

Abstract
The development of antibodies that specifically detect histidine-phosphorylated proteins is a recent achievement and allows potential roles of histidine phosphorylated proteins in pathological and physiological conditions to be characterized. Immunohistochemical analyses enable the detection of proteins in tissues and can reveal alterations to the quantity and/or localization of these proteins through comparisons of normal and diseased specimens. However, the sensitivity of phosphohistidine modifications to phosphatases, acidic pH, and elevated temperatures poses unique challenges to the detection process and requires a protocol that bypasses traditional procedures utilizing paraffin-embedding and antigen-retrieval methods. Here, we detail a method for a brief fixation by 4% (v/v) paraformaldehyde on freshly collected tissues in the presence of PhosSTOP to block phosphatase activity, followed by a float on sucrose to protect the tissue prior to freezing. Specimens are then embedded in a cryopreservation medium in molds and frozen using an isoflurane, dry ice bath to best preserve the tissue morphology and phosphohistidine signal. We validate this technique in normal mouse liver using SC44-1, a monoclonal anti-3-pHis antibody used to uncover a role for a protein histidine phosphatase as a tumor suppressor in the liver. Furthermore, we demonstrate that the antibody signal can be eliminated by preincubating SC44-1 with a peptide treated with phosphoramidate to phosphorylate histidine residues. Thus, we present an IHC protocol suitable for specific detection of 3-phosphohistidine proteins in mouse liver tissue, and suggest that this can be used as a starting point for optimization of IHC using other phosphohistidine antibodies or in other tissue types, generating information that will enhance our understanding of phosphohistidine in models of disease.
Keywords: Immunohistochemistry, IHC, Phosphohistidine, pHis, Histidine phosphorylation, pHis antibodies, Histidine phosphorylation antibodies
1. Introduction
Given the recent, successful development of antibodies that specifically recognize protein histidine phosphorylation [1–4], and the reported roles for histidine phosphorylated (pHis) proteins in mammalian disease and physiology [5–11], it is of interest to adapt traditional immunohistochemistry (IHC) protocols to better understand where pHis modifications occur within tissues, and to determine whether there are disease-mediated phenotypic changes in pHis. Immunohistochemistry (IHC) is the process by which antibodies detecting specific proteins or protein modifications are applied to tissues to determine the localization of these factors amongst the varied cell types in a tissue environment. Although IHC can be used in combination with immunofluorescent (IF) detection methods, the workflow outlined here includes a secondary antibody conjugated to horseradish peroxidase (HRP) for the chromogenic detection of antigens through the conversion of 3,3′-diamino benzidine (DAB) to a brownish precipitate that remains permanently detectable on slides by light microscopy. Due to the longevity of the signal on slides, this protocol permits the comparison of tissues collected from multiple sources over time.
To our knowledge, there are no published studies using the recently developed pHis antibodies for IHC or for any microscopic analysis of tissue. However, Fuhs et al. used immunocytochemistry (ICC) with IF detection to study pHis protein localization in cultured cells [1]. For this study, cells were fixed by one of two methods, methanol or 4% (v/v) paraformaldehyde (PFA); boiling slides for 5 min in 100 mM citrate buffer served as a negative control, due to the acid and heat sensitivity of histidine phosphorylation. A detailed protocol for ICC analysis with IF detection is included in a separate chapter (see Chapter 14).
Consequently, in the development of an IHC protocol, we selected methanol and 4% (v/v) PFA as two methods for comparison in the fixation of fresh, frozen, mouse liver tissue. Both methods avoided the requirement for antigen retrieval which proved too harsh for pHis detection based on the Fuhs et al. analysis [1]. For both methods, pieces of liver from C57BL/6 mice, freshly isolated, were embedded in a cryopreservative medium in plastic molds and frozen on an isopentane–dry ice bath and stored at −80 °C prior to sectioning. For fixation by methanol, frozen sections (prepared from unfixed, frozen tissue) were fixed for 2 min after cryosectioning and processed for staining afterward. For the 4% (v/v) PFA fixation, pieces of liver, processed immediately after removing from dissected mice, were fixed on ice for 3.5 h in the presence of PhosSTOP phosphatase inhibitors, washed in TBS at 4 °C, then floated overnight at 4 °C on 30% (w/v) sucrose, and embedded in cryoprotectant and frozen in plastic molds in the morning. After developing slides in parallel for each technique, we only observed signal that could be blocked by coincubation of the monoclonal antibody (mAb) with histidine phosphorylated peptide for liver samples prepared by 4% (v/v) PFA fixation (in the presence of PhosSTOP), highlighting the importance of phosphatase inhibitors in preserving pHis signal in tissue. Thus, we present this protocol, which specifically detects 3-pHis using SC44-1 mAb, in formaldehyde-fixed, fresh, frozen mouse liver tissue, as a starting point for future studies of pHis protein localization in other tissues.
