

Abstract

An additive-free, visible light-driven annulation between N-aryl amino acids and maleimide to form tetrahydroquinolines (THQs) is disclosed. Photochemical activation of an electron donor–acceptor (EDA) complex between amino acids and maleimides drives the reaction, and aerobic oxygen acts as the terminal oxidant in the net oxidative process. A range of N-aryl amino acids and maleimides have been investigated as substrates to furnish the target THQ in good to excellent yield. Mechanistic investigations, including titration and UV–vis studies, demonstrate the key role of the EDA complex as the photoactive species.
Introduction
α-Functionalization of amines is an invaluable synthetic method and has been used frequently in the synthesis of complex molecular structures, natural products, and biologically active compounds.1−3 Recently, radical approaches to α-functionalization have received widespread attention due to the development of photoredox catalysis, which has realized a range of visible light-driven methods.3−6 Typically, a photoredox catalyst in its excited state acts as an electron acceptor to oxidize the amine via a single electron transfer (SET). The formed amine radical cation can then undergo α-deprotonation to form a reactive alkyl radical that can be reacted with a suitable radical acceptor. Alternatively, the α-aminoalkyl radical can be further oxidized to form the iminium ion and coupled with a nucleophilic species. However, the use of photoredox catalysts can be troublesome and typically relies on expensive and rare transition metals or complex organic dyes. Separation and recycling of the catalyst pose another major problem.7 A way to circumvent the use of a catalyst is to take advantage of an electron donor–acceptor (EDA) complex. An EDA complex is a weak molecular aggregate that can form between a nucleophilic donor and an electrophilic acceptor.8,9 Associated with the formed complex is the emergence of a new electronic transition in the electromagnetic spectrum, absent in either the donor or acceptor. Excitation of the complex at this energy induces a net SET from the donor to the acceptor.9,10 The radicals formed after the SET can subsequently be used in a range of reactions.8,11−17 The fact that the new electronic transition is of lower energy than needed to excite any of the individual species separately very often results in that visible light can be used to excite the EDA complex which is attractive as UV light can be circumvented.
The use of amines as potent electron donors in EDA complexes has been widely documented.8,11−13 Recently, we have shown that tertiary anilines can act as donors in EDA complexes in combination with activated alkenes for the visible light-driven synthesis of tetrahydroquinolines (THQs), circumventing the use of complex and expensive photoredox catalysts.18−20 However, whereas this catalyst-free approach works well for N,N-dialkylated anilines, secondary anilines have not been suitable substrates most likely due to back electron transfer (BET). Thus, a major challenge for applications of EDA complexes in organic synthesis is to overcome the BET that can occur after the photoinitiated SET, suppressing the formation of radical ions.11,21 This is especially prominent for the generation of secondary α-aminoalkyl radicals as the BET from the amine radical cation back to the oxidant is faster than any forward process (Scheme 1A).22 Therefore, developing ways of achieving α-functionalization of secondary anilines is highly desirable.
Scheme 1. Challenges Associated with the Synthesis of N-H–THQs and Biologically Important Targets That Can be Derived from the N-H–THQ Core Structure.

(A) General principle of the generation of secondary α-aminoalkyl radicals from N-alkyl anilines; (B) previous methodologies for the synthesis of N-H–THQ; (C) this work; and (D) numbering of the THQ scaffold and examples of biologically active THQ and quinolines.
Different approaches have been developed to circumvent this problem.22 One strategy relies on installing a good leaving group in the α-position of the aniline which, after the SET, results in the rapid fragmentation of the substrate, driving the reaction forward. Examples include silyl and carboxyl groups. Several reports of the use of N-aryl glycines as the α-aminoalkyl radical precursor in combination with photocatalysts have been reported in the literature.23−49 Previous methods providing α-aminoalkyl radicals from amino acids, using an EDA complex approach, include the use of activated esters as acceptors.14,50,51
Generation of secondary α-aminoalkyl radicals and reacting them with alkenes would potentially give access to N-H–THQ (Scheme 1A), a useful core structure that can be used for the synthesis of a range of biologically active compounds including cytotoxins, γ-sectretase inhibitors, and caspase inhibitors (Scheme 1D).52−55N-H–THQs are typically accessible via Povarov-type reactions between imines and electron-rich alkenes.56 However, synthesis of N-H–THQs bearing electron-withdrawing groups is limited to reverse polarity cycloadditions catalyzed by iridium complexes or via deprotection of the N-Bn–THQ derivative (Scheme 1B).57−59
Inspired by our earlier findings on the synthetic power of aniline–alkene EDA complexes, we postulated that an N-aryl amino acid could serve as an electron donor in combination with an activated alkene as the electron acceptor.20,60 A photoinitiated charge transfer would lead to an α-carboxyl amine radical cation that could fragment irreversibly, via decarboxylation, to give an α-amino radical.61 Herein, we present a novel synthesis where photo-generated secondary α-amino radicals react with maleimides under aerobic conditions to form N-H–THQs (Scheme 1C).
Results and Discussion
To initiate the study, a model system based on N-phenyl glycine (1) and N-phenyl maleimide (2) was chosen. When mixed in solution, an increased light absorption in the visible range compared to that of the individual species was observed (Figure 1). This result, indicating the formation of an EDA complex between the two reactants, prompted the investigation of the photoproducts upon irradiation.
Figure 1.
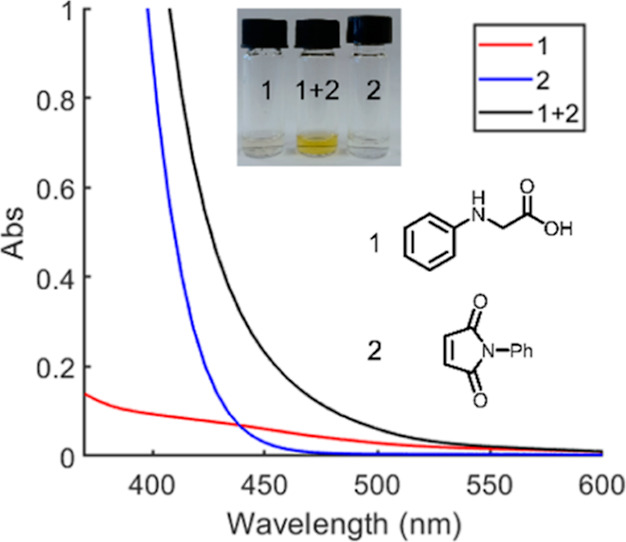
Indication of formation of an EDA complex between 1 and 2.
A solvent screen revealed that methanol with water as the co-solvent and a 440 nm light-emitting diode (LED) as the light source gave the best reaction outcome in terms of yield and purity of the product. Under our optimal conditions, the desired product 3 was formed in 95% yield after 3 h irradiation time (Table 1, entry 1).
Table 1. Effect of Reaction Conditions.

| entry | deviation from standard conditionsa | yield 3b |
|---|---|---|
| 1 | None | 95 (90c) |
| 2 | EtOAc as the solvent | 15 |
| 3 | ACN as the solvent | 52 |
| 4 | 1,4-dioxane as the solvent | 85 |
| 5 | MeOH as the solvent | 90 |
| 6 | DMSO as the solvent | 41 |
| 7 | green LED (525 nm), 3 h | 37 |
| 8 | green LED (525 nm), 18 h | 90 |
| 9 | under an O2 atmosphere | 96 |
| 10 | under a N2 atmosphere | 3 |
| 11 | in the dark | 0 |
| 12 | UV–CFL (370 nm), 3 h | 34 |
Standard conditions: 1 (0.1 mmol), 2 (4 equiv) in MeOH/H2O (2:1). 3 mL was irradiated for 3 h using a 440 nm 40 W LED under an ambient atmosphere.
Determined by GC–FID using chlorobenzene as the internal standard.
Isolated yield.
Omitting the addition of water, using pure methanol as the solvent, decreased the yield of the reaction slightly, whereas other polar aprotic solvents such as acetonitrile, 1,4-dioxane, or dimethyl sulfoxide (DMSO) resulted in significantly lower yields (Table 1, entries 3 and 4). These results are in line with the literature, and the beneficial impact of water on the decarboxylation of N-aryl glycines has been observed previously.23,48,62 Less polar solvents, like ethyl acetate, were not suitable as reaction medium (Table 1, entry 2), and other non-polar solvents could not dissolve the reactants, suppressing the reaction completely. The absorption of the reaction mixture in different solvents was carried out (Supporting Information), and it was observed that methanol and methanol–water resulted in the most red-shifted absorption, compared to less polar systems. Using a green LED (525 nm) resulted in a lower reaction rate with only 37% yield after 3 h (Table 1, entry 7). Increasing the reaction time to 18 h, however, resulted in comparable yields as those with the blue LED (Table 1, entry 8). The lower reaction rate can be related to the significantly lower absorption of the reaction mixture above 500 nm compared to that above 440 nm, see Figure 1. If run under an atmosphere of oxygen, the reaction proceeds smoothly, whereas the exclusion of the oxidant results in diminished conversion (Table 1, entries 9 and 10), showing that oxygen has an important role in this transformation. For discussion on the mechanism, see below.
With our optimal reaction conditions developed, different maleimides as acceptors in the reaction were investigated (Scheme 2). Different N-aryl maleimides were generally well tolerated in the reaction, leading to the products 3–14 in moderate to high yields. Electron-withdrawing halogens in the para position resulted in higher yields than the electron-rich p-OMe substituent (Scheme 2, 4–7). Installation of the bulky t-Bu group in the ortho position of the N-phenyl maleimide decreased the yield significantly to 50% (Scheme 2, entry 8). The product 8 was obtained in an excellent diastereomeric ratio due to the steric hindrance and restricted rotation.
Scheme 2. Scope of Maleimides.

Conditions: substituted maleimide (0.1 mmol) and 1 (4 equiv) in MeOH/H2O (2:1). 3–4 mL was irradiated for 3–6 h using a 440 nm 40 W LED under an ambient atmosphere. For full conditions, see the Experimental Section part.
Next, different N-alkyl-substituted maleimides were investigated as substrates in the reaction leading to products 10–14 in 60–95% yield. For example, the bulky N-cyclohexyl maleimide is a potent substrate in this transformation, and the corresponding N-H–THQ 19 could be isolated in 90% yield indicating that bulky groups on the maleimide do not interfere with the formation of the EDA complex. Unsubstituted maleimide was also a suitable substrate in the reaction leading to the product 9 in 75% yield.
The aromatic substitution on the amine reaction partner was proven to have a significant impact on the reaction outcome (Scheme 3). Electron-donating groups such as methyl or mildly withdrawing groups such as bromide were tolerated giving the products 17–18 in 77–87% yield. However, the strongly electron-withdrawing cyano group resulted in only a trace amount of the product formed (Scheme 3, entry 19). Installation of a methyl group in the ortho position of the amine resulted in a significantly lower yield, and product 20 could be isolated in 59% yield as compared to compound 17 isolated in 87% yield lacking the ortho-methyl substitution. This might be due to the less planar o-substituted aniline interfering with the EDA complex formation. Installation of a methyl group in the less sterically demanding meta position did not affect the yield but gave a mixture of regioisomers 21 and 2′ in a 1.3:1 ratio.
Scheme 3. Scope of N-Aryl Amino Acids.

Conditions: 2 (0.1 mmol), amino acid derivative (4 equiv) in MeOH/H2O (2:1). 3–4 mL was irradiated for 3–6 h using a 440 nm 40 W LED under an ambient atmosphere. For full conditions, see the Experimental Section part.
Introduction of α-substituents of the N-aryl glycine reaction partner led to the formation of products 22–24 (Scheme 3). Generally, the yield was lower compared to that of unsubstituted 1, presumably due to the increased steric hindrance, and the products were obtained as mixtures of diastereomers. When N-phenyl-N-methyl glycine was used as the reactant, product 25 was observed as the only product, indicating that the decarboxylation is significantly faster than α-deprotonation of the amine radical cation. The benzyl- and phenyl-substituted 26 and 27 were also tried as substrates in the reaction; however, in these cases, the amine decomposed upon exposure to the reaction conditions, and no desired product could be obtained.
To show the synthetic potential of the reported method, a reaction was performed on the gram scale to provide the desired product 3 in a yield of 60% (Scheme 4).
Scheme 4. Scale Up of the Annulation Reaction.

The formed product could then easily be transformed to different derivatives (Scheme 5). The N-acylated products 28 and 29 could be obtained in high yields by treating 3 with acyl or toluoyl chloride under basic conditions. The corresponding N-mesyl derivative 30 could also easily be furnished. Under oxidative conditions, 3 could be transformed to the quinoline analogue 31 in a facile manner.
