

Abstract
Phenylalanine hydroxylase (PAH) deficient phenylketonuria (PKU) is rightfully considered the paradigm treatable metabolic disease. Dietary substrate restriction (i.e. phenylalanine (Phe) restriction) was applied >60 years ago and remains the primary PKU management means. The traditional model of PKU neuropathophysiology dictates blood Phe over-representation directs asymmetric blood:brain barrier amino acid transport through the LAT1 transporter with subsequent increased cerebral Phe concentration and low concentrations of tyrosine (Tyr), tryptophan (Trp), leucine (Leu), valine (Val), and isoleucine (Ile). Low Tyr and Trp concentrations generate secondary serotonergic and dopaminergic neurotransmitter paucities, widely attributed as drivers of PKU neurologic phenotypes. White matter disease, a central PKU characteristic, is ascribed to Phe-mediated tissue toxicity. Impaired cerebral protein synthesis, by reduced concentrations of non-Phe large neutral amino acids, is another cited pathological mechanism. The PKU amino acid transport model suggests Phe management should be more efficacious than is realized, as even early identified, continuously treated patients that retain therapy compliance into adulthood, demonstrate neurologic disease elements. Reduced cerebral metabolism was an early-recognized element of PKU pathology. Legacy data (late 1960’s to mid-1970s) determined the Phe catabolite phenylpyruvate inhibits mitochondrial pyruvate transport. Respirometry of Pahenu2 cerebral mitochondria have attenuated respiratory chain complex 1 induction in response to pyruvate substrate, indicating reduced energy metabolism. Oxidative stress is intrinsic to PKU and Pahenu2 brain tissue presents increased reactive oxygen species. Phenylpyruvate inhibits glucose-6-phosphate dehydrogenase that generates reduced niacinamide adenine dinucleotide phosphate the obligatory cofactor of glutathione reductase. Pahenu2 brain tissue metabolomics identified increased oxidized glutathione and glutathione disulfide. Over-represented glutathione disulfide argues for reduced glutathione reductase activity secondary to reduced NADPH. Herein, we review evidence of energy and oxidative stress involvement in PKU pathology. Data suggests energy deficit and oxidative stress are features of PKU pathophysiology, providing intervention-amenable therapeutic targets to ameliorate disease elements refractory to standard of care.
Keywords: Phenylketonuria, Energy, Respiratory chain complex 1, Oxidative stress, Phenylpyruvate
1. Introduction
Prominent presentations of PAH deficient PKU are neurologic. An incompletely penetrant osteopenia phenotype manifests in a patient subset, as do emerging, incompletely penetrant cardiac and ocular phenotypes [1–4]. While expression of the PAH enzyme is restricted to liver and kidney, disease presents in tissues (brain, bone) that neither express PAH nor hydroxylate Phe. Liver and kidney tissue, sites of deficient PAH activity, display neither structural nor functional pathology. PAH deficiency leads to systemic over-representation of Phe, Phe catabolites, and Phe conjugates [5,6]. PKU pathologies are secondary to altered small molecule homeostasis, effecting susceptible cells in affected tissue [6].
The predominant mechanism attributed to PKU neurologic disease is asymmetric blood:brain barrier amino acid transport, driven by Phe over-representation. Were this the singular neurologic disease mechanism, adherence to Phe restriction should have greater effect to limit neurologic presentation. Newborn screening identifies patients enabling early intervention effectively excluding neurologic devastation. However, affected patients present residual early onset neurologic disease (loss of IQ, executive function deficit) and a plethora of late onset phenotypes (seizures, neuropsychiatric, Parkinsonism).
Over sixty years ago, PKU mitochondria involvement was recognized [7]. More recently, appreciation that oxidative stress is intrinsic to PKU has renewed interest in mitochondria involvement [8–12]. Contemporary studies in our laboratory and others have compiled evidence that mitochondrial energy deficit and oxidative stress participate in neurologic phenotypes and osteopenia. Moreover, characterization of mitochondrial involvement presents interventional opportunities. This review focuses on PKU oxidative stress and mitochondrial involvement with consideration of means whereby alternative pathway energy support and oxidative stress reduction may augment Phe restriction to ameliorate residual disease.
Traditional PKU Neuropathophysiology. The defining PKU characteristic is an elevated blood Phe concentration. Plasma Phe is central to PKU diagnostics and subsequently serves as the biochemical metric to monitor substrate-reducing management. Fig. 1 schematically relates hepatic PAH deficiency, blood Phe over-representation, amino acid interaction with the blood brain barrier LAT1 transporter, and consequences of asymmetric amino acid transport. Dopamine and serotonin paucities occur in PKU models and patients, hypothesized as a primary driver of neurologic phenotypes [13–15]. However, mental retardation is the primary consequence of untreated classical PKU while neurotransmitter deficiencies present with seizures [16]. It is unlikely neurotransmitter deficit fully explains PKU neurologic findings.
Fig. 1.
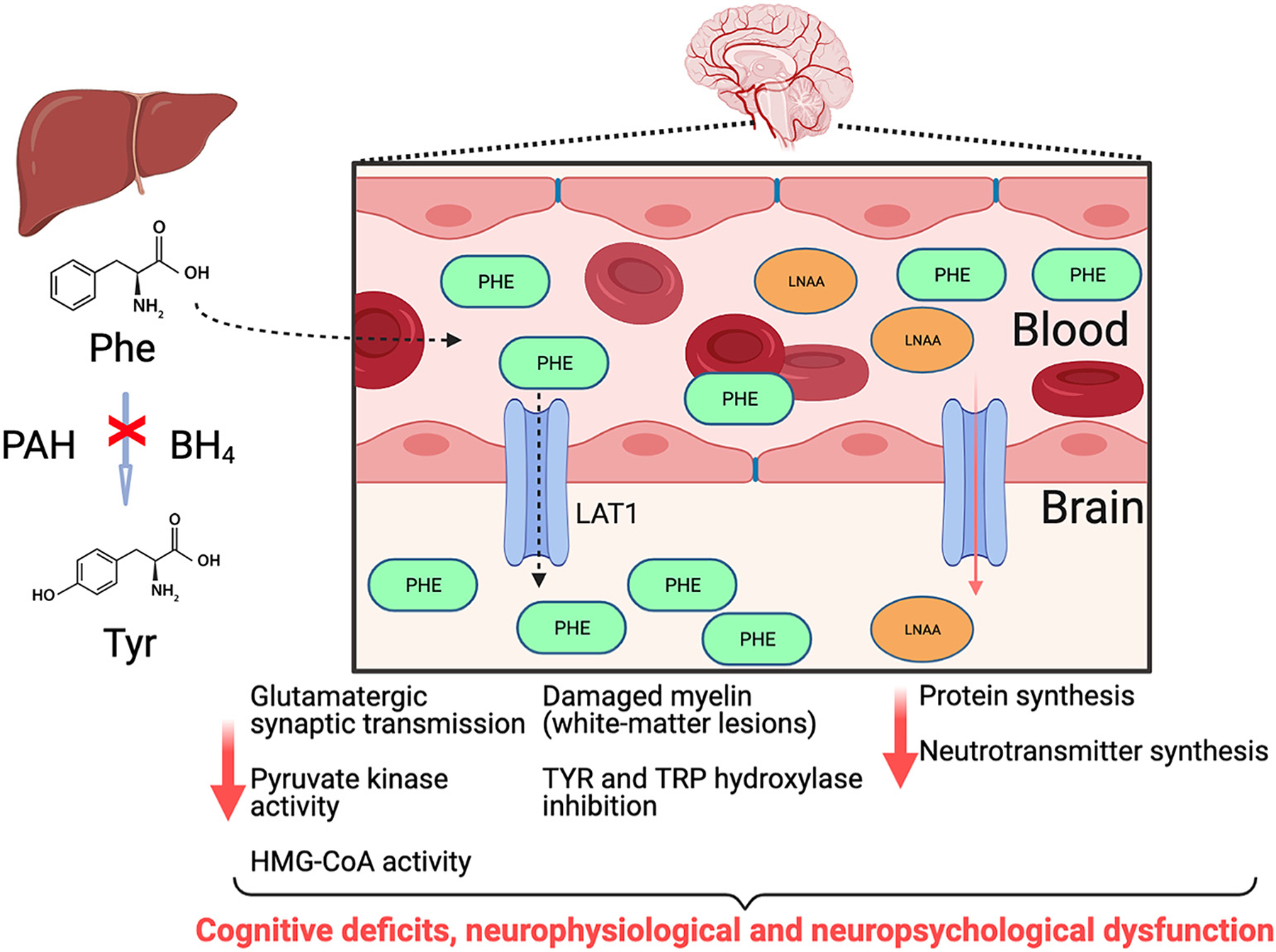
Traditional PKU pathology where hepatic PAH deficiency leads to Phe over-representation in blood. Phe over-representation leads to asymmetric amino acid transport by the large neutral amino acid transporter (LAT1, SLC7A5 gene product), with the ultimate consequence being cerebral Phe over representation and Tyr, Trp, Val, Ile, and Leu under-representation. LNAA: large neutral amino acids Tyr, Trp, Val, Ile, Leu; BH4: tetrahydrobiopterin.