2. Materials
Use sterile, ultrapure water (MilliQ water is sufficient) and analytical grade reagents to prepare solutions. Equilibrate all solutions to the indicated temperatures prior to performing the experiment. Tissue, chemical and biohazard waste disposal must all be performed in accordance with local regulations. Specifically, any research involving animals or human subjects must be planned well in advance with protocols approved by the appropriate organizations according to your institution’s rules.
2.1. Tissue Fixation and Cryopreservation in Sucrose
Tris-buffered saline (TBS; 10×): 1.5 M NaCl, 0.1 M Tris–HCl, pH 7.5. Dilute enough 10× solution to 1× in water for washing samples three times in 15 mL of TBS. Chill wash buffer to 4 °C.
Sucrose: 30% (w/v) in 1× TBS. Dissolve slowly with a stir bar in room temperature water. This can be prepared one day before the experiment; prepare at least 20 mL per sample. Chill to 4 °C.
Paraformaldehyde (PFA): 4% (v/v) PFA in TBS. Use ultrapure, electron microscopy grade PFA (such as 16% (w/v) formaldehyde, Polysciences, Inc., #18814). The day of fixation, in a chemical fume hood, dilute an entire 10 mL ampule of 16% (w/v) formaldehyde into a small glass bottle (with a cap) containing 4 mL 10× TBS and 26 mL of water (prechilled at 4 °C). Dissolve 1 tablet of PhosSTOP (Roche) for every 10 mL of solution (4 tablets for 40 mL) (see Note 1). Parafilm the top of the bottle or tube and store on ice in the chemical fume hood until use.
Labeled 15 mL conical tubes (Falcon) for each tissue sample that will be analyzed; prechill at 4 °C, then place on ice in the chemical fume hood.
6-Well plates filled with TBS for washing individual tissue specimens.
Dissecting scissors and forceps.
Tissue specimen(s) (see Note 2).
2.2. Tissue Cryo Embedding
Disposable embedding molds, such as Polysciences, Inc., #18986. Label in advance for each sample.
Optimal cutting temperature (OCT) compound, such as TissueTek Optimal Cutting Temperature Compound (OCT), #4583.
Forceps: 4–5″ long for handling tissue; 10″ long if needed for securing molds while freezing.
P200 pipet tips to secure tissue in OCT.
Styrofoam container with dry ice for transporting cryo molds to storage in −80 °C.
Dry ice–isopentane (2-methylbutane, Fisher #O3551-4) bath (in Styrofoam container) for freezing embedded tissue in molds.
Ziploc bags sufficient to accommodate individual molds. Label in advance and freeze at −80 °C prior to making cryo molds.
2.3. Blocking of Antibody with pHis Peptide
Peptide to phosphorylate: 10 mg/mL in water, pH 7–9 (see Note 3).
Phosphoramidate: 81 mg/mL in water (see Note 4).
Diluent solution: 10 mM Tris–HCl, pH 8.8.
Nitrocellulose.
Dot blot blocking buffer: 0.2× Tris Buffered Saline (TBS) with 0.1% (w/v) casein.
TBST: 1× TBS containing 0.1% (v/v) Tween 20.
Anti-pHis antibodies: such as SC44-1 anti-3-pHis monoclonal rabbit antibody diluted to 0.5 μg/mL [1] (see Note 5).
Secondary antibody to detect the primary pHis antibody for dot blot (see Note 6).
TBST: 1× TBS containing 0.1% (v/v) Tween 20.
Blocking buffer: 5% (v/v) normal goat serum in TBST.
Rotator at room temperature that will accommodate 1.5 mL Eppendorf tubes.
2.4. Staining and Mounting of Coverslips to Cryosection Slides
Slide staining apparatus: staining racks and jars sufficient to process slides and complete washes.
Hydrophobic barrier pen for IHC.
Humid chamber.
TBST: 1× TBS containing 0.1% (v/v) Tween 20.
Blocking buffer: 5% (v/v) normal goat serum in TBST.
IHC detection reagent (HRP), such as SignalStain Boost, rabbit, CST #8114, a polymer-based HRP-conjugated antibody.
3,3′-diaminobenzidine (DAB) peroxidase (HRP) substrate (e.g., ImmPACT (SK-4105, Vector Labs) prepared according to manufacturer’s instructions immediately prior to use (see Note 7).
Hematoxylin, such as Fisher #CA401-1D.
Slide staining jars that accommodate glass slide racks, prepared for serial processing of slides in the hood, and placed in this order: 50% (v/v) ethanol, 50% (v/v) ethanol, 75% (v/v) ethanol, 95% (v/v) ethanol, 100% (v/v) ethanol, 100% (v/v) ethanol, 100% (v/v) xylene, 100% (v/v) xylene, 100% (v/v) xylene. Ethanol reagents are diluted into ultrapure water (see Note 8).
Mounting media, such as Sigma DPX #06522 mounting media.
Coverslips of sufficient size to cover mounted tissue on slides (see Note 9).
