

Abstract
Cyanine dyes carrying quinoline moieties are an important class of organic molecules that are of great interest for applications in many fields like medicine, pharmacology, and engineering. Despite their exceptional properties, such as stability, high molar extinction coefficients, and high pH-sensitivity, this class of dyes has been less analyzed and reviewed in the last few decades. Therefore, this review article focuses on discussing the history of quinoline compounds, various synthetic routes to prepare quinolinium salts and symmetrical and asymmetrical mono-, di-, tri-, penta- and heptamethine cyanine dyes, containing quinoline moieties, together with their optical properties and applications as photosensitizers in photodynamic therapy, probes in biomolecules for labeling of nucleic acids, as well as imaging agents.
Keywords: cyanine dyes, lepidine, NIR, quinaldine, quinoline
Introduction to Cyanine Dyes
Cyanine dyes are a class of organic functional dyes, distinguished by two nitrogen centers. The general structure of the dyes is shown in Figure 1. Cyanine dyes have conjugated systems represented by alternating single and double bonds in a chain. The chain is constituted of sp2-hybridized carbon atoms forming a conjugated bond. Conjugation between groups results in the displacement of π electrons and positive charge on the nitrogen atom.[1]
Figure 1.
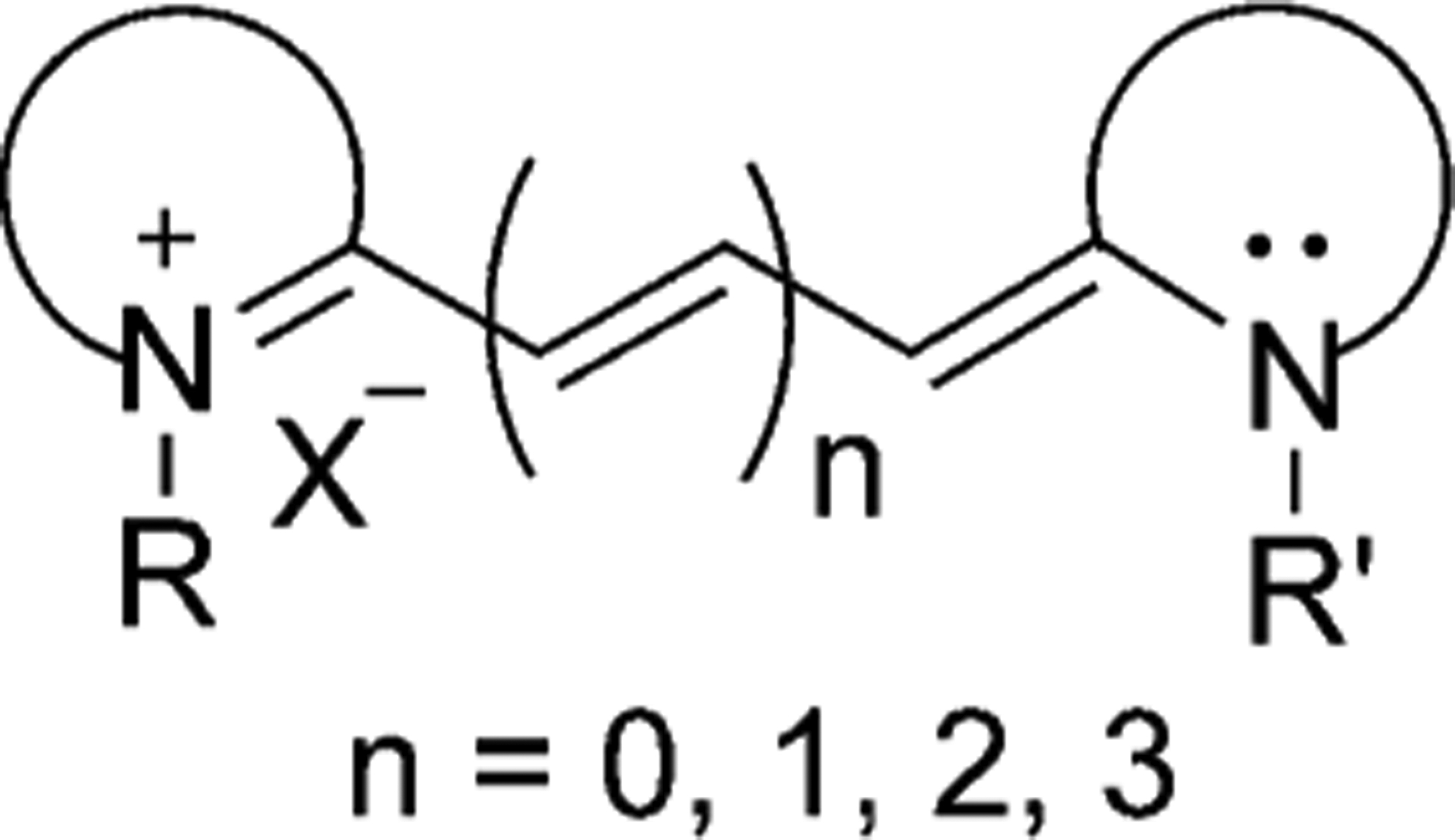
General structure of cyanine dyes.
The carbon chain makes cyanine dyes unique, giving them a more extensive absorption wavelength than the other known dyes. With the addition of each covalent bond in the chain, the absorption of the dye is increased by about 100 nm. As a result, cyanine dyes can display absorption and fluorescence in the range of the electromagnetic spectrum from the visible to infrared (Figure 2). The strong absorption of light at various wavelengths causes solutions of the cyanine molecules to become brightly colored. The stronger the absorption, the higher influence the molecule will have towards applications in various research areas.[2–4] These dyes also tend to aggregate, which gives rise to a narrower absorption of light.
Figure 2.
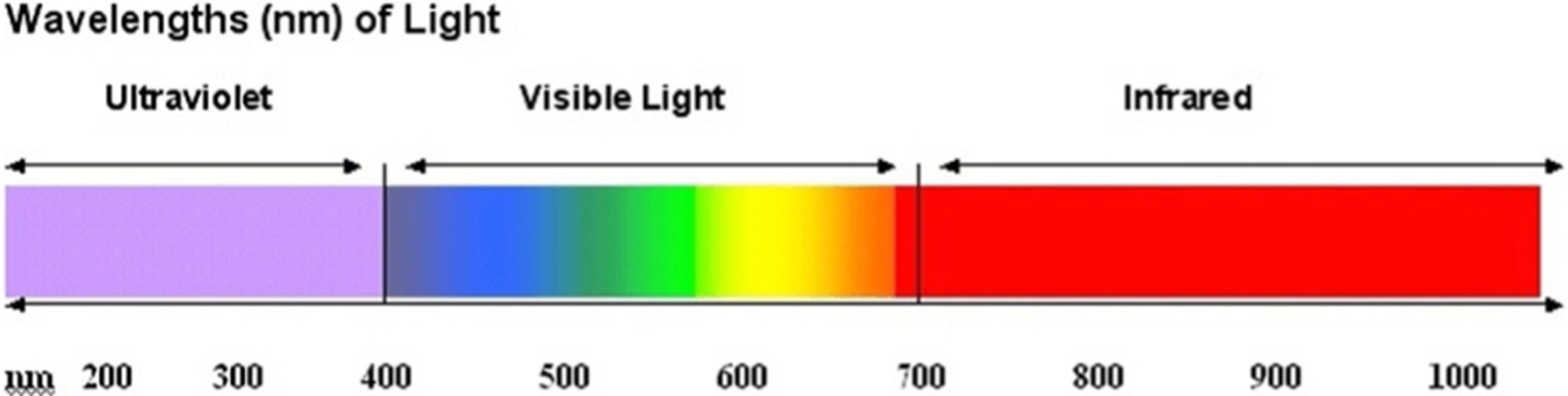
Electromagnetic spectrum.[5]
The name of the dye is specific to every compound, from the length of the chain between the two nitrogen atoms, and the number methine groups involved. If one group is present in the chain, the dye is called a monomethine cyanine. The dyes with 2, 3, 5, and 7 methine groups are called dimethine, trimethine, pentamethine, and heptamethine cyanine dyes accordingly.[6]
Cyanine dyes can be structurally classified based on the nature of the end groups. Compounds with two heterocycles are called closed-chain cyanines, those with one terminal heterocyclic group and a non-cyclic end group are hemicyanines, and those without heterocyclic groups are defined as streptocyanines. These three classes of cyanine dyes are presented in Figure 3.
Figure 3.
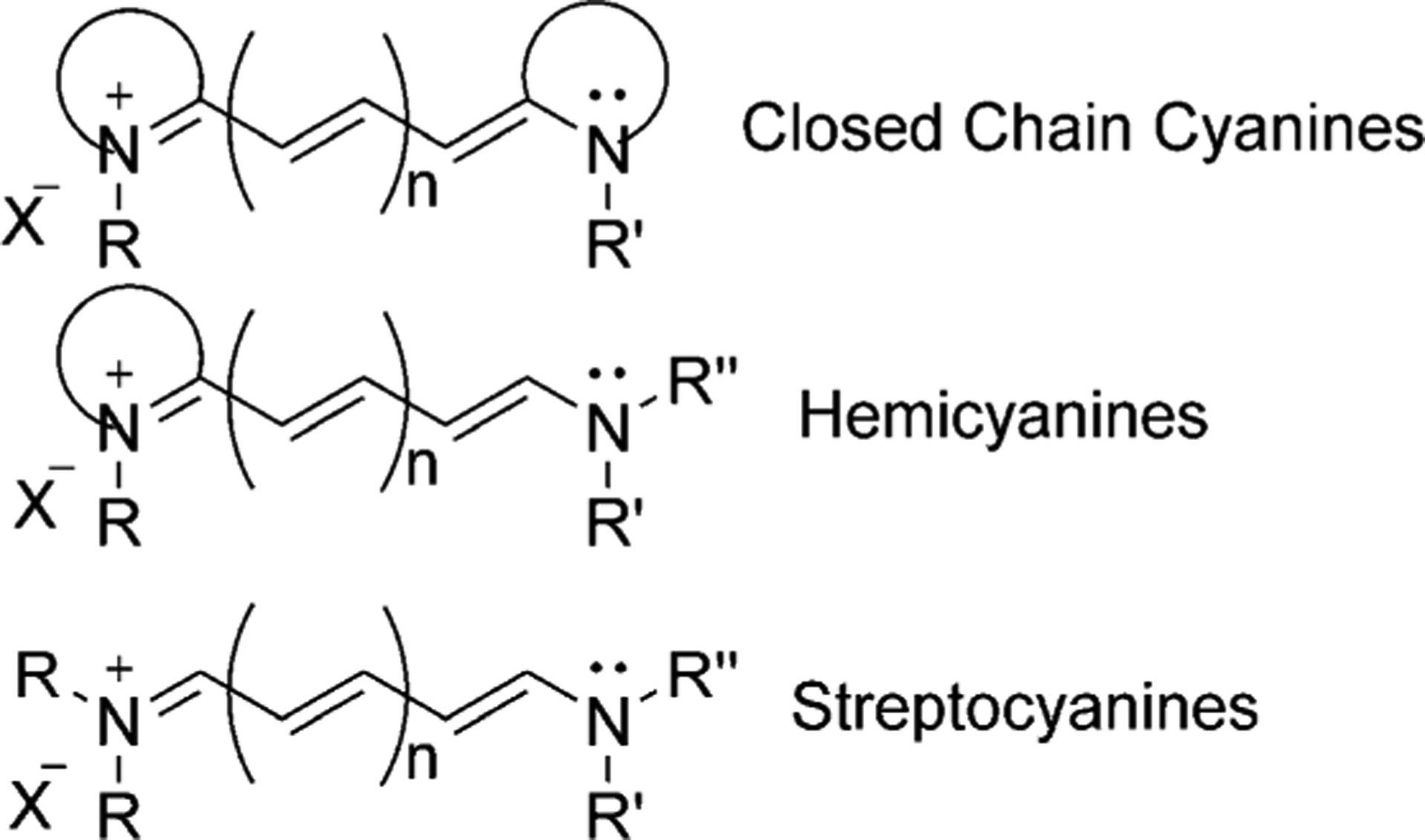
Classification of cyanine dyes.
Each nitrogen atom is an independent part of the heterocyclic moieties, taking the form of indole, benzoxazole, benzothiazole, and quinoline. A variety of commonly used heterocyclic groups is illustrated in Figure 4.
Figure 4.
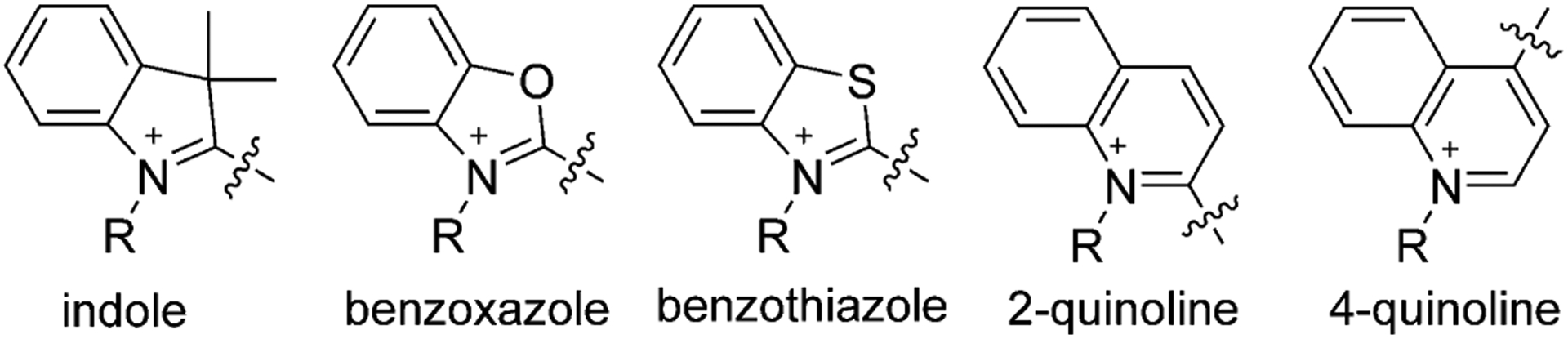
Examples of heterocyclic moieties used to synthesize cyanine dyes.
