

Abstract
The androgen receptor (AR) remains to be a key target for the treatment of prostate cancer, including the majority of castration-resistant prostate cancer (CRPC). AR is stabilized in CRPC and the ubiquitin-proteasome system (UPS) plays a major role in AR degradation. Targeting AR for degradation provides a potential approach to overcome the resistance of CRPC to current AR antagonists, including the next generation AR signaling inhibitors. Different types of AR degraders have been developed, including the proteolysis-targeting chimeras (PROTACs), selective AR degraders (SARDs), and novel AR degraders, with several AR PROTACs currently in clinical trials. The present mini-review discusses the regulation of AR degradation by the UPS, the potential role of a novel nuclear degradation signal in AR, and different types of AR degraders.
Keywords: Prostate cancer, androgen receptor, degradation, proteasome, nuclear degradation signal
Introduction
The androgen receptor (AR) is a steroid hormone receptor that is crucial for the development and progression of prostate cancer [1]. As such, the AR has emerged as the major therapeutic target for treating prostate cancer [2]. Upon binding with high affinity to dihydrotestosterone (DHT), AR is translocated to the nucleus to function as a transcription factor [3]. Current therapies have been effective in targeting the AR-signaling axis by inhibiting the biosynthesis, the amount, and the binding of DHT to the AR [4-6]. Since AR directed therapies have been successful in treating prostate cancer, it is critically important to understand the molecular mechanisms that regulate AR level and activity. Degradation of AR is one such major mechanism that regulates AR activity. Degradation of AR occurs predominantly in the nucleus [7] and is primarily regulated by the ubiquitin proteasome pathway [8]. Importantly, targeting AR degradation has recently shown promise as a therapeutic strategy for prostate cancer, particularly in patients who have developed resistance to existing therapies [9]. Understanding the mechanisms regulating AR degradation could lead to the development of better therapies.
AR stabilization in CRPC
The standard of care for patients with prostate cancer is androgen deprivation therapy (ADT). Patients initially respond favorably to this treatment, but inevitably develop resistance to this treatment and their tumors progress to castration-resistant prostate cancer (CRPC) [10,11]. There has been a concentrated effort to better understand the mechanisms regulating this therapy resistance. An increase in the protein level of AR is one of the mechanisms that confers resistance to ADT. This increase of AR level is caused by both an increase in the gene amplification/expression of AR and through the increased stability of AR [12,13]. AR in CRPC cell lines have a 2-4 fold increase in half-life compared to AR in hormone-sensitive prostate cancer cell lines [13]. A hallmark of CRPC is hypersensitivity to low levels of androgen, allowing for transcriptionally active AR with castrate levels of androgen. The increased stability of AR may contribute to AR hypersensitivity.
Although it is known that AR is stabilized in CRPC, the mechanisms conferring this stabilization are not fully understood. Recent studies have indicated that the degradation of AR happens predominantly in the nucleus and that nuclear AR is not exported back into the cytoplasm. Moreover, the localization of AR in CRPC cells is more nuclear than in hormone-sensitive cells [7]. This finding implies that the increased stability of AR in CRPC is due to the decreased rate of nuclear degradation. Since AR can only act as a transcription factor in the nucleus, this elevated AR in the nucleus of CRPC cells provides a potential explanation for resistance to therapies. More work is needed to better understand the factors that regulate AR stability. These factors could be promising therapeutic targets for preventing the transition to CRPC.
AR degradation via ubiquitin-proteasome system (UPS)
The degradation of many proteins, including AR, is regulated by the ubiquitin-proteasome system (UPS). Ubiquitination involves a variety of ubiquitination enzymes [14]. UPS exists both in the cytoplasm and nucleus, and coordinates the degradation and recycling of numerous proteins. During ubiquitination, the small (8.6 kDa) protein ubiquitin covalently links to the target protein through a peptide bond. The ubiquitinated target protein is then shuttled to a protease complex called the 26S proteasome for proteolysis. UPS includes ubiquitin, ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), ubiquitin ligases (E3s), and the 26S proteasome. As shown in Figure 1, the attachment of ubiquitin or ubiquitin chains to substrates involves several enzymatic reactions. Before AR is hydrolyzed by the proteasome, the protein undergoes a cascade of ubiquitination reactions. The first step is the activation reaction, in which ubiquitins are activated by the ubiquitin-activating enzyme E1. Activation occurs after the formation of a covalent linkage between the carboxy terminus of ubiquitin and a cysteine residue present on E1 in the form of a thioester bond (E1-Ub). The second step is the conjugation reaction, where E1-activated ubiquitin is presented to E2 ubiquitin-conjugating enzymes to form E2-Ub. The third step is the ligation reaction. The E3 ligase, which has the function of recognizing its substrate, transfers ubiquitin from E2 to the substrate, leading to a chain reaction, resulting in the polyubiquitination of the substrate. In the UPS system, E3 ligases play an important role in the regulation of substrate degradation. Since E3 ligases are responsible for recognizing and binding different protein substrates, nearly 600 E3 ligases are involved in the ubiquitination of different proteins. Several of these E3 ligases can target AR.
Figure 1.
