

Abstract
Background and aims:
Shortened duration therapy for acute and recent hepatitis C virus (HCV) infection has been shown to be highly effective in several small non-randomised studies with direct-acting antiviral regimens, however large randomised studies are lacking.
Methods:
REACT was an NIH-funded multicentre international, open-label, randomised, phase 4 non-inferiority trial examining the efficacy of short course (6 weeks) versus standard course (12 weeks) therapy with sofosbuvir-velpatasvir for recent HCV infection (estimated duration of infection <= 12 months). Randomisation occurred at week 6. The primary endpoint was SVR12 in the intention-to treat (ITT) population. A total of 250 participants were planned for enrolment. On advice of the data safety and monitoring board the study was halted early.
Results:
Primary analysis population consisted of 188 randomised participants at termination of study enrolment; short arm (n=93), standard arm (n=95). Ninety seven percent were male and 69% HIV positive. ITT SVR12 was 76/93, 81.7% (95% CI 72.4–89.0) in the short arm and 86/95, 90.5% (95% CI 82.7–95.6) in the standard arm. The difference between the arms was −8.8 (95% CI: −18.6, 1.0). By modified ITT analysis in which non-virological reasons for failure were excluded (death, reinfection, lost to follow-up) SVR12 was 76/85, 89.4% (95% CI 80.8–95.0) in the short arm and 86/88, 97.7% in the standard arm (95% CI 92.0–99.7; difference −8.3%, p=0.025).
Conclusions:
In this randomised study in recent HCV infection, 6 weeks sofosbuvir-velpatasvir did not meet the criteria for non-inferiority to standard 12 weeks duration.
Keywords: HCV, treatment, direct-acting antivirals, recently acquired, acute, short duration
Lay summary
In this randomised trial one hundred and eighty people with recently acquired hepatitis C infection were randomly assigned to treatment using either a short 6-week course (93 people) or standard 12-week course (95 people) of the hepatitis C treatment sofosbuvir/velpatasvir. There were nine cases of relapse after treatment in the short course and two using the standard course. A shortened course of 6 weeks therapy for hepatitis C infection was considered not as effective as a standard twelve week course in people with recently acquired hepatitis C infection.
Graphical abstract:
End of treatment and SVR12 outcomes by intent-to-treat (ITT) analysis
INTRODUCTION
Individuals identified in the ‘acute’ phase of hepatitis C virus (HCV) infection have historically responded better to therapy than individuals with chronic HCV infection. Several studies in the interferon based-therapy era confirmed that duration of therapy, if commenced early, could be shortened by as much as half, with equivalent or higher sustained virological response (SVR) or ‘cure’. (1–3) This was demonstrated irrespective of whether the infection was considered acute (within the prior six months) or recent (within the prior one year) at therapy commencement (4), and was true across at-risk populations including people who inject drugs (PWID)(5) and people with HIV. (6, 7)
With the advent of direct acting antiviral (DAA) therapies, the paradigm of shortened treatment for those with acute or recent HCV infection has been further examined. Although studies with initial regimens (including sofosbuvir and ribavirin) were disappointing, (8, 9) several single arm studies with more potent regimens demonstrated encouraging results. (10–12) One of the largest studies, the Dutch Acute HCV in HIV (DAHHS2) study, reported an SVR of 99% in 80 individuals with genotype 1 or 4 using a shortened duration of 8 weeks grazoprevir-elbasvir. (13) Most recently, the first pan-genotypic study in recent HCV infection (TARGET3D) demonstrated an SVR of 96% (per-protocol) in 30 individuals using six weeks of gleceprevir-pibrentasvir.(14) Although encouraging, these studies are limited by the lack of control group and small sample sizes, reflecting the difficulties of identifying and recruiting large numbers of individuals in early HCV infection.
Recruiting through a large international network, the Recently Acquired HCV Infection Trial (REACT) aimed to test the hypothesis that six weeks (short) of sofosbuvir-velpatasvir is non-inferior to 12 weeks (standard) of sofosbuvir-velpatasvir among people with recent HCV infection.
METHODS
Study design and randomisation
In this open-label international multicentre phase 3 trial, adults with recent HCV were randomly assigned (1:1) to receive sofosbuvir-velpatasvir 400mg-100mg once daily for six or 12 weeks. Randomisation was undertaken using permutated block design with computer random number generator using fixed block size of four, stratified according to site and HIV status, and occurred between week five and six on treatment. Block size was known only to the study statistician and clinical trial manager. Participation was capped for HCV/HIV coinfection enrolled to the study at 70% of the total study population, additionally, the number of participants with HCV reinfection enrolled to the study was originally capped at 20% of the total study population (although subsequently revised to uncapped). Participants randomised to the short arm completed therapy at the end of six weeks, whilst those in the standard arm continued for a further six weeks (total of 12 weeks).
Participants
Participants were screened and enrolled at 24 sites: Australia (n=5), Canada (n=4), Germany (n=4), Netherlands (n=1), New Zealand (n=1), Switzerland (n=3), United Kingdom (n=4), and United States (n=2). Study recruitment was conducted through a network of tertiary viral hepatitis clinics (n=18), and primary care clinics (n=6)
Adults (age ≥18 years) with recent HCV infection as defined below, (genotypes 1–6) and HCV RNA ≥10,000 IU/mL at screening were eligible. Individuals with acute or chronic hepatitis B co-infection were excluded. Full eligibility criteria are provided in the study protocol, available in the Supplementary Material.
Sites were instructed to observe participants for four to 12 weeks between screening and baseline, providing an opportunity to assess for HCV spontaneous clearance. The timing of treatment initiation was made by the investigator on an individual basis at site level.
Study definitions
Recent primary HCV infection was defined as initial detection of anti-HCV antibody and/or HCV RNA within six months of enrolment and either: (i) documented recent HCV seroconversion (anti-HCV antibody negative result in the 18 months prior to enrolment) or (ii) acute clinical hepatitis (jaundice or alanine aminotransferase [ALT] greater than 10 times the upper limit of normal [ULN]) within the previous 12 months with the exclusion of other causes of acute hepatitis or (iii) acute asymptomatic hepatitis (acute rise in ALT >5x ULN) within the previous 12 months with the exclusion of other causes of acute hepatitis. Recent HCV reinfection was defined as new detectable HCV RNA within six months of enrolment and evidence of prior spontaneous or treatment-induced clearance (previous positive anti-HCV antibody and undetectable HCV RNA on ≥2 occasions).
The presentation of recent HCV infection at the time of diagnosis was classified as either acute clinical or asymptomatic infection. Acute clinical infection included participants with a documented clinical history of symptomatic seroconversion illness (including, but not limited to, the presence of jaundice, nausea/vomiting, abdominal pain, fever and hepatomegaly) and those without clinical symptoms, but with a documented peak ALT greater than ten times ULN within the 12 months prior to diagnosis. Asymptomatic infection included participants with anti-HCV antibody seroconversion or reinfection, but no acute clinical symptoms or documented peak ALT less than ten times ULN.
In addition to these definitions of recent primary HCV and recent HCV reinfection, estimated duration of HCV infection less than 12 months at screening was required for inclusion. The estimated date of HCV infection in those with acute clinical infection was calculated as six weeks before the onset of seroconversion illness or six weeks before the first ALT greater than ten times ULN. The estimated date of HCV infection in those with asymptomatic infection was calculated as the midpoint between the last negative anti-HCV antibody or HCV RNA and the first positive anti-HCV antibody or HCV RNA. For participants who were anti-HCV antibody negative and HCV RNA positive at screening, the estimated date of infection was six weeks before enrolment, regardless of symptom status.