If the heterocyclic moieties on both sides of the chain have the same structure, then the dye is called symmetrical. The electron density distribution of such dyes is also symmetrical. If the heterocyclic terminal groups differ from one another, then the dye is called asymmetrical.[7]
There is one more type of cyanines in which two nitrogen groups are directly linked and have no methine groups, these are called apocyanine dyes. The only known compounds of this type of dyes are red Erythroapocyanine and Xanthoapo-cyanine as shown in Figure 5.[8]
Figure 5.
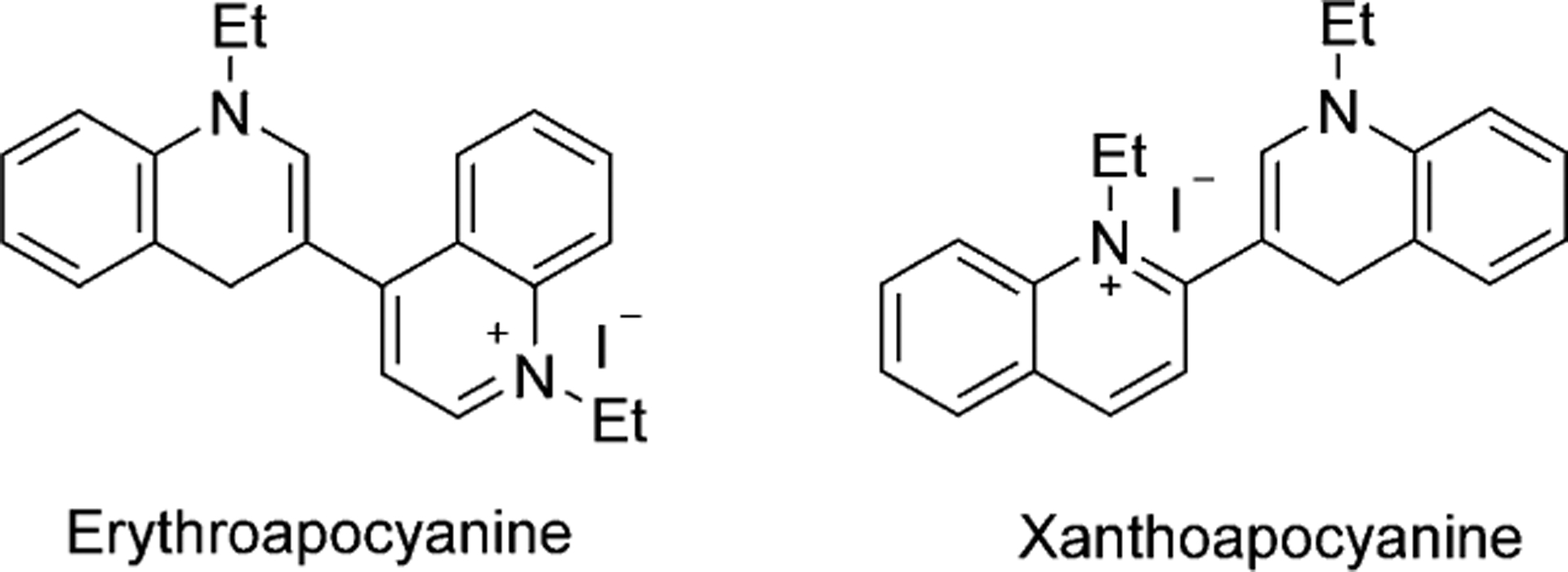
Structures of apocyanine dyes.
Charles Hanson Greville Williams synthesized the first cyanine dye in 1856. Williams obtained quinoline by distillation of cinchonine and heated the distillate with amyl iodide and an excess of ammonia, which resulted in “a magnificent blue color” compound, called Quinoline Blue, which is illustrated in Figure 6.[9–10]
Figure 6.
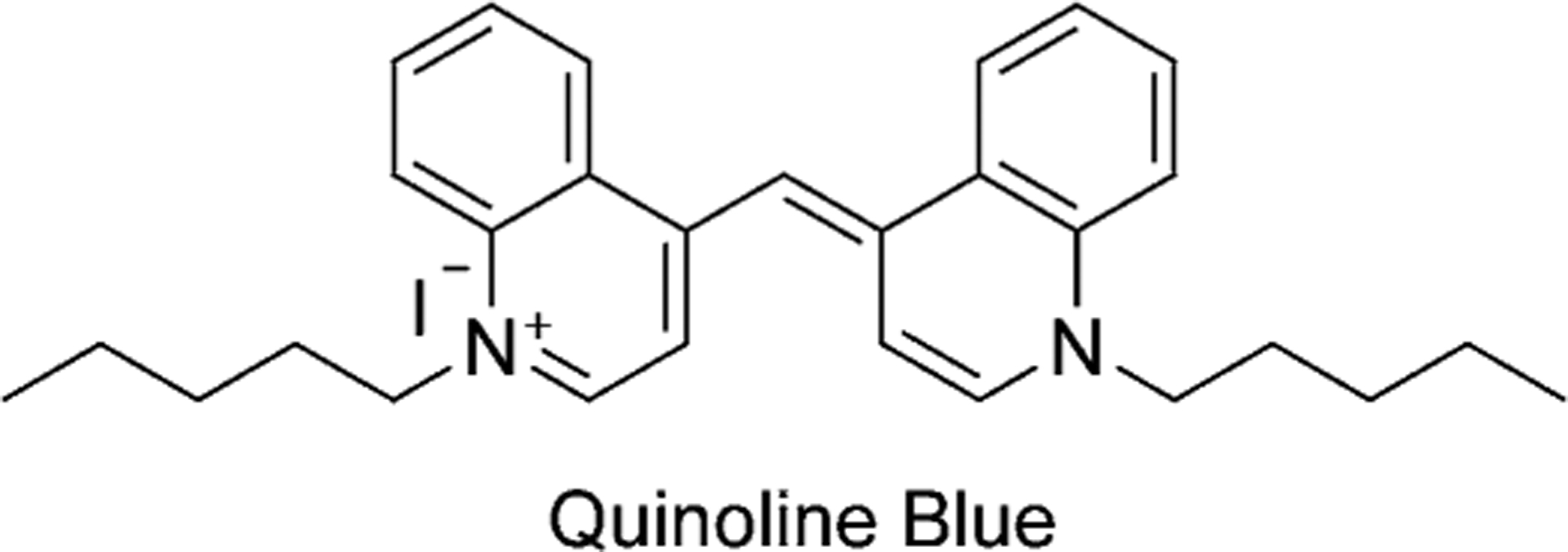
The structure of the first synthesized cyanine dye.
Williams noted that quinolinium salts tend to give intense colors on heating with silver oxide. But it was useless for dyeing fabrics because of its photo instability. Later in 1873, H. W. Vogel started to use them as photosensitizers in photography.[11] This led to the synthesis of a series of photographic sensitizers, including Ethyl Red and Sensitol Red (Pynacyanole), shown in Figure 7. Since then, the class of cyanine dyes has grown exponentially and they have been used in a wide range of applications. Another important dye, pseudoisocyanine (PIC; Figure 7), synthesized in 1936 by Jelley and Scheibe, was the first dye, possessing unique J-aggregation properties, that showed two absorption maxima.[12]
Figure 7.
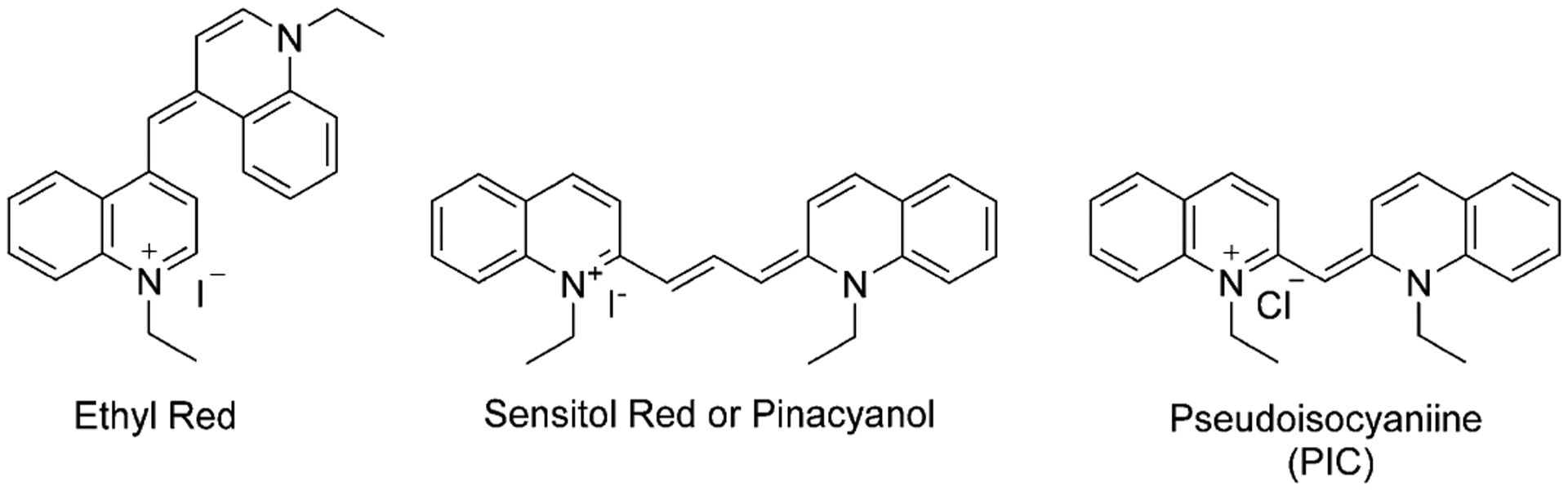
Dyes used as photosensitizers in photography.
In this review article, we focus on closed-chain cyanine dyes, where at least one heterocyclic moiety contains quinoline. The history, synthesis, optical properties, and applications of symmetrical and asymmetrical mono-, di-, tri-, penta-, and heptamethine cyanines with quinoline moieties are discussed in detail.
Introduction to Cyanine Dyes Containing Quinoline Moieties
In the 1940s, acridine compounds were reported to have antibacterial results. Some simple aminoacridines had a wide range of applications before penicillin and became easily accessible to the armed forces in World War II. Moreover, in 1981, it was found that aminoacridines’ affinity for DNA caused the poisonous effect on yeasts in culture.[13] To understand the reasons for the acridine compounds’ performance, the activity of recognizable moieties was examined. The quinoline and pyridine fractions were identified by the inspection of the acridine molecule. The cyanine fragment from the acriflavine molecule was also explored as an antibacterial agent and looked promising for in vivo studies (Figure 8).[14]
Figure 8.
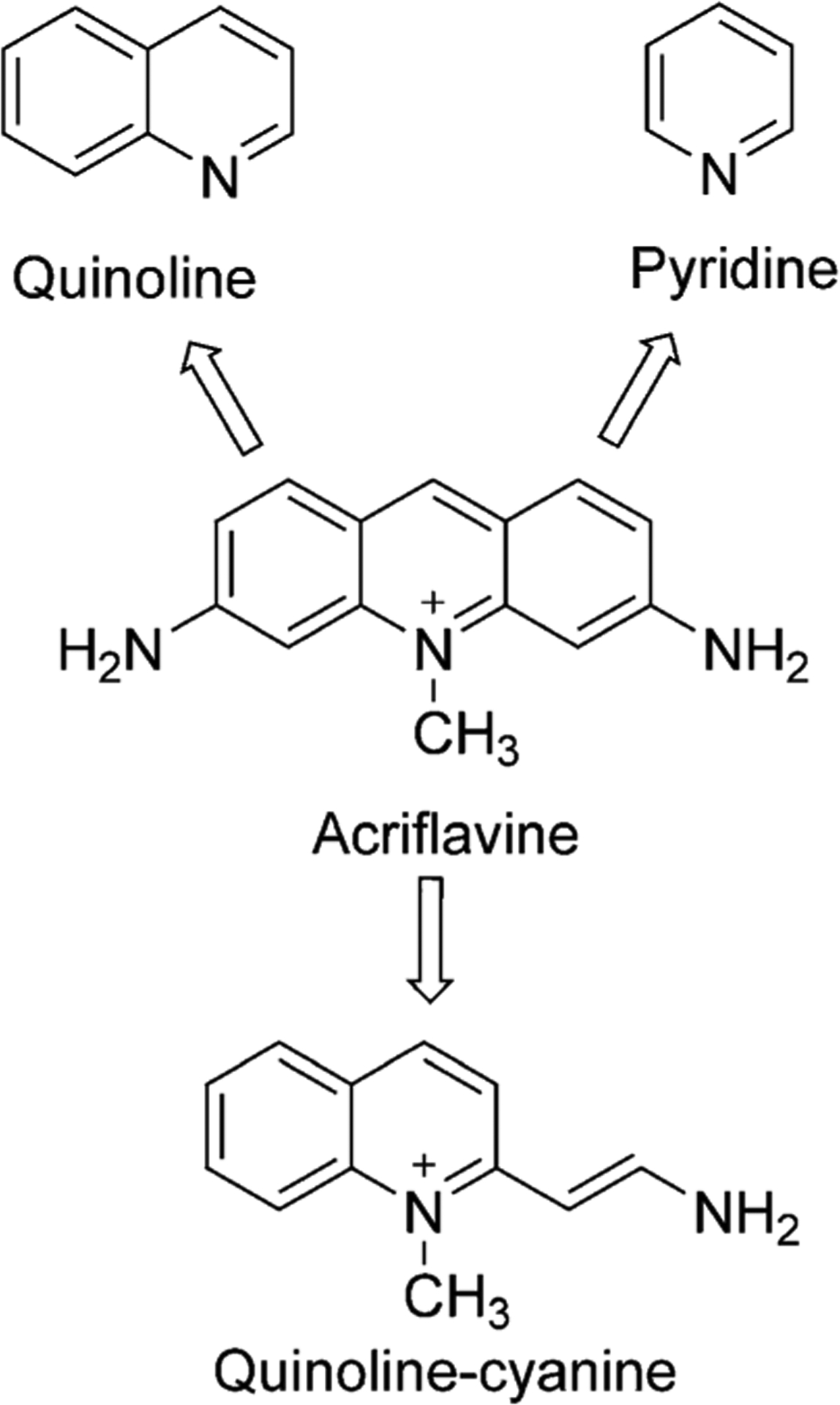
Historic cyanine development from Acriflavine.
