. J., Scholes F. H., Watkins S. E., Subbiah J., Jones D. J., Kim D. Y., Vak D., Adv. Mater. 2015, 27, 1241–1247. [DOI] [PubMed] [Google Scholar]
- 8. Chang N. L., Ho-Baillie A. W. Y., Vak D., Gao M., Green M. A., Egan R. J., Sol. Energy Mater. Sol. Cells 2018, 174, 314–324. [Google Scholar]
- 9. Daem N., Dewalque J., Lang F., Maho A., Spronck G., Henrist C., Colson P., Stranks S. D., Cloots R., Solar RRL 2021, 5, 2100422. [Google Scholar]
- 10. Barrows A. T., Pearson A. J., Kwak C. K., Dunbar A. D. F., Buckley A. R., Lidzey D. G., Energy Environ. Sci. 2014, 7, 2944–2950. [Google Scholar]
- 11. Hawash Z., Ono L. K., Qi Y., Adv. Mater. Interfaces 2018, 5, 1700623. [Google Scholar]
- 12. Shlenskaya N. N., Belich N. A., Grätzel M., Goodilin E. A., Tarasov A. B., J. Mater. Chem. A 2018, 6, 1780–1786. [Google Scholar]
- 13. Lamberti F., Gatti T., Cescon E., Sorrentino R., Rizzo A., Menna E., Meneghesso G., Meneghetti M., Petrozza A., Franco L., Chem 2019, 5, 1806–1817. [Google Scholar]
- 14. Lamberti F., Schmitz F., Chen W., He Z., Gatti T., Solar RRL 2021, 5, 2100514. [Google Scholar]
- 15. Hajikhanmirzaei L., Shahroosvand H., Pashaei B., Monache G. D., Nazeeruddin M. K., Pilkington M., J. Mater. Chem. C 2020, 8, 6221–6227. [Google Scholar]
- 16. Fagiolari L., Bella F., Energy Environ. Sci. 2019, 12, 3437–3472. [Google Scholar]
- 17. Chu Q. Q., Sun Z., Ding B., sik Moon K., Yang G. J., Wong C. P., Nano Energy 2020, 77, 105110. [Google Scholar]
- 18. Chen R., Feng Y., Jing L., Wang M., Ma H., Bian J., Shi Y., J. Mater. Chem. C 2021, 9, 3546–3554. [Google Scholar]
- 19. Guo M., Liu J., Yuan Y., Zhang Z., Yin S., Leng J., Huang N., J. Photochem. Photobiol. A 2020, 403, 112843. [Google Scholar]
- 20. Wu Y., Wang S., Ouyang T., Li W., Chen M., Lu Y., Qi P., Tang Y., Nanotechnology 2021, 32, 225701. [DOI] [PubMed] [Google Scholar]
- 21. Zhang G., Xie P., Huang Z., Yang Z., Pan Z., Fang Y., Rao H., Zhong X., Adv. Funct. Mater. 2021, 31, 2011187. [Google Scholar]
- 22. Liu H., Xie Y., Wei P., Wang W., Chen H., Geng C., Qiang Y., J. Alloys Compd. 2020, 842, 155851. [Google Scholar]
- 23. Zhang C., Wang S., Zhang H., Feng Y., Tian W., Yan Y., Bian J., Wang Y., Jin S., Zakeeruddin S. M., Grätzel M., Shi Y., Energy Environ. Sci. 2019, 12, 3585–3594. [Google Scholar]
- 24. Wu C., Wang K., Jiang Y., Yang D., Hou Y., Ye T., Han C. S., Chi B., Zhao L., Wang S., Deng W., Priya S., Adv. Funct. Mater. 2021, 31, 2006803. [Google Scholar]
- 25. Li M., Li H., Fu J., Liang T., Ma W., J. Phys. Chem. C 2020, 124, 27251–27266. [Google Scholar]
- 26. Su P., Liu Y., Zhang J., Chen C., Yang B., Zhang C., Zhao X., J. Phys. Chem. Lett. 2020, 11, 2812–2817. [DOI] [PubMed] [Google Scholar]