Virological definitions
HCV virological suppression was defined as HCV RNA below the lower limit of quantification (LLoQ). An end-of-treatment response (ETR) was defined as HCV RNA below the LLoQ (target not detected or target detected, not quantifiable) at the end of treatment (date of treatment cessation). SVR12 was defined as HCV RNA below the LLoQ at or after 12 weeks post cessation of treatment. Treatment failure was defined as either virologic failure (HCV RNA above the LLoQ at 12 weeks post cessation of treatment with reinfection excluded on sequencing) or non-virologic failure (including reinfection, death, premature treatment discontinuation, loss to follow up or missing HCV RNA values). Reinfection was defined as HCV RNA above the LLoQ after end of treatment with detection of infection with an HCV strain that was distinct from the primary infecting strain, confirmed as heterologous virus on sequencing. SVR12 assessment was nominally set at day 84 post treatment, with a lower limit of day 70 post-treatment.
Study assessments
In the short duration arm, scheduled study visits were undertaken at baseline, treatment weeks one, two, four and six (end of treatment), and post-treatment weeks four and 12. In the standard duration arm, scheduled study visits were undertaken at treatment weeks one, two, four, six, eight, 10 and 12 (end of treatment), and post-treatment weeks four and 12. Randomisation occurred during week five on treatment (prior to the week six study visit). Study drug was dispensed at all scheduled visits (except week one) between baseline and end of treatment (14-day supply). Study drug adherence was assessed by pill count and self-reported adherence questionnaires at each scheduled visit between week two and end of treatment.
The presence of HCV RNA in plasma was assessed at all scheduled study visits using Aptima HCV Quant Dx assay, version 2.15.5 (LLoQ 10IU/mL; Hologic, Inc., Marlborough, MA, USA), with centralised testing performed at St Vincent’s Centre for Applied Medical Research (Sydney, NSW, Australia). Sequencing was conducted on HCV RNA extracted from plasma using published methods. Briefly, reverse transcription of HCV RNA was performed with random hexamers using the Invitrogen™ Superscript™ IV VILO™ Master Mix (ThermoFischer Scientific), and the Core-E2, NS5A and NS3 HCV regions were amplified by polymerase chain reaction.(15, 16) Sanger sequencing was performed at the Australian Genome Research Facility on the Applied Biosystems™ 3730xl DNA Analyzer. Sequence curation was performed using RECall.(17) The presence of polymorphisms in NS3 and NS5A at baseline (and at virological failure when occurring) was evaluated using Geno2PhenoHCV. (18)
Study endpoints
The primary endpoint was undetectable HCV RNA below the LLoQ at 12 weeks following the completion of sofosbuvir-velpatasvir treatment (SVR12). Secondary endpoints included treatment adherence, and treatment-emergent adverse events.
Statistical analysis and sample size
A total of 250 participants (1:1 randomisation) were planned for enrolment, randomisation and evaluation as the intention-to-treat (ITT) population. Given a sample size of 250 people, an assumption that the proportion achieving SVR12 would be 90% in the 12 week arm, and a lower confidence bound for an SVR12 difference (six week arm minus 12 week arm) greater than −12%, the study had approximately 80% power to demonstrate non-inferiority of the six week arm as compared to the 12 week arm. The non-inferiority margin of 12% was selected in accordance with the principles outlined in guidance on conducting non-inferiority trials (19) (20) with the choice of margin also taking into account the clinical significance of SVR12 in relation to stage of infection. A narrower non-inferiority margin would be justified in the setting of chronic HCV infection, particularly more advanced liver disease. In contrast, given that early intervention is being considered in the context of potential HCV treatment as prevention and elimination strategies, a broader margin was considered pragmatic and appropriate.
A Data Safety and Monitoring Board (DSMB) was established prior to trial commencement consisting of a blinded external statistician and three clinicians. An initial DSMB review was pre-determined for when the first 50 participants in each arm reached the SVR12 visit. At this review, the DSMB requested a further analysis after a total of 60 participants reached SVR4. Following this second review, recruitment was halted in May 2019 given concerns regarding efficacy in the six-week arm. Participants in screening or on treatment but prior to randomisation continued in the study and received 12 weeks of sofosbuvir and velpatasvir but were not randomised or included in the primary analysis population.
Primary efficacy and safety data were analysed in the ITT population (which included all randomized participants), with loss to follow-up deemed treatment failure. The modified intention-to-treat (mITT) population included participants in the ITT population, but excluded those with non-virological reasons for treatment failure (including death and loss to follow up) and reinfection. The per-protocol (PP) population included participants who received >90% of scheduled treatment for >90% of the scheduled treatment period with follow-up virologic data at SVR12 (excluding reinfection and retreatments).
Categorical parameters were summarised as number and proportion. Continuous variables were summarised by either mean and standard deviation (SD) or median and interquartile range (IQR), as appropriate. For all efficacy endpoints, means and proportions with two-sided 95% confidence intervals (CI) were determined. On-treatment adherence was calculated by subtracting the number of missed doses from the total number of doses prescribed for therapy duration and dividing by the total number of doses prescribed for therapy duration. Sofosbuvir-velpatasvir adherence was calculated by pill count and self-reported questionnaire. In calculating adherence, pill count took precedence over self-report if discrepancies were noted. A participant was considered adherent if that individual received ≥95% of scheduled doses for ≥95% of the scheduled treatment period. Analysis was performed using SAS (Version 9.4, SAS Institute Inc, Carey, NC, USA) and STATA (version 15.0; StataCorp, College Station, TX).
Study oversight
All participants provided written informed consent before study procedures. The study protocol was approved by Royal Adelaide Hospital Human Research Ethics Committee (primary study committee), as well as through local ethics committees at all study sites, and was conducted according to the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice (ICH/GCP) guidelines. The study was registered with clinicaltrials.gov (NCT02625909).
Role of the Funding source
The study was funded by National Institutes of Health (NIDA division). Study medication was provided by Gilead Sciences Inc. The sponsor (The Kirby Institute, UNSW Sydney) collected the data, managed study samples, monitored study conduct, performed the statistical analysis, and drafted the manuscript.
RESULTS
Baseline characteristics
Between March 2017 and July 2019, 277 individuals were assessed for eligibility. Of these 196 were enrolled and randomised, 97 individuals into the short arm and 99 individuals into the standard arm (Figure 1). Fifty-five individuals were excluded at screening, 38 (69%) of whom did not meet eligibility criteria, with most not meeting inclusion criteria for HCV RNA >10,000 IU/ml (Supplementary Table 1). Twenty-six individuals were enrolled, but not randomised at week six. In the majority (n=21/26, 80%), the reason for non-randomisation was the participant being before week six at time of DSMB recommendation to halt short arm. These participants were immediately extended to 12 weeks. Five individuals were not randomised due to lost to follow-up between baseline and week six. Of 196 people randomised, four individuals in the short arm were at the week six timepoint when the DSMB recommendation was made and were also immediately extended to 12 weeks therapy and excluded from the primary analysis. A further four participants correspondingly randomised to the standard arm at the time of the DSMB recommendation were also excluded from the primary analysis population. The final population for primary analysis thus consists of 188 participants, 93 in the short arm and 95 in the standard arm.
Figure 1: Participant disposition.

*These 4 participants were randomised to 12 weeks within the same timeframe as the 4 participants in short arm who were at randomisation and extended following DSMB advice and therefore excluded from the analyses.
The demographic and clinical characteristics of participants were similar between the two arms (Table 1). Mean age was 44 years, and majority male (98%) and white (84%). Seventy-four percent identified as gay and 69% were HIV positive (100% on ART, median CD4 605 [IQR 474–798 cells/mm3]). Sixty-three percent had primary HCV infection whilst just over a third (37%) presented with an HCV reinfection episode. The genotype distribution included 65% (n=122) genotype 1 (1a n=115; 1b n=6; 1, no subtype, n=1), 2% (n=4) genotype 2, 17% (n=32) genotype 3, and 16% (n=30) genotype 4. Median baseline HCV RNA was 5.6 log10 IU/mL (IQR 4.6, 6.5), with baseline HCV RNA >1,000,000 IU/ml (>6 log10) in 38% (n=72) and > 10,000,000 IU/ml (>7log10) in 10% (n=18). Median duration of HCV infection at baseline was 26 weeks (IQR 17, 35).
