

Abstract
Disinfectants are routinely used in human environments to control and prevent the transmission of microbial disease, and this is particularly true during the current COVID-19 crisis. However, it remains unclear whether the increased disinfectant loadings to wastewater treatment plants facilitate the dissemination of antibiotic resistance genes (ARGs) in sewage sludge microbiomes. Here, we investigated the impacts of benzalkonium chlorides (BACs), widely used disinfectants, on ARGs profiles and microbial community structures in sewage sludge by using high-throughput quantitative PCR and Illumina sequencing. A total of 147 unique ARGs and 39 mobile genetic elements (MGEs) were detected in all sewage sludge samples. Our results show that exposure to BACs disinfectants at environmentally relevant concentrations significantly promotes both the diversity and absolute abundance of ARGs in sludge microbiomes, indicating the co-selection of ARGs by BACs disinfectants. The enrichment of ARGs abundance varied from 2.15-fold to 3.63-fold compared to controls. In addition, BACs exposure significantly alters bacterial and protistan communities, resulting in dysbiosis of the sludge microbiota. The Mantel test and Procrustes analysis confirm that bacterial communities are significantly correlated with ARGs profiles under BACs treatments. The structural equation model explains 83.8 % of the total ARGs variation and further illustrates that the absolute abundance of MGEs exerts greater impacts on the variation of absolute abundance of ARGs than microbial communities under BACs exposure, suggesting BACs may promote antibiotic resistance by enhancing the horizontal gene transfer of ARGs across sludge microbiomes. Collectively, our results provide new insights into the proliferation of antibiotic resistance through disinfectant usage during the pandemic and highlight the necessity to minimize the environmental release of disinfectants into the non-target environment for combating antibiotic resistance.
Keywords: Benzalkonium chlorides disinfectants, Antibiotic resistance genes, Co-selection, Sewage sludge
Graphical abstract
1. Introduction
The emergence and dissemination of antibiotic resistance pose a major threat to human health worldwide, and have been considered as a “silent pandemic”. It was reported that antibiotic-resistant pathogens claimed 4.95 million lives around the world in 2019 (Murray et al., 2022). The selective pressures from anthropogenic activities are thought to be the causes of antibiotic resistance genes (ARGs) propagation in the environment. In general, antibiotics abuse is recognized as the leading and direct reasons for ARGs enrichment. However, increasing numbers of studies reported that various non-antibiotic chemicals (e.g., disinfectants, non-antibiotic pharmaceuticals, nanomaterials, and herbicides etc.) could impose antibiotic-like effects to facilitate the spread of ARGs (Liao et al., 2021; Maier et al., 2018; Qiu et al., 2012; Wang et al., 2022; Zhang et al., 2017).
During the current pandemic of COVID-19, disinfectants containing benzalkonium chlorides (BACs) are extensively used to prevent and control the transmission of SARS-CoV-2 in communal spaces, such as hospitals, schools, shopping malls, households, workplaces, and industrial facilities (Hora et al., 2020). As chemical disinfectants, BACs are highly broad-spectrum disinfectants attributed to the alteration of essential enzymes, inactivation of outer membrane proteins, and disruption of membrane bilayers (Chen et al., 2021; Pereira et al., 2019). However, whether exposure to BACs can exacerbate the issue of antibiotic resistance is highly debatable because of the different modes of action between disinfectants and antibiotics. Previous studies have reported that BACs were implicated in antibiotic resistance by mechanisms of co-selection (Hegstad et al., 2010; Mahoney et al., 2021). After BACs exposure, microbial communities increased resistance to clinically relevant antibiotics (e.g., penicillin G, tetracycline, ciprofloxacin) via either degrading the antibiotics per se or upregulating the activity of efflux pumps (Tandukar et al., 2013). Kim et al. (Kim et al., 2018) demonstrated that exposure to BACs co-selected antibiotic-resistant bacteria in river sediments, and multiple adaptive pathways were observed in isolates of Pseudomonas aeruginosa, e.g., mutations conferring polymyxin resistance and overexpression of multidrug efflux pump genes. In contrast, several studies found that antibiotic susceptibility was not significantly affected under BACs exposure in multiple pathogenic bacterial strains, such as P. aeruginosa and Listeria monocytogenes (Loughlin et al., 2002; Ortiz et al., 2014; Voumard et al., 2020). Moreover, most of studies regarding BACs exposure could promote antibiotic resistance focused on aerobic bioreactors (Kim et al., 2018; Tandukar et al., 2013), freshwater (Yang and Wang, 2018), agricultural soils (Zeng et al., 2022), and bacterial isolates (Kim et al., 2018; Tandukar et al., 2013). It was reported that most (~ 75 %) of the applied BACs were eventually released into wastewater treatment plants (WWTPs) (Zhang et al., 2015). However, the potential impacts of elevated usage of BACs on antibiotic resistance of sewage sludge microbiomes are largely uncharacterized.
WWTPs are considered significant reservoirs for ARGs (Su et al., 2017), and most ARGs can spread into natural environments through sewage sludge application (Chen et al., 2016). Before the pandemic, BACs have been commonly detected in municipal WWTPs with a concentration of 0.09–191 mg·kg−1 dry-weight sludge in China (Ruan et al., 2014). The increased consumption of BACs during the current pandemic will inevitably lead to increased BACs loads to WWTPs (Barber and Hartmann, 2022; Hora et al., 2020; Zhang et al., 2020). Albeit BACs are considered biodegradable in activated sludge systems, high concentrations of BACs would adversely impact the performance of activated sludge systems by altering the sludge microbiota. In particular, protists play critical roles in micropollutants purification in activated sludge treatment systems (Nicolau et al., 2005), and can regulate bacterial community structures via predation activities so as to affect the resistome of environments (Nguyen et al., 2020a). It is therefore necessary to investigate the influence of BACs on the resistome of sewage sludge by exploring the interactions of multitrophic communities.
In the present study, we set out to investigate the impacts of increased BACs concentrations on the ARGs profiles in sewage sludge. By combining high throughput quantitative PCR (HT-qPCR) with 384 validated primers targeting almost all major classes of ARGs and mobile genetic elements (MGEs) and Illumina sequencing of both bacterial and protistan communities, our objectives were to (1) provide an integrated evaluation of the impact of elevated BACs concentrations on the profiles of ARGs and MGEs in sewage sludge; (2) characterize the community structures of bacteria and protists in sewage sludge under BACs exposure; (3) explore the potential factors accounting for ARGs variation in sewage sludge under BACs disinfectants stress.