Reduced cerebral protein synthesis has been hypothesized to drive PKU neurologic disease owing to non-Phe large neutral amino acid (Leu, Ile, Val, Tyr, Trp) under-representation. Some evidence supports reduced protein synthesis, namely labeled amino acid incorporation in mouse and rat PKU models [17–18]. In patients, PET-scans with labeled amino acids suggested reduced protein synthesis [19]. Reduced protein synthesis is equated to white matter abnormalities primarily demyelination [17]. Alternatively, some evidence suggests demyelination owes to cholesterol deficiency through reduced activity of 3-hydroxy-3-methylglutryl coenzyme A reductase [20]. There are late-identified classical PKU patients that retain cognition despite years to decades of hyperphenylalaninemia and assumed protein synthesis reduction, these patients argue against a protein synthesis defect being a primary causal element [21–24].
PKU and Energy Deficit. The earliest data supporting energy deficit assessed “phenylpyruvic oligophrenia”, patients where oxygen and sugar content of cerebral arterial and venous blood were determined [25]. PKU patients displayed reduced sugar and oxygen utilization in blood having traversed the brain, interpreted to indicate diminished cerebral metabolism [25]. Early biochemical studies (1969–1976), relating PKU cerebral energy, focused on the Phe catabolite phenylpyruvate and inhibition of energy pathway enzymes. Originally, phenylpyruvate was determined to inhibit hexokinase, which creates glucose-6-phosphate being a glycolysis and pentose phosphate pathway analyte [7]. Oxidative steps of the pentose phosphate pathway produce a large portion of cerebral reduced niacinamide adenine dinucleotide phosphate (NADPH), relevant to oxidative stress as NADPH is the cofactor of glutathione reductase (see below PKU and Oxidative Stress). Moreover, the pentose phosphate pathway also produces glycolysis analytes (fructose-6-phosphate, 3-phosphoglycerate) to support energy production. The group of Dr. M.S. Patel published three manuscripts demonstrating phenylpyruvate inhibition of pyruvate carboxylase [26–28]. The reaction product of pyruvate carboxylase is the Kreb cycle analyte oxaloacetate, which upon condensation with acetyl-CoA forms citrate to drive creation of reducing equivalents (NADH, FADH2) relevant to oxidative energy production. Phenylpyruvate is also described as an inhibitor of mitochondrial pyruvate transport, pyruvate kinase, and glucose-6-phosphate dehydrogenase [29,30]. Broad inhibitory activity of phenylpyruvate appears somewhat over-stated; while veracity of these data are not in question, complete relevance to PKU pathology is unclear.
The PKU rat was extensively applied in cerebral energy studies. The rat model injects newborn animals with Phe and the PAH inhibitor α-methylphenylalanine where a daily regimen maintains hyperphenylalaninemia over initial weeks of life. PKU rat studies, by Rech et al and Dimer et al, identified activity deficits in mitochondria respiratory chain complexes [31,32]. A study contradicting these found no respiratory chain deficit using hyperphenylalaninemic conditions with a human astrocyte cell line [33]. The same authors applied similar means to PKU patient blood mononuclear cells wherein respiratory complex activity was equivalent to controls [34]. These contradictory studies made assumptions limiting their utility: 1. Cell culture will replicate the in vivo brain biochemical milieu; 2. Leukocytes will share functional consequences with PKU affected tissue (e.g. brain). A recent study in succinic semialdehyde dehydrogenase deficiency determined affected tissue is the most relevant source of pathological analytes, while peripheral blood was an unreliable source of the same analytes [35]. Our PKU comparative metabolomic study (see below) made similar observations that extended to respiratory complex 1 functional deficit [6]. It is emerging that affected tissue (e.g. PKU brain) verses a non-affected tissue (e.g. PKU blood), do not elicit equivalent response including pathway dysregulation and analyte production. Most patient studies are limited to accessible samples (blood, urine, CSF, skin biopsy); while much has been discerned from accessible sample sources, their utility to reflect outcomes realized in affected tissue may be limited.
Additional PKU rat studies sought to replete cerebral energetics. The driving force behind these studies is the group of Dr. C.M. Wannmacher at the Instituto de Ciências Básicas da Saúde, Porto Alegre, Brazil. Among their initial observations was hyperphenylalaninemia-induced, down-regulation of cerebral creatine kinase activity [36]. The role of creatine in cerebral energy homeostasis is established [37]. Studies in the PKU rat assessed concurrent application of creatine and pyruvate where follow-up included behavior characteristics, dendritic spine formation, and phosphotransferase network functionality [38–40]. They also applied innovative creatine nanoliposome delivery to demonstrate improved respiratory complex activities [41]. These studies characterize cerebral energy deficit, but more importantly, define the PKU cerebral energy deficit as treatable.