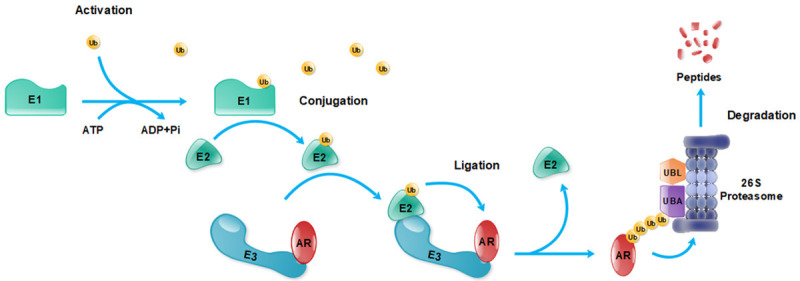
Ubiquitination and degradation of AR by the ubiquitin-proteasome system (UPS). Abbreviations: AR, androgen receptor; E1, ubiquitin ligase E1; E2, ubiquitin ligase E2; E3, ubiquitin ligase E3; Ub, ubiquitin; UBA, ubiquitin-associated domains; UBL, ubiquitin-like domains.
One of the AR E3 ligases is MDM2, a RING finger protein which was found to regulate p53 tumor suppressor via promoting p53 ubiquitination and degradation. MDM2 has been shown to ubiquitinate AR at K311 and K845/K847 [15,16]. MDM2 is required for Akt-induced, phosphorylation-dependent AR ubiquitination and degradation, because MDM2 deletion impairs Akt effects on AR ubiquitination and degradation [17]. Akt phosphorylates AR at S210/213 and S790 [18]. Phosphorylation is a critical step in Akt-dependent AR degradation since phosphorylation-resistant AR mutants prevented Akt-induced AR degradation. As expected, Akt-induced AR ubiquitylation requires AR phosphorylation. Since MDM2 is required for Akt induction of AR ubiquitination, Lin and colleagues showed MDM2, Akt, and AR could form a complex and AR phosphorylation is essential for MDM2 binding to AR [17]. As expected, MDM2-mediated AR degradation suppressed AR transcriptional activity in transient transfection assays. Taken together, these findings led to a hypothesis that Akt phosphorylates AR, facilitating MDM2 binding to AR that leads to AR ubiquitylation, degradation, and reduced activity. MDM2 could also recognize phosphorylated Serine 213 of AR-v7 to induce AR-v7 ubiquitination and degradation, similar to FL-AR [19]. In addition to Akt, Pim1S kinase was reported to bind to AR and phosphorylate AR at S210/213 that led to AR destabilization via MDM2 recruitment [20,21]. These findings suggest that multiple signals could impact AR ubiquitination, degradation, and activity via modulating AR phosphorylation and subsequent MDM2 recruitment.
SPOP [22,23] is another important E3 ligase for AR. SPOP mutations were identified in up to 15% of human prostate cancers [24]. SPOP binds to AR in the hinge domain, via a perfectly matched SPOP-binding motif (645ASSTT649). The SPOP-CUL3-RBX1 complex induces AR ubiquitination and subsequent degradation. Deletion of the 645ASSTT649 motif in AR completely abolished AR binding to SPOP and SPOP-mediated polyubiquitination of AR [22]. Wild-type SPOP can suppress AR activity and prostate cancer growth whereas prostate cancer-associated mutants of SPOP do not bind to AR and are unable to induce AR degradation. Consistently, prostate tumor xenografts with mutant SPOP grew faster and expressed higher AR protein levels than tumors expressing wild-type SPOP. Furthermore, deletion of SPOP enhanced AR protein levels in the mouse prostate [23]. There is also a strong correlation of SPOP signature score with AR activity score in prostate cancer patient cohorts in public human prostate cancer datasets. In primary prostate cancer, AR is highly active in prostate cancer tumors with SPOP mutations as compared to other molecular subtypes of tumors [25]. In a clinical study, SPOP mutations correlated with improved outcomes to ADT plus androgen receptor axis-targeted therapies (ARAT) in patients with de novo metastatic castration-sensitive prostate cancer (mCSPC), suggesting that SPOP mutations could be used as a predictive biomarker for treatment selection for mCSPC patients [26]. These findings also highlight the importance of AR stability in prostate cancer progression.
C-terminal Hsp-interacting protein (CHIP) interacts with molecular chaperones Hsp70 and Hsp90 via a tetratricopeptide domain and inhibits chaperone-dependent protein folding in vitro. CHIP was identified as an AR E3 ligase [27,28] that can bind to AR in a sequence-specific, phosphorylation- and sequence-dependent manner. This provides a potential mechanism regulating the degradation of CHIP substrates [29]. CHIP can also act as an E3 ubiquitin ligase through its U box domain. In transient transfection assays, CHIP overexpression caused a marked decrease in AR steady state levels and enhanced AR ubiquitination. CHIP effects on AR were partially reversed by proteasome inhibitors, suggesting that proteasome-mediated degradation was partially responsible for CHIP-mediated AR degradation. CHIP could also cause AR-v7 protein degradation via inducing AR-v7 ubiquitination [30].
SKP2 is another important E3 ligase that can effectively induce AR degradation via ubiquitination. Skp2 was shown to co-localize and co-immunoprecipitate with AR protein in prostate cancer cells, suggesting a direct interaction between Skp2 and AR [31]. Overexpression of Skp2 enhanced AR ubiquitination and reduced the level and activity of AR. In contrast, Skp2 knockdown suppressed AR ubiquitination and enhanced AR protein level and activity. K847 appears to be the recognition site for Skp2-mediated ubiquitination because AR mutant K847R resisted Skp2-mediated ubiquitination of AR. These findings provided strong evidence for an essential role of Skp2 in regulating AR degradation.