2. Materials and methods
2.1. Sample collection
Sewage sludge samples were collected from a municipal WWTP with a treatment capacity of 1 × 106 m3 d−1 in Xiamen, China. The sampled plant utilizes an anaerobic-anoxic-oxic (AAO) process and serves an area of about 3.5 × 107 m2. The sewage sludge sample was thickened to a solid content of about 2.5 %, shipped immediately to the laboratory on ice and processed within 2 h for further experiment.
2.2. Microcosm experiments of BACs exposure
The BACs exposure experiment of sewage sludge was carried out in bioreactors with five biological replicates. In this study, the BACs concentrations were set at 0 mg·L−1 (CK), 1 mg·L−1 (B1), 10 mg·L−1 (B10), and 100 mg·L−1 (B100) according to the environmental BACs concentrations reported (Pereira et al., 2019; Ruan et al., 2014). In detail, 1 L sewage sludge was added separately in glass beakers, mixed evenly with different concentrations of BACs (CAS: 68424–85-1, Aladdin, Shanghai, China). Given that microbial communities are sensitive to BACs disinfectants and may response really quickly to BACs exposure (Yang and Wang, 2018), batch microcosms were incubated for 5 days in the dark and aerobic conditions at 28 ± 2 °C. During the exposure experiment, a proper amount of sterile distilled water was sprayed into each reactor every day to supplement the evaporated moisture. After incubation, the soluble chemical oxygen demand (sCOD), total nitrogen (TN), ammonia (NH4 +-N), and nitrate (NO3 −-N) of the sewage sludge were analyzed using standard methods (APHA, 2017). The sludge pH was measured using a pH meter (Thermo Scientific, USA) at room temperature.
2.3. DNA extraction
After incubation, 1 mL of treated sewage sludge from each reactor of five biological replicates was centrifuged at 10,000 ×g for 5 min at 4 °C, respectively. Solid pellets (~ 0.3 g) were retained for DNA extraction using the FastDNA Spin Kit for Soil (MP Biomedicals, USA), and the final elution volume was 80 μL. DNA samples were checked for quality using NanoDrop ND-1000 (Nanodrop, USA), and quantified by QuantiFluor dsDNA Assay kit (Promega, USA) with the Qubit 3.0 Fluorometer (Thermo Fisher Scientific, USA). DNA samples were stored at −20 °C until further analysis.
2.4. High-throughput quantitative PCR (HT-qPCR)
To characterize the profiles of ARGs and MGEs in sewage sludge, HT-qPCR was performed using the SmartChip Real-time PCR system (Wafergen Inc., USA) (Yang et al., 2020). A total of 384 primers were used to target 327 ARGs conferring resistance to major classes of antibiotics, 7 taxonomic genes, 49 MGEs, and one 16S rRNA gene (Table S1). Briefly, 100-nL HT-qPCR mixtures containing each primer, DNA template, LightCycler 480 SYBR Green I Master (Roche Applied Sciences, IN), and nuclease-free water was dispensed into each well of the chip, and then real-time qPCR was performed employing the conditions: 95 °C for 10 min, 40 cycles (95 °C for 30 s, 60 °C for 30 s) (Yang et al., 2020). Technical triplicates and one negative control were set in one chip for quality control. The raw results were preprocessed with SmartChip qPCR Software as described previously (Yang et al., 2020). The gene copy number was calculated using the equation: Gene copy number = 10(31−CT)/(10/3). Where CT is the threshold cycle, the detection limit CT (31) was taken as a replacement for the genes with no amplification. Only samples with all three replicates amplified were considered as positively detected. The absolute quantification of 16S rRNA gene was performed using a SYBR Green approach on a Roche 480 LightCycler (Roche, Germany) (Zhu et al., 2017). More details were described in Text S1 of Supplementary material.
2.5. Illumina sequencing and data processing
To explore the taxonomic profiles of bacterial communities and protistan communities, the V4-V5 region of 16S rRNA and 18S rRNA genes were PCR-amplified using primer pairs 515F/907R and 616*F/1132R, respectively (Oliverio et al., 2018; Yang et al., 2020). The amplicons of 16S rRNA and 18S rRNA genes were sequenced on an Illumina Hiseq 2500 platform (Novogene, Beijing, China). Detailed information about library preparation and data processing is provided in Text S2 of Supplementary material. The sequencing data was archived at the NCBI SRA database under accession no. PRJNA799835.
2.6. Detection of Cellular Reactive Oxygen Species (ROS)
The cellular ROS production induced by BACs exposure was detemined by ROS assay kit (Beyotime Biotechnology). Briefly, bacterial suspensions of two model strains Escherichia coli MG1655 and Staphylococcus aureus RN4220 were incubated with the dye 2′7’-dichlorofluorescein diacetate (DCFH-DA, final concentration at 20 μM) for 30 min at 37 °C in the dark. The bacterial suspensions were washed twice with 0.1 M PBS to remove the remaining dye. Bacterial cells were then exposed to different concentrations (0, 1, 10, and 100 mg·L−1) of BACs for 2 h at 37 °C in the dark. H2O2 was set as positive control, and PBS was set as negative control. The fluorescence intensity was measured by a Guava easyCyte 5 flow cytometer (Gernsheim, Germany) with excitation at 488 nm and emission at 525 nm. All measurements were performed in biological triplicates.
2.7. Statistical analysis
The alpha-diversity indexes and rarefaction curves were determined and visualized using “vegan”, “ggplot2” and “plyr” packages in R (3.5.3 version) (Hadley Wickham, 2009; Jari et al., 2019; R Core Team, 2017). Statistical significances among samples were conducted using one-way analysis of variance (ANOVA), and P < 0.05 was considered significant. Nonmetric multidimensional scaling (NMDS), multivariate analysis of similarity (ANOSIM), Mantel test, and Procrustes test were performed using “vegan” package (Jari et al., 2019). The differential ARGs (LDA > 2.0, P < 0.05) and bacterial taxa (LDA > 4.0, P < 0.01) were identified using linear discriminant analysis effect size (LEfSe) method with the Kruskall-Wallis test (Segata et al., 2011). The differential ARGs identified by LEfSe were clustered using the complete method and visualized using “pheatmap” package. Correlation matrixes were constructed by “psych” package with the threshold (ρ > 0.8 and P < 0.01), and the correlations were visualized in the network graphs by using Gephi (0.9.2 version) software. To evaluate the direct and indirect effects of communities (protists and bacteria), BACs concentration, sludge physicochemical properties, and absolute abundance of MGEs on the absolute abundance of ARGs, the SEM was constructed using the lavaan package in R environment (Rosseel, 2012).