The quinoline ring system has been widely used in medicinal and industrial chemistry.[15–18] Quinoline has become an attractive scaffold for the development of new fluorescent reagents owing to its small molecular size and nitrogen’s ability to form hydrogen bonds. A lot of drugs and natural compounds have had this moiety in their structures. For these reasons, quinoline-containing dyes are valuable and show great potential for various studies.[19]
Quinolines (also known as benzo[b]pyridines,1-azanaphthalenes, and benzazines) are heterocyclic aromatic compounds with one nitrogen atom. It consists of a benzene ring condensed with a pyridine ring at the 2,3-positions. First, quinoline was synthesized from coal tar in 1834 by a German chemist Friedlieb Ferdinand Runge. Coal tar remains the main source of commercial quinoline.[20] The prime sources of quinoline are petroleum, coal processing, wood preservation, and shale oil.
In 1881, Skraup presented the classic quinoline synthesis by heating aniline with glycerol and with concentrated sulfuric acid with the subsequent addition of nitrobenzene (Scheme 1). The use of inexpensive starting materials in the synthesis of quinolines makes this method important despite the low yields and other practical shortcomings.[21]
Scheme 1.
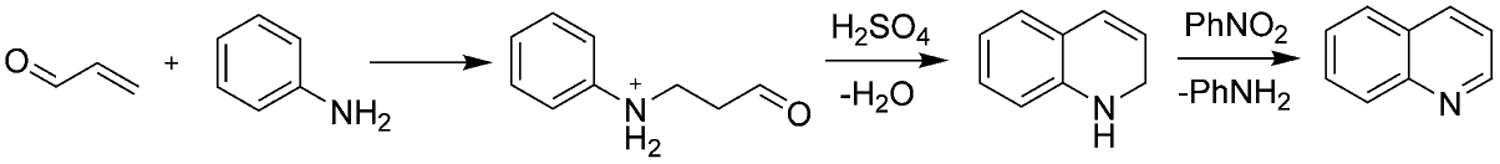
Skraup synthesis of quinolines.
Alkylquinolines can be obtained from petroleum, coal tar, shale oil, soya bean, and green coffee.[22] The quinoline derivatives can be found in various natural products like tecleabine, tecleoxine, quinine, ciprofloxacine, which are interesting for medicinal chemistry and biomedical uses, are obtained by the thermal degradation of natural organic compounds. For instance, furoquinoline alkaloids tecleoxine and tecleabine can be isolated from the aerial parts of the Teclea nobitis plant, which is used in traditional medicine in Africa as an analgesic and antipyretic and to treat gonorrhea.[23]
Toddaquinoline can be separated from the root bark of formosan Toddalia asiatica, which is used as a folk medicine in Asia.[24] The bark of the cinchona tree was used to isolate quinine in 1820 to treat malaria and babesiosis.[25] Quinoline-containing drugs have been used to treat malaria for a long time, and some of them are presented in Figure 9. The highly cost-effective chloroquine was widely used in antimalarial therapy during World War II.[26]
Figure 9.
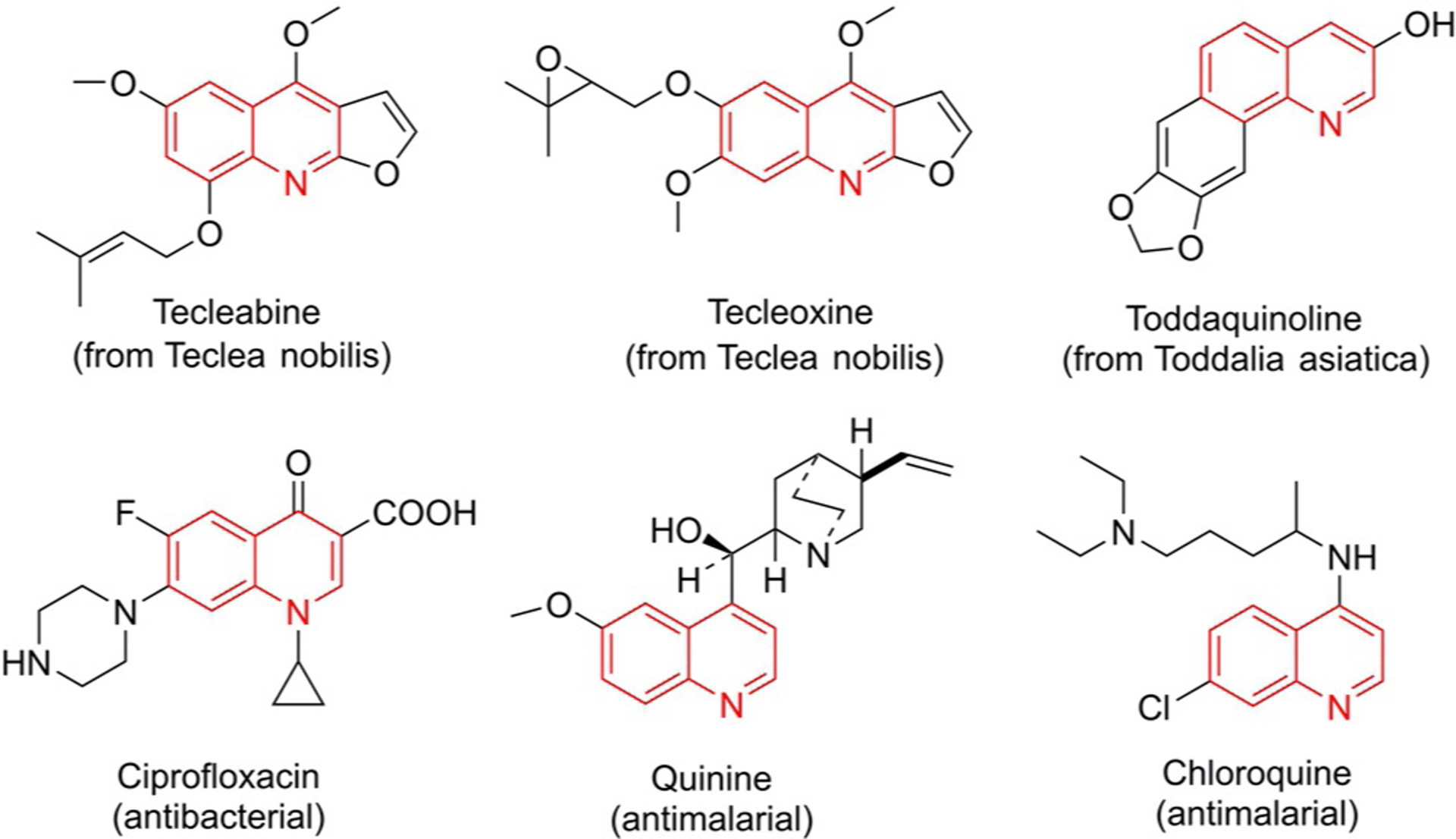
Natural products containing quinoline nucleus.
The most frequently used quinoline compounds are the 2- and 4-methyl derivatives, called quinaldine and lepidine, respectively.
Lepidine (4-methyl quinoline) is used in medicine, synthesis of dyes, and as a food additive. Lepidine can be obtained from the highest-boiling fraction of the coal-tar quinoline bases through addition of o-cresol compound. It can also be formed by reaction of aniline with methyl vinyl ketone in the presence of hydrochloric acid in ethanol or ethanolic ferric chloride or with 3-oxobutanal with a catalytic amount of concentrated sulfuric acid (Scheme 2).[27]
Scheme 2.
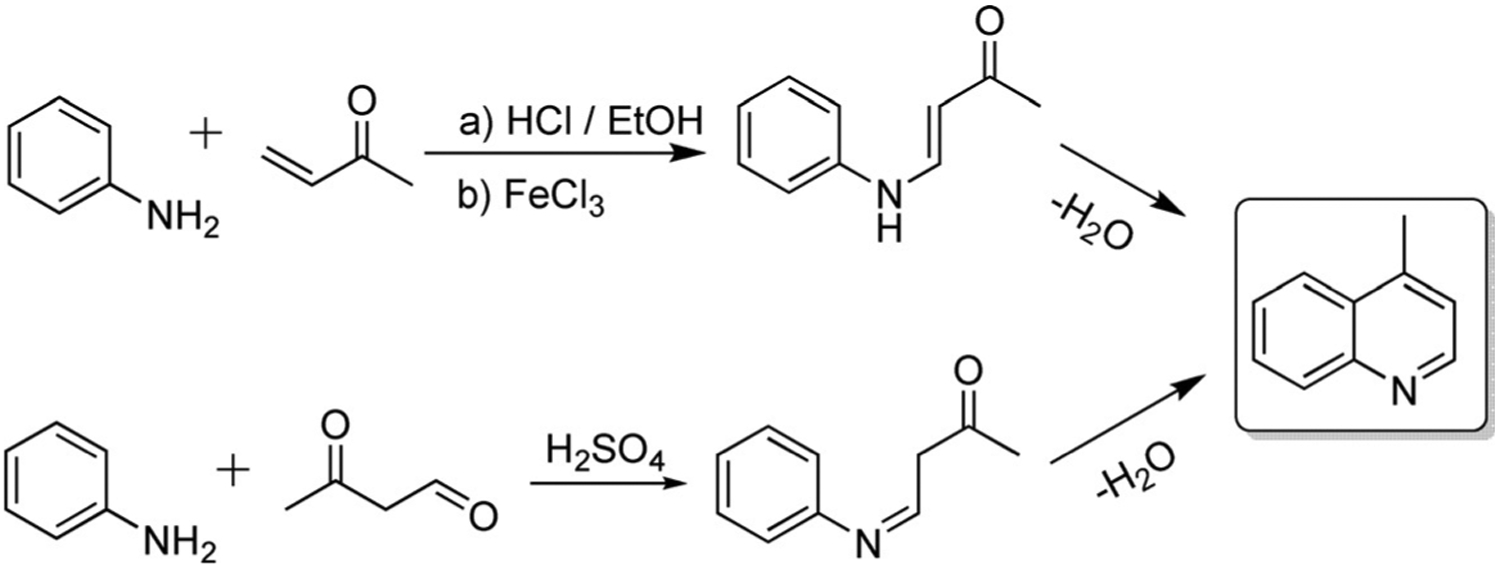
Lepidine (4-methylquinoline) synthetic routes.
Quinaldine (2-methyl quinoline, Scheme 3) is a useful ingredient in antimalarial drugs, in manufacturing dyes, food colorants, pharmaceuticals, and pH indicators. Quinaldine can be produced from aniline by various synthetic methods:
The condensation of aniline with acetaldehyde under the influence of hydrochloric acid (Doebner–Miller synthesis). This reaction may also be completed in the microwave within several mins;[27]
The reaction of aniline with an aldehyde and pyruvic acid (Doebner synthesis);[28]
The optimized method—an aerobic oxidative aromatization of simple aliphatic alcohols and anilines by using a catalytic system containing 2,4,6-collidine, Pd(OAc)2, and trifluoroacetic acid (TFA) with 93 % yield.[29]
Scheme 3.