- 27. Wang M., Wang W., Ma B., Shen W., Liu L., Cao K., Chen S., Huang W., in Lead-Free Perovskite Materials for Solar Cells, Springer Singapore, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McClure E. T., Ball M. R., Windl W., Woodward P. M., Chem. Mater. 2016, 28, 1348–1354. [Google Scholar]
- 29. Du K. Z., Meng W., Wang X., Yan Y., Mitzi D. B., Angew. Chem. Int. Ed. 2017, 56, 8158–8162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8270–8274. [Google Scholar]
- 30. Igbari F., Wang R., Wang Z. K., Ma X. J., Wang Q., Wang K. L., Zhang Y., Liao L. S., Yang Y., Nano Lett. 2019, 19, 2066–2073. [DOI] [PubMed] [Google Scholar]
- 31. Bartesaghi D., Slavney A. H., Gélvez-Rueda M. C., Connor B. A., Grozema F. C., Karunadasa H. I., Savenije T. J., J. Phys. Chem. C 2018, 122, 4809–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kentsch R., Scholz M., Horn J., Schlettwein D., Oum K., Lenzer T., J. Phys. Chem. C 2018, 122, 25940–25947. [Google Scholar]
- 33. Savory C. N., Walsh A., Scanlon D. O., ACS Energy Lett. 2016, 1, 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schileo G., Grancini G., J. Mater. Chem. C 2021, 9, 67–76. [Google Scholar]
- 35. Peng Y., Huq T. N., Mei J., Portilla L., Jagt R. A., Occhipinti L. G., MacManus-Driscoll J. L., Hoye R. L. Z., Pecunia V., Adv. Energy Mater. 2021, 11, 2002761. [Google Scholar]
- 36. Pecunia V., Occhipinti L. G., Chakraborty A., Pan Y., Peng Y., APL Mater. 2020, 8, 100901. [Google Scholar]
- 37. Jin Z., Zhang Z., Xiu J., Song H., Gatti T., He Z., J. Mater. Chem. A 2020, 8, 16166–16188. [Google Scholar]
- 38. Li J., Meng X., Wu Z., Duan Y., Guo R., Xiao W., Zhang Y., Li Y., Shen Y., Zhang W., Shao G., Adv. Funct. Mater. 2022, 32, 2112991. [Google Scholar]
- 39. Xiao B., Tan Y., Yi Z., Luo Y., Jiang Q., Yang J., ACS Appl. Mater. Interfaces 2021, 13, 37027–37034. [DOI] [PubMed] [Google Scholar]
- 40. Li J., Duan J., Du J., Yang X., Wang Y., Yang P., Duan Y., Tang Q., ACS Appl. Mater. Interfaces 2020, 12, 47408–47415. [DOI] [PubMed] [Google Scholar]
- 41. Li J., Yan F., Yang P., Duan Y., Duan J., Tang Q., Solar RRL 2021, 6, 2100791. [Google Scholar]
- 42. Sirtl M. T., Hooijer R., Armer M., Ebadi F. G., Mohammadi M., Maheu C., Weis A., van Gorkom B. T., Häringer S., Janssen R. A. J., Mayer T., Dyakonov V., Tress W., Bein T., Adv. Energy Mater. 2022, 12, 2103215. [Google Scholar]
- 43. Greul E., Petrus M. L., Binek A., Docampo P., Bein T., J. Mater. Chem. A 2017, 5, 19972–19981. [Google Scholar]
- 44. Chu Q. Q., Ding B., Qiu Q., Liu Y., Li C. X., Li C. J., Yang G. J., Fang B., J. Mater. Chem. A 2018, 6, 8271–8279. [Google Scholar]
- 45. Li J., Meng X., Wu Z., Duan Y., Guo R., Xiao W., Zhang Y., Li Y., Shen Y., Zhang W., Shao G., Adv. Funct. Mater. 2022, 32, 2112991. [Google Scholar]
- 46. Formela K., Adv. Ind. Eng. Polym. Res. 2021, 4, 209–222. [Google Scholar]
