

Abstract
Many lung diseases are caused by an excessive inflammatory response, and inflammatory lung diseases are often modeled using lipopolysaccharide (LPS) in mice. Cyclooxygenase-2 (COX-2) encoded by the Ptgs2 gene is induced in response to inflammatory stimuli including LPS. The objective of this study was to test the hypothesis that mice deficient in COX-2 (Ptgs2-/-) will be protected from LPS-induced lung injury. Wild-type (WT; CD1 mice) and Ptgs2-/- mice (on a CD1 background) were treated with LPS or vehicle for 24 h. LPS treatment resulted in histological evidence of lung injury, which was attenuated in the Ptgs2-/- mice. LPS treatment increased the mRNA levels for tumor necrosis factor-α, interleukin-10, and monocyte chemoattractant protein-1 in the lungs of WT mice, and the LPS-induced increases in these levels were attenuated in the Ptgs2-/- mice. The protein levels of active caspase-3 and caspase-9 were lower in the LPS-treated lungs of Ptgs2-/- mice than in LPS-treated WT mice, as were the number of terminal deoxynucleotide transferase dUTP nick end labeling-positive cells in lung sections. LPS exposure resulted in a greater lung wet-to-dry weight ratio (W/D) in WT mice, suggestive of pulmonary edema, while in LPS-treated Ptgs2-/- mice, the W/D was not different from controls and less than in LPS-treated WT mice. These results demonstrate that COX-2 is involved in the inflammatory response to LPS and suggest that COX-2 not only acts as a downstream participant in the inflammatory response, but also acts as a regulator of the inflammatory response likely through a feed-forward mechanism following LPS stimulation.
Keywords: cytokines, inflammation, prostaglandins, pulmonary edema, pulmonary function
INTRODUCTION
Lung inflammation is central to many lung diseases, particularly diseases associated with acute lung injury (ALI) like acute respiratory distress syndrome (1, 2). The regulation of the innate immune response is complex and includes a number of signal transduction networks, some of which are redundant and some of which have unique cellular effects. Gaining a more complete understanding of the regulation of the complex signal transduction pathways leading to ALI is vital for the future development of targeted pharmacotherapies for inflammatory lung injury for which there are currently no targeted pharmacotherapies. Cyclooxygenase-2 (COX-2), the inducible form of cyclooxygenase, is encoded by the Ptgs2 gene and has been shown to play a key role in inflammatory signaling in the lung (3–5). We have previously found that treatment of adult mice with lipopolysaccharide (LPS) results in robust induction of COX-2 in the lung (6), as does treatment with >95% oxygen for 60 h (7). Furthermore, we observed that neonatal mice exposed to 85% oxygen had robust induction of COX-2 but not COX-1 in the lung (8). In 12-wk-old mice we have found that LPS induces robust COX-2 expression in epithelial cells (9). While Robertson, et al. (10) found in a murine model of ventilation-induced lung injury that COX-2 protein expression was abundant in epithelial cells, as well as alveolar and interstitial monocytes/macrophages. Furthermore, therapies that decrease COX-2 expression or activity have been shown to be beneficial in murine models of ALI (11–13). However, the role of COX-2 in the regulation of LPS-induced inflammation, apoptosis and the resultant ALI have not been examined using COX-2 deficient mice. We hypothesized that COX-2 would have an important positive regulator role in the pulmonary LPS response, such that mice deficient in COX-2 would exhibit less LPS-induced cytokine and/or chemokine expression and have attenuated indices of ALI when compared with wild-type (WT) mice. To test this hypothesis, we utilized COX-2 knockout mice (Ptgs2-/-) on a CD1 background and treated Ptgs2-/- or WT (CD1) mice with vehicle or LPS. We examined the lung levels of selected inflammatory cytokines and chemokines; activated caspase-3, caspase-8, and caspase-9; measured lung wet-to-dry weights; and assessed pulmonary function.
METHODS
Mice
Mice with deletion of the gene encoding COX-2 (Ptgs2tm1Jed) were reestablished on a CD-1 genetic background (Charles River Laboratories, Wilmington, MA) and maintained by periodic outcross (14–16). Heterozygous pairings were bred in-house to yield Ptgs2-/- and WT mice. The mice were genotyped using PCR. The Ptgs2 WT primer set was as follows: forward: ACC TCT GCG ATG CTC TTC C; and reverse: CAC CAT AGA ATC CAG TCC GG. The Ptgs2 KO primer set was as follows: forward: ATC GCC TTC TTG ACG AGT TC; and reverse: CAC CAT AGA ATC CAG TCC GG. The wild-type Ptgs2 band is at 850 bp, the knockout Ptgs2 band is at 450 bp, and heterozygous mice have both bands. A representative PCR gel is shown in Fig. 2A from 10 animals.
Figure 2.

LPS-induced increases in lung mRNA levels for TNF-α, monocyte chemoattractant protein-1 (MCP-1), and IL-10 were significantly attenuated in the cyclooxygenase-2 (COX-2)-deficient mice. Levels of mRNA in lungs harvested 24 h after vehicle or LPS treatment in the wild-type (WT; open boxes; n = 6–8 for each condition) and Ptgs2-/- mice (gray boxes; n = 5–8 for each condition) are shown. A: representative genotyping PCR gel done on 10 animals; lane 1 is the 100-bp DNA ladder (New England Biolabs, Ipswich, MA); lanes 2, 4-6, 8, and 11 are Ptgs2-/- mice; lanes 7 and 10 are WT mice; and lanes 3 and 9 are heterozygous mice (not used in the experiments). B: TNF-α mRNA levels normalized to 18S rRNA. By two-way ANOVA there was a difference in genotype (P < 0.05) and with LPS (P < 0.001) and a significant interaction (P < 0.05). Post hoc analysis identified the following differences: *P < 0.05, different from Ptgs2-/- control; ***P < 0.001, different from the WT control; #P < 0.005, different from WT LPS. C: MCP-1 mRNA normalized to 18S rRNA. By two-way ANOVA, there was a difference in genotype (P < 0.001) and with LPS (P < 0.001) and a significant interaction (P < 0.001). Post hoc analysis: *P < 0.001, different from the WT control; #P < 0.001, different from WT LPS. D: IL-10 mRNA levels normalized to 18S rRNA. By two-way ANOVA, there was a difference in genotype (P < 0.05) and with LPS (P < 0.001) and a significant interaction (P < 0.05). Post hoc analysis: *P < 0.001, different from the WT control; #P < 0.01, different from WT LPS. E: levels of CXCL-2 mRNA normalized to 18S rRNA. By two-way ANOVA, there was a difference in genotype (P < 0.001) and with LPS (P < 0.001) and a significant interaction (P < 0.001). Post hoc analysis: *P < 0.05. LPS different from control same genotype; #P < 0.001, different from WT LPS.
The mice were essentially split evenly between males and females. The mice were between 2 and 4 mo of age and weighed between 23 and 37 g for females and 27 and 42 g for males. All mice were maintained at 24°C with a relative humidity between 30% and 70% on a 12:12-h light-dark cycle. The mice were fed Harlan Teklad irradiated diet (Harlan Sprague Dawley, Indianapolis, IN) ad libitum. All animals received humane care in accordance with the guidelines of the National Institutes of Health. The study protocol was approved by the Institutional Animal Care and Use Committee of the Abigail Wexner Research Institute at Nationwide Children’s Hospital.