Our recent study in the Pahenu2 mouse further characterize energy dysregulation and oxidative stress [6]. We applied metabolomics to Paheun2 blood plasma, liver tissue, and brain tissue. While liver is the principal site of PAH activity, abnormal elements of the liver analyte profile was limited to over-representation of Phe, Phe catabolites (phenylpyruvate, phenyllactate, phenylacetate, 2-hydroxyphenylacetate), and Phe-conjugates (N-acetylphenylalanine, N-formylphenylalanine, γ-glutamylphenyalanine, 1-carboxyethylphenylalanine). Liver showed no evidence of energy dysregulation. Abnormal blood analytes in Pahenu2 were similar to liver with Phe over-representation in addition to Phe catabolites, and Phe conjugates. No evidence of energy dysregulation was observed in Pahenu2 blood. The metabolomic profile of Pahenu2 brain tissue was unique providing evidence of energy pathway disruption with over-representation of glycolysis analytes and an increased NADH/NAD ratio. This pattern suggests a deficit in respiratory complex 1 with compensatory glycolysis up regulation. Respirometry with mitochondria prepared from Pahenu2 brain tissue and Pahenu2 liver tissue evidenced differential susceptibility to energy dysregulation. Respirometry with Pahenu2 and control liver mitochondria generated identical rates of oxygen consumption and response to substrates inducing complex 1 (pyruvate, glutamate) and complex 2 (succinate). Whereas mitochondria from Pahenu2 brain tissue showed oxygen consumption deficit in response to pyruvate substrate; however, response to glutamate substrate was similar to controls albeit beginning at a lower baseline. Pahenu2 and control brain tissue respond similarly to complex 2 induction with succinate substrate. Attenuated complex 1 response to pyruvate substrate is consistent with published data showing phenylpyruvate inhibition of mitochondria pyruvate transport [29]. Pahenu2 brain tissue analyte profile provided evidence of energy dysregulation while Pahenu2 liver tissue provided no evidence of energy dysregulation. These data demonstrate biochemical insult affects one or more cell populations in the Pahenu2 brain precipitating energy dysregulation. Cell populations in the Pahenu2 liver are refractory to biochemical insult relating to energy disruption. Prior studies support finding in Pahenu2 brain tissue [31,32]. We suggest standard of care management inadequately remediates cerebral energy deficit, which may be contributory to residual neurologic disease.
2. PKU and oxidative stress
Oxidative stress is defined by over-representation of reactive oxygen species being in excess of intrinsic anti-oxidative buffering capacity (e.g. glutathione). Several neurologic disorders identify oxidative stress participates in disease pathology [42,43]. Low plasma selenium concentration in treated PKU patients attenuated glutathione peroxidase activity inducing oxidative stress [44–46]. PKU oxidative stress was near simultaneously described in brain tissue of the Pahenu2 mouse and PKU rat [8,10]. Shortly thereafter, oxidative stress was identified in PKU patient plasma [12,13]. When PKU oxidative stress was characterized, there was legacy data [26–30] but little contemporary evidence of PKU mitochondria involvement, making this an unanticipated finding. The manuscript of Sirtorri et al 2005 was highly relevant as multiple lines of evidence (thiobarbituric-acid reactive species, antioxidant reactivity, glutathione peroxidase activity) demonstrated persistent oxidative stress among managed and therapy non-complaint patients [12]. Later, Kumru et al showed Phe management moderated oxidative stress; however, even among managed patients oxidative stress persisted [47]. Systemic impact of oxidative stress was demonstrated to include DNA damage [48,49]. Assessing oxidative damage in patient brain tissue has been elusive, while surrogates for cerebral oxidative stress are identified; clear utility of these surrogates in management is not established [50–53]. Our Pahenu2 metabolomic investigation provide insight to tissue-specific anti-oxidative response. Pahenu2 brain tissue demonstrates anti-oxidative responses through glutathione and homocarnosine pathways. Oxidized glutathione and cysteine-glutathione disulfide are over-represented in Pahenu2 brain tissue indicating anti-oxidative response [6]. Relevant to glutathione anti-oxidative response efficacy is Pahenu2 cerebral representation of reduced and oxidized niacinamide adenine dinucleotide phosphate (NADP, NADPH). We determined NADP is over-represented in Pahenu2 brain while NADPH is under-represented. NAPDH is the obligatory cofactor of glutathione reductase that cleaves cysteine-glutathione disulfide into glutathione monomers driving continued anti-oxidative response. Rosa et al demonstrated the Phe catabolite phenylpyruvate inhibits glucose-6-phosphate dehydrogenase (G6PD) a critical source of cerebral NADPH [30]. It is possible phenylpyruvate is central to cerebral energy deficit and oxidative stress by inhibiting mitochondria pyruvate transport reducing oxidative energy production and G6PD activity denying NADPH cofactor to glutathione reductase reducing enzyme processivity. Fig. 2 diagrams how the Phe catabolite phenylpyruvate may have a central role in dysregulation of oxidative energy production and glutathione anti-oxidative response. Notable was Pahenu2 brain over-representation of homocarnosine and homocarnosine pathway analytes. Homocarnosine synthesis condenses histidine (His) and γ-aminobutyrate. His was over-represented in Panenu2 brain with chronic over-representation evidenced by increased His conjugates (N-acetylhistidine, 1-carboxyethylhistidine, γ-glutamylhistidine) and the catabolite histamine. Interestingly, similar Phe conjugates (N-acetyl, 1-carboxyethyl, γ-glutamyl) are observed in Pahenu2 tissues [6]. Neuroprotective and antioxidant properties of homocarnosine are emerging making this element of PKU cerebral response a possible route to leverage therapeutically [54].
Fig. 2.
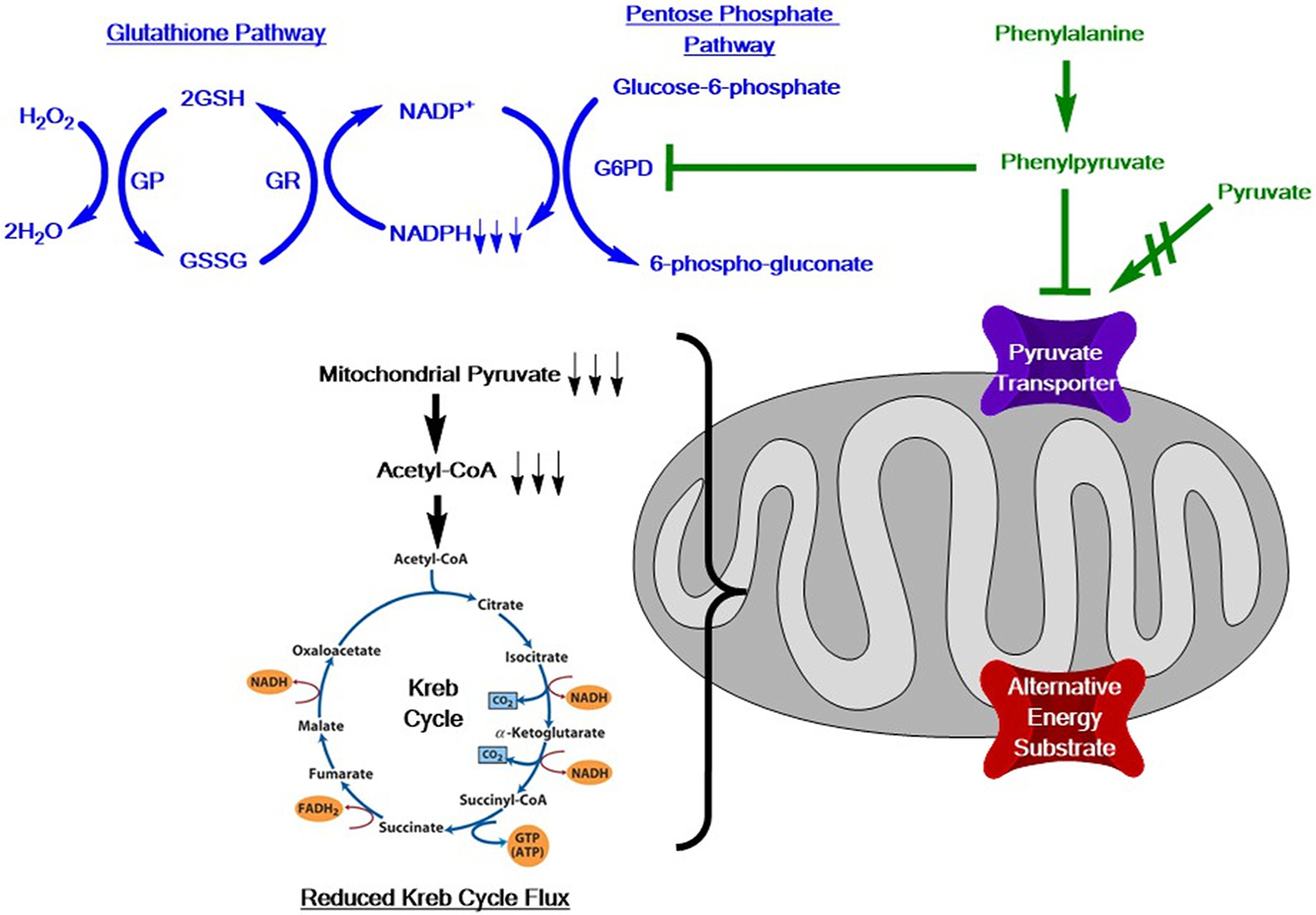
In green, Phe catabolism generates phenylpyruvate that inhibits mitochondria pyruvate transport [29] and glucose-6-phosphate dehydrogenase (G6PD) [30]. Respirometry in mitochondria from Pahenu2 mesenchymal stem cells [60] and brain tissue [6] show reduced oxygen consumption in response to pyruvate substrate to reduce acetyl-CoA, limiting Kreb cycle flux, and reduce mitochondria function [60]. We suggest alternative energy substrates (transporter in red), using means independent of pyruvate transporters, will replete energy production. In blue Pahenu2 brain tissue demonstrates reduced NADPH [6] which limits activity of glutathione reductase (GR) resulting in over-representation of glutathione disulfide (GSSG) [6]. GSH = glutathione; GP = glutathione peroxidase, NADPH = reduced niacinamide adenine dinucleotide phosphate; NADP = oxidized niacinamide adenine dinucleotide phosphate.
