

Keywords: excretion, homeostasis, metal, transporter, ZnT10
Abstract
The activity of the manganese (Mn) efflux transporter SLC30A10 in the liver and intestines is critical for Mn excretion and preventing Mn toxicity. Homozygous loss-of-function mutations in SLC30A10 are a well-established cause of hereditary Mn toxicity. But, the relationship between more common SLC30A10 polymorphisms, Mn homeostasis, and disease is only recently emerging. In 2021, the first coding SNP in SLC30A10 (T95I) was associated with liver disease raising the hypothesis that the T95I substitution may induce disease by inhibiting the Mn efflux function of SLC30A10. Here, we test this hypothesis using structural, viability, and metal quantification approaches. Analyses of a predicted structure of SLC30A10 revealed that the side chain of T95 pointed away from the putative Mn-binding cavity, raising doubts about the impact of the T95I substitution on SLC30A10 function. In HeLa or HepG2 cells, overexpression of SLC30A10-WT or T95I resulted in comparable reductions of intracellular Mn levels and protection against Mn-induced cell death. Furthermore, ΔSLC30A10 HepG2 cells, generated using CRISPR/Cas9, exhibited elevated Mn levels and heightened sensitivity to Mn-induced cell death, and these phenotypic changes were similarly rescued by expression of SLC30A10-WT or T95I. Finally, turnover rates of SLC30A10-WT or T95I were also comparable. In summary, our results indicate that the Mn transport activity of SLC30A10-T95I is essentially comparable to the WT protein. Our findings imply that SLC30A10-T95I either has a complex association with liver injury that extends beyond the simple reduction in SLC30A10 activity or alternatively the T95I mutation lacks a causal role in liver disease.
NEW & NOTEWORTHY This study demonstrates that the T95I polymorphism in the manganese transporter SLC30A10, which has been associated with liver disease in human GWAS studies, does not impact transporter function in cell culture. These findings raise doubts about the causal relationship of the T95I polymorphism with human disease and highlight the importance of validating GWAS findings using mechanistic approaches.
INTRODUCTION
Manganese (Mn) is an essential metal, but at elevated levels, Mn accumulates in the brain and induces severe neurotoxicity (1–5). Mn toxicity presents itself as Parkinsonism in adults, and neurobehavioral and fine motor deficits in children (6–11). Currently, Mn toxicity remains an important public health issue due to the risk of Mn overexposure in occupational settings (welding and steel manufacture) and environmental exposure through drinking water and air (6–11). Furthermore, excretion of Mn by the liver and intestines is essential for regulating blood and brain Mn levels, and even in the absence of Mn overexposure, patients with chronic liver disease may retain excessive levels of Mn in the body and develop Mn toxicity (12–15). Overall, the extent of Mn exposure from the environment, and biological factors affecting Mn excretion are both important determinants of the risk for Mn toxicity.
Over the last decade, the discovery of hereditary forms of Mn toxicity among carriers of homozygous loss-of-function mutations in SLC30A10 or SLC39A14 has further emphasized the role of Mn excretion in maintaining Mn homeostasis (16–18). Patients harboring these mutations exhibit elevations in brain and blood Mn levels, and Mn-associated Parkinsonism in the absence of documented Mn overexposure (16–18). We reported that SLC30A10 is a Mn-specific efflux transporter that localizes to the cell surface and reduces cellular Mn levels (19–24). At the organism level, activity of SLC30A10 in the liver and intestines mediates Mn excretion, reduces whole body Mn levels, and prevents the accumulation of Mn in the blood and brain (21) [note that activity of SLC30A10 in the brain also plays a role in directly reducing brain Mn levels and providing neuroprotection (21)]. Other studies revealed that the in vivo function of SLC39A14 is to import Mn from blood into the liver and intestines for subsequent excretion via SLC30A10 (18, 19, 25–27). Thus, SLC30A10 and SLC39A14 cooperatively mediate Mn excretion, and loss-of-function of either transporter induces Mn toxicity by inhibiting Mn excretion, which results in the retention of Mn in the blood and brain (18–28) [in the case of loss-of-function of SLC30A10, inhibition of Mn efflux in the brain also contributes to the disease (21)]. Furthermore, since SLC30A10 exports Mn from the liver into bile and SLC39A14 imports blood Mn into the liver, loss-of-function of SLC30A10, but not SLC39A14, is also characterized by the accumulation of Mn in the liver and subsequent liver disease (16–18, 24).
More recently, SNPs in SLC30A10 have been associated with modest changes in human blood Mn levels and neurobehavioral or hepatic symptoms in candidate gene and GWAS studies (29–33). But the causal relationship between these SNPs and SLC30A10 function has yet to be examined. Among the SLC30A10 SNPs associated with human neurological function or hepatic disease, only one is in the SLC30A10 coding region and results in a T95I substitution (33). The potential role of the SLC30A10-T95I SNP in disease emerged from a GWAS study in 2021, which identified SLC30A10-T95I as the locus associated with an increase of the liver injury biomarkers alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (33). In addition, T95I was associated with other aspects of hepatic damage (e.g., liver MRI cT1, decreased albumin, increased apolipoprotein A), some symptoms of inherited Mn toxicity (e.g., increased hematocrit), and bile duct cancer and other biliary disorders (e.g., cholangitis and cholecystitis) (33). Notably, neurological symptoms of Mn toxicity were not associated with the T95I variant carriers in the initial study (33) but were subsequently associated in a second study (34). The phenotypes associated with SLC30A10-T95I SNP and the location of the missense residue in the primary sequence of SLC30A10 raised the hypothesis that the T95I substitution may induce disease by inhibiting the Mn efflux activity of SLC30A10 [note that the impact of T95I on Mn levels was not evaluated in the 2021 study (33)]. Direct testing of this hypothesis is critical because of the novelty of the T95I SNP as the first coding SNP in SLC30A10 associated with human disease. Here, we use structural, viability, and metal measurement approaches to assay for the impact of the T95I mutation on SLC30A10 function. Our results show that, in contrast with expectations from the human studies, the Mn efflux activity of SLC30A10-T95I is comparable to the WT protein, raising questions about the role of this mutation in disease.