SIAH2 [32] directly binds to the ligand-binding domain (LBD) of AR and is an AR E3 ligase that targets a select pool of NCOR1-bound, transcriptionally-inactive AR for ubiquitin-dependent degradation. This leads to elevated expression of a subset of AR target genes that are involved in lipid metabolism, cell motility, and proliferation. Siah2 is elevated in CRPC human specimens and appears to be essential for prostate cancer cell growth under androgen-deprivation conditions in vitro and in vivo. Siah2 knockdown not only inhibited the growth of CRPC C4-2 xenograft tumors, but also sensitized the tumors to castration [32]. Taken together, Siah2 is likely to play an important role in CRPC progression by selective induction of AR transcriptional activity via the removal of the transcriptionally-inactive AR from chromatin.
AR ubiquitination by RNF6, appears to mainly impact AR transcriptional activity. RNF6, a RING domain-containing E3 ligase, was identified as an AR-associated protein by proteomics. RNF6 induces AR ubiquitination at K845/K847 in the LBD and promotes AR transcriptional activity [33]. Inactivation of RNF6 by knockdown or mutation at RNF6 ubiquitination acceptor sites on AR selectively altered the expression of a subset of AR target genes and reduced recruitment of AR and its coactivators to AREs in these target genes. RNF6 is overexpressed in CRPC specimens and is required for prostate cancer proliferation under androgen-depleted conditions. These findings suggest that the major effect of RNF6-induced AR ubiquitination is to modulate AR transcriptional activity and specificity via influencing cofactor recruitment.
Additional potential AR E3 ligases also include NEDD4 [34] and ARNIP [35]. AR ubiquitination could be regulated by multiple E3 ligases in coordination and some of the AR E3 ligases may be yet to be identified. Different E3 ligases may influence AR differently and how they are regulated or dysregulated may vary in prostate cancer cells. Further investigation of the roles of various AR E3 ligases in prostate cancer cells and their coordinated regulation of AR level and activity may lead to new approaches to suppress AR activity in prostate cancer cells.
The nuclear degradation signal in AR
A novel nuclear degradation signal (NDSAR) was discovered when our lab was investigating the mechanisms regulating AR nuclear localization. The AR contains an N-terminal domain (NTD), DNA binding domain and hinge (DBDH) region, and LBD (Figure 2). The nuclear localization of AR is partially driven by a bipartite nuclear localization signal (NLS) in the DBDH of the AR [36]. Upon ligand binding, the NLS is unshielded and AR is actively transported into the nucleus [37]. Previously, the classical model of AR trafficking involved bidirectional trafficking of AR into and out of the nucleus. The export of AR was thought to be regulated through ligand withdrawal, which exposed a nuclear export signal (NESAR) in the LBD of AR [38]. Recent evidence has shown that this signal does not induce the export of nuclear AR back into the cytoplasm. Instead, the NESAR actually stimulates proteasome-mediated nuclear specific degradation of AR [7,39]. As such, the name of the NESAR has been proposed to be changed to the nuclear degradation signal NDSAR (Figure 2). Since the NDSAR is localized in the LBD, it is likely that androgen binding to the LBD enhances the stability of AR via inhibiting NDSAR activity [40]. The evidence of the NDSAR as a regulator of nuclear degradation establishes that nuclear degradation is a key step regulating AR level, particularly nuclear AR. Since AR is required to be in the nucleus to act as a transcription factor, understanding the mechanisms of NDSAR mediated nuclear degradation could be of critical importance. It is still unclear how the NDSAR relates to the dysfunctional degradation of AR in CRPC. More research is needed to understand the mechanisms for how the NDSAR regulates AR nuclear degradation.
Figure 2.
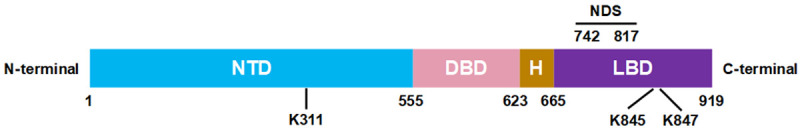
Structure of the androgen receptor (AR). The AR contains a N-terminal domain (NTD), DNA binding domain (DBD), hinge region (H), and ligand binding domain (LBD). The nuclear degradation signal (NDS) regulates AR nuclear proteasomal degradation in a hormone-dependent manner. K311, K845, and K847 are known ubiquitination sites on AR.
The presence of constitutively active splice variants is a common mechanism in which CRPC evades conventional treatments [41]. These AR variants (ARvs) lack the LBD and thus do not have the NDSAR. Consistent with this observation, AR-v7 has been reported to have a longer half-life than full-length AR [42]. Despite not having the NDSAR, ARvs are degraded by the proteasome pathway [19]. This suggests that other regions of AR can also regulate AR degradation. Interestingly, some molecules can selectively cause degradation of AR-v7, but not full-length AR [43-45]. This indicates that there may be some different regulators of degradation of ARvs and full-length AR. Interestingly, full-length AR and AR-v7 protein levels are negatively correlated, raising questions whether the stability of AR and AR-v7 are related [46]. Despite this, targeting the degradation of full-length AR alone may be sufficient to target ARvs since some research has suggested that ARvs require full-length AR [47,48].
While the underlying mechanisms governing the increased nuclear stability of AR are not fully understood, the E3 ligase MDM2 is believed to play a role as MDM2 has been shown to catalyze AR polyubiquitination [49]. MDM2 in hormone-sensitive prostate cancer cells appears to be higher than in CRPC cells and is only able to interact with AR in the nucleus, despite both proteins being present in both the cytoplasm and nucleus [7]. Taken together, these findings suggest that MDM2 may be a major player for regulating both the nuclear specific degradation of AR and the increased stability of AR in CRPC. It is unclear if MDM2 induction of nuclear AR ubiquitination is mediated through NDSAR, which will need to be clarified in future studies.
