

Abstract
In the presence of infection, neutropenia is considered to be a marker of poor prognosis; conversely, neutrophilia may not be a determinant of a better prognosis. Since apoptotic neutrophils are compromised functionally, we evaluated the effect of infection on neutrophil apoptosis. The rate of apoptosis was greater for neutrophils isolated from patients with infection than for healthy controls. Escherichia coli did not directly modulate the rate of neutrophil apoptosis. However, sera from infected patients promoted (P < 0.001) neutrophil apoptosis. Interestingly, the sera of patients with different types of infection (gram negative, gram positive, or culture negative) exerted a more or less identical response on neutrophil apoptosis. Sera of infected patients showed a fivefold greater content of FasL compared to controls. Moreover, anti-FasL antibody partly attenuated the infected-serum-induced neutrophil apoptosis. In in vitro studies, E. coli enhanced monocyte FasL expression. Moreover, conditioned media prepared from activated macrophages from control mice showed enhanced apoptosis of human as well as mouse neutrophils. On the contrary, conditioned media prepared from activated macrophages isolated from FasL-deficient mice induced only a mild degree of neutrophil apoptosis. These results suggest that neutrophils in patients with infection undergo apoptosis at an accelerated rate. Infection not only promoted monocyte expression of FasL but also increased FasL content of the serum. Because the functional status of apoptotic cells is compromised, a significant number of neutrophils may not be participating in the body's defense. Since neutrophils play the most important role in innate immunity, their compromised status in the presence of infection may transfer the host defense burden from an innate response to acquired immunity. The present study provides some insight into the lack of correlation between neutrophilia and the outcome of infection.
Neutrophilia is a common finding in patients with infections. Polymorphonuclear cells are considered to be important players in the host defense system and are the first cells to arrive at the site of injury. Their mobility to sites of infection or inflammation is mediated through the emanation of chemotactic substances, such as formylmethionylpeptides, by neutrophils or macrophages at the site of invasion or by activation of serum complement (19). Neutrophils can be produced at a rate of 100 billion cells daily for an average adult (37), indicating a huge defense potential. Therefore, neutrophils have been considered to function as the primary defenders in acute bacterial infections. However, this defense is compromised in patients with neutropenia, leukemia, or various congenital abnormalities affecting neutrophil structure or function (13, 22, 38).
Infection, directly or indirectly through sympathetic stimulation, induces an increase in blood neutrophil count. Since infection also stimulates the bone marrow to release neutrophils, the appearance of immature neutrophils in the peripheral smear has long been considered a marker of infection. Because neutrophils phagocytize and kill bacteria, one may be tempted to presume that an increased number of neutrophils is associated with a better host defense and an improved outcome. However, there is no relationship between increased neutrophil count and outcome of infection. It is possible that increased neutrophil count in the face of infection may be contributed to by functionally compromised neutrophils. Since apoptotic neutrophils have been reported to lose their function (35), we asked whether infection could promote apoptosis of neutrophils.
Mature neutrophils have a relatively short life span (32, 37). They die through a process of apoptosis (23, 27, 28). It has been demonstrated that interleukin-2, gamma interferon, tumor necrosis factor (TNF) alpha, granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, and glucocorticoids increase the life span of neutrophils (4–6, 14, 36). On the contrary, nitric oxide generation, both endogenous and exogenous, may accelerate neutrophil apoptosis (8, 29).
Neutropenia and/or neutrophil dysfunction is often associated with certain microbial agents, such as Staphylococcus aureus and Pseudomonas aeruginosa. In addition, neutropenia in the presence of infection is considered to be a poor prognostic marker.
Proapoptotic factors, such as cell surface expression of Fas and FasL, have been reported to play an important role in the initiation of the apoptotic process for the maintenance of cell population in both physiologic and pathologic states (10–12). Upregulation of monocyte FasL expression has been studied by using nonparticulate stimuli, such as soluble immune complexes, superantigen, phytohemagglutinin, and antibody-mediated cross-linking of monocytes (16, 26). Various investigators have reported that monocytes can induce apoptosis in bystander cells by modulating the expression of FasL and by the release of soluble FasL (10, 16, 21, 26). Cells expressing Fas receptors, including neutrophils, eosinophils, monocytes, and lymphocytes, may be susceptible to apoptosis induced by cross-linking with soluble FasL (3, 7, 16, 21, 26).
In the present study we evaluated the occurrence of neutrophil apoptosis in patients with infection and in uninfected subjects. In addition, we evaluated the molecular mechanism of infection-induced neutrophil apoptosis.
MATERIALS AND METHODS
Study subjects.