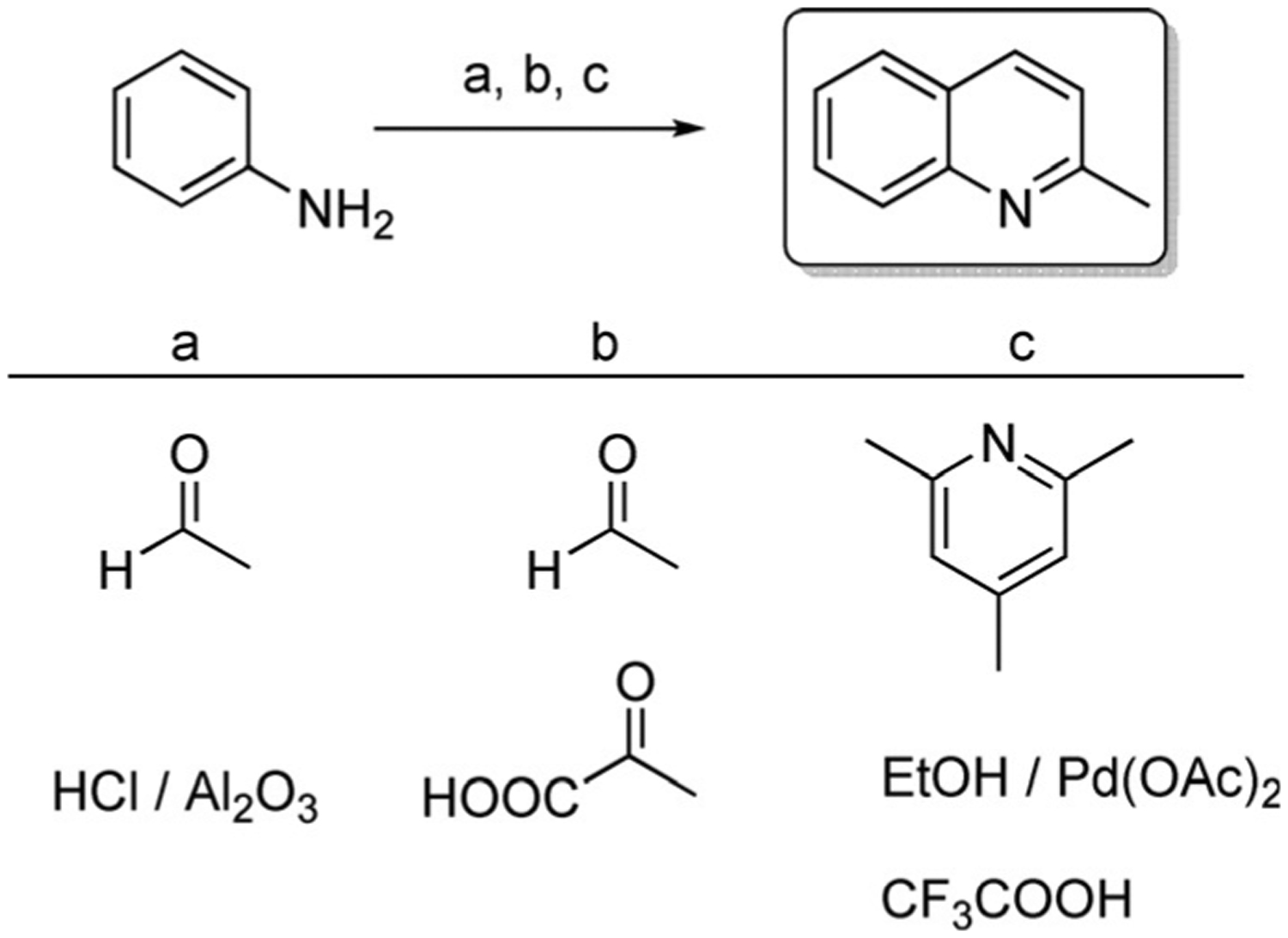
Quinaldine (2-methylquinoline) synthetic routes.
Quinoline is a weakly basic compound, as the lone pair of electrons on nitrogen does not participate in the formation of a delocalized π molecular orbital. As a result, quinolines can undergo nucleophilic substitution reactions and form various salts. These salts are an essential step in the synthesis of cyanine dyes. A lot of different types of salts have been synthesized and various methods have been optimized.[30–34] There are over 180 published salts synthesized by various methods from quinaldine and almost 450 salts from lepidine.[28]
The common methods for the synthesis of some lepidine- and quinaldine-based salts available in the literature are presented in Scheme 4 and Scheme 5. The identical salts are shown to compare the conditions used for the preparation of quinoline-containing salts. Most of them are synthesized from lepidine and quinaldine directly, for example, salts 1–6 (Scheme 4) and 8–13 (Scheme 5), whereas some of them require a different initial reagent, such as diphenylamine for the synthesis of salts 7 (Scheme 4) and 14 (Scheme 5).[30, 31, 33–48]
Scheme 4.
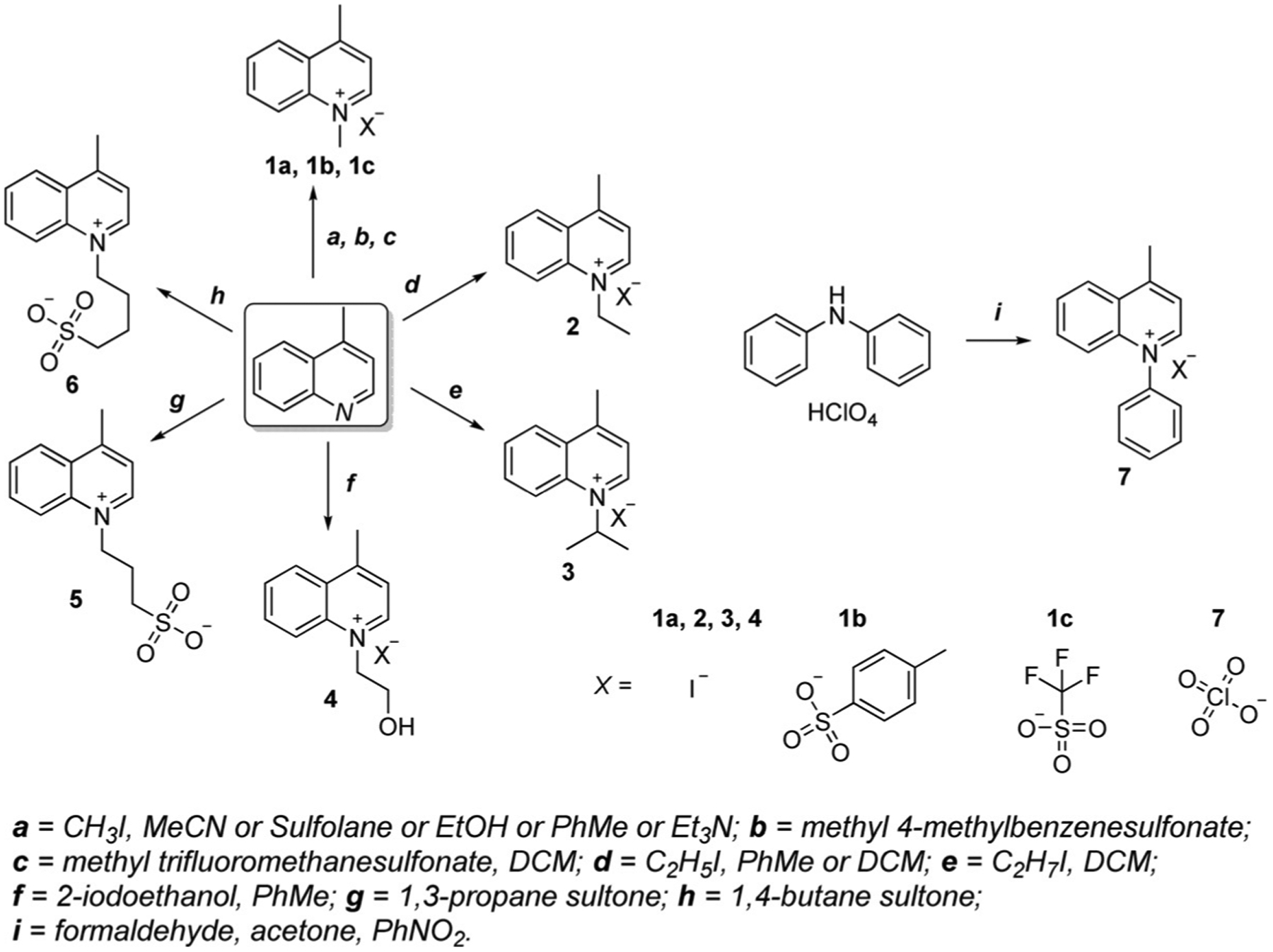
Synthetic routes of various lepidine salts.
Scheme 5.
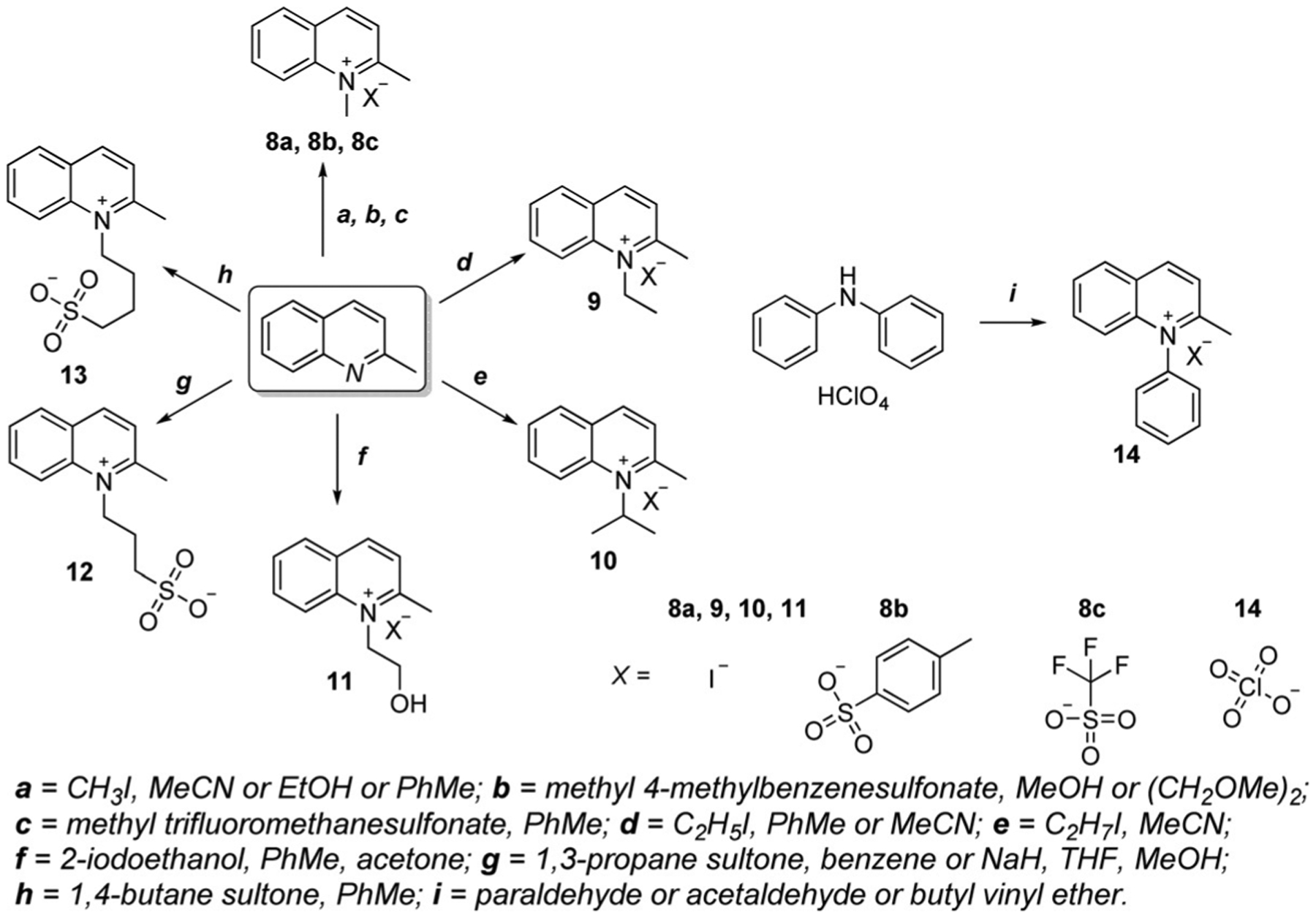
Synthetic routes of various quinaldine salts.
Winstead’s group presented an alternate synthetic route for salts 1a and 2 in 2013. This technique uses a microwave and allows the reaction time to be decreased to 5 min and production of the target quinolinium salts with no purification necessary and with higher yields above 70 %.[42]
Further, the detailed synthesis of each class of cyanine dyes containing quinoline moieties, their optical properties, and applications in biomedicine were discussed.
Synthesis of Monomethine Cyanine Dyes Containing Quinoline Moieties
Monomethine dyes have only one methine unit in the conjugated chain between heterocycles. They absorb in the visible region at 450–520 nm. Commonly, monomethine cyanine dyes are synthesized by the condensation of two heterocyclic quaternary salts with a basic reagent.[49, 50]
One of the methods to synthesize symmetrical monomethine cyanines is carried out by the condensation of sulfobetaines from N-alkylheterocyclic compounds with quaternary salts of heterocyclic 2- or 4-methyl compounds in the presence of triethylamine. Another method is performed upon heating of 2-methylthioquinolinium salt and quinaldine salts in pyridine at reflux. Both routes are shown in Equation (1).[51]
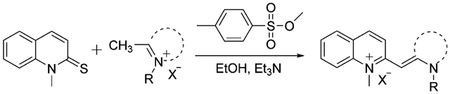
The majority of synthesized monomethine dyes are found to be asymmetrical, so the heterocyclic moieties are usually different. One of them should have a good leaving group, and the reaction should be run in the presence of a base, such as triethylamine or pyridine. Asymmetrical monomethine cyanines have shown very high binding constants, high molar absorptivities and quantum yields, and the generation of high fluorescence signals upon binding. General synthesis of monomethine cyanine dyes containing quinoline, where X is a leaving group, and Y is an anion is shown in Equation (2).