Regulation of AR by NDSAR-associated proteins, DHX15 and Prp8
To investigate the mechanisms of NDSAR action, we used yeast genetics to identify factors that can influence NDSAR function. NDSAR was shown to promote cytoplasmic localization in yeast Saccharomyces cerevisiae, suggesting that the yeast has the necessary cellular factors for NDSAR function [50]. This led to the development of a yeast genetic screen and identification of 7 yeast mutants that do not support NDSAR function [51,52]. Two of these mutants had a single point mutation in PRP8 and PRP43 genes, with a change of Gly to Ser at a.a. 1772 in PRP8 and Glu to Asp in a.a. 304 of PRP43.
PRP8 is a splicing factor that is highly conserved between humans and yeast, sharing 61% sequence identity [53]. Using co-immunoprecipitation coupled with deletion mutagenesis, PRP8 was shown to interact with AR via the NDSAR [51]. PRP8 knockdown induced nuclear accumulation of GFP-tagged AR in PC3 cells, suggesting PRP8 could suppress GFP-AR protein stability in the nucleus. However, it remains to be tested if PRP8 knockdown could influence endogenous AR stability in prostate cancer cells. Prp8 knockdown enhanced AR transcriptional activity as measured by western blot analysis of prostate-specific antigen (PSA) and PSA promoter-driven luciferase assay. PRP8 knockdown increased polyubiquitination of endogenous AR in LNCaP and C4-2 prostate cancer cells, which may represent a possible mechanism for PRP8 to modulate AR activity. PRP8 mRNA expression is down-regulated in high Gleason human prostate cancer specimens and in castration-resistant patient derived LuCaP35 prostate tumor xenografts. These findings suggest that PRP8 down-regulation could enhance AR signaling in prostate cancer progression. However, the mechanisms of PRP8 down-regulation and its regulation of AR ubiquitination, stability, and activity are still unclear.
DHX15 is the mammalian ortholog of PRP43 and is an ATP-dependent DEAH-box helicase that is also involved in RNA splicing. Knockdown of DHX15 suppressed some, but not all, AR-target genes under both androgen-dependent and castration-resistant cell models. Specifically, a ChIP analysis showed DHX15 knockdown reduced AR binding to the AREs of PSA and TMPRSS2 in C4-2 cells [52]. Further analysis using a PSA promoter-driven luciferase assay showed DHX15 knockdown suppressed, whereas DHX15 overexpression enhanced, AR transcriptional activity. Transfection of ATPase-deficient DHX15 mutants, DHX15-K166A and DHX15-D260A, also resulted in activation of luciferase reporter comparable to the effect of wild-type DHX15, suggesting that DHX15 stimulation of AR is independent of its ATPase activity. As an NDSAR-associated factor capable of stimulating AR activity, DHX15 may modulate AR ubiquitination, since NDSAR is a potent ubiquitination signal and AR transcriptional activity can be modulated by ubiquitination. DHX15 knockdown reduced AR ubiquitination in both LNCaP and C4-2 cells. In contrast, DHX15 overexpression enhanced ubiquitination of transfected AR. K166A mutation did not block DHX15 induction of AR ubiquitination, indicating that DHX15 stimulation of AR ubiquitination is independent of its ATPase activity. DHX15 regulation of AR activity appears to be mediated through Siah2, an AR E3 ligase. DHX15 knockdown caused a dramatic reduction in the protein level of Siah2 in LNCaP and C4-2 cells. In contrast, co-transfection of DHX15 stabilized Siah2 and increased its E3 ligase activity. Knockdown analysis suggested that DHX15 regulation of AR activity and ubiquitination requires Siah2. In co-immunoprecipitation assays, both transfected AR and endogenous AR were shown to interact with DHX15. The GFP-tagged AR deletion constructs containing NDSAR, but not those without NDSAR, co-precipitated with DHX15, suggesting that the NDSAR region mediated the interaction between AR and DHX15. In clinical specimens, DHX15 immunostaining exhibited a positive correlation with Gleason scores and PSA recurrence, which suggested an important role for DHX15 in prostate cancer progression.
Identification and characterization of PRP8 and DHX15 suggest that these two NDSAR-associated factors are involved in NDSAR-mediated AR ubiquitination and activation in prostate cancer cells. The identification of additional NDSAR-associated factors in future studies will provide new insights into how NDSAR modulates AR ubiquitination and function.
Targeting AR degradation - AR degraders
Since AR plays a key role in prostate cancer, AR antagonists and the next generation AR signaling targeting agents have been developed for the treatment of metastatic prostate cancer, including CRPC. However, the treated prostate cancer will develop resistance to AR antagonists and the next generation AR signaling targeting agents, mainly due to reactivation of the AR signaling. Developing novel agents that can induce AR degradation provides a potentially effective approach to suppress the reactivated AR signaling. Multiple small molecules capable of causing AR degradation have been identified and characterized.
A major class of AR degraders are heterobifunctional small molecules based on the proteolysis-targeting chimera (PROTAC) technology, which was initially developed in the laboratories of Deshaies and Crews in 2001 [54]. A PROTAC molecule consists of both a ligand for the target protein and a ligand for an E3 ligase such as MDM2 and Von Hippel-Lindau (VHL), connected by a linker. Thus, a PROTAC molecule can recruit the E3 ligase to cause ubiquitination and subsequent proteosome-dependent degradation of the target protein. Recent rapid progress has led to the development of a large number of PROTACs for many different proteins, including AR [55]. The first PROTAC for AR used the AR antagonist bicalutamide to directly bind to AR and a MDM2 inhibitor as the E3 ligase ligand and was capable of causing cellular AR protein degradation at micromolar concentrations. The subsequent AR PROTACs are much more potent, capable of degrading AR in prostate cancer cells in the nanomolar ranges. Some of the AR PROTACs such as ARV-110 exhibited an excellent pharmacokinetic profile and oral bioavailability, which led to the effective reduction of AR protein and suppression of AR-regulated gene expression in tumor tissues with oral administration. AR PROTACs effectively inhibited prostate tumor growth without observable toxicity in mice. Several AR targeting PROTACs including ARV-110 are now in clinical trials. These AR antagonist-based PROTACs target the LBD of AR and will not affect AR splice variants that lack LBD.