Scheme 5. Derivatization of the Model Product 3 and Methodology Application.
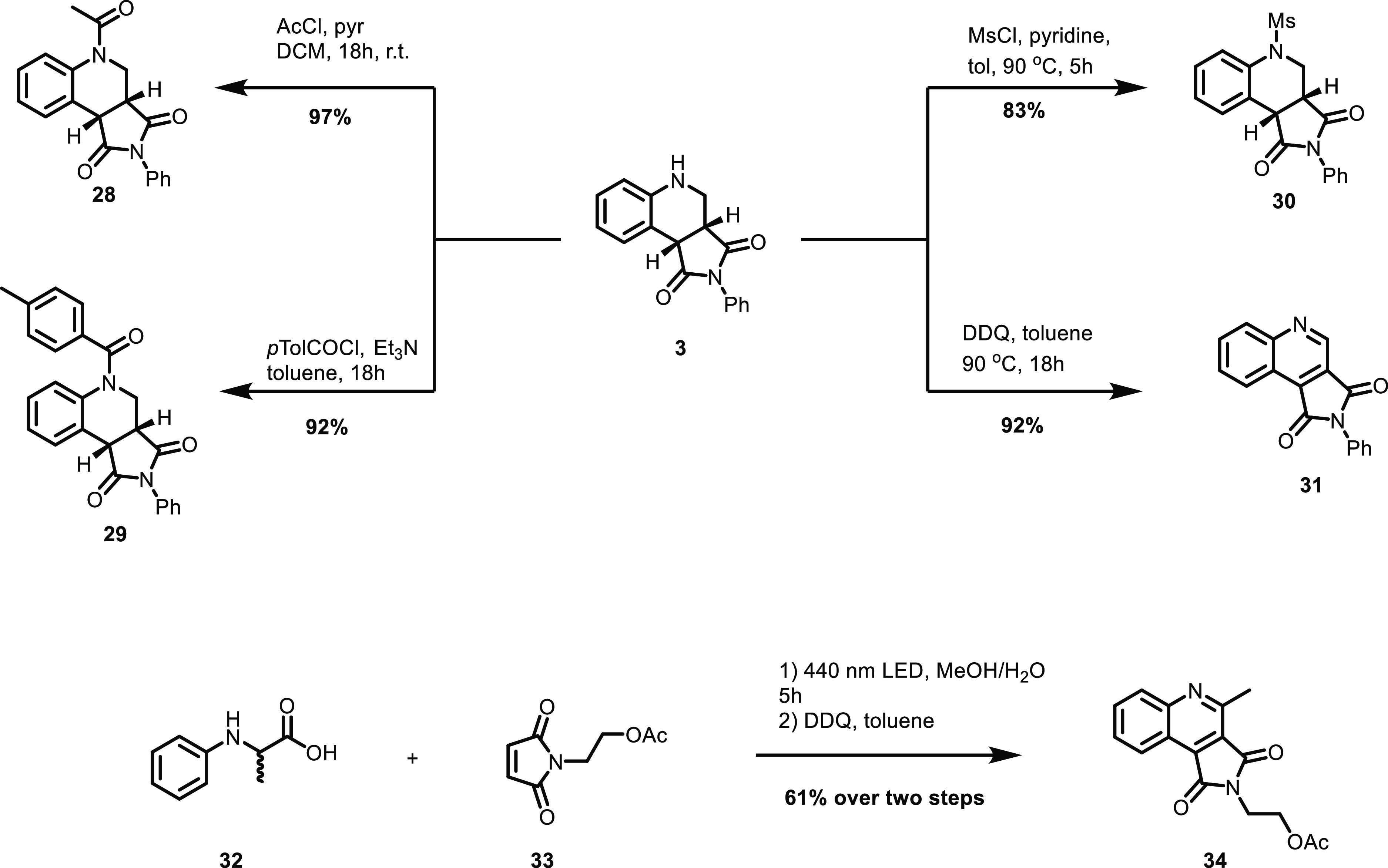
To further exemplify the usefulness of the developed protocol, quinoline 34 was synthesized in 61% yield in a two-step procedure from amino acid 32 and maleimide 33. Quinolines with the core structure of 34, that has been shown to be active as potent caspase-3 inhibitors, are typically synthesized from isatins using a multistep procedure.53−55 The present protocol showcases the possibilities for a fast and facile synthesis of a large library of the compound class.
To elucidate the mechanism of the reaction, a series of control experiments were conducted. First, the necessity of oxygen for a successful reaction outcome was demonstrated as only trace amounts of the product could be observed when the reaction was run under an atmosphere of nitrogen (Table 1, entry 10). Light irradiation was also shown to be needed to drive the reaction (Table 1, entry 11). As demonstrated in Figure 1, the formation of a red-shifted absorbance in the UV–vis spectrum upon mixing of the two reactants points to the formation of an EDA complex which could be vital to the reaction outcome. It has previously been shown that excitation of N-H–maleimide in the presence of N-phenyl glycine can lead to formation of an annulation product.63 The proposed mechanism involves the excitation of maleimide and a SET from ground-state N-phenyl glycine to the excited-state maleimide. However, UVA light was used, and the reactivity was limited to unsubstituted maleimides. The fact that our system works smoothly with a wide range of N-substituted maleimides, is activated under visible light irradiation, and is run under an open atmosphere suggests that another reaction mechanism is operating. When varying the irradiation wavelength, the reaction outcome is significantly changed. As a reflection of the low absorbance of the reaction mixture above 500 nm, using a 525 nm LED as the irradiation source lowered the reaction rate, and only after 18 h of irradiation the product yield could reach 90% (Table 1, entry 8). Furthermore, UVA light (370 nm) was shown to be ineffective in driving the reaction, giving a yield of 34% after 3 h, albeit the significant absorbance of the reaction mixture in this region (Table 1, entry 12). These results are compatible with the notion that an EDA complex between the glycines and the maleimides acts as an important photoactive intermediate. To establish the formation of such a complex in solution, titration experiments were carried out (Supporting Information). The association constant KEDA for the complexes between N-(4′-OMe–phenyl) glycine 35 and either phenyl maleimide 2 or methyl maleimide 36 was determined (Scheme 6).
Scheme 6. Determination of the Association Constant of the EDA Complex.

The association constants for the donor–acceptor pairs were observed to be 0.05 M–1 for phenyl maleimide and 0.2 M–1 for methyl maleimide (see the Supporting Information). These values are comparable to those of previous studies involving EDA complexes dimethyl aniline and maleimides.20 The higher association constant for the amine–methyl maleimide complex compared to that for the phenyl maleimide could be attributed to the higher steric demand of the phenyl group.
To investigate the influence of the electronic properties of the amine, a series of competition experiments were carried out (Figure 2).
Figure 2.
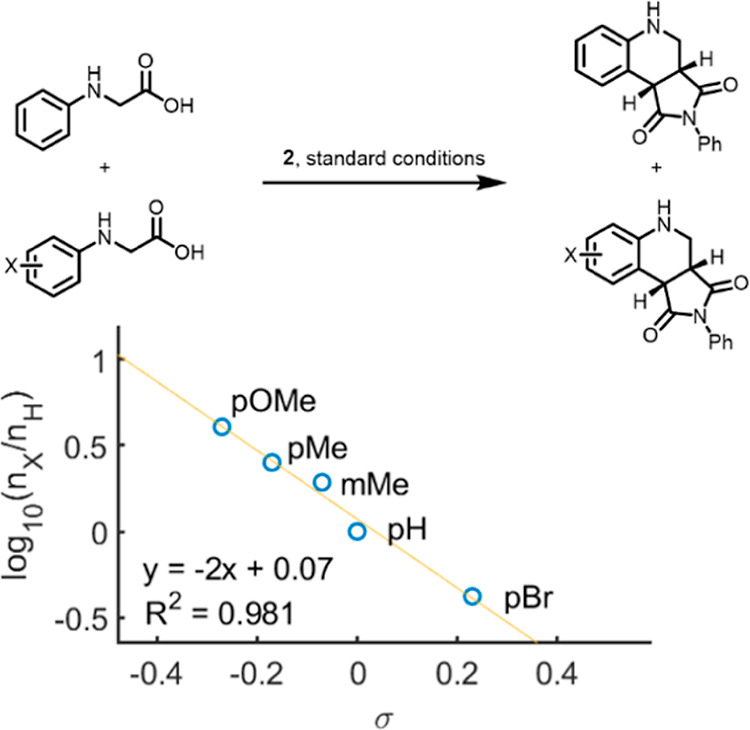
Influence of the electronic properties of the amino acid on the relative reaction rate.
A significant effect of the electronic properties on the relative rate of product formation was observed. Amines with electron-donating groups installed on the aromatic ring reacted faster, whereas electron-withdrawing groups resulted in a decrease in the reaction rate. The large electronic effect is consistent with a build-up of positive charge on the amine, likely due to a single electron oxidation.
The quantum yield of the reaction at 456 nm irradiation was determined to be 5% (Supporting Information), suggesting that the reaction proceeds without any significant contribution from radical chain propagations.
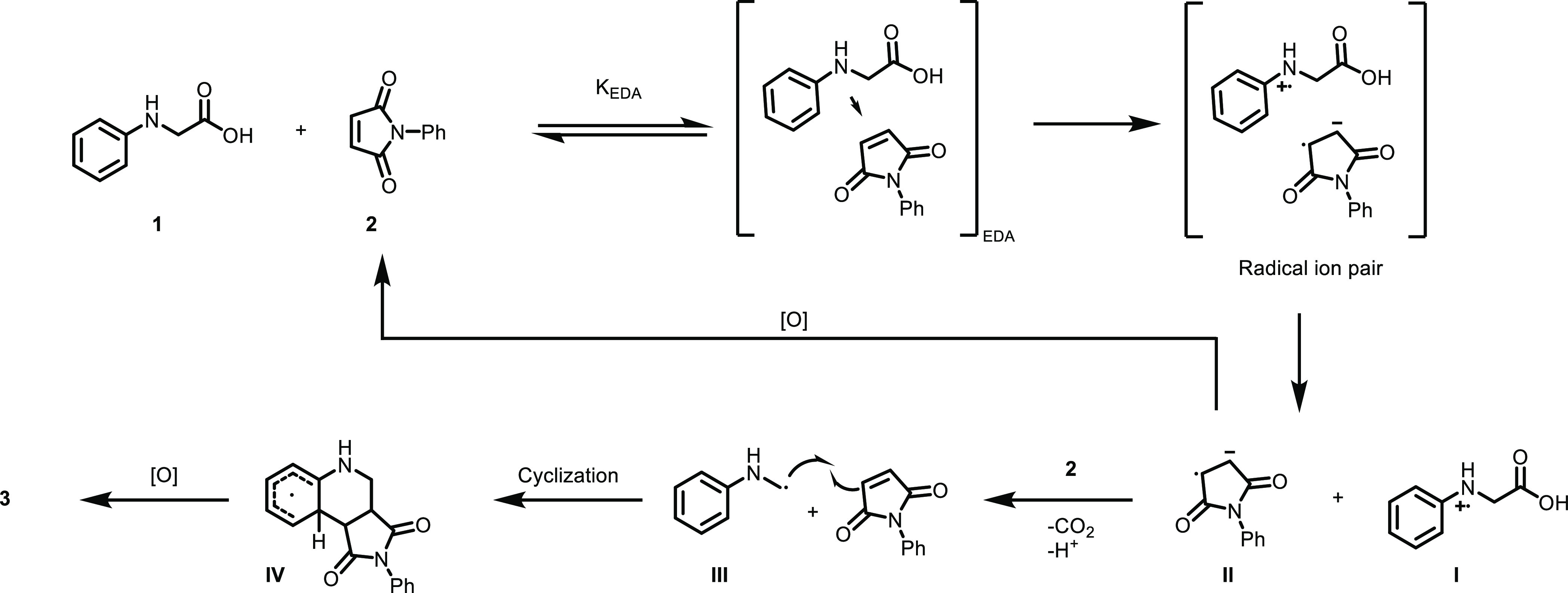
With the background of these observations, a plausible reaction mechanism can be postulated (Scheme 7). Initially, the amine 1 and maleimide 2 associate in solution to form an EDA complex which after photoexcitation leads to a radical ion pair. Deprotonation and decarboxylation of radical anion I generate the α-aminoalkyl radical (III).23,61 The radical anion II is oxidized by molecular oxygen to reform the maleimide. In the final step, radical III reacts with maleimide 2 to form the intermediate IV, which after oxidation yields the final product 3. Since 2 absorbs light above 400 nm (Figure 1), an alternative route that could be operating to some degree is the direct excitation of 2 followed by a SET from ground-state 1 to excited-state 2 resulting in species I and II.63
Scheme 7. Plausible Reaction Mechanism.