Table 1:
Participant demographic and clinical enrolment characteristics
| Short duration 6 weeks | Standard duration 12 weeks | Total | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| N=93 | % | N=95 | % | 188 | % | |
|
| ||||||
| Gender | ||||||
| Female | 2 | 2.2 | 4 | 4.2 | 6 | 3.2 |
| Male | 91 | 97.8 | 91 | 95.8 | 182 | 96.8 |
| Recent HCV infection | ||||||
| Primary infection | 59 | 63.4 | 60 | 63.2 | 119 | 63.3 |
| Reinfection | 34 | 36.6 | 35 | 36.8 | 69 | 36.7 |
| Race | ||||||
| Caucasian/White | 79 | 84.9 | 78 | 82.1 | 157 | 83.5 |
| Asian | 4 | 4.3 | 4 | 4.2 | 8 | 4.3 |
| Black or African American | 0 | 0.0 | 2 | 2.1 | 2 | 1.1 |
| Other | 9 | 9.7 | 8 | 8.4 | 17 | 9.0 |
| Unknown or not reported | 1 | 1.1 | 3 | 3.2 | 4 | 2.1 |
| Ethnicity | ||||||
| Hispanic or Latino | 3 | 3.2 | 8 | 8.4 | 11 | 5.9 |
| Not Hispanic or Latino | 89 | 95.7 | 87 | 91.6 | 176 | 93.6 |
| Unknown or not reported | 1 | 1.1 | 0 | 0.0 | 1 | 0.5 |
| HIV positive | 65 | 69.9 | 65 | 68.4 | 130 | 69.1 |
| Age (mean, SD) | 44.2 | 10.3 | 43.4 | 10.2 | 43.8 | 10.2 |
| Baseline HCV RNA, log10 IU/mL (median, IQR) | 5.6 | 4.8−6.5 | 5.4 | 4.3−6.3 | 5.6 | 4.6−6.5 |
| HCV genotype | ||||||
| 1a | 58 | 62.4 | 57 | 60.0 | 115 | 61.2 |
| 1b | 4 | 4.3 | 2 | 2.1 | 6 | 3.2 |
| 1 unknown subtype | 1 | 1.1 | 0 | 0 | 1 | 0.5 |
| 2 | 0 | 0.0 | 4 | 4 | 4 | 2.1 |
| 3 | 15 | 16.1 | 17 | 17.9 | 32 | 17.0 |
| 4 | 15 | 16.1 | 15 | 15.8 | 30 | 15.9 |
| Mode of HCV exposure # | ||||||
| Injecting drug use | 18 | 19.4 | 22 | 22.2 | 40 | 21.3 |
| Sexual exposure with person(s) of opposite sex | 3 | 3.2 | 4 | 4.2 | 7 | 3.7 |
| Sexual exposure with person(s) of same sex | 69 | 74.2 | 66 | 69.5 | 135 | 71.8 |
| Occupational (needle stick or other exposure) | 0 | 0.0 | 1 | 1.1 | 1 | 0.5 |
| Use of non-injectable recreational drugs | 1 | 1.1 | 0 | 0.0 | 1 | 0.5 |
| Other, specify* | 2 | 2.2 | 2 | 2.1 | 4 | 2.1 |
| Max ALT (median, IQR) | 364 | 152−799 | 360 | 155−871 | 362 | 154−847 |
| Baseline ALT (median, IQR) | 114 | 56−257 | 128 | 69−222 | 126 | 62−250 |
| Symptomatic presentation | 16 | 17 | 14 | 15 | 30 | 16 |
|
| ||||||
| Estimated duration of infection to baseline (weeks, median, IQR) | 26.1 | 17−33.8 | 25.0 | 17−35.4 | 25.8 | 17−35.2 |
|
| ||||||
| Injecting drug use characteristics: | ||||||
| Total respondents | 88 | 89 | 177 | |||
| Injecting drug use, n (%) | ||||||
| Never | 38 | 43.2 | 40 | 44.9 | 78 | 44.1 |
| Ever (total of the groups below) | 50 | 56.8 | 49 | 55.1 | 99 | 55.9 |
| Not recent (Last injected >6 months ago)1 | 10 | 20 | 14 | 28.5 | 24 | 24.2 |
| Recent (Last injected between 1–6 months ago)1 | 14 | 28 | 14 | 28. | 28 | 28.3 |
| Current (Last injected within 30 days)1 | 26 | 52 | 21 | 42.9 | 47 | 47.5 |
| In those reporting injecting drug use: | Median | IQR | Median | IQR | Median | IQR |
| Age at first injecting, median (range) | 34.5 | (24−43) | 32 | (19−40) | 33 | (21–42) |
| If injected in the previous 1 month, frequency (n, %) 2 : | N | % | N | % | N | % |
| >3x most days | 0 | 0 | 0 | 0 | 0 | 0 |
| 2–3x most days | 3 | 11.5 | 2 | 9.5 | 5 | 10.6 |
| Daily | 2 | S7.7 | 1 | 4.8 | 3 | 6.4 |
| More than weekly, but less than daily | 4 | 15.4 | 4 | 19.0 | 8 | 17.0 |
| Less than weekly | 9 | 34.6 | 9 | 42.9 | 18 | 38.3 |
| Missing | 2 | 7.7 | 2 | 9.5 | 4 | 8.5 |
| Drug injected most in last month, n (%) 2 | ||||||
| Heroin | 0 | 0.0 | 5 | 23.8 | 5 | 10.6 |
| Cocaine | 1 | 3.8 | 0 | 0.0 | 1 | 2.1 |
| Methamphetamines (ice, base, speed, meth crystal) | 12 | 46.2 | 10 | 47.6 | 22 | 46.8 |
| Morphine | 1 | 3.8 | 1 | 4.8 | 2 | 4.3 |
| Other | 3 | 11.5 | 0 | 0.0 | 3 | 6.4 |
| Fentanyl | 1 | 3.8 | 0 | 0.0 | 1 | 2.1 |
| Missing | 8 | 30.8 | 5 | 23.8 | 13 | 27.7 |
| Opioid substitution therapy, n (%) 3 | ||||||
| Never | 73 | 83.0 | 76 | 85.4 | 149 | 84.2 |
| Ever: | 12 | 13.6 | 8 | 9.0 | 20 | 11.3 |
| Current | 3 | 3.4 | 5 | 5.6 | 8 | 4.5 |
| Not current | 9 | 10.2 | 3 | 3.4 | 12 | 6.8 |
Jail, unknown, accidental needle stick, nasal drug use (also reported sexual exposure with other known to be HCV positive and use of non-injectable rec drugs)
Mode of exposure determined by clinician
Denominators: 1 – ever injected; 2 – current (injected in last 30 days); 3 – all survey respondents
The likely mode of HCV acquisition was deemed sexual exposure in 142 (76%), injecting drug use in 40 (21%), non-injecting drug use in 1 (0.5%), occupational exposure in 1 (0.5%), and other or unknown in 4 (2%). Of the sexual acquisitions, the majority (n=135/142, 95%) were same sex male-male exposures.
Risk behavioural characteristics
Most participants were in full-time employment (55%) and living in either privately owned (31%) or rental (58%) accommodation. Seventy-nine percent had undergone screening for sexually transmitted infection (STI) within the prior 12 months with 53% reporting a positive STI diagnosis (predominantly syphilis). A history of incarceration was reported in 29 participants (15%). Although IDU was reported as most likely mode of HCV acquisition in only 40 participants, 99 of 177 (56%) reported ever having injected drugs, and of these 48% injected in last 30 days, 28% in last 1–6 months, and 24% longer than 6 months ago. The most common drug used was methamphetamine (47%). Detailed description of risk behaviour characteristics is given in Supplementary Table 2.