In addition to LBD-targeting AR PROTACs, AR PROTACs can target other domains of the AR. An NTD-targeting AR PROTAC, MTX-23, was reported that can degrade AR-v7 and AR-full length (AR-FL) at 0.37 and 2 μM of the degradation concentration 50%, respectively [56]. MTX-23 can bind to both the AR’s DNA binding domain (DBD) and the VHL E3 ubiquitin ligase simultaneously. MTX-23 inhibited cellular proliferation and enhanced apoptosis only in AR-positive prostate cancer cells, including those resistant to four FDA-approved next generation AR targeting agents-abiraterone, enzalutamide, apalutamide, and darolutamide. Furthermore, MTX-23 inhibited enzalutamide-resistant xenograft tumor growth. A novel peptide-based PROTAC targeting DBD of AR, Au-AR DBD PROTAC, was also recently developed [57]. Au-AR DBD PROTAC is highly potent against both AR-FL and AR-v7 and effectively inhibits both enzalutamide-sensitive and -resistant prostate tumors in animal models. Thus, MTX-23 and Au-AR DBD PROTAC could be effective for the treatment of CRPC that are resistant to the next generation AR targeting agents by degrading both AR-v7 and AR-FL.
Selective AR degraders (SARDs) are small molecules capable of inducing specific degradation of AR as compared to the other nuclear receptors, which provides another approach to degrade AR [58]. These small molecules can also induce AR degradation via UPS. SARDs are expected to exhibit better pharmacokinetic properties as compared to AR PROTACs because SARDs have concise structure composition. Galeterone and galeterone derivatives can act as SARDs because they can form a complex of AR, MDM2, and AKT that impacts the balance between AR ubiquitination and deubiquitination [59,60]. UT series compounds, UT-69 and UT-155, were initially developed from a library of rational drug designs based on the structure of the LBD of AR [61]. Both UT-69 and UT-155 exhibited affinity to the LBD and could suppress AR expression at the protein level, but not the mRNA level. Further studies showed that UT-155 also inhibited AR-v7 protein level, possibly through its direct binding to AF-1 of the NTD domain of AR. UT-155 was able to inhibit AR-v7-positive prostate cancer cells both in vitro and in tumor xenografts. Darolutamide derivatives represent another type of SARDs, which could down-regulate both AR-FL and AR-v7 [62]. Recently, novel nuclear AR degraders were identified through a high throughput high content screen. These nuclear AR degraders are capable of inhibiting CRPC, including those resistant to enzalutamide, both in vitro and in tumor xenografts [7,63-66]. These findings suggest that small molecules can also cause AR degradation efficiently, providing potential new approaches for the treatment of CRPC.
There are many other agents that can degrade AR [58]. Some examples are ASC-J9, 17-AAG and Niclosamide. ASC-J9 binding to AR could disrupt HSP90 association with AR. Since HSP90 is required for stabilization and function of AR [67], ASC-J9 leads to ubiquitination and degradation of the unprotected AR [68]. ASC-J9 also causes dissociation of AR from its coregulators such as ARA55 or ARA70, inducing association between AR and MDM2 to degrade AR via the UPS [69]. 17-AAG is a Geldanamycin-derived HSP90 inhibitor. 17-AAG therefore inhibits HSP90 interaction with AR and results in AR degradation. Niclosamide, an FDA-approved antihelminthic drug, degrades AR-v7 through UPS [45]. The mechanisms of these novel AR degraders are in general not well defined. Defining the mechanisms of action by these novel AR degraders in future studies should facilitate the development of more effective analogs for these agents.
Perspectives
Targeting AR for degradation appears to be an attractive approach for the treatment of patients with CRPC since AR plays an essential role in CRPC. AR degraders are being actively developed, which may lead to highly effective AR degraders eventually. The UPS plays a major role in regulating AR degradation and the mechanisms regulating UPS-dependent AR degradation appears to be complex, involving multiple E3 ligases and factors that can regulate these E3 ligases. Understanding the detailed mechanisms regulating AR degradation and the potential dysregulation of these mechanisms in CRPC will be helpful for the development of various types of AR degraders. Defining how AR is stabilized and/or degraded in CRPC may lead to the identification of new targets to enhance AR degradation.
Acknowledgements
This work was supported in part by NIH grant R01 CA265897 (ZW) and by the Department of Urology, University of Pittsburgh School of Medicine.
Disclosure of conflict of interest
Z Wang is listed as a co-inventor on a US patent held by the University of Pittsburgh (US 10004730, “Small molecules targeting androgen receptor nuclear localization and/or level in prostate cancer”).