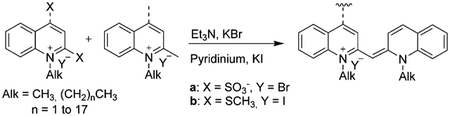 |
(1) |
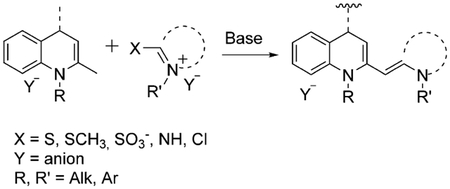 |
(2) |
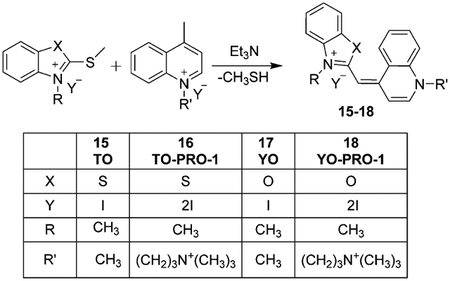
Most approaches towards the preparation of these monomethine dyes function under the principle where quaternary salts of heterocyclic compounds are catalyzed by heat in the presence of a basic compound. Two well-known asymmetrical dyes Thiazole Orange (TO) and Oxazole Yellow (YO) are synthesized by the reaction of 2-methylthiobenzothiazole (TO) or 2-methylthiobenzoxazole (YO) salts with quaternized heterocyclic salts containing an active methyl group in the presence of triethylamine.[37, 49] The various TO and YO related monomethine dyes containing different side chains and anions 15–18 are shown in Equation (3). The alternate method of synthesis of TO monomethine dyes uses microwave-assisted synthesis, decreasing the reaction time to 10 min and improving the yield to 90 %.[37] But both methods have some disadvantages owing to the toxicity of released methyl thiol.
In 1995, Deligeorgiev and co-workers offered another method of TO and YO monomethine dyes synthesis, which avoided the evolution of methyl thiol.[52] The new synthetic route is based on the condensation of 2-iminobenzothiazolines with 1-alkyl-4(or 2)-methylquinolinium salts with no solvents and are presented in Scheme 6.[51] In 1999, he also offered an option to heat an N-alkylheterocyclic compound together with the quaternary salt of a heterocyclic 4-methyquinoline either in solvents or by melting of the starting compounds under vacuum at 150–220°C. Dyes 19 and 20 are examples of compounds synthesized by these methods with good yields above 60 %.[52]
 |
(3) |
Scheme 6.
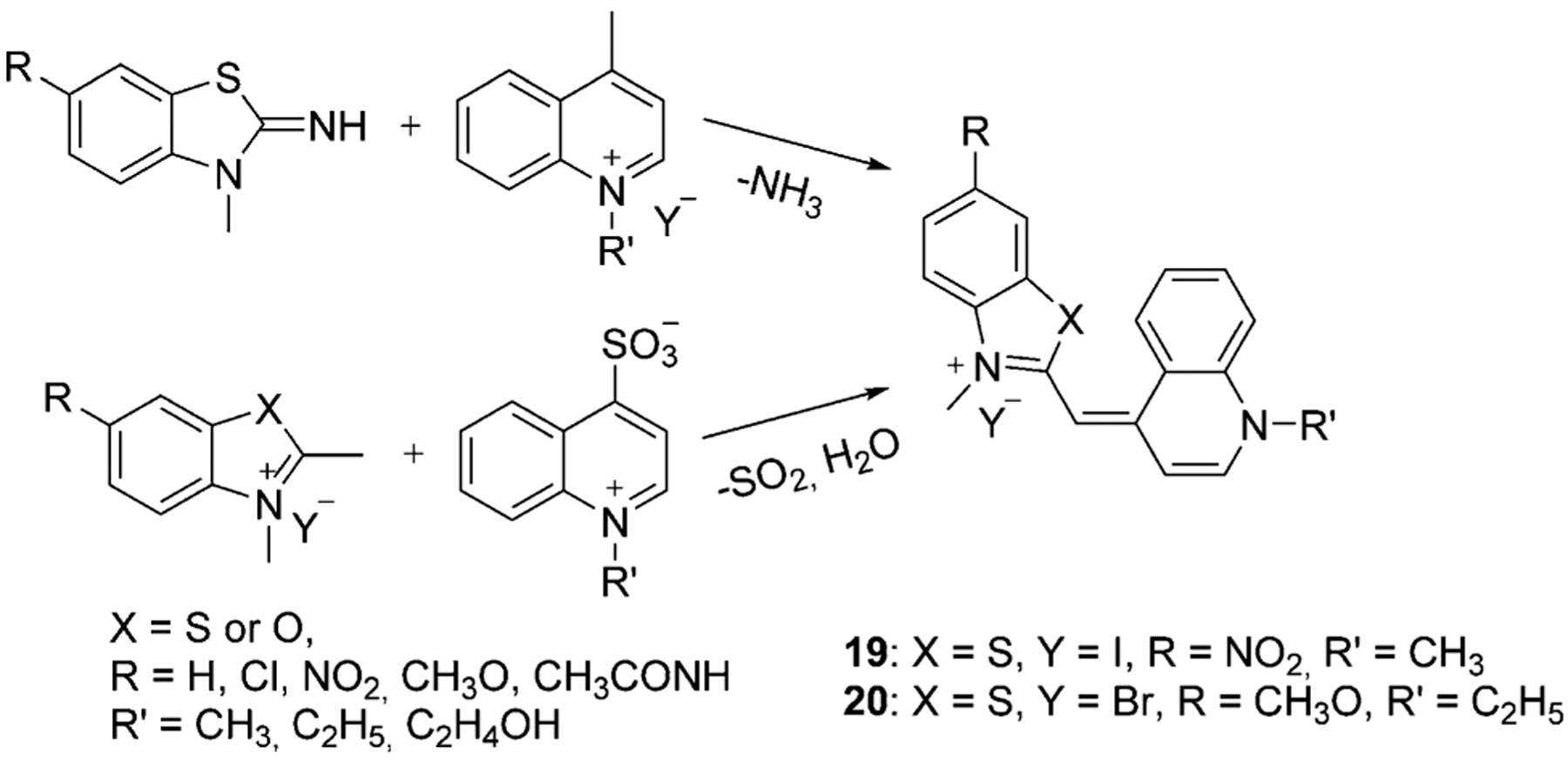
Synthesis of TO and YO monomethine cyanine dyes.
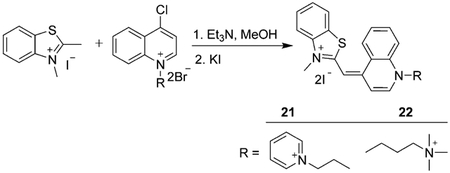
Another pathway for the production of a monomethine dye involves the reaction of N-alkyl-2-methylbenzothiazolium salt with 4-chloroquinolinium salt. This method also avoids the formation of toxic volatile byproducts. This synthetic route can be used for the preparation of dicationic benzothiazole cyanine dyes 21 and 22 [Eq. (4)]. The goal of increased cationic charge is to increase the water solubility of the dyes.[52]
Fei et al. offered a new method of synthesis TO and its derivatives by using solid-phase resin support. The schematic representation of the preparation of TO and its derivatives is shown in Scheme 7. The first step is the attachment of 2-mercapto-benzothiazole to the Merrifield resin, and then the formed intermediate 23 reacts with 4-methylbenzenesulfonate to form N-methyl-benzothiazole salt 24. The final step involves the condensation of the N-methyl-benzothiazole salt with lepidine derivatives forming cyanine dyes 15, 25–27 (Scheme 7) in high yields above 95 % and with excitation wavelengths at around 570–600 nm.[53]
 |
(4) |
Scheme 7.

Solid-phase synthesis of monomethine cyanines.
The majority of published monomethine dyes are synthesized from lepidine salts. However, various asymmetrical monomethine dyes have been synthesized from quinaldine and thiazole salts or two quinolinium salts connected at the 2- and 4-positions. Asymmetrical compounds 28–34 in Figure 10 are synthesized by the condensation of two salts in the presence of methyl p-toluenesulfonate and triethylamine as shown in Equation (5).[54]
 |
(5) |
Figure 10.
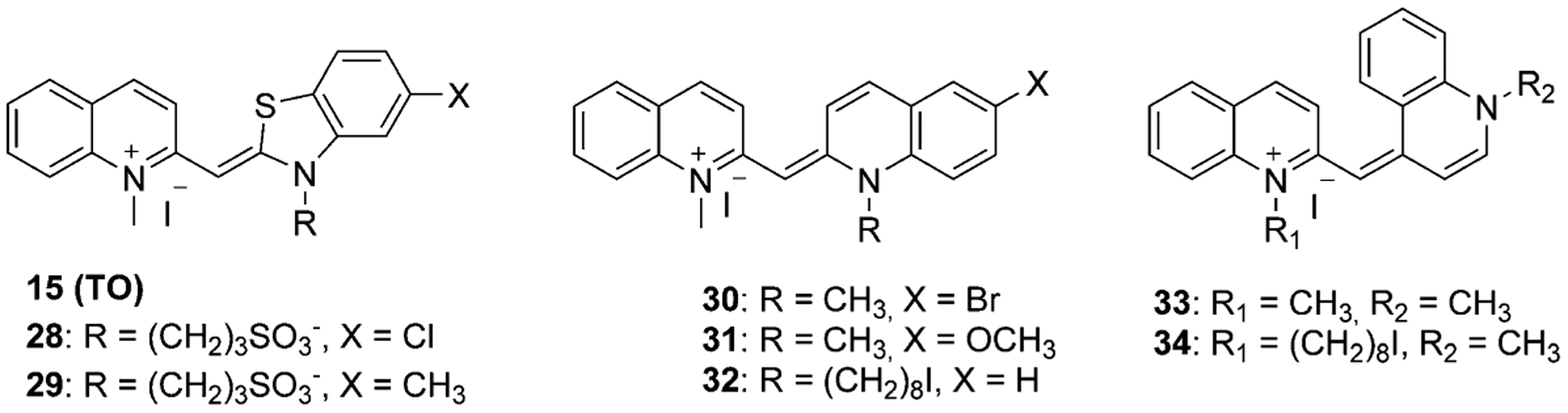
Compounds 28–34 (Figure 10) were synthesized in yields range from 18 to 50 %. Dyes 32 and 34, which differ only by the bridge attachment position were synthesized in 19 % and 45 % yields and showed excitation wavelengths at 522 nm and 557 nm, respectively.[54] Dye 35 was excited at the same wavelength as dye 34.[55]
Synthesis of Dimethine Cyanine Dyes Containing Quinoline Moieties
Dimethine cyanine dyes have two methine carbons in a chain and absorb in the visible range at 450–620 nm. As well as monomethine cyanines, dimethine cyanines have been found to be mostly asymmetrical and do not differ much in terms of synthetic methods. Compared with monomethine cyanine dyes, dimethine and other polymethine cyanine dyes are more environmentally friendly to synthesize, avoiding the formation of thiol molecules.[15]
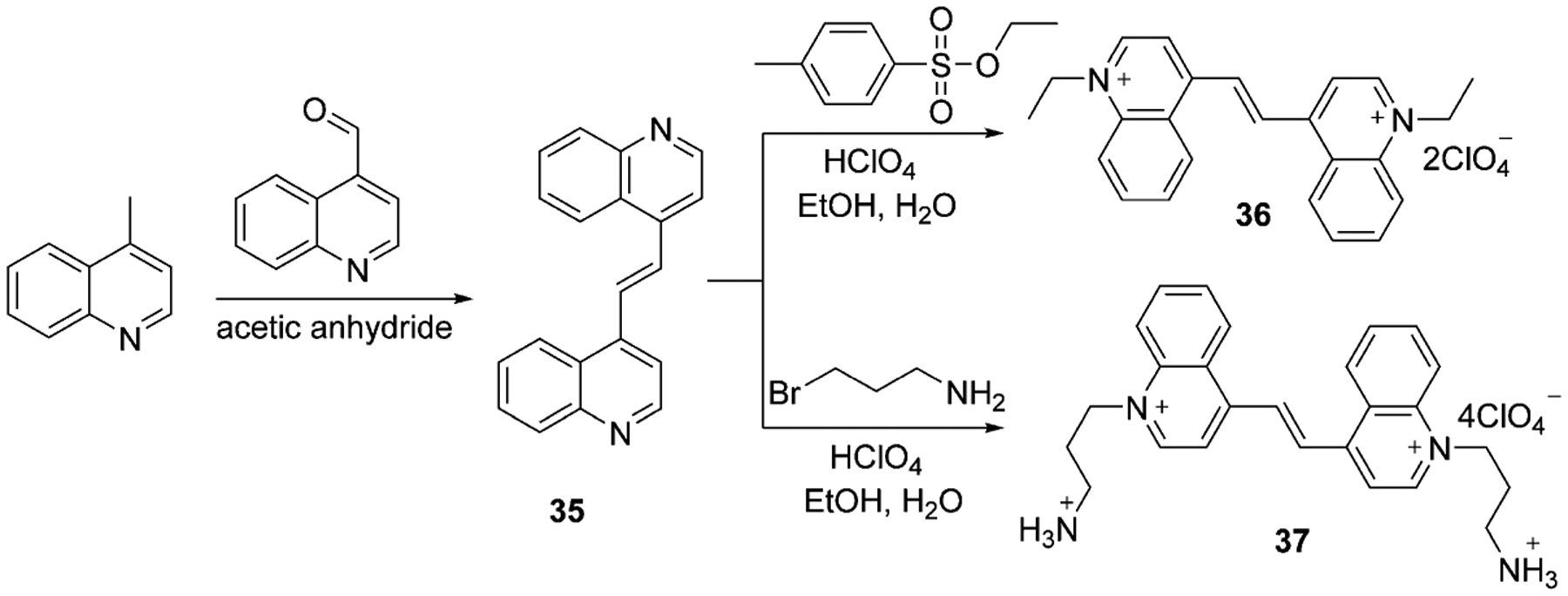
Only two available symmetrical dimethine cyanine dyes were found, which were synthesized by Kuz’mina et al. in 2006.[56, 57] Dyes 36 and 37 contain several cations and anions and are prepared in two steps with the formation of intermediate 35, which is subsequently quaternized by ethyl 4-methylbenzenesulfonate or 3-bromopropylamine hydrobromide and treated by concentrated perchloric acid. The synthesis is presented in Scheme 8.