Remediating PKU oxidative stress is investigated. Carnitine has anti-oxidative properties. While carnitine is synthesized in vivo, a substantial portion is of dietary origin. As PKU management restricts carnitine rich food (meat, fish), assessing carnitine in patients and effect of carnitine supplements was determined. Well-managed patients show reduced plasma carnitine, while noncompliant or poorly compliant patients have higher carnitine concentrations, which are assumed to derive from diet [55]. While both well managed and therapy noncompliant patients showed oxidative stress, a negative correlation was demonstrated between thiobarbituric-acid reactive species and carnitine concentration [55]. Subsequently, it was shown that carnitine supplements reduce PKU patient oxidative stress [56]. Lipoic acid is an enzyme cofactor but also has anti-oxidative properties and reduces oxidative stress in the PKU rat [57]. Pyruvate and creatine were discussed above regarding energy augmentation; however, this strategy also reduced oxidative stress [40]. Other investigations have applied melatonin, vitamin E and vitamin C as PKU antioxidant regimens [58,59]. We treated Pahenu2 mesenchymal stem cells with resveratrol, which normalized reactive oxygen species, increased mitochondria mass, and improved osteoblast differentiation [60]. While PKU formula contains antioxidants, oxidative stress remediation has demonstrated refractivity to standard of care; normalizing the oxidative burden in PKU patients may require aggressive anti-oxidative regimens.
3. PKU osteopenia
PKU osteopenia was described in the 1960s [61,62]. Originally, PKU osteopenia was deemed secondary to diet therapy whereby calcium, phosphorous, and other bone forming material are reduced or rendered biologically unavailable; however, this is disproven as osteopenia is observed in patients that never received diet therapy and young patients after short-term therapy. The relationship between Phe and bone status remains unclear as conflicting studies indicate PHE correlates with osteopenia [63–70] while, others show no correlation [71,72]. The majority of PKU osteopenia literature is clinical and descriptive.
The first PKU osteopenia mechanistic studies assessed imbalanced bone formation and bone resorption. Osteoclastogenesis in PKU patient peripheral blood was greater in patient mixed leukocyte cultures, assayed by tartate resistant acid phosphatase staining [73,74]. In the recent decade, there has been no further published evidence substantiating these findings.
Our group hypothesized PKU osteopenia owed to a mesenchymal stem cell (MSC) developmental deficit in the osteoblast lineage. While osteopenia is not fully penetrant in patients, the Pahenu2 mouse is universally osteopenic. Our initial Pahenu2 MSC study showed an osteoblast developmental deficit, defined at cellular and molecular levels [75]. The follow-up study demonstrated Pahenu2 MSCs contain increased superoxide reactive oxygen species, deficient mitochondria functional metrics, and a deficit in pyruvate induction of respiratory complex 1 [76]. Similar to the brain, bone is an “affected tissue” wherein oxidative stress and energy deficit are identified. Unique to PKU MSC involvement is direct circulatory exposure mediates small molecule insult without selection as mediated by the blood:brain barrier in brain tissue. Glycolysis is the primary energy source of resting and proliferating MSCs [77]. Demonstrated in mouse and human MSCs, osteoblast differentiation requires upregulation of oxidative ATP production to support differentiation and extracellular matrix synthesis [78]. Based on our data showing mitochondria dysfunction in Pahenu2 MSCs, we hypothesize energy deficit is an osteopenia contributing factor. MSCs are the first cell population identified where energy deficit and oxidative stress contribute to a PKU clinical phenotype. MSCs have preferred energy substrates. Providing preferred substrates may support MSC differentiation to improve bone density. Data published by Dr. Denise Ney’s group [79] and confirmed by our group [80], showed glycomacropeptide (GMP) improves Pahenu2 bone density. Moreover, the Ney group determined Pahenu2 animals managed with GMP showed increased plasma representation of glutamate (Glu) and glutamine (Gln), being MSC preferred energy substrates [81]. We interpret these data to suggest increased Glu and Gln, secondary to GMP diet, provide energy substrates to support MSC osteoblast differentiation improving bone density. Notable is Pahenu2 Phe homeostasis with GMP is ~750 μM. Pahenu2 provided amino acid defined diet have Phe homeostasis of ~200 μM, yet bone density is indistinguishable from untreated animals [80]. Plasma Phe is the singular metric defining PKU management for >50 years; however, these data suggest energy augmentation may enable superior outcomes in the context of higher Phe homeostasis.
4. Conclusions
While PKU is the paradigm treatable genetic disease, residual neurologic phenotypes are common. Early identified, continuously treated patients present lower IQ than unaffected siblings, executive function deficit, neuropsychiatric issues, increased anxiety, and increased depression [82–85]. Attention deficit-hyperactivity disorder is common among PKU patients as ~25% of the children with early-treated PKU receive stimulant medication for attention deficit-hyperactivity disorder compared to 7% of children with diabetes and 6% of children in the general population [86]. PKU is unequivocally treatable; however, the need for means with greater efficacy is obvious.
Defined pathophysiology enables evidence-based intervention. We put forward the entirety of PKU pathophysiology is unrealized. Moreover, currently recognized consequences of asymmetric blood:brain barrier amino acid transport (Fig. 1) is unlikely to represent all relevant neuropathological mechanisms, in part explaining residual neurologic disease. While reducing Phe homeostasis will remain central to PKU intervention, characterizing pathophysiology will identify unappreciated/under-appreciated interventional routes. This review engages legacy data to that recently reported, regarding two elements of PKU pathology: 1. Oxidative stress and 2. Energy involvement. We contend intervention targeting affected tissue to support energy and reduce oxidative stress will augment Phe reduction to improve outcomes.