3. Results
3.1. Diversity and abundance of ARGs and MGEs
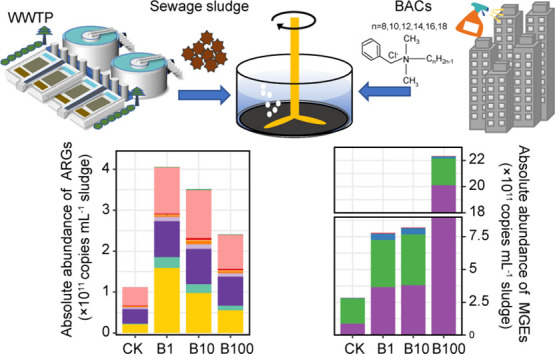
A total of 147 unique ARGs and 39 MGEs were detected among all sludge samples (Fig. 1 ), which conferred resistance to almost all major classes of antibiotics, including aminoglycosides, β-lactams, chloramphenicols, fluoroquinolones, macrolide-lincosamide-streptogramin B (MLSB), rifamycins, sulfonamides, tetracycline, trimethoprim, and vancomycin. Although not statistically significant (P > 0.05), the mean percentage of efflux pump mechanisms increased with BACs concentrations from 15.32 % to 17.07 % (Fig. S1). The detected number of ARGs in sludge samples after BACs exposure ranged from 61 to 94 with the highest number detected in B10, much higher than that in CK ranged from 43 to 61 (Fig. 1A). In addition, the detected number of MGEs after BACs treatment ranged from 21 to 33 with the highest number detected in B10, higher than that in control ranged from 15 to 21 (Fig. 1B).
Fig. 1.

Number of detected ARGs (A) and MGEs (B). Absolute abundance of detected ARGs (C) and MGEs (D). (E) NMDS analysis of ARGs (including MGEs) based on the Bray-Curtis distance. The detected ARGs were classified into 12 subtypes based on antibiotic classes to which they conferred resistance. MLSB, macrolide-lincosamide-streptogramin B. IS, insertional sequence.
The absolute abundance of ARGs and MGEs were significantly increased under BACs exposure (P < 0.05, Fig. 1C and D). The ARGs abundance was 1.12 × 1011, 4.06 × 1011, 3.51 × 1011 and 2.41 × 1011 copies mL−1 sludge in CK, B1, B10 and B100 treatment, respectively (Fig. 1C). The dominated ARG subtypes were conferring resistance to multidrug, aminoglycosides and sulfonamides (Fig. 1C and S2A). The MGEs abundance was 2.84 × 1011, 7.80 × 1011, 8.21 × 1011, 2.23 × 1012 copies mL−1 sludge in CK, B1, B10 and B100, respectively (Fig. 1D). The dominated MGE subtypes were transposon and insertional sequence (IS) (Fig. 1D and S2B). The absolute abundance of ARGs was positively correlated with that of MGEs (R2 = 0.368, P = 0.0027, Fig. S3). Furthermore, BACs exposure significantly altered the profiles of ARGs and MGEs as revealed by NMDS analysis based on the Bray-Curtis distance (stress = 0.03; ANOSM, R = 0.88, P = 0.001, Fig. 1E).
The LEfSe analysis further illustrated the main differences in the absolute abundance of ARGs and MGEs. A total of 58 unique ARGs and 19 MGEs exhibited significant differences among samples (P < 0.05, LDA > 2.0; Table S2 and Fig. 2 ). These genes could be divided into three clusters: (A) the ARGs and MGEs that persist in all samples, (B) the ARGs and MGEs that were not detected in CK, (C) the elevated ARGs and MGEs after BACs exposure. In addition, the ARGs in clusters B and C mainly encoded enzyme inactivation, efflux pump, and ribosome protective protein.
Fig. 2.

Heatmap presenting the absolute abundances of differential ARGs and MGEs (P < 0.05, LDA > 2.0) in sludge samples. Each column is labeled with the sample name with numbers representing replicates. Each row is labeled with the name and classification of ARGs and MGEs. Values plotted are the natural logarithm transformed proportion of the absolute abundances of each ARG and MGE. MLSB, macrolide-lincosamide-streptogramin B. IS, insertional sequence.
3.2. Co-occurrence patterns of ARG and MGE subtypes
To reveal the different ARG patterns of each BACs treatment, the co-occurrence networks were created by using correlation matrices between ARGs (including MGEs). The networks were clearly parsed into 7–9 major modules based on the modularity class and exhibited differences in the number of nodes and edges (Fig. S4 and Table S3). The nodes with ≥5 correlations (edges) were selected to generate the bipartite network (Fig. S5). Among these selected core genes, 106 were unique in BACs exposed treatments, accounting for 67.09 % of total genes. Interestingly, dfra21, tetW, aadA6, aac(3)-Via, bla TEM, aph6ic and msr(E) were shared by B1, B10 and B100 (Fig. S5), and their abundance were significantly increased after BACs exposure (Fig. 2).
3.3. Microbial community compositions and structures
BACs exposure adversely affected the performance of sludge microcosms through significantly increasing the pH, and sCOD, NH4 +-N, while significantly decreasing TN and NO3 −-N of sewage sludge (Table S4). To reveal the corresponding alteration in taxonomic profiles of microbial communities, amplicon-based high-throughput sequencing was perfomed. A total of 1,086,135 high-quality sequences were obtained to generate 1019 bacterial amplicon sequence variants (ASVs) at the 3 % dissimilarity level. Rarefaction curves of bacterial ASVs at the sequencing depth of 22,263 were shown in Fig. S6. The richness, Chao, and Shannon indexes were higher in the treatments of BACs, while the Simpson index was higher in the control (Fig. S6). The NMDS analysis showed that the overall pattern of bacterial communities was significantly altered by BACs exposure (stress = 0.08; ANOSIM, R = 0.98, P = 0.001; Fig. 3A). The absolute abundance of bacterial 16S rRNA genes increased after exposure to BACs (Fig. 3B). Proteobacteria, Chloroflexi, Bacteroidetes, Planctomycetes, Acidobacteria, and Actinobacteria were the top six dominant bacterial phyla, accounting for over 80 % of the total bacterial ASVs in all samples (Fig. 3C). The LEfSe analysis was further used to illustrate the main differences in bacterial communities among treatments (Fig. S7). It was observed that Actinobacteria, Myxococcota, γ-proteobacteria, and α-proteobacteria were enriched following the BACs exposure, while the relative abundance of Bacteroidetes and Chloroflexi were reduced (LDA > 4.0, P < 0.01; Fig. S7).