Fifteen patients with infections were selected from the general medical floor at the Long Island Jewish Medical Center, New Hyde Park, N.Y. All patients had an elevated white blood cell count and predominant neutrophilia secondary to bacterial infection. Active infection was characterized by the presence of fever in the preceding 24 h and microbiological or radiological evidence of infection. Eight of the patients were male and seven were female. The patients ranged in age from 30 to 90, with a mean age of 69.8 years. Twelve patients had either gram-negative (Escherichia coli, two patients; P. aeruginosa, two patients; Proteus mirabilis, one patient; Bacteroides fragilis, one patient) or gram-positive (S. aureus, two patients; Staphylococcus epidermidis, one patient; Staphylococcus haemolyticus, one patient; Streptococcus pneumoniae, two patients) infections, and three patients were culture negative. Seven of the patients had radiographic evidence of pneumonia, two had positive urine cultures, one patient had a wound infection, and five of the patients had positive blood cultures without a clear focus of infection. The measured white blood cell counts on the day of sample collection ranged from 13 × 109 to 34.2 × 109 cells/mm3 (a normal white blood cell count is 3.5 × 109 to 11 × 109 cells/mm3). Based on the fact that they required hospital admission to have antibiotics administered intravenously, all of the patients were considered to have moderate to severe infections. Controls (six subjects) were healthy volunteers from the institution's staff who were on no medications and were free of infection. The absence of infection was confirmed by following the controls for a period of 2 weeks, during which none of the controls developed a febrile illness and all had a white blood cell count within the normal range.
E. coli.
A clinical pathologic isolate of E. coli (BH5, a derivative of strain TN 675; Takeda Chemical Industries Ltd., Osaka, Japan) (9) was seeded in agar plates and incubated at 37°C overnight. One colony was picked up with a sterile pipette tip and added to a 50-ml tube containing medium (RPMI 1640 with 4.5 g of glucose, 1 mM sodium pyruvate, 2 g of NaHCO3/liter) and kept at 37°C for 6 h. Bacteria were killed by being incubated at 70°C for 60 min and then were opsonized. We evaluated the direct effect of bacteria on neutrophils as well as monocytes.
Neutrophil isolation.
Ten milliliters of peripheral blood was collected from each donor in heparinized tubes. Neutrophils were separated by a standard technique of density-gradient centrifugation over Ficoll-Hypaque (29).
Collection of sera.
Five milliliters of peripheral blood was collected at the time of admission (before initiation of antibiotic administration) from each patient, and sera were separated.
Apoptosis studies.
To evaluate the occurrence of apoptosis and necrosis in neutrophils, we used Hoechst (H)-33342 (Molecular Probes, Eugene, Oreg.) and propidium iodide (Sigma, St. Louis, Mo.). H-33342 stains the nuclei of live cells and identifies apoptotic cells by increased fluorescence, whereas propidium iodide stains the necrosed cells pink (30, 31). We observed a significantly greater rate of apoptosis in neutrophils incubated in media containing serum-free media or media with less than 10% serum; therefore, we included 10% serum in the incubation media.
To determine the rate of neutrophil apoptosis in infected patients and healthy subjects, equal numbers of neutrophils (105) harvested either from infected patients or healthy subjects were incubated in medium (RPMI 1640) containing 10% fetal calf serum (FCS) for 6, 12, and 24 h.
To determine the direct effect of bacteria on neutrophil apoptosis, equal numbers of neutrophils were incubated in media (RPMI plus 10% FCS) containing either variable concentrations of E. coli (serotype O127:B8) (105 to 5 × 105 bacteria/ml) or lipopolysaccharide (LPS) (a bacterial endotoxin, Sigma CN# L3129) (10, 50, and 100 ng/ml) for 6 and 24 h.
To determine the effect of infection-induced soluble factors on neutrophil apoptosis, we evaluated the effect of sera on apoptosis of neutrophils isolated from healthy subjects. Equal numbers of neutrophils were incubated in medium (RPMI) containing 10% sera of patients with either gram-negative, gram-positive, or culture-negative infections or containing sera of healthy subjects for 24 h.
To confirm the role of FasL in the infection-induced neutrophil apoptosis, we examined the effect of anti-FasL antibody-neutralized pooled infected-serum-induced neutrophil apoptosis. Equal numbers of neutrophils were incubated in media containing either 10% pooled healthy sera or 10% pooled anti-FasL antibody-neutralized infected sera (10% infected sera were preincubated with anti-FasL antibody, 1 μg/ml, for 60 min at 37°C) for 24 h.
At the end of the incubation period, cells were stained with H-33342 and propidium iodide as described previously (30, 31). The percentages of live, apoptotic, and necrosed cells were recorded in eight random fields by two observers unaware of the experimental conditions.
DNA isolation and gel electrophoresis.