In summary, a simple, additive-free, and visible light-driven protocol for the synthesis of N-H–THQs using simple amino acids and maleimides as starting materials is provided. The reaction is thought to proceed via the photoactivation of a novel EDA complex, with aerobic oxygen as the terminal oxidant. The reaction tolerates a range of aromatic and α-substituents on the amine reaction partner and a range of N-substituted maleimides to provide the desired THQ in moderate to high yields.
Experimental Section
General Information
All reagents and solvents were purchased from Sigma-Aldrich and Alfa Aesar and used without any further purification unless specified. Synthesis of maleimides has been described elsewhere.19 Purifications were performed using an automated column chromatography Biotage Isolera Spektra One with a Biotage SNAP-10 g KP-silica column together with a 1 g sample cartridge using n-heptane or petroleum ether (40–60 °C)/ethyl acetate as the solvent mixture unless otherwise noted. 1H (400 MHz) and 13C (101 MHz) NMR spectra were acquired on an Agilent NMR machine at 25 °C. The chemical shifts for 1H and 13C NMR spectra are reported in parts per million (ppm) relative to the residual peak from solvent CDCl3 as the internal standard; 1H NMR at δ 7.26 ppm and 13C NMR at δ 77.16 ppm for CDCl3. All coupling constants (J) are reported in Hertz (Hz), and multiplicities are indicated by s (singlet), d (doublet), dd (doublet of doublet), td (triplet of doublet), ddd (doublet of doublets of doublets), triplet (t), dt (doublet of triplet), and m (multiplet). Structural assignments were made with additional information from gradient correlation spectroscopy, g heteronuclear single quantum coherence, and g heteronuclear multiple bond correlation experiments. Infrared (IR) spectra were recorded on a Bruker ATR FT-IR spectrometer and are reported in wavenumber (cm–1). High-resolution mass spectrometry (HRMS) measurements were performed by CMSI service at Chalmers University of Technology. An Agilent QTOF 6520 equipped with an electrospray interface operated in the positive ionization mode. UV–vis absorption spectra were recorded on a Cary 4000 UV/vis spectrometer, using 1 × 1 cm or 1 × 0.2 cm quartz cuvettes. Light-promoted reactions were carried out in Biotage microwave vials (2–5 mL) under irradiation with a commercial compact fluorescent lamp (Narva Scandinavia, UV light bulb, 15 W, λmax 370 nm), 40 W Kessil PR160L-440 LED lamp (λmax 440 nm), or 40 W Kessil PR160L-525 LED lamp (λmax 525 nm). Gas chromatographic studies were performed using an Agilent 7820A equipped with a flame ionization detector and an Agilent HP-5 19091J-413 column. The emission spectrum of light sources was measured using an AvaSpec-2048-2.
General Procedure A for the Synthesis of N-Aryl Amino Acids
The synthesis was carried out according to a modified published procedure.64 Ethyl bromoacetate (11 mmol, 1 equiv), appropriate aniline (18 mmol, 1.7 equiv), and potassium carbonate (22 mmol, 2 equiv) were stirred in acetonitrile (15 mL) at 60 °C for 18 h. After cooling the mixture to room temperature, the mixture was filtered, and the filtrate was concentrated in vacuo. The residue was partitioned between ethyl acetate and water, and the aqueous phase was extracted three times with ethyl acetate. The combined organic layers were dried over sodium sulphate and were concentrated under reduced pressure. The crude product was dissolved in methanol (10 mL), and an aqueous solution of sodium hydroxide (500 mg) was added under strong stirring. The mixture was stirred at room temperature until full conversion as determined by thin layer chromatography (TLC), and the methanol was then removed under reduced pressure. The aqueous phase was washed with ethyl acetate three times and was then acidified to pH 2 with concentrated hydrochloric acid. Ethyl acetate was then used to extract the product three times, and the combined organic layers were dried over sodium sulphate and concentrated in vacuo. The crude product was recrystallized from hot ethanol twice to give the desired N-aryl glycine.
General Procedure B for the Synthesis of N-Aryl Amino Acids
Following a published procedure,65 the appropriate amino acid (3.2 mmol, 1 equiv), copper iodide (61 mg, 0.32 mmol, 0.1 equiv), potassium carbonate (660 mg, 4.8 mmol, 1.5 equiv), and bromobenzene (500 mg, 3.2 mmol, 1 equiv) were added to a 10–20 mL Biotage microwave vial followed by dimethylacetamide (4 mL). The vial was capped and was purged with nitrogen flow for 10 min. The mixture was then heated to 90 °C for 18 h. Water was added followed by concentrated hydrochloric acid until pH 2. The product was extracted with ethyl acetate three times, and the combined organic layers were dried over sodium acetate, and the solvent was removed under reduced pressure. The solid residue was recrystallized from boiling ethanol twice to yield the desired N-aryl amino acid.
p-Toulylglycine (S1)
General procedure A was used to furnish compound S1 as a slightly yellow solid (1.2 g, 68%). Spectroscopic data are in accordance with the literature;661H NMR (400 MHz, DMSO-d6): δ; 1H NMR (400 MHz, DMSO): δ 6.89 (d, J = 7.9 Hz, 2H), 6.53–6.46 (m, 2H), 3.75 (d, J = 1.1 Hz, 2H), 2.14 (s, 3H) ppm; 13C{1H} (201 MHz, DMSO-d6): δ 172.7, 145.6, 129.3, 124.9, 112.5, 45.1, 20.1 ppm.
(2,4-Dimethylphenyl)glycine (S2)
General procedure A was used to furnish compound S2 as a slightly white solid (820 mg, 43%). Spectroscopic data are in accordance with the literature;671H NMR (400 MHz, DMSO-d6): δ 6.82–6.76 (m, 2H), 6.29–6.23 (m, 1H), 3.78 (s, 2H), 2.13 (s, 3H), 2.06 (s, 3H); 13C{1H} (201 MHz, DMSO-d6): δ 172.9, 143.6, 130.6, 126.9, 124.5, 121.7, 109.3, 45.1, 20.0, 17.4 ppm.
(4-Methylphenyl)glycine (S3)
General procedure A was used to furnish compound S3 as a slightly yellow solid (700 mg, 39%). Spectroscopic data are in accordance with the literature;681H NMR (400 MHz, DMSO-d6): δ 6.95 (t, J = 7.6 Hz, 1H), 6.42–6.31 (m, 3H), 3.76 (d, J = 0.7 Hz, 2H), 2.17 (s, 3H); 13C{1H} (201 MHz, DMSO-d6): δ 172.7, 148.1, 137.8, 128.7, 117.3, 112.9, 109.6, 44.8, 21.4 ppm.
N-Phenylvaline (S4)
General procedure B was used to furnish compound S4 as a slightly yellow solid (320 mg, 52%). Spectroscopic data are in accordance with the literature;651H NMR (400 MHz, DMSO-d6): δ 7.09–6.99 (m, 2H), 6.61 (d, J = 8.0 Hz, 2H), 6.56–6.50 (m, 1H), 3.61 (d, J = 6.8 Hz, 1H), 2.04 (h, J = 6.8 Hz, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 6.7 Hz, 2H) ppm; 13C{1H} (201 MHz, DMSO-d6): δ 174.8, 148.3, 128.8, 116.2, 112.5, 61.9, 30.4, 19.2, 19.0 ppm.
N-Phenylleucine (S5)
General procedure B was used to furnish compound S5 as a slightly yellow solid (195 mg, 30%). Spectroscopic data are in accordance with the literature;651H NMR (800 MHz, DMSO-d6): δ 7.10–7.01 (m, 2H), 6.57–6.49 (m, 3H), 3.83 (dd, J = 8.8, 5.7 Hz, 1H), 1.78 (dt, J = 13.5, 6.8 Hz, 1H), 1.68–1.49 (m, 2H), 0.94 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H) ppm; 13C{1H} (201 MHz, DMSO-d6): δ 175.9, 148.1, 128.9, 116.1, 112.2, 54.1, 41.1, 24.4, 22.8, 21.8 ppm.
Synthesis of Maleimide 33(69)
Following a procedure modified from the literature, maleic anhydride (3.0 g, 31 mmol, 1.5 equiv), ethanolamine (1.3 g, 20 mmol, 1 equiv) in acetic acid (38 mL), and toluene (12 mL) were stirred at 120 °C for 18 h. The solvents were then removed under reduced pressure, and the oily residue was taken up in ethyl acetate and was washed in order with water, saturated sodium bicarbonate solution, and brine. The organic layer was then dried over sodium sulphate and was concentrated under reduced pressure to yield a solid that was recrystallized from ethanol to furnish the desired product as a white solid (1.5 g, 40%).
2-(2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl Acetate (33)
Compound 33 was afforded as a white solid (1.5 g, 40%). Spectroscopic data are in accordance with the literature;691H NMR (400 MHz, chloroform-d): δ 6.73 (s, 1H), 3.86–3.70 (m, 2H), 2.02 (s, 1H); 13C{1H} (101 MHz, chloroform-d): δ 171.0, 170.6, 134.4, 61.6, 37.0, 20.9 ppm.
General Procedure Oxidative Annulation Reaction
To a 2–5 mL Biotage microwave vial were added N-substituted maleimide (0.1 mmol, 1 equiv) and N-phenyl glycine (0.4 mmol, 4 equiv). Methanol (2 mL) and water (1 mL) were then added, and the mixture was stirred until it turned homogeneous. The reaction mixture was then stirred for 3 h under an open atmosphere irradiated with a 440 nm Kessil LED, at a distance of 10 cm. A fan was used to ensure a stable temperature <25 °C. When the reaction was complete, as determined by TLC (SiO2, 25% ethyl acetate in n-heptane), the methanol was removed in vacuo, and the residue was taken up in 10 mL of ethyl acetate. The organic phase was then washed with saturated sodium hydrogen carbonate solution (2 × 5 mL) and was then dried and evaporated in vacuo. If further purification was needed, the product was loaded on a silica column and eluted with a mixture of ethyl acetate in petroleum ethers to afford the desired THQ product.
5-Methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3)23
On a 1.15 mmol scale, compound 3 was afforded as a yellow solid (290 mg, 90%). For the gram-scale version of the reaction, 2 (708 mg, 4.1 mmol, 1 equiv) and 1 (1.7 g, 11.6 mmol, 3 equiv) were dissolved in 70 mL of methanol–water 2:1 in a 100 mL round bottom flask. The reaction mixture was then stirred under an ambient atmosphere at room temperature for 18 h under irradiation with two 40 W Kessil blue LEDs at a distance of 10 cm. The methanol was then removed under reduced pressure, and ethyl acetate was used to dissolve the product. The organic phase was washed with sodium bicarbonate solution and was then dried over magnesium sulphate. The solvent was then removed, and the crude product was purified using flash chromatography (SiO2, 0–20% ethyl acetate in petroleum ethers), to yield 3 as a yellow solid (680 mg, 60%). Spectroscopic data are in accordance with the literature;231H NMR (400 MHz, chloroform-d): δ 7.55 (d, J = 7.8 Hz, 1H), 7.47–7.39 (m, 2H), 7.39–7.34 (m, 1H), 7.27 (d, J = 5.9 Hz, 2H), 7.15–7.09 (m, 1H), 6.87 (td, J = 7.5, 1.3 Hz, 1H), 6.63 (dd, J = 8.0, 1.3 Hz, 1H), 4.17 (d, J = 9.3 Hz, 1H), 3.78 (dd, J = 11.3, 3.2 Hz, 1H), 3.55 (ddd, J = 9.3, 4.3, 3.1 Hz, 1H), 3.33 (dd, J = 11.3, 4.3 Hz, 1H) ppm; 13C{1H} (101 MHz, chloroform-d): δ 177.6, 175.9, 146.1, 132.1, 130.6, 129.1, 128.7, 128.5, 126.5, 120.2, 116.9, 115.8, 43.4, 41.7, 41.6 ppm.