Exposures
Adult WT and Ptgs2-/- mice were given either vehicle or 10 mg/kg lipopolysaccharide (LPS) via intraperitoneal injection. The mice were monitored for predetermined end point criteria, and no mice met the predetermined end point criteria during the 24-h treatment period. We chose 24 h following LPS challenge as the time point because in our models of LPS-induced injury there is lung injury demonstrable in the LPS-treated mice at 24 h, while mortality from LPS starts after 24 h (17). By avoiding later time points we hoped to avoid the potential confounder of premorbid changes in lung function, histology, etc.
Measurement of Pulmonary Function
Mice were anesthetized with an intraperitoneal injection of ketamine (65 mg/kg, Hospira, Lake Forest, IL)/xylazine (10 mg/kg, Lloyd Laboratories, Shenandoah, IA) to suppress spontaneous breathing. The trachea was cannulated via a tracheostomy, and the cannula was connected to a FlexiVent system (SCIREQ Scientific Respiratory Equipment, Montreal, QC, Canada) under the controlled mechanical ventilation mode with a tidal volume of 10 mL/kg at a frequency of 150 breaths/min and a positive end-expiratory pressure of 3 cmH2O as previously described (7, 18). Total lung capacity perturbation was applied first to standardize volume history. Snapshot perturbation was then performed to measure resistance (RRS) and elastance (ERS) of the respiratory system (airways, lung, and chest wall). All perturbations were repeated three times with acceptable measurements (coefficient of determination >0.95) and the average was calculated for each individual subject. After these measurements were performed, pressure-volume loops were generated to measure quasistatic compliance (CRS).
Tissue Preparation
On the day of study, mice were euthanized with an intraperitoneal injection of ketamine (90 mg/kg)/xylazine (4.5 mg/kg). The right lungs were removed, weighed (wet weight), and then dried in the oven at 60°C. The weight was checked each day, and the lungs remained in the oven until the weight stabilized (dry weight). The lung wet-to-dry weight ratio (W/D) was calculated as a measure of pulmonary edema formation, with greater lung W/D indicative of edema formation. The left lungs were removed, weighed, freeze clamped in liquid nitrogen, and kept at −80°C for further analyses.
Lung Histology and Lung Injury Scoring
The lungs are inflation fixed using 4% neutral-buffered formalin for 24–48 h and then transferred to PBS as previously described (7). The tissue was then embedded in paraffin, cut into 4-µm-thick sections, and stained with hematoxylin and eosin (H&E) for histological evaluation. Images were taken using an Aperio AT-2 scanner (Leica Biosystems, Inc., Bufflo Grove, IL). Histological changes were evaluated by an observer blinded to the treatment or genotype. The severity of lung injury was graded from 0 (minimal) to 4 (maximal) for alveolar congestion, intra-alveolar infiltrates, inflammation, and hemorrhage as described by Murakami et al. (19). In each animal, four lung sections were graded to generate the mean score. The lung injury score was calculated by adding the scores in each category.
Real-Time PCR
Total RNA was isolated from the frozen lungs using TRIzol (Invitrogen, Carlsbad, CA) following the procedure recommended by the manufacturer’s instruction. Reverse transcription-coupled real-time PCR for TNF-α, IL-10, monocyte chemoattractant protein-1 (MCP-1), cxcl-2, and IL-12 was performed as previously described (7, 20, 21). Briefly, all RNA samples were treated with RNase-free DNase (Promega, Madison, WI). Complementary DNA (cDNA) was synthesized using GoScript reverse transcriptase (Promega), and cDNAs were analyzed by real-time PCR using PowerUp SYBR Green Master Mix (Applied Biosystems, Austin, TX). Primers were synthesized by Invitrogen using the following sequences: for mouse TNF-α: 5′-CTA CTC CCA GGT TCT CTT CAA TCG-3′ (forward) and 5′-GCA GAG AGG TTG ACT TTC-3′ (reverse); mouse IL-10: 5′-ACA GCC GGG AAG ACA ATA AC-3′ (forward) and 5′-CAG CTG GTC CTT TGT TTG AAA G-3′ (reverse); mouse MCP-1: 5′-AGC AGG TGT CCC AAA GAA GCT GTA-3′ (forward) and 5′-AAA GGT GCT GAA GAC CTT AGG GCA-3′ (reverse); mouse CXCL-2: 5′-TAA GCA CCG AGG AGA GTA GAA-3′ (forward) and 5′-GTC CAA GGG TTA CTC ACA ACA-3′ (reverse); and mouse IL-12: 5′-CCT CCT CAC ACA GAT AGG AAA C-3′ (forward) and 5′-GAG ATG AGA TGT GAT GGG AGA AC-3′ (reverse). 18S was amplified as an internal control using the forward primer 5′-CCA GAG CGA AAG CAT TTG CCA AGA-3′ and the reverse primer 5′-TCG GCA TCG TTT ATG GTC GGA ACT-3′. For each reaction, negative controls containing reaction mixture and primers without cDNA were performed to verify that primers and reaction mixtures were free of template contamination. Relative RNA amounts were normalized to 18S expression using the ΔΔCT method. All samples were analyzed in duplicate. Data are shown as fold change relative to vehicle-treated WT mice.
Protein Isolation
Protein was isolated as previously described (7, 20, 21). Lung tissues were homogenized with PBS/EDTA buffer containing protease inhibitor and phosphatase inhibitor and centrifuged at 14,000 rpm for 15 min at 4°C. The supernatant was stored at −80°C for subsequent Western blot analysis. Total protein concentration was determined by the Bradford method using a commercially available assay (Bio-Rad, Hercules, CA).
Immunoblotting
Cell lysates were assayed for active (cleaved) caspase-3, active (cleaved) caspase-8, total caspase-8, active (cleaved) caspase-9, total caspase-9, intercellular adhesion molecule 1 (ICAM-1; also known as CD54), myeloperoxidase (MPO), and β-actin using Western blot analysis as previously described (7, 20, 21). Aliquots of cell lysate were diluted with appropriate amounts of 10× NuPAGE reducing agent, 4× NuPAGE LDS sample buffer, and deionized water. The samples were then heated to 95°C for 5 min, and then separated using SDS-PAGE. The proteins were transferred to polyvinylidene difluoride membranes and blocked in Tris-buffered saline with 0.1% Tween (TBS-T) containing 10% skim milk for 1 h. The membranes were then washed with TBS-T and incubated with primary antibody against cleaved caspase-3 (Cell Signaling Technology, Danvers, MA, catalog no. 9664, lot no. 20), cleaved caspase-8 (Cell Signaling Technology, catalog no. 9429, lot no. 2), total caspase-8 (Cell Signaling Technology, catalog no. 4790, lot no. 2), cleaved caspase-9 (Cell Signaling Technology, catalog no. 9509, lot no. 3), total caspase-9 (Cell Signaling Technology, catalog no. 9508, lot no. 2), ICAM-1 (Cell Signaling Technology, catalog no. 4915, lot no. 1), or MPO (Cell Signaling Technology, catalog no. 4162, lot no. 1) for 1 h. The membranes were washed three times with TBS-T and incubated with goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (1:15,000; Bio-Rad) for 1 h. Then the membranes were washed three times with TBS-T. The bands for specific protein were visualized using chemiluminescence (Amersham ECL, Piscataway, NJ) and quantified using densitometry (Total Lab gel analysis; Biosystematica, Mountain Hall, Wales, UK). To control for protein loading for the cleaved capase-3 and ICAM-1, the blots were stripped using a stripping buffer (62.5 mM Tris·HCl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol) and reprobed for β-actin using a monoclonal antibody (1:10,000; Sigma, catalog no. A1978-200UL, control no. 010M4816). To control for protein loading for cleaved caspase-8 and cleaved caspase-9, the respective Western blots were stripped and reprobed for total caspase-8 or total caspase-9.