References
- 1.Perner S, Cronauer MV, Schrader AJ, Klocker H, Culig Z, Baniahmad A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget. 2015;6:35542–55. doi: 10.18632/oncotarget.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharifi N, Farrar WL. Androgen receptor as a therapeutic target for androgen independent prostate cancer. Am J Ther. 2006;13:166–70. doi: 10.1097/00045391-200603000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J Cell Biochem. 2006;99:333–44. doi: 10.1002/jcb.20794. [DOI] [PubMed] [Google Scholar]
- 4.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA Jr. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 5.Davis ID, Martin AJ, Stockler MR, Begbie S, Chi KN, Chowdhury S, Coskinas X, Frydenberg M, Hague WE, Horvath LG, Joshua AM, Lawrence NJ, Marx G, McCaffrey J, McDermott R, McJannett M, North SA, Parnis F, Parulekar W, Pook DW, Reaume MN, Sandhu SK, Tan A, Tan TH, Thomson A, Tu E, Vera-Badillo F, Williams SG, Yip S, Zhang AY, Zielinski RR, Sweeney CJ ENZAMET Trial Investigators and the Australian and New Zealand Urogenital and Prostate Cancer Trials Group. Enzalutamide with standard first-line therapy in metastatic prostate cancer. N Engl J Med. 2019;381:121–31. doi: 10.1056/NEJMoa1903835. [DOI] [PubMed] [Google Scholar]
- 6.Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294:238–44. doi: 10.1001/jama.294.2.238. [DOI] [PubMed] [Google Scholar]
- 7.Lv S, Song Q, Chen G, Cheng E, Chen W, Cole R, Wu Z, Pascal LE, Wang K, Wipf P, Nelson JB, Wei Q, Huang W, Wang Z. Regulation and targeting of androgen receptor nuclear localization in castration-resistant prostate cancer. J Clin Invest. 2021;131:e141335. doi: 10.1172/JCI141335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaworski T. Degradation and beyond: control of androgen receptor activity by the proteasome system. Cell Mol Biol Lett. 2006;11:109–31. doi: 10.2478/s11658-006-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salami J, Alabi S, Willard RR, Vitale NJ, Wang J, Dong H, Jin M, McDonnell DP, Crew AP, Neklesa TK, Crews CM. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol. 2018;1:100. doi: 10.1038/s42003-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higano CS, Crawford ED. New and emerging agents for the treatment of castration-resistant prostate cancer. Urol Oncol. 2011;29(Suppl):S1–8. doi: 10.1016/j.urolonc.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T, Kallioniemi OP. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–9. [PubMed] [Google Scholar]
- 13.Gregory CW, Johnson RT Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–8. [PubMed] [Google Scholar]
- 14.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31:137–55. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 15.McClurg UL, Cork DMW, Darby S, Ryan-Munden CA, Nakjang S, Mendes Côrtes L, Treumann A, Gaughan L, Robson CN. Identification of a novel K311 ubiquitination site critical for androgen receptor transcriptional activity. Nucleic Acids Res. 2017;45:1793–804. doi: 10.1093/nar/gkw1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen S, Niu Y, Huang H. Posttranslational regulation of androgen dependent and independent androgen receptor activities in prostate cancer. Asian J Urol. 2020;7:203–18. doi: 10.1016/j.ajur.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002;21:4037–48. doi: 10.1093/emboj/cdf406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, Hung MC. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000;60:6841–5. [PubMed] [Google Scholar]
- 19.Li Y, Xie N, Gleave ME, Rennie PS, Dong X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget. 2015;6:33743–54. doi: 10.18632/oncotarget.5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ha S, Iqbal NJ, Mita P, Ruoff R, Gerald WL, Lepor H, Taneja SS, Lee P, Melamed J, Garabedian MJ, Logan SK. Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene. 2013;32:3992–4000. doi: 10.1038/onc.2012.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linn DE, Yang X, Xie Y, Alfano A, Deshmukh D, Wang X, Shimelis H, Chen H, Li W, Xu K, Chen M, Qiu Y. Differential regulation of androgen receptor by PIM-1 kinases via phosphorylation-dependent recruitment of distinct ubiquitin E3 ligases. J Biol Chem. 2012;287:22959–68. doi: 10.1074/jbc.M111.338350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657–69. doi: 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M, Bond R, He B, Coarfa C, Mitsiades N. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014;74:5631–43. doi: 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blattner M, Lee DJ, O’Reilly C, Park K, MacDonald TY, Khani F, Turner KR, Chiu YL, Wild PJ, Dolgalev I, Heguy A, Sboner A, Ramazangolu S, Hieronymus H, Sawyers C, Tewari AK, Moch H, Yoon GS, Known YC, Andrén O, Fall K, Demichelis F, Mosquera JM, Robinson BD, Barbieri CE, Rubin MA. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia. 2014;16:14–20. doi: 10.1593/neo.131704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swami U, Graf RP, Nussenzveig RH, Fisher V, Tukachinsky H, Schrock AB, Li G, Ross JS, Sayegh N, Tripathi N, Mathew Thomas V, Oxnard GR, Antonarakis ES, Agarwal N. SPOP mutations as a predictive biomarker for androgen receptor axis-targeted therapy in de novo metastatic castration-sensitive prostate cancer. Clin Cancer Res. 2022;28:4917–25. doi: 10.1158/1078-0432.CCR-22-2228. [DOI] [PubMed] [Google Scholar]