3 mL disposable plastic transfer pipettes, at least 2.
Cardboard of sufficient size to hold all slides for drying.
3. Methods
We recommend that immunoblots are performed on the tissue prior to IHC, using anti-pHis antibodies of interest (see Chapter 12). Immunoblots involve fewer steps from tissue isolation to lysis in phosphatase inhibitors, and results should be easier to interpret, clearly revealing the number and size of proteins detected, which will aid in interpreting IHC results. Immunoblotting can be used to probe a tissue model individually with multiple pHis antibodies to discern which, if any, produces a pattern of interest. If pHis proteins cannot be detected by immunoblot in the tissue of interest with the pHis antibody of choice, it is unlikely that IHC will be successful. Fig. 1 outlines the steps detailed below.
Fig. 1.

Workflow for IHC. Steps and important timings are in bold; italic font refers to sections detailed in the text. Photographs and schematics are provided to better illustrate certain steps or materials
3.1. Tissue Fixation and Cryopreservation in Sucrose
Fill a sufficient number of 6-wells of a multiwell plate with TBS to rinse each piece of isolated tissue. Immediately after isolating fresh tissue, use dissecting forceps and scissors to cut a piece for processing (~3–5 mm thick, no larger than 1–2 cm across). Holding the tissue with forceps, immerse the piece in TBS to remove blood, fur, etc. from your sample.
Immediately place the rinsed tissue in a labeled, 15 mL conical tube containing 4% (v/v) PFA in TBS containing PhosSTOP. We use 5 mL of solution for pieces of liver tissue that are about 1 cm × 1 cm × 0.5 cm (0.5 cm3) (see Note 10). Leave on ice in the chemical fume hood for 3–4 h.
When incubation is complete, discard PFA solution and, with forceps, transfer the pieces of tissue to a 15 mL conical tube containing 15 mL of prechilled TBS. Place on a rocker at 4 °C and wash for 15 min. Discard the buffer, and repeat the wash two more times.
After the final wash, discard the buffer, removing all residual buffer using a P1000 tip. Add 15 mL of prechilled 30% (w/v) sucrose in TBS. Invert the tube twice. The tissue will float on top. Cap the tube and place on ice at 4 °C overnight.
In the morning, invert the tube 2× again. The tissue should now have sunk to the bottom of the tube. If so, the sucrose has impregnated the tissue sufficiently to preserve it during the freezing process and you can proceed (see Note 11).
3.2. Tissue Cryo Embedding
Remove all but 1 mL of the sucrose solution from the 15 mL tube containing the tissue. Add 1 mL of OCT to the tube. Place on a rocker at 4 °C for 30 min.
While the tissue is equilibrating in OCT, prepare two Styrofoam containers with dry ice. One container must contain sufficient space to hold all of the cryo molds to transport these to the −80 °C freezer for storage. The other will contain dry ice for the isopentane bath, which can be used to freeze 2–3 molds at a time. Wearing safety glasses for protection, pour isopentane over the dry ice, wait until bubbling and vaporization of isopentane ceases, then repeat. Continue until there is enough volume of liquid isopentane to sufficiently cover the dry ice such that cryomolds can be balanced on top of the dry ice in the isopentane bath, with the liquid covering the base of the mold without inundating the sample in isopentane.
Once tissue pieces have equilibrated for 30 min in the OCT/sucrose mixture, place a sufficient volume of OCT at the base of a labeled, disposable cryomold such that it can fully cover the tissue piece. Use a P200 pipette tip as a tool to lift the OCT media and wrap it in a circular motion around the piece of tissue. Take care to avoid the introduction of air bubbles. Gently press the tissue to the bottom, again avoiding bubbles (see Note 12). Carefully, squeeze out additional OCT from the bottle to slowly cover the tissue piece, filling the mold to the crease where the mold widens (see Note 13). The tissue should be entirely covered by OCT and without bubbles in proximity of the tissue.
Avoiding the introduction of bubbles, move the mold to the isopentane bath. Balance the mold carefully on the dry ice making sure that the bottom is level (see Note 14).
Continue processing samples and adding to the isopentane bath as space allows, but samples can be moved to the Styrofoam container containing dry ice once the OCT is opaque white.
Once all samples have been processed and placed on dry ice, store these at −80 °C in labeled Ziploc bags prior to cryosectioning and staining.
Prepare 5 μm sections on poly-l-lysine–coated glass slides using a cryostat (see Note 15).
3.3. Blocking of Antibody with pHis Peptide
One night before staining and mounting the cryosections onto slides, prepare a reaction to phosphorylate your chosen peptide overnight in phosphoramidate (see Notes 16 and 17). Label three 1.5 mL Eppendorf tubes: one will contain phosphoramidate (PA), one will serve as a control to contain unphosphorylated peptide and have water instead of PA, and one will contain PA and no peptide. Place these on ice.