PKU patients and animal models present oxidative stress. Defined roles for oxidative stress in neurologic diseases (Alzheimer’s, amyotrophic lateral sclerosis) give precedent to the oxidative stress role in PKU. Comparative metabolomics identified Pahenu2 cerebral anti-oxidative responses. Brain susceptibility to oxidative damage is recognized [87]. We suggest oxidative stress participates in both early onset and late-onset PKU neurologic phenotypes; however, mechanisms of oxidative involvement may differ. Developmental susceptibility mediates severe early-onset PKU neurologic disease, as persistent high-level hyperphenylalaninemia that would devastate a newborn brain does not elicit similar outcomes in adolescent/adult patients that digress to therapy noncompliance. It is likely the immature brain has reduced oxidative tolerance and anti-oxidative responses may differ from the mature brain. Metabolic codependence between brain cellular components (neurons, glia, pericyte, endothelial) and developmental processes in the immature brain (e.g. proliferation, arborization, myelination, establish neural connectivity) may alter anti-oxidative response. In PKU, clear need exists to characterize both oxidative stress and oxidative stress response relative to brain development. Late-onset PKU neurologic phenotypes (e.g. neuropsychiatric, Parkinsonism) may share elements with adult onset neurologic diseases. Age alters brain energy metabolism, hypothesized as participatory in age-related neurologic decline and classical neurologic disease (e.g. amyotrophic lateral sclerosis). PKU alters cerebral biochemical homeostasis and energy utilization in addition to increasing oxidative stress. We suggest oxidative stress participates in early onset and late onset neurologic phenotypes. Aggressive regimens with anti-oxidants able to cross the blood:brain barrier may protect against oxidative damage to blunt residual neurologic disease elements.
Diseases involving energy deficit present diverse clinical phenotypes. We posit energy deficit is a driver of PKU neurologic phenotypes and osteopenia. Primary energy deficits, forms of Leigh syndrome providing an excellent example, respond poorly to treatment. However, long chain fatty acid oxidation defects respond to alternative energy repletion as triheptanoin elegantly demonstrates [88]. Respirometry in Pahenu2 brain tissue and in MSCs during osteoblast differentiation present a common deficit in complex 1 induction with pyruvate substrate [6,76]. Dr. A.P. Halestrap demonstrated the Phe catabolite phenylpyruvate inhibits mitochondrial pyruvate transport [29]. Fig. 2 provides a potential mechanism, supported by previously published data, whereby phenylpyruvate is a mediator of oxidative energy deficit and oxidative stress. We vision energy deficit in PKU affected tissue involves partial pyruvate transport inhibition to reduce acetyl-CoA production with secondary Kreb cycle effect. It is logical that alternative energy substrate, entering the mitochondria independent of elements that transport pyruvate, will replete energy to improve clinical phenotypes. Fig. 2 provides a generic mitochondrial “alternative energy substrate” channel to facilitate anaplerosis. Creatine alternative energy substrate studies by C.M. Wannmacher provide evidence for efficacy of energy repletion strategies [36,38–40]. Denise Ney showed GMP improved Pahenu2 bone density that we suggest occurs by providing MSCs their preferred Gln and Glu energy substrates [76,79,80]. We believe ongoing investigation will identify energy anaplerosis roles to improve PKU management.
It is clear PKU standard of care management counters early-onset mental retardation. Equally clear is residual neurologic disease presents in a large majority of early identified, continuously treated patients. Osteopenia is incompletely penetrant in PKU; however, it too occurs in early identified, continuously treated patients. Energy deficit and oxidative stress are under-appreciated PKU pathophysiological elements. We hypothesize energy and oxidative stress represent interventional targets whose remediation will improve neurologic outcomes and osteopenia. MSCs and cellular populations within the brain have preferred energy substrates (e.g. Gln for MSCs) and flexibility to utilize alternative substrates (e.g. lactate via astrocyte neuron exchange). Therapeutically providing energy substrate alternatives may support neural function or bone development. Energy utilization in PKU effected tissue is poorly characterized. Further investigation is required to clarify PKU energy dysregulation and oxidative stress to identify potential interventional modalities.
References
- [1].Serfozo C, Barta AG, Horvath E, Sumanszki C, Csakany B, Resch M, Nagy ZZ, Reismann P, Altered visual functions, macular ganglion cell and papillary retinal nerve fiber layer thickness in early-treated adult PKU patients, Mol. Genet. Metab. Rep 22 (25) (2020) 100649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hopf S, Saccadic reaction time and ocular findings in phenylketonuria, Orphanet J. Rare Dis 15 (2020) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Azabdaftari A, van der Giet M, Schuchardt M, Hennermann JB, Plöckinger U, Querfeld U, The cardiovascular phenotype of adult patients with phenylketonuria, Orphanet J. Rare Dis 14 (2019) 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dobrowolski SF, Tourkova IL, Sudano CR, Larrouture QC, Blair HC, A new view of bone loss in phenylketonuria, Organogenesis. 17 (2021) 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wild J, Shanmuganathan M, Hayashi M, Potter M, Britz-McKibbin P, Metabolomics for improved treatment monitoring of phenylketonuria: urinary biomarkers for non-invasive assessment of dietary adherence and nutritional deficiencies, Analyst. 144 (2019) 6595–6608. [DOI] [PubMed] [Google Scholar]
- [6].Dobrowolski SF, Phua YL, Sudano C, Spridik K, Zinn PO, Wang Y, Bharathi S, Vockley J, Goetzman E, Comparative metabolomics in the Pahenu2 classical PKU mouse identifies cerebral energy pathway disruption and oxidative stress, Mol. Genet. Metab (2022) in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Weber G, Inhibition of human brain pyruvate kinase and hexokinase by phenylalanine and phenylpyruvate: possible relevance to phenylketonuric brain damage, Proc. Natl. Acad. Sci. U. S. A 63 (1969) 1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kienzle Hagen ME, Pederzolli CD, Sgaravatti AM, Bridi R, Wajner M, Wannmacher CM, Wyse AT, Dutra-Filho CS, Experimental hyperphenylalaninemia provokes oxidative stress in rat brain, Biochim. Biophys. Acta 1586 (2002) 344–352. [DOI] [PubMed] [Google Scholar]