MATERIALS AND METHODS
Cell Culture, Transient Transfections, and Lentiviral Infections for Overexpression of SLC30A10-WT or T95I
HeLa and HepG2 cells were grown in minimum essential medium (MEM) supplemented with 10% fetal bovine serum, 100 IU/mL penicillin-G, and 100 µg/mL streptomycin as described by us previously (20, 35). Transient transfections were performed using JET-PEI reagent (VWR) as described by us previously (20). Lentivirus production in HEK293T cells and lentiviral infections of HeLa cells were exactly as described by us previously (36). Packaging and envelope plasmids, but no transfer plasmid, were used for mock infections. For lentiviral infections of HepG2 cells, the only difference between the protocols used for HeLa cells (36) was that ∼300,000 HepG2 cells were plated in 10 cm dishes the day before the collection of lentiviral media, and the lentivirus-polybrene mixture was then filtered and dispensed to the 10 cm dishes the next day. Forty-eight hours after infection, 2 µg/mL puromycin was added to the media to select for infected cells. Single-cell clones were subsequently selected by limiting dilution in the continued presence of 2 µg/mL puromycin in the media.
Generation and Validation of ΔSLC30A10 HepG2 Cells
ΔSLC30A10 HepG2 cells were generated by targeting exon 1 of SLC30A10 in HepG2 cells using a lentiviral CRISPR/Cas9 expression system that we have previously described (36). The lentiviral CRISPR/Cas9 transfer plasmid pL-CRISPR.EFS.PAC was from Addgene (plasmid No. 57828; http://n2t.net/addgene:57828; RRID: Addgene_57828). The guide RNA sequence was 5′-GGUCAUCGAUGCGCUCGGGCCGG-3′. Lentivirus generation, infection, and clonal cell selection were conducted as described above for overexpression of SLC30A10 variants in HepG2 cells. Knockout of SLC30A10 was confirmed essentially as described by us previously (36). Briefly, from each cell clone, the SLC30A10 exon 1 genomic DNA was amplified using PCR (forward: 5′- TGACAAGCTTGCTACTCTGGCAAGACGTGC-3′ and reverse: 5′- ATACGGATCCGCTTC CTTTCCACCGAGGTC-3′) and subcloned into the HindIII and BamHI restriction sites of pEGFP-N3 vector. At least eight bacterial clones per cell clone were sequenced. One of the cell clones provided only two independent genomic DNA sequences with mutations that introduced a stop codon in SLC30A10 exon 1. This was indicative of two independent mutations in the two chromosomes. We further validated this clone using RT-PCR as described below to assay for loss of SLC30A10 mRNA expression.
RT-PCR, qRT-PCR, Antibodies, and Immunoblots
Protocols for RT-PCR, qRT-PCR and immunoblot analyses, and antibodies against human SLC30A10 [rabbit; custom generated by us (19)], FLAG (mouse; Sigma no. F3165) and tubulin (mouse; Sigma no. T5168) were described by us previously (19, 20, 35). To summarize salient features here, for immunoblots, cells were lysed with RIPA lysis buffer [50 mM Tris·HCl, pH 7.4; 0.25% sodium deoxycholate; 1% Triton; 0.1% SDS; 150 mM NaCl; 1 mM EDTA; protease inhibitor cocktail (Thermo A32953)]. Immunoblotting was performed as described by us previously (20). Blots were quantified using ImageJ Fiji (37). After Process-Subtract Background Function was used, band intensity was measured by outlining with the Rectangle tool and using the Analyze-Measure tool. Primers to validate loss of SLC30A10 mRNA in ΔSLC30A10 HepG2 cells using RT-PCR were forward: 5′-CATCGATGACCCCGAGCTG-3′ and reverse: 5′-CATGTCTTCTGGCTCATTCTGG-3′. For qRT-PCR, SLC30A10 transcript levels were quantified using the ΔΔCT method with TATA-binding protein (TBP) as the internal control. Primers used for qRT-PCR for SLC30A10 and qRT-PCR/RT-PCR for TBP were described by us previously (20, 38). CFX96 Touch Real-Time PCR Detection System (Bio-Rad) was used.
Constructs
FLAG-tagged SLC30A10-WT plasmid used for transient transfections as well as the FLAG-SLC30A10 transfer plasmid used for lentiviral infections were described by us previously (20, 39). T95I point mutation was introduced in the FLAG-SLC30A10 transfer plasmid using QuikChange site-directed mutagenesis kit (Agilent Technologies). The lentivirus envelope and packaging plasmids were described previously (36), and the CRISPR/Cas9 plasmid is described above.
SLC30A10 Predicted Structure
The predicted structure of SLC30A10 was generated by the algorithm of AlphaFold Protein Structure Database (40, 41). T95I mutation was introduced in the predicted structure and protein structure images were generated using PyMOL (The PyMOL Molecular Graphics System, Version 2.3.4 Schrödinger, LLC).
Viability Assays in HeLa or HepG2 Cells
Cell viability was assessed using methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay essentially as described by us previously (20). Cells were plated in 24-well plates (25,000 cells/well) overnight. The next day, cells were exposed to the indicated concentrations of Mn for 16 h, and viability was assayed using the MTT assay as described by us previously (20, 23, 36, 39).
Intracellular Metal Measurements in HeLa and HepG2 Cells
These were performed essentially as described by us previously (20) with some minor modifications. Cells were plated in 35-mm dishes (300,000 cells/well) overnight. The next day, cells were exposed to 125 µM (HepG2) or 250 µM (HeLa) Mn for 16 h. After Mn treatment, cells were washed with phosphate-buffered saline (PBS) and collected after 5 min incubation in trypsin-EDTA at 37°C. Then, trypsin was neutralized with serum-containing MEM, and cells were collected by centrifuging at 2,500 rpm for 2 min. After the supernatant was discarded, the cell pellets were washed twice with PBS containing 10 mM EDTA, and the cells were collected after each wash by centrifuging at 1,500 rpm for 3 min at 4°C. After another 1-mL PBS wash, 50 µL of the sample was used for counting cells, and the remainder was collected by centrifuging at 5,000 rpm for 5 min. The cell pellets were resuspended in 20-µL PBS, and digested in acid-washed glass vials containing 200 µL of 70% metal-free nitric acid. For digestion, cells were incubated at 85°C for 1 day, followed by incubation at room temperature for another day. Digested samples were diluted to 2% nitric acid with UltraPure distilled water (Invitrogen). Intracellular Mn, Zn, Fe, and Cu amounts were measured using an Agilent 7500ce quadrupole inductively coupled plasma mass spectrometer.