Add 2.5 μL of 10 mg/mL peptide (or water for the PA only control) at pH 7–9 and 10 μL of 81 mg/mL PA (or water for the unphosphorylated peptide control) to the appropriate tubes on ice. This can be scaled up as needed for the experiment. The amount of PA may need to be increased to conserve the molar excess of PA if your peptide is much larger than 1 kDa (see Note 18).Incubate overnight on ice in the cold room to phosphorylate the peptide (see Note 19).
After incubation, remove 1 μL from each reaction and pipet as a spot onto a small piece of nitrocellulose labeled in pencil to indicate +PA, −PA, −peptide. Allow to dry on a flat, clean surface for 5 min. Holding the blot with forceps, blow compressed air on the nitrocellulose to complete the drying process, or allow the spots to dry at room temperature for longer.
Process the dot blot as an immunoblot using the anti-pHis antibody (see Note 20).
Incubate the dot blot for 30 min in dot blot blocking buffer.
Incubate the dot blot in primary antibody to detect phosphohistidine for 2 h.
Wash the dot blot for 3 × 5 min in TBST.
Incubate the dot blot for 50 min in secondary antibody conjugated with a substrate to detect the phosphohistidine antibody used (see Note 21).
Wash the dot blot for 3 × 5 min in TBST.
Detect the pHis signals. An example of a dot blot from a histidine phosphorylation experiment is presented in Fig. 1 (see also Chapter 12).
Dilute the remaining phosphorylated and unphosphorylated peptide reactions (to reduce the levels of PA present) by adding 189 μL of 10 mM Tris pH 8.8 to the remainder of the reaction (which should be about 11.5 μL).
Prepare three 1.5 mL Eppendorf tube containing anti-pHis antibody diluted to 2 μg/mL in 0.5 mL of blocking buffer and label for pHis peptide, unphosphorylated peptide control (should not block the antibody), and a no peptide control (to verify that the peptide itself does not interfere with the signal). Add 5 μL of the appropriate reactions (now diluted in 200 μL) to the pHis peptide and unphosphorylated peptide tubes. Reactions can be scaled up as needed for IHC staining.
Wrap Parafilm around the caps to seal, and place on a rotator at room temperature for 1–2 h. Remove Parafilm and place on ice (see Note 22).
3.4. Staining and Mounting of Coverslips to Cryosection Slides
If you do not already have solutions prepared to fill the tubs of xylene and ethanol needed in the dehydration and coverslip mounting steps, prepare these as indicated in Subheading 2.4.
Thaw cryosection slides at 4 °C for at least 2 h.
Prepare a humid chamber while slides thaw: soak Whatman 3MM paper in deionized water to cover the bottom of a tray (with a cover) sufficient to hold slides. Create an area for slides to sit that is not on the damp surface, either using pieces of Parafilm, or by place pieces of plastic pipettes vertically to sit at the left and right ends of slides and act as supports.
Wash slides at 4 °C with TBS for 3 × 5 min each using a staining rack and tub.
Move slides to room temperature, leaving them on the rack in the final TBS wash. Remove slides one-by-one to avoid drying of the tissue on the slides. Tap the edges of the slide on a stack of dry paper towels to remove excess TBS, being cautious not to touch the pieces of tissue mounted on the front of slide.
Once residual liquid has been removed, outline the pieces of tissue (the shape of a circle or rectangle is usually best) with a hydrophobic barrier pen. Lay the slide in the humid chamber, either on a strip of Parafilm or on top of vertically placed pipets that hold the slides at the left and right sides. Add sufficient blocking buffer to cover the piece of tissue outlined by the hydrophobic barrier pen. This should form a bubble over the piece of tissue that will not dry during incubation (see Note 23).
Repeat steps 4–5 for each individual slide until all slides have bubbles of blocking solution and are sitting in the humid chamber. Place a cover on the humid chamber and incubate for at least 45 min at room temperature, ensuring that it is not touched.
Add the primary antibody solutions that were prepared in Subheading 3.3: pHis peptide blocked, unphosphorylated peptide control, and no peptide control. Working with slides one-by-one as in step 4, remove the blocking buffer by tapping the slide on a stack of paper towels, then place flat in the humid chamber. Add a sufficient volume to each tissue outline of slides to cover for an overnight incubation. Be sure to record which primary antibody solution is added to each tissue section. Incubate at 4 °C overnight with the humid chamber closed and ensure that it will not be touched or moved during the incubation.
In the morning, equilibrate to room temperature the IHC detection reagent (such as SignalStain Boost, rabbit) designed for the host species of your antibody. Move the humid chamber carefully to room temperature.
Using a tub that holds a slide staining rack, wash each slide in TBST for 3 × 5 min, using a sufficient volume of TBST to cover the slides placed in the tub. As in step 4, process slides one-by-one, tapping off the antibody and placing on the staining rack in the tub of TBST. Repeat this process until all slides have been moved to the rack immersed in the tub of TBST.