- 27.Cardozo CP, Michaud C, Ost MC, Fliss AE, Yang E, Patterson C, Hall SJ, Caplan AJ. C-terminal Hsp-interacting protein slows androgen receptor synthesis and reduces its rate of degradation. Arch Biochem Biophys. 2003;410:134–40. doi: 10.1016/s0003-9861(02)00680-x. [DOI] [PubMed] [Google Scholar]
- 28.Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–8. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rees I, Lee S, Kim H, Tsai FT. The E3 ubiquitin ligase CHIP binds the androgen receptor in a phosphorylation-dependent manner. Biochim Biophys Acta. 2006;1764:1073–9. doi: 10.1016/j.bbapap.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Moon SJ, Jeong BC, Kim HJ, Lim JE, Kwon GY, Kim JH. DBC1 promotes castration-resistant prostate cancer by positively regulating DNA binding and stability of AR-V7. Oncogene. 2018;37:1326–39. doi: 10.1038/s41388-017-0047-5. [DOI] [PubMed] [Google Scholar]
- 31.Li B, Lu W, Yang Q, Yu X, Matusik RJ, Chen Z. Skp2 regulates androgen receptor through ubiquitin-mediated degradation independent of Akt/mTOR pathways in prostate cancer. Prostate. 2014;74:421–32. doi: 10.1002/pros.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi J, Tripathi M, Mishra R, Sahgal N, Fazli L, Ettinger S, Placzek WJ, Claps G, Chung LW, Bowtell D, Gleave M, Bhowmick N, Ronai ZA. The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell. 2013;23:332–46. doi: 10.1016/j.ccr.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu K, Shimelis H, Linn DE, Jiang R, Yang X, Sun F, Guo Z, Chen H, Li W, Chen H, Kong X, Melamed J, Fang S, Xiao Z, Veenstra TD, Qiu Y. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell. 2009;15:270–82. doi: 10.1016/j.ccr.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Xu LL, Masuda K, Raymundo E, McLeod DG, Dobi A, Srivastava S. A feedback loop between the androgen receptor and a NEDD4-binding protein, PMEPA1, in prostate cancer cells. J Biol Chem. 2008;283:28988–95. doi: 10.1074/jbc.M710528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beitel LK, Elhaji YA, Lumbroso R, Wing SS, Panet-Raymond V, Gottlieb B, Pinsky L, Trifiro MA. Cloning and characterization of an androgen receptor N-terminal-interacting protein with ubiquitin-protein ligase activity. J Mol Endocrinol. 2002;29:41–60. doi: 10.1677/jme.0.0290041. [DOI] [PubMed] [Google Scholar]
- 36.Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J Biol Chem. 1994;269:13115–23. [PubMed] [Google Scholar]
- 37.Ni L, Llewellyn R, Kesler CT, Kelley JB, Spencer A, Snow CJ, Shank L, Paschal BM. Androgen induces a switch from cytoplasmic retention to nuclear import of the androgen receptor. Mol Cell Biol. 2013;33:4766–78. doi: 10.1128/MCB.00647-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saporita AJ, Zhang Q, Navai N, Dincer Z, Hahn J, Cai X, Wang Z. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J Biol Chem. 2003;278:41998–2005. doi: 10.1074/jbc.M302460200. [DOI] [PubMed] [Google Scholar]
- 39.Gong Y, Wang D, Dar JA, Singh P, Graham L, Liu W, Ai J, Xin Z, Guo Y, Wang Z. Nuclear export signal of androgen receptor (NESAR) regulation of androgen receptor level in human prostate cell lines via ubiquitination and proteasome-dependent degradation. Endocrinology. 2012;153:5716–25. doi: 10.1210/en.2012-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Astapova O, Seger C, Hammes SR. Ligand binding prolongs androgen receptor protein half-life by reducing its degradation. J Endocr Soc. 2021;5:bvab035. doi: 10.1210/jendso/bvab035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakazawa M, Antonarakis ES, Luo J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm Cancer. 2014;5:265–73. doi: 10.1007/s12672-014-0190-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferraldeschi R, Welti J, Powers MV, Yuan W, Smyth T, Seed G, Riisnaes R, Hedayat S, Wang H, Crespo M, Nava Rodrigues D, Figueiredo I, Miranda S, Carreira S, Lyons JF, Sharp S, Plymate SR, Attard G, Wallis N, Workman P, de Bono JS. Second-generation HSP90 inhibitor onalespib blocks mRNA splicing of androgen receptor variant 7 in prostate cancer cells. Cancer Res. 2016;76:2731–42. doi: 10.1158/0008-5472.CAN-15-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Yu C, Shao Z, Xia X, Hu T, Kong W, He X, Sun W, Deng Y, Liao Y, Huang H. Selective degradation of AR-V7 to overcome castration resistance of prostate cancer. Cell Death Dis. 2021;12:857. doi: 10.1038/s41419-021-04162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao Y, Liu Y, Xia X, Shao Z, Huang C, He J, Jiang L, Tang D, Liu J, Huang H. Targeting GRP78-dependent AR-V7 protein degradation overcomes castration-resistance in prostate cancer therapy. Theranostics. 2020;10:3366–81. doi: 10.7150/thno.41849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014;20:3198–210. doi: 10.1158/1078-0432.CCR-13-3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krause WC, Shafi AA, Nakka M, Weigel NL. Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol. 2014;54:49–59. doi: 10.1016/j.biocel.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–65. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P, McCollum BL, Wilder-Romans K, Apel IJ, Cao X, Speers C, Wang S, Chinnaiyan AM. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia. 2020;22:111–9. doi: 10.1016/j.neo.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaughan L, Logan IR, Neal DE, Robson CN. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005;33:13–26. doi: 10.1093/nar/gki141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nguyen MM, Harmon RM, Wang Z. Characterization of karyopherins in androgen receptor intracellular trafficking in the yeast model. Int J Clin Exp Pathol. 2014;7:2768–79. [PMC free article] [PubMed] [Google Scholar]