Fig. 3.

(A) NMDS analysis of bacterial communities based on the Bray-Curtis distance. (B) Absolute abundance of 16S rRNA gene in different sludge samples. Significant differences between 16S rRNA gene abundances among different treatments were tested (ANOVA, LSD test, P < 0.001). (C) Relative abundance of bacterial phyla. Others contains the taxa with an abundance of <0.01 in samples. (D) Procrustes test depicting the significant correlation between the ARG pattern (including MGEs) and bacterial ASV composition based on the Bray-Curtis dissimilarity metrics (sum of squares M2 = 0.4626, r = 0.73, P < 0.001; 9999 permutations).
A total of 1,139,451 high-quality sequences were obtained to generate 269 protistan ASVs at the 3 % dissimilarity level. Rarefaction curves of protistan ASVs at the sequencing depth of 6237 were shown in Fig. S8. The richness, Chao, and Shannon index decreased in the treatments of B1 and B100 (Fig. S8). The NMDS analysis showed that the protistan community compositions differed significantly between the control and BACs exposed samples (stress = 0.177; ANOSIM, R = 0.37, P = 0.001; Fig. S9). Intramacronucleata was the dominant protistan phylum with percentages ranging from 21.26 % to 37.88 % in all samples (Fig. S10). Compared to the control, the relative abundance of SAR (stramenoplies, alveolates, and rhizaria), and Amoebozoa were higher in the BACs treatments (Fig. S10). Procrustes test showed that community structures of protists were significantly correlated with that of bacteria (M 2 = 0.198, r = 0.46, P < 0.001; Fig. S11).
3.4. Correlation between ARGs and bacterial communities
A Mantel test showed that ARGs patterns (including MGEs) were significantly correlated to bacterial communities (r = 0.71, P < 0.0001). Procrustes analysis further indicated that ARGs and 16S rRNA gene ASVs were well clustered in each BACs exposure concentration, showing a goodness-of-fit test (M 2 = 0.4626, r = 0.73, P < 0.001; Fig. 3D). To elucidate the impacts of BACs on the antibiotic resistance of bacterial communities, the co-occurrence patterns among the resistant genes and bacterial genera were conducted by using correlation networks. All networks could be parsed into modules based on the modularity (Fig. 4 ). Compared with CK, the hub (node with the most dense connection) in the modules of the network and their related co-occurring ARGs or bacteria were different under BACs treatment (Fig. 4). BACs exposure increased the frequency of co-occurrence between bacteria and ARGs (Table S3), indicating BACs might promote the spread of ARGs among sludge microbiomes.
Fig. 4.

Network analysis showing the patterns of co-occurrence among ARGs, MGEs and bacterial genera. The nodes were colored according to ARG types, MGEs and bacteria. The size of each node was weighted according to the degree. A connection stands for a strong (Spearman's correlation coefficient ρ > 0.8) and significant (adj. P < 0.01) correlation. The green lines indicate co-occurrence (positive) and yellow lines for mutualistic exclusion (negative). MLSB, macrolide-lincosamide-streptogramin B. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The SEM analysis was constructed to further reveal the possible drivers of ARGs abundance in sludge microbiome. The results showed that 83.8 % of the variance of ARGs abundance was successfully explained (Fig. 5 ). BACs concentration could directly affect the absolute abundance of ARGs or could do so indirectly by strongly affecting the absolute abundance of MGEs (Fig. 5A, P < 0.05). BACs concentrations significantly and negatively affected the absolute abundance of ARGs and sludge properties (P < 0.05). In addition, BACs concentrations significantly and positively affected the absolute abundance of MGEs (Fig. 5, P < 0.05). The absolute abundance of MGEs was significantly and positively correlated with the absolute abundance of ARGs (P < 0.05). Sludge properties was significantly and negatively correlated with the absolute abundance of ARGs and MGEs (Fig. 5, P < 0.05). Bacterial and protistan communities had nonsignificant direct impacts on the absolute abundance of ARGs (P > 0.05).
Fig. 5.

(A) SEM showing the effects of multiple variables on the abundance of ARGs in sewage sludge. Numbers adjacent to pathway arrows are standardized path coefficient. Orange lines indicate negative effects and blue lines indicate positive effects. Solid or dashed pathways indicate significant (P < 0.05) or non-significant relationships. R2 represents the explanatory rate of variance in ARGs and MGEs abundance by the model. Chi-square (χ2) = 0.217, degrees of freedom (df) = 3, P = 0.857, comparative fit index (CFI) = 0.956, goodness-of-fit index (GFI) = 0.999, root mean square error of approximation (RMSEA) = 0.02. (B) Standardized total effects (sum of direct and indirect effects) on the abundance of ARGs derived from the SEM. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5. Cellular ROS level induced by BACs exposure
To further verify whether BACs exposure can induce the overproduction of cellular ROS, the ROS level of two model strains under different BACs concentrations was determined by using a flow cytometer with DCFH-DA dye staining. Our results showed that compared with the corresponding control, low BACs concentration (1 mg L−1) induced less ROS production, with an increase of only 1.36- (S. aureus, P > 0.05) and 1.72-fold (E. coli, P > 0.05), respectively. However, at higher exposure concentrations (i.e., 10 and 100 mg L−1), BACs increased the ROS production significantly, from 2.72- (E. coli at 10 mg L−1 of BACs, P < 0.01) to 7.93-fold increase (S. aureus, at 10 mg L−1 of BACs, P < 0.001, Fig. S12).