Scheme 8.
The commonly used synthesis of asymmetrical dimethine dyes includes the reaction of a heterocyclic quaternary salt with an active methyl group at the 2- or 4-position with an aromatic heterocyclic aldehyde. Ethanol with piperidine has become the most often used conditions for making dimethine cyanine dyes [Eq. (6)], showing the highest yielding results.
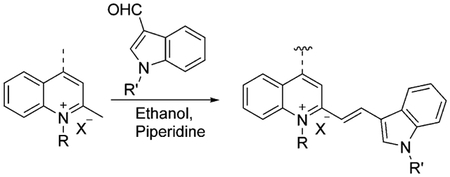 |
(6) |
The method, shown in Equation (6), has been improved by microwave irradiation with no solvent used. This approach results in shorter reaction times and simple product isolation procedures with no toxicity or harm.[32] There are some examples of dimethine cyanine dyes 38–48 available in the literature in which they are synthesized in ethanol with piperidine (Figure 11).[15, 32, 58–61] Compounds 42–45 were synthesized by using a microwave in yields above 90 % in less than 5 min and with absorption maxima in the presence of DNA at 483–499 nm.[32]
Figure 11.
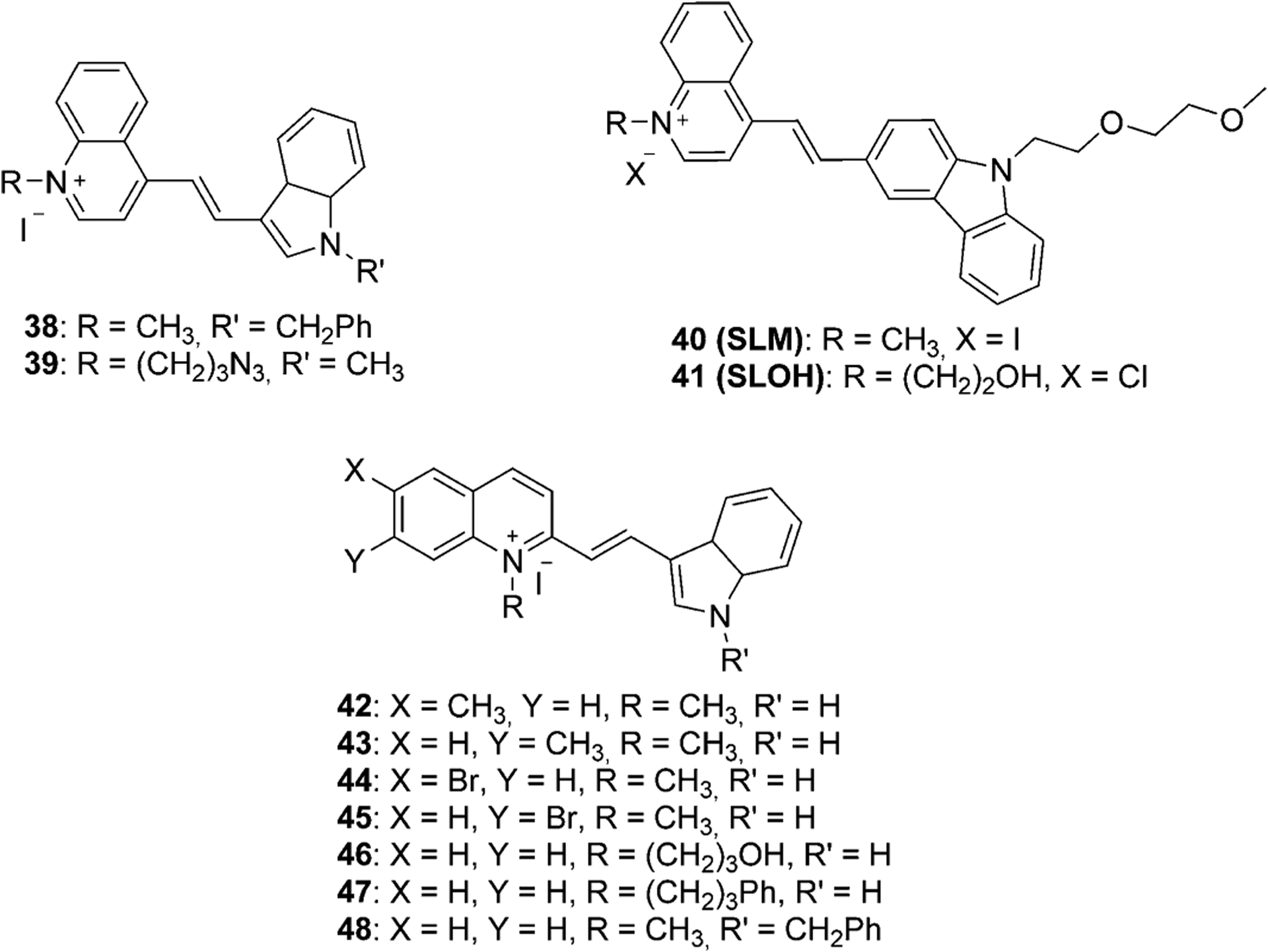
Structures of some known dimethine cyanine dyes, synthesized from lepidine and quinaldine.[15, 32, 58–61]
Synthesis of Trimethine Cyanine Dyes Containing Quinoline Moieties
Trimethine cyanine dyes have three methine carbons in the bridge and absorb in the visible range at 450–620 nm. The first trimethine quinoline-containing cyanine dye was symmetrical and described in 1920 by Mills and Pope and by Wise, Adams, Stewart, and Lund. Symmetrical dye 1,1-diethyl-2,2-carbocyanine iodide, also known as Pinacyanol or Sensitol Red (Figure 12), was prepared from two molecules of quinaldine ethyl iodide and formaldehyde. The same method was used to synthesize another symmetrical 1,1-diethyl-4,4-carbocyanine iodide, called kryptocyanine. Another synthesis method uses ethyl orthoformate in the presence of acetic anhydride instead of formaldehyde.[9]
Figure 12.
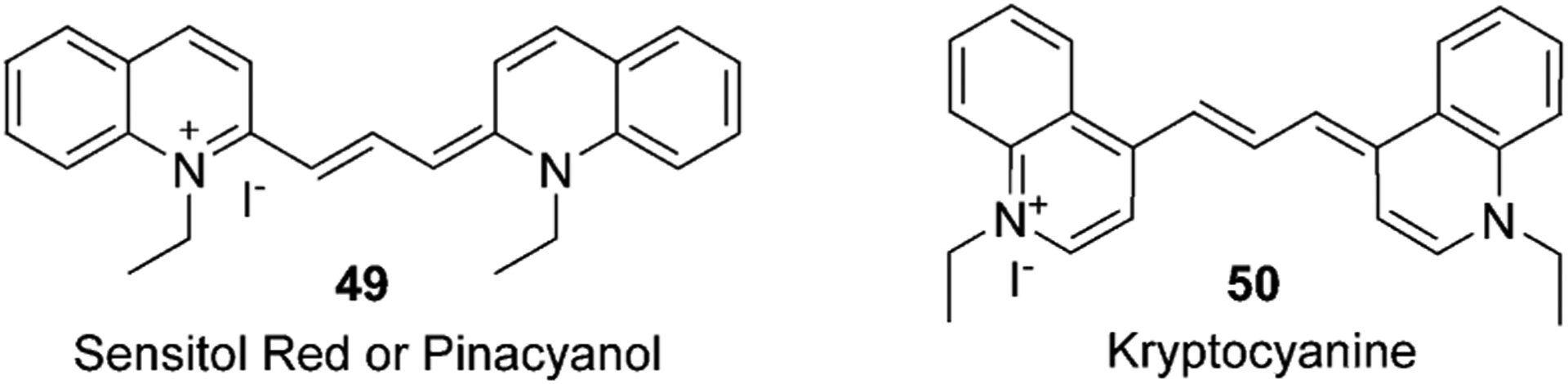
Structures of first symmetrical trimethine cyanine dyes containing quinoline moieties.
One more synthetic route to synthesize symmetrical trimethine cyanines includes the reaction of lepidine salts with triethoxymethane. The intermediates 51 and 52 were obtained in 26 % and 19 % yields. The resulting cyanine iodide 51 and its tosylate 52 were treated with lithium bis(perfluorobutylsulfonyl)-imide, forming new trimethine cyanine dyes 53 and 54 (Scheme 9).[62]
Scheme 9.
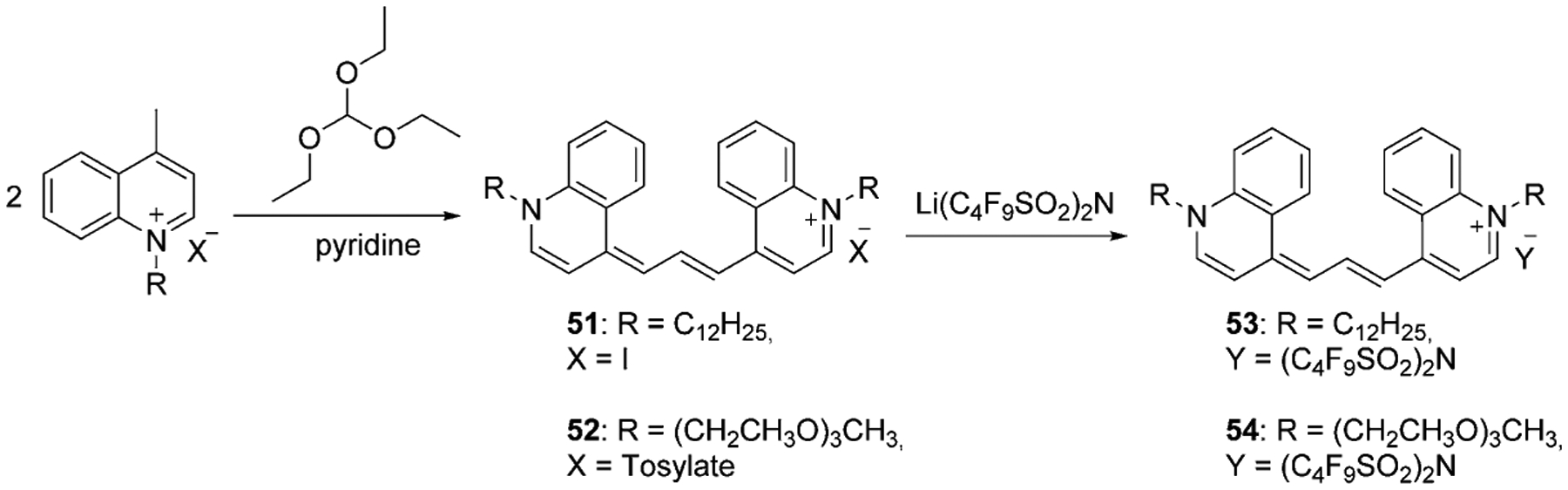
Synthesis of symmetrical trimethine cyanine dyes.
Usually, for the synthesis of asymmetrical trimethine cyanine dyes, one molecule of quaternary salt is used to form a hemicyanine intermediate, which then reacts with another salt molecule to gain the asymmetrical dye. Triethylamine and pyridine are the most used bases. Two of the most famous trimethine asymmetrical cyanines are TO-3 (55) based on Thiazole Orange (TO) and YO-3 (56) based on oxazole yellow (YO) with three carbons in a chain, and their dicationic derivatives TO-PRO-3 (57) and YO-PRO-3 (58) are presented in Figure 13.
Figure 13.
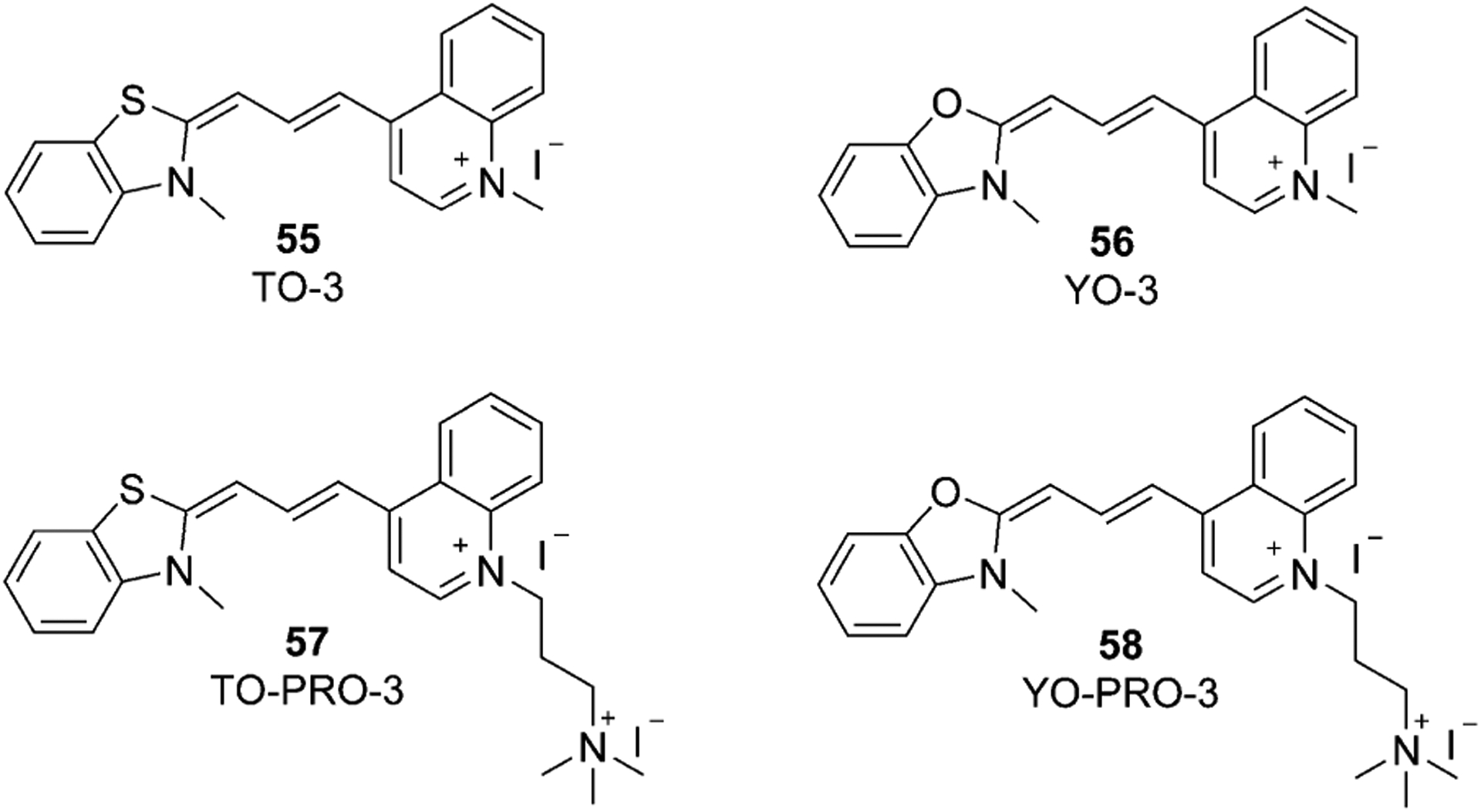
Chemical structures of TO-3, YO-3, TO-PRO-3, and YO-PRO-3.