- 51.Wang D, Nguyen MM, Masoodi KZ, Singh P, Jing Y, O’Malley K, Dar JA, Dhir R, Wang Z. Splicing factor Prp8 interacts with NES(AR) and regulates androgen receptor in prostate cancer cells. Mol Endocrinol. 2015;29:1731–42. doi: 10.1210/me.2015-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jing Y, Nguyen MM, Wang D, Pascal LE, Guo W, Xu Y, Ai J, Deng FM, Masoodi KZ, Yu X, Zhang J, Nelson JB, Xia S, Wang Z. DHX15 promotes prostate cancer progression by stimulating Siah2-mediated ubiquitination of androgen receptor. Oncogene. 2018;37:638–50. doi: 10.1038/onc.2017.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hodges PE, Jackson SP, Brown JD, Beggs JD. Extraordinary sequence conservation of the PRP8 splicing factor. Yeast. 1995;11:337–42. doi: 10.1002/yea.320110406. [DOI] [PubMed] [Google Scholar]
- 54.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–9. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiang W, Wang S. Therapeutic strategies to target the androgen receptor. J Med Chem. 2022;65:8772–97. doi: 10.1021/acs.jmedchem.2c00716. [DOI] [PubMed] [Google Scholar]
- 56.Lee GT, Nagaya N, Desantis J, Madura K, Sabaawy HE, Kim WJ, Vaz RJ, Cruciani G, Kim IY. Effects of MTX-23, a novel PROTAC of androgen receptor splice variant-7 and androgen receptor, on CRPC resistant to second-line antiandrogen therapy. Mol Cancer Ther. 2021;20:490–9. doi: 10.1158/1535-7163.MCT-20-0417. [DOI] [PubMed] [Google Scholar]
- 57.Ma B, Fan Y, Zhang D, Wei Y, Jian Y, Liu D, Wang Z, Gao Y, Ma J, Chen Y, Xu S, Li L. De novo design of an androgen receptor DNA binding domain-targeted peptide PROTAC for prostate cancer therapy. Adv Sci (Weinh) 2022;9:e2201859. doi: 10.1002/advs.202201859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ha S, Luo G, Xiang H. A comprehensive overview of small-molecule androgen receptor degraders: recent progress and future perspectives. J Med Chem. 2022;65:16128–16154. doi: 10.1021/acs.jmedchem.2c01487. [DOI] [PubMed] [Google Scholar]
- 59.Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015;6:27440–60. doi: 10.18632/oncotarget.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bastos DA, Antonarakis ES. Galeterone for the treatment of advanced prostate cancer: the evidence to date. Drug Des Devel Ther. 2016;10:2289–97. doi: 10.2147/DDDT.S93941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ponnusamy S, Coss CC, Thiyagarajan T, Watts K, Hwang DJ, He Y, Selth LA, McEwan IJ, Duke CB, Pagadala J, Singh G, Wake RW, Ledbetter C, Tilley WD, Moldoveanu T, Dalton JT, Miller DD, Narayanan R. Novel selective agents for the degradation of androgen receptor variants to treat castration-resistant prostate cancer. Cancer Res. 2017;77:6282–98. doi: 10.1158/0008-5472.CAN-17-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu J, Zhou P, Hu M, Yang L, Yan G, Xu R, Deng Y, Li X, Chen Y. Discovery and biological evaluation of darolutamide derivatives as inhibitors and down-regulators of wild-type AR and the mutants. Eur J Med Chem. 2019;182:111608. doi: 10.1016/j.ejmech.2019.111608. [DOI] [PubMed] [Google Scholar]
- 63.Cole RN, Chen W, Pascal LE, Nelson JB, Wipf P, Wang Z. (+)-JJ-74-138 is a novel noncompetitive androgen receptor antagonist. Mol Cancer Ther. 2022;21:483–92. doi: 10.1158/1535-7163.MCT-21-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang Z, Wang D, Johnson JK, Pascal LE, Takubo K, Avula R, Chakka AB, Zhou J, Chen W, Zhong M, Song Q, Ding H, Wu Z, Chandran UR, Maskrey TS, Nelson JB, Wipf P, Wang Z. A novel small molecule targets androgen receptor and its splice variants in castration-resistant prostate cancer. Mol Cancer Ther. 2020;19:75–88. doi: 10.1158/1535-7163.MCT-19-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Masoodi KZ, Eisermann K, Yang Z, Dar JA, Pascal LE, Nguyen M, O’Malley K, Parrinello E, Feturi FG, Kenefake AN, Nelson JB, Johnston PA, Wipf P, Wang Z. Inhibition of androgen receptor function and level in castration-resistant prostate cancer cells by 2-[(isoxazol-4-ylmethyl)thio]-1-(4-phenylpiperazin-1-yl)ethanone. Endocrinology. 2017;158:3152–61. doi: 10.1210/en.2017-00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Z, Wang K, Yang Z, Pascal LE, Nelson JB, Takubo K, Wipf P, Wang Z. A novel androgen receptor antagonist JJ-450 inhibits enzalutamide-resistant mutant AR(F876L) nuclear import and function. Prostate. 2020;80:319–28. doi: 10.1002/pros.23945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fang Y, Fliss AE, Robins DM, Caplan AJ. Hsp90 regulates androgen receptor hormone binding affinity in vivo. J Biol Chem. 1996;271:28697–702. doi: 10.1074/jbc.271.45.28697. [DOI] [PubMed] [Google Scholar]
- 68.Yang Z, Chang YJ, Yu IC, Yeh S, Wu CC, Miyamoto H, Merry DE, Sobue G, Chen LM, Chang SS, Chang C. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med. 2007;13:348–53. doi: 10.1038/nm1547. [DOI] [PubMed] [Google Scholar]
- 69.Lai KP, Huang CK, Chang YJ, Chung CY, Yamashita S, Li L, Lee SO, Yeh S, Chang C. New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am J Pathol. 2013;182:460–73. doi: 10.1016/j.ajpath.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]