4. Discussions
4.1. Selectively enrichment of ARGs and MGEs under BACs exposure
To control and prevent the pandemic of COVID-19, BACs disinfectants have been widely used (Hora et al., 2020). However, it remains elusive which ARGs could be enriched due to the increased release of disinfectant into WWTPs. In present study, we conducted BACs exposure experiments to evaluate the impact of increased BACs usage on the ARG profiles in sewage sludge microbiomes. By using HT-qPCR targeting almost all major classes of ARGs and MGEs, our results indicated that BACs exposure significantly promotes the diversity and abundance of ARGs and MGEs at all concentrations investigated (Fig. 1). Compared with previous studies that targeted a limited number (<15) of ARGs from environmental samples (Chacón et al., 2021; Han et al., 2019), our work provided comprehensive profiles of ARGs under BACs exposure. It should be acknowledged that our results were based on a short-term and single BACs exposure experiment, which was different from previous studies that were conducted for a long time (Tandukar et al., 2013). However, both studies suggested that exposure to BACs clearly boosted antibiotic resistance in microbial community. The increased antibiotic resistance may be driven by the co-selection of BACs through cross-resistance and co-resistance. Cross-resistance is the phenomenon in which the same genetic mechanisms responsible for simultaneous resistance to both BACs and antibiotics, such as efflux pumps (Hegstad et al., 2010; Ye et al., 2017). For example, BACs treatments significantly enriched ARGs conferring resistance to multidrug (qacF_H, qacH_351, mdtg, msr(E), and mtrD), these genes conferred resistance to BACs and cross-resistance to multidrug through efflux pump (Buffet-Bataillon et al., 2012). These results were consistent with other previous studies that BACs can selectively enrich multidrug resistance genes (Chacón et al., 2021; Han et al., 2019). In addition, co-resistance occurs when the genes conferring resistance to BACs and antibiotics are present on the same genetic units, such as the same integron, transposon, and plasmid (Baker-Austin et al., 2006; Ye et al., 2017). It is documented qac gene alleles (such as qacF_H and qacH_351), responsible for resistance to BACs, were physically located on the same MGEs with other ARGs. For example, previous studies have reported that qac gene alleles and other resistance cassettes (conferring resistance to aminoglycosides, beta-lactams, trimethoprims, and sulfonamides) were frequently located in integrons (An et al., 2018; Gillings et al., 2009). Our results showed that BACs exposure significantly increased the absolute abundance of MGEs, such as integrons (int1 and int3), transposons (IS613, IS6100, IS26, IS1247, and tnpA-3), IS (ISSm2- Xanthob, and ISPps1-pseud), and plasmids (trb-C and IncP_oriT) (Fig. 1C and 2). Therefore, the enrichment of aminoglycosides (aac(3)-Via, aac(6′)-ig, aph6ic, aadA6, and aadA16), beta-lactams (bla OXY-1, bla TEM), trimethoprims (dfra12, dfra14, dfra17, dfra21, and dfra27) and sulfonamides (sul1, sul2, and sulA_folP) (Fig. 1C and 2) in the present study could be explained by the co-resistance of these ARGs embedded in gene cassettes under BACs selective pressure. This hypothesis was further confirmed by the significant correlations between the absolute abundance of ARGs and MGEs as well (P < 0.05, Table S7). Taken together, our results demonstrated that increased BACs exposure can selectively promote ARGs in sludge microbiomes and emphasized the potential risks for ARGs dissemination mediated by MGEs in the environment.
4.2. Effects of BACs on microbial communities
It was reported that bacteria can survive and thrive under BACs treatment through tolerance or resistance to BACs (Nordholt et al., 2021; Weber et al., 2007). Interestingly, we observed that the alpha diversity (Observed richness, Chao) of bacterial communities was significantly increased after BACs exposure at low concentrations (1 and 10 mg·L−1), and then decreased at high concentration (100 mg·L−1) (Fig. S6). Previous reports have shown that BACs could be used as an available carbon source by certain aerobic bacteria in activated sludge treatment systems (Ertekin et al., 2016; Tandukar et al., 2013), which could promote the proliferation of these bacteria. BACs exposure selective enriched Actinobacteria, Myxococcia, α-proteobacteria, γ-proteobacteria, and Thermoleophilia in sludge bacterial communities (Fig. S7), indicating these bacteria may be either BACs resistant species or BACs degraders. It was reported that Pseudomonas spp. affiliated with the class γ-proteobacteria play important roles in the biodegradation of BACs (Tandukar et al., 2013). These degraders may decrease the selection pressure of BACs on the surrounding sensitive species, thus alleviating antibiotic resistance development in microbial communities (Bottery et al., 2021). In addition, some enriched species were potential ARGs hosts in the sludge microbiomes. For instance, Actinobacteria were well-known as antibiotic-producing bacteria and often detected with multiple ARGs, which could contribute to the elevated abundance of ARGs in sludge samples (D'Costa et al., 2006). Kim et al. (2018) recovered the opportunistic pathogen P. aeruginosa from BAC-fed bioreactors, that were more resistant to polymyxin B, a “last resort” antibiotic, than the counterparts in control bioreactors (Kim et al., 2018). Moreover, BACs exposure may stimuli the proliferation of specific BACs degraders, which is possible to cause a cascading dissemination of ARGs through conjugation across sludge microbiomes.
Furthermore, protists are important components of microbial organisms in the activated sludge treatment systems and major bioindicators for assessing WWTP performance (Nicolau et al., 2005). It was reported protists were sensitive to anthropogenic activities and environmental changes (Gad et al., 2020; Simon et al., 2015; Zhu et al., 2020; Zou et al., 2021). To the best of our knowledge, this is the first work to report the changes in protistan communities in sewage sludge under BACs treatments. In addition, our results indicated that BACs exposure significantly enriched SAR (stramenoplies, alveolates, and rhizaria) and Amoebozoa in sludge protistan communities (Fig. S10). Protists are considered key regulators of bacterial community compositions and evolution through their predation activities (Nguyen et al., 2020b; Nicolau et al., 2005). BACs pressures might increase the abundance of certain bacterial species, which could be potential food sources and facilitate the development of these above protistan consumers. Moreover, recent studies have reported that the interactions between protists and bacteria could affect the abundance and diversity of ARGs (Nguyen et al., 2020a; Zhou et al., 2021). Further studies are required to fully elucidate the roles of protists on the dissemination of ARGs in the sludge microbiomes under BACs exposure.