DNA isolation and gel electrophoresis are a simple technique to confirm the occurrence of apoptosis (30, 31). It provides morphologic evidence of DNA fragmentation. In apoptosis, activation of an endogenous DNase (specific for internucleosomal DNA) may trigger degradation of DNA into multiple integers of 180 to 200 base pairs. To confirm the occurrence of neutrophil apoptosis in infected patients, equal numbers of neutrophils harvested from either infected patients or healthy subjects were lysed in DNA lysis buffer. DNA was extracted and run on a 1.8% agarose gel and electrophoresed.
Protein extraction and Western blotting.
Equal amounts of sera were collected from either infected patients or healthy subjects and were mixed with lysis buffer followed by protein assessment using a bicinchoninic acid kit (Pierce, Rockford, Ill.). The proteins (20 μg/lane) extracted from sera were separated on a 4 to 20% gradient polyacrylamide gel and probed for FasL by using rabbit polyclonal anti-FasL antibody. Two sets of experiments were performed.
To determine the FasL kinetics for infected patients, 5 ml of peripheral blood was collected on days 1 (before antibiotic administration), 2, 5, and 10 in two patients with gram-negative infection. Proteins were extracted from sera and separated on a 4 to 20% gradient polyacrylamide gel and probed for FasL using mouse monoclonal anti-FasL antibody (Pharmingen, San Diego, Calif.).
Northern blotting and identification of mRNA of FasL.
Since neutrophils are reported not to express FasL, the source of FasL in serum may be from monocytes or lymphocytes. We evaluated the effect of E. coli or S. aureus on monocyte expression of FasL. Equal numbers of monocytes harvested from healthy subjects were incubated in medium (Dulbecco's modified Eagle's medium plus 10% FCS) containing either buffer or E. coli or S. aureus (5 × 105 bacteria/ml) for 6 h. Total RNA was extracted. A cDNA probe specific for FasL was used for hybridization after [32P]dCTP labeling by a random-primed method. The membranes were stripped to remove the hybridized probe and were reprobed with a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe to ascertain that similar amounts of RNA were applied to the gel.
Evaluation of the role of FasL in induction of neutrophil apoptosis.
To evaluate the role of FasL in the induction of neutrophil apoptosis, we studied the effect of monocyte/macrophage secretory products of control (C57BL/6J, stock no. 000664; Jackson Laboratories, Bar Harbor, Maine) and FasL-deficient (B6Smn.C3H-FasLgld, stock no. 001021; the background of this strain is statistically 96.9% C57BL/6-like; Jackson Laboratories) mice on neutrophil apoptosis. Peritoneal macrophages were isolated from control mice and FasL-deficient mice as described previously (24). Macrophages were characterized by uniform labeling with mouse anti-CD14 antibody (Becton Dickinson, San Jose, Calif.).
To prepare macrophage secretory products (MS), equal numbers of MøC (macrophages derived from control mice) and MøFLKO (macrophages derived from FasL-deficient mice) were incubated with either media alone, aggregated immunoglobulin G (IgG) (100 μg/ml), or E. coli (5 × 105 bacteria/ml) for 2 h at 37°C. At the end of the incubation period, supernatants (MøC-MS; MøFLKO-FLKO-MS; MøC plus IgG-IgG-MS; MøFLKO plus IgG-FLKO-IgG-MS; MøC plus E. coli; E. coli-MS; MøFLKO plus E. coli-FLKO-E. coli-MS) were collected, passed through a 0.2-μm-pore-size filter, and stored at −70°C. Then we examined the effect of the macrophage supernatants derived from control and FasL-deficient mice on apoptosis of neutrophils isolated from control mice as well as healthy subjects.
Statistical analysis.
For comparison of mean values between groups, the unpaired t test was used. To compare values between multiple groups, analysis of variance was applied and a Newman-Keuls multiple range test was used to calculate a q value. All values are means ± standard errors of the means (SEM) except where otherwise indicated. Statistical significance was defined as P < 0.05.
RESULTS
The fluorescence microscopic evaluation of apoptosis was reproducible. The occurrence of apoptosis was further confirmed by a DNA fragmentation assay. At 0 h, the neutrophils from neither infected patients nor healthy subjects showed morphologic evidence of apoptosis. However, as neutrophils aged in vitro (Fig. 1 and 2), the number of apoptotic neutrophils increased for infected patients as well as for controls (control: 6 h, 0.2% ± 0.2%; 12 h, 0.5% ± 0.2%; 24 h, 1.3% ± 0.7%; 48 h, 3.6% ± 1.2%; patients: 6 h, 4.9% ± 1.1%; 12 h, 16.3% ± 3.3%; 24 h, 44.2% ± 5.2%; 48 h, 59.6% ± 11.9% apoptotic cells/field). Nevertheless, the rate of apoptosis was many times higher for infected patients than for controls for the same time points (Fig. 2). Neutrophils harvested from infected patients showed DNA fragmentation into multiple integers of 180 bp (data not shown).