Treatment efficacy
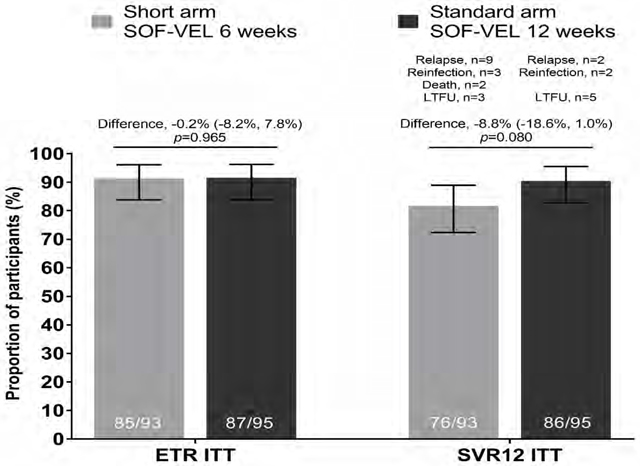
In the ITT population (n=188), the ETR was 85/93, 91.4% (95% CI 83.8–96.2) in the short arm and 87/95, 91.6% (95% CI 84.1–96.3) in the standard arm (an absolute difference in proportions of 0.2, p=0.965). SVR12 in the short arm was 76/93, 81.7% (95% CI 72.4–89.0) and 86/95, 90.5% (95% CI 82.8–95.6) in the standard arm (Figure 2). The difference between the arms was −8.81 (95% CI: −18.6, 1.0) with the 95% lower confidence bound for the difference falling below the pre-specified level of 12%. Criteria for non-inferiority was therefore not met. Although non-inferiority was not shown, the difference in SVR12 rates between the arms in the ITT analysis was not significant (p=0.080). A variety of reasons for not achieving SVR12 were observed. Overall, two participants died (both in short arm), eight were lost to follow-up (three in short arm, five in standard arm), five were re-infected (three in short arm, two in standard arm) and 11 had virological relapse (nine in short arm, two in standard arm) (Suppl Table 3).
Figure 2: Virological outcomes at end of treatment and SVR12.


Figure 2A. End of treatment response (ETR) and SVR12 outcomes by intent-to-treat (ITT) analysis Proportion of patients achieving ETR and SVR12 are given in solid bars with 95% CI for each outcome represented by line bars. Difference in proportions between the two arms (represented by horizontal line with 95% CI) at ETR p=0.965 and SVR12 ITT p=0.080 (test for equality of proportions)
Figure 2B. SVR12 outcomes by modified ITT (mITT) and per-protocol (PP) analyses Proportion of patients achieving SVR12 by modified ITT (mII) and per protocol (PP) analyses are given in solid bars with 95% CI for each outcome represented by line bars. Difference in proportions between the two arms (represented by horizontal line with 95% CI) at SVR12 (mITT) p=0.025 and SVR12 (PP) p=0.020 (test for equality of proportions)
By modified ITT (mITT) analysis in which participants with non-virological failure were excluded (including death, reinfection, lost to follow-up), SVR12 was 76/85, 89.4% (95%CI 80.8–95.0) in the short arm and 86/88 97.7% in the standard arm (95%CI 92.3–99.7; difference - 8.3, p=0.025).
In a further per-protocol analysis which included only participants who were >90% adherent and attended an SVR12 visit (excluding participants who were reinfected or retreated), SVR12 was 93.2% (95%CI 84.9–97.8) in the short arm and 100% (95%CI 95.5–100.0) in the standard arm (p=0.020).
Sixteen participants did not achieve ETR by ITT analysis of which eight participants were in each arm. In the standard arm all of eight participants were considered ETR failures due to missing data (4 of these achieved subsequent SVR12, 3 remained missing data and one patient had virological failure at SVR12). In the short arm two were due to missing data and six had detectable virus measured at ETR. All but one of these six patients with detectable virus subsequently achieved SVR12. Thus only one patient with documented detectable virus at ETR subsequently had virological failure.
Virological recurrence
Sixteen participants within the study experienced virological recurrence at or before SVR12, 12 in the short arm and four in the standard arm. Sequencing from baseline and time of recurrence was performed in all 16. Five participants were identified as having reinfection – four on the basis of a genotype switch (1a to 3a, 1a to 4d, 3a to 1a, 4d to 1a) and one with the same genotype (1a) at both timepoints but with a genetic distance that indicated heterologous virus at relapse (11.4% in core-E2, 9.4% in NS5A). Eleven (5.8%) participants were classified as relapse (9 [9.6%] in short arm, two [2.0%] in standard arm). All had homologous virus at time of relapse with a genetic distance in core-E2 of < 1.5% compared to baseline. Characteristics of the participants with reinfection and relapse are given in Table 2. Although limited by small numbers, no clear association with baseline characteristics was observed, although those with relapse did have a higher median baseline HCV RNA (6.7 log10 IU/mL, 6.8 log10 IU/mL for just those in 6 weeks arm) and longer estimated duration of infection (30 weeks across both arms) than those who did not (5.5 log10 IU/mL and 25 weeks, respectively). The proportion with baseline HCV RNA > 7 log10 IU/mL was 8% in the study population overall but 36% in the small group who relapsed. All but one of the participants with relapse was >95% adherent to therapy. One participant in the standard arm took only four weeks of treatment and subsequently relapsed. Of the eleven participants with virological failure, eight were detected as virological relapse by SVR4, one at SVR12 having been not detected at SVR4, and two did not attend for an SVR4 visit.
Table 2:
Characteristics of participants with virological failure at SVR12
| Subject | Reason for Viral recurrence at SVR 12 | Gender | Recent infection status at BL | Initial GT | GT at recurrence | Baseline HCV RNA (log IU/ml) | HCV Mode of exposure | HIV positive | Adherent (>95%) | Estimated duration of infection to BL - days | Age (years) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Standard Arm | |||||||||||
| 1 | RELAPSE | Male | Reinfection | 1b | 1b | 5.96 | IDU | No | No | 221 | 57 |
| 2 | REINFECTION | Male | Reinfection | 1a | 3a | 5.00 | IDU | No | Yes | 380 | 27 |
| 3 | REINFECTION | Male | Reinfection | 1a | 4d | 6.77 | Sexual | Yes | Yes | 154 | 52 |
| 4 | RELAPSE | Male | Primary | 4d | 4d | 6.92 | Sexual | Yes | Yes | 134 | 55 |
| Shortened Arm | |||||||||||
| 1 | RELAPSE | Male | Primary | 1a | 1a | 7.43 | Sexual | Yes | Yes | 178 | 57 |
| 2 | RELAPSE | Male | Reinfection | 3a | 3a | 5.58 | IDU | No | Yes | 228 | 56 |
| 3 | RELAPSE | Male | Primary | 1a | 1a | 6.14 | Sexual | Yes | Yes | 208 | 32 |
| 4 | RELAPSE | Male | Reinfection | 1a | 1a | 6.53 | Sexual | Yes | Yes | 245 | 46 |
| 5 | RELAPSE | Male | Reinfection | 1a | 1a | 6.81 | Sexual | Yes | Yes | 249 | 47 |
| 6 | REINFECTION | Male | Reinfection | 1a | 1a | 5.74 | Sexual | Yes | Yes | 237 | 51 |
| 7 | RELAPSE | Male | Primary | 1a | 1a | 7.10 | Sexual | Yes | Yes | 228 | 45 |
| 8 | RELAPSE | Male | Primary | 1a | 1a | 7.22 | Sexual | Yes | Yes | 175 | 57 |
| 9 | RELAPSE | Male | Primary | 4d | 4d | 7.20 | Sexual | Yes | Yes | 85 | 43 |
| 10 | REINFECTION | Male | Reinfection | 3a | 1a | 3.78 | Sexual | Yes | Yes | 140 | 45 |
| 11 | REINFECTION | Male | Primary | 4d | 1a | 5.40 | Sexual | Yes | Yes | 229 | 46 |
| 12 | RELAPSE | Male | Reinfection | 1a | 1a | 6.69 | IDU | Yes | Y | 183 | 53 |
BL: Baseline; IDU: Injecting dug use; GT: Genotype
Adherence
Overall adherence within the study was good, although was higher in the short versus standard arm. Adherence at a level of >80% and >95% was observed in 97% and 95% of participants in the short arm, and 91% and 84% of those in the standard arm (p=0.13 and 0.031 for >80% and >95% comparisons, respectively)(Table 3).