4.3. Potential factors accounting for ARGs variation under BACs exposure
Several potential factors may have contributed to the overall ARGs variation, that is, BACs concentration, sludge physicochemical properties, bacterial and protistan communities, and MGEs abundance. The SEM analysis showed that BACs concentrations were positively correlated with the absolute abundance of MGEs, which could simultaneously affect the absolute abundance of ARGs (Fig. 5). For instance, resistance genes for both antibiotics and BACs are always physically co-located on the same plasmid, which could promote the spread of ARGs embedded in gene cassettes via HGT under BACs exposure (Chen et al., 2021). Of note, certain subtypes of ARGs, such as multidrug efflux pumps, were enriched under BACs stress, emphasizing the potential risks of excessive application of disinfectants. It was reported efflux pump can expel both antibiotics and BACs, thus exhibited not only BACs resistance but also multidrug resistance because of cross-resistance (Pereira et al., 2019). Although the compositions of bacterial communities were positively correlated with ARGs patterns (Fig. 3D and Fig. 5A), which were in agreement with previous studies (Chen et al., 2016; Su et al., 2015; Yang et al., 2020; Zhu et al., 2018). In this study, our results indicated that MGEs appeared to play a more important role in accounting for ARGs variation than microbial composition under BACs pressure (Fig. 5). A plausible explanation was that BACs exposure induced over-production of ROS (Fig. S12) (Han et al., 2019), which could facilitate the acquisition of ARGs through horizontal gene transfer mediated by MGEs. As chemical disinfectants, BACs could disrupt microbial membrane properties and function, resulting in the loss of cytoplasmic constituents and eventual cell death (Tezel and Pavlostathis, 2015; Zhang et al., 2015). When at environmentally relevant concentrations, BACs can alter bacterial cell membrane permeability and mobility and stimulate the overproduction of ROS (Han et al., 2019; Tezel and Pavlostathis, 2015), thereby inducing the horizontal transfer of MGEs that harbor both BACs resistance genes and ARGs. Moreover, the increased concentration of BACs promoted the frequency of co-occurrence between bacteria and ARGs (Table S8), indicating that more bacterial genera could act as the possible ARGs hosts under BACs exposure. This result was also supported by the co-occurrence network analysis of bacterial genera and ARGs, which suggested that BACs exposure extended the interactions among microbial taxa and ARGs (Fig. 4). These results collectively strengthened the evidences that BACs exposure increased the horizontal transfer of ARGs into pathogenic hosts in sewage sludge microbiomes.
5. Conclusions
BACs disinfectants have been extensively used during the pandemic of COVID-19. In this study, our results showed that BACs exposure significantly increased the absolute abundance of ARGs in sewage sludge, as well as altering community compositions of protists and bacteria. The SEM further demonstrated that the abundance of MGEs exert greater impacts on the variation of ARGs abundance than microbial communities under BACs exposure, suggesting BACs may promote antibiotic resistance by enhancing the horizontal transfer of ARGs across sludge microbiomes. This study provides experimental evidences that excessive application of disinfectants may result in the enrichment of ARGs in sewage sludge and highlights the potential ecological risks of spreading ARGs into the ambient environment through biosolids application. As the current COVID-19 pandemic continues, the environmental residue levels of BACs would likely be elevated, thus further studies are needed in the “One Health” framework to fully elucidate the impact of persisting input of disinfectants on microbial ecology during the pandemic.
CRediT authorship contribution statement
K. Y.: experiment and visualization, supervision, funding acquisition, review and revision of manuscript. M.-L. C.: experiment and data analyses, visualization, and writing the original manuscript. D. Z.: funding acquisition and revision of manuscript. All authors read and approved the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank Professor Yong-Guan Zhu for illuminating discussions and suggestions. We thank Li-Juan Li and Xin-Rong Huang (from Zhu lab) for helping with sample collection. This work was supported by the National Natural Science Foundation of China (22193062, 22206184, 42021005, 21936006); the Alliance of International Science Organizations (ANSO-PA-2020-18).
Editor: Fang Wang
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scitotenv.2023.161527.
Appendix A. Supplementary data
Supplementary material
Data availability
Data will be made available on request.
References
- An X.-L., Chen Q.-L., Zhu D., Zhu Y.-G., Gillings M.R., Su J.-Q. Impact of wastewater treatment on the prevalence of integrons and the genetic diversity of integron gene cassettes. Appl. Environ. Microbiol. 2018;84 doi: 10.1128/AEM.02766-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APHA . American Public Health Association; Washington, DC: 2017. Standard Methods for the Examination of Water and Wastewater. [Google Scholar]
- Baker-Austin C., Wright M.S., Stepanauskas R., McArthur J.V. Co-selection of antibiotic and metal resistance. Trends Microbiol. 2006;14:176–182. doi: 10.1016/j.tim.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Barber O.W., Hartmann E.M. Benzalkonium chloride: a systematic review of its environmental entry through wastewater treatment, potential impact, and mitigation strategies. Crit. Rev. Environ. Sci. Technol. 2022;52:2691–2719. [Google Scholar]