TO analogs have attracted researchers’ attention and different modifications and incorporations of TO-3 have been synthesized. Firstly, either quinoline or thiazole salt is connected to the linker. The formed hemicyanine 59 condenses with another quaternary salt and gains various alternatives of TO-3. Sun et al. synthesized the dye 61 with two positive charges, forming first the intermediate bromo-version 60 in 64 % yield. Then, the bromine atom in dye 60 is converted into a quaternary ammonium in dye 61 (53 % yield), as shown in Scheme 10.[63]
Scheme 10.
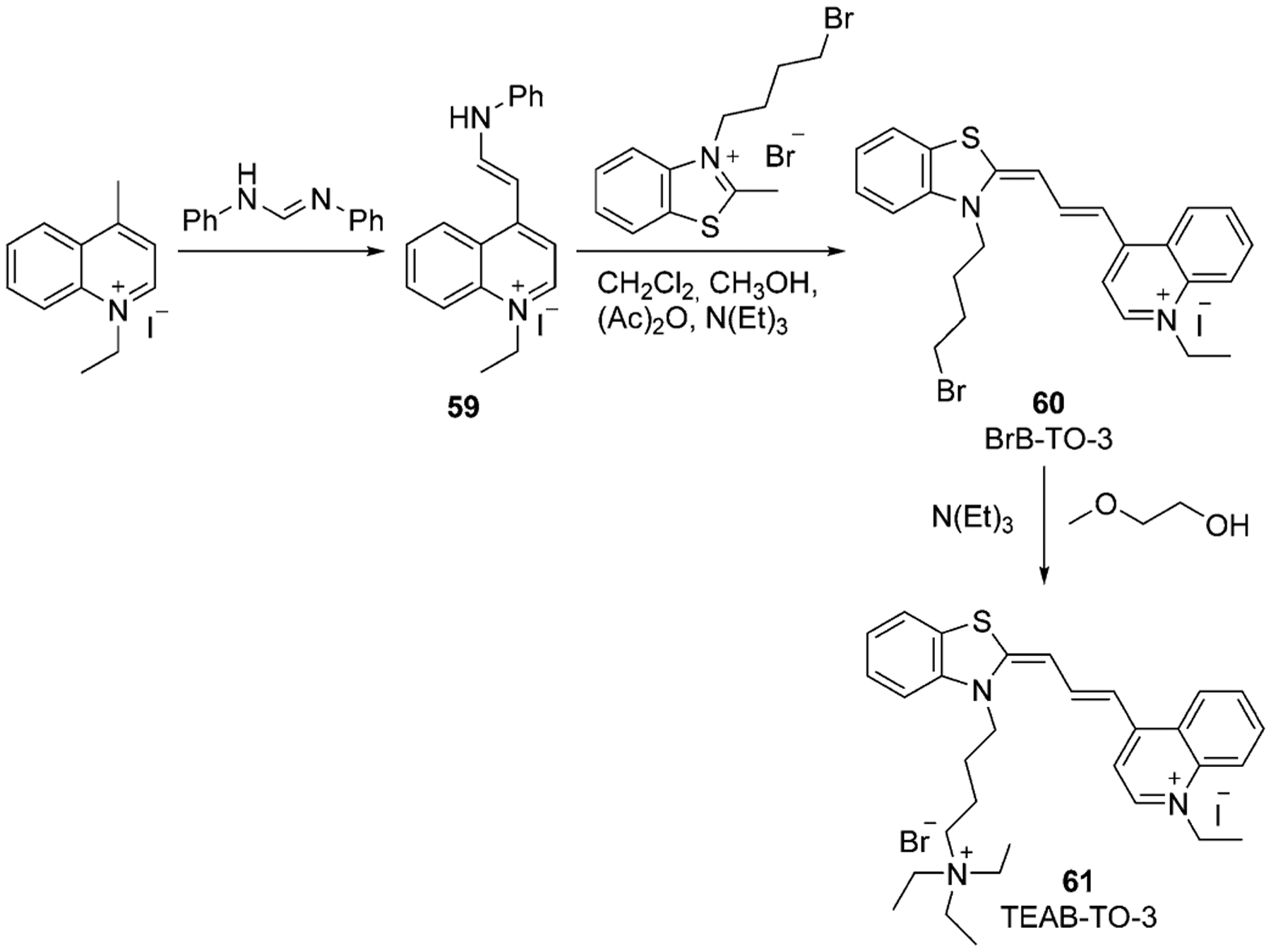
Synthesis of trimethine cyanine TEAB-TO-3.[63]
The indole moiety is often used instead of thiazole in asymmetrical cyanine dyes and may form Dimethyl Indole Red (DIR) 63. The first step of 63 synthesis includes quaternary salt preparation by alkylation of lepidine with propanesultone, which is subsequently condensed with N,N-diphenylformamidine and forms hemicyanine dye 62 (40 % yield). The last step includes the formation of trimethine dye 63 (23 % yield) by condensation of the hemi-dye with methyl indoline bromide under basic conditions, as illustrated in Scheme 11.[64]
Scheme 11.
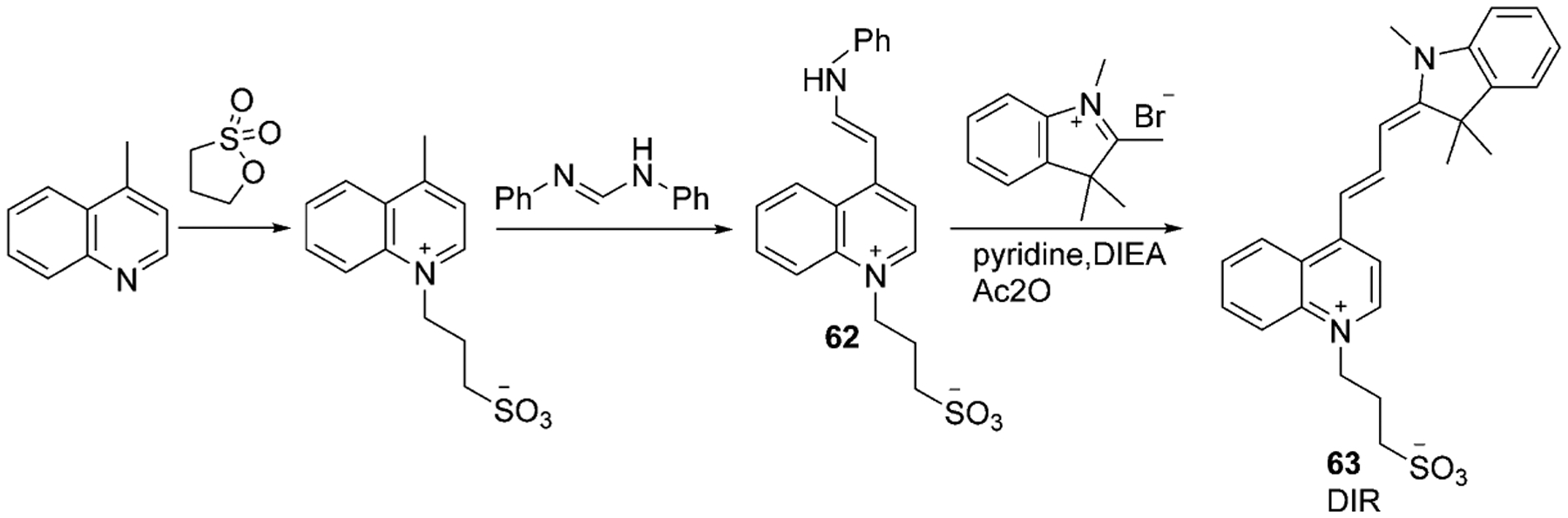
Synthesis of dimethyl indole red (DIR).[64]
Another approach to the synthesis of asymmetrical trimethine cyanine dyes with an indole moiety was accomplished by Fei’s group (Scheme 12). Herein, the hemicyanine 64 is formed from the methyl indoline derivative, which then reacts with lepidine salt under basic conditions to form the asymmetrical cyanine dyes 65–68.[65]
Scheme 12.
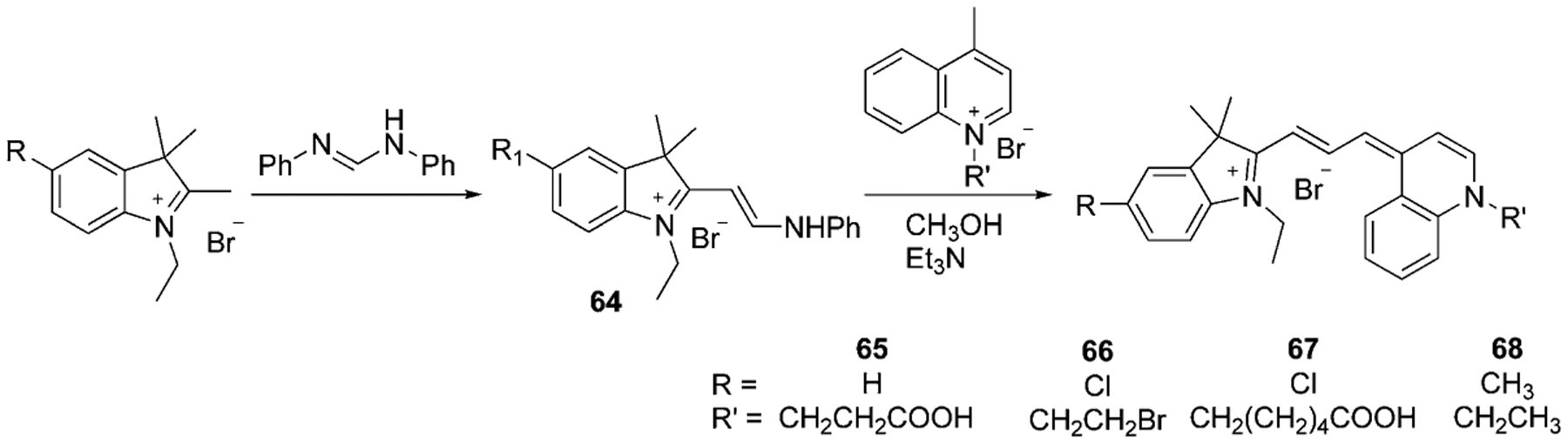
Synthesis of trimethine indoline quinoline dyes.
The synthetic approach, presented in Scheme 12, has some disadvantages, such as the formation of intermediates and difficulties with the purification of dyes. To avoid these drawbacks, in 2005, Mason et al. offered some new approaches to synthesize quinoline-containing trimethine cyanine dyes, 72–76.[66] The synthesis of carbocyanine dye 72 is presented in Scheme 13 and involves two synthetic routes. The first approach includes the formation of hemicyanine 69, which then is activated by sulfonylation with sulfonyl chloride polystyrene, forming 70, which then reacts with quinolinium salt 2. Unreacted hemicyanine remains on the resin, and the pure trimethine dye can be isolated. The second method involves the synthesis of polystyrene-bound immediate 71 from immobilized aniline, which is subsequently used to react with quinolinium salt 2 to form the dye 72 in 19 % yield.[66] Figure 14 shows various dyes synthesized by this method; including both quinaldine (73 and 75) and lepidine, (74 and 76) based fluorophores.
Scheme 13.