Statistical Analysis
Data are presented as box plots with the median, interquartile range, and 5–95% confidence intervals. Two-way ANOVA was used to compare data among treatment groups within WT and Ptgs2-/- mice. To identify differences between groups, an all pairwise multiple comparison procedure using the Holm-Sidak method was done as a post hoc test and the group differences are shown as various symbols in the figures. Differences were considered significant when P < 0.05.
RESULTS
Deficiency of COX-2 Attenuated Histological Lung Injury
In WT mice, LPS treatment caused alveolar congestion, hemorrhage, intra-alveolar fibrin, and intra-alveolar infiltrates as shown in the representative lung section in Fig. 1A. In the WT mice, LPS caused a significant increase in the lung injury score (Fig. 1B) and in intra-alveolar infiltrates (Fig. 1C) compared with control WT mice. In the Ptgs2-/- mice, LPS also caused a significantly greater histological evidence of lung injury (Fig. 1, A), with greater lung injury score (Fig. 1B) and intra-alveolar infiltrates (Fig. 1C) than in control Ptgs2-/- mice. However, in the LPS-treated Ptgs2-/- mice the lung injury score and intra-alveolar infiltrates were significantly less than in the LPS-treated WT mice (Fig. 1, B and C).
Figure 1.

Histological evidence of LPS-induced lung injury was attenuated in the cyclooxygenase-2 (COX-2)-deficient mice. Lung tissue was fixed in 4% neutral-buffered formalin, later embedded in paraffin, cut into 4-µm-thick sections, and stained with hematoxylin and eosin (H&E) for histological evaluation. Images were taken with an Aperio AT-2 scanner. A: representative images at ×100 magnification. Histological changes were evaluated by an observer blinded to the treatment. B: the severity of lung injury was graded from 0 (minimal) to 4 (maximal) for alveolar congestion, intra-alveolar infiltrates, inflammation, and hemorrhage in a lung injury score (19). In each animal, 4 lung sections were graded to generate the mean score. There were 6 animals in the wild-type (WT) control and 5 animals in the Ptgs2-/- control, WT LPS, and Ptgs2-/- LPS groups. The animals were evenly split between female and male mice. The lung injury score was calculated by adding the scores in each category. By two-way ANOVA, the lung injury scores were significantly different for both LPS (P < 0.001) and genotype (P < 0.001), and there was a significant interaction between LPS and genotype (P < 0.001). By post hoc analysis, LPS increased the lung injury score in both genotypes, but the lung injury score in the LPS-treated Ptgs2-/- mice was significantly lower than in LPS-treated WT mice. C: intra-alveolar infiltrates were scored on a scale of 0 to 4 (19). By two-way ANOVA, alveolar infiltrates were significantly different for both LPS (P < 0.001) and genotype (P = 0.022), and there was a significant interaction between LPS and genotype (P = 0.006). By post hoc analysis and similar to the lung injury score, LPS-treated Ptgs2-/- mice had significantly fewer intra-alveolar infiltrates than LPS-treated WT mice. *P < 0.01, LPS-treated Ptgs2-/- different from Ptgs2-/- control; #P < 0.001, LPS-treated Ptgs2-/- different from LPS-treated WT.
Deficiency of COX-2 Attenuated the LPS-Induced Increase in TNF-α, MCP-1, and IL-10 mRNA Expression in Mouse Lungs
In WT mice, LPS treatment induced significantly greater TNF-α, MCP-1, and IL-10 mRNA levels in the lungs than did vehicle treatment at 24 h (Fig. 2). The LPS-induced levels of TNF-α, MCP-1, and IL-10 mRNA expression were significantly lower in the lungs of the LPS-treated Ptgs2-/- mice than in LPS-treated WT mice (Fig. 2, B–D). By two-way ANOVA there was a significant interaction between LPS and genotype for all three cytokines/chemokine mRNAs. In contrast, although LPS also induced a significant increase in the C-X-C motif chemokine ligand 2 (CXCL-2) expression in the WT mouse lungs, the LPS-induced CXCL-2 mRNA levels were significantly greater in the Ptgs2-/- mice than in the LPS-treated WT (Fig. 2E). By two-way ANOVA, there was a significant interaction between LPS and genotype for CXCL-2. There was no difference in IL-12 mRNA levels in the lungs of vehicle and LPS-treated mice of either genotype 24 h posttreatment (data not shown).
To better understand the effects of COX-2 deficiency on mechanisms of leukocyte recruitment to areas of injury, we also examined the effect of LPS on ICAM-1 protein expression in WT and Ptgs2-/- mice. ICAM-1 levels were robustly induced in WT mice by treatment with LPS (Fig. 3, A and B). The LPS-induced induction of ICAM-1 protein levels was significantly lower in the lungs from Ptgs2-/- mice compared with the LPS-treated WT mice (Fig. 3B). We then examined the effects of LPS and genotype on myeloperoxidase (MPO) protein levels in the lungs of WT and Ptgs2-/- mice as a marker for leukocyte infiltration. The protein levels of MPO were significantly greater in the lungs from LPS-treated WT mice than from control WT mice (Fig. 3, C and D). The LPS-induced lung MPO protein levels were significantly attenuated in the LPS-treated Ptgs2-/- mice (Fig. 3D).
Figure 3.