(3aR,9bS)-2-(4-Fluorophenyl)-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (4)
Compound 4 was afforded as a yellow solid after purification on a silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (16.2 mg, 54%); mp °C; 1H NMR (400 MHz, chloroform-d): δ 7.53 (dd, J = 7.9, 1.3 Hz, 1H), 7.30–7.23 (m, 2H), 7.11 (dddd, J = 8.2, 6.7, 3.3, 1.8 Hz, 3H), 6.88 (td, J = 7.5, 1.3 Hz, 1H), 6.63 (dd, J = 8.0, 1.2 Hz, 1H), 4.16 (d, J = 9.3 Hz, 1H), 3.87–3.73 (m, 2H), 3.54 (ddd, J = 9.3, 4.2, 3.0 Hz, 1H), 3.31 (dd, J = 11.2, 4.3 Hz, 1H) ppm; 13C{1H} (101 MHz, chloroform-d): δ 177.5, 175.8, 162.2 (d, 1JC–F = 248.3 Hz), 146.1, 130.6, 128.6, 128.4 (d, 3JC–F = 8.8 Hz), 127.9 (d, 4JC–F = 3.3 Hz), 120.4, 116.8, 116.3, 116.1, 115.9, 43.4, 41.7, 41.6 ppm; 19F{1H} NMR (659 MHz, chloroform-d): δ −112.37 ppm; FTIR (ATR) ν: 1369, 1708, 1605, 1509, 1389, 1223, 1773, 820, 757 cm–1; HRMS (ESI) m/z: calcd C17H14FN2O2 [M + H]+, 297.1039; found, 297.1039.
(3aR,9bS)-2-(4-Chlorophenyl)-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (5)
Compound 5 was afforded as a yellow solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (31.5 mg, 87%); mp 188.0–191.5 °C; 1H NMR (400 MHz, chloroform-d): δ 7.53 (dt, J = 7.8, 1.1 Hz, 1H), 7.44–7.37 (m, 2H), 7.26–7.19 (m, 2H), 7.12 (td, J = 7.7, 1.5 Hz, 1H), 6.88 (td, J = 7.5, 1.3 Hz, 1H), 6.63 (dd, J = 8.0, 1.2 Hz, 1H), 4.17 (d, J = 9.3 Hz, 1H), 3.77 (dd, J = 11.3, 3.1 Hz, 1H), 3.54 (ddd, J = 9.3, 4.3, 3.1 Hz, 1H), 3.32 (dd, J = 11.3, 4.3 Hz, 1H); 13C{1H} (101 MHz, chloroform-d): δ 177.3, 175.6, 146.1, 134.4, 130.6, 130.5, 129.4, 128.7, 127.7, 120.4, 116.7, 115.9, 43.5, 41.7, 41.6 ppm; FTIR (ATR) ν: 3371, 1710, 1603, 1491, 1387, 1360, 1278, 1195, 1176, 1130, 1089, 1016, 878, 815, 762, 725, 649, 598 cm–1; HRMS (ESI) m/z: calcd C17H14ClN2O2 [M + H]+, 313.0744; found, 313.0745.
(3aR,9bS)-2-(4-Bromophenyl)-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (6)
Compound 6 was afforded as a yellow solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (30 mg, 81%); mp 182.5–184.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.61–7.49 (m, 3H), 7.22–7.16 (m, 2H), 7.16–7.08 (m, 1H), 6.88 (td, J = 7.5, 1.2 Hz, 1H), 6.63 (dd, J = 8.0, 1.2 Hz, 1H), 4.17 (d, J = 9.4 Hz, 1H), 3.79 (d, J = 3.0 Hz, 1H), 3.76 (t, J = 3.0 Hz, 1H), 3.62–3.51 (m, 1H), 3.32 (dd, J = 10.8, 4.3 Hz, 1H); 13C{1H} (101 MHz, chloroform-d): δ 177.3, 175.6, 146.1, 132.3, 131.0, 130.6, 128.7, 128.0, 122.5, 120.4, 116.7, 115.9, 43.5, 41.7, 41.6 ppm; FTIR (ATR) ν: 1708, 1601, 1487, 1392, 1353, 1308, 1264, 1196, 1178, 1112, 1069, 1013, 809, 753, 722, 638, 600, 559 cm–1; HRMS (ESI) m/z: calcd C17H14BrN2O2 [M + H]+, 357.0239; found, 357.0237.
(3aR,9bS)-2-(4-Methoxyphenyl)-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (7)
Compound 7 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (20 mg, 58%); mp 184.5–186.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.54 (d, J = 7.6 Hz, 1H), 7.20–7.15 (m, 2H), 7.11 (dddd, J = 7.9, 7.3, 1.5, 0.6 Hz, 1H), 6.96–6.91 (m, 2H), 6.87 (td, J = 7.5, 1.2 Hz, 1H), 6.63 (dd, J = 8.0, 1.2 Hz, 1H), 4.15 (d, J = 9.3 Hz, 1H), 3.86–3.73 (m, 5H), 3.56–3.44 (m, 1H), 3.38–3.26 (m, 1H); 13C{1H} (176 MHz, chloroform-d): δ 177.8, 176.1, 159.6, 146.1, 130.7, 128.5, 127.8, 124.8, 120.3, 117.0, 115.9, 114.5, 55.6, 43.3, 41.8, 41.6; FTIR (ATR) ν: 3367, 1707, 1606, 1511, 1392, 1358, 1301, 1250, 1165, 1109, 1029, 819, 757, 582 cm–1; HRMS (ESI) m/z: calcd C18H17N2O3 [M + H]+, 309.1239; found, 309.1254.
(3aR,9bS)-2-[2-(tert-Butyl)phenyl]-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (8)
Compound 8 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (18.1 mg, 50%); mp 125.0–126.5 °C; 1H NMR (400 MHz, chloroform-d): δ 7.56 (dd, J = 8.1, 1.4 Hz, 1H), 7.52 (dt, J = 7.8, 1.1 Hz, 1H), 7.35 (ddd, J = 8.1, 7.3, 1.5 Hz, 1H), 7.22–7.16 (m, 1H), 7.13 (dddd, J = 7.9, 7.4, 1.5, 0.5 Hz, 1H), 6.87 (td, J = 7.5, 1.2 Hz, 1H), 6.68 (dd, J = 7.8, 1.5 Hz, 1H), 6.65 (dd, J = 8.0, 1.2 Hz, 1H), 4.16 (d, J = 9.4 Hz, 1H), 3.83 (s, 1H), 3.76 (dt, J = 11.4, 2.6 Hz, 1H), 3.53 (ddd, J = 9.4, 4.4, 3.1 Hz, 1H), 3.30 (dd, J = 11.2, 4.4 Hz, 1H), 1.35 (s, 9H) ppm; 13C{1H} (101 MHz, chloroform-d): δ 178.9, 176.9, 147.9, 146.3, 131.0, 130.9, 130.6, 129.8, 128.8, 128.5, 127.5, 120.3, 117.1, 115.8, 43.7, 42.0 (2C), 35.8, 31.8 ppm; FTIR (ATR) ν: 3364, 1699, 1489, 1446, 1378, 1318, 1261, 1160, 1122, 882, 810, 750, 728, 633, 582, 555, 541, 529, 501 cm–1; HRMS (ESI) m/z: calcd C21H23N2O2 [M + H]+, 335.1760; found, 335.1761.
(3aR,9bS)-3a,4,5,9b-Tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (9)
Compound 9 was afforded as a white solid after purification on the silica column using a gradient of 0–50% ethyl acetate in petroleum ethers (26.3 mg, 75%). Spectroscopic data are in accordance with the literature;631H NMR (400 MHz, acetone-d6): δ 10.00 (s, 1H), 7.37 (dt, J = 7.5, 1.2 Hz, 1H), 7.02 (dddd, J = 7.9, 7.2, 1.5, 0.6 Hz, 1H), 6.77–6.65 (m, 2H), 4.04 (d, J = 9.2 Hz, 1H), 3.54 (dd, J = 11.3, 3.1 Hz, 1H), 3.49 (ddd, J = 9.3, 4.4, 3.0 Hz, 1H), 3.14 (ddd, J = 11.2, 4.4, 0.8 Hz, 1H) ppm; 13C{1H} NMR (101 MHz, acetone-d6): δ 179.8, 178.1, 148.0, 131.1, 128.5, 119.5, 118.5, 116.1, 45.1, 43.4, 41.8 ppm.
(3aR,9bS)-2-Methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (10)
Compound 10 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (25.6 mg, 60%). Spectroscopic data are in accordance with the literature;701H NMR (400 MHz, chloroform-d): δ 7.49 (dt, J = 7.7, 1.0 Hz, 1H), 7.08 (td, J = 7.7, 1.5 Hz, 1H), 6.85 (td, J = 7.5, 1.2 Hz, 1H), 6.58 (dd, J = 8.0, 1.2 Hz, 1H), 4.00 (d, J = 9.1 Hz, 1H), 3.69 (dd, J = 11.3, 2.9 Hz, 1H), 3.36 (ddd, J = 9.2, 4.3, 2.9 Hz, 1H), 3.23 (dd, J = 11.3, 4.3 Hz, 1H), 2.98 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.7, 176.9, 146.1, 130.5, 128.4, 120.2, 117.1, 115.8, 43.4, 41.54, 41.48, 25.4 ppm.
(3aR,9bS)-2-Ethyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (11)
Compound 11 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (17.4 mg, 67%). Spectroscopic data are in accordance with the literature;231H NMR (400 MHz, chloroform-d): δ = 7.50 (dt, J = 7.7, 1.1 Hz, 1H), 7.14–7.07 (m, 1H), 6.86 (td, J = 7.5, 1.2 Hz, 1H), 6.59 (dd, J = 8.0, 1.2 Hz, 1H), 3.98 (d, J = 9.2 Hz, 1H), 3.73 (d, J = 3.0 Hz, 1H), 3.66 (dt, J = 11.2, 3.0 Hz, 1H), 3.55 (qd, J = 7.2, 5.8 Hz, 2H), 3.35 (dddd, J = 8.9, 4.2, 3.1, 1.0 Hz, 1H), 3.29–3.21 (m, 1H), 1.14 (t, J = 7.2 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.4, 176.6, 146.1, 130.5, 128.3, 120.2, 117.2, 115.8, 43.3, 41.7, 41.4, 34.3, 13.1 ppm.
(3aR,9bS)-2-Propyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (12)
Performed on a 0.19 mmol scale. Compound 12 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (33.9, 74%); mp 105.5–107.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.53–7.47 (m, 1H), 7.13–7.05 (m, 1H), 6.86 (td, J = 7.5, 1.2 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 3.98 (d, J = 9.1 Hz, 1H), 3.66 (dd, J = 11.3, 3.2 Hz, 1H), 3.53–3.39 (m, 2H), 3.38–3.32 (m, 1H), 3.24 (dd, J = 11.3, 4.4 Hz, 1H), 1.56 (h, J = 7.3 Hz, 2H), 0.81 (t, J = 7.4 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.6, 176.9, 145.9, 130.5, 128.4, 120.3, 117.4, 115.8, 43.2, 41.7, 41.4, 40.9, 21.0, 11.2 ppm; FTIR (ATR) ν: 3332, 1771, 1686, 1604, 1489, 1434, 1398, 1339, 1310, 1257, 1204, 1138, 1105, 813, 759, 731, 687, 606, 508 cm–1; HRMS (ESI) m/z: calcd C14H17N2O2 [M + H]+, 245.1290; found, 245.1293.
(3aR,9bS)-2-Cyclohexyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (13)
Compound 13 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (27.5 mg, 90%); mp 150.5–152 °C; 1H NMR (400 MHz, chloroform-d): δ 7.48 (dt, J = 7.7, 1.1 Hz, 1H), 7.08 (td, J = 7.7, 1.5 Hz, 1H), 6.85 (td, J = 7.5, 1.2 Hz, 1H), 6.58 (dd, J = 8.0, 1.2 Hz, 1H), 4.03–3.86 (m, 2H), 3.72 (s, 1H), 3.61 (dd, J = 11.0, 3.1 Hz, 1H), 3.34–3.11 (m, 2H), 2.23–1.98 (m, 2H), 1.87–1.72 (m, 2H), 1.67–1.45 (m, 3H), 1.35–1.12 (m, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.6, 176.9, 146.0, 130.6, 128.9, 120.1, 117.4, 115.7, 52.3, 42.9, 41.9, 41.2, 29.0, 28.9, 25.94, 25.89, 25.2 ppm; FTIR ν: 1734, 1700, 1653, 1635, 1559, 1540, 1521, 1506, 1473, 1457, 1374, 668 cm–1; HRMS (ESI) m/z: calcd C17H21N2O2 [M + H]+, 285.1603; found, 285.1606.