Table 3.
On treatment adherence
| N adherent | % | L95%CI | L95%CI | p-value diff (Arm 1 vs 2) | |
|---|---|---|---|---|---|
|
| |||||
| 80% adherence | 0.136 | ||||
| Short Arm | 90 | 96.77 | 90.86 | 99.33 | |
| Standard Arm | 86 | 90.52 | 82.78 | 95.58 | |
| 90% adherence | 0.250 | ||||
| Short Arm | 89 | 95.70 | 89.35 | 98.82 | |
| Standard Arm | 86 | 90.52 | 82.78 | 95.58 | |
| 95% adherence | 0.031 | ||||
| Short Arm | 88 | 94.62 | 87.90 | 98.23 | |
| Standard Arm | 80 | 84.21 | 75.30 | 90.81 | |
| 100% adherence | 0.025 | ||||
| Short Arm | 78 | 83.87 | 74.80 | 90.68 | |
| Standard Arm | 66 | 69.47 | 59.18 | 78.53 | |
Difference in proportion – Fisher’s exact p value
Resistance and retreatment
All 11 participants with virological relapse were sequenced for the development of NS3 and NS5A resistance associated substitutions (RAS). Six participants had no evidence of resistance with wild type virus at baseline and relapse. Three participants had RAS at baseline that remained unchanged at relapse (M31L, n=1 (GT4d); Q30H + Y93H (Gt1a), n=1; 62T, n=1 (Gt3a)). One participant had Y93H at baseline (GT4d) that had reverted to wild type at relapse and one participant had no RAS at baseline and L31M at relapse (GT1a, short arm). Thus, only one of 11 participants with virological relapse potentially had treatment emergent resistance following short-course treatment (L31M). Of the 11 participants with virological relapse, nine were retreated. Retreatment regimens included sofosbuvir-velpatasvirvoxilaprevir (12 weeks, n=4), sofosbuvir-velpatasvir (12 weeks, n=1) and glecaprevir-pibrentasvir (8 weeks, n=4). All achieved SVR12 apart from one whose outcome was unknown due to loss to follow-up.
Safety
In the randomised population of 188 participants, two deaths occurred, both in the short arm and occurred following the SVR4 timepoint (at which both participants had undetectable HCV RNA). The cause of death was illicit drug use plus ischaemic heart disease in one and unknown in the other; neither were considered treatment related given they occurred at least 1 month following treatment cessation. Overall, 55% of participants experienced at least one adverse event (AE), but only 23% experienced a treatment-related AE (22 in short arm; 21 in standard arm). Ninety-eight per cent of treatment-related AEs were Grade 1–2 with only one Grade 3 treatment related AE and no Grade 4 events. The only AE occurring in > 10% of the study population was fatigue, reported in 11.2% of people. Serious adverse events (SAEs) were reported in 6 participants – one in the short arm and five in the standard arm (Supplementary Table 4); only one was considered a possible treatment related SAE. This participant experienced an episode of rhabdomyolysis (rash and raised creatinine kinase) one week after commencing medication and was briefly hospitalised. Although the SAE was considered possibly treatment-related and the participant was advised to stop taking treatment, medication was continued, and the episode spontaneously resolved with full completion of therapy. Therefore, no participant discontinued treatment due to adverse events. Six participants discontinued treatment for non-safety reasons, five were lost to follow-up during the treatment period (all in the second half of therapy in the standard arm), two of whom subsequently returned for subsequent visits; one decided not to continue treatment and was subsequently lost to follow-up by SVR12 (also in standard arm).
DISCUSSION
In this randomised study of shortened treatment duration for individuals with recent HCV, sofosbuvir-velpatasvir for six weeks failed to meet the pre-specified criteria for non-inferiority and the study was terminated early following the second DSMB review. The sub-optimal efficacy in the short arm was driven largely by a higher post-treatment relapse, observed in 10% (n=9) of participants compared with 2% (n=2) of participants in the standard arm. The REACT study thus found a six-week course of the pan-genotypic regimen sofosbuvir-velpatasvir did not reach non-inferiority against the standard 12-week duration in the setting of recently acquired HCV infection and cannot be considered as effective as the 12-week course.
Although virological relapse was higher in the short compared to the standard arm, there were only 11 participants in total with this endpoint, thus limiting power to detect associations. Those with relapse were slightly older, had higher baseline HCV RNA and marginally longer duration of HCV. Although none ofthese factors could be definitively associated with failure it is of note that the median baseline HCV RNA was 1.3log higher in the 6 week relapsers than in patients who achieved SVR12, and the median duration of infection was 5 weeks longer. This may suggest that patients without these negative prognostic factors may indeed do well with shortened therapy. Failure did not appear linked to the presence of RAS at baseline. Seven of the 11 participants with relapse had wild type at baseline and only one gained a RAS (L31M) at the failure timepoint. This participant was successfully retreated with sofosbuvir-velpatasvir-voxilaprevir for 12 weeks. Retreatment was left to the discretion of the site investigator and it is noteworthy that almost all participants were retreated and achieved SVR12. These findings are encouraging for salvage from short course therapy and confirm previous findings from studies in chronic HCV infection.(21)
Short course therapy (4–6 weeks) for both acute and chronic HCV infection has been explored using a number of DAA regimens with varying success.(9, 12, 14, 22, 23) In established chronic HCV infection results have generally been sub-optimal, one exploratory study reporting SVR rates in the region of 20–40% with three and four DAA agent regimens(23). In a small study (n=16) of young PWID with early liver disease (age <50 years, LSM < 8.0kPa) in Denmark using four weeks of sofosbuvir-ledipasvir plus ribavirin, SVR12 was higher at 93% by per protocol analysis with just one case of relapse (SVR12 ITT 75%).(24) A subsequent study by the same group and in the same target population demonstrated similar results using a combination of glecaprevir-pibrentasvir plus ribavirin with an ITT SVR of 75%.(25) A lower ITT SVR of 59% was observed in 17 patients treated with four weeks glecaprevir-pibrentasvir alone. Results with short course DAA therapy in the setting of acute and recent HCV infection have been more encouraging, although again somewhat varied. Two studies from Germany, one in HIV negative and one in HIV positive participants, evaluated sofosbuvir-ledipasvir for six weeks for acute HCV infection with genotype 1 or 4 demonstrating an ITT SVR of 100% and 77%, respectively.(10, 11) A further larger study in genotype 1 and 4 using 8 weeks of grazoprevir-elbasvir, the DAHHS2 study, confirmed extremely high SVR with only one virological failure in 80 HIV positive participants.(13) TARGET3D, the only study to date using a pan-genotypic regimen, contained only one virological failure in a smaller group of 30 individuals treated with six weeks glecaprevir-pibrentasvir.(14) None of these studies was randomised and a variety of different definitions were used to characterise the often-heterogeneous study populations. In contrast, REACT is a randomised study evaluating the pan-genotypic regimen of sofosbuvir-velpatasvir; although the results appear conclusive, the study was not powered to look at differences within sub-populations and results cannot be extrapolated to all regimens and settings.