MG132 Treatment of HepG2 Cells
Proteasome inhibitor MG132 (Sigma) was dissolved in DMSO and used at an effective concentration of 0.5 µM.
Statistical Analyses
Prism 9 software (GraphPad Software, San Diego, CA) was used. All experiments were independently repeated three or more times. P < 0.05 was considered to be statistically significant and represented with asterisks in the figures.
RESULTS
Analysis of Predicted Structure of SLC30A10 Raises Doubts about the Role of T95I in Mn Efflux
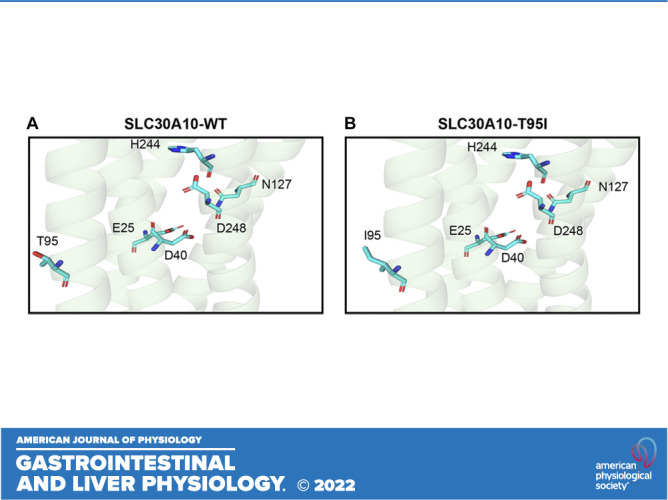
We previously performed structure-function analyses of SLC30A10 using a predicted SLC30A10 structure generated by PHYRE 2.0 (23, 39, 42). In the current study, we improved the structural prediction using the algorithm of AlphaFold Protein Structure Database (40, 41). The T95 residue is located in the third transmembrane domain of SLC30A10 (Fig. 1A). Although T95 appears to be in the proximity of residues that are required for Mn efflux in the first (E25), second (D40), fourth (N127), and fifth (H244 and D248) transmembrane domains (23), the side chain of T95 points away from the putative Mn binding cavity formed by the side chains of the required residues within the transmembrane domain (Fig. 1A). Moreover, after introducing the T95I mutation, the side chain of the most likely rotamer of the T95I variant still points away from the other required residues and the putative ion binding cavity in the transmembrane domain (Fig. 1B). These results raised doubts about the role of the T95 residue and the T95I mutation in the Mn transport activity of SLC30A10.
Figure 1.

Structural prediction of SLC30A10-WT or SLC30A10-T95I. A and B: cartoon representation of the predicted structure of SLC30A10-WT (A) or SLC30A10-T95I (B) is depicted. Amino acid residues are shown as cyan sticks with nitrogen atoms colored in blue and oxygen atoms colored in red. WT, wild type.
Mn Efflux Activity of SLC30A10-T95I Is Comparable to the WT Protein in HeLa Cells
To directly assay for the impact of the T95I substitution on the Mn efflux activity of SLC30A10, we performed metal measurement and cell viability assays. For the first set of experiments, we utilized HeLa cells, which we have used in extensive previous studies to characterize the Mn efflux activity of various SLC30A10 mutants (20, 23). We detected SLC30A10-WT or T95I using a custom rabbit polyclonal antibody against the COOH-terminus of the human protein, which we previously generated and validated (19, 21, 35, 39). For this study, we reconfirmed the specificity of this antibody by performing immunoblots in HeLa cells transiently transfected with or without FLAG-tagged SLC30A10-WT. Consistent with our prior work, we did not detect endogenous SLC30A10 in HeLa cells (Fig. 2A). In cells expressing the SLC30A10-WT construct, the SLC30A10 antibody detected a specific signal at the expected molecular weight of ∼50 kDa (Fig. 2A). A similar signal was also detected with a FLAG antibody (Fig. 2A). Both SLC30A10 and FLAG antibodies detected a nonspecific band below ∼70 kDa (Fig. 2A; note that subsequent immunoblots are cropped to depict the specific band at ∼50 kDa). We then used a lentiviral expression system to stably express SLC30A10-WT or T95I in HeLa cells and selected clones of SLC30A10-WT or T95I expressing cells for metal analyses and viability experiments. We preferred stable infections to transient transfections for these assays because results would otherwise be confounded by independent transfections in each experimental replicate. Our goal was to perform the efflux assays (in Fig. 2 as well as subsequent experiments) with infected cells expressing identical levels of SLC30A10-WT or T95I, but this was not always possible. Since expression of SLC30A10-T95I at a level higher than SLC30A10-WT could lead us to erroneously overinterpret the Mn efflux capacity of the mutant, in situations where we did not obtain identical expression of the two SLC30A10 constructs, we performed assays with cells in which expression of SLC30A10-T95I was slightly (less than onefold) lower than the WT protein. For the selected HeLa cell clones, immunoblots revealed that expression of SLC30A10-T95I was modestly (∼35%) lower than the WT protein (Fig. 2B). Despite the lower expression of the mutant, intracellular Mn levels of cells expressing SLC30A10-WT or T95I were comparable and lower than infection control cells (Fig. 2C). Levels of other metals [iron (Fe), copper (Cu), and zinc (Zn)] were not affected by the expression of either SLC30A10 construct (Fig. 2, D–F). In viability assays, expression of SLC30A10-WT or T95I robustly protected against Mn-induced cell death (Fig. 2G). Protection by SLC30A10-WT at the higher Mn dose used in the assay was stronger than the T95I mutant (Fig. 2G), but this is likely a consequence of the slightly higher expression of the WT protein in the selected clone. Overall, results in HeLa cells indicate that the Mn efflux activity of SLC30A10-T95I is comparable to that of SLC30A10-WT.
Figure 2.