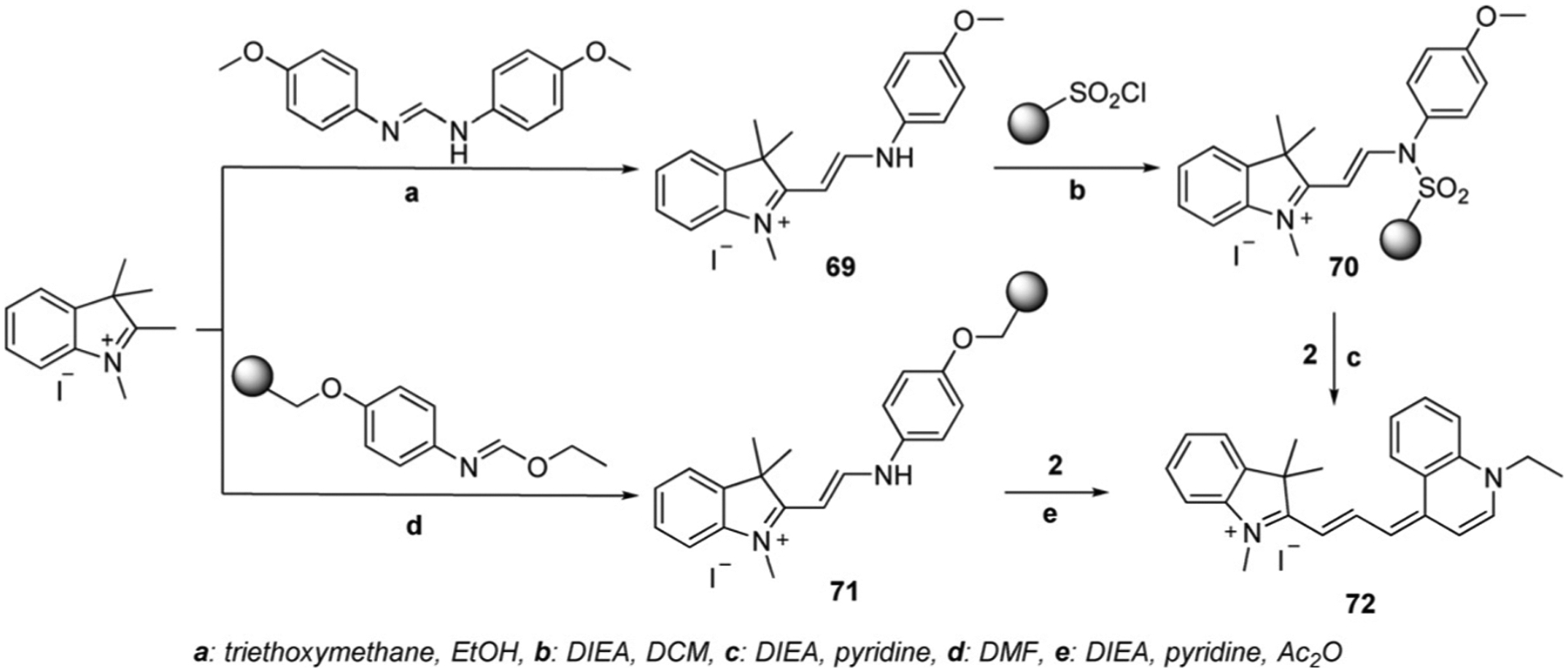
Solid-phase synthesis of trimethine cyanine dyes.[66]
Figure 14.
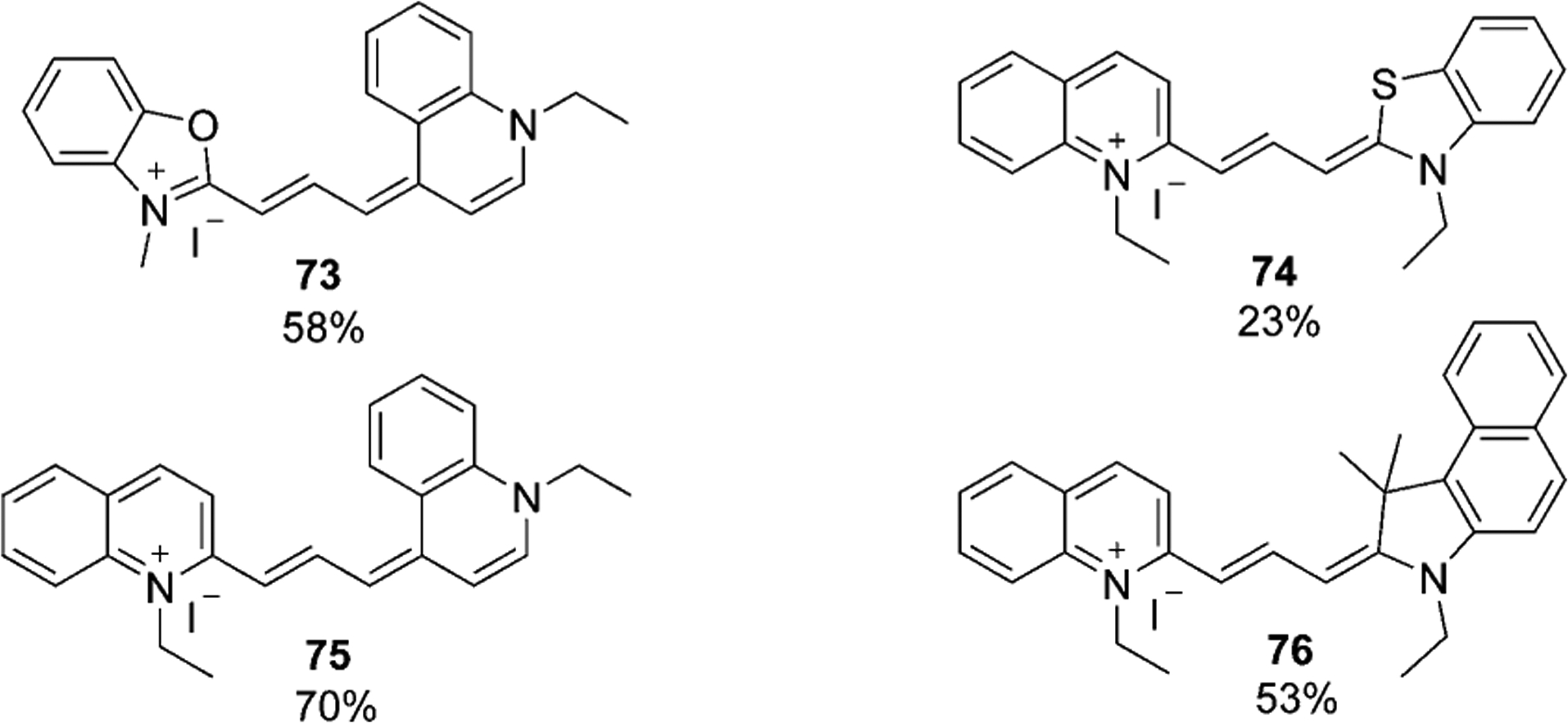
Trimethine cyanine dyes synthesized by solid-phase method.
Synthesis of Pentamethine Cyanine Dyes Containing Quinoline Moieties
Pentamethine cyanine dyes commonly absorb in the visible and near-infrared (NIR) region at 650–850 nm. The general synthetic procedure of symmetrical pentamethine dyes involves the use of malonaldehyde bis(phenylimine) derivatives under basic conditions and two moles of quinoline-containing salts as exemplified in Equation (7). Pentamethine fluorophores often have a precursor at the meso-position to improve their photophysical properties.[67] Meso-substituents in the pentamethine chain usually belong to proton or halogen atoms.
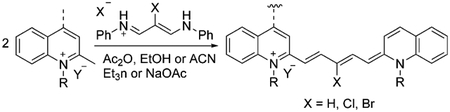 |
(7) |
The presented method in Equation 7 can be used to synthesize symmetrical pentamethine dyes 77–83 in good yields, which are illustrated in Figure 15.[16, 67, 68]
Figure 15.
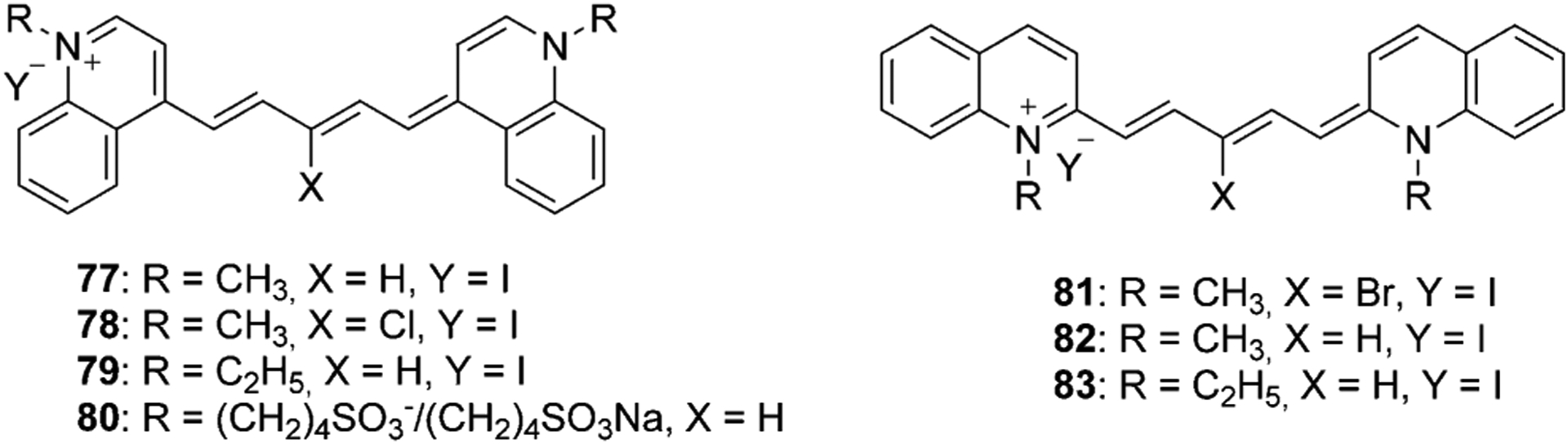
Examples of symmetrical pentamethine cyanine dyes.[16, 67, 68]
To synthesize asymmetrical pentamethine cyanine dyes, the first step is to prepare the hemicyanine intermediate, which then reacts with quinolinium salts under basic conditions. The same method as described earlier for trimethine synthesis was used by Mason et al. for the synthesis of pentamethine cyanines 86–90, based on the use of solid-phase resin support (Scheme 14). The first step involves the reaction of malonaldehyde diacetal, forming the intermediate precursor 84. The second step is the reaction of the received precursor with methyl indolinium salt to form hemicyanine 85, which generates the pentamethine cyanine dye 86 in 60 % yield by subsequent reaction with quinolinium salts.[66] The described method was used for the synthesis of dyes 87–90, which are illustrated in Figure 16.
Scheme 14.
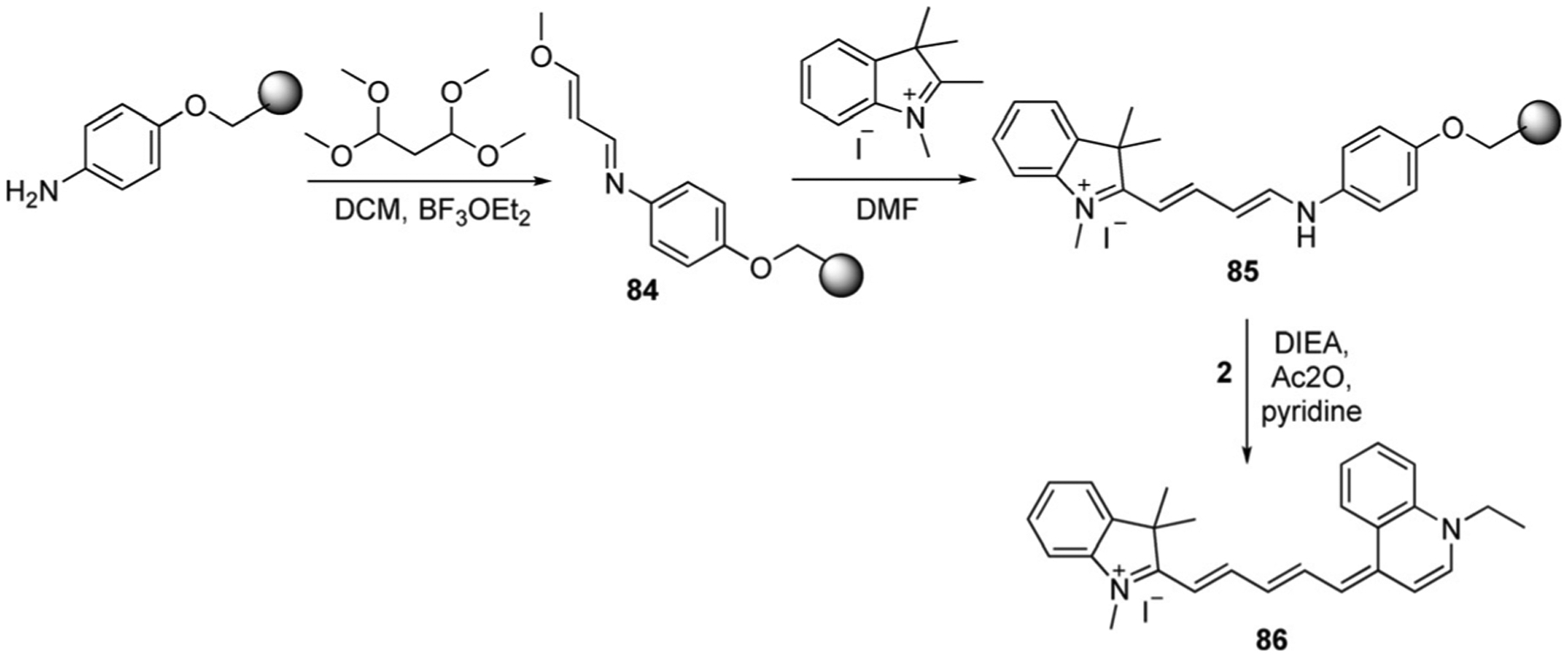
Solid-phase synthesis of pentamethine cyanine dyes.[66]
Figure 16.