- Bottery M.J., Pitchford J.W., Friman V.-P. Ecology and evolution of antimicrobial resistance in bacterial communities. ISME J. 2021;15:939–948. doi: 10.1038/s41396-020-00832-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffet-Bataillon S., Tattevin P., Bonnaure-Mallet M., Jolivet-Gougeon A. Emergence of resistance to antibacterial agents: the role of quaternary ammonium compounds—a critical review. Int. J. Antimicrob. Agents. 2012;39:381–389. doi: 10.1016/j.ijantimicag.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Chacón L., Arias-Andres M., Mena F., Rivera L., Hernández L., Achi R., et al. Short-term exposure to benzalkonium chloride in bacteria from activated sludge alters the community diversity and the antibiotic resistance profile. J. Water Health. 2021;19:895–906. doi: 10.2166/wh.2021.171. [DOI] [PubMed] [Google Scholar]
- Chen Q., An X., Li H., Su J., Ma Y., Zhu Y.-G. Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil. Environ. Int. 2016;92–93:1–10. doi: 10.1016/j.envint.2016.03.026. [DOI] [PubMed] [Google Scholar]
- Chen B., Han J., Dai H., Jia P. Biocide-tolerance and antibiotic-resistance in community environments and risk of direct transfers to humans: unintended consequences of community-wide surface disinfecting during COVID-19? Environ. Pollut. 2021;283 doi: 10.1016/j.envpol.2021.117074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Costa V.M., McGrann K.M., Hughes D.W., Wright G.D. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- Ertekin E., Hatt J.K., Konstantinidis K.T., Tezel U. Similar microbial consortia and genes are involved in the biodegradation of benzalkonium chlorides in different environments. Environ. Sci. Technol. 2016;50:4304–4313. doi: 10.1021/acs.est.5b05959. [DOI] [PubMed] [Google Scholar]
- Gad M., Hou L., Li J., Wu Y., Rashid A., Chen N., et al. Distinct mechanisms underlying the assembly of microeukaryotic generalists and specialists in an anthropogenically impacted river. Sci. Total Environ. 2020;748 doi: 10.1016/j.scitotenv.2020.141434. [DOI] [PubMed] [Google Scholar]
- Gillings M.R., Xuejun D., Hardwick S.A., Holley M.P., Stokes H.W. Gene cassettes encoding resistance to quaternary ammonium compounds: a role in the origin of clinical class 1 integrons? ISME J. 2009;3:209–215. doi: 10.1038/ismej.2008.98. [DOI] [PubMed] [Google Scholar]
- Han Y., Zhou Z.-C., Zhu L., Wei Y.-Y., Feng W.-Q., Xu L., et al. The impact and mechanism of quaternary ammonium compounds on the transmission of antibiotic resistance genes. Environ. Sci. Pollut. Res. 2019;26:28352–28360. doi: 10.1007/s11356-019-05673-2. [DOI] [PubMed] [Google Scholar]
- Hegstad K., Langsrud S., Lunestad B.T., Scheie A.A., Sunde M., Yazdankhah S.P. Does the wide use of quaternary ammonium compounds enhance the selection and spread of antimicrobial resistance and thus threaten our health? Microb. Drug Resist. 2010;16:91–104. doi: 10.1089/mdr.2009.0120. [DOI] [PubMed] [Google Scholar]
- Hora P.I., Pati S.G., McNamara P.J., Arnold W.A. Increased use of quaternary ammonium compounds during the SARS-CoV-2 pandemic and beyond: consideration of environmental implications. Environ. Sci. Technol. Lett. 2020;7:622–631. doi: 10.1021/acs.estlett.0c00437. [DOI] [PubMed] [Google Scholar]
- Jari O., F. Guillaume B., Michael F., Roeland K., Pierre L., Dan M., et al. 2019. vegan: Community Ecology Package. [Google Scholar]
- Kim M., Weigand M.R., Oh S., Hatt J.K., Krishnan R., Tezel U., et al. Widely used benzalkonium chloride disinfectants can promote antibiotic resistance. Appl. Environ. Microbiol. 2018;84:e01201–e01218. doi: 10.1128/AEM.01201-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H., Li X., Yang Q., Bai Y., Cui P., Wen C., et al. Herbicide selection promotes antibiotic resistance in soil microbiomes. Mol. Biol. Evol. 2021;38:2337–2350. doi: 10.1093/molbev/msab029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin M.F., Jones M.V., Lambert P.A. Pseudomonas aeruginosa cells adapted to benzalkonium chloride show resistance to other membrane-active agents but not to clinically relevant antibiotics. J. Antimicrob. Chemother. 2002;49:631–639. doi: 10.1093/jac/49.4.631. [DOI] [PubMed] [Google Scholar]
- Mahoney A.R., Safaee M.M., Wuest W.M., Furst A.L. The silent pandemic: emergent antibiotic resistances following the global response to SARS-CoV-2. iScience. 2021;24 doi: 10.1016/j.isci.2021.102304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier L., Pruteanu M., Kuhn M., Zeller G., Telzerow A., Anderson E.E., et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555:623. doi: 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C.J.L., Ikuta K.S., Sharara F., Swetschinski L., Robles Aguilar G., Gray A., et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 2022;399:629–655. doi: 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B.-A.T., Chen Q.-L., He J.-Z., Hu H.-W. Microbial regulation of natural antibiotic resistance: understanding the protist-bacteria interactions for evolution of soil resistome. Sci. Total Environ. 2020;705 doi: 10.1016/j.scitotenv.2019.135882. [DOI] [PubMed] [Google Scholar]
- Nguyen B.-A.T., Chen Q.-L., He J.-Z., Hu H.-W. Oxytetracycline and ciprofloxacin exposure altered the composition of protistan consumers in an agricultural soil. Environ. Sci. Technol. 2020;54:9556–9563. doi: 10.1021/acs.est.0c02531. [DOI] [PubMed] [Google Scholar]
- Nicolau A., Martins M.J., Mota M., Lima N. Effect of copper in the protistan community of activated sludge. Chemosphere. 2005;58:605–614. doi: 10.1016/j.chemosphere.2004.08.096. [DOI] [PubMed] [Google Scholar]
- Nordholt N., Kanaris O., Schmidt S.B.I., Schreiber F. Persistence against benzalkonium chloride promotes rapid evolution of tolerance during periodic disinfection. Nat. Commun. 2021;12:6792. doi: 10.1038/s41467-021-27019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliverio A.M., Power J.F., Washburne A., Cary S.C., Stott M.B., Fierer N. The ecology and diversity of microbial eukaryotes in geothermal springs. ISME J. 2018;12:1918–1928. doi: 10.1038/s41396-018-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz S., López P., López V., Martínez-Suárez J.V. Antibiotic susceptibility in benzalkonium chloride-resistant and -susceptible Listeria monocytogenes strains. Foodborne Pathog. Dis. 2014;11:517–519. doi: 10.1089/fpd.2013.1724. [DOI] [PubMed] [Google Scholar]