Overexpression of SLC30A10-WT or SLC30A10-T95I in HeLa cells reduces Mn levels and protects against Mn toxicity. A: HeLa cells were either left untransfected (−) or transiently transfected with FLAG-SLC30A10-WT construct (+) and harvested 48 h after transfection. Immunoblots were performed to detect SLC30A10 (using the custom anti-SLC30A10 antibody), FLAG or tubulin. B: immunoblots were performed to detect SLC30A10 (using the anti-SLC30A10 antibody) or tubulin from HeLa clones expressing SLC30A10-WT or SLC30A10-T95I. Relative expression of SLC30A10-WT or T95I, normalized to tubulin, respectively is 1.00 ± 0.008 and 0.655 ± 0.096 (means ± SE; n = 3; P < 0.05 by t test). C–F: mock-infected HeLa cells (control) or clonal HeLa cells expressing SLC30A10-WT or SLC30A10-T95I were treated with 250 µM Mn for 16 h. Intracellular levels of Mn (C), Fe (D), Cu (E), or Zn (F) were measured by inductively coupled plasma-mass spectrometry (ICP-MS) and normalized to total cell counts (means ± SE; n = 4–5; *P < 0.05 and n.s. denotes not significant for indicated comparisons by one-way ANOVA and Tukey’s post hoc test). G: viability of cells infected as described in C–F was assessed 16 h after treatment with indicated Mn doses. For each infection condition, viability at 0 mM Mn was independently set to 100 (means ± SE; n = 4, *P < 0.05 by two-way ANOVA and Tukey’s post hoc test with a and b indicating differences in comparison with control or SLC30A10-WT infection conditions, respectively, at each Mn concentration). WT, wild type.
Validation of the Mn Efflux Activity of SLC30A10-T95I in HepG2 Cells
Since the phenotype of the SLC30A10-T95I SNP is associated with liver injury (33), we repeated the metal measurement and viability assays in HepG2 cells that model hepatocytes and express endogenous SLC30A10 (19, 35). We previously demonstrated that Mn-induced homeostatic control of SLC30A10 in HepG2 cells is similar to that observed in the mouse liver or intestines (35). We used the same lentiviral system as HeLa to overexpress SLC30A10-WT or T95I in HepG2 cells and initially performed experiments using a mixed population of cells. Consistent with results in HeLa cells, intracellular Mn levels of SLC30A10-WT or T95I expressing cells were similar and lower than infection control (Fig. 3A). Levels of other metals were not affected by SLC30A10 expression (Fig. 3, B–D). In viability assays, SLC30A10-WT or T95I strongly protected against Mn-induced cell death, but similar to HeLa cells, the protection conferred by SLC30A10-WT was modestly higher than T95I at two of the doses tested (Fig. 3E). To determine whether the difference in the viability of cells expressing the WT or T95I mutant was a consequence of differences in infection efficiency and transgene expression [qRT-PCR analyses indicated that expression of SLC30A10-WT was higher than the T95I mutant in the mixed population (Fig. 3F)], we repeated the above experiment after selecting clones expressing SLC30A10-WT or T95I. Immunoblots revealed that expression of SLC30A10-T95I was ∼50% lower than WT in the clonal cells (Fig. 4A). Under these conditions, intracellular Mn levels of cells expressing SLC30A10-WT or T95I were again lower than infection control and comparable to each other (Fig. 4B). As expected, there was no impact of SLC30A10-WT or T95I expression on other metals (Fig. 4, C–E). Furthermore, SLC30A10-WT and T95I provided comparable, and almost complete, protection against Mn at all doses (Fig. 4F). Thus, similar to HeLa cells, the Mn efflux activity of SLC30A10-T95I is comparable to WT in HepG2 cells as well.
Figure 3.

Effect of overexpression of SLC30A10-WT or SLC30A10-T95I in HepG2 cells on intracellular Mn levels and Mn-induced cell death is essentially similar. A–D: intracellular Mn (A), Fe (B), Cu (C), or Zn (D) levels were measured in mock-infected HepG2 cells (control) or mixed population of HepG2 cells infected with SLC30A10-WT or SLC30A10-T95I after 16 h of 125 µM Mn treatment. Values were normalized to total cell counts (means ± SE; n = 7–8, *P < 0.05 and n.s. denotes not significant for indicated comparisons by one-way ANOVA and Tukey’s post hoc test). E: cell viability was analyzed after 16 h treatment with indicated Mn doses. Infection conditions are identical to A–D. For each infection condition, viability at 0 mM Mn was independently set to 100 (means ± SE; n = 5, *P < 0.05 using two-way ANOVA and Tukey’s post hoc test with a and b indicating differences in comparison with control or SLC30A10-WT infection conditions, respectively, at each Mn concentration). F: qRT-PCR analyses were performed under infection conditions identical to A–D. Relative expression of SLC30A10 in the control infection group was normalized to 1 (n = 3, *P < 0.05 using one-way ANOVA and Tukey’s post hoc test for indicated comparisons). WT, wild type.
Figure 4.

In clonal HepG2 cells, overexpression of SLC30A10-WT or SLC30A10-T95I has a comparable effect on intracellular Mn levels and Mn-induced cell death. A: immunoblot analyses were performed to detect SLC30A10 (using the custom anti-SLC30A10 antibody) or tubulin in HepG2 clones expressing SLC30A10-WT or SLC30A10-T95I. Relative expression of SLC30A10-WT or T95I, normalized to tubulin, respectively is 1.00 ± 0.136 and 0.541 ± 0.021 (means ± SE; n = 3, P < 0.05 by t test). Intracellular Mn (B), Fe (C), Cu (D), or Zn (E) levels were assayed in mock-infected HepG2 cells (control) or HepG2 clones expressing SLC30A10-WT or SLC30A10-T95I after 16 h treatment with 125 µM Mn. Metal values were normalized to total cell counts (means ± SE; n = 7–8, *P < 0.05 and n.s. denotes not significant for indicated comparisons by one-way ANOVA and Tukey’s post hoc test). F: viability of cells infected as described in B–E was assayed after 16 h treatment with indicated Mn doses. For each infection condition, viability at 0 mM Mn was set to 100 (means ± SE; n = 5, *P < 0.05 using two-way ANOVA and Tukey’s post hoc test with a indicating differences in comparison with the control group at each Mn dose. There were no differences between SLC30A10-WT or SLC30A10-T95I infection groups at any of the Mn doses by two-way ANOVA (P > 0.05). WT, wild type.