(3aR,9bS)-2-tert-Butyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (14)
Compound 14 was afforded as a white solid after simple extraction of excess amino acids and drying (26.2 mg, 95%); mp 163.0–164.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.46 (dt, J = 7.6, 1.1 Hz, 1H), 7.12–7.05 (m, 1H), 6.84 (td, J = 7.5, 1.2 Hz, 1H), 6.59 (dd, J = 7.9, 1.2 Hz, 1H), 3.83 (d, J = 8.8 Hz, 1H), 3.63–3.56 (m, 1H), 3.25–3.16 (m, 2H), 1.54 (s, 9H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 179.5, 178.0, 145.9, 130.6, 128.3, 120.0, 117.5, 115.7, 58.9, 42.9, 41.8, 41.6, 28.5 ppm; FTIR (ATR) ν: 1695, 1602, 1488, 1364, 1340, 1265, 1218, 1165, 1124, 1105, 1047, 813, 751, 721, 692, 651, 616, 563, 519, 471 cm–1; HRMS (ESI) m/z: calcd C15H19N2O2 [M + H]+, 259.1447; found, 259.1449.
(3aR,9bS)-2-Benzyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (15)
Compound 15 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (25.9 mg, 81%). Spectroscopic data are in accordance with the literature;231H NMR (400 MHz, chloroform-d): δ 7.47 (dt, J = 7.6, 1.1 Hz, 1H), 7.32–7.19 (m, 5H), 7.07 (td, J = 7.6, 1.5 Hz, 1H), 6.84 (td, J = 7.5, 1.2 Hz, 1H), 6.57 (dd, J = 8.0, 1.2 Hz, 1H), 4.63 (q, J = 14.3 Hz, 2H), 3.97 (d, J = 9.1 Hz, 1H), 3.75 (s, 1H), 3.62 (dt, J = 11.2, 3.0 Hz, 1H), 3.33 (dt, J = 9.6, 3.8 Hz, 1H), 3.21 (dd, J = 11.3, 4.4 Hz, 1H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.2, 176.5, 146.1, 135.7, 130.5, 128.7, 128.5, 128.4, 127.9, 120.2, 117.0, 115.8, 43.4, 42.9, 41.7, 41.5 ppm.
(3aR,9bS)-8-Methoxy-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (16)
Compound 16 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (27.6 mg, 75%); mp 136.0–138.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.47–7.40 (m, 2H), 7.39–7.33 (m, 1H), 7.30–7.24 (m, 2H), 7.12 (d, J = 2.8 Hz, 1H), 6.73 (dd, J = 8.7, 2.8 Hz, 1H), 6.58 (d, J = 8.7 Hz, 1H), 4.16–4.07 (m, 1H), 3.78 (s, 3H), 3.74 (dd, J = 11.3, 3.2 Hz, 1H), 3.54–3.47 (m, 1H), 3.27 (dd, J = 11.4, 4.3 Hz, 1H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.6, 175.8, 153.7, 140.0, 132.1, 129.2, 128.7, 126.5, 117.7, 116.8, 115.4, 114.9, 55.9, 43.3, 42.4, 41.9 ppm; FTIR (ATR) ν: 3356, 1711, 1509, 1386, 1250, 1190, 1033, 827, 694 cm–1; HRMS (ESI) m/z: calcd C18H17N2O3 [M + H]+, 309.1239; found, 309.1251.
(3aR,9bS)-8-Methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (17)
Compound 17 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (35 mg, 87%). Spectroscopic data are in accordance with the literature;231H NMR (400 MHz, chloroform-d): δ 7.47–7.39 (m, 2H), 7.39–7.31 (m, 2H), 7.30–7.22 (m, 2H), 6.93 (dt, J = 7.9, 1.5 Hz, 1H), 6.55 (d, J = 8.1 Hz, 1H), 4.13 (d, J = 9.3 Hz, 1H), 3.76 (dd, J = 11.1, 3.1 Hz, 1H), 3.70 (s, 1H), 3.52 (ddd, J = 9.0, 4.2, 3.1 Hz, 1H), 3.33–3.25 (m, 1H), 2.28 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.7, 176.0, 143.8, 132.1, 130.8, 129.5, 129.2, 129.1, 128.6, 126.5, 116.8, 115.8, 43.4, 42.0, 41.6, 20.7 ppm.
(3aR,9bS)-8-Bromo-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (18)
Compound 18 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (41.2 mg, 77%); mp 182.5–183.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.70–7.65 (m, 1H), 7.47–7.41 (m, 2H), 7.39–7.33 (m, 1H), 7.29–7.23 (m, 2H), 7.20 (dd, J = 8.5, 2.2 Hz, 1H), 6.53 (d, J = 8.5 Hz, 1H), 4.11 (d, J = 9.3 Hz, 1H), 3.76 (dd, J = 11.4, 3.2 Hz, 1H), 3.53 (ddd, J = 9.3, 4.4, 3.2 Hz, 1H), 3.31 (dd, J = 11.4, 4.4 Hz, 1H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.1, 175.2, 145.1, 133.1, 131.9, 131.4, 129.2, 128.8, 126.5, 118.6, 117.4, 111.9, 43.0, 41.5, 41.3 ppm; FTIR (ATR) ν: 3391, 1699, 1496, 1381, 1319, 1287, 1267, 1193, 1162, 1134, 883, 806, 749, 698, 622, 568, 537, 510, 492 cm–1; HRMS (ESI) m/z: calcd C17H14BrN2O2 [M + H]+, 357.0239; found, 357.0241.
(3aR,9bS)-6,8-Dimethyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (20)
Compound 20 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (27.6 mg, 59%). mp 198.5–200.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.46–7.39 (m, 2H), 7.39–7.32 (m, 1H), 7.30–7.23 (m, 3H), 6.88–6.82 (m, 1H), 4.14 (d, J = 9.3 Hz, 1H), 3.81 (d, J = 11.3 Hz, 1H), 3.73 (s, 1H), 3.52 (ddd, J = 9.3, 4.2, 3.1 Hz, 1H), 3.27 (dd, J = 11.3, 4.3 Hz, 1H), 2.26 (s, 3H), 2.11 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.8, 176.1, 142.0, 132.1, 130.6, 129.1(2C), 128.7, 128.6, 126.5, 122.7, 116.2, 43.6, 42.0, 41.9, 20.7, 16.9 ppm; FTIR (ATR) ν: 3423, 1768, 1699, 1501, 1389, 1332, 1257, 1194, 1177, 1161, 862, 745, 732, 705, 691, 528 cm–1; HRMS (ESI) m/z: calcd C19H19N2O2 [M + H]+, 307.1447; found, 307.1447.
(3aR,9bS)-9-Methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (21) and (3aR,9bS)-7-Methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (21′)
Compounds 21 and 21′ as an inseparable mixture were obtained as a colorless oil after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (30.4 mg, 95%). mp 152.0–156.0 °C; 1H NMR (400 MHz, chloroform-d): δ 7.47–7.40 (m, 6H), 7.39–7.32 (m, 2H), 7.30–7.24 (m, 5H), 7.01 (t, J = 7.7 Hz, 1H), 6.77 (dt, J = 7.4, 1.0 Hz, 1H), 6.70 (ddd, J = 7.8, 1.7, 0.8 Hz, 1H), 6.55–6.48 (m, 1H), 6.45 (t, J = 1.2 Hz, 1H), 4.50 (d, J = 9.6 Hz, 1.33H, major), 4.13 (d, J = 9.3 Hz, 1H, minor), 3.84–3.67 (m, 5H), 3.54 (ddt, J = 12.6, 7.7, 2.6 Hz, 2H), 3.30 (dd, J = 10.7, 4.3 Hz, 1H), 3.16 (dd, J = 11.1, 4.6 Hz, 1H), 2.59 (s, 4H, major), 2.26 (s, 3H, minor) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.4, 177.7, 176.1, 175.7, 147.7, 146.0, 139.4, 138.6, 132.2, 132.1, 130.4, 129.2, 129.1, 128.7, 128.6, 128.1, 126.6, 126.5, 123.0, 121.3, 117.9, 116.4, 114.1, 11 4.0, 44.9, 43.8, 43.4, 41.8, 41.4, 39.4, 21.3, 20.3 ppm; FTIR (ATR) ν: 1699, 1592, 1479, 1455, 1381, 1273, 1240, 1178, 1037, 873, 789, 747, 691, 620, 605, 570, 540, 510, 485, 470 cm–1; HRMS (ESI) m/z: calcd C18H17N2O2 [M + H]+, 293.1290; found, 293.1291.
(3aR,9bS)-4-Methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (22)
Compound 22 was afforded as an inseparable mixture of diastereomers after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (24.1 mg, 79%). Spectroscopic data are in accordance with the literature;591H NMR (400 MHz, chloroform-d): δ 7.66–7.59 (m, 1H), 7.56 (dd, J = 7.7, 2.1 Hz, 1H), 7.49–7.30 (m, 6H), 7.30–7.19 (m, 5H), 7.12 (td, J = 7.6, 1.8 Hz, 2H), 6.90–6.81 (m, 2H), 6.62 (dt, J = 7.9, 1.6 Hz, 2H), 4.20–4.15 (m, 1H), 4.13–4.08 (m, 1H), 3.72–3.63 (m, 1H), 3.62–3.53 (m, 1H), 3.50–3.42 (m, 1H), 3.17 (ddd, J = 8.7, 6.1, 1.8 Hz, 1H), 1.60 (dd, J = 6.7, 1.8 Hz, 4H), 1.43 (dd, J = 6.5, 1.8 Hz, 3H) ppm; 13C{1H} (101 MHz, chloroform-d): δ 176.5, 175.86, 175.85, 175.8, 146.2, 143.8, 132.0, 130.4, 129.2, 129.1, 128.7, 128.6, 128.5, 126.6, 126.5, 120.2, 119.7, 117.0, 115.77, 115.75, 115.6, 49.2, 47.8, 47.3, 47.1, 43.0, 40.6, 20.2, 18.2 ppm.
(3aR,9bS)-4-Isobutyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (23)
Compound 23 as an inseparable mixture of diastereomers was afforded as a colorless oil after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (16.8 mg, 42%). 1H NMR (400 MHz, chloroform-d): δ 7.57–7.49 (m, 2H), 7.46–7.37 (m, 5H), 7.37–7.29 (m, 2H), 7.27–7.18 (m, 6H), 7.14–7.06 (m, 3H), 6.84 (dtd, J = 15.2, 7.5, 1.2 Hz, 3H), 6.62 (dd, J = 7.9, 1.2 Hz, 2H), 6.56 (dd, J = 8.0, 1.2 Hz, 1H), 4.18 (d, J = 9.1 Hz, 1H), 4.08 (d, J = 9.0 Hz, 1H), 3.90–3.81 (m, 2H), 3.69 (s, 1H), 3.52 (dd, J = 9.1, 3.8 Hz, 2H), 3.44 (ddd, J = 7.8, 6.3, 3.8 Hz, 2H), 3.26 (dd, J = 9.0, 3.5 Hz, 1H), 1.96 (dt, J = 12.9, 6.5 Hz, 1H), 1.90–1.73 (m, 3H), 1.73–1.35 (m, 4H), 0.98 (d, J = 6.4 Hz, 11H), 0.92 (d, J = 6.4 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.2, 176.0 (2C), 175.87, 146.4, 143.2, 132.04, 131.99, 130.4, 130.2, 129.2, 129.1, 128.8, 128.63, 128.61, 128.5, 126.6, 126.5, 120.2, 119.7, 117.3, 116.2, 116.1, 115.8, 51.9, 48.6, 47.3, 46.1, 43.3, 41.5, 40.6, 40.3, 25.2, 24.9, 23.5, 23.0, 22.6, 21.8 ppm; FTIR (ATR) ν: 3365, 2955, 1776, 1704, 1602, 1497, 1378, 1257, 1177, 910, 808, 750, 711, 690, 623, 498 cm–1; HRMS (ESI) m/z: calcd C21H22N2O2 [M + H]+, 335.1760; found, 335.1760.
(3aR,4S,9bS)-4-Isopropyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (24, Less Polar Isomer)
Compound 24 was afforded as a colorless oil after purification on the silica column using a gradient of 0–5% ethyl acetate in petroleum ethers (10.3 mg, 29%). 1H NMR (400 MHz, chloroform-d): δ 7.54 (d, J = 7.7 Hz, 1H), 7.43–7.36 (m, 2H), 7.36–7.29 (m, 1H), 7.24–7.18 (m, 2H), 7.16–7.07 (m, 1H), 6.86 (td, J = 7.4, 1.1 Hz, 1H), 6.65 (d, J = 8.0 Hz, 1H), 4.20 (d, J = 9.1 Hz, 1H), 3.73 (dd, J = 9.2, 3.1 Hz, 1H), 2.87 (dd, J = 10.1, 3.1 Hz, 1H), 2.72–2.57 (m, 1H), 1.20 (d, J = 6.6 Hz, 3H), 1.12 (d, J = 6.6 Hz, 3H) ppm; 13C{1H} NMR (176 MHz, chloroform-d): δ 175.9, 175.6, 146.8, 132.0, 130.2, 129.0, 128.6, 128.5, 126.6, 120.4, 117.2, 115.9, 61.0, 44.8, 44.0, 28.6, 20.8, 19.9 ppm; FTIR (ATR) ν: 2962, 1710, 1604, 1498, 1381, 1182, 1134, 751, 691, 624 cm–1; HRMS (ESI) m/z: calcd C20H21N2O2 [M + H]+, 321.1603; found, 321.1615.