- [9].Martinez-Cruz F, Pozo D, Osuna C, Espinar A, Marchante C, Guerrero JM, Oxidative stress induced by phenylketonuria in the rat: prevention by melatonin, vitamin E, and vitamin C, J. Neurosci. Res 69 (2002) 550–558. [DOI] [PubMed] [Google Scholar]
- [10].Ercal N, Aykin-Burns N, Gurer-Orhan H, McDonald JD, Oxidative stress in a phenylketonuria animal model, Free Radic. Biol. Med 32 (2002) 906–911. [DOI] [PubMed] [Google Scholar]
- [11].Sitta A, Barschak AG, Deon M, Terroso T, Pires R, Giugliani R, Dutra-Filho CS, Wajner M, Vargas CR, Investigation of oxidative stress parameters in treated phenylketonuric patients, Metab. Brain Dis 21 (2006) 287–296. [DOI] [PubMed] [Google Scholar]
- [12].Sirtori LR, Dutra-Filho CS, Fitarelli D, Sitta A, Haeser A, Barschak AG, Wajner M, Coelho DM, Llesuy S, Belló-Klein A, Giugliani R, Deon M, Vargas CR, Oxidative stress in patients with phenylketonuria, Biochim. Biophys. Acta 2005. (1740) 68–73. [DOI] [PubMed] [Google Scholar]
- [13].Burlina AB, Bonafé L, Ferrari V, Suppiej A, Zacchello F, Burlina AP, Measurement of neurotransmitter metabolites in the cerebrospinal fluid of phenylketonuric patients under dietary treatment, J. Inherit. Metab. Dis 23 (2000) 313–316. [DOI] [PubMed] [Google Scholar]
- [14].Pascucci T, Ventura R, Puglisi-Allegra S, Cabib S, Deficits in brain serotonin synthesis in a genetic mouse model of phenylketonuria, Neuroreport. 13 (2002) 2561–2564. [DOI] [PubMed] [Google Scholar]
- [15].Pascucci T, Giacovazzo G, Andolina D, et al. , In vivo catecholaminergic metabolism in the medial prefrontal cortex of ENU2 mice: an investigation of the cortical dopamine deficit in phenylketonuria, J. Inherit. Metab. Dis 35 (2012) 1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pearl PL, Capp PK, Novotny EJ, Gibson KM, Inherited disorders of neurotransmitters in children and adults, Clin. Biochem 38 (2005) 1051–1058. [DOI] [PubMed] [Google Scholar]
- [17].Berger R, Springer J, Hommes FA, Brain protein and myelin metabolism in hyperphenylalaninemic rats cell, Mol. Biol 26 (1980) 31–36. [PubMed] [Google Scholar]
- [18].Smith CB, Kang J, Cerebral protein synthesis in a genetic mouse model of phenylketonuria proc, Natl. Acad. Sci. USA 97 (2000) 11014–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hoeksma M, Reijngoud D-J, Pruim J, de Valk HW, Paans AM, van Spronsen FJ FJ., Phenylketonuria: high plasma phenylalanine decreases cerebral protein synthesis Mol, Genet. Metab 96 (2009) 177–182. [DOI] [PubMed] [Google Scholar]
- [20].Shefer S, Tint GS, Jean-Guillaume D, Daikhin E, Kendler A, Nguyen LB, Yudkoff M, Dyer CA, Is there a relationship between 3-hydroxy-3-methylglutaryl coenzyme a reductase activity and forebrain pathology in the PKU mouse? J. Neurosci. Res 61 (2000) 549–563. [DOI] [PubMed] [Google Scholar]
- [21].Culley PD, Another population of phenylketonuria? Studies on atypical phenylketonurics, Dev. Med. Child Neurol 11 (6) (1969) 718–729. [DOI] [PubMed] [Google Scholar]
- [22].Armstrong MD, Carlisle JW, Low NL, Phenylketonuria; two unusual cases, Lancet. 271 (1956) 917–918. [PubMed] [Google Scholar]
- [23].Gostomzyk JG, Dressler F, Phenylketonuria with normal IQ, Klin. Wochenschr 45 (1967) 793–794. [DOI] [PubMed] [Google Scholar]
- [24].van Vliet D, van Wegberg AMJ, Ahring K, Bik-Multanowski M, Blau N, Bulut FD, Casas K, Didycz B, Djordjevic M, Federico A, Feillet F, Gizewska M, Gramer G, Hertecant JL, Hollak CEM, Jørgensen JV, Karall D, Landau Y, Leuzzi V, Mathisen P, Moseley K, Mungan NÖ, Nardecchia F, Õunap K, Powell KK, Ramachandran R, Rutsch F, Setoodeh A, Stojiljkovic M, Trefz FK, Usurelu N, Wilson C, van Karnebeek CD, Hanley WB, van Spronsen FJ, Can untreated PKU patients escape from intellectual disability? A systematic review, Orphanet J. Rare Dis 29 (13) (2018) 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Himwich HE, Fazekas JF, Cerebral metabolism in mongolian idiocy and phenylpyruvic oligophrenia, Arch. NeurPsych 44 (1940) 1213–1218. [Google Scholar]
- [26].Patel MS, The effect of Phenylpyruvate on pyruvate metabolism in rat brain, Biochem. J 128 (1972) 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Patel MS, Grover WD, Auerbach VH, Pyruvate metabolism by homogenates of human brain: effects of phenylpyruvate and implications for the etiology of the mental retardation in phenylketonuria, J. Neurochem 20 (1973) 289–296. [DOI] [PubMed] [Google Scholar]
- [28].Patel MS, Arinze IJ, Phenylketonuria: metabolic alterations induced by phenylalanine and phenylpyruvate, Am. J. Clin. Nutr 28 (1975) 183–188. [DOI] [PubMed] [Google Scholar]
- [29].Halestrap AP, The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors, Biochem. J 148 (1975) 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rosa AP, Jacques CE, Moraes TB, Wannmacher CM, Dutra Ade M, Dutra-Filho CS, Phenylpyruvic acid decreases glucose-6-phosphate dehydrogenase activity in rat brain, Cell. Mol. Neurobiol 32 (2012) 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rech VC, Feksa LR, Dutra-Filho CS, Wyse AT, Wajner M, Wannmacher CM, Inhibition of the mitochondrial respiratory chain by phenylalanine in rat cerebral cortex, Neurochem. Res 27 (2002) 353–357. [DOI] [PubMed] [Google Scholar]
- [32].Dimer NW, Ferreira BK, Agostini JF, Gomes ML, Kist LW, Malgarin F, Carvalho-Silva M, Gomes LM, Rebelo J, Frederico MJS, Silva FRMB, Rico EP, Bogo MR, Streck EL, Ferreira GC, Schuck PF, Brain bioenergetics in rats with acute hyperphenylalaninemia, Neurochem. Int 117 (2018) 188–203. [DOI] [PubMed] [Google Scholar]
- [33].Hargreaves IP, Heales SJ, Briddon A, Land JM, Lee PJ, Blood mononuclear cell coenzyme Q10 concentration and mitochondrial respiratory chain succinate cytochrome-c reductase activity in phenylketonuric patients, J. Inherit. Metab. Dis 25 (2002) 673–679. [DOI] [PubMed] [Google Scholar]
- [34].Kyprianou N, Murphy E, Lee P, Hargreaves I, Assessment of mitochondrial respiratory chain function in hyperphenylalaninaemia, J. Inherit. Metab. Dis 32 (2009) 289–296. [DOI] [PubMed] [Google Scholar]
- [35].Walters DC, Lawrence R, Kirby T, Ahrendsen JT, Anderson MP, Roullet JB, Murphy EJ, Gibson KM, SSADH deficiency investigators consortium (SDIC). Postmortem analyses in a patient with succinic Semialdehyde dehydrogenase deficiency (SSADHD): II. Histological, lipid, and gene expression outcomes in regional brain tissue, J. Child Neurol 9 (2021. Feb) 883073820987742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Costabeber E, Kessler A, Severo Dutra-Filho C, de Souza Wyse AT, Wajner M, Wannmacher CM, Hyperphenylalaninemia reduces creatine kinase activity in the cerebral cortex of rats, Int. J. Dev. Neurosci 21 (2003) 111–116. [DOI] [PubMed] [Google Scholar]