- 47. Dabic-Miletic S., Simic V., Karagoz S., Environ. Sci. Pollut. Res. Int. 2021, 28, 68053–68070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sathiskumar C., Karthikeyan S., Sustain. Mater. Technol. 2019, 22, e00125. [Google Scholar]
- 49. Schmitz F., Guo K., Horn J., Sorrentino R., Conforto G., Lamberti F., Brescia R., Drago F., Prato M., He Z., Giovanella U., Cacialli F., Schlettwein D., Meggiolaro D., Gatti T., J. Phys. Chem. Lett. 2020, 11, 8893–8900. [DOI] [PubMed] [Google Scholar]
- 50. Ganesan K., Ghosh S., Gopala Krishna N., Ilango S., Kamruddin M., Tyagi A. K., Phys. Chem. Chem. Phys. 2016, 18, 22160–22167. [DOI] [PubMed] [Google Scholar]
- 51. Morgan D. J., C 2021, 7, 51, https://www.mdpi.com/journal/carbon. [Google Scholar]
- 52. Ferrari A. C., Robertson J., Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar]
- 53. Schuepfer D. B., Badaczewski F., Guerra-Castro J. M., Hofmann D. M., Heiliger C., Smarsly B., Klar P. J., Carbon 2020, 161, 359–372. [Google Scholar]
- 54. Puech P., Dabrowska A., Ratel-Ramond N., Vignoles G. L., Monthioux M., Carbon 2019, 147, 602–611. [Google Scholar]
- 55. Fan H., Yang Z., Ren X., Yin M., Gao F., Liu S., AIP Adv. 2016, 6, 15314. [Google Scholar]
- 56. Dong L., Sun S., Deng Z., Li W., Wei F., Qi Y., Li Y., Li X., Lu P., Ramamurty U., Comput. Mater. Sci. 2018, 141, 49–58. [Google Scholar]
- 57. Mott N. F., Gurney R. W., in Electronic Processes in Ionic Crystals, Clarendon Press, 1948. [Google Scholar]
- 58. Varadwaj P. R., Nanomater. 2020, 10, 973. [Google Scholar]
- 59. Fujisawa J., Eda T., Hanaya M., Chem. Phys. Lett. 2017, 685, 23–26. [Google Scholar]
- 60. Ilie A., Hart A., Flewitt A. J., Robertson J., Milne W. I., J. Appl. Phys. 2000, 88, 6002–6010. [Google Scholar]
- 61. Sandberg O. J., Kurpiers J., Stolterfoht M., Neher D., Meredith P., Shoaee S., Armin A., Adv. Mater. Interfaces 2020, 7, 2000041. [Google Scholar]
- 62. Denhoff M. W., Drolet N., Sol. Energy Mater. Sol. Cells 2009, 93, 1499–1506. [Google Scholar]
- 63. Wu C., Zhang Q., Liu Y., Luo W., Guo X., Huang Z., Ting H., Sun W., Zhong X., Wei S., Wang S., Chen Z., Xiao L., Adv. Sci. 2018, 5, 1700759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pantaler M., Cho K. T., Queloz V. I. E., García Benito I., Fettkenhauer C., Anusca I., Nazeeruddin M. K., Lupascu D. C., Grancini G., ACS Energy Lett. 2018, 3, 1781–1786. [Google Scholar]
- 65. Sokel R., Hughes R. C., J. Appl. Phys. 1982, 53, 7414–7424. [Google Scholar]
- 66. Sherkar T. S., Momblona C., Gil-Escrig L., Ávila J., Sessolo M., Bolink H. J., Koster L. J. A., ACS Energy Lett. 2017, 2, 1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ebadi F., Aryanpour M., Mohammadpour R., Taghavinia N., Sci. Rep. 2019, 9, 11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pecunia V., Occhipinti L. G., Hoye R. L. Z., Adv. Energy Mater. 2021, 11, 2100698. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.