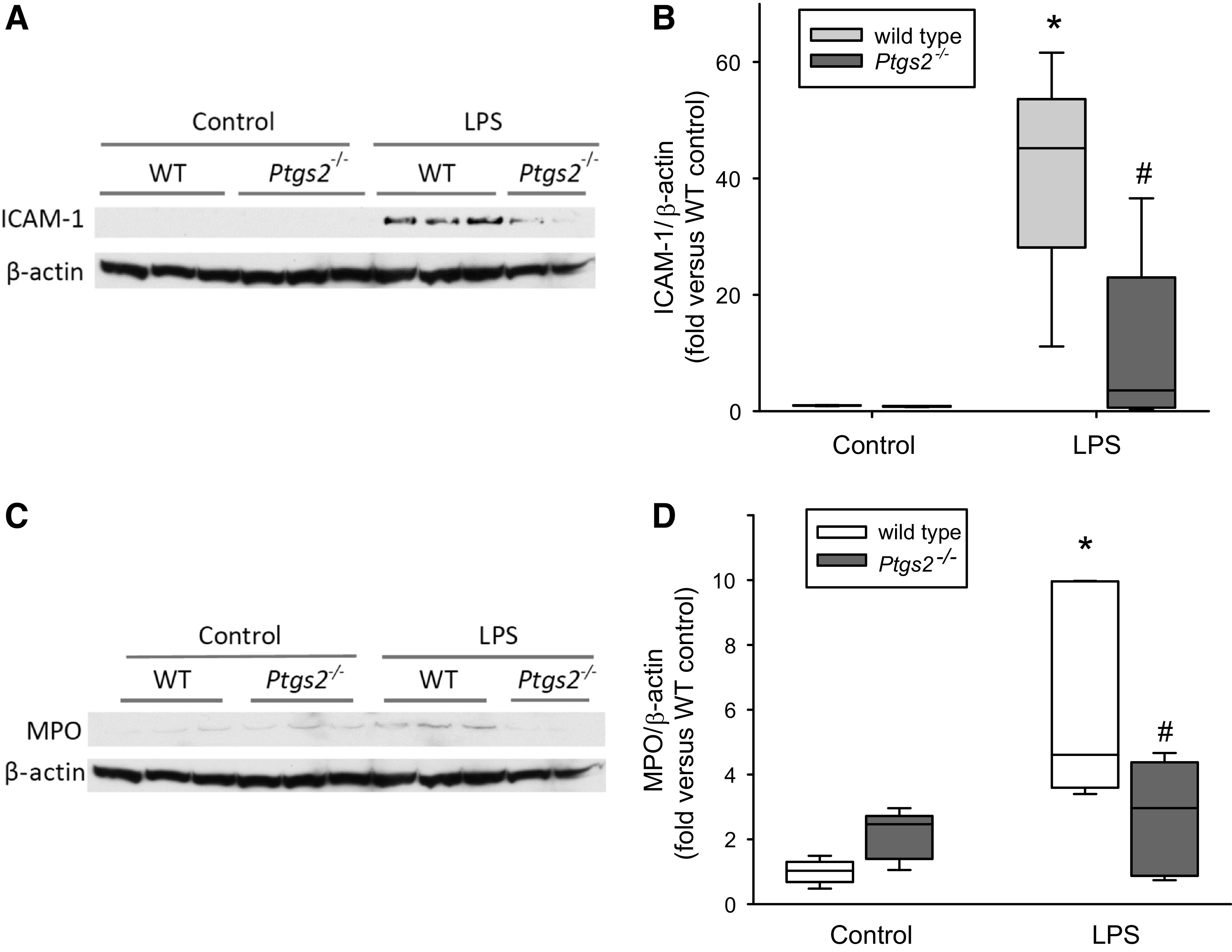
LPS-induced ICAM-1 and myeloperoxidase (MPO) protein levels were attenuated in cyclooxygenase-2 (COX-2)-deficient mice. A: representative Western blots for ICAM-1 from the lungs of wild-type (WT) and Ptgs2-/- mice treated with either vehicle (control) or LPS for 24 h. B: densitometry of Western blots showing lung ICAM-1 protein levels normalized to β-actin for WT (white scaled boxes; n = 6–8 for each condition) and Ptgs2-/- (gray boxes; n = 6 for each condition) mice. By two-way ANOVA, there was a difference in genotype (P < 0.005) and with LPS (P < 0.001) and a significant interaction (P < 0.005). Post hoc analysis identified the following differences: *P < 0.001, different from the WT control; #P < 0.001, different from WT LPS. C: representative Western blots for MPO from the lungs of WT and Ptgs2-/- mice treated with either vehicle (control) or LPS for 24 h. D: densitometry of Western blots showing lung MPO protein levels normalized to β-actin for WT (white box; n = 5–7 for each condition) and Ptgs2-/- (gray boxes; n = 5 for each condition) mice. By two-way ANOVA, there was a difference with LPS (P < 0.005) and a significant interaction between genotype and LPS (P < 0.05). Post hoc analysis identified the following differences: *P < 0.001, WT LPS different from the WT control; #P < 0.05, Ptgs2-/- LPS different from WT LPS.
LPS-Induced Increase in Cleaved Caspase-3 Expression Was Attenuated in COX-2-Deficient Mice
In WT mice, LPS treatment induced substantially greater levels of cleaved caspase-3 protein in the lungs than in vehicle-treated WT mice (Fig. 4A). However, the LPS-induced cleaved caspase-3 induction in the lungs was significantly lower in Ptgs2-/- mice than in the LPS-treated WT mice (Fig. 4A). LPS induced cleaved caspase-9 protein expression in the lungs from the WT mice, whereas the protein levels of cleaved caspase-9 in the lungs from LPS-treated Ptgs2-/- mice were not different from the levels in vehicle-treated Ptgs2-/- mice and significantly lower than the levels in the lungs from WT LPS-treated mice (Fig. 4B). LPS induced moderate cleaved caspase-8 protein expression in both WT and Ptgs2-/- mice, and there was no difference between the genotypes (Fig. 4C).
Figure 4.

LPS-induced caspase-3 activation was attenuated in cyclooxygenase-2 (COX-2)-deficient mice. Representative Western blot and densitometry data for each caspase of interest in the lungs of wild-type (WT; white boxes, n = 6 for each condition) and Ptgs2-/- (gray boxes, n = 6 for WT and n = 5 for Ptgs2-/-) mice are shown. A: cleaved caspase-3. By two-way ANOVA, there was a difference in genotype (P < 0.05) and with LPS (P < 0.001) and a significant interaction (P < 0.05). Post hoc analysis identified the following differences: *P < 0.005, different from control same genotype; #P < 0.05, LPS-treated Ptgs2-/- different from LPS-treated WT. B: cleaved caspase-9. By two-way ANOVA there was a difference with LPS (P < 0.005) and a significant interaction (P < 0.05). Post hoc analysis identified the following differences: *P < 0.001, WT LPS different from WT control; #P < 0.05, Ptgs2-/- LPS different from WT LPS. C: cleaved caspase-8. By two-way ANOVA, there was a difference with LPS (P < 0.001) but no significant interaction (P = 0.92). Post hoc analysis identified the following differences: *P < 0.05, LPS different from control same genotype.
Deficiency of COX-2 Attenuated Apoptosis in the Lung
In WT mice, LPS treatment resulted in substantially more terminal deoxynucleotide transferase dUTP nick end labeling (TUNEL)-positive cells per field of view than in control WT mice (Fig. 5). In Ptgs2-/- mice, LPS treatment also resulted in significantly more TUNEL-positive cells per field of view than in control Ptgs2-/- mice (Fig. 5). However, the LPS-induced increase in TUNEL-positive cells was significantly less in the Ptgs2-/- mice than in the WT mice (Fig. 5).
Figure 5.

LPS increased the number of terminal deoxynucleotide transferase dUTP nick end labeling (TUNEL)-positive cells, which was attenuated in Ptgs2-/- mice. Mice were exposed to either vehicle [wild type (WT): n = 6; Ptgs2-/-: n = 5] or 10 mg/kg LPS (WT: n = 5; Ptgs2-/-: n = 5) for 24 h, and then euthanized. The lungs were fixed and embedded in paraffin, and 5-µm sections were cut for each sample. The slides were assayed for TUNEL using immunohistochemistry. LPS treatment resulted in significantly more TUNEL-positive cells than in control mice in both genotypes. The number of TUNEL-positive cells per visual field were significantly lower in the lungs from LPS treated Ptgs2-/- mice compared with LPS-treated WT lungs. By two-way ANOVA, TUNEL staining was significantly different for both LPS (P < 0.001) and genotype (P < 0.001), and there was a significant interaction between LPS and genotype (P < 0.001). *P < 0.005, LPS different from control same genotype; #P < 0.001, LPS-treated Ptgs2-/- mice different from LPS-treated WT mice.