SLC30A10-T95I or WT Comparably Rescue ΔSLC30A10 HepG2 Cells
For further validation of the Mn efflux activity of SLC30A10-T95I, we performed a rescue experiment in ΔSLC30A10 HepG2 cells, in which endogenous SLC30A10 expression was depleted using CRISPR/Cas9. We generated the knockout cells using a lentiviral CRISPR/Cas9 construct targeting exon 1 of SLC30A10 and selecting clones from the infected cells. In the knockout clone selected, genomic sequencing revealed that premature stop codons were introduced in exon 1, and expression of SLC30A10 mRNA was abolished (Fig. 5, A and B). The ΔSLC30A10 cells also exhibited significantly elevated intracellular levels of Mn, but not other metals, and enhanced sensitivity to Mn-induced cell death (Fig. 5, C–G). To assay for rescue, we stably expressed SLC30A10-WT or T95I in the ΔSLC30A10 cells (Fig. 5H) and performed metal measurement and viability assays. In the first set of rescue experiments, we did not select clones of SLC30A10-WT or T95I expressing rescue constructs. In these mixed population cells, we observed that expression of SLC30A10-WT or T95I in the ΔSLC30A10 cells robustly lowered Mn levels (Fig. 5C). Notably, Mn levels of rescue cells expressing WT or T95I were comparable to each other and to infection control cells (Fig. 5C). Levels of other metals were essentially unaffected (Fig. 5, D–F). In addition, expression of either SLC30A10-WT or T95I also strongly rescued the enhanced sensitivity of ΔSLC30A10 cells to Mn-induced cell death (Fig. 5G). We did observe that viability of SLC30A10-WT expressing ΔSLC30A10 cells at one Mn concentration (1 mm Mn) was higher than SLC30A10-T95I (Fig. 5G). To determine whether this difference in viability was reflective of differences in infection efficiency, we selected clones of SLC30A10-WT or T95I-expressing ΔSLC30A10 cells. Immunoblots revealed that expression of SLC30A10-WT and T95I were comparable (Fig. 6A). Expression of either SLC30A10-WT or T95I significantly reduced Mn levels compared with the ΔSLC30A10 cells and the infection control (Fig. 6B). Modest changes in Fe or Cu, but not Zn, between groups were evident (Fig. 6, C–E), but the biological relevance of these changes is unclear. Importantly, under these conditions, the viability of ΔSLC30A10 cells expressing SLC30A10-WT or T95I was comparable at all Mn concentrations tested, and significantly higher than ΔSLC30A10 cells (Fig. 6F). In totality, the rescue assays provide further evidence to support the conclusion that the T95I substitution does not impact the Mn efflux activity of SLC30A10.
Figure 5.

Expression of SLC30A10-WT or SLC30A10-T95I in ΔSLC30A10 HepG2 cells rescues changes in Mn levels and cell viability. A: amino acid sequence of the wild-type (WT) SLC30A10 protein or the SLC30A10 protein sequence translated from the genomic DNA sequence of the ΔSLC30A10 HepG2 clone. There are two sequences (Seq1 and 2) for the ΔSLC30A10 clone because CRISPR/Cas9 introduced two independent mutations in the corresponding genomic DNA, likely one for each chromosome. Differences between the sequences obtained from the ΔSLC30A10 clone and the WT protein are shown in red. Numbers indicate amino acid residue. B: RT-PCR analyses were performed to detect SLC30A10 or TBP mRNA in control or ΔSLC30A10 HepG2 cells. C–F: intracellular metal measurements were performed after 125 µM Mn treatment for 16 h in mock-infected HepG2 cells (control) or ΔSLC30A10 HepG2 cells that underwent a second mock, SLC30A10-WT or SLC30A10-T95I infection (ΔSLC30A10 + control; ΔSLC30A10+SLC30A10-WT; or ΔSLC30A10+SLC30A10-T95I, respectively). Clonal selection was not performed after SLC30A10-WT or SLC30A10-T95I infection. Metal values were normalized to total cell counts (means ± SE; n = 8, *P < 0.05 and n.s. denotes not significant for indicated comparisons by one-way ANOVA and Tukey’s post hoc test). G: viability of cells infected as described in C–F was assayed after 16 h of treatment with indicated Mn doses. For each infection condition, viability at 0 mM Mn was set to 100 (means ± SE; n = 5, *P < 0.05 using two-way ANOVA and Tukey’s post hoc test with a, b, and c indicating differences in comparison with control, ΔSLC30A10 + control, or ΔSLC30A10+SLC30A10-T95I infection groups, respectively at each Mn concentration). H: RT-PCR analyses were performed in cells infected as described in C–F to detect SLC30A10 or TBP mRNA. Cont., control.
Figure 6.

After clonal selection of SLC30A10-WT or SLC30A10-T95I expression in ΔSLC30A10 HepG2 cells, rescue activity of the SLC30A10-T95I mutant is comparable to SLC30A10-WT. A: immunoblot analyses were performed to detect SLC30A10 (using the custom anti-SLC30A10 antibody) or tubulin in ΔSLC30A10 HepG2 cells infected a second time with SLC30A10-WT or SLC30A10-T95I. Cells were clonally selected after the second infection. Relative expression of SLC30A10-WT or T95I, normalized to tubulin, respectively is 1.00 ± 0.239 and 0.835 ± 0.085 (means ± SE; n = 3; P > 0.05 by t test). B–E: intracellular metal measurements were performed in mock-infected HepG2 cells (control) or ΔSLC30A10 HepG2 cells that underwent a second mock, SLC30A10-WT or SLC30A10-T95I infection (ΔSLC30A10 + control; ΔSLC30A10+SLC30A10-WT; or ΔSLC30A10+SLC30A10-T95I, respectively). ΔSLC30A10 cells infected with SLC30A10-WT or SLC30A10-T95I underwent clonal selection. Cells were treated with 125 µM Mn for 16 h before analyses. Metal levels were normalized to total cell counts (means ± SE; n = 5–7, *P < 0.05 and n.s. denotes not significant by one-way ANOVA and Tukey’s post hoc test). F: viability of cells infected and clonally selected as described in B–E was assayed after 16 h treatment with indicated Mn doses. For each cell line, viability at 0 mM Mn was set to 100 (means ± SE; n = 5, *P < 0.05 using two-way ANOVA and Tukey’s post hoc test with a and b indicating differences in comparison with control or ΔSLC30A10 + control groups, respectively at each Mn dose. There were no differences between ΔSLC30A10+SLC30A10-WT or ΔSLC30A10+SLC30A10-T95I groups at any Mn concentration by two-way ANOVA). WT, wild type.