(3aR,4R,9bS)-4-Isopropyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (24′, More Polar Isomer)
Compound 24′ was afforded as a colorless oil after purification on the silica column using a gradient of 0–5% ethyl acetate in petroleum ethers (4.5 mg, 12%). 1H NMR (400 MHz, chloroform-d): δ 7.50 (d, J = 7.7 Hz, 1H), 7.45–7.39 (m, 2H), 7.38–7.31 (m, 1H), 7.26 (s, 2H), 7.10 (td, J = 7.8, 1.5 Hz, 1H), 6.81 (t, J = 7.5 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H), 4.09 (d, J = 9.1 Hz, 1H), 3.58 (dd, J = 9.1, 2.8 Hz, 1H), 3.45 (dd, J = 9.1, 2.8 Hz, 1H), 2.02–1.89 (m, 1H), 1.06 (d, J = 6.7 Hz, 3H), 0.97 (d, J = 6.7 Hz, 3H) ppm; 13C{1H} NMR (201 MHz, chloroform-d): δ 177.7, 176.0, 143.3, 132.1, 130.0, 129.1, 128.8, 128.6, 126.5, 119.7, 116.8, 116.0, 56.8, 45.0, 40.8, 29.2, 20.0, 19.0 ppm; FTIR (ATR) ν: 2961, 1707, 1602, 1497, 1382, 1260, 1178, 911, 810, 735, 712, 691, 624, 483 cm–1; HRMS (ESI) m/z: calcd C20H21N2O2 [M + H]+, 321.1603; found, 321.1610.
(3aR,9bS)-5-Methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (25)
Compound 25 was afforded as a white solid after purification on the silica column using a gradient of 0–10% ethyl acetate in petroleum ethers (28 mg, 76%). Spectroscopic data are in accordance with the literature;201H NMR (400 MHz, chloroform-d): δ 7.53 (dt, J = 7.5, 1.3 Hz, 1H), 7.47–7.39 (m, 2H), 7.39–7.33 (m, 1H), 7.31–7.20 (m, 3H), 6.91 (td, J = 7.5, 1.2 Hz, 1H), 6.75 (dd, J = 8.2, 1.2 Hz, 1H), 4.16 (d, J = 9.6 Hz, 1H), 3.62 (dd, J = 11.4, 2.8 Hz, 1H), 3.54 (ddd, J = 9.6, 4.4, 2.7 Hz, 1H), 3.13 (dd, J = 11.5, 4.4 Hz, 1H), 2.84 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.8, 175.9, 148.7, 132.1, 130.5, 129.1, 128.8, 128.6, 126.5, 119.8, 118.7, 112.7, 50.8, 43.7, 42.3, 39.6 ppm.
Synthesis of N-Acylated Product 28
Following a modified literature procedure,71 to a Biotage 2–5 mL microwave vial were added 3 (32.1 mg, 0.12 mmol, 1 equiv) and dichloromethane (DCM) (2 mL) followed by pyridine (12 μL, 0.15 mmol, 1.2 equiv). Acetyl chloride (17.2 mg, 0.21 mmol, 1.9 equiv) in 1 mL of DCM was then added dropwise. The reaction mixture was stirred at 23 °C until full conversion (ca 2 h). The reaction mixture was then washed with aqueous saturated ammonium chloride, and the solvent was removed under reduced pressure. The crude product was purified using flash chromatography (SiO2, 0–5% methanol in DCM).
(3aR,9bS)-5-Acetyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (28)
Compound 28 was afforded as a yellow solid (36.0 mg, 97%). mp 142.5–144 °C; 1H NMR (800 MHz, chloroform-d): δ 7.61–7.57 (m, 1H), 7.43 (t, J = 7.7 Hz, 2H), 7.40–7.34 (m, 2H), 7.34–7.28 (m, 1H), 7.24–7.11 (m, 3H), 5.13 (s, 1H), 4.22 (d, J = 9.2 Hz, 1H), 3.62 (s, 1H), 3.33 (s, 1H), 2.10 (s, 3H); 13C{1H} NMR (101 MHz, chloroform-d): δ 176.4, 175.0, 169.5, 140.5, 131.7, 130.8, 129.3, 129.0, 128.8, 128.7, 127.2, 126.4, 124.9, 44.4, 43.6, 29.8, 22.2 ppm. FTIR (ATR) ν: 1708, 1652, 1582, 1492, 1458, 1382, 1307, 1185, 1158, 1120, 1060, 817, 779, 760, 691, 629, 616, 524 cm–1; HRMS (ESI) m/z: calcd C19H17N2O3 [M + H]+, 321.1239; found, 321.1254.
Synthesis of N-Benzoylated Product 29
Following a modified literature procedure,72 to a Biotage 2–5 mL microwave vial were added 3 (29.1 mg, 0.10 mmol, 1 equiv) and toluene (2 mL) followed by triethyl amine (30 μL, 0.2 mmol, 2.1 equiv) and toluoyl chloride (20 mg, 0.13 mmol, 1.2 equiv). The reaction mixture was stirred at 40 °C for 18 h. The solvent was then removed under reduced pressure, and the crude was purified using flash chromatography (SiO2, 0–40% EtOAc in n-pentane).
(3aR,9bS)-5-(4-Methylbenzoyl)-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (29)
Compound 29 was afforded as a white solid (38.0 mg, 92%). mp 87.0–89.5 °C; 1H NMR (400 MHz, chloroform-d): δ 7.63–7.56 (m, 1H), 7.44–7.29 (m, 3H), 7.23–7.12 (m, 5H), 7.07–6.99 (m, 3H), 6.64 (d, J = 8.1 Hz, 1H), 5.27 (d, J = 13.1 Hz, 1H), 4.32 (d, J = 9.4 Hz, 1H), 3.72 (ddd, J = 9.4, 5.2, 2.1 Hz, 1H), 3.42 (dd, J = 13.2, 5.2 Hz, 1H), 2.30 (s, 3H); 13C{1H} NMR (101 MHz, chloroform-d): δ 176.3, 175.2, 169.7, 141.6, 140.8, 131.7, 131.6, 130.7, 129.4, 129.1, 129.0, 128.98, 128.2, 126.4, 126.0, 125.2, 124.5, 44.6, 44.1, 43.5, 21.6 ppm. FTIR (ATR) ν: 1712, 1636, 1375, 1176, 1148, 1077, 901, 831, 814, 753, 744, 729, 610, 526 cm–1; HRMS (ESI) m/z: calcd C25H21N2O3 [M + H]+, 397.1552; found, 397.1559.
Reaction of 3 with Mesyl Chloride
Following a published procedure,733 (29.3 mg, 0.105 mmol, 1 equiv) was dissolved in toluene (1 mL) in a 2–5 mL Biotage microwave vial. Pyridine (15 μL, 0.19 mmol, 1.8 equiv) and mesyl chloride (30 μL, 0.39 mmol, 3.7 equiv) were then added, and the vial was then capped and stirred at 90 °C until full conversion of 3 (as determined by TLC, 5 h reaction time). The solvent was then removed under reduced pressure, and the product was isolated after flash chromatography (SiO2, 10–50% EtOAc in petroleum ethers).
(3aR,9bS)-5-(Methylsulfonyl)-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (30)
Compound 30 was afforded as a yellow oil (31.0 mg, 83%). 1H NMR (400 MHz, chloroform-d): δ 7.71 (ddd, J = 7.6, 1.8, 0.9 Hz, 1H), 7.58 (dd, J = 8.1, 1.3 Hz, 1H), 7.48–7.42 (m, 2H), 7.42–7.32 (m, 2H), 7.31–7.26 (m, 3H), 4.39 (dd, J = 13.8, 4.2 Hz, 1H), 4.24 (d, J = 9.5 Hz, 1H), 3.79 (dd, J = 13.8, 5.6 Hz, 1H), 3.67 (ddd, J = 9.7, 5.5, 4.2 Hz, 1H), 3.02 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 175.7, 174.7, 137.5, 131.7, 131.2, 129.4, 129.0, 128.9, 126.40, 126.35, 124.2, 122.9, 45.4, 43.2, 41.9, 40.7 ppm. FTIR (ATR) ν: 1708, 1491, 1457, 1385, 1328, 1150, 1039, 993, 960, 910, 838, 798, 726, 693, 648 cm–1; HRMS (ESI) m/z: calcd C18H17N2O4S [M + H]+, 357.0909; found, 357.0914.
Oxidation of 3 to Quinoline 31
Following a modified published procedure,593 (32.1 mg, 0.115 mmol, 1 equiv) and 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) (55.2 mg, 0.24 mmol, 2.1 equiv) were dissolved in toluene (2 mL) in a 2–5 mL Biotage microwave vial and heated at 90 °C for 18 h using a heating block. The solvent was then removed under reduced pressure, and the crude was passed through a short silica plug and eluted with 100% DCM to yield the quinoline 31.
2-Phenyl-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (31)
Compound 31 was afforded as white crystals (29.1 mg, 92%). Spectroscopic data are in accordance with the literature;741H NMR (400 MHz, chloroform-d): δ 9.45 (s, 1H), 8.89 (ddd, J = 8.4, 1.4, 0.7 Hz, 1H), 8.29 (dt, J = 8.6, 0.9 Hz, 1H), 7.96 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 7.82 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.60–7.51 (m, 2H), 7.51–7.42 (m, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 167.3, 166.8, 152.3, 143.9, 135.2, 133.0, 131.3, 130.5, 130.4, 129.4, 128.6, 126.7, 125.2, 123.5, 121.6 ppm.
Synthesis of Quinoline 34
Following the general procedure, maleimide 33 (55.4 mg, 0.30 mmol, 1 equiv) and amino acid 32 (168.6 mg, 1.0 mmol, 3.4 equiv) were dissolved in a mixture of 1 mL of water and 2 mL of methanol and irradiated for 18 h. The reaction mixture was then mixed with ethyl acetate (15 mL) and the organic layer washed with saturated sodium bicarbonate solution (2 × 10 mL) to remove the excess amino acid. The organic layer was then dried over sodium sulphate and concentrated under reduced pressure to yield 33 that was used without further purification. In the next step, 33 was dissolved in 1 mL of toluene, and DDQ (90.4 mg, 0.40 mmol) was added, and the mixture was stirred at 90 °C for 18 h. The solvent was then removed under reduced pressure, and the product was isolated using flash chromatography (SiO2, 0–10% EtOAc in petroleum ethers).
2-(4-Methyl-1,3-dioxo-1,3-dihydro-2H-pyrrolo[3,4-c]quinolin-2-yl)ethyl Acetate (34)
Compound 34 was afforded as off-white crystals (54.8 mg, 61% over two steps). Spectroscopic data are in accordance with the literature;751H NMR (400 MHz, chloroform-d): δ 8.79 (ddd, J = 8.4, 1.5, 0.7 Hz, 1H), 8.13 (d, J = 8.6 Hz, 1H), 7.88 (ddd, J = 8.6, 6.9, 1.5 Hz, 1H), 7.71 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 4.36 (dd, J = 5.8, 4.8 Hz, 2H), 4.01 (dd, J = 5.8, 4.8 Hz, 2H), 3.05 (s, 3H), 2.03 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 171.1, 168.3, 168.1, 155.1, 151.6, 136.1, 132.9, 129.3, 129.1, 125.0, 122.0, 120.7, 61.6, 37.3, 22.2, 20.9 ppm.
Acknowledgments
This work was supported by grants from the Swedish research council FORMAS (2019-00699) and from Wilhelm och Martina Lundgrens vetenskapliga stiftelse.
Data Availability Statement
The data underlying this study are available in the published article and its online Supporting Information material.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c02448.