In the context of treatment of recent HCV infection among high-risk populations, there is clearly subsequent risk for HCV reinfection. Reinfections were observed in five (2.6%) participants at or prior to SVR12, with four identified by genotype switch and one with same genotype confirmed through sequencing and genetic diversity evaluation. The study population had high levels of sexual and injecting risk. Although IDU was identified as the most likely route of HCV in only 21%, a history of injecting drugs was reported by 56%, 75% of whom had injected in the prior six months. Methamphetamine was overwhelmingly the commonest drug injected. In relation to sexual risk, over half (53%) of participants reported an STI in the prior 12 months and just under half (48%) of the HIV negative population were receiving pre-exposure prophylaxis (PrEP). New HCV acquisitions among gay and bisexual men using PrEP appear to be increasing in several European countries,(26–28) and HCV testing should be considered as part of a routine sexual health check among those taking PrEP. Post-SVR12, HCV RNA testing was performed three-monthly for up to 24 months in the REACT population and will provide important further insights regarding populations at risk for reinfection.
Within the REACT cohort, no safety issues were identified and adherence to therapy and protocol were high. Adverse events reported were mild and no participant had to discontinue therapy due to side effects. Adherence was high and not a factor in most treatment failures. Ninety-three per cent of participants took more than 80% of doses and 90% took more than 95% of doses. Interestingly, adherence was higher in the short arm, suggesting that short duration therapy may have advantages in terms of treatment completion. Lost to follow-up rates were also low – only eight (4%) participants were lost before SVR12. This may in part reflect that in some REACT sites, treatment for recent HCV could only be accessed through clinical trial protocols providing additional motivation for protocol adherence, and in many sites participants were already enrolled in regular HIV care and/or opioid substitution programs. It is also true that some participants (n=5) were lost on-treatment prior to randomisation and are not included in this analysis.
Although this was a relatively large randomised trial, it does have a number of limitations impacting on our ability to make broad generalisations. Firstly despite attempts to include sites likely to see a variety of individuals with HCV acquisition, including the addition of extra sites during the study period, the study population was overwhelmingly male, limiting its generalisability to females. Similarly, most participants were HIV positive and not infected through IDU. This group may be different in engagement to HIV negative people who inject drugs, particularly in terms of patterns of drug use. Relatively few (17%) were injecting opioids although this is now the greatest source of new HCV infections across the United States. (29) Future studies should address this expanding epidemic and the role of short course therapy in this population. Third, the study population included a heterogeneous group of patients with recently acquired infection, including acute patients and those with symptomatic and asymptomatic infection. Although the small number of overall failures limited the ability to draw conclusions within sub-populations there was only one relapse patient in each arm with duration of infection less than six months at treatment commencement while most participants had a duration of infection > 6 months (median 26 weeks). Finally, sofosbuvir-velpastasvir is the only regimen evaluated in this study.
Engagement of individuals early in HCV infection is crucial for HCV elimination efforts. Despite higher relapse in the short arm, REACT confirms that treatment initiated early in infection is safe, feasible and highly effective. Even with a relapse rate of 10% in the short arm, generation of resistance was limited and almost all participants were able to be quickly and successfully retreated. Nevertheless, acknowledging the caveats on generalizability above, REACT clearly demonstrates that at least in this predominantly HIV positive population 6 weeks of sofosbuvir/velpatasvir is suboptimal and patients should be treated with at least 8 weeks duration (30) REACT provides important data on HCV therapeutic intervention outcomes for recent infection among a high-risk population, with implications for individual care and HCV elimination strategies.
Supplementary Material
Highlights.
REACT is a randomised study of short course DAA therapy for recently acquired HCV
188 participants were treated with either 6 or 12 weeks sofosbuvir/velapatasvir
Study population was predominantly cis-male with a high proportion living with HIV
The study was stopped early due to high rate of virological relapse in the short arm.
Six weeks of sofosbuvir/velapatasvir cannot be considered non-inferior to 12 weeks.
Acknowledgements
The REACT study group includes members of the Protocol Steering Committee; Coordinating Centre, The Kirby Institute, UNSW Sydney; and Study Site Principal Investigators.
Protocol Steering Committee – Marc van der Valk (Amsterdam University medical Centers, , The Netherlands), Margaret Hellard (The Alfred Hospital and Burnet Institute, Melbourne, Australia), Ed Gane (Auckland City Hospital, Auckland, New Zealand), Andri Rauch (Bern Inselspital, Bern, Switzerland), Julie Bruneau (Centre Hospitalier de l’Université de Montréal, Montréal, Canada), Arthur Kim (Massachusetts General Hospital, Boston, USA), Sanjay Bhagani (Royal Free Hospital, London, UK), Greg Dore (St Vincent’s Hospital, Sydney, Australia), Pip Marks (The Kirby Institute, Sydney, Australia), Gail Matthews (The Kirby Institute, Sydney, Australia), Jason Grebely (The Kirby Institute, Sydney, Australia), Kathy Petoumenos (The Kirby Institute, Sydney, Australia), Marianne Martinello (The Kirby Institute, Sydney, Australia), Tanya Applegate (The Kirby Institute, Sydney, Australia), Jordan Feld (Toronto General Hospital, Toronto, Canada), Jürgen Rockstroh (University Clinic Bonn, Bonn, Germany).
Coordinating Centre, The Kirby Institute, UNSW Sydney, Sydney, Australia – Gail Matthews (Coordinating Principle Investigator), Pip Marks (Clinical Trials Manager), Sophia Amjad (Study Co-ordinator), Elise Tu (Study Co-ordinator), Kathy Petoumenos (Statistician) and Mahshid Tamaddoni (Systems and Data Manager).
Site Principal Investigators – Marc van der Valk (Amsterdam University medical Centers, , The Netherlands), Margaret Hellard (The Alfred Hospital and Burnet Institute, Melbourne, Australia), Ed Gane (Auckland City Hospital, Auckland, New Zealand), Maria Christine Thurnheer (Bern Inselspital, Bern, Switzerland), Yvonne Gilleece (Brighton and Sussex University Hospitals, Brighton, UK), Julie Bruneau (Centre Hospitalier de l’Université de Montréal, Montréal, Canada), Mark Nelson (Chelsea and Westminster Hospital, London, UK), Chris Fraser (Cool Aid Community Health Centre, Victoria, Canada), Alberto Moriggia (Fondazione Epatocentro Ticino, Lugano, Switzerland), Thomas Lutz (Infektio-Research GmbH, Frankfurt, Germany), Juhi Moon(Johns Hopkins University, Baltimore, USA), Phillip Read (Kirketon Road Centre, Sydney, Australia), Arthur Y Kim (Massachusetts General Hospital, Boston, USA), Andrew Ustianowski (Pennine Acute Hospitals, Manchester, UK), Christiane Cordes (Praxis Dr Cordes, Berlin, Germany), David Shaw (Royal Adelaide Hospital, Adelaide, Australia), Sanjay Bhagani (Royal Free Hospital, London, UK), Joe Sasadeusz (Royal Melbourne Hospital, Melbourne, Australia), Mark Hull (St Paul’s Hospital, Vancouver, Canada), Greg Dore (St Vincent’s Hospital, Sydney, Australia), Jordan Feld (Toronto General Hospital, Toronto, Canada), Jürgen Rockstroh (University Clinic Bonn, Bonn, Germany), Dominique Braun (University Hospital Zurich, Zurich, Switzerland), Patrick Ingiliz (Zentrum für Infektiologie Berlin-Prenzlauer Berg, Berlin, Germany).
The authors would like to thank the study participants for their contribution to the research. The authors would also like to acknowledge the work undertaken by the Study Site Co-ordinators.