Deficiency of COX-2 Prevented the LPS-Induced Increase in Lung W/D
In the WT mice, LPS treatment resulted in a significantly greater lung W/D when compared with vehicle treatment (Fig. 6). However, in the Ptgs2-/- mice there was no significant difference in lung W/D between vehicle treatment and LPS treatment (Fig. 6). The LPS-treated WT mice had a significantly greater W/D than did the LPS-treated Ptgs2-/- mice (Fig. 6).
Figure 6.

LPS induced an increase in the wet-to-dry lung weight ratio (W/D) in wild-type (WT) mice, but not in cyclooxygenase-2 (COX-2)-deficient mice. Mice were treated with vehicle (open boxes; n = 6–8 for each condition) or LPS (gray boxes; n = 6–7 for each condition), and 24 h later the right lung was harvested and weighed (wet weight). The right lungs were placed in an oven and weighed daily until the weight stabilized (dry weight). The wet-to-dry lung weight ratio was calculated. By two-way ANOVA, there was a significant effect of genotype (P < 0.001) and LPS (P < 0.001) and there was a significant interaction between the 2 (P < 0.05). Post hoc analysis identified the following differences: *P < 0.001, WT LPS different from WT control; #P < 0.001, different from WT LPS.
Deficiency of COX-2 Attenuated the LPS-Induced Increase in Resistance
We measured pulmonary function in the mice after exposure to either vehicle or LPS. After 24 h of exposure to either vehicle or LPS, the mice were anesthetized and connected to the FlexiVent® system and RRS and ERS were measured. In the WT mice, treatment with LPS resulted in a significantly higher RRS than that seen after vehicle treatment (Fig. 7A). In the Ptgs2-/- mice the LPS-induced increase in RRS was significantly less than that in the LPS-treated WT mice (Fig. 7A). LPS treatment resulted in a significantly greater ERS in both the WT and Ptgs2-/- mice compared with their respective controls and there was no difference between genotypes (Fig. 7B). We also measured quasi-static compliance (since elastance is the reciprocal of compliance) in the anesthetized mice and found that LPS treatment resulted in significantly lower static compliance than in the respective controls and that there was no difference between genotypes (Fig. 7C).
Figure 7.
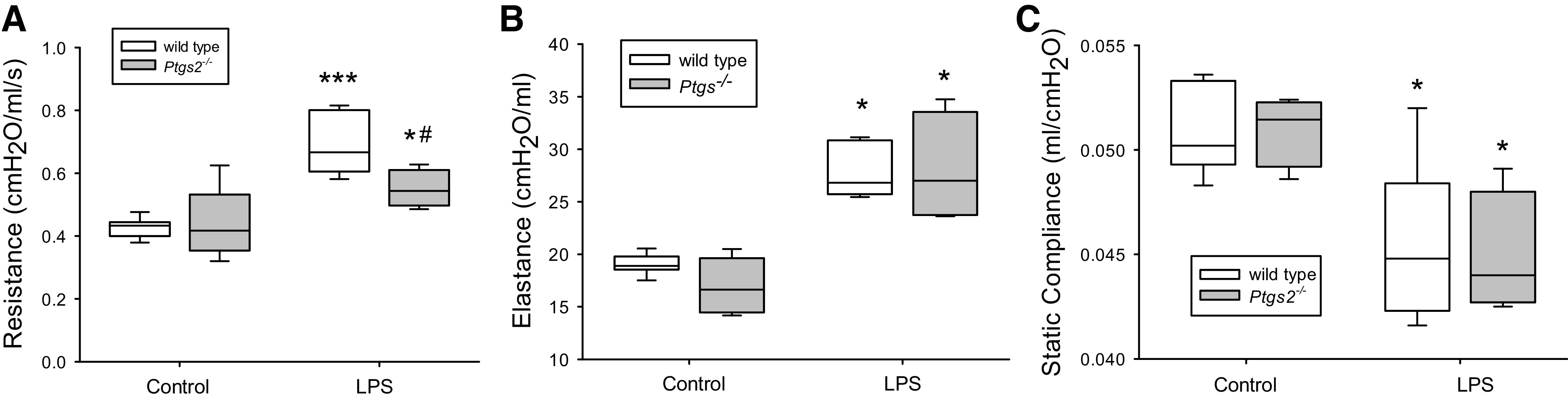
LPS caused an increase in the resistance in the respiratory system (RRS) in wild-type (WT) mice, which was significantly attenuated in the cyclooxygenase-2 (COX-2)-deficient mice. A: RRS in the WT (open boxes; n = 6–8 for each condition) and Ptgs2-/- mice (gray boxes; n = 4–7 for each condition) 24 h after vehicle or LPS treatment. By two-way ANOVA there was a difference in genotype (P = 0.05) and with LPS (P < 0.001) and a significant interaction (P < 0.05). Post hoc analysis identified the following differences: *P < 0.05, different from Ptgs2-/- control; ***P < 0.001, different from the WT control; #P < 0.01, different from WT LPS. B: respiratory system elastance (ERS) in the WT (open boxes, n = 6–8 for each condition) and Ptgs2-/- mice control (gray bars; n = 4–6 for each condition) 24 h after vehicle or LPS treatment. By two-way ANOVA, there was no difference in genotype (P = 0.44), but a significant difference with LPS (P < 0.001), although there was no significant interaction (P = 0.34). Post hoc analysis identified the following differences: *P < 0.001, LPS different from control same genotype. C: quasistatic compliance in the WT (open boxes; n = 5–7 for each condition) and Ptgs2-/- mice (gray bars; n = 4 for each condition) 24 h after vehicle or LPS treatment. By two-way ANOVA, there was no difference in genotype (P = 0.98), but a significant difference with LPS (P < 0.001), and there was no significant interaction (P = 0.81). Post hoc analysis identified the following differences: *P < 0.01, LPS different from the control same genotype.
DISCUSSION
The main findings of this study were that COX-2 played an important positive regulatory role in LPS-induced inflammation and the resultant ALI. Specifically, mice deficient in COX-2 demonstrated attenuation of the following LPS-induced responses: 1) expression of TNF-α, MCP-1, and IL-10; 2) expression of ICAM-1 and MPO; 3) activation of caspase-3 and caspase-9; 4) apoptosis as assessed by TUNEL staining; 5) lung W/D; and 6) RRS. On the other hand, CXCL-2 mRNA expression was greater in the lungs from LPS-treated Ptgs2-/- mice than in the lungs from LPS-treated WT mice. To the best of our knowledge, this is the first description of the effect of deficiency of the Ptgs2 gene on LPS-induced inflammation and resultant ALI in the mouse. Our findings are generally consistent with a previous study in Ptgs2-/- mice demonstrating attenuation of the lung injury caused by the induction of acute pancreatitis (22). Interestingly, Ejima et al. (23) reported that COX-2-deficient mice were resistant to mortality induced by a one-time injection of 100 mg/kg LPS. Our findings suggest that COX-2 plays an important role in inflammatory signaling in LPS-induced ALI, not only as a downstream effector of the inflammatory response, but also as an active participant in the amplification and perpetuation of the inflammatory response.