UV–vis studies, emission spectrum of the irradiation source, mechanistic investigations, and NMR spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Campos K. R. Direct Sp3 C–H Bond Activation Adjacent to Nitrogen in Heterocycles. Chem. Soc. Rev. 2007, 36, 1069–1084. 10.1039/B607547A. [DOI] [PubMed] [Google Scholar]
- Mitchell E. A.; Peschiulli A.; Lefevre N.; Meerpoel L.; Maes B. U. W. Direct α-Functionalization of Saturated Cyclic Amines. Chem.—Eur. J. 2012, 18, 10092–10142. 10.1002/chem.201201539. [DOI] [PubMed] [Google Scholar]
- Deb M. L.; Saikia B. S.; Borpatra P. J.; Baruah P. K. Progress of Metal-Free Visible-Light-Driven α-C–H Functionalization of Tertiary Amines: A Decade Journey. Asian J. Org. Chem. 2022, 11, e202100706 10.1002/ajoc.202100706. [DOI] [Google Scholar]
- Nakajima K.; Miyake Y.; Nishibayashi Y. Synthetic Utilization of α-Aminoalkyl Radicals and Related Species in Visible Light Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1946–1956. 10.1021/acs.accounts.6b00251. [DOI] [PubMed] [Google Scholar]
- Hu J.; Wang J.; Nguyen T. H.; Zheng N. The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. 10.3762/bjoc.9.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Xia W. Photoredox Functionalization of C–H Bonds Adjacent to a Nitrogen Atom. Chem. Soc. Rev. 2012, 41, 7687–7697. 10.1039/C2CS35203F. [DOI] [PubMed] [Google Scholar]
- Cole-Hamilton D. J. Homogeneous Catalysis--New Approaches to Catalyst Separation, Recovery, and Recycling. Science 2003, 299, 1702–1706. 10.1126/science.1081881. [DOI] [PubMed] [Google Scholar]
- Lima C. G. S.; de M. Lima T.; Duarte M.; Jurberg I. D.; Paixão M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]
- Rosokha S. V.; Kochi J. K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41, 641–653. 10.1021/ar700256a. [DOI] [PubMed] [Google Scholar]
- Kochi J. K. Electron Transfer and Charge Transfer: Twin Themes in Unifying the Mechanisms of Organic and Organometallic Reactions. Angew. Chem., Int. Ed. Engl. 1988, 27, 1227–1266. 10.1002/anie.198812273. [DOI] [Google Scholar]
- Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasnim T.; Ayodele M. J.; Pitre S. P. Recent Advances in Employing Catalytic Donors and Acceptors in Electron Donor–Acceptor Complex Photochemistry. J. Org. Chem. 2022, 87, 10555–10563. 10.1021/acs.joc.2c01013. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Majumder S.; Yang M.; Guo S. Recent Advances in Catalyst-Free Photochemical Reactions via Electron-Donor-Acceptor (EDA) Complex Process. Tetrahedron Lett. 2020, 61, 151506. 10.1016/j.tetlet.2019.151506. [DOI] [Google Scholar]
- Fu M.-C.; Shang R.; Zhao B.; Wang B.; Fu Y. Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide. Science 2019, 363, 1429–1434. 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Wang G.-Z.; Shang R. Catalyst-Free Decarboxylation and Decarboxylative Giese Additions of Alkyl Carboxylates through Photoactivation of Electron Donor-Acceptor Complex. Adv. Synth. Catal. 2019, 361, 4500–4505. 10.1002/adsc.201900803. [DOI] [Google Scholar]
- Fu M.-C.; Wang J.-X.; Shang R. Triphenylphosphine-Catalyzed Alkylative Iododecarboxylation with Lithium Iodide under Visible Light. Org. Lett. 2020, 22, 8572–8577. 10.1021/acs.orglett.0c03173. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Wu S.; Melchiorre P. Tetrachlorophthalimides as Organocatalytic Acceptors for Electron Donor–Acceptor Complex Photoactivation. J. Am. Chem. Soc. 2022, 144, 8914–8919. 10.1021/jacs.2c03546. [DOI] [PubMed] [Google Scholar]
- Runemark A.; Zacharias S. C.; Sundén H. Visible-Light-Driven Stereoselective Annulation of Alkyl Anilines and Dibenzoylethylenes via Electron Donor–Acceptor Complexes. J. Org. Chem. 2021, 86, 1901–1910. 10.1021/acs.joc.0c02819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runemark A.; Sundén H. Aerobic Oxidative EDA Catalysis: Synthesis of Tetrahydroquinolines Using an Organocatalytic EDA Active Acceptor. J. Org. Chem. 2022, 87, 1457–1469. 10.1021/acs.joc.1c02776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C.-W.; Sundén H. α-Aminoalkyl Radical Addition to Maleimides via Electron Donor–Acceptor Complexes. Org. Lett. 2018, 20, 2051–2054. 10.1021/acs.orglett.8b00597. [DOI] [PubMed] [Google Scholar]
- McClain E. J.; Monos T. M.; Mori M.; Beatty J. W.; Stephenson C. R. J. Design and Implementation of a Catalytic Electron Donor–Acceptor Complex Platform for Radical Trifluoromethylation and Alkylation. ACS Catal. 2020, 10, 12636–12641. 10.1021/acscatal.0c03837. [DOI] [Google Scholar]
- Zhao H.; Leonori D. Minimization of Back-Electron Transfer Enables the Elusive Sp3 C–H Functionalization of Secondary Anilines. Angew. Chem., Int. Ed. 2021, 60, 7669–7674. 10.1002/anie.202100051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Chao C. S.; Pan Y.; Dong S.; Teo Y. C.; Wang J.; Tan C.-H. Amphiphilic Methyleneamino Synthon through Organic Dye Catalyzed-Decarboxylative Aminoalkylation. Org. Biomol. Chem. 2013, 11, 5922–5925. 10.1039/C3OB41091A. [DOI] [PubMed] [Google Scholar]
- Luo C.; Zhou T.; Wang W.; Han P.; Jing L. An Efficient Approach to Access 2,2-Diarylanilines via Visible-Light-Promoted Decarboxylative Cross-Coupling Reactions. Asian J. Org. Chem. 2021, 10, 2342–2346. 10.1002/ajoc.202100458. [DOI] [Google Scholar]
- Zhou C.; Li M.; Sun J.; Cheng J.; Sun S. Photoredox-Catalyzed α-Aminomethyl Carboxylation of Styrenes with Sodium Glycinates: Synthesis of γ-Amino Acids and γ-Lactams. Org. Lett. 2021, 23, 2895–2899. 10.1021/acs.orglett.1c00536. [DOI] [PubMed] [Google Scholar]
- Huang X.-L.; Cheng Y.-Z.; Zhang X.; You S.-L. Photoredox-Catalyzed Intermolecular Hydroalkylative Dearomatization of Electron-Deficient Indole Derivatives. Org. Lett. 2020, 22, 9699–9705. 10.1021/acs.orglett.0c03759. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-Z.; Huang X.-L.; Zhuang W.-H.; Zhao Q.-R.; Zhang X.; Mei T.-S.; You S.-L. Intermolecular Dearomatization of Naphthalene Derivatives by Photoredox-Catalyzed 1,2-Hydroalkylation. Angew. Chem., Int. Ed. 2020, 59, 18062–18067. 10.1002/anie.202008358. [DOI] [PubMed] [Google Scholar]
- Yang H.; Wei G.; Jiang Z. Access to Isoxazolidines through Visible-Light-Induced Difunctionalization of Alkenes. ACS Catal. 2019, 9, 9599–9605. 10.1021/acscatal.9b03567. [DOI] [Google Scholar]
- Zhou Z.; Nie X.; Harms K.; Riedel R.; Zhang L.; Meggers E. Enantioconvergent Photoredox Radical-Radical Coupling Catalyzed by a Chiral-at-Rhodium Complex. Sci. China: Chem. 2019, 62, 1512–1518. 10.1007/s11426-019-9584-x. [DOI] [Google Scholar]
- Li J.; Gu Z.; Zhao X.; Qiao B.; Jiang Z. Asymmetric Aerobic Decarboxylative Povarov Reactions of N-Aryl α-Amino Acids with Methylenephthalimidines via Cooperative Photoredox and Chiral Brønsted Acid Catalysis. Chem. Commun. 2019, 55, 12916–12919. 10.1039/C9CC07380A. [DOI] [PubMed] [Google Scholar]
- Liu X.; Yin Y.; Jiang Z. Photoredox-Catalysed Formal [3+2] Cycloaddition of N-Aryl α-Amino Acids with Isoquinoline N-Oxides. Chem. Commun. 2019, 55, 11527–11530. 10.1039/C9CC06249A. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Tang Y.; Fan L.; Lefebvre Q.; Hou H.; Rueping M. Heterogeneous Visible-Light Photoredox Catalysis with Graphitic Carbon Nitride for α-Aminoalkyl Radical Additions, Allylations, and Heteroarylations. ACS Catal. 2018, 8, 9471–9476. 10.1021/acscatal.8b02937. [DOI] [Google Scholar]
- Yin Y.; Dai Y.; Jia H.; Li J.; Bu L.; Qiao B.; Zhao X.; Jiang Z. Conjugate Addition–Enantioselective Protonation of N-Aryl Glycines to α-Branched 2-Vinylazaarenes via Cooperative Photoredox and Asymmetric Catalysis. J. Am. Chem. Soc. 2018, 140, 6083–6087. 10.1021/jacs.8b01575. [DOI] [PubMed] [Google Scholar]
- Millet A.; Lefebvre Q.; Rueping M. Visible-Light Photoredox-Catalyzed Giese Reaction: Decarboxylative Addition of Amino Acid Derived α-Amino Radicals to Electron-Deficient Olefins. Chem.—Eur. J. 2016, 22, 13464–13468. 10.1002/chem.201602257. [DOI] [PubMed] [Google Scholar]
- Shao T.; Yin Y.; Lee R.; Zhao X.; Chai G.; Jiang Z. Sequential Photoredox Catalysis for Cascade Aerobic Decarboxylative Povarov and Oxidative Dehydrogenation Reactions of N-Aryl α-Amino Acids. Adv. Synth. Catal. 2018, 360, 1754–1760. 10.1002/adsc.201800135. [DOI] [Google Scholar]
- Pan S.; Jiang M.; Zhong G.; Dai L.; Zhou Y.; Wei K.; Zeng X. Visible-Light-Induced Selectivity Controllable Synthesis of Diamine or Imidazoline Derivatives by Multicomponent Decarboxylative Radical Coupling Reactions. Org. Chem. Front. 2020, 7, 4043–4049. 10.1039/D0QO01028F. [DOI] [Google Scholar]
- Yang J.; Song M.; Zhou H.; Qi Y.; Ma B.; Wang X.-C. Visible-light-promoted decarboxylative addition cyclization of N-aryl glycines and azobenzenes to access 1,2,4-triazolidines. Green Chem. 2021, 23, 5806–5811. 10.1039/D1GC02272E. [DOI] [Google Scholar]
- Pan S.; Jiang M.; Hu J.; Xu R.; Zeng X.; Zhong G. Synthesis of 1{,}2-Amino Alcohols by Decarboxylative Coupling of Amino Acid Derived α-Amino Radicals to Carbonyl Compounds via Visible-Light Photocatalyst in Water. Green Chem. 2020, 22, 336–341. 10.1039/C9GC03470F. [DOI] [Google Scholar]
- Chen Y.; Lu P.; Wang Y. 3-Amino-Fluorene-2,4-Dicarbonitriles (AFDCs) as Photocatalysts for the Decarboxylative Arylation of α-Amino Acids and α-Oxy Acids with Arylnitriles. Org. Lett. 2019, 21, 2130–2133. 10.1021/acs.orglett.9b00443. [DOI] [PubMed] [Google Scholar]
- Li H.-H.; Li J.-Q.; Zheng X.; Huang P.-Q. Photoredox-Catalyzed Decarboxylative Cross-Coupling of α-Amino Acids with Nitrones. Org. Lett. 2021, 23, 876–880. 10.1021/acs.orglett.0c04101. [DOI] [PubMed] [Google Scholar]