FIG. 1.

Morphologic evaluation of neutrophil apoptosis. Neutrophils were isolated from healthy subjects and infected patients and were incubated in medium containing 10% pooled human sera for 24 h. At the end of the incubation period, cells were stained with H-33342 and propidium iodide. (A) Neutrophils isolated from healthy subjects; (B) neutrophils isolated from infected subjects. Apoptotic neutrophils show bright fluorescence, whereas necrosed cells are stained pink.
FIG. 2.

Time course effect on aging neutrophils in vitro. Neutrophils were isolated from healthy subjects (open triangles) and infected patients (open squares) and were incubated in medium containing 10% FCS serum for 6, 12, 24, and 48 h. At the end of the incubation period, cells were stained with H-33342 and propidium iodide. Results (means ± SEM) are from six series of experiments, each carried out in triplicate.
To determine the direct effect of bacteria on neutrophil apoptosis, we studied the effect of E. coli as well as LPS (bacterial endotoxin) on neutrophils. Neither E. coli nor LPS modulated apoptosis in neutrophils (control, 1.3% ± 0.7%; E. coli, 0.2% ± 0.1%; LPS, 0.4% ± 0.1% neutrophils/field after 24 h).
To evaluate whether it is a soluble factor in the sera of infected patients which is playing a role in the induction of neutrophil apoptosis, we evaluated the effect of sera of infected patients and healthy subjects on neutrophil apoptosis. As shown in Fig. 3, sera of patients with gram-negative infections accelerated neutrophil apoptosis compared to sera of healthy controls as well as FCS. These results suggest that sera have a factor(s) which promotes neutrophil apoptosis. To determine whether intensity of this factor differs in patients with gram-negative, gram-positive, or culture-negative infections, we evaluated the sera of these patients on the basis of neutrophil apoptosis. As shown in Fig. 4, sera from infected patients accelerated neutrophil apoptosis compared to control sera. However, the rate of neutrophil apoptosis was greater for neutrophils treated with gram-negative, culture-negative, or pooled sera than that for gram-positive sera. Neutrophils treated with gram-negative and gram-positive sera showed DNA fragmentation in the form of a ladder pattern, further confirming the presence of an apoptotic factor in the sera of infected patients (data not shown).
FIG. 3.

Effect of sera on neutrophil apoptosis. Neutrophils isolated from healthy subjects were incubated in media containing either 10% FCS (FCS-control), 10% HS (sera from healthy subjects) (HS control) or 10% gram-negative HS (sera from gram-negative-infected patients) for 6, 12, and 24 h. At the end of the incubation period, cells were stained with H-33342 and propidium iodide. Results (means ± SEM) are from six series of experiments, each carried out in triplicate.
FIG. 4.

Effect of gram-negative and gram-positive sera on neutrophil apoptosis. Neutrophils isolated from healthy subjects were incubated in media containing either 10% HS (sera from healthy subjects, control), 10% gram-negative sera (sera from gram-negative infected patients), 10% gram-positive sera (sera from gram-positive infected patients), 10% culture-negative sera (sera from infected culture-negative patients), or 10% pooled sera (infected patients) for 6, 12, and 24 h. At the end of the incubation period, cells were stained with H-33342 and propidium iodide. Results (means ± SEM) are from three series of experiments, each carried out in triplicate. ∗, P < 0.001 compared with the respective control; ∗∗, P < 0.01 compared with the respective control; ∗∗∗, P < 0.001 compared with the respective control.
Since monocyte upregulation of FasL has been found to promote bystander lymphocyte apoptosis (24), we evaluated the effect of E. coli on monocyte and neutrophil expression of FasL. Neutrophils did not express FasL under basal or E. coli-treated states (data not shown), whereas E. coli treatment increased monocyte expression of FasL (Fig. 5). Similarly, monocytes treated with S. aureus enhanced expression of FasL (data not shown). In addition, the sera of infected patients showed an abundance of FasL protein compared to the sera of healthy subjects (Fig. 6).
FIG. 5.

Effect of E. coli on monocyte FasL expression. The upper lane shows monocyte mRNA expression of FasL in control and E. coli-treated conditions. The lower lane shows monocyte mRNA expression of GAPDH under identical conditions.
FIG. 6.
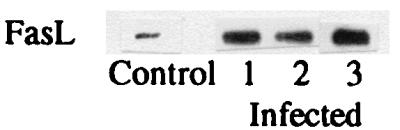
Western blots showing FasL content in serum for control and infected patients. Sera of infected patients showed a fivefold increase of FasL compared with control sera.
To determine the role of FasL in the induction of neutrophil apoptosis, we evaluated the effect of activated macrophages (as a source of FasL) derived from normal mice and FasL-deficient mice on murine and human neutrophil apoptosis. As shown in Fig. 7, FLKO-IgG-MS induced apoptosis in a lower percentage of murine neutrophils than IgG-MS. Similarly, E. coli-MS triggered apoptosis in a greater percentage of murine neutrophils than FLKO-E. coli-MS. This effect of macrophage supernatants was dose dependent, indicating the presence of an apoptotic factor. The effect of macrophage supernatants on human neutrophils is shown in Fig. 8. IgG-MS induced apoptosis in a greater percentage of human neutrophils than FLKO-IgG-MS.
FIG. 7.

Effect of activated peritoneal macrophage (derived from either normal mice or FasL-deficient mice) supernatants on murine neutrophil apoptosis. Results (means ± SEM) are from four sets of experiments, each carried out in triplicate. FLKO-IgG-MS induced apoptosis in a lower percentage of murine neutrophils than IgG-MS. Similarly, E. coli-MS triggered apoptosis in a greater percentage of murine neutrophils than FLKO-E. coli-MS. This effect of macrophage supernatants was dose dependent, indicating the presence of an apoptotic factor. ∗, P < 0.01 compared with the respective control; ∗∗, P < 0.001 compared with the respective control; ∗∗∗, P < 0.001 compared with IgG-MS; ∗∗∗∗, P < 0.001 compared with E. coli-MS.
FIG. 8.

The effect of activated peritoneal macrophage (derived from either normal mice or FasL-deficient mice) supernatants (10, 30, and 50%) on human neutrophils. Results (means ± SEM) are from four sets of experiments, each carried out in triplicate. IgG-MS induced apoptosis in a greater percentage of human neutrophils than FLKO-IgG-MS. ∗, P < 0.001 compared with respective MS, FLKO-MS, and FLKO-IgG-MS.
To confirm the role of FasL, we evaluated the effect of anti-FasL antibody-neutralized infected sera on neutrophil apoptosis. Neutralized infected sera showed only a mild effect on neutrophil apoptosis compared with infected sera. Anti-FasL antibody attenuated the effect of infected sera on apoptosis in neutrophils (control, 3.9% ± 0.4%; infected sera, 16.2% ± 0.8%; neutralized infected sera, 6.1% ± 0.5% apoptotic neutrophils/field; for infected sera, P < 0.001 compared with control and neutralized sera).
To determine the kinetics of FasL in infected patients, FasL content in serum was measured in two infected patients on days 1, 2, 5, and 10. As shown in Fig. 9, FasL content in serum decreased with time as well as with cure of infection.
FIG. 9.

Serum FasL kinetics in two patients with gram-negative infection. Sera were collected from two patients on days 1, 2, 5, and 10 and FasL contents were measured. Results (means ± SEM) are from two patients. A representative Western blot showing FasL content in serum at different time periods is shown at the top.
DISCUSSION
The present study demonstrates that neutrophils have an accelerated rate of apoptosis in patients with infection. E. coli did not promote neutrophil apoptosis directly; nevertheless, E. coli and S. aureus enhanced monocyte expression of FasL. Moreover, sera (irrespective of coming from gram-positive or gram-negative patients) promoted neutrophil apoptosis, indicating the presence of a circulating factor in sera. Interestingly, anti-FasL antibody attenuated infected-serum-induced neutrophil apoptosis. Infected patients also showed an increased content in serum of FasL. Conditioned media from activated macrophages derived from FasL-deficient mice induced less apoptosis in human and murine neutrophils than conditioned media of control macrophages. These results suggest that infection enhances neutrophil apoptosis through FasL which may have been released in the sera from microorganism-activated monocytes.
Neutrophils are the most abundant of all leukocytes. The life of a neutrophil is spent in three environments: marrow, blood, and tissues. Proliferation and maturation take place in the marrow over a period of 13 to 15 days (32), after which mature neutrophils enter the blood stream. This period can be shortened during times of stress. Under steady-state conditions, death and clearance in the tissues balance daily production of neutrophils. Neutrophils are programmed to die by apoptosis at the time of differentiation. Although inevitable, the exact timing of neutrophil death is subject to regulation by external factors (4).
Host defense in response to bacterial invasion utilizes both the innate and acquired immune response. Neutrophils appear to be the important mediators of innate immunity. This is exemplified by the occurrence of recurrent infections in patients with neutropenia (38). Moreover, the susceptibility of humans with neutrophil dysfunction to a wide array of microbial infections would also appear to be good evidence of the importance of neutrophils in microbial clearance in the early stage of infection. With this background, it appears that increased apoptosis of the neutrophils during infection may be an important step in the transition of the host response from its initial stage (which relies predominantly on the components of the innate response, such as neutrophils) to its intermediate and later stages (which rely more heavily on the components of acquired immunity, such as monocytes and antibodies). The present study suggests that neutrophilia may not always mean increased numbers of functioning neutrophils. Moreover, the promotion of apoptosis by infection may transfer host dependency from an innate response to an acquired immunity.
In vivo, apoptotic neutrophils are rapidly cleared by neighboring phagocytes (3). This mechanism of cell death and clearance of neutrophils has been postulated to represent an injury-limiting process (3). Death by necrosis would result in widespread release of neutrophil cytoplasmic enzymes and subsequent extensive tissue destruction. Apoptosis is a highly regulated and genetically directed process. It is induced by intracellular cues, such as DNA damage or osmotic stress, and extracellular cues, including growth factor withdrawal, matrix detachment, and direct cytokine-mediated killing (15, 17, 21, 22). Two main pathways, caspase proteases or the mitochondrial pathway, are involved in apoptotic cell death (15, 17). However, at every level the action of proapoptotic molecules is opposed by a set of inhibitors. A number of signals such as interaction of TNF-FasL with its respective cognate receptor, TNF-R–Fas, induce trimerization of the receptors (TNF-R1, Fas, DR3, DR4, DR5, and DR6 all contain an intracellular “death domain”), recruiting adapter proteins such as FADD to the death domain (17, 25). These adapter molecules then recruit and activate caspase 8. In the mitochondrial pathway, in response to apoptotic signals, proapoptotic bcl-2 family members translocate to and alter the permeability of the mitochondrial membrane, cytochrome c release, and the production of reactive oxygen species. Antiapoptotic members of the bcl-2 family, such as bcl-2, reside in the mitochondrial membrane and may counter these effects. Both caspase and mitochondrial pathways are interconnected; i.e., caspase 8 cleaves Bid (of the bcl-2 family) to produce tBid; in turn, tBid in cooperation with Bad (of the Bcl-2 family) can trigger cytochrome c release, inducing the caspase adapter Apaf-1 to activate caspases 9 and 3. Finally, activation of caspase 3 either through cleavage of caspase 8 or through the mitochondrial pathway causes degradation of proteins. In the present study, Fas (APO-1; CD95)-FasL interaction has triggered apoptosis of neutrophils. We believe that the downstream signaling of infection-induced neutrophil apoptosis involves recruitment of the adapter protein, FADD, followed by caspase 8 and caspase 3 activation. Since phagocytization of microorganisms by neutrophils is associated with the generation of reactive oxygen species, the mitochondrial pathway also seems to be operative. Whether Fas-FasL interaction and generation of reactive oxygen species are interrelated and/or have additive or synergistic effects on neutrophil apoptosis requires future studies. We plan to probe infection-induced neutrophil downstream signaling in future studies.
Human immunodeficiency virus type 1 gp120 envelope protein has been shown to increase FasL expression in monocytes and to enhance the expression of FasL gene transcription via the FasL gene enhancer-promoter region upregulation (26). Upregulation of FasL mRNA and the associated increase in apoptosis of Fas-susceptible targets have been reported previously (2). Interestingly, macrophage-associated FasL has been shown to trigger selective apoptosis of Fas-susceptible CD4 but not CD8 T cells from human immunodeficiency virus-positive patients (1).
Recently, Brown and Savill demonstrated that exposure of monocytes/macrophages to opsonized zymosan induced the release of soluble FasL (3). The conditioned supernatants containing FasL led to Fas-mediated apoptosis of bystander monocytes and FasL-negative neutrophils. Although macrophages phagocytizing latex beads produced soluble FasL, it did not show proapoptotic effects on neutrophils (3). For the occurrence of bystander cell apoptosis, these investigators hypothesized a requirement of additional soluble factors besides soluble FasL (3).
Recently it was reported that E. coli directly triggers macrophage apoptosis (33). To determine the molecular mechanism of E. coli-induced macrophage apoptosis, in the present study we evaluated the effect of E. coli on monocyte mRNA expression of FasL. Interestingly, E. coli promoted monocyte mRNA expression of FasL. Since activated monocytes have been demonstrated to express Fas, we propose that E. coli-induced monocyte FasL expression and subsequent Fas-FasL cross-linking may trigger autocrine cell death. Neutrophils have also been demonstrated to express Fas. Interaction between infection-induced increase of FasL (released from circulating monocytes) in serum and neutrophil Fas receptors may accelerate neutrophil apoptosis. In the present study, bacteria did not promote apoptosis of neutrophils; however, sera of infected patients enhanced neutrophil apoptosis, indicating the presence of a soluble factor. That factor seems to be FasL.
We conclude that patients with infection show accelerated neutrophil apotosis. This effect of infection on neutrophils seems to be mediated through FasL. We propose that microorganisms may be compromising the innate response and may thus make the host more dependent on acquired immunity. Since microorganisms are capable of promoting apoptosis in neutrophils and monocytes, a transfer of the host defense burden from innate to acquired immunity may be a forced rather than a sound strategy on the part of the host defense system and thus may not always be associated with a positive outcome. The present study provides a plausible explanation for the lack of correlation between neutrophilia and the outcome of infection.
ACKNOWLEDGMENT
This work was supported by grant R01 DA12111 from the National Institutes of Health.
REFERENCES
- 1.Badley A D, Dockrell D, Simpson M, Schut R, Lynch D H, Leibson P, Paya C V. Macrophage-dependent apoptosis of CD4+ T lymphocytes from HIV-infected individuals is mediated by FasL and tumor necrosis factor. J Exp Med. 1997;185:55–64. doi: 10.1084/jem.185.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badley A D, McElhinny J A, Leibson P J, Lynch D H, Alderson M R, Paya C V. Upregulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J Virol. 1996;70:199–206. doi: 10.1128/jvi.70.1.199-206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown S B, Savill J. Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander neutrophils. J Immunol. 1999;162:480–485. [PubMed] [Google Scholar]
- 4.Colotta F, Polentarutti N, Sozzani S, Montovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012–2020. [PubMed] [Google Scholar]
- 5.Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- 6.Dalhoff K, Hansen F, Dromann D, Schaaf B, Aries S P, Braun J. Inhibition of neutrophil apoptosis and modulation of the inflammatory response by granulocyte colony-stimulating factor in healthy and ethanol- treated human volunteers. J Infect Dis. 1998;178:891–895. doi: 10.1086/515350. [DOI] [PubMed] [Google Scholar]
- 7.Druilhe A, Cai Z, Haile S, Chouaib S, Pretolani M. Fas-mediated apoptosis in cultured human eosinophils. Blood. 1996;87:2822–2830. [PubMed] [Google Scholar]
- 8.Foretenberry J D, Owens M L, Brown M R, Atkinson D, Brown L A. Exogenous nitric oxide enhances neutrophil cell death and DNA fragmentation. Am J Respir Cell Mol Biol. 1998;8:421–428. doi: 10.1165/ajrcmb.18.3.2875. [DOI] [PubMed] [Google Scholar]
- 9.Iwahi T Y A, Nakao M, Imada A, Tsuchiya K. Role of type 1 fimbriae in the pathogenesis of ascending urinary tract infection induced by Escherichia coli in mice. Infect Immun. 1983;39:1307–1315. doi: 10.1128/iai.39.3.1307-1315.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwai K, Miyawaki T, Takizawa T, Konno A, Ohta K, Yachie A, Seki H, Taniguchi N. Differential expression of bcl-2 and susceptibility to anti-Fas-mediated cell death in peripheral blood lymphocytes, monocytes and neutrophils. Blood. 1994;84:1201–1208. [PubMed] [Google Scholar]
- 11.Kiener P A, Davis P M, Starling G C, Mehlin C, Klebanoff S J, Ledbetter J A, Liles W C. Differential induction of apoptosis by Fas-Fas ligand interaction in human monocytes and macrophages. J Exp Med. 1997;185:1511–1516. doi: 10.1084/jem.185.8.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiener P A, Davis P M, Rankin B M, Klebanoff S J, Starling G C, Liles W C. Human monocytic cells contain high levels of intracellular Fas ligand: rapid release following cellular activation. J Immunol. 1997;159:1594–1598. [PubMed] [Google Scholar]
- 13.Klebanoff S J, Clark R A. The neutrophil: function and clinical disorders. Amsterdam, The Netherlands: Elsevier; 1978. [Google Scholar]
- 14.Klebanoff S J, Vadas M A, Harlan J M, Sparks L H, Gamble J R, Agosti J M, Waltersdorph A M. Stimulation of neutrophils by tumor necrosis factor. J Immunol. 1986;136:4220–4225. [PubMed] [Google Scholar]
- 15.Kroemer G, Reed J C. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 16.Liles W C, Kiener P A, Ledbetter J A, Aruffo A, Klebanoff S J. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med. 1996;184:429–440. doi: 10.1084/jem.184.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lincz L F. Deciphering the apoptosis pathway: all roads lead to death. Immun Cell Biol. 1998;76:1–19. doi: 10.1046/j.1440-1711.1998.00712.x. [DOI] [PubMed] [Google Scholar]
- 18.Majno G, Joris J. Apoptosis, oncosis, and necrosis: an overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 19.Marasco W A, Phan S A, Kruzsch H. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J Biol Chem. 1984;259:5430–5439. [PubMed] [Google Scholar]
- 20.Martin S J, Green D R, Cotter T G. Dicing with death: dissecting the components of apoptosis machinery. Trends Biol Sci. 1994;19:26–30. doi: 10.1016/0968-0004(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto K, Schiemer R P, Saito H, Likura Y, Bochner B S. Induction of apoptosis in human eosinophils by anti-Fas antibody treatment in vitro. Blood. 1995;86:1437–1443. [PubMed] [Google Scholar]
- 22.Muzyka B C. Host factors affecting disease transmission. Dent Clin N Am. 1996;40:263–275. [PubMed] [Google Scholar]
- 23.Newman S L, Henson J E, Henson P M. Phagocytosis of senescent neutrophils by human monocyte-derived macrophages and rabbit inflammatory macrophages. J Exp Med. 1982;156:430–442. doi: 10.1084/jem.156.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oyaizu N, Adachi Y, Hashimoto F, McClosky T W, Hosaka N, Kayagaki N, Yagita N, Pahawa S. Monocytes express Fas ligand upon CD4 cross-linking and induce CD4+ T cells apoptosis: a possible mechanism of bystander cell death in HIV infection. J Immunol. 1997;158:2456–2463. [PubMed] [Google Scholar]
- 25.Pan G. An antagonist decoy receptor and death domain-containing receptor for TRAIL. Science. 1997;277:815–818. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 26.Richardson B C, Lalwani N D, Johnson K J, Marks R M. Fas ligation triggers apoptosis in macrophages but not in endothelial cells. Eur J Immunol. 1994;24:2640–2646. doi: 10.1002/eji.1830241111. [DOI] [PubMed] [Google Scholar]
- 27.Savill J S, Wyllie A H, Henson J E, Walport M J, Henson P M, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. J Clin Investig. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savill J S, Henson P M, Haslett C. Phagocytosis of aged neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Investig. 1989;84:1518–1527. doi: 10.1172/JCI114328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singhal P C, Patel P, Nahar N, Reddy K, Kapasi A, Nwakoby I E, Franki N. Ethanol-induced neutrophil apoptosis is mediated through nitric oxide. J Leukoc Biol. 1999;66:930–936. doi: 10.1002/jlb.66.6.930. [DOI] [PubMed] [Google Scholar]
- 30.Singhal P C, Reddy K, Ding G, Kapasi A, Franki N, Ranjan R, Nwakoby I E, Gibbons N. Ethanol-induced macrophage apoptosis: the role of TGF-ß. J Immunol. 1999;162:3031–3036. [PubMed] [Google Scholar]
- 31.Singhal P C, Sharma P, Kapasi A A, Reddy K, Franki N, Gibbons N. Morphine enhances macrophage apoptosis. J Immunol. 1998;160:1886–1893. [PubMed] [Google Scholar]
- 32.Squier M K T, Sehert A J, Cohen J J. Apoptosis in leukocytes. J Leukoc Biol. 1995;57:2–10. doi: 10.1002/jlb.57.1.2. [DOI] [PubMed] [Google Scholar]
- 33.Stravodimos K G, Singhal P C, Sharma S, Reddy K, Smith A D. Escherichia coli promotes macrophage apoptosis. J Endourol. 1999;13:273–277. doi: 10.1089/end.1999.13.273. [DOI] [PubMed] [Google Scholar]
- 34.Wade B H, Mandell G L. Polymorphonuclear leukocytes: dedicated professional phagocytes. Am J Med. 1983;74:686–93. doi: 10.1016/0002-9343(83)91028-8. [DOI] [PubMed] [Google Scholar]
- 35.Whyte M K B, Meagher L C, MacDermot J, Haslett C. Impairment of function in aging neutrophils is associated with apoptosis. J Immunol. 1993;150:5124–5134. [PubMed] [Google Scholar]
- 36.Williams G T, Smith C A, Spooner E, Dexter T M, Taylor D R. Hematopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature. 1990;343:76–79. doi: 10.1038/343076a0. [DOI] [PubMed] [Google Scholar]
- 37.Williams W J, Beutler E, Ersler A J, Lichtman M J. Hematology. 4th ed. New York, N.Y: McGraw-Hill; 1990. [Google Scholar]
- 38.Wright D G. The neutrophil as a secretory organ of host defense. In: Gallin J I, Fauci A S, editors. Advances in host defense mechanisms. New York, N.Y: Raven Press; 1982. p. 212. [Google Scholar]