Site Co-ordinators and Site Co-investigators – Hadassa Porretta, Martine Peters and Jeltje Helder (Amsterdam University medical Centers, The Netherlands), Michelle Hagenauer (The Alfred Hospital, Melbourne, Australia), Victoria Oliver and Genevieve
Morris (Auckland City Hospital, Auckland, New Zealand), Melanie Lacalamita, Pia Scherler, Daniela Hirter, Christine Bruelisauer and Manuela Manz (Bern Inselspital, Bern, Switzerland), Tanya Adams (Brighton and Sussex University Hospitals, Brighton, UK), Barbara Kotsoros and Rachel Bouchard (Centre Hospitalier de l’Université de Montréal, Montréal, Canada), Thomas Morrish, Orla Thunder, Lester Macabodbod and Serge Fedele (Chelsea and Westminster Hospital, London, UK), Marion Selfridge (Cool Aid Community Health Centre, Victoria, Canada), Paola Messina and Selma Calcagnile (Fondazione Epatocentro Ticino, Lugano, Switzerland), Christina Appelhans and Annette Haas (Infektio-Research GmbH, Frankfurt, Germany), Mark Sulkowski, Stacey Reece and Stephanie Katz (Johns Hopkins University, Baltimore, USA), Karen Chronister and Rosie Gilliver (Kirketon Road Centre, Sydney, Australia), Jenna Gustafson and Fiona Evans (Massachusetts General Hospital, Boston, USA), Gabriella Lindergard and Valerie George (Pennine Acute Hospitals, Manchester, UK), Reinhold Schröder (Praxis Dr Cordes, Berlin, Germany), Catherine Ferguson (Royal Adelaide Hospital, Adelaide, Australia), Anne Carroll, Parizade Raymode and Eric Witele (Royal Free Hospital, London, UK), Joanne Patterson (Royal Melbourne Hospital, Melbourne, Australia), Marianne Harris and Bruce Ganase (St Paul’s Hospital, Vancouver, Canada), Alison Sevehon (St Vincent’s Hospital, Sydney, Australia), Orlando Cerocchi (Toronto General Hospital, Toronto, Canada), Brigitta Becker, Angelika Saidi and Karina Mohrmann (University Clinic Bonn, Bonn, Germany), Christina Grube (University Hospital Zurich, Zurich, Switzerland), Christoph Gerlach and Christin Monnich (Zentrum für Infektiologie Berlin-Prenzlauer Berg, Berlin, Germany).
Funding:
National Institutes of Health (NIDA)
Disclosure of Interest Statement:
The Kirby Institute is funded by the Australian Government Department of Health and Ageing. The views expressed in this publication do not necessarily represent the position of the Australian Government. Research reported in this publication was supported by NIH and Gilead Sciences Inc (study medication).
Footnotes
Author conflict of interest statement:
GVM: grants from Gilead Sciences and AbbVie Inc, outside the submitted work
SB: grants from Gilead Sciences, outside the submitted work; personal fees for Advisory Boards and lectures/presentations from Gilead Sciences, outside the submitted work
MvDW: grants and personal fees from AbbVie, grants and personal fees from Gilead, grants and personal fees from Johnson & Johnson, grants and personal fees from MSD, grants and personal fees from ViiV, outside the submitted work
JR: personal fees from Gilead Sciences, Janssen, Merck, Theratechnologies and ViiV, outside the submitted work
JF: grants and personal fees from Gilead Sciences, grants and personal fees from AbbVie, personal fees from GSK, personal fees from Roche, grants from Janssen, grants from Eiger, grants and personal fees from Enanta, personal fees from Arubutus, outside the submitted work
AR: Advisory boards: MSD, Gilead Sciences, Travel grants: Gilead Sciences, Pfizer, AbbVie; Research support: Investigator initiated trial grant from Gilead Sciences. All remuneration went to his home institution and not to Dr. Rauch personally.
CT: Gilead Advisory board (Remdesivir) 2020; MSD Advisory board (HCV) 2018; educational grants from Gilead and AbbVie (Annual Preceptorship on HCV in PWID), outside the submitted work.
JB: grants from NIH, during the conduct of the study; personal fees from AbbVie, grants and personal fees from Gilead, outside the submitted work
AK: grants from PCORI, grants from NIH/NIAID, grants from NIH/NIA, grants from UpToDate, Inc., personal fees from Biomarin, Inc., personal fees for lectures/presentations: CME companies, Clinical Care Options companies, Mentor Planning and Practice Point, personal fees from DKBMed for communications, personal fees for academic work from Geisinger Health Systems and St. Luke’s/Roosevelt, personal fees from Ken Krayesek Law Offices, personal fees from Duke University, outside the submitted work
MH: grants from Gilead Sciences, grants from AbbVie, outside the submitted work
EG: personal fees from Gilead Scientific Advisory Board, personal fees from AbbVie Scientific Advisory Board, personal fees from Janssen Scientific Advisory Board, outside the submitted work
MN: grants, personal fees and non-financial support from AbbVie, grants, personal fees and non-financial support from MSD, grants, personal fees and non-financial support from BMS, grants, personal fees and non-financial support from Gilead Sciences, payment or honoraria: Gilead, AbbVie, BMS and MSD, travel support: Gilead, AbbVie, BMS and MSD, personal fees from MBS DMSB or equivalent, outside the submitted work
PI: grants and personal fees from Gilead Sciences , personal fees from AbbVie, personal fees from ViiV, outside the submitted work
JG: grants and personal fees from AbbVie, grants and personal fees from Gilead Sciences, grants and personal fees from Merck , grants and personal fees from Cepheid, grants from Hologic, grants from Indivior, payment or honoraria: AbbVie, Gilead Sciences and Cepheid, travel support: AbbVie, Gilead Sciences and Cepheid, receipt of testing equipment and cartridges from Cepheid, receipt of testing reagents from Hologic, outside the submitted work
KP: grants from Gilead sciences Australia and ViiV Healthcare Australia, outside the submitted work
GD: grants, personal fees and non-financial support from Gilead Sciences, AbbVie and Merck, grants from Bristol-Myers Squibb, outside the submitted work.
DS, MM, TA and PM: nothing to disclose
Trial Registration: clinicaltrials.gov Identifier: NCT02625909
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement
Due to the sensitive nature of some of the data, including that related to injecting drug use, data included in this manuscript has not been placed in an open access database. However, data is available to be shared on request to the protocol steering committee.