- Li J.; Kong M.; Qiao B.; Lee R.; Zhao X.; Jiang Z. Formal Enantioconvergent Substitution of Alkyl Halides via Catalytic Asymmetric Photoredox Radical Coupling. Nat. Commun. 2018, 9, 2445. 10.1038/s41467-018-04885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Y.-F.; Sun K.; Chen X.-L.; Fu X.-Y.; Liu Y.; Zeng F.-L.; Shi T.; Qu L.-B.; Yu B. Arylaminomethyl Radical-Initiated Cascade Annulation Reaction of Quinoxalin-2(1H)-Ones Catalyzed by Recyclable Photocatalyst Perovskite. Org. Lett. 2020, 22, 6960–6965. 10.1021/acs.orglett.0c02518. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Liu X.; Li J.; Zhao X.; Qiao B.; Jiang Z. Catalytic Enantioselective Radical Coupling of Activated Ketones with N-Aryl Glycines. Chem. Sci. 2018, 9, 8094–8098. 10.1039/C8SC02948B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.; Wang J.-T.; Liu L.-L.; Ma N.; Yang C.; Gao Y.; Xia W. Synthesis of Carbonylated Heteroaromatic Compounds via Visible-Light-Driven Intramolecular Decarboxylative Cyclization of o-Alkynylated Carboxylic Acids. Chem. Commun. 2017, 53, 8533–8536. 10.1039/C7CC04813K. [DOI] [PubMed] [Google Scholar]
- Si Y.-F.; Chen X.-L.; Fu X.-Y.; Sun K.; Song X.; Qu L.-B.; Yu B. Divergent G-C3N4-Catalyzed Reactions of Quinoxalin-2(1H)-Ones with N-Aryl Glycines under Visible Light: Solvent-Controlled Hydroaminomethylation and Annulation. ACS Sustainable Chem. Eng. 2020, 8, 10740–10746. 10.1021/acssuschemeng.0c02289. [DOI] [Google Scholar]
- Fan L.; Jia J.; Hou H.; Lefebvre Q.; Rueping M. Decarboxylative Aminomethylation of Aryl- and Vinylsulfonates through Combined Nickel- and Photoredox-Catalyzed Cross-Coupling. Chem.—Eur. J. 2016, 22, 16437–16440. 10.1002/chem.201604452. [DOI] [PubMed] [Google Scholar]
- Shi T.; Sun K.; Chen X.-L.; Zhang Z.-X.; Huang X.-Q.; Peng Y.-Y.; Qu L.-B.; Yu B. Recyclable Perovskite as Heterogeneous Photocatalyst for Aminomethylation of Imidazo-Fused Heterocycles. Adv. Synth. Catal. 2020, 362, 2143–2149. 10.1002/adsc.201901324. [DOI] [Google Scholar]
- Duan Y.; Zhang M.; Ruzi R.; Wu Z.; Zhu C. The Direct Decarboxylative Allylation of N-Arylglycine Derivatives by Photoredox Catalysis. Org. Chem. Front. 2017, 4, 525–528. 10.1039/C6QO00711B. [DOI] [Google Scholar]
- del Río-Rodríguez R.; Westwood M. T.; Sicignano M.; Juhl M.; Fernández-Salas J. A.; Alemán J.; Smith A. D. Isothiourea-Catalysed Enantioselective Radical Conjugate Addition under Batch and Flow Conditions. Chem. Commun. 2022, 58, 7277–7280. 10.1039/D2CC02432B. [DOI] [PubMed] [Google Scholar]
- Wang J.-X.; Wang Y.-T.; Zhang H.; Fu M.-C. Visible-Light-Induced Iodine-Anion-Catalyzed Decarboxylative/Deaminative C–H Alkylation of Enamides. Org. Chem. Front. 2021, 8, 4466–4472. 10.1039/D1QO00660F. [DOI] [Google Scholar]
- Bosque I.; Bach T. 3-Acetoxyquinuclidine as Catalyst in Electron Donor–Acceptor Complex-Mediated Reactions Triggered by Visible Light. ACS Catal. 2019, 9, 9103–9109. 10.1021/acscatal.9b01039. [DOI] [Google Scholar]
- Probst G.; Aubele D. L.; Bowers S.; Dressen D.; Garofalo A. W.; Hom R. K.; Konradi A. W.; Marugg J. L.; Mattson M. N.; Neitzel M. L.; Semko C. M.; Sham H. L.; Smith J.; Sun M.; Truong A. P.; Ye X. M.; Xu Y.; Dappen M. S.; Jagodzinski J. J.; Keim P. S.; Peterson B.; Latimer L. H.; Quincy D.; Wu J.; Goldbach E.; Ness D. K.; Quinn K. P.; Sauer J.-M.; Wong K.; Zhang H.; Zmolek W.; Brigham E. F.; Kholodenko D.; Hu K.; Kwong G. T.; Lee M.; Liao A.; Motter R. N.; Sacayon P.; Santiago P.; Willits C.; Bard F.; Bova M. P.; Hemphill S. S.; Nguyen L.; Ruslim L.; Tanaka K.; Tanaka P.; Wallace W.; Yednock T. A.; Basi G. S. Discovery of (R)-4-Cyclopropyl-7,8-difluoro-5-(4-(trifluoromethyl)phenylsulfonyl)-4,5-dihydro-1H-pyrazolo[4,3-c]quinoline (ELND006) and (R)-4-Cyclopropyl-8-fluoro-5-(6-(trifluoromethyl)pyridin-3-ylsulfonyl)-4,5-dihydro-2H-pyrazolo[4,3-c]quinoline (ELND007): Metabolically Stable γ-Secretase Inhibitors that Selectively Inhibit the Production of Amyloid-β over Notch. J. Med. Chem. 2013, 56, 5261–5274. 10.1021/jm301741t. [DOI] [PubMed] [Google Scholar]
- Kravchenko D. V.; Kuzovkova Y. A.; Kysil V. M.; Tkachenko S. E.; Maliarchouk S.; Okun I. M.; Balakin K. V.; Ivachtchenko A. V. Synthesis and Structure–Activity Relationship of 4-Substituted 2-(2-Acetyloxyethyl)-8-(Morpholine- 4-Sulfonyl)Pyrrolo[3,4-c]Quinoline- 1,3-Diones as Potent Caspase-3 Inhibitors. J. Med. Chem. 2005, 48, 3680–3683. 10.1021/jm048987t. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Ravichandran V.; Jain P. K.; Mourya V. K.; Agrawal R. K. Prediction of Caspase-3 Inhibitory Activity of 1,3-Dioxo-4-Methyl-2,3-Dihydro-1h-Pyrrolo[3,4-c] Quinolines: QSAR Study. J. Enzyme Inhib. Med. Chem. 2008, 23, 424–431. 10.1080/14756360701652476. [DOI] [PubMed] [Google Scholar]
- Kravchenko D. V.; Kysil V. V.; Ilyn A. P.; Tkachenko S. E.; Maliarchouk S.; Okun I. M.; Ivachtchenko A. V. 1,3-Dioxo-4-Methyl-2,3-Dihydro-1H-Pyrrolo[3,4-c]Quinolines as Potent Caspase-3 Inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1841–1845. 10.1016/j.bmcl.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Sridharan V.; Suryavanshi P. A.; Menéndez J. C. Advances in the Chemistry of Tetrahydroquinolines. Chem. Rev. 2011, 111, 7157–7259. 10.1021/cr100307m. [DOI] [PubMed] [Google Scholar]
- Leitch J. A.; Fuentes de Arriba A. L.; Tan J.; Hoff O.; Martínez C. M.; Dixon D. J. Photocatalytic Reverse Polarity Povarov Reaction. Chem. Sci. 2018, 9, 6653–6658. 10.1039/C8SC01704B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Bai Z.-Q.; Yuan P.-F.; Wu L.-Z.; Liu Q. Highly Efficient Iridium-Based Photosensitizers for Thia-Paternò–Büchi Reaction and Aza-Photocyclization. ACS Catal. 2021, 11, 446–455. 10.1021/acscatal.0c05005. [DOI] [Google Scholar]
- Hwang J. Y.; Ji A. Y.; Lee S. H.; Kang E. J. Redox-Selective Iron Catalysis for α-Amino C–H Bond Functionalization via Aerobic Oxidation. Org. Lett. 2020, 22, 16–21. 10.1021/acs.orglett.9b03542. [DOI] [PubMed] [Google Scholar]
- Yang X.; Zhu Y.; Xie Z.; Li Y.; Zhang Y. Visible-Light-Induced Charge Transfer Enables Csp3-H Functionalization of Glycine Derivatives: Access to 1,3-Oxazolidines. Org. Lett. 2020, 22, 1638–1643. 10.1021/acs.orglett.0c00234. [DOI] [PubMed] [Google Scholar]
- Su Z.; Mariano P. S.; Falvey D. E.; Yoon U. C.; Oh S. W. Dynamics of Anilinium Radical α-Heterolytic Fragmentation Processes. Electrofugal Group, Substituent, and Medium Effects on Desilylation, Decarboxylation, and Retro-Aldol Cleavage Pathways. J. Am. Chem. Soc. 1998, 120, 10676–10686. 10.1021/ja981541f. [DOI] [Google Scholar]
- Yoshimi Y.; Hayashi S.; Nishikawa K.; Haga Y.; Maeda K.; Morita T.; Itou T.; Okada Y.; Ichinose N.; Hatanaka M. Influence of Solvent, Electron Acceptors and Arenes on Photochemical Decarboxylation of Free Carboxylic Acids via Single Electron Transfer (SET). Molecules 2010, 15, 2623–2630. 10.3390/molecules15042623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley D. W.; Mills A.; O’Rourke C.; Slawin A. M. Z.; Walton J. C. Catalyst-Free Photoredox Addition–Cyclisations: Exploitation of Natural Synergy between Aryl Acetic Acids and Maleimide. Chem.—Eur. J. 2014, 20, 5492–5500. 10.1002/chem.201304929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng L.; Ready J. M. Photocatalytic α-Alkylation of Amines with Alkyl Halides. ACS Catal. 2020, 10, 13196–13201. 10.1021/acscatal.0c04519. [DOI] [Google Scholar]
- Ma D.; Zhang Y.; Yao J.; Wu S.; Tao F. Accelerating Effect Induced by the Structure of α-Amino Acid in the Copper-Catalyzed Coupling Reaction of Aryl Halides with α-Amino Acids. Synthesis of Benzolactam-V8. J. Am. Chem. Soc. 1998, 120, 12459–12467. 10.1021/ja981662f. [DOI] [Google Scholar]
- Specklin S.; Decuypere E.; Plougastel L.; Aliani S.; Taran F. One-Pot Synthesis of 1,4-Disubstituted Pyrazoles from Arylglycines via Copper-Catalyzed Sydnone–Alkyne Cycloaddition Reaction. J. Org. Chem. 2014, 79, 7772–7777. 10.1021/jo501420r. [DOI] [PubMed] [Google Scholar]
- Ikeda S.; Murata S. Photolysis of N-Phenylglycines Sensitized by Polycyclic Aromatic Hydrocarbons: Effects of Sensitizers and Substituent Groups and Application to Photopolymerization. J. Photochem. Photobiol., A 2002, 149, 121–130. 10.1016/s1010-6030(01)00646-3. [DOI] [Google Scholar]
- Lakeland C. P.; Watson D. W.; Harrity J. P. A. Exploiting Synergistic Catalysis for an Ambient Temperature Photocycloaddition to Pyrazoles. Chem.—Eur. J. 2020, 26, 155–159. 10.1002/chem.201904210. [DOI] [PubMed] [Google Scholar]
- Shelar S. V.; Argade N. P. Facile synthesis of indolizinoindolone, indolylepoxypyrrolooxazole, indolylpyrrolooxazolone and isoindolopyrazinoindolone heterocycles from indole and imide derivatives. Org. Biomol. Chem. 2021, 19, 6160–6169. 10.1039/D1OB00754H. [DOI] [PubMed] [Google Scholar]
- Green A. I.; Burslem G. M. Photochemical Synthesis of an Epigenetic Focused Tetrahydroquinoline Library. RSC Med. Chem. 2021, 12, 1780–1786. 10.1039/D1MD00193K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose A.; Mal P. Using Weak Interactions to Control C–H Mono-Nitration of Indolines. Chem. Commun. 2017, 53, 11368–11371. 10.1039/C7CC06267B. [DOI] [PubMed] [Google Scholar]
- Abrams R.; Jesani M. H.; Browning A.; Clayden J. Triarylmethanes and Their Medium-Ring Analogues by Unactivated Truce–Smiles Rearrangement of Benzanilides. Angew. Chem., Int. Ed. 2021, 60, 11272–11277. 10.1002/anie.202102192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashad A. A.; Jones A. J.; Avery V. M.; Baell J.; Keller P. A. Facile Synthesis and Preliminary Structure–Activity Analysis of New Sulfonamides Against Trypanosoma Brucei. ACS Med. Chem. Lett. 2014, 5, 496–500. 10.1021/ml400487t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K.; Das B.; Gogoi P. Synthesis of Pyrrolo[3,4-c]Quinoline-1,3-Diones: A Sequential Oxidative Annulation Followed by Dehydrogenation and N-Demethylation Strategy. New J. Chem. 2018, 42, 18894–18905. 10.1039/c8nj04443k. [DOI] [Google Scholar]
- Hwang J. Y.; Ji A. Y.; Lee S. H.; Kang E. J. Redox-Selective Iron Catalysis for α-Amino C–H Bond Functionalization via Aerobic Oxidation. Org. Lett. 2020, 22, 16–21. 10.1021/acs.orglett.9b03542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its online Supporting Information material.