

Abstract
A strategy for the photosensitized cycloaddition of alkenylboronates and allylic alcohols by a temporary coordination is presented. The process allows for the synthesis of a diverse range of cyclobutylboronates. Key to development of these reactions is the temporary coordination of the allylic alcohol to the Bpin unit. This not only allows for the reaction to proceed in an intramolecular manner but also allows for high levels of stereo and regiocontrol. A key aspect of these studies is the utility of the cycloadducts in the synthesis of complex natural products artochamin J and piperarborenine B.
Graphical Abstract

Cycloaddition reactions are one of the most useful strategies to generate rings.1 Among these processes, [2 + 2]-cycloaddition of alkenes is regarded as the premier approach for the synthesis of cyclobutanes.2 The majority of these reactions are photochemical and therefore proceed via high-energy, and short-lived, excited-state intermediates. Consequently, to achieve reasonable reaction rates and minimize secondary photochemical reactions, researchers have extensively developed intramolecular [2 + 2]-cyclo-additions. Due to constraints with the ring forming event, stereochemical control can often be achieved (Scheme 1A). However, the substrates must be carefully designed to generate a ring and therefore this necessarily limits the scope of products that can be generated to polycycles. In addition, laborious synthetic sequences can be necessary to prepare the substrate. Intermolecular [2 + 2]-cycloadditions are also known, but to achieve reasonable reaction rates, both substrates typically need activating groups (arenes, dienes, ester, etc.) (Scheme 1A). Since these reactions are bimolecular, a range of cyclobutanes can be synthesized. However, the requirement that both coupling alkenes need activating groups remains a limitation for intermolecular [2 + 2]-cycloadditions. In addition, unlike concerted [4 + 2]-cycloadditions in which the regio- and stereochemistry can be controlled by FMOs (frontier molecular orbitals),1 in many cases, it is difficult to control the stereo- and regiochemical outcome of intermolecular [2 + 2]-cycloaddition reactions.
Scheme 1.

Approaches Toward [2 + 2]-Cycloadditions
In this manuscript, an alternative approach is disclosed in which by virtue of a temporary association between the reactants, a biomolecular reaction can be achieved in an intramolecular fashion (Scheme 1B). In this approach, stereo and regiochemical control can be achieved while retaining the benefits of diversity from an intermolecular [2 + 2]-cycloaddition of readily available components. To implement this approach, we designed a reaction between an allylic alcohol and an alkeneylboronate (Scheme 1C). This process would allow simple unactivated alkenes to participate in [2 + 2]-cycloadditions in the form of allylic alcohols and lead to the formation of diverse borylated cyclobutanes.3
Borylated carbocycles and heterocycles are useful intermediates in the construction of complex molecules. This is primarily due to the ease with which the C–B bond can be converted to other functional groups.4 In recent years, an emphasis has been placed on the synthesis of rigid carbocycles and heterocycles to enable drug development. Consequently, the preparation of cyclobutylboronates is regarded as an important goal. While several strategies are known for cyclobutylboronate synthesis,5–9 [2 + 2]-cycloadditions retain their own importance because (1) the reactions are convergent, (2) the alkene starting materials are generally widely available, and (3) products can be prepared that would be inaccessible by other approaches.10,11 With respect to photochemical [2 + 2]-cycloaddition,12,13 prior work has demonstrated that alkene triplet excited states can be captured with alkenylboronates. More recently, our lab has demonstrated that alkenylboronates can be photosensitized and undergo reaction with a variety of activated alkenes (e.g., styrenes, dienes).14 In the latter case, the Bpin unit acted as an activating group to allow for the cycloaddition to proceed in good yield. In this study, the Bpin unit is utilized as a coordinating group to direct the cycloaddition to occur with allylic alcohol derivatives (Scheme 1C). Cycloadditions of this type are valuable because (1) allyl alcohol derivatives are widely available yet underutilized in this field and (2) the product contains multiple functional groups for further elaboration to enable the synthesis of complex molecules, such as artochamin J and piperarborenine B.