- [37].Lowe MT, Kim EH, Faull RL, Christie DL, Waldvogel HJ, Dissociated expression of mitochondrial and cytosolic creatine kinases in the human brain: a new perspective on the role of creatine in brain energy metabolism, J. Cereb. Blood Flow Metab 33 (2013) 1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reis EA, Rieger E, Souza S, Rasia-Filho AA, Wannmacher CM, Effects of a co-treatment with pyruvate and creatine on dendritic spines in rat hippocampus and posterodorsal medial amygdala in a phenylketonuria animal model, Metab. Brain Dis 28 (2013) 509–517. [DOI] [PubMed] [Google Scholar]
- [39].Berti SL, Nasi GM, Garcia C, Castro FL, Nunes ML, Rojas DB, Moraes TB, Dutra-Filho CS, Wannmacher CM, Pyruvate and creatine prevent oxidative stress and behavioral alterations caused by phenylalanine administration into hippocampus of rats, Metab. Brain Dis 27 (2012) 79–89. [DOI] [PubMed] [Google Scholar]
- [40].Bortoluzzi VT, Brust L, Preissler T, de Franceschi ID, Wannmacher CMD, Creatine plus pyruvate supplementation prevents oxidative stress and phosphotransfer network disturbances in the brain of rats subjected to chemically-induced phenylketonuria, Metab. Brain Dis 34 (2019) 1649–1660. [DOI] [PubMed] [Google Scholar]
- [41].Mezzomo NJ, Becker Borin D, Ianiski F, Dotto Fontana B, Diehl de Franceschi I, Bolzan J, Garcez R, Grings M, Parmeggiani B, da Silva Fernandes L, de Almeida Vaucher R, Leipnitz G, Duval Wannmacher CM, Cielo Rech V, Creatine nanoliposome reverts the HPA-induced damage in complex II-III activity of the rats’ cerebral cortex, Mol. Biol. Rep 46 (2019) 5897–5908. [DOI] [PubMed] [Google Scholar]
- [42].Cunha-Oliveira T, Montezinho L, Mendes C, Firuzi O, Saso L, Oliveira PJ, Silva FSG, Oxidative stress in amyotrophic lateral sclerosis: pathophysiology and opportunities for pharmacological intervention, Oxidative Med. Cell. Longev 2020 (2020) 5021694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bhatia V, Sharma S, Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease, J. Neurol. Sci 421 (2021) 117253. [DOI] [PubMed] [Google Scholar]
- [44].Lombeck I, Kasperek K, Feinendegen LE, Bremer HJ, Serum-selenium concentrations in patients with maple-syrup-urine disease and phenylketonuria under diet-therapy. Clinica chimica acta, Int. J. Clin. Chem 64 (1975) 57–61. [DOI] [PubMed] [Google Scholar]
- [45].Lombeck I, Kasperek K, Feinendegen LE, Bremer HJ, Trace element disturbances in dietetically treated patients with phenylketonuria and maple syrup urine disease, Monogr. Hum. Genet 9 (1978) 114–117. [DOI] [PubMed] [Google Scholar]
- [46].Reilly C, Barrett JE, Patterson CM, Tinggi U, Latham SL, Marrinan A, Trace element nutrition status and dietary intake of children with phenylketonuria, Am. J. Clin. Nutr 52 (1990) 159–165. [DOI] [PubMed] [Google Scholar]
- [47].Kumru B, Kaplan DS, Oztürk Hismi B, Celik H, Effect of blood phenylalanine levels on oxidative stress in classical Phenylketonuric patients, Cell. Mol. Neurobiol 38 (2018) 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schulpis KH, Tsakiris S, Traeger-Synodinos J, Papassotiriou I, Low total antioxidant status is implicated with high 8-hydroxy-2-deoxyguanosine serum concentrations in phenylketonuria, Clin. Biochem 38 (2005) 239–242. [DOI] [PubMed] [Google Scholar]
- [49].Sitta A, Manfredini V, Biasi L, Tremea R, Schwartz IV, Wajner M, Vargas CR, Evidence that DNA damage is associated to phenylalanine blood levels in leukocytes from phenylketonuric patients, Mutat. Res 679 (2009) 13–16. [DOI] [PubMed] [Google Scholar]
- [50].Keshavarzi F, Rastegar M, Vessal M, Dehbidi GR, Khorsand M, Ganjkarimi AH, Takhshid MA, Serum ischemia modified albumin is a possible new marker of oxidative stress in phenylketonuria, Metab. Brain Dis 33 (2018) 675–680. [DOI] [PubMed] [Google Scholar]
- [51].Rausell D, García-Blanco A, Correcher P, Vitoria I, Vento M, Cháfer-Pericás C, Newly validated biomarkers of brain damage may shed light into the role of oxidative stress in the pathophysiology of neurocognitive impairment in dietary restricted phenylketonuria patients, Pediatr. Res 85 (2019) 242–250. [DOI] [PubMed] [Google Scholar]
- [52].Tavana S, Amini S, Hakhamaneshi MS, Andalibi P, Hajir MS, Ardalan A, Abdi M,Fathollahpour A, Prooxidant-antioxidant balance in patients with phenylketonuria and its correlation to biochemical and hematological parameters, J. Pediatr. Endocrinol. Metab 29 (2016) 675–680. [DOI] [PubMed] [Google Scholar]
- [53].Dick S, Funchal C, Pelaez Pde L, Loureiro SO, Vivian L, Pessutto FD, Almeida LM, Wannmacher CM, Pessoa-Pureur R, Cytoskeleton of human mononuclear cells as a possible peripheral marker for phenylalanine neurotoxicity in PKU, Neurochem. Res 12 (2002) 1569–1576. [DOI] [PubMed] [Google Scholar]
- [54].Aldini G, de Courten B, Regazzoni L, Gilardoni E, Ferrario G, Baron G, Altomare A,D’Amato A, Vistoli G, Carini M, Understanding the antioxidant and carbonyl sequestering activity of carnosine: direct and indirect mechanisms, Free Radic. Res 11 (2020) 1–10. [DOI] [PubMed] [Google Scholar]
- [55].Sitta A, Vanzin CS, Biancini GB, Manfredini V, de Oliveira AB, Wayhs CA, Ribas GO, Giugliani L, Schwartz IV, Bohrer D, Garcia SC, Wajner M, Vargas CR, Evidence that l-carnitine and selenium supplementation reduces oxidative stress in phenylketonuric patients, Cell. Mol. Neurobiol 31 (2011) 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Deon M, Landgraf SS, Lamberty JF, Moura DJ, Saffi J, Wajner M, Vargas CR, Protective effect of l-carnitine on phenylalanine-induced DNA damage, Metab. Brain Dis 30 (2015) 925–933. [DOI] [PubMed] [Google Scholar]
- [57].Moraes TB, Zanin F, da Rosa A, de Oliveira A, Coelho J, Petrillo F, Wajner M, Dutra-Filho CS, Lipoic acid prevents oxidative stress in vitro and in vivo by an acute hyperphenylalaninemia chemically-induced in rat brain, J. Neurol. Sci 292 (2010) 89–95. [DOI] [PubMed] [Google Scholar]
- [58].Martinez-Cruz F, Pozo D, Osuna C, Espinar A, Marchante C, Guerrero JM, Oxidative stress induced by phenylketonuria in the rat: prevention by melatonin, vitamin E, and vitamin C, J. Neurosci. Res 69 (2002) 550–558. [DOI] [PubMed] [Google Scholar]
- [59].Martinez-Cruz F, Osuna C, Guerrero JM, Mitochondrial damage induced by fetal hyperphenylalaninemia in the rat brain and liver: its prevention by melatonin, vitamin E, and vitamin C, Neurosci. Lett 392 (2006) 1–4. [DOI] [PubMed] [Google Scholar]
- [60].Dobrowolski SF, Sudano C, Phua YL, Tourkova IL, Spridik K, Goetzman ES, Vockley J, Blair HC, Mesenchymal stem cell energy deficit and oxidative stress contribute to osteopenia in the Pahenu2 classical PKU mouse, Mol. Genet. Metab 132 (2021) 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Feinberg SB, Fisch RO, Roentgenologic findings in growing long bones in phenylketonuria. Preliminary study, Radiology 78 (1962) 394–398. [DOI] [PubMed] [Google Scholar]