The cytokines/chemokines that were examined 24 h after LPS treatment demonstrated that the effect of COX-2 deficiency was somewhat variable. We found that the expression of the cytokines TNF-α and IL-10 as well as the chemokine MCP-1 was attenuated in the LPS-treated Ptgs2-/- mice when compared with the LPS-treated WT mice. In contrast, the chemokine CXCL-2 was augmented in the LPS-treated Ptgs2-/- mice when compared with the LPS-treated WT mice. Furthermore, we found that COX-2 deficiency had no effect on the LPS-induced response of IL-12. In a neonatal mouse model of hyperoxia-induced lung injury, Olave et al. (24) reported that the selective COX-2 antagonist nimesulfide significantly attenuated hyperoxia-induced TNF-α expression in lung homogenates, but it had little effect on hyperoxia-induced IL-10 expression levels. We have previously shown in a neonatal mouse model of hyperoxia-induced lung injury that Ptgs2-/- mice had attenuated MCP-1 levels compared with WT mice (8). In a model of sepsis using cecal ligation and perforation, a dual inhibitor of COX-2 and LOX-5 attenuated the increase in lung TNF-α 18 h after CLP but augmented the increase in IL-10 and improved survival following CLP (25). In a study where a large dose (100 mg/kg) of LPS was given intraperitoneally to WT and Ptgs2-/- mice, Ptgs2-/- mice had improved survival and had greater LPS-induced production of IL-10 with no differences in TNF-α in harvested peritoneal macrophages, which led the authors to speculate that the increase in IL-10 may have been responsible for the improvement in survival (23). It may be that in some models of sepsis that the augmentation of IL-10 expression seen with either COX-2 deficiency or COX-2 inhibitors may be involved in improvements in survival. However, in our study, which lasted only 24 h, we did not seen any mortality in any of the groups studied. Furthermore, we did not find an augmentation of LPS-induced IL-10 expression in the lung 24 h after LPS, in fact LPS-induced IL-10 expression was attenuated, and yet we did find improvements in indices of ALI. Thus, it is likely that the protective effects of COX-2 deficiency observed are more complex than the augmentation of any specific anti-inflammatory cytokine or chemokine.
The lung injury caused by LPS is associated with the influx of activated leukocytes including macrophages and neutrophils, and this is consistent with our findings of increased lung inflammation by histology and greater levels of lung MPO protein after LPS treatment in WT mice. Adhesion molecules like ICAM-1 are important for the influx of these activated leukocytes. For example, Liu et al. (26) reported in a rat model of paclitaxel-induced lung injury that the selective COX-2 inhibitor parecoxib attenuated ICAM-1 protein expression and the number of leukocytes in the lung, and improved indexes of lung injury. Similarly, Cuzzocrea et al. (27) in a carrageenan-induced lung injury model in rats found that celecoxib attenuated ICAM-1 expression and cellular infiltration in the lung. It has also been shown that antibodies directed at ICAM-1 attenuate lung injury in various animal models (28). We found that LPS-induced ICAM-1 protein expression and the expression of MPO protein in the lungs were attenuated in the Ptgs2-/- mice. The lower levels of LPS-induced ICAM-1 expression could potentially be a part of the mechanism resulting in the attenuation of lung injury and lower levels of lung MPO protein in the Ptgs2-/- mice, although further studies are needed to delineate the exact role of decreased ICAM-1 expression in the attenuation of LPS-induced ALI in the Ptgs2-/- mice.
The ALI associated with LPS involves apoptosis of cells in the lung, and we have previously shown that mice treated with LPS have significant apoptosis and caspase-3 activation in the lung (18). In this study, we found that LPS activated caspase-3 and caspase-9 in the lungs of the WT mice and that COX-2 deficiency attenuated the LPS-induced activation of caspase-3 and caspase-9. We also found that LPS activated caspase-8 but that there was no difference between the WT and Ptgs2-/- mice. These findings suggest that the attenuation of caspase-3 activation in the Ptgs2-/- mice occurred through the attenuation of the mitochondrial apoptotic pathway (caspase-9). This is consistent with a study in a mouse transgenic model wherein COX-2 was overexpressed in the liver and it was found that the transgenic mice had a more robust induction of liver caspase-9 and caspase-3 after LPS treatment than did WT mice (29). We also found that LPS-induced apoptosis in the lung was attenuated as assessed by TUNEL staining of lung sections. Our finding of COX-2 deficiency attenuating apoptosis in the lungs of mice is different from what has been described in various cancer cells wherein selective COX-2 inhibitors actually induced apoptosis (30–32). For example, new selective COX-2 inhibitors have been recently shown to induce apoptosis through the mitochondrial pathway in cancerous lymphoblasts obtained from patients with ALL but not in normal lymphoblasts obtained from healthy donors (33, 34). Thus the effect of inhibiting COX-2 on apoptosis is different in cancerous cells versus noncancerous cells.
In WT mice we found a significantly greater lung W/D after LPS treatment, whereas in the Ptgs2-/- mice there was no significant difference in lung W/D in vehicle or LPS treatment. This suggests that deficiency in COX-2 protected mice from LPS-induced changes in endothelial barrier function that leads to pulmonary edema. In a recent study in a double hit model of lung injury with LPS and injurious mechanical ventilation, the selective COX-2 inhibitor parecoxib was found to significantly attenuate the ventilator-induced increase in lung W/D (35). We have previously found that LPS treatment resulted in an increase in lung W/D and an increase in RRS and a decrease in CRS in mice (18). In this study we found similar LPS-induced changes in lung W/D and lung mechanics following LPS, although in the Ptgs2-/- mice we found an attenuation of the LPS-induced changes in lung W/D and RRS. However, we found no statistically significant difference in ERS between LPS-treated WT and Ptgs2-/- mice. The lack of attenuation in the LPS-induced increase in ERS despite attenuation of the LPS-induced lung W/D in the Ptgs2-/- mice was a surprise, so we also measured quasi-static compliance and found the same result, i.e., no difference between the genotypes after LPS treatment. These findings may have to do with the magnitude of the difference in lung W/D in this study between the vehicle-treated WT and LPS-treated WT, which was smaller than we saw in our previous study using 129/Sv mice (18). We speculate that the smaller change in lung W/D following LPS challenge could be attributed to differences in mouse strains used in the two studies (CD1 in this study and 129/Sv in our previous study). In has been found that the genetic background of the mouse strain has a considerable effect on the degree of pulmonary edema following lung injury (36). Strain differences have also been shown in cigarette smoke priming of LPS-induced ALI (37). Interestingly, the study by Sakhatskyy et al. (37) also found that significant increases in lung W/D were not always associated with significant decreases in lung compliance. Thus, although we were surprised that the attenuation in LPS-induced increases in lung W/D in the Ptgs2-/- mice were not associated with attenuated ERS or quasistatic compliance, we did find evidence (lung W/D and RRS) of attenuation of LPS-induced lung injury in COX-2-deficient mice. Interestingly, in a bleomycin model of lung fibrosis, it was found that COX-2-deficient mice had exacerbated changes in ERS and static compliance, although there were no differences in the extent of fibrosis between COX-2-deficient and wild-type mice (38). The authors (38) also found an increase in cysteinyl leukotriene levels in the bronchoalveolar lavage fluid from COX-2-deficient mice, suggesting that the changes in respiratory function may have been related to an increase in leukotriene synthesis in COX-2-deficient mice. These findings suggest the role of COX-2 in lung injury may be dependent on the stimulus.