Leave slides on the rack in the tub with the final TBST wash. Process slides again as in step 4, one-by-one, to tap away residual liquid. Add sufficient IHC detection reagent to cover each tissue section (usually 100–300 μl), and place on the humid chamber as for the blocking buffer. Once all slides have been transferred to the humid chamber, cover and incubate for 45 min at room temperature, ensuring that the humid chamber is not touched during this incubation.
Wash the slides in TBST for 3 × 5 min each, as in step 9.
Remove residual TBST from slides by tapping the edges of each slide on a stack of paper towels, then place flat in the humid chamber. Process a few slides at a time, preferably those that are being directly compared, and add sufficient DAB peroxidase HRP substrate to cover the outlined tissue sections, working quickly to avoid drying of tissue sections. Using a timer, monitor the color change on the tissue sections as the substrate is converted to a brownish precipitate. This is usually observed by 10 min (see Note 24).
Once the color has developed to the required shade, place the slides on the slide staining rack in the tub of ultrapure water. Incubate for 2 min once all slides are on the rack.
Place the slide staining rack in a tub of hematoxylin and incubate for up to 5 min, to achieve the required shade, if you wish to counterstain nuclei.
Place the slide staining rack in TBS for 30 s then move to ultrapure water for 8 min.
Dehydrate slides by incubating the slide staining rack for 3 min each in tubs of ethanol of progressively higher percentages (v/v): 50%, 50%, 75%, 95%, 100%, 100%.
Continue dehydration using three tubs of xylene (working in a chemical fume hood), incubating the slide staining rack for 5 min in each. While washing, move a stack of paper towels, coverslips, mounting media, two 3 mL disposable transfer pipettes, and a piece of cardboard of suitable size for holding all of the mounted slides to the hood.
Leave the slide staining rack in the final xylene wash tub, and remove one slide at a time to mount coverslips. Tap the edges of the slide on a stack of paper towels. Holding the slide above the paper towels and level to a flat surface, add a couple of drops of mounting media to the slide (using the transfer pipette) on the face that contains the tissue section(s) (see Note 25). Add three drops of xylene from the final wash to the tissue section face of the slide while still holding it level. Mount the coverslip from one side to allow air bubbles to be released. Place the mounted slide on the cardboard in the hood, and leave for 24 h to cure.
View the cured slides by light microscopy. The slides can be stored in a slide storage box; the color is stable for long-term storage. Examples of results from our validation of SC44-1 anti-3-pHis signal in liver tissue are found in Fig. 2.
Fig. 2.
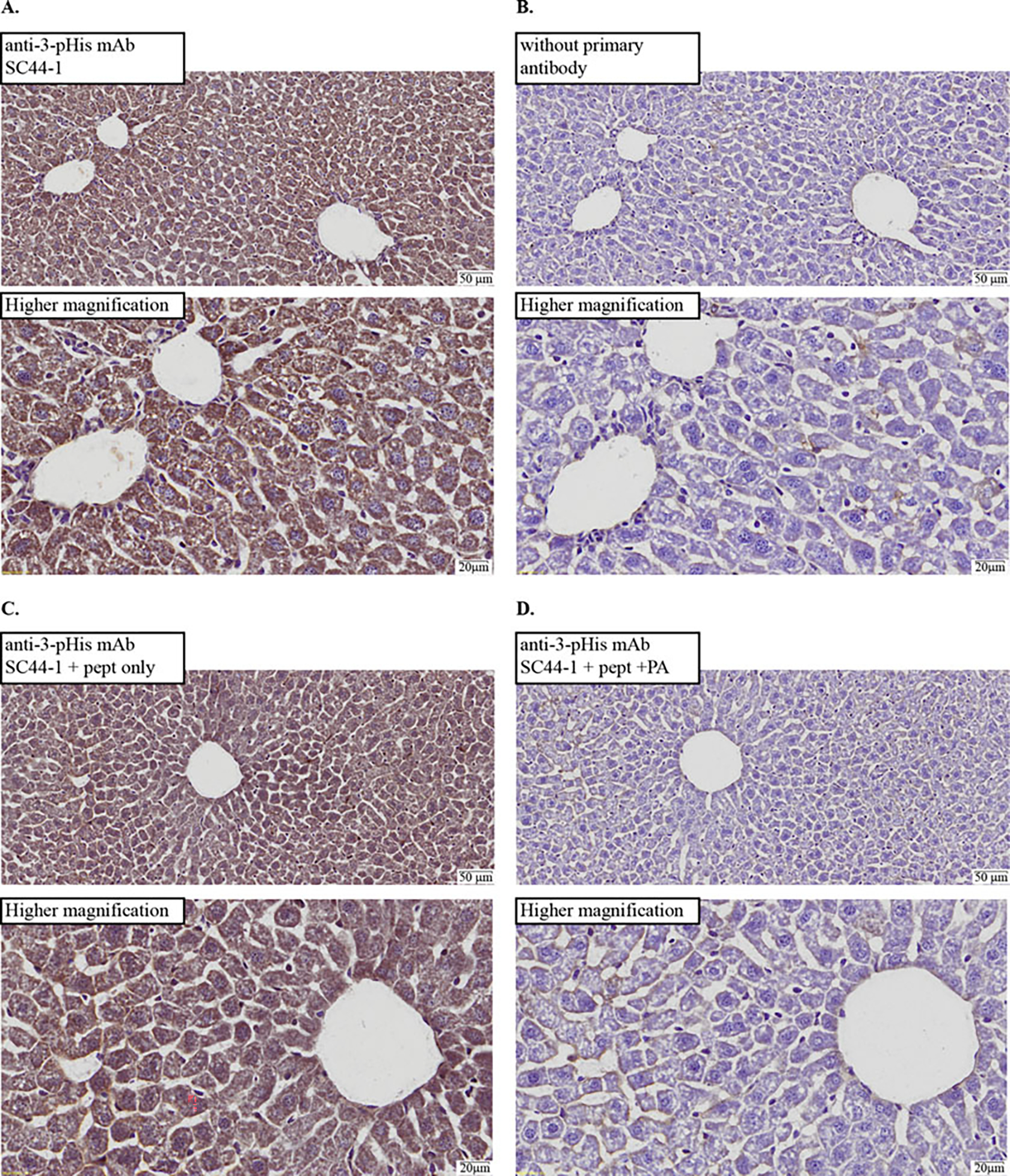
Chromogenic detection of SC44-1 anti-3-phosphohistidine mAb signals in cryosections from formaldehyde-fixed, cryoprotected mouse liver. IHC images of slides for serial cryosections of C57BL/6 normal mouse liver. To best compare controls to experimental conditions, two separate slides were processed and analyzed: one was mounted with both (a) and (b) cryosections and another mounted with both (c) and (d) cryosections. Low magnification (top) and higher magnification (bottom) images with a scale bar are shown for each. Histidine phosphorylated peptides or controls were used in primary antibody incubations to probe phosphorylation-dependence of the IHC signal: (a) antibody (SC44-1) incubated without peptide (pept); (b) blocking buffer alone (no antibody) used in primary incubation; (c) antibody incubated with pept, untreated with phosphoramidate (PA); (d) antibody incubated with pept treated with PA (i.e., phosphorylated). Results support the phosphorylation dependence of IHC signal
4. Notes
You can aliquot 10 mL of the 4% (v/v) PFA solution to a 15 mL conical tube (Falcon) placed on ice to reduce the number of PhosSTOP tablets used. PhosSTOP is needed to inhibit protein histidine phosphatase activity from enzymes such as PP1, PP2A, and PP2B [12], and possibly protein-tyrosine phosphatases.
Normal mouse C57BL/6 liver has been validated with this method, but other tissues can be optimized using this protocol as a starting point. Isolate fresh immediately prior to fixation.
We used the H2N-Ala-Gly-Ala-Gly-His-Ala-Gly-Ala-Gly-NH2 peptide used previously to validate pHis antibodies [1]. In selecting the sequence for a peptide to phosphorylate, it is important to note that phosphoramidate produces phosphoramidate (P-N) bonds on histidine, lysine and possibly on arginine residues [13]. For example, a study characterizing PHPT1 activity against lysine phosphoramidate bonds selected a histone H1 variant for incubation with phosphoramidate that did not contain histidine residues [14]. For this reason, in your peptide sequence, you might choose to substitute lysine and possibly arginine residues within a sequence of interest containing phosphohistidine to uniquely analyze the contribution of the phosphohistidine.
This can be purchased or synthesized [15] in a chemical lab equipped and approved for this process.
This antibody was validated for IHC in normal mouse liver using the SC44-1 anti-3-pHis rabbit monoclonal antibody, which appears to demonstrate a partial bias for 3-pHis on GHAGA sequences [1].
Secondary antibody for the dot blot should be coupled to the appropriate substrate for the detection system desired, such as goat anti-rabbit IgG with Alexa 680 conjugate for Li-Cor Odyssey detection.
Prepare DAB HRP substrate during the final TBST wash after secondary antibody incubation (Subheading 3.4, step 11).
For 50% (v/v) ethanol, 95% (v/v) ethanol can be diluted into ultrapure water. For other ethanol dilutions, use absolute ethanol in the dilution. We reuse ethanol and xylene solutions for IHC staining, but do not use xylene from deparaffinization steps in other protocols in this protocol. Solutions used in tubs in the chemical fume hood during the protocol should be stored prior to and afterward in tightly capped containers to prevent evaporation and placed in an appropriate storage area for flammable liquids in accordance with regulations.
We used 22 mm × 50 mm No. 1 thickness cover glass from Fisher.
The volume of 4% (v/v) PFA needed for each piece of tissue will depend on its size. We recommend a volume of 10–20 times the tissue volume, but you might need to optimize this if your antibody fails to recognize overfixed antigens or if under-fixation affects tissue morphology.
If the tissue does not sink, you can try a gradient of sucrose concentrations: 10% (w/v), 20% (w/v), and 30% (w/v), incubating the tissue at 4 °C until it sinks or for 24 h in each. However, there might not be any difference in the morphology even if the tissue does not sink, and it is unclear how longer incubation times might affect the pHis signal detected. We recommend analysing a portion of the tissue that does not sink in 30% (w/v) sucrose to decide whether the gradient is necessary. In the future, verify that your sucrose solution is freshly made at the appropriate concentration, use a larger volume of solution to tissue, and agitate periodically during the incubation to prevent the formation of a gradient.
The idea is to coat the surface of the tissue with OCT such that air bubbles are not trapped between the tissue and the OCT media. If it proves challenging to prevent the introduction of bubbles to the tissue, transfer the tissue from the 15 mL conical tube to a Petri dish containing enough OCT to cover the piece. Use a P200 tip to “wrap” the tissue in OCT and eliminate bubbles, then move the tissue to the bottom of the cryo mold.
The molds that we use are tapered at the bottom and broaden to the widest point, which is visible as a creased line around the mold (as shown in Fig. 1).
To balance the cryo molds in the isopentane–dry ice bath, you can use forceps that are longer than the depth of the bath. Insert these to stand as a support for the mold, arms down in the bath, placing the mold against the forceps’ arms (as shown in Fig. 1). You will need more than one set of forceps if you have multiple molds to process and wish to progress rapidly. You can also simply hold the mold using forceps in the bath to a depth that immerses the portion of the mold containing the OCT-embedded tissue without allowing isopentane to touch the inside of the mold. This takes more time since only one sample is processed at a time but may better ensure that molds do not spill or become immersed in isopentane.
Due to the challenging nature of cryosectioning, we rely on the expertise of a core facility to prepare 5 μm sections on poly-l-lysine–coated glass slides using a cryostat. We typically have serial sections cut, with two sections mounted per slide. In this manner, we can make the following comparisons in parallel: anti-pHis antibody alone vs. no primary antibody and anti-pHis antibody blocked with peptide vs. anti-pHis antibody blocked with phosphorylated peptide.
If you are using anti-1-pHis antibodies in your study, rather than anti-3-pHis antibodies as we have in our optimization of the IHC method, you will likely need a different peptide control.
PA-phosphorylated peptides contain a mixture of the 1- and 3-pHis isomers. Due to the relatively greater thermodynamic instability of 1-pHis as compared to 3-pHis [1], this isomer is poorly retained in the final reaction that is used to block the anti-pHis antibodies and hence may not effectively block enough antibody to observe a decrease in IHC signal in validation experiments. Thus, if you are using anti-1-pHis antibodies for IHC, we recommend blocking the anti-1-pHis antibodies using peptides synthesized to incorporate a stable pHis mimetic (1-pTza) at the histidine residue rather than PA-phosphorylated peptides. This approach was utilized to validate anti-pHis antibody staining (ICC/IF) in the Fuhs et al. publication [1].
These amounts represent a greater than 100-fold molar excess of phosphorylated peptide (≅700 Da, 0.6 μg added to blocking reaction) to immunoglobulin protein (IgG ≅ 150 kDa, 1 μg used in blocking reaction). It is important to conserve at least this ratio, particularly since peptides are not phosphorylated to 100%. If you use more or less antibody (i.e., if the 2 μg/mL concentration of antibody is not ideal for your samples), you can alter the amount of peptide that you prepare accordingly. This protocol utilizes a greater than 200-fold molar excess of PA to peptide. You can attempt to use less PA than this, but due to the instability of pHis, you might encounter difficulty in generating enough pHis peptide to sufficiently block the anti-pHis antibody in validation experiments.
You can also phosphorylate peptides using PA by incubating the reaction for 2 h at room temperature if you are unable to perform the reaction overnight.
It is best to process the dot blot rapidly in parallel with the remaining steps of the protocol and use the peptide reactions to block the anti-pHis antibody (steps 11–13, Subheading 3.3) prior to observing the dot blot results. It is possible to incubate the dot blot in primary antibody overnight while completing the protocol, skipping steps 7–10 of Subheading 3.3, and completing these steps of the dot blot protocol the following day. However, the levels of pHis available to bind the anti-pHis antibody will likely be reduced and yield lower (possibly undetectable) signal on the dot blot. Nevertheless, even if the signal on your dot blot is weak, you may still have enough phosphorylated peptide to block the anti-pHis antibody.
Secondary antibodies conjugated to dye for Li-Cor Odyssey detection, or conjugated to HRP for chemiluminescent detection can be used.
If you observe background in peptide incubations with antibody, you can clear the antibody blocking reactions after the 1 h incubation by spinning at 10,000 × g for 5 min and using the supernatant.
It is useful to try the pen on a piece of paper towel before applying to the slide to ensure that the media is flowing well. Hold the slide with the tissue section facing you to apply the outline; placing it flat and outlining with the pen is likely to release too much media and smudge. Any discontinuity in the outline will allow your solutions to leak away from the tissue. It is worthwhile practicing ahead of time with the pen to prepare you for how to outline the tissue sections effectively.
To make appropriate comparisons, we examine the tissue treated with anti-pHis antibody alone first, incubating the other controls for at least as much time as this slide.
It is best to put the mounting media near but not on the tissue section(s), and to avoid introducing bubbles. Air bubbles on the tissue will pose problems in viewing the tissue by microscopy. This process is diagrammed in Fig. 1.
References
- 1.Fuhs SR, Meisenhelder J, Aslanian A, Ma L, Zagorska A, Stankova M, Binnie A, Al-Obeidi F, Mauger J, Lemke G, Yates JR 3rd, Hunter T (2015) Monoclonal 1- and 3-phosphohistidine antibodies: new tools to study histidine phosphorylation. Cell 162 (1):198–210. 10.1016/j.cell.2015.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kee JM, Oslund RC, Couvillon AD, Muir TW (2015) A second-generation phosphohistidine analog for production of phosphohistidine antibodies. Org Lett 17(2):187–189. 10.1021/ol503320p [DOI] [PubMed] [Google Scholar]
- 3.Kee JM, Oslund RC, Perlman DH, Muir TW (2013) A pan-specific antibody for direct detection of protein histidine phosphorylation. Nat Chem Biol 9(7):416–421. 10.1038/nchembio.1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kee JM, Villani B, Carpenter LR, Muir TW (2010) Development of stable phosphohistidine analogues. J Am Chem Soc 132 (41):14327–14329. 10.1021/ja104393t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hindupur SK, Colombi M, Fuhs SR, Matter MS, Guri Y, Adam K, Cornu M, Piscuoglio S, Ng CKY, Betz C, Liko D, Quagliata L, Moes S, Jenoe P, Terracciano LM, Heim MH, Hunter T, Hall MN (2018) The protein histidine phosphatase LHPP is a tumour suppressor. Nature 555(7698):678–682. 10.1038/nature26140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai X, Srivastava S, Surindran S, Li Z, Skolnik EY (2014) Regulation of the epithelial Ca(2) (+) channel TRPV5 by reversible histidine phosphorylation mediated by NDPK-B and PHPT1. Mol Biol Cell 25(8):1244–1250. 10.1091/mbc.E13-04-0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan I, Steeg PS (2018) The relationship of NM23 (NME) metastasis suppressor histidine phosphorylation to its nucleoside diphosphate kinase, histidine protein kinase and motility suppression activities. Oncotarget 9 (12):10185–10202. 10.18632/oncotarget.23796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panda S, Srivastava S, Li Z, Vaeth M, Fuhs SR, Hunter T, Skolnik EY (2016) Identification of PGAM5 as a mammalian protein histidine phosphatase that plays a central role to negatively regulate CD4(+) T cells. Mol Cell 63 (3):457–469. 10.1016/j.molcel.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivastava S, Li Z, Soomro I, Sun Y, Wang J, Bao L, Coetzee WA, Stanley CA, Li C, Skolnik EY (2018) Regulation of KATP channel trafficking in pancreatic beta-cells by protein histidine phosphorylation. Diabetes 67 (5):849–860. 10.2337/db17-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava S, Panda S, Li Z, Fuhs SR, Hunter T, Thiele DJ, Hubbard SR, Skolnik EY (2016) Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. eLife 5. 10.7554/eLife.16093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava S, Zhdanova O, Di L, Li Z, Albaqumi M, Wulff H, Skolnik EY (2008) Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1. Proc Natl Acad Sci U S A 105 (38):14442–14446. 10.1073/pnas.0803678105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matthews HR, MacKintosh C (1995) Protein histidine phosphatase activity in rat liver and spinach leaves. FEBS Lett 364(1):51–54 [DOI] [PubMed] [Google Scholar]
- 13.Kowalewska K, Stefanowicz P, Ruman T, Fraczyk T, Rode W, Szewczuk Z (2010) Electron capture dissociation mass spectrometric analysis of lysine-phosphorylated peptides. Biosci Rep 30(6):433–443. 10.1042/BSR20090167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ek P, Ek B, Zetterqvist O (2015) Phosphohistidine phosphatase 1 (PHPT1) also dephosphorylates phospholysine of chemically phosphorylated histone H1 and polylysine. Ups J Med Sci 120(1):20–27. 10.3109/03009734.2014.996720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei YF, Matthews HR (1991) Identification of phosphohistidine in proteins and purification of protein-histidine kinases. Methods Enzymol 200:388–414 [DOI] [PubMed] [Google Scholar]