- Pereira B.M.P., Tagkopoulos I., Vieille C. Benzalkonium chlorides: uses, regulatory status, and microbial resistance. Appl. Environ. Microbiol. 2019;85 doi: 10.1128/AEM.00377-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z., Yu Y., Chen Z., Jin M., Yang D., Zhao Z., et al. Nanoalumina promotes the horizontal transfer of multiresistance genes mediated by plasmids across genera. Proc. Natl. Acad. Sci. U. S. A. 2012;109:4944–4949. doi: 10.1073/pnas.1107254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2017. R: A Language and Environment for Statistical Computing. [Google Scholar]
- Rosseel Y. lavaan: an R package for structural equation modeling. J. Stat. Softw. 2012;48:1–36. [Google Scholar]
- Ruan T., Song S., Wang T., Liu R., Lin Y., Jiang G. Identification and composition of emerging quaternary ammonium compounds in municipal sewage sludge in China. Environ. Sci. Technol. 2014;48:4289–4297. doi: 10.1021/es4050314. [DOI] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M., López-García P., Deschamps P., Moreira D., Restoux G., Bertolino P., et al. Marked seasonality and high spatial variability of protist communities in shallow freshwater systems. ISME J. 2015;9:1941–1953. doi: 10.1038/ismej.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.-Q., Wei B., Ou-Yang W.-Y., Huang F.-Y., Zhao Y., Xu H.-J., et al. Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ. Sci. Technol. 2015;49:7356–7363. doi: 10.1021/acs.est.5b01012. [DOI] [PubMed] [Google Scholar]
- Su J.-Q., An X.-L., Li B., Chen Q.-L., Gillings M.R., Chen H., et al. Metagenomics of urban sewage identifies an extensively shared antibiotic resistome in China. Microbiome. 2017;5:84. doi: 10.1186/s40168-017-0298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandukar M., Oh S., Tezel U., Konstantinidis K.T., Pavlostathis S.G. Long-term exposure to benzalkonium chloride disinfectants results in change of microbial community structure and increased antimicrobial resistance. Environ. Sci. Technol. 2013;47:9730–9738. doi: 10.1021/es401507k. [DOI] [PubMed] [Google Scholar]
- Tezel U., Pavlostathis S.G. Quaternary ammonium disinfectants: microbial adaptation, degradation and ecology. Curr. Opin. Biotechnol. 2015;33:296–304. doi: 10.1016/j.copbio.2015.03.018. [DOI] [PubMed] [Google Scholar]
- Voumard M., Venturelli L., Borgatta M., Croxatto A., Kasas S., Dietler G., et al. Adaptation of Pseudomonas aeruginosa to constant sub-inhibitory concentrations of quaternary ammonium compounds. Environ. Sci.: Water Res.Technol. 2020;6:1139–1152. [Google Scholar]
- Wang Y., Yu Z., Ding P., Lu J., Klümper U., Murray A.K., et al. Non-antibiotic pharmaceuticals promote conjugative plasmid transfer at a community-wide level. Microbiome. 2022;10:124. doi: 10.1186/s40168-022-01314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber D.J., Rutala W.A., Sickbert-Bennett E.E. Outbreaks associated with contaminated antiseptics and disinfectants. Antimicrob. Agents Chemother. 2007;51:4217–4224. doi: 10.1128/AAC.00138-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham Hadley. Springer-Verlag; New York: 2009. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Yang Y., Wang W. Benzyldimethyldodecyl ammonium chloride shifts the proliferation of functional genes and microbial community in natural water from eutrophic lake. Environ. Pollut. 2018;236:355–365. doi: 10.1016/j.envpol.2018.01.059. [DOI] [PubMed] [Google Scholar]
- Yang K., Chen Q.-L., Chen M.-L., Li H.-Z., Liao H., Pu Q., et al. Temporal dynamics of antibiotic resistome in the plastisphere during microbial colonization. Environ. Sci. Technol. 2020;54:11322–11332. doi: 10.1021/acs.est.0c04292. [DOI] [PubMed] [Google Scholar]
- Ye J., Rensing C., Su J., Zhu Y.-G. From chemical mixtures to antibiotic resistance. J. Environ. Sci. 2017;62:138–144. doi: 10.1016/j.jes.2017.09.003. [DOI] [PubMed] [Google Scholar]
- Zeng J., Li Y., Jin G., Su J.-Q., Yao H. Short-term benzalkonium chloride (C12) exposure induced the occurrence of wide-spectrum antibiotic resistance in agricultural soils. Environ. Sci. Technol. 2022;56:15054–15063. doi: 10.1021/acs.est.2c04730. [DOI] [PubMed] [Google Scholar]
- Zhang C., Cui F., Zeng G.-m., Jiang M., Yang Z.-z., Yu Z.-g., et al. Quaternary ammonium compounds (QACs): a review on occurrence, fate and toxicity in the environment. Sci. Total Environ. 2015;518–519:352–362. doi: 10.1016/j.scitotenv.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Gu A.Z., He M., Li D., Chen J. Subinhibitory concentrations of disinfectants promote the horizontal transfer of multidrug resistance genes within and across genera. Environ. Sci. Technol. 2017;51:570–580. doi: 10.1021/acs.est.6b03132. [DOI] [PubMed] [Google Scholar]
- Zhang H., Tang W., Chen Y., Yin W. Disinfection threatens aquatic ecosystems. Science. 2020;368:146–147. doi: 10.1126/science.abb8905. [DOI] [PubMed] [Google Scholar]
- Zhou S.-Y.-D., Lin C., Yang K., Yang L.-Y., Yang X.-R., Huang F.-Y., et al. Discarded masks as hotspots of antibiotic resistance genes during COVID-19 pandemic. J. Hazard. Mater. 2021;127774 doi: 10.1016/j.jhazmat.2021.127774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.-G., Zhao Y., Li B., Huang C.-L., Zhang S.-Y., Yu S., et al. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2017;2:16270. doi: 10.1038/nmicrobiol.2016.270. [DOI] [PubMed] [Google Scholar]
- Zhu D., An X.-L., Chen Q.-L., Yang X.-R., Christie P., Ke X., et al. Antibiotics disturb the microbiome and increase the incidence of resistance genes in the gut of a common soil collembolan. Environ. Sci. Technol. 2018;52:3081–3090. doi: 10.1021/acs.est.7b04292. [DOI] [PubMed] [Google Scholar]
- Zhu W., Qin C., Ma H., Xi S., Zuo T., Pan W., et al. Response of protist community dynamics and co-occurrence patterns to the construction of artificial reefs: a case study in Daya Bay,China. Sci. Total Environ. 2020;742 doi: 10.1016/j.scitotenv.2020.140575. [DOI] [PubMed] [Google Scholar]
- Zou K., Wang R., Xu S., Li Z., Liu L., Li M., et al. Changes in protist communities in drainages across the Pearl River Delta under anthropogenic influence. Water Res. 2021;200 doi: 10.1016/j.watres.2021.117294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Data Availability Statement
Data will be made available on request.