In these studies, we did not find differences between sexes in the responses to LPS (data not shown). We studied essentially 50% males and 50% females, and males and females were the same ages and weight. Although there are likely not large differences in LPS responses between the sexes, we may not have detected smaller differences due to the relatively small numbers studied when the mice were subgrouped by sex. Sex differences in inflammatory responses and lung injury have been previously reported in Wistar rats following the intraperitoneal injection of LPS at a dose of 1.5 mg/kg (39); however, the sex differences disappeared with a 10-fold larger dose of LPS (15 mg/kg). In our study, we used a LPS dose (10 mg/kg) that was closer to the high-dose LPS in that study by Kosyreva et al. (39). In neonatal ICR mice, Nguyen et al. (40) reported no differences between males and females in LPS response as measured by impaired lung development, LPS-induced pulmonary gene expression, or activation of canonical NF-κB signaling.
Perspectives and Significance
Mice deficient in COX-2 had attenuation of the LPS-induced inflammatory response and the resultant acute lung injury. COX-2 deficiency had variable effects on LPS-induced levels of cytokines/chemokines, which underscores the complex nature of their cellular regulation in the lung in response to inflammatory stimuli. We did find that COX-2 clearly has a key role in the LPS-induced upregulation of TNF-α, IL-10, MCP-1, and cxcl-2 expression in the lung. The Ptgs2-/- mice also had lower protein levels of ICAM-1 and MPO following LPS than did WT mice, which may represent another important COX-2-mediated mechanism for LPS-induced ALI. Furthermore, the mitochondrial apoptosis pathway was attenuated in the lungs from LPS-treated Ptgs2-/- mice leading to lower lung levels of activated caspase-3, which likely underlies, at least in part, the attenuated pulmonary edema formation seen in the COX-2 deficient mice. Overall, our findings are consistent with a central role of COX-2 in the initiation and augmentation of LPS-induced ALI. Although further studies are needed to better understand specific mechanistic pathways, we speculate that COX-2 may represent a therapeutic target in ALI.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.D.N., Y.L., L.K.R., and J.R. conceived and designed research; Y.J. performed experiments; L.D.N. and Y.J. analyzed data; L.D.N., B.C., Y.L., L.K.R., and J.R. interpreted results of experiments; L.D.N., and Y.J. prepared figures; L.D.N. and Y.J. drafted manuscript; L.D.N., Y.J., B.C., Y.L., L.K.R., and J.R. edited and revised manuscript; L.D.N., Y.J., B.C., Y.L., L.K.R., and J.R. approved final version of manuscript.
REFERENCES
- 1.Chopra M, Reuben JS, Sharma AC. Acute lung injury: apoptosis and signaling mechanisms. Exp Biol Med (Maywood) 234: 361–371, 2009. doi: 10.3181/0811-MR-318. [DOI] [PubMed] [Google Scholar]
- 2.Galani V, Tatsaki E, Bai M, Kitsoulis P, Lekka M, Nakos G, Kanavaros P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): an up-to-date cell-specific review. Pathol Res Pract 206: 145–150, 2010. doi: 10.1016/j.prp.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Lee IT, Yang CM. Inflammatory signaling involved in airway and pulmonary diseases. Mediators Inflamm 2013: 791231, 2013. doi: 10.1155/2013/791231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mancini AD, Di Battista JA. The cardinal role of the phospholipase A(2)/cyclooxygenase-2/prostaglandin E synthase/prostaglandin E(2) (PCPP) axis in inflammostasis. Inflamm Res 60: 1083–1092, 2011. doi: 10.1007/s00011-011-0385-7. [DOI] [PubMed] [Google Scholar]
- 5.Rumzhum NN, Ammit AJ. Cyclooxygenase 2: its regulation, role and impact in airway inflammation. Clin Exp Allergy 46: 397–410, 2016. doi: 10.1111/cea.12697. [DOI] [PubMed] [Google Scholar]
- 6.Frazier WJ, Wang X, Wancket LM, Li XA, Meng X, Nelin LD, Cato AC, Liu Y. ncreased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J Immunol 183: 7411–7419, 2009. doi: 10.4049/jimmunol.0804343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Y, Liu Y, Nelin LD. Deficiency of cationic amino acid transporter-2 protects mice from hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol 316: L598–L607, 2019. doi: 10.1152/ajplung.00223.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Britt RD, Velten M, Tipple TE, Nelin LD, Rogers LK. Cyclooxygenase-2 in newborn hyperoxic lung injury. Free Radic Biol Med 61: 502–511, 2013. doi: 10.1016/j.freeradbiomed.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Britt RD, Locy ML, Tipple TE, Nelin LD, Rogers LK. Lipopolysaccharide-induced cyclooxygenase-2 expression in mouse transformed clara cells. Cell Physiol Biochem 29: 213–222, 2012. doi: 10.1159/000337602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson JA, Sauer D, Gold JA, Nonas SA. The role of cyclooxygenase-2 in mechanical ventilation-induced lung injury. Am J Respir Cell Mol Biol 47: 387–394, 2012. doi: 10.1165/rcmb.2011-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park EJ, Kim YM, Kim HJ, Chang KC. Luteolin activates ERK1/2- and Ca(2+)-dependent HO-1 induction that reduces LPS-induced HMGB1, iNOS/NO, and COX-2 expression in RAW264.7 cells and mitigates acute lung injury of endotoxin mice. Inflamm Res 67: 445–453, 2018. doi: 10.1007/s00011-018-1137-8. [DOI] [PubMed] [Google Scholar]
- 12.Shaikh SB, Prabhakar Bhandary Y. Effect of curcumin on IL-17A mediated pulmonary AMPK kinase/cyclooxygenase-2 expressions via activation of NFkappaB in bleomycin-induced acute lung injury in vivo. Int Immunopharmacol 85: 106676, 2020. doi: 10.1016/j.intimp.2020.106676. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Luo Z, Bi A, Yang W, An W, Dong X, Chen R, Yang S, Tang H, Han X, Luo L. Compound edaravone alleviates lipopolysaccharide (LPS)-induced acute lung injury in mice. Eur J Pharmacol 811: 1–11, 2017. doi: 10.1016/j.ejphar.2017.05.047. [DOI] [PubMed] [Google Scholar]