References
- 1.Dore GJ, Hellard M, Matthews GV, Grebely J, Haber PS, Petoumenos K, et al. Effective treatment of injecting drug users with recently acquired hepatitis C virus infection. Gastroenterology. 2010;138(1):123–35 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Rosa FG, Bargiacchi O, Audagnotto S, Garazzino S, Cariti G, Calleri G, et al. Twelve-week treatment of acute hepatitis C virus with pegylated interferon- alpha −2b in injection drug users. Clin Infect Dis. 2007;45(5):583–8. [DOI] [PubMed] [Google Scholar]
- 3.Santantonio T, Fasano M, Sinisi E, Guastadisegni A, Casalino C, Mazzola M, et al. Efficacy of a 24-week course of PEG-interferon alpha-2b monotherapy in patients with acute hepatitis C after failure of spontaneous clearance. J Hepatol. 2005;42(3):329–33. [DOI] [PubMed] [Google Scholar]
- 4.Martinello M, Hellard M, Shaw D, Petoumenos K, Applegate T, Grebely J, et al. Short duration response-guided treatment is effective for most individuals with recent hepatitis C infection: the ATAHC II and DARE-C I studies. Antivir Ther. 2016;21(5):465. [DOI] [PubMed] [Google Scholar]
- 5.Grebely J, Petoumenos K, Matthews GV, Haber P, Marks P, Lloyd AR, et al. Factors associated with uptake of treatment for recent hepatitis C virus infection in a predominantly injecting drug user cohort: The ATAHC Study. Drug Alcohol Depend. 2010;107(2–3):244–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthews GV, Hellard M, Haber P, Yeung B, Marks P, Baker D, et al. Characteristics and treatment outcomes among HIV-infected individuals in the Australian Trial in Acute Hepatitis C. Clin Infect Dis. 2009;48(5):650–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dominguez S, Ghosn J, Valantin MA, Schruniger A, Simon A, Bonnard P, et al. Efficacy of early treatment of acute hepatitis C infection with pegylated interferon and ribavirin in HIV-infected patients. AIDS. 2006;20(8):1157–61. [DOI] [PubMed] [Google Scholar]
- 8.Naggie S, Marks KM, Hughes M, Fierer DS, Macbrayne C, Kim A, et al. Sofosbuvir Plus Ribavirin Without Interferon for Treatment of Acute Hepatitis C Virus Infection in HIV-1-Infected Individuals: SWIFT-C. Clin Infect Dis. 2017;64(8):1035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinello M, Gane E, Hellard M, Sasadeusz J, Shaw D, Petoumenos K, et al. Sofosbuvir and ribavirin for 6 weeks is not effective among people with recent hepatitis C virus infection: The DARE-C II study. Hepatology. 2016;64(6):1911–21. [DOI] [PubMed] [Google Scholar]
- 10.Deterding K, Spinner CD, Schott E, Welzel TM, Gerken G, Klinker H, et al. Ledipasvir plus sofosbuvir fixed-dose combination for 6 weeks in patients with acute hepatitis C virus genotype 1 monoinfection (HepNet Acute HCV IV): an open-label, single-arm, phase 2 study. Lancet Infect Dis. 2017;17(2):215–22. [DOI] [PubMed] [Google Scholar]
- 11.Rockstroh JK, Bhagani S, Hyland RH, Yun C, Dvory-Sobol H, Zheng W, et al. Ledipasvir-sofosbuvir for 6 weeks to treat acute hepatitis C virus genotype 1 or 4 infection in patients with HIV coinfection: an open-label, single-arm trial. Lancet Gastroenterol Hepatol. 2017;2(5):347–53. [DOI] [PubMed] [Google Scholar]
- 12.Naggie S, Fierer DS, Hughes MD, Kim AY, Luetkemeyer A, Vu V, et al. Ledipasvir/Sofosbuvir for 8 Weeks to Treat Acute Hepatitis C Virus Infections in Men With Human Immunodeficiency Virus Infections: Sofosbuvir-Containing Regimens Without Interferon for Treatment of Acute HCV in HIV-1 Infected Individuals. Clin Infect Dis. 2019;69(3):514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boerekamps A, De Weggheleire A, van den Berk GE, Lauw FN, Claassen MAA, Posthouwer D, et al. Treatment of acute hepatitis C genotypes 1 and 4 with 8 weeks of grazoprevir plus elbasvir (DAHHS2): an open-label, multicentre, single-arm, phase 3b trial. Lancet Gastroenterol Hepatol. 2019;4(4):269–77. [DOI] [PubMed] [Google Scholar]
- 14.Martinello M, Orkin C, Cooke G, Bhagani S, Gane E, Kulasegaram R, et al. Short-Duration Pan-Genotypic Therapy With Glecaprevir/Pibrentasvir for 6 Weeks Among People With Recent Hepatitis C Viral Infection. Hepatology. 2019. [DOI] [PubMed] [Google Scholar]
- 15.Lamoury FM, Jacka B, Bartlett S, Bull RA, Wong A, Amin J, et al. The Influence of Hepatitis C Virus Genetic Region on Phylogenetic Clustering Analysis. PLoS One. 2015;10(7):e0131437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindstrom I, Kjellin M, Palanisamy N, Bondeson K, Wesslen L, Lannergard A, et al. Prevalence of polymorphisms with significant resistance to NS5A inhibitors in treatment-naive patients with hepatitis C virus genotypes 1a and 3a in Sweden. Infect Dis (Lond). 2015;47(8):555–62. [DOI] [PubMed] [Google Scholar]
- 17.Woods CK, Brumme CJ, Liu TF, Chui CK, Chu AL, Wynhoven B, et al. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J Clin Microbiol. 2012;50(6):1936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalaghatgi P, Sikorski AM, Knops E, Rupp D, Sierra S, Heger E, et al. Geno2pheno[HCV] - A Web-based Interpretation System to Support Hepatitis C Treatment Decisions in the Era of Direct-Acting Antiviral Agents. PLoS One. 2016;11(5):e0155869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Group. IEW. ICH Harmonised tripartite guideline: Statistical principles for clinical trials E9. 1998. [PubMed] [Google Scholar]
- 20.FDA administration. Non inferiority trials to establish effectiveness: Guidance for Industry. US Department of Health and Human Services. 2016. [Google Scholar]
- 21.Wilson EM, Kattakuzhy S, Sidharthan S, Sims Z, Tang L, McLaughlin M, et al. Successful Retreatment of Chronic HCV Genotype-1 Infection With Ledipasvir and Sofosbuvir After Initial Short Course Therapy With Direct-Acting Antiviral Regimens. Clin Infect Dis. 2016;62(3):280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawitz E, Poordad F, Gutierrez JA, Wells JT, Landaverde CE, Evans B, et al. Short-duration treatment with elbasvir/grazoprevir and sofosbuvir for hepatitis C: A randomized trial. Hepatology. 2017;65(2):439–50. [DOI] [PubMed] [Google Scholar]
- 23.Kohli A, Kattakuzhy S, Sidharthan S, Nelson A, McLaughlin M, Seamon C, et al. Four-Week Direct-Acting Antiviral Regimens in Noncirrhotic Patients With Hepatitis C Virus Genotype 1 Infection: An Open-Label, Nonrandomized Trial. Ann Intern Med. 2015;163(12):899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ovrehus ALH, Krarup H, Birkemose I, Holm DK, Mossner B, Ernst A, et al. Four weeks of ledipasvir/sofosbuvir and ribavirin with or without pegylated interferon for chronic hepatitis C in non-cirrhotic people who inject drugs. A randomized trial. J Hepatol. 2018;68(4):840–2. [DOI] [PubMed] [Google Scholar]
- 25.Madsen L, Ovrehaus A, Gerstoft J, al. e. THU-193-4 week treatment for hepatitis C. A randomised controlled trial (4RIBC). Journal of Hepatology. 2019;70. [Google Scholar]
- 26.Hoornenborg E, Coyer L, Boyd A, Achterbergh RCA, Schim van der Loeff MF, Bruisten S, et al. High incidence of HCV in HIV-negative men who have sex with men using pre-exposure prophylaxis. J Hepatol. 2020;72(5):855–64. [DOI] [PubMed] [Google Scholar]
- 27.Cotte L, Cua E, Reynes J, Raffi F, Rey D, Delobel P, et al. Hepatitis C virus incidence in HIV-infected and in preexposure prophylaxis (PrEP)-using men having sex with men. Liver Int. 2018. [DOI] [PubMed] [Google Scholar]
- 28.Bradshaw D, Vasylyeva TI, Davis C, Pybus OG, Theze J, Thomson EC, et al. Transmission of hepatitis C virus in HIV-positive and PrEP-using MSM in England. J Viral Hepat. 2020;27(7):721–30. [DOI] [PubMed] [Google Scholar]
- 29.Suryaprasad AG, White JZ, Xu F, Eichler BA, Hamilton J, Patel A, et al. Emerging epidemic of hepatitis C virus infections among young nonurban persons who inject drugs in the United States, 2006–2012. Clin Infect Dis. 2014;59(10):1411–9. [DOI] [PubMed] [Google Scholar]
- 30.EASL. EASL recommendations on treatment of hepatitis C: Final update of the series. Journal of Hepatology. 2020;73:1170–218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Due to the sensitive nature of some of the data, including that related to injecting drug use, data included in this manuscript has not been placed in an open access database. However, data is available to be shared on request to the protocol steering committee.