The initial reaction optimization was conducted with allyl alcohol (1) and E-styrenylBpin (2). The choice to use E-styrenylBpin (2) stems from the fact that the sytrenyl component could be sensitized with common visible-light-activated sensitizers,14,15 thus avoiding the use of high-energy UV light at this stage of development. Treatment of allyl alcohol (1) and E-styrenylBpin (2) in the presence of fac-Ir(ppy)3 did not lead to product formation. It is likely that association of alcohol and the Bpin unit was minimal, and dissociation occurs before cycloaddition (Scheme 2A, entry 1). Therefore, bases were evaluated to shift the position of the equilibrium to the borate complex (Scheme 2A, entries 2–4). It was identified that use of KOt-Bu allowed for formation of 3a (Scheme 2A, entry 4). Continued optimization along this line demonstrated that use of a sensitizer with a higher triplet energy, in conjunction with a more nonpolar solvent, led to formation of [2 + 2] cycloadduct 3a in 89% yield (Scheme 2A, entry 8). The use of the higher-energy sensitizer is likely necessary to increase the rate of quenching, whereas the use of toluene likely drives the formation of the borate complex. Finally, styrenylBneop and styrenylBdmpd work in the reaction, albeit with reduced yields (see the Supporting Information for details).
Scheme 2.

Reaction Optimization
Studies were also conducted to probe the importance of the alcohol-Bpin coordination (Scheme 2B). It was found that reaction of bis-allyl boronate 4 was not productive when irradiated in the presence of 450 nm LEDs and an Ir-sensitizer. However, the addition of KOt-Bu allowed for the reaction to proceed to generate 3b. This is likely the result of the formation of tetravalent borate, which constricts the angle (compare I to II) to be like III and thus allows for cycloaddition to occur.
Under the optimized reaction conditions, the scope of the cycloaddition was investigated (Scheme 3). 1,2-Disubstituted alkenes allowed for product formation; however, a mixture of diastereomers was formed (products 6–8). With trisubstituted alkenes, the formation of penta-substituted cyclobutanes could be achieved (products 9–15). Reactions with cyclic trisubstituted alkenes allowed for the formation of bicyclic structures (products 11–15). In a similar vein to reactions of 1,2-disubstituted alkenes, reactions of unsymmetric trisubstituted alkenes gave rise to diastereomers (product 10). In one example, a tetrasubstituted alkene was tolerated to generate a highly substituted cyclobutane (product 16). At this stage of development, use of homoallylic alcohols of any substitution patterns did not result in product formation.
Scheme 3.
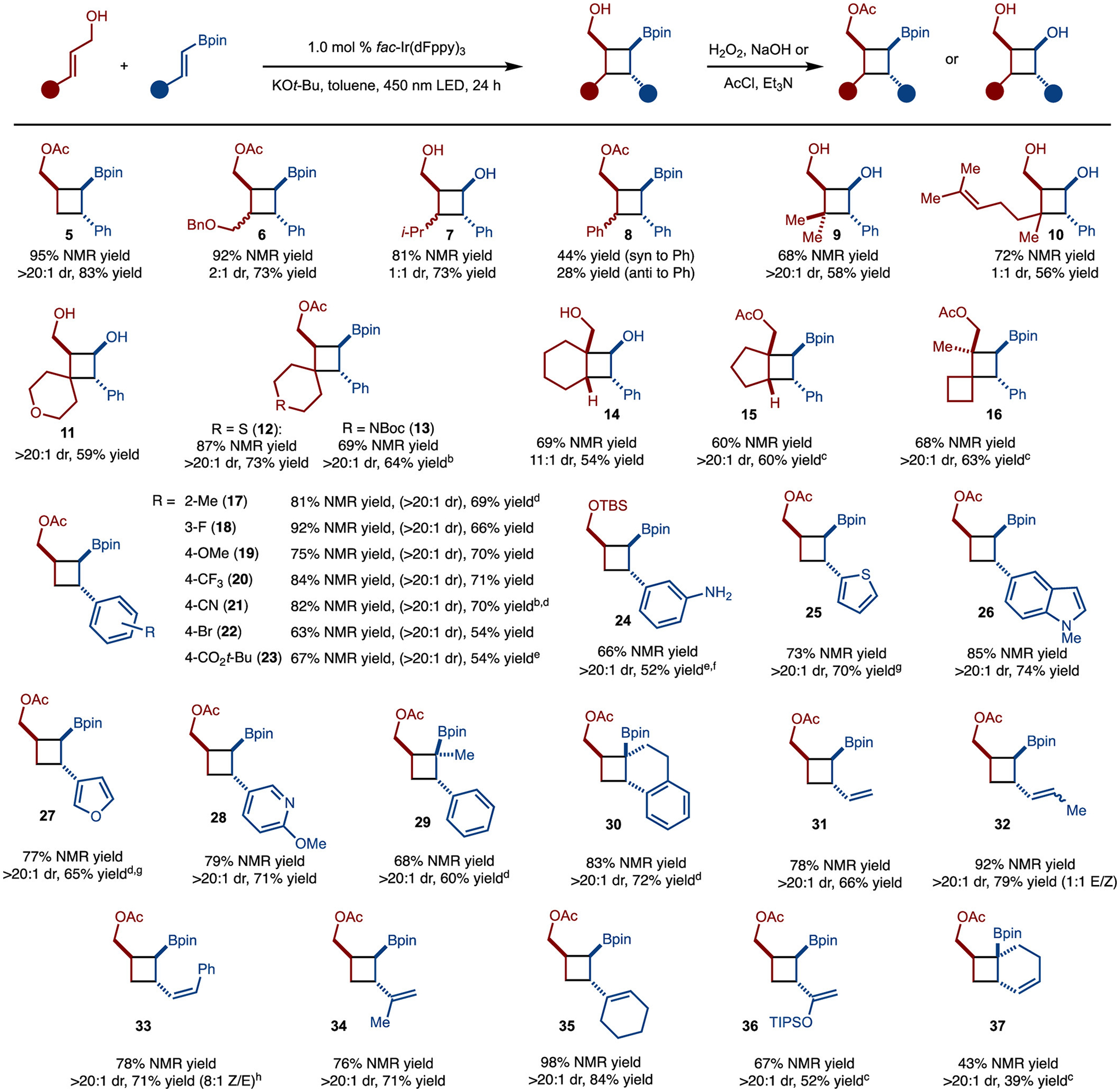
Substrate Scope
aNMR yield refers to yield determined by 1H NMR analysis of the unpurified reaction mixture of the product after functionalization of the Bpin unit. Diastereomeric ratio (dr) determined of the unpurified reaction mixture by 1H NMR analysis. Yield is of isolated, purified product. bChlorobenzene was used as solvent. cPenn PhD Photoreactor M2 was used. d48 h. eDCM was used as solvent. fThe alcohol was protected using TBSCl and imidazole. g2.0 mol % fac-Ir(dFppy)3. h1.0 mol % fac-Ir(ppy)3 was used as catalyst.
In addition to a variety of allylic alcohols, many diverse alkenyl boronates participated in the reaction. The aryl unit could be substituted with various functional groups such as Br (product 22), unprotected amine (product 24), and CN (product 21). In addition, electron-withdrawing (product 20, 21, 23), electron-donating (product 19), and sterically demanding (product 17) substitutions were well-tolerated. Several classes of heteroaromatic compounds could be used such as furan (product 27), thiophene (product 25), indole (product 26), and pyridine (product 28). The alkenylboronate could also be substituted at the α position to form sterically congested products (29-30). Finally, dienylboronates of several substitution patterns were also tolerated (products 31–37). When substrates are 1,4-substituted dienes, a secondary photoisomerization was also observed (products 32 and 33).16 To achieve better E/Z selectivity, we envisioned using a lower-energy sensitizer that could still enable the [2 + 2]-cycloaddition to synthesize 33 efficiently (given that the substrate is highly conjugated and likely has a lower triplet energy) yet allow for a more selective photoisomerization. Indeed, fac-Ir(ppy)3 catalyzed the reaction to form 33 with 8:1 Z/E selectivity in good yield. The synthesis of 36 is also notable as the silyl enol ether can be used as a functional handle. Notably, dienylboronates are known to undergo [4 + 2]-cycloadditions with allylic alcohols,3e our method demonstrated an alternative reactivity. Finally, under the reaction conditions, use of alkyl-substituted alkenyl boronates did not allow for product formation.
With respect to the allylic alcohol unit, both secondary and tertiary examples were tolerated. In the case of secondary alcohols, the reactions led to the formation of single observable diastereomers (products 39 and 40) (Scheme 4A). It is likely that the high levels of stereocontrol were observed because the reaction proceeded via IV, in which the R-group was positioned in the pseudoequatorial position. In the case of tertiary alcohols, the reaction occurred smoothly and led to the synthesis of sterically congested products 42–45 (Scheme 4B). In the cases of 44 and 45, low levels of diastereoselectivity were observed due to the relatively small size difference between substituents as compared to 39 and 40.
Scheme 4.

Additional Examples
The reaction of dieneol 46 is interesting because under different sets of conditions, alternate products could be made (Scheme 5). For example, under the conditions described in this study, the alcohol directs the cycloaddition to occur at the proximal double bond to generate 47 (Scheme 5A). The reaction likely occurred by coordination of the alcohol to the Bpin unit followed by triplet energy transfer to generate VI. Radical addition allows for formation of VII, which upon intersystem crossing (ISC) and radical recombination, leads to formation of the product. On the other hand, if the conditions are modified such that the Bpin acts as an activating group and not a coordinating group (as previously described),14 product 48 is generated (Scheme 5B). Here, triplet energy transfer occurs with 2 to form VIII. The initial bond formation takes place at the terminal position of the diene. After ISC and radical recombination, product 48 is formed.
Scheme 5.

Divergent Reactivity
It is well known that organoboronates can undergo Pd-catalyzed cross coupling with aryl bromides.17 However, alkylboronates are significantly more reluctant to undergo cross coupling compared to their aryl or alkenyl counterparts.18 To overcome this issue, the Morken group developed a cross coupling of γ-hydroxyl alkylBpin.19 In these examples, the hydroxyl group facilitates the cross-coupling reaction.
As illustrated in Scheme 6A, this method can be applied to the cycloadducts to generate diverse cyclobutanes. The reactions proceed with retention of stereochemistry and function for aryl (product 49), heteroaryl (products 50–52), and alkenyl bromides/triflates (products 53 and 54).
Scheme 6.

Synthesis of Complex Molecules
Moreover, this sequence has been instrumental in the synthesis of the cyclobutane natural products artochamin J20 and piperaborenine B (Scheme 6B, C).21 In the case of artochamin J, the synthesis commenced with cycloaddition of allylic alcohol 56 and alkenylBpin 55 (Scheme 6B). The crude material was then subjected to the cross-coupling sequence to generate 58 as a single observable diastereomer on gram scale. At this stage, cyclization of the primary alcohol with the neighboring electron-rich arene was desired. However, even after extensive investigations, the cyclization could not be achieved. Therefore, a sequence was devised that involved oxidation to the acid and Friedel–Crafts cyclization promoted by TFAA to generate 59. Clemmensen reduction22 and deprotection with TBAF allowed for the synthesis of artochamin J.
A brief synthesis of piperaborenine B was also enabled by this methodology (Scheme 6C). Starting with alkenylboronic ester 60 and allylic alcohol 61, [2 + 2]-cycloaddition and cross coupling allowed for the synthesis of 63 in 66% yield and 3.3:1 dr. Oxidative cleavage of the alkene resulted in the formation of aldehyde 64.23 Exhaustive oxidation under Fe-catalyzed aerobic oxidation24 led to formation of a dicarboxylic acid, which upon sequential treatment with oxalyl chloride and dihydropyridinone led to formation of piperaborenine B. Our method complements the reported routes, which rely on a directed C–H activation method to install the arene(s).21 Without additional steps to install and remove the directing group, only six steps were required to prepare piperaborenine B. Finally, it is important to note that the approaches described here are modular and thus amendable to the synthesis of derivatives.
In conclusion, a new strategy to access synthetically versatile cyclobutylboronates is presented. By taking advantage of a temporary tether between an alcohol and boronate, a region- and stereoselective reaction can be achieved. Due to the functional groups on the cyclobutane, facile diversification can be carried out to allow for the efficient synthesis of complex natural products.
Supplementary Material
ACKNOWLEDGMENTS
We thank Indiana University and the NIH (R35GM131755) for financial support. This project was partially funded by the Vice Provost for Research through the Research Equipment Fund and the NSF MRI program, CHE-1726633 and CHE-1920026. We thank Dr. Maren Pink and Dr. Veronica Carta of the IU Molecular Structure Center for acquisition of X-ray crystal structure data. Y.L. thanks Dr. Sarah Braley for helpful discussion. Support for the acquisition of the Bruker Venture D8 diffractometer through the Major Scientific Research Equipment Fund from the President of Indiana University and the Office of the Vice President for Research is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c08777.
Experimental procedures and analytical data for all new compounds (PDF)
Accession Codes
CCDC 2083387 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c08777
Contributor Information
Yanyao Liu, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
Dongshun Ni, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
M. Kevin Brown, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
REFERENCES
- (1).(a) Kobayashi S; Jørgensen KA Cycloaddition Reactions in Organic Synthesis; Wiley, 2002. [Google Scholar]; (b) Carruthers W Cycloaddition Reactions in Organic Synthesis; Elsevier, 2013. [Google Scholar]; (c) Nicolaou KC; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels–Alder Reaction in Total Synthesis. Angew. Chem., Int. Ed 2002, 41, 1668–1698. [DOI] [PubMed] [Google Scholar]
- (2).(a) Poplata S; Tröster A; Zou Y-Q; Bach T Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed. Engl 2015, 54, 3872–3890. [DOI] [PubMed] [Google Scholar]; (c) Xu Y; Conner ML; Brown MK Cyclobutane and Cyclobutene Synthesis: Catalytic Enantioselective [2 + 2] Cycloadditions. Angew. Chem., Int. Ed 2015, 54, 11918–11928. [DOI] [PubMed] [Google Scholar]
- (3).(a) For boron tethered Diels–Alder reactions, see: Kirkham JD; Butlin RJ; Harrity JPA A Mild Benzannulation through Directed Cycloaddition Reactions. Angew. Chem., Int. Ed 2012, 51, 6402–6405. [DOI] [PubMed] [Google Scholar]; (b) Feng C; Wang H; Xu L; Li P N–B Dative Bond-Induced [3.3.0] Bicyclic Boronate-Tethered exo-Selective Intramolecular Diels–Alder Reaction. Org. Biomol. Chem 2015, 13, 7136–7139. [DOI] [PubMed] [Google Scholar]; (c) Scholl K; Dillashaw J; Timpy E; Lam Y-H; DeRatt L; Benton TR; Powell JP; Houk KN; Morgan JB Quinine-Promoted, Enantioselective Boron-Tethered Diels–Alder Reaction by Anomeric Control of Transition-State Conformation. J. Org. Chem 2018, 83, 5756–5765. [DOI] [PubMed] [Google Scholar]; (d) Batey RA; Thadani AN; Lough AJ Alkenylboronate Tethered Intramolecular Diels-Alder Reactions. J. Am. Chem. Soc 1999, 121, 450–451. [Google Scholar]; (e) Batey RA; Thadani AN; Lough AJ Diels–Alder Reactions of Dienylboron Compounds with Unactivated Dienophiles: an Application of Boron Tethering for Substituted Cyclohexenol Synthesis. Chem. Commun 1999, 475–476. [Google Scholar]
- (4).(a) Armstrong RJ; Aggarwal V 50 Years of Zweifel Olefination: a Transition-Metal-Free Coupling. Synthesis 2017, 49, 3323–3336. [Google Scholar]; (b) Sandford C; Aggarwal VK Stereospecific Functionalizations and Transformations of Secondary and Tertiary Boronic Esters. Chem. Commun 2017, 53, 5481–5494. [DOI] [PubMed] [Google Scholar]
- (5).(a) For hydroboration/protoboration reactions, see: Clement HA; Boghi M; McDonald RM; Bernier L; Coe JW; Farrell W; Helal CJ; Reese MR; Sach NW; Lee JC; Hall DG High-Throughput Ligand Screening Enables the Enantioselective Conjugate Borylation of Cyclobutenones to Access Synthetically Versatile Tertiary Cyclobutylboronates. Angew. Chem., Int. Ed. Engl 2019, 58, 18405–18409. [DOI] [PubMed] [Google Scholar]; (b) Mercer JAM; Cohen CM; Shuken SR; Wagner AM; Smith MW; Moss FR III; Smith MD; Vahala R; Gonzalez-Martinez A; Boxer SG; Burns NZ Chemical Synthesis and Self-Assembly of a Ladderane Phospholipid. J. Am. Chem. Soc 2016, 138, 15845–15848. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guisán-Ceinos M; Parra A; Martín-Heras V; Tortosa M Enantioselective Synthesis of Cyclobutylboronates via a Copper-Catalyzed Desymmetrization Approach. Angew. Chem., Int. Ed 2016, 55, 6969. [DOI] [PubMed] [Google Scholar]; (d) Brener L; Brown HC Hydroboration. 47. Unique Stereospecificity of the Hydroboration of 1, 3-Dimethylcycloalkenes with 9-Borabicyclo [3.3.1] Nonane. J. Org. Chem 1977, 42, 2702. [Google Scholar]
- (6).(a) For carboboration reactions, see: Hancock EN; Kuker EL; Tantillo DJ; Brown MK Lessons in Strain and Stability: Enantioselective Synthesis of (+)-[5]-Ladderanoic Acid. Angew. Chem., Int. Ed 2020, 59, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Logan KM; Brown MK Catalytic Enantioselective Arylboration of Alkenylarenes. Angew. Chem., Int. Ed 2017, 56, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) For example, see: Silvi M; Aggarwal VK Radical Addition to Strained σ-Bonds Enables the Stereocontrolled Synthesis of Cyclobutyl Boronic Esters. J. Am. Chem. Soc 2019, 141, 9511–9515. [DOI] [PubMed] [Google Scholar]; (b) Fawcett A; Biberger T; Aggarwal VK Carbopalladation of C–C σ-Bonds Enabled by Strained Boronate Complexes. Nat. Chem 2019, 11, 117–122. [DOI] [PubMed] [Google Scholar]; (c) Hari DP; Abell JC; Fasano V; Aggarwal VK Ring-Expansion Induced 1,2-Metalate Rearrangements: Highly Diastereoselective Synthesis of Cyclobutyl Boronic Esters. J. Am. Chem. Soc 2020, 142 (12), 5515–5520. [DOI] [PubMed] [Google Scholar]
- (8).Giustra ZX; Yang X; Chen M; Bettinger HF; Liu S-Y Accessing 1,2-Substituted Cyclobutanes Through 1,2-Azaborine Photoisomerization. Angew. Chem., Int. Ed. Engl 2019, 58, 18918–18922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For example, see: He J; Shao Q; Wu Q; Yu J-Q Pd(II)-Catalyzed Enantioselective C(Sp3)-H Borylation. J. Am. Chem. Soc 2017, 139, 3344–3347. [DOI] [PubMed] [Google Scholar]; (b) Murakami R; Tsunoda K; Iwai T; Sawamura M Stereoselective C-H Borylations of Cyclopropanes and Cyclobutanes with Silica-Supported Monophosphane-Ir Catalysts. Chem.—Eur. J 2014, 20, 13127–13131. [DOI] [PubMed] [Google Scholar]
- (10).Parsutkar MM; Pagar VV; RajanBabu TV Catalytic Enantioselective Synthesis of Cyclobutenes From Alkynes and Alkenyl Derivatives. J. Am. Chem. Soc 2019, 141, 15367–15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Conner ML; Brown MK Synthesis of 1,3-Substituted Cyclobutanes by Allenoate-Alkene [2 + 2] Cycloaddition. J. Org. Chem 2016, 81, 8050–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) For selected examples, see: Murray PRD; Bussink WMM; Davies GHM; van der Mei FW; Antropow AH; Edwards JT; D’Agostino LA; Ellis JM; Hamann LG; Romanov-Michailidis F; Knowles RR Intermolecular Crossed [2 + 2] Cycloaddition Promoted by Visible-Light Triplet Photosensitization: Expedient Access to Polysubstituted 2-Oxaspiro[3.3]Heptanes. J. Am. Chem. Soc 2021, 143 (10), 4055. [DOI] [PubMed] [Google Scholar]; (b) Scholz SO; Kidd JB; Capaldo L; Flikweert NE; Littlefield RM; Yoon TP Construction of Complex Cyclobutane Building Blocks by Photosensitized [2 + 2] Cycloaddition of Vinyl Boronate Esters. Org. Lett 2021, 23, 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Demchuk OP; Hryshchuk OV; Vashchenko BV; Kozytskiy AV; Tymtsunik AV; Komarov IV; Grygorenko OO Photochemical [2 + 2] Cycloaddition of Alkenyl Boronic Derivatives: an Entry Into 3-Azabicyclo[3.2.0]Heptane Scaffold. J. Org. Chem 2020, 85, 5927–5940. [DOI] [PubMed] [Google Scholar]; (d) Coote SC; Bach T Enantioselective Intermolecular [2 + 2] Photocycloadditions of Isoquinolone Mediated by a Chiral Hydrogen-Bonding Template. J. Am. Chem. Soc 2013, 135, 14948–14951. [DOI] [PubMed] [Google Scholar]; (e) Kleinnijenhuis RA; Timmer BJJ; Lutteke G; Smits JMM; de Gelder R; van Maarseveen JH; Hiemstra H Formal Synthesis of Solanoeclepin a: Enantioselective Allene Diboration and Intramolecular [2 + 2] Photocycloaddition for the Construction of the Tricyclic Core. Chem.—Eur. J 2016, 22, 1266–1269. [DOI] [PubMed] [Google Scholar]; (f) Hollis WG Jr; Lappenbusch WC; Everberg KA; Woleben CM The Use of Alkenylboronate Esters in [2 + 2] Enone-Olefin Photocycloadditions. Tetrahedron Lett 1993, 34, 7517. [Google Scholar]
- (13).(a) For reviews regarding cycloadditions of alkenylboron, see: Welker ME Recent Advances in Syntheses and Reaction Chemistry of Boron and Silicon Substituted 1,3-Dienes. Tetrahedron 2008, 64 (51), 11529–11539. [Google Scholar]; (b) Eberlin L; Tripoteau F; Carreaux F; Whiting A; Carboni B Boron-Substituted 1,3-Dienes and Heterodienes as Key Elements in Multicomponent Processes. Beilstein J. Org. Chem 2014, 10, 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pyziak J; Walkowiak J; Marciniec B Recent Advances in Boron-Substituted 1,3-Dienes Chemistry: Synthesis and Application. Chem. – Eur. J 2017, 23 (15), 3502–3541. [DOI] [PubMed] [Google Scholar]; (d) Grygorenko OO; Moskvina VS; Hryshchuk OV; Tymtsunik AV Cycloadditions of Alkenylboronic Derivatives. Synthesis 2020, 52 (19), 2761–2780. [Google Scholar]
- (14).Liu Y; Ni D; Stevenson BG; Tripathy V; Braley SE; Raghavachari K; Swierk JR; Brown MK Photosensitized [2 + 2]-Cycloadditions of Alkenylboronates and Alkenes. Angew. Chem., Int. Ed 2022, 61, No. e202200725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) For reviews regarding energy transfer, see: Strieth-Kalthoff F; Glorius F Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem 2020, 6, 1888. [Google Scholar]; (b) Zhou Q-Q; Zou Y-Q; Lu L-Q; Xiao W-J Visible-Light-Induced Organic Photochemical Reactions Through Energy-Transfer Pathways. Angew. Chem., Int. Ed 2019, 58, 1586–1604. [DOI] [PubMed] [Google Scholar]; (c) Strieth-Kalthoff F; James MJ; Teders M; Pitzer L; Glorius F Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]
- (16).Singh K; Staig SJ; Weaver JD Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc 2014, 136, 5275–5278. [DOI] [PubMed] [Google Scholar]; Nevesely T; Wienhold M; Molloy JJ; Gilmour R Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts. Chem. Rev 2022, 122, 2650–2694. [DOI] [PubMed] [Google Scholar]
- (17).(a) For selected reviews on Suzuki reaction, see: Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organo-boron Compounds. Chem. Rev 1995, 95, 2457–2483. [Google Scholar]; (b) Beletskaya IP; Alonso F; Tyurin V The Suzuki-Miyaura reaction after the Nobel prize. Coord. Chem. Rev 2019, 385, 137–173. [Google Scholar]; (c) Biffis A; Centomo P; Del Zotto A; Zecca M Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev 2018, 118, 2249–2295. [DOI] [PubMed] [Google Scholar]; (d) Partyka DV Transmetalation of Unsaturated Carbon Nucleophiles from Boron-Containing Species to the Mid to Late d-Block Metals of Relevance to Catalytic C-X Coupling Reactions (X = C, F, N, O, Pb, S, Se, Te). Chem. Rev 2011, 111, 1529–1595. [DOI] [PubMed] [Google Scholar]
- (18).(a) For selected reviews on Suzuki coupling of alkyl boronates, see: Jana R; Pathak TP; Sigman MS Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev 2011, 111, 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leonori D; Aggarwal VK Stereospecific Couplings of Secondary and Tertiary Boronic Esters. Angew. Chem., Int. Ed 2015, 54, 1082–1096. [DOI] [PubMed] [Google Scholar]; (c) Cherney AH; Kadunce NT; Reisman SE Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C–C Bonds. Chem. Rev 2015, 115, 9587–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Blaisdell TP; Morken J Hydroxyl-Directed Cross-Coupling: A Scalable Synthesis of Debromohamigeran E and Other Targets of Interest. J. Am. Chem. Soc 2015, 137, 8712–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Nicolaou KC; Lister T; Denton RM; Gelin C Cascade Reactions Involving Formal [2 + 2] Thermal Cycloadditions: Total Synthesis of Artochamins F, H, I, and J. Angew. Chem., Int. Ed 2007, 46, 7501–7505. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC; Lister T; Denton RM; Gelin CF Total Synthesis of Artochamins F, H, I, and J through Cascade Reactions. Tetrahedron 2008, 64, 4736–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) For previous synthesis of piperaborenine B, see: Gutekunst WR; Baran PS Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation. J. Am. Chem. Soc 2011, 133, 19076–19079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutekunst WR; Baran PS Applications of C–H Functionalization Logic to Cyclobutane Synthesis. J. Org. Chem 2014, 79, 2430–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Panish RA; Chintala SR; Fox JM A Mixed-Ligand Chiral Rhodium (II) Catalyst Enables the Enantioselective Total Synthesis of Piperarborenine B. Angew. Chem., Int. Ed 2016, 55, 4983–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hu J-L; Feng L-W; Wang L; Xie Z; Tang Y; Li X Enantioselective Construction of Cyclobutanes: A New and Concise Approach to the Total Synthesis of (+)-Piperarborenine B. J. Am. Chem. Soc 2016, 138, 13151–13154. [DOI] [PubMed] [Google Scholar]
- (22).Xu S; Toyama T; Nakamura J; Arimoto H One-pot Reductive Cleavage of exo-Olefin to Methylene with a Mild Ozonolysis-Clemmensen Reduction Sequence. Tetrahedron Lett 2010, 51, 4534–4537. [Google Scholar]
- (23).Nicolaou KC; Adsool VA; Hale CRH An Expedient Procedure for the Oxidative Cleavage of Olefinic Bonds with PhI(OAc) 2, NMO, and Catalytic OsO4. Org. Lett 2010, 12, 1552–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jiang X; Zhang J; Ma S Iron Catalysis for Room-Temperature Aerobic Oxidation of Alcohols to Carboxylic Acids. J. Am. Chem. Soc 2016, 138, 8344–8347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.