- [62].Murdoch MM, Holman GH, Roentgenologic bone changes in phenylketonuria. Relationship to dietary phenylalanine and serum alkaline phosphatase, Am. J. Dis. Child 107 (1964) 523–532. [PubMed] [Google Scholar]
- [63].Greeves LG, Carson DJ, Magee A, Patterson CC, Fractures and phenylketonuria, Acta Paediatr. 86 (1997) 242–244. [DOI] [PubMed] [Google Scholar]
- [64].de Groot MJ, Hoeksma M, van Rijn M, Slart RH, van Spronsen FJ, Relationships between lumbar bone mineral density and biochemical parameters in phenylketonuria patients, Mol. Genet. Metab 105 (2012) 566–570. [DOI] [PubMed] [Google Scholar]
- [65].Lage S, Bueno M, Andrade F, Prieto JA, Delgado C, Legarda M, Sanjurjo P, Aldámiz-Echevarría LJ, Fatty acid profile in patients with phenylketonuria and its relationship with bone mineral density, J. Inherit. Metab. Dis 33 (Suppl. 3) (2010) S363–S371. [DOI] [PubMed] [Google Scholar]
- [66].Zeman J, Bayer M, Stepán J, Bone mineral density in patients with phenylketonuria, Acta Paediatr. 88 (1999) 1348–1351. [DOI] [PubMed] [Google Scholar]
- [67].Allen JR, Humphries IR, Waters DL, Roberts DC, Lipson AH, Howman-Giles RG, Gaskin KJ, Decreased bone mineral density in children with phenylketonuria, Am. J. Clin. Nutr 59 (1994) 419–422. [DOI] [PubMed] [Google Scholar]
- [68].Hillman L, Schlotzhauer C, Lee D, Grasela J, Witter S, Allen S, Hillman R, Decreased bone mineralization in children with phenylketonuria under treatment, Eur. J. Pediatr 155 (Suppl. 1) (1996) S148–S152. [DOI] [PubMed] [Google Scholar]
- [69].Modan-Moses D, Vered I, Schwartz G, Anikster Y, Abraham S, Segev R, Efrati O, Peak bone mass in patients with phenylketonuria, J. Inherit. Metab. Dis 30 (2007) 202–208. [DOI] [PubMed] [Google Scholar]
- [70].Adamczyk P, Morawiec-Knysak A, Płudowski P, Banaszak B, Karpe J, Pluskiewicz W, Bone metabolism and the muscle-bone relationship in children, adolescents and young adults with phenylketonuria, J. Bone Miner. Metab 29 (2011) 236–244. [DOI] [PubMed] [Google Scholar]
- [71].Barat P, Barthe N, Redonnet-Vernhet I, Parrot F, The impact of the control of serum phenylalanine levels on osteopenia in patients with phenylketonuria, Eur. J. Pediatr 161 (2002) 687–688. [DOI] [PubMed] [Google Scholar]
- [72].Nagasaka H, Tsukahara H, Takatani T, Sanayama Y, Takayanagi M, Ohura T, Sakamoto O, Ito T, Wada M, Yoshino M, Ohtake A, Yorifuji T, Hirayama S, Miida T, Fujimoto H, Mochizuki H, Hattori T, Okano Y, Cross-sectional study of bone metabolism with nutrition in adult classical phenylketonuric patients diagnosed by neonatal screening, J. Bone Miner. Metab 29 (2011) 737–743. [DOI] [PubMed] [Google Scholar]
- [73].Porta F, Roato I, Mussa A, Repici M, Gorassini E, Spada M, Ferracini R, Increased spontaneous osteoclastogenesis from peripheral blood mononuclear cells in phenylketonuria, J. Inherit. Metab. Dis 31 (Suppl. 2) (2008) S339–S342. [DOI] [PubMed] [Google Scholar]
- [74].Roato I, Porta F, Mussa A, D’Amico L, Fiore L, Garelli D, Spada M, Ferracini R, Bone impairment in phenylketonuria is characterized by circulating osteoclast precursors and activated T cell increase, PLoS One 5 (2010), e14167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dobrowolski SF, Tourkova IL, Robinson LJ, Secunda C, Spridik K, Blair HC, A bone mineralization defect in the Pahenu2 model of classical phenylketonuria involves compromised mesenchymal stem cell differentiation, Mol. Genet. Metab 125 (2018) 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Dobrowolski SF, Sudano C, Phua YL, Tourkova IL, Spridik K, Goetzman ES, Vockley J, Blair HC, Mesenchymal stem cell energy deficit and oxidative stress contribute to osteopenia in the Pahenu2 classical PKU mouse, Mol. Genet. Metab 132 (2021) 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Shum LC, White NS, Mills BN, Bentley KL, Eliseev RA, Energy metabolism in mesenchymal stem cells during osteogenic differentiation, Stem Cells Dev. 25 (2016) 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Guntur AR, Le PT, Farber CR, Rosen CJ, Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass, Endocrinology. 155 (2014) 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Solverson P, Murali SG, Litscher SJ, Blank RD, Ney DM, Low bone strength is a manifestation of phenylketonuria in mice and is attenuated by a glycomacropeptide diet, PLoS One 7 (2012), e45165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Dobrowolski SF, Tourkova IL, Blair HC, PKU osteopenia involves a mesenchymal stem cell developmental defect, Mol. Genet. Metab 126 (2019) 296. [Google Scholar]
- [81].Solverson P, Murali SG, Brinkman AS, Nelson DW, Clayton MK, Yen E, Ney DM, Glycomacropeptide, a low-phenylalanine protein isolated from cheese whey, supports growth and attenuates metabolic stress in the murine model of phenylketonuria, Am. J. Physiol. Endocrinol. Metab 302 (2012) E885–E895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Moyle JJ, Meta-analysis of neuropsychological symptoms of adolescents and adults with PKU, Neuropsychol. Rev 17 (2007) 91–101. [DOI] [PubMed] [Google Scholar]
- [83].Hoedt AE, High phenylalanine levels directly affect mood and sustained attention in adults with phenylketonuria: a randomised, double-blind, placebo-controlled, crossover trial, J. Inherit. Metab. Dis 34 (2011) 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huijbregts SC, Sustained attention and inhibition of cognitive interference in treated phenylketonuria: associations with concurrent and lifetime phenylalanine concentrations, Neuropsychologia. 40 (2002) 7–15. [DOI] [PubMed] [Google Scholar]
- [85].Welsh MC, Neuropsychology of early-treated phenylketonuria: specific executive function deficits, Child Dev. 61 (1990) 1697–1713. [PubMed] [Google Scholar]
- [86].Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, Mitchell J, Smith WE, Thompson BH, Berry SA, American College of Medical Genetics and Genomics Therapeutics Committee, Phenylalanine hydroxylase deficiency: diagnosis and management guideline, Genet. Med 16 (2014) 188–200. [DOI] [PubMed] [Google Scholar]
- [87].Cobley JN, Fiorello M, Bailey DM, 13 reasons the brain is susceptible to oxidative stress, Redox Biol. 15 (2018) 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Sklirou E, Alodaib AN, Dobrowolski SF, Mohsen AA, Vockley J, Physiological perspectives on the use of Triheptanoin as Anaplerotic therapy for long chain fatty acid oxidation disorders, Front. Genet 11 (2021) 598760. [DOI] [PMC free article] [PubMed] [Google Scholar]