- 14.McKnight K, Hoang HD, Prasain JK, Brown N, Vibbert J, Hollister KA, Moore R, Ragains JR, Reese J, Miller MA. Neurosensory perception of environmental cues modulates sperm motility critical for fertilization. Science 344: 754–757, 2014. doi: 10.1126/science.1250598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reese J, Paria BC, Brown N, Zhao X, Morrow JD, Dey SK. Coordinated regulation of fetal and maternal prostaglandins directs successful birth and postnatal adaptation in the mouse. Proc Natl Acad Sci U S A 97: 9759–9764, 2000. doi: 10.1073/pnas.97.17.9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reese J, Anderson JD, Brown N, Roman C, Clyman RI. Inhibition of cyclooxygenase isoforms in late- but not midgestation decreases contractility of the ductus arteriosus and prevents postnatal closure in mice. Am J Physiol Regul Integr Comp Physiol 291: R1717–R1723, 2006. doi: 10.1152/ajpregu.00259.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med 203: 131–140, 2006. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trittmann JK, Jin Y, Liu Y, Nelin LD. Differential effects of the Src family tyrosine kinases Yes and Fyn on lipopolysaccharide-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 321: L392–L403, 2021. [Erratum in Am J Physiol Lung Cell Mol Physiol 321: L988, 2021]. doi: 10.1152/ajplung.00181.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murakami K, McGuire R, Ram C, Jodoin JM, Bjertnaes LJ, Katahira J, Traber LD, Schmalstieg FC, Kawkins HK, Herndon DN, Traber DL. Heparin nebulization attenuates acute lung injury in sepsis following smoke inhalation in sheep. Shock 18: 236–241, 2002. doi: 10.1097/00024382-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Kim VY, Batty A, Li J, Kirk SG, Crowell SA, Jin Y, Tang J, Zhang J, Rogers LK, Deng HX, Nelin LD, Liu Y. Glutathione reductase promotes fungal clearance and suppresses inflammation during systemic candida albicans infection in mice. J Immunol 203: 2239–2251, 2019. doi: 10.4049/jimmunol.1701686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Talavera MM, Kralik N, Jin Y, Chen B, Liu Y, Nelin LD. Mitogen-activated protein kinase phosphatase-1 prevents lipopolysaccharide-induced apoptosis in immature rat intestinal epithelial cells. Pediatr Res 78: 128–136, 2015. doi: 10.1038/pr.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song AM, Bhagat L, Singh VP, Van Acker GG, Steer ML, Saluja AK. Inhibition of cyclooxygenase-2 ameliorates the severity of pancreatitis and associated lung injury. Am J Physiol Gastrointest Liver Physiol 283: G1166–G1174, 2002. doi: 10.1152/ajpgi.00370.2001. [DOI] [PubMed] [Google Scholar]
- 23.Ejima K, Layne MD, Carvajal IM, Kritek PA, Baron RM, Chen YH, Vom Saal J, Levy BD, Yet SF, Perrella MA. Cyclooxygenase-2-deficient mice are resistant to endotoxin-induced inflammation and death. FASEB J 17: 1325–1327, 2003. doi: 10.1096/fj.02-1078fje. [DOI] [PubMed] [Google Scholar]
- 24.Olave N, Lal CV, Halloran B, Bhandari V, Ambalavanan N. Iloprost attenuates hyperoxia-mediated impairment of lung development in newborn mice. Am J Physiol Lung Cell Mol Physiol 315: L535–L544, 2018. doi: 10.1152/ajplung.00125.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bitto A, Minutoli L, David A, Irrera N, Rinaldi M, Venuti FS, Squadrito F, Altavilla D. Flavocoxid, a dual inhibitor of COX-2 and 5-LOX of natural origin, attenuates the inflammatory response and protects mice from sepsis. Crit Care 16: R32, 2012. doi: 10.1186/1364-8535-16-R32. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Liu WJ, Zhong ZJ, Cao LH, Li HT, Zhang TH, Lin WQ. Paclitaxel-induced lung injury and its amelioration by parecoxib sodium. Sci Rep 5: 12977, 2015. doi: 10.1038/srep12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuzzocrea S, Mazzon E, Sautebin L, Dugo L, Serraino I, De Sarro A, Caputi AP. Protective effects of Celecoxib on lung injury and red blood cells modification induced by carrageenan in the rat. Biochem Pharmacol 63: 785–795, 2002. doi: 10.1016/S0006-2952(01)00908-X. [DOI] [PubMed] [Google Scholar]
- 28.Zhao YJ, Yi WJ, Wan XJ, Wang J, Tao TZ, Li JB, Wang JF, Deng XM. Blockade of ICAM-1 improves the outcome of polymicrobial sepsis via modulating neutrophil migration and reversing immunosuppression. Mediators Inflamm 2014: 195290, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han C, Li G, Lim K, DeFrances MC, Gandhi CR, Wu T. Transgenic expression of cyclooxygenase-2 in hepatocytes accelerates endotoxin-induced acute liver failure. J Immunol 181: 8027–8035, 2008. doi: 10.4049/jimmunol.181.11.8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai F, Chen M, Zha D, Zhang P, Zhang X, Cao N, Wang J, He Y, Fan X, Zhang W, Fu Z, Lai Y, Hua ZC, Zhuang H. Curcumol potentiates celecoxib-induced growth inhibition and apoptosis in human non-small cell lung cancer. Oncotarget 8: 115526–115545, 2017. doi: 10.18632/oncotarget.23308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim B, Kim J, Kim YS. Celecoxib induces cell death on non-small cell lung cancer cells through endoplasmic reticulum stress. Anat Cell Biol 50: 293–300, 2017. doi: 10.5115/acb.2017.50.4.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Chen J, Cheng C, Li P, Cai F, Xu H, Lu Y, Cao N, Liu J, Wang J, Hua ZC, Zhuang H. Aspirin potentiates celecoxib-induced growth inhibition and apoptosis in human non-small cell lung cancer by targeting GRP78 activity. Ther Adv Med Oncol 12: 1758835920947976, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pirahmadi N, Zarghi A, Salimi A, Arefi H, Pourahmad J. Beta-lactam structured, 4-(4-(methylsulfonyl)phenyl)-1-pentyl-3-phenoxyazetidin-2-one: selectively targets cancerous b lymphocyte mitochondria. Anticancer Agents Med Chem 17: 1292–1301, 2017. [DOI] [PubMed] [Google Scholar]
- 34.Salimi A, Aghvami M, Azami Movahed M, Zarei MH, Eshghi P, Zarghi A, Pourahmad J. Evaluation of cytotoxic potentials of novel cyclooxygenase-2 inhibitor against ALL lymphocytes and normal lymphocytes and its anticancer effect through mitochondrial pathway. Cancer Invest 38: 463–475, 2020. doi: 10.1080/07357907.2020.1808898. [DOI] [PubMed] [Google Scholar]
- 35.Zhang C, Hu S, Zosky GR, Wei X, Shu S, Wang D, Chai X. Paracoxib alleviates ventilator-induced lung injury through functional modulation of lung-recruited CD11bloLy6Chi monocytes. Shock 55: 236–243, 2021. doi: 10.1097/SHK.0000000000001591. [DOI] [PubMed] [Google Scholar]
- 36.Dodd-O JM, Hristopoulos ML, Welsh-Servinsky LE, Tankersley CG, Pearse DB. Strain-specific differences in sensitivity to ischemia-reperfusion lung injury in mice. J Appl Physiol (1985) 100: 1590–1595, 2006. doi: 10.1152/japplphysiol.00681.2005. [DOI] [PubMed] [Google Scholar]
- 37.Sakhatskyy P, Wang Z, Borgas D, Lomas-Neira J, Chen Y, Ayala A, Rounds S, Lu Q. Double-hit mouse model of cigarette smoke priming for acute lung injury. Am J Physiol Lung Cell Mol Physiol 312: L56–L67, 2017. doi: 10.1152/ajplung.00436.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Card JW, Voltz JW, Carey MA, Bradbury JA, Degraff LM, Lih FB, Bonner JC, Morgan DL, Flake GP, Zeldin DC. Cyclooxygenase-2 deficiency exacerbates bleomycin-induced lung dysfunction but not fibrosis. Am J Respir Cell Mol Biol 37: 300–308, 2007. doi: 10.1165/rcmb.2007-0057OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kosyreva AM, Makarova OV, Kakturskiy LV, Mikhailova LP, Boltovskaya MN, Rogov KA. Sex differences of inflammation in target organs, induced by intraperitoneal injection of lipopolysaccharide, depend on its dose. J Inflamm Res 11: 431–445, 2018. doi: 10.2147/JIR.S178288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen L, Castro O, De Dios R, Sandoval J, McKenna S, Wright CJ. Sex-differences in LPS-induced neonatal lung injury. Sci Rep 9: 8514, 2019. doi: 10.1038/s41598-019-44955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]