Stability of SLC30A10-T95I Is Also Comparable to the Wild-Type Protein
We previously reported that several loss-of-function SLC30A10 mutants were trapped in the endoplasmic reticulum and underwent enhanced proteasome-mediated degradation (20). As the T95 residue was located in a transmembrane domain, it was possible that the T95I mutation impacted the stability of SLC30A10 and induced its proteasomal degradation. To test this possibility, we performed a time-course experiment after treatment of HepG2 cells overexpressing SLC30A10-WT or T95I with the proteasomal inhibitor MG132 (20). Notably, the accumulation of SLC30A10-T95I was comparable to the WT protein after MG132 treatment (Fig. 7, A and B). Thus, the T95I substitution also does not affect the stability or turnover rate of SLC30A10.
Figure 7.

Turnover of SLC30A10-WT and SLC30A10-T95I is comparable. A: HepG2 clones overexpressing SLC30A10-WT or SLC30A10-T95I (identical to Fig. 4) were treated with 0.5 µM MG132 for indicated times. Cell lysates were collected and SLC30A10 or tubulin were detected by immunoblot. The custom antibody against SLC30A10 was used. B: quantification of SLC30A10 levels from A. At each time point, SLC30A10 expression is normalized to tubulin. For each infection condition, normalized SLC30A10 expression at 0 h is set to 1 (n = 3; there were no differences between SLC30A10-WT or SLC30A10-T95I groups at any time-point using two-way ANOVA and Sidak’s post hoc test). WT, wild type.
DISCUSSION
Previously, we utilized a mutational approach guided by a predicted structure of SLC30A10 to identify several transmembrane residues, including E25 and D40, which are required for Mn efflux activity (23, 39). Cell viability and intracellular Mn measurements after Mn treatment were shown to be biologically relevant and accurate measures for the Mn transport activity of SLC30A10 because three SLC30A10 mutations located at or adjacent to these critical residues (D40A, L26P, ΔS41) were later reported to cause hereditary Mn toxicity in humans (43). In this study, we used the same techniques to show that the SLC30A10 missense variant T95I, which is associated with increased serum levels of liver injury biomarkers ALT and AST (33), retains Mn transport activity comparable to the WT protein. Thus, the association of the T95I mutant with liver injury cannot be simply explained by reduced activity of SLC30A10.
In addition to our findings, doubts about the causal role of the SLC30A10-T95I mutation in liver disease also come from close analyses of the available human literature, including a second recent study [by Seidelin et al. (34)] that further examined the association between this variant and liver injury markers, Mn toxicity symptoms, and risk of biliary tract cancer in a different cohort. In the newer study (34), some findings strengthened the association between T95I and increased ALT, hepatic inflammation, and hematocrit. But, other findings were more aligned with a lack of an effect of the T95I mutation on liver injury or Mn homeostasis (34). As an example, plasma Mn levels did not change in T95I carriers (34). Furthermore, the first study of the T95I variant [Ward et al. (33)] did not find an association between neurological symptoms and the T95I mutation in their cohort (UK Biobank). In contrast, Seidelin et al. (34) identified an association between neurological function and T95I in the cohort also obtained from the UK Biobank, but not in another cohort (Copenhagen). Association with biliary tract cancer was also reported in the UK Biobank cohorts by Ward et al. and Seidelin et al. but not in the Copenhagen cohort by Seidelin et al. (33, 34). Thus, the associations between T95I and human disease appear to not be robustly reproducible across cohorts.
In this discussion, it is important to note a few caveats about the interpretation of our data. In our studies, SLC30A10-WT or T95I was expressed at a substantially higher level than the endogenous protein. Although protein expression level has not been an issue in our previous studies of inactive SLC30A10 mutants (20, 23, 39), it remains possible that the T95I variant introduces a subtle deficit in SLC30A10 function that was masked by high expression of the mutant. It is also possible that deficits in the Mn transport activity of SLC30A10-T95I function may emerge under physiological or pathological conditions or in the context of gene-environment interactions that are challenging to model in cell culture (e.g., long-term exposure to Mn through increasing age, inflammation etc.). Indeed, the T95I SNP is reported to be in a strong linkage with two common noncoding SNPs, rs1776029 and rs2275707 (33). Recently, the minor allele of rs2275707 has been associated with modest elevation in blood Mn level and lower SLC30A10 transcript level in adult cohorts in Argentina, Italy, and Bangladesh (32). Furthermore, Mendelian randomization study of rs1776029 showed that the minor allele associated with increased blood Mn level and poorer cognition and increased neurodevelopmental problems in children (30). In addition, bioinformatic analysis revealed that rs1776029 is located at a genomic region with potential regulatory activity (DNAse I hypersensitivity, strong H3K27 acetylation), suggesting a regulatory role for the SNPs in SLC30A10 gene expression (30, 31). Therefore, the effect of the T95I mutation may depend on the allele of rs1776029 and/or rs2275707. A related issue is that SLC30A10 has also been reported to be expressed in human cholangiocytes (17). Although unlikely, the possibility that the T95I SNP influences Mn transport function of SLC30A10 in cholangiocytes in a manner that impacts biliary disease independent of liver function cannot be eliminated. Finally, the T95I mutant may also potentially impact an as yet undiscovered function of SLC30A10 that influences liver and bile duct biology, but not Mn transport. Mouse models with knock-in of the T95I mutation in the SLC30A10 open reading frame are now necessary to determine whether the T95I mutation has any biological role in human health and disease. The significance of our current studies is that the epidemiological association between SLC30A10-T95I and liver disease cannot be simply explained by deficits in Mn transporter activity. Further, our studies show the importance of functional validation of SNPs identified via GWAS.
In conclusion, the T95I variant of the Mn efflux transporter SLC30A10 retains Mn efflux activity comparable to SLC30A10-WT in cell-based assays suggesting that either the T95I variant does not directly cause the associated symptoms, or that this variant has a very minor deficiency in activity that presents itself as liver injury in conjunction with other risk factors.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This study was supported by National Institute of Environmental Health Sciences Grant R01 ES031574 (to S.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.C.G. and S.M. conceived and designed research; K.C.G. and D.L. performed experiments; K.C.G. analyzed data; K.C.G. and S.M. interpreted results of experiments; K.C.G. prepared figures; K.C.G. drafted manuscript; K.C.G., K.B., and S.M. edited and revised manuscript; S.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Nathan Miller (UT Austin) for performing ICP-MS analyses.
REFERENCES
- 1. Aschner M, Erikson KM, Herrero Hernández E, Tjalkens R. Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med 11: 252–266, 2009. [Erratum in Neuromolecular Med 11: 267, 2009]. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balachandran RC, Mukhopadhyay S, McBride D, Veevers J, Harrison FE, Aschner M, Haynes EN, Bowman AB. Brain manganese and the balance between essential roles and neurotoxicity. J Biol Chem 295: 6312–6329, 2020. doi: 10.1074/jbc.REV119.009453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kwakye GF, Paoliello MM, Mukhopadhyay S, Bowman AB, Aschner M. Manganese-induced Parkinsonism and Parkinson's disease: shared and distinguishable features. Int J Environ Res Public Health 12: 7519–7540, 2015. doi: 10.3390/ijerph120707519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bowman AB, Aschner M. Considerations on manganese (Mn) treatments for in vitro studies. Neurotoxicology 41: 141–142, 2014. doi: 10.1016/j.neuro.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guilarte TR, Gonzales KK. Manganese-induced Parkinsonism is not idiopathic Parkinson's disease: environmental and genetic evidence. Toxicol Sci 146: 204–212, 2015. doi: 10.1093/toxsci/kfv099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oulhote Y, Mergler D, Barbeau B, Bellinger DC, Bouffard T, Brodeur MÈ, Saint-Amour D, Legrand M, Sauvé S, Bouchard MF. Neurobehavioral function in school-age children exposed to manganese in drinking water. Environ Health Perspect 122: 1343–1350, 2014. doi: 10.1289/ehp.1307918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouchard MF, Sauvé S, Barbeau B, Legrand M, Brodeur MÈ, Bouffard T, Limoges E, Bellinger DC, Mergler D. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ Health Perspect 119: 138–143, 2011. doi: 10.1289/ehp.1002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bouchard M, Laforest F, Vandelac L, Bellinger D, Mergler D. Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ Health Perspect 115: 122–127, 2007. doi: 10.1289/ehp.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Racette BA, Criswell SR, Lundin JI, Hobson A, Seixas N, Kotzbauer PT, Evanoff BA, Perlmutter JS, Zhang J, Sheppard L, Checkoway H. Increased risk of parkinsonism associated with welding exposure. Neurotoxicology 33: 1356–1361, 2012. doi: 10.1016/j.neuro.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhang SY, Cho SC, Kim JW, Hong YC, Shin MS, Yoo HJ, Cho IH, Kim Y, Kim BN. Relationship between blood manganese levels and children's attention, cognition, behavior, and academic performance–a nationwide cross-sectional study. Environ Res 126: 9–16, 2013. doi: 10.1016/j.envres.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 11. Lucchini RG, Guazzetti S, Zoni S, Donna F, Peter S, Zacco A, Salmistraro M, Bontempi E, Zimmerman NJ, Smith DR. Tremor, olfactory and motor changes in Italian adolescents exposed to historical ferro-manganese emission. Neurotoxicology 33: 687–696, 2012. doi: 10.1016/j.neuro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pomier-Layrargues G, Spahr L, Butterworth RF. Increased manganese concentrations in pallidum of cirrhotic patients. Lancet 345: 735, 1995. doi: 10.1016/s0140-6736(95)90909-5. [DOI] [PubMed] [Google Scholar]
- 13. Rose C, Butterworth RF, Zayed J, Normandin L, Todd K, Michalak A, Spahr L, Huet PM, Pomier-Layrargues G. Manganese deposition in basal ganglia structures results from both portal-systemic shunting and liver dysfunction. Gastroenterology 117: 640–644, 1999. doi: 10.1016/s0016-5085(99)70457-9. [DOI] [PubMed] [Google Scholar]
- 14. Spahr L, Butterworth RF, Fontaine S, Bui L, Therrien G, Milette PC, Lebrun LH, Zayed J, Leblanc A, Pomier-Layrargues G. Increased blood manganese in cirrhotic patients: relationship to pallidal magnetic resonance signal hyperintensity and neurological symptoms. Hepatology 24: 1116–1120, 1996. doi: 10.1002/hep.510240523. [DOI] [PubMed] [Google Scholar]
- 15. Butterworth RF. Parkinsonism in cirrhosis: pathogenesis and current therapeutic options. Metab Brain Dis 28: 261–267, 2013. doi: 10.1007/s11011-012-9341-7. [DOI] [PubMed] [Google Scholar]
- 16. Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Chong WK, Mills PB. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet 90: 457–466, 2012. [Erratum in Am J Hum Genet 99: 521, 2016] doi: 10.1016/j.ajhg.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 90: 467–477, 2012. doi: 10.1016/j.ajhg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun 7: 11601, 2016. doi: 10.1038/ncomms11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, Dray BK, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J Biol Chem 292: 16605–16615, 2017. doi: 10.1074/jbc.M117.804989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci 34: 14079–14095, 2014. doi: 10.1523/JNEUROSCI.2329-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taylor CA, Hutchens S, Liu C, Jursa T, Shawlot W, Aschner M, Smith DR, Mukhopadhyay S. SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity. J Biol Chem 294: 1860–1876, 2019. doi: 10.1074/jbc.RA118.005628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem 292: 9760–9773, 2017. doi: 10.1074/jbc.M117.783605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zogzas CE, Aschner M, Mukhopadhyay S. Structural elements in the transmembrane and cytoplasmic domains of the metal transporter SLC30A10 are required for its manganese efflux activity. J Biol Chem 291: 15940–15957, 2016. doi: 10.1074/jbc.M116.726935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gurol KC, Aschner M, Smith DR, Mukhopadhyay S. Role of excretion in manganese homeostasis and neurotoxicity: a historical perspective. Am J Physiol Gastrointest Liver Physiol 322: G79–G92, 2022. doi: 10.1152/ajpgi.00299.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jenkitkasemwong S, Akinyode A, Paulus E, Weiskirchen R, Hojyo S, Fukada T, Giraldo G, Schrier J, Garcia A, Janus C, Giasson B, Knutson MD. SLC39A14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice. Proc Natl Acad Sci USA 115: E1769–E1778, 2018. [Erratum in Proc Natl Acad Sci USA 115: E4730, 2018]. doi: 10.1073/pnas.1720739115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aydemir TB, Kim MH, Kim J, Colon-Perez LM, Banan G, Mareci TH, Febo M, Cousins RJ. Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J Neurosci 37: 5996–6006, 2017. doi: 10.1523/JNEUROSCI.0285-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aydemir TB, Thorn TL, Ruggiero CH, Pompilus M, Febo M, Cousins RJ. Intestine-specific deletion of metal transporter Zip14 (Slc39a14) causes brain manganese overload and locomotor defects of manganism. Am J Physiol Gastrointest Liver Physiol 318: G673–G681, 2020. [Erratum in Am J Physiol Gastrointest Liver Physiol 320: G557, 2021]. doi: 10.1152/ajpgi.00301.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercadante CJ, Prajapati M, Conboy HL, Dash ME, Herrera C, Pettiglio MA, Cintron-Rivera L, Salesky MA, Rao DB, Bartnikas TB. Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity. J Clin Invest 129: 5442–5461, 2019. doi: 10.1172/JCI129710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broberg K, Taj T, Guazzetti S, Peli M, Cagna G, Pineda D, Placidi D, Wright RO, Smith DR, Lucchini RG, Wahlberg K. Manganese transporter genetics and sex modify the association between environmental manganese exposure and neurobehavioral outcomes in children. Environ Int 130: 104908, 2019. doi: 10.1016/j.envint.2019.104908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wahlberg KE, Guazzetti S, Pineda D, Larsson SC, Fedrighi C, Cagna G, Zoni S, Placidi D, Wright RO, Smith DR, Lucchini RG, Broberg K. Polymorphisms in manganese transporters SLC30A10 and SLC39A8 are associated with children's neurodevelopment by influencing manganese homeostasis. Front Genet 9: 664, 2018. doi: 10.3389/fgene.2018.00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wahlberg K, Arora M, Curtin A, Curtin P, Wright RO, Smith DR, Lucchini RG, Broberg K, Austin C. Polymorphisms in manganese transporters show developmental stage and sex specific associations with manganese concentrations in primary teeth. Neurotoxicology 64: 103–109, 2018. doi: 10.1016/j.neuro.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wahlberg K, Kippler M, Alhamdow A, Rahman SM, Smith DR, Vahter M, Lucchini RG, Broberg K. Common polymorphisms in the solute carrier SLC30A10 are associated with blood manganese and neurological function. Toxicol Sci 149: 473–483, 2016. doi: 10.1093/toxsci/kfv252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ward LD, Tu HC, Quenneville CB, Tsour S, Flynn-Carroll AO, Parker MM, Deaton AM, Haslett PAJ, Lotta LA, Verweij N, Ferreira MAR; Regeneron Genetics Center, Geisinger-Regeneron DiscovEHR Collaboration, Baras A, Hinkle G, Nioi P. GWAS of serum ALT and AST reveals an association of SLC30A10 Thr95Ile with hypermanganesemia symptoms. Nat Commun 12: 4571, 2021. doi: 10.1038/s41467-021-24563-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seidelin A-S, Nordestgaard BG, Tybjærg-Hansen A, Yaghootkar H, Stender S. A rare genetic variant in the manganese transporter SLC30A10 and elevated liver enzymes in the general population. Hepatol Int 16: 702–711, 2022. doi: 10.1007/s12072-022-10331-w. [DOI] [PubMed] [Google Scholar]
- 35. Liu C, Jursa T, Aschner M, Smith DR, Mukhopadhyay S. Up-regulation of the manganese transporter SLC30A10 by hypoxia-inducible factors defines a homeostatic response to manganese toxicity. Proc Natl Acad Sci USA 118: e2107673118, 2021. doi: 10.1073/pnas.2107673118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Selyunin AS, Iles LR, Bartholomeusz G, Mukhopadhyay S. Genome-wide siRNA screen identifies UNC50 as a regulator of Shiga toxin 2 trafficking. J Cell Biol 216: 3249–3262, 2017. doi: 10.1083/jcb.201704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682, 2012. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li D, Mukhopadhyay S. Functional analyses of the UDP-galactose transporter SLC35A2 using the binding of bacterial Shiga toxins as a novel activity assay. Glycobiology 29: 490–503, 2019. doi: 10.1093/glycob/cwz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zogzas CE, Mukhopadhyay S. Putative metal binding site in the transmembrane domain of the manganese transporter SLC30A10 is different from that of related zinc transporters. Metallomics 10: 1053–1064, 2018. doi: 10.1039/c8mt00115d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589, 2021. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, Žídek A, Green T, Tunyasuvunakool K, Petersen S, Jumper J, Clancy E, Green R, Vora A, Lutfi M, Figurnov M, Cowie A, Hobbs N, Kohli P, Kleywegt G, Birney E, Hassabis D, Velankar S. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50: D439–D444, 2022. doi: 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4: 363–371, 2009. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 43. Zaki MS, Issa MY, Elbendary HM, El-Karaksy H, Hosny H, Ghobrial C, El Safty A, El-Hennawy A, Oraby A, Selim L, Abdel-Hamid MS. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: six novel SLC30A10 mutations and further phenotype delineation. Clin Genet 93: 905–912, 2018. doi: 10.1111/cge.13184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.