

Abstract
Immunoblotting is a ubiquitous immunological technique that aids in detecting and quantifying proteins (including those of lower abundance) and their posttranslational modifications such as phosphorylation, acetylation, ubiquitylation, and sumoylation. The technique involves electrophoretically separating proteins on an SDS-PAGE gel, transferring them onto a PVDF (or nitrocellulose) membrane and probing with specific antibodies. Here we describe an immunoblotting technique for detecting cellular phosphohistidine, a labile posttranslational modification, by optimizing experimental conditions such that the labile phosphohistidine signal is conserved throughout the experiment.
Keywords: Posttranslational modification, Histidine phosphorylation, pHis antibodies, Western blotting, Immunoblotting, Phosphoramidate bond
1. Introduction
Western blotting, also referred to as immunoblotting or protein blotting, is a technique that has been widely used to identify, characterize, and quantify proteins since its inception in the 1970s [1]. This versatile technique can be modified and adapted to a variety of sample preparations and experimental conditions. Western blotting is carried out in three steps: (1) SDS-PAGE, (2) blotting, and (3) immunostaining. In the first step, proteins are typically separated on an SDS-polyacrylamide gel based on their molecular weight. The separated proteins are then transferred electrophoretically onto a membrane (PVDF or nitrocellulose). The final part of the technique is protein detection by immunostaining, which involves use of protein or posttranslational modification (PTM)-specific antibodies. Antibodies against many stable PTMs such as O-linked phosphorylation (Ser, Thr, and Tyr), acetylation, ubiquitylation, and sumoylation [2–4] have been commercially available for a number of years, but only recently have these become available for labile N-linked phosphorylation. Consequently, proteins carrying N-linked phosphate are comparatively understudied [5]. Histidine undergoes N-linked phosphorylation on the imidazole ring forming a phosphoramidate (N-P) bond whose lability is conferred by the relatively high free energy of hydrolysis (−12 to −13 kcal/mol) [6]. Hydrolysis of the phosphoramidate bond is further accelerated by low pH and high temperature [7], common treatment conditions during biochemical analyses.
Recently, Fuhs et al. developed monoclonal antibodies (mAbs) against two stereoisomers of phosphohistidine (pHis; 1-pHis and 3-pHis) by incorporating the relatively stable pHis analogs, 1-pTza and 3-pTza (phosphoryltriazolylalanine), in degenerate peptides. These mAbs display the properties of pHis isoform stereospecificity, are sequence independent and exhibit submicromolar affinity to peptides and proteins which contain histidine phosphorylation [8]. Adapting standard immunoblotting techniques to preserve the phosphohistidine signal, in conjunction with the application of these newly developed pHis antibodies is helping unveil the mechanistic aspects of the pHis-containing proteins in normal and disease physiology [9–11].
In this chapter, we describe a modified immunoblotting technique which can be employed to detect histidine phosphorylated proteins in cell and/or tissue extracts using pHis mAbs. We demonstrate its utility using IMR-32 and HeLa cell lines established from neuroblastoma and cervical adenocarcinoma, respectively. This protocol can be further exploited to ascertain phosphohistidine levels in tissue samples and other cell lines.
2. Materials
All solutions should be prepared with deionized ultrapure water with resistivity of 18.2 MΩ cm and analytical grade reagents. All reagents should be handled and disposed of according to local safety guidelines.
2.1. Cell Culture and Lysate Preparation
Cell line: IMR-32 (human neuroblastoma); HeLa (cervical adenocarcinoma) cell lines.
Cell growth medium: RPMI-1640 (Rosewell Park Memorial Institute Medium) supplemented with 10% FBS (fetal bovine serum) for IMR-32 cell line; DMEM supplemented with 10% FBS for HeLa cell line and 100× Penicillin-Streptomycin solution (Purchase from Corning, Product number 30-002-cI; see Note 1).
Phosphate buffered saline (PBS; 1×): 0.8 mM sodium phosphate dibasic, 1.47 mM sodium phosphate monobasic, 137 mM NaCl, and 2.5 mM KCl.
Lysis buffer: 25 mM Tris–HCl pH 8.5, 140 mM NaCl, 0.1% (v/v) Tween 20.
Protease and phosphatase inhibitors: Protease inhibitor cocktail tablets (Roche), PhosSTOP tablets (Roche) and 1 mM PMSF (phenyl methyl sulfonyl fluoride) prepared in ethanol (see Note 2).
CO2 incubator.
10 cm2 cell culture dishes
Probe sonicator.
2.2. SDS-Polyacrylamide Gel
30% (w/v) acrylamide solution: Weigh 30 g of acrylamide and add to 60 mL of water. After the dissolution of the acrylamide, filter the solution through a 0.45 μM filter and store at 4 °C in a brown bottle (see Note 3).
1% (w/v) bis-acrylamide solution: Weigh 1 g of bis-acrylamide and add to a cylinder with 100 mL of water. Filter through a 0.45 μM filter and store at 4 °C in a brown bottle.
Resolving and stacking gel buffer: 3 M Tris–HCl, pH 8.8. Weigh 363.4 g of Tris Base and add to 900 mL of water in a beaker. Adjust the pH to 8.8 with HCl. Make up the volume to 1 L before storing at 4 °C (see Note 4).
20% (w/v) sodium dodecyl sulfate: Add 20 g of SDS to 80 mL of water and warm gently to dissolve SDS (see Note 5). Make up the volume to 100 mL after dissolution.
Ammonium persulfate: Prepare 10% (w/v) solution in water (see Note 6).
N,N,N′,N′-tetramethylethane-1,2-diamine (TEMED): Store at 4 °C.
SDS-PAGE running buffer (1×): 25 mM Tris–HCl pH 8.5, 0.1% SDS, and 192 mM glycine, precooled to 4 °C (see Note 7).
SDS-PAGE sample buffer (5×): 250 mM Tris–HCl pH 8.8, 10% SDS, 50 mM EDTA, 500 mM 1,4 dithiothreitol (DTT), 50% glycerol, and 0.02% bromophenol blue. Store the sample buffer at −20 °C (see Note 8).
Polyacrylamide gel electrophoresis equipment: Gel plates, electrophoresis chamber unit appropriate for gel plates, electric wires, and power supply with programmable voltage and current settings.
2.3. Immunoblotting
0.45 μM PVDF membrane wetted in methanol for at least 2 min (see Note 9).
Transfer buffer (1×): 25 mM Tris–HCl pH 8.5, 0.1% SDS, 192 mM Glycine and 20% methanol (see Note 10).
Tris–NaCl buffer (10×): 500 mM Tris–HCl pH 8.5 and 1.5 M NaCl.
Blocking buffer: 0.2× Tris–NaCl buffer (pH 8.5), 0.1% casein (see Note 11).
Antibody dilution buffer: Blocking buffer with 0.1% Tween 20.
Wash buffer: 1× Tris–NaCl buffer with 0.1% Tween 20.
Wet transfer equipment: Wet transfer tank, two gel holder cassettes, filter paper, Whatman 3MM chromatography filter paper, foam pads, electric wires, and power supply with programmable voltage and current settings (see Note 12).
2.4. Immunostaining
Primary antibodies: Anti-N1-pHis (1-pHis) rabbit monoclonal antibodies purified from clone SC1-1 (Purchase from Millipore, product number MABS1330) and purified anti-N3-pHis (3-pHis) rabbit monoclonal antibodies from SC44-1 hybridoma cells [8] (see Note 13).
Antibody for loading control: anti-β-actin monoclonal antibody raised in mouse.
Secondary antibodies: goat-anti rabbit IgG with Alexa Fluor 680 conjugate, goat-anti mouse IgG with DyLight 800 conjugate.
Prestained protein molecular weight markers.
3. Methods
Detecting the pHis signal is pH and temperature dependent; hence, most of the solutions should be adjusted to a pH between 7 and 9 and stored at 4 °C, unless otherwise mentioned.
3.1. Whole Cell Lysate Preparation
Except for growing the cells, all steps should be carried out 4 °C. An alternative lysis strategy for tissue is provided in Note 14.
Grow the IMR-32 cells in RPMI medium and HeLa cells in DMEM medium, at 37 °C with 5% CO2 in 10 cm2 dishes until confluency reaches 70–90% (see Note 15).
Wash the cells twice with 5 mL of cold 1× PBS kept at 4 °C (see Note 16). Scrape the cells off the plate into 250 μL lysis buffer containing 1× protease and phosphatase inhibitor cocktail solution (see Note 2).
Mount the samples onto a probe sonicator for cell lysis. Keep the cells on ice during sonication to avoid sample heating. Program the sonicator to deliver 10 s bursts with a 10 s interval for three times at 40% amplitude (see Note 17).
Remove the cell debris by centrifugation at 14,000 × g for 10 min at 4 °C. Estimate the protein concentration in the clarified lysate by colorimetric assay and load around 30 μg of protein onto the SDS-PAGE gel for analysis (see Note 18). Do not heat the samples before loading on the gel (see Note 19).
3.2. 12% Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis
It is imperative to carry out all the steps at 4 °C except for the polymerization of the gel.
Prepare the 12.5% resolving gel by mixing 2 mL of resolving gel buffer with 6.8 mL of 30% acrylamide and 1.66 mL of 1% bis-acrylamide. Make up the volume to 16 mL by adding 5.8 mL of water. To this mixture add 80 μL of 20% SDS, 60 μL of 10% ammonium persulfate (APS) and 10 μL of TEMED. Gently mix the solution before pouring into the gel cassette with 0.2 mm thickness. Fill two-thirds of the gel cassette with the mixture before layering isopropanol carefully on top (see Note 20). After the gel polymerizes remove the isopropanol and gently wash the gel with water to completely remove isopropanol.
Prepare the 4% stacking gel by adding 5.04 mL of water to 1.06 mL of 30% acrylamide, 0.8 mL of 1% bis-acrylamide and 1.0 mL of resolving buffer. To this mixture add 40 μL of 20% SDS, 30 μL of 10% APS and 5 μL of TEMED. Immediately insert the comb (10 well Teflon) without trapping air (see Note 21).
Mount the gel on the gel-running apparatus then fill it with SDS-PAGE running buffer precooled to 4 °C (see Note 22). To evaluate, the fidelity of the phosphohistidine signal, divide the samples into two tubes (Fig. 1). Keep one tube on ice at 4 °C. Add acid (0.01% acetic acid or formic acid) to the second tube till the pH drops to 4. Incubate for one minute before restoring the pH of the acidified sample to 8.5. After which boil the second tube for 15–30 min at 90 °C (see Note 23). Mix the protein samples with the 5× sample buffer. Load the protein samples (~30 μg of protein; see Note 18) into the wells along with a protein marker. Apply 100 V for 1–2 h for electrophoresis of the samples. This step should be carried out in a cold room or chamber at 4 °C.
Fig. 1.
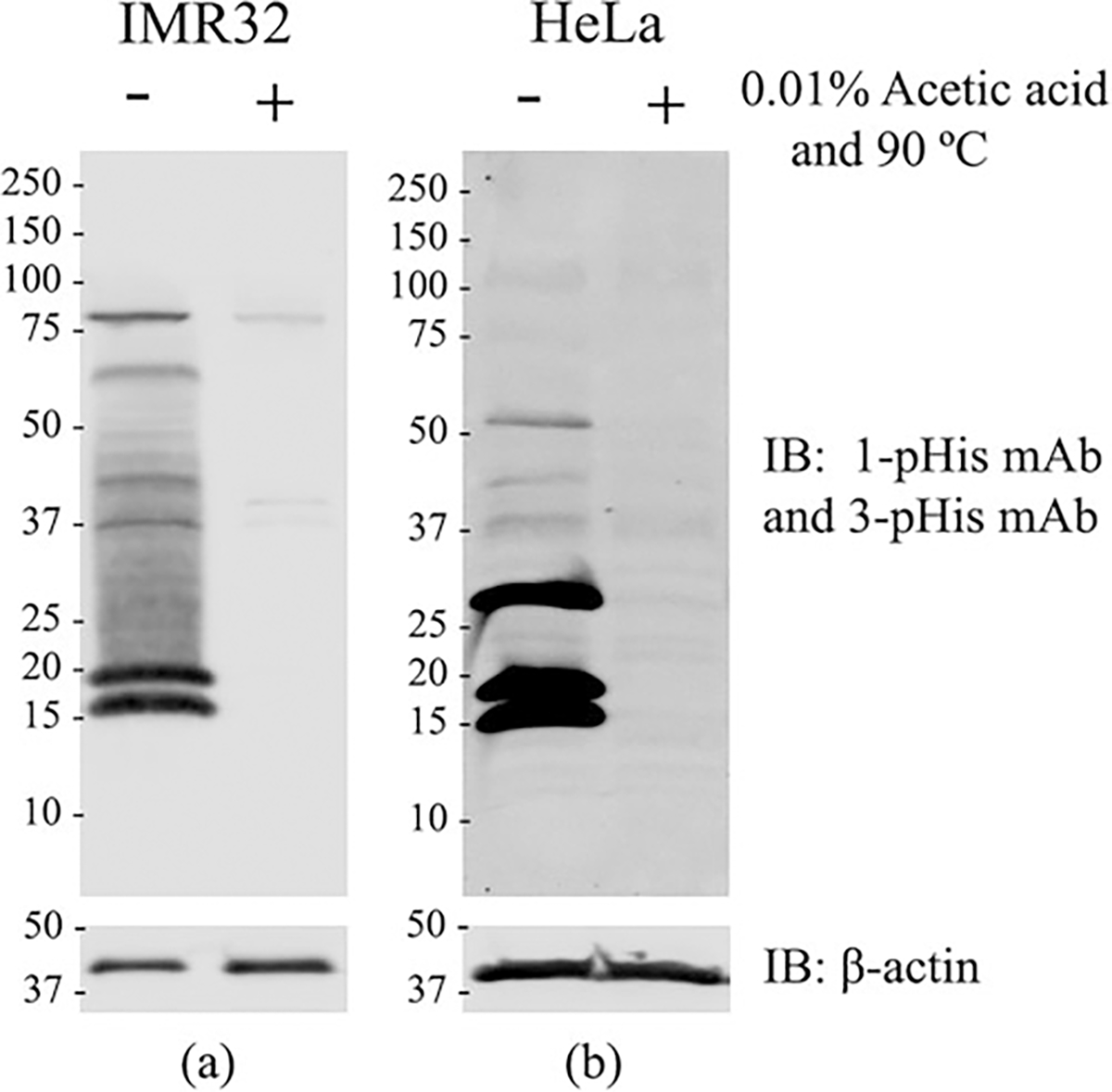
Western blot detection of phosphohistidine proteins: Proteins containing 1- or 3-pHis isomers from either IMR-32 (a) and HeLa (b) cell lysates were detected using a combination of SC1-1 (1-pHis mAb) and SC44-1 (3-pHis mAb) monoclonal antibodies. Heat and acid treatment of the lysate prior to electrophoresis resulted in a considerable decrease in the phosphohistidine signal. β-actin is used as the loading control
3.3. Immunoblotting
All the buffers should be precooled and all steps performed at 4 °C, unless otherwise stated.
When the dye-front reaches bottom of the gel plates, switch off the current and open the gel plates using a spatula or similar (see Note 24). Carefully remove the stacking gel. Place the gel in the transfer buffer.
Cut the PVDF membrane and two Whatman 3MM filter papers to the size of the gel and make a small cut at the right top of the membrane to mark the orientation of the gel. Equilibrate the membrane in methanol for 20 s before equilibration in cold transfer buffer (see Note 25).
Soak the SDS-PAGE gel, foam pads and Whatman 3MM filter paper in transfer buffer along with the membrane. Place the membrane over the gel and sandwich with two Whatman 3MM filter papers followed by two foam pads on either side in a gel cassette (see Note 26).
Place the cassette in the tank so that the membrane is toward the anode (+) and the SDS-PAGE gel is toward the cathode (−). Fill the transfer buffer to the level indicated on the tank. Apply a voltage of 30 V for 12–18 h, or 75 V for 2 h at 4 °C (see Note 27).
3.4. Immunostaining
Precool all buffers and perform all the steps at 4 °C unless otherwise specified.
Immediately after the transfer, place the membrane in the blocking solution and incubate for 2 h at 4 °C or 45–60 min at room temperature on a shaking platform.
Dilute the primary antibody (1- or 3-pHis antibody) to a final concentration of 0.5 μg/mL in antibody dilution buffer. Immediately after removal of the blocking solution, incubate the membrane for 1–4 h at 4 °C or 1 h at room temperature (see Note 28). Wash the membrane 3× with 10 mL of wash buffer for 5 min each.
Dilute the secondary antibody (anti-rabbit IgG) 1:20,000 in antibody dilution buffer with 0.01% SDS. Incubate the membrane for 1–4 h at 4 °C, or 1 h at room temperature. Wash the membrane 3× with 10 mL of wash buffer for 10 min each.
Develop the membrane using a LI-COR Odyssey Infrared imaging system with 700 and 800 nm filters (Fig. 1) (see Note 29).
Repeat steps 2–4 using primary and secondary antibodies against β-actin which is used as the protein loading control (see Note 30).
4. Notes
Filter FBS through 0.22 μM filter before adding to the RPMI or DMEM medium. The final concentration of penicillin-streptomycin antibiotic solution should be 1× in the media. Store the media at 4 °C after addition of FBS and antibiotics.
-
It is recommended to add protease and phosphatase inhibitors during lysis to prevent the degradation of proteins and loss of the phosphate signal. 10× solution of protease and phosphatase inhibitors can be prepared by dissolving one tablet each in 1 mL of lysis buffer separately and added to the sample immediately prior to sonication. The 10× stock solution can be stored at −20 °C for long term storage and 4 °C for short term storage. Protease and phosphatase inhibitor tablets can be replaced by making an in-house cocktail with appropriate concentrations of various inhibitors such as trypsin, chymotrypsin, thermolysin, papain, and pronase.
PMSF is sparingly soluble and highly unstable in aqueous solutions hence it should be prepared immediately prior to use. Alternatively, it can be prepared in organic solvents such as ethanol or DMF where it is highly soluble and stable up to 2 years when stored at −20 °C.
Acrylamide is a potential neurotoxin. Hence wear gloves and mask while weighing and dissolving acrylamide.
Resolving and stacking gels have the same pH albeit the percentage acrylamide used will likely differ (depending on the size of the proteins to be resolved). The classic Laemmli’s stacking gel pH is maintained at 6.8, which helps to focus the proteins at the separating gel interface. However, the current protocol requires the stacking gel pH to be at 8.8. This modification prevents hydrolysis of the phosphohistidine signal that could occur at the more acidic pH of a conventional stacking gel.
SDS is an irritant. Wear gloves and mask while weighing. Prevent shaking or agitation which leads to inaccurate volume measurement due to frothing, instead dissolve SDS by keeping the buffer on hot plate with magnetic stirrer and slowly add SDS to it.
Ammonium persulfate solution should be filtered and stored at −20 °C as aliquots.
SDS-PAGE running buffer can be prepared as 10× native buffer without SDS and stored at 4 °C. Weigh 30 g of Tris base and 144 g of glycine and add 950 mL water before adjusting the pH to 8.5 using HCl. Make up the volume to 1000 mL. This 10× stock solution can be stored at 4 °C. To make a 1× solution, dilute 100 mL of the native 10× buffer into 890 mL of water kept 4 °C. Then add 10 mL of 10% (w/v) SDS solution to the buffer just before each experiment. SDS should be added at the end to prevent frothing and precipitation at 4 °C.
DTT is unstable in presence of metal ions, alkaline pH and higher temperatures. Addition of EDTA and storage at −20 °C improves the half-life of DTT in buffers. As SDS precipitates at lower temperatures, allow the sample buffer to reach room temperature before adding to the protein sample.
Nitrocellulose membrane can also be used for detecting pHis signals. 0.22 μM pore size membranes should be used for transferring smaller peptides and proteins below 15 kDa while 0.45 μM membranes can be used for proteins above 15 kDa.
The composition of the transfer buffer is similar to the SDS-PAGE running buffer except for the addition of 20% methanol. Prepare 1× buffer by diluting 100 mL of 10× native buffer into 690 mL of water kept at 4 °C. Add 10 mL of 10% (w/v) SDS solution along with 200 mL of methanol. The transfer buffer pH is not usually adjusted, but it is within the acceptable pH range for conserving pHis signal.
Casein can be replaced with serum free BSA as a blocking agent. Though casein is a phosphoprotein, no cross-reactivity with the pHis antibodies has been observed.
Semi-dry transfer is another popular western blotting technique used to transfer proteins from the gel to a membrane. Less heat convection occurs during the transfer, so semi-dry transfer is not used for immunoblotting pHis proteins.
There are other anti-pHis antibodies which also can be used for detection of phosphoproteins by immunoblotting which include SC50-3 (Purchase from Millipore, product number MABS1341) for 1-pHis detection and SC39–6 and SC56-2-(Purchase from Millipore, products numbers MABS1351 and MABS1352, respectively) for 3-pHis detection. Though the antibodies are sequence independent, SC44-1 appears to demonstrate a partial bias for 3-pHis on GHAGA sequences (Fig. 1) [8].
For the preparation of tissue lysates, homogenize tissue in a manner that minimizes freeze–thaw, such as grinding in liquid nitrogen, focusing on keeping mortar, pestle, and any apparatus cold by adding liquid nitrogen throughout the entire process. For instance, liver powder is resuspended in a lysis buffer containing 10 mM Tris base pH 8.8, 0.1% SDS, 1% sodium deoxycholate, 0.5 mM EDTA, and 150 mM NaCl, but other lysis buffers of pH 8–8.8 should be sufficient. Protease inhibitor tablets (Roche), PhosSTOP tablets (Roche) (see Note 2), and 1 mM PMSF are added fresh to lysis buffer and chilled prior to adding to tissue. Sonication must be performed on ice or maintained at 4 °C. Clear tissue lysates at 14,000 × g for 10 min. All other steps should follow the cell culture protocol outlined herein.
If the cells are retrieved for growth from liquid nitrogen, change the media after one day to remove DMSO.
Be gentle while washing adherent cells with the PBS otherwise cells will be washed off with the buffer.
Sonicator amplitude should be optimized for individual cell lines and tissue samples based on the volume and viscosity of the samples. As an alternative to sonication, the complete volume of lysate can be passed through a 23-G needle fitted to a syringe of sufficient volume. Repeat this process five times, keeping the lysate at a temperature of 4 °C (on ice or in a cold room).
DC assay (Detergent Compatible assay from Bio-Rad) can be used to estimate protein concentration in the lysate. Tween 20 present in the lysis buffer does not interfere with DC colorimetric assay. Serial dilutions of bovine serum albumin (BSA) can be used to generate a standard curve, based on which protein concentration in the lysate can be estimated.
It is imperative not to heat samples in sample buffer before loading on the gel to avoid pHis hydrolysis. If the samples are viscous, add small amounts of 8 M urea or 6 M guanidinium chloride (prepared in lysis buffer to maintain desirable pH) and vortex briefly before loading.
Layering isopropanol on top of the resolving gel speeds up the gel polymerization by preventing contact with oxygen. It can be replaced by isobutanol, 95% (v/v) ethanol or water.
Insert the comb into the stacking gel so that the depth of the well is not more than half of the stacking gel length. This ensures that there is sufficient room for the stacking of the proteins before entering the resolving gel. If air is trapped in the stacking gel while inserting the comb, add a small amount of stacking gel mixture to the gel cassette along the sides of the comb which will displace the trapped air.
After the gel solidifies keep it at 4 °C for at least 10 min before loading the protein.
Ensure that the pH of the acidified sample does not drop below pH 3 which may precipitate the proteins and make loading onto the SDS polyacrylamide gel difficult. Restore the pH of the acidified sample to pH 8.5 before boiling to avoid gel running issues. Heating should follow acidification for complete elimination of pHis signal.
Allow the dye front to just exit the gel, otherwise it stains the membrane.
Handle the PVDF membrane at the corners with tweezers only. If nitrocellulose membrane is used instead of PVDF for blotting, skip the methanol treatment step.
While placing the membrane over gel for transfer it is important to ensure that no air is trapped. Air bubbles can be removed by gently rolling a pipet over the membrane. Repeat this step after sandwiching with the Whatman paper and foam pads.
Electrophoretic transfer from the gel to the membrane at 30 V is highly recommended to prevent the buffer system from heating up and to conserve the pHis signals. If a higher voltage is required to reduce transfer time, the system should be maintained at 4 °C. To distribute the heat generated during electrophoretic transfer evenly across the tank and decrease localized effects of heat on the gel, place the gel tank on a magnetic stirrer with a magnetic bead inside the tank.
Membranes can be left in the primary antibody solution for up to 18 h at 4 °C.
Develop the membrane based on the secondary antibody conjugate. We observe consistent phosphohistidine signals across experiments with the LI-COR Odyssey Infrared imaging detection system.
Secondary antibodies that are used to detect phosphohistidine proteins and actin on the same blot should be tagged with two fluorophores with different emission wavelengths. Steps 2–4 (Subheading 3.4) can be carried out with a mixture of primary and secondary antibodies against phosphohistidine proteins and actin simultaneously, or can be done in tandem. However, if performed sequentially, the phosphohistidine blot should be developed first due to diminution of the signal.
References
- 1.Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to NC sheets: procedure and applications. Proc Natl Acad Sci U S A 76:4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glenney JR, Zokas L, Kamps MP (1988) Monoclonal antibodies to phosphotyrosine. J Immunol Methods 109:277–285 [DOI] [PubMed] [Google Scholar]
- 3.Heffetz D, Fridkin M, Zick Y (1989) Antibodies directed against phosphothreonine residues as potent tools for studying protein phosphorylation. Eur J Biochem 182:343–348 [DOI] [PubMed] [Google Scholar]
- 4.Fujimuro M, Sawada H, Yokosawa H (1994) Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett 349 (2):173–180 [DOI] [PubMed] [Google Scholar]
- 5.Kee JM, Muir TW (2012) Chasing phosphohistidine, an elusive sibling in the phosphoaminoacid family. ACS Chem Biol 7(1):44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attwood PV, Piggott MJ, Zu XL, Besant PG (2007) Focus on phosphohistidine. Amino Acids 32(1):145–156 [DOI] [PubMed] [Google Scholar]
- 7.Wei YF, Matthews HR (1991) Identification of phosphohistidine in proteins and purification of protein-histidine kinases. Methods Enzymol 200:388–414 [DOI] [PubMed] [Google Scholar]
- 8.Fuhs SR, Meisenhelder J, Aslanian A, Ma L, Zagorska A, Stankova M et al. (2015) Monoclonal 1- and 3-phosphohistidine antibodies: new tools to study histidine phosphorylation. Cell 162:198–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuhs SR, Hunter T (2017) pHisphorylation: the emergence of histidine phosphorylation as a reversible regulatory modification. Curr Opin Cell Biol 45:8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hindupur SK, Colombi M, Fuhs SR, Matter MS, Adam K (2018) The protein histidine phosphatase LHPP is a tumour suppressor. Nature 555(7698):678–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava S, Panda S, Li Z, Fuhs SR, Hunter T, Thiele J (2016) Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. elife 5:pii:e16093. [DOI] [PMC free article] [PubMed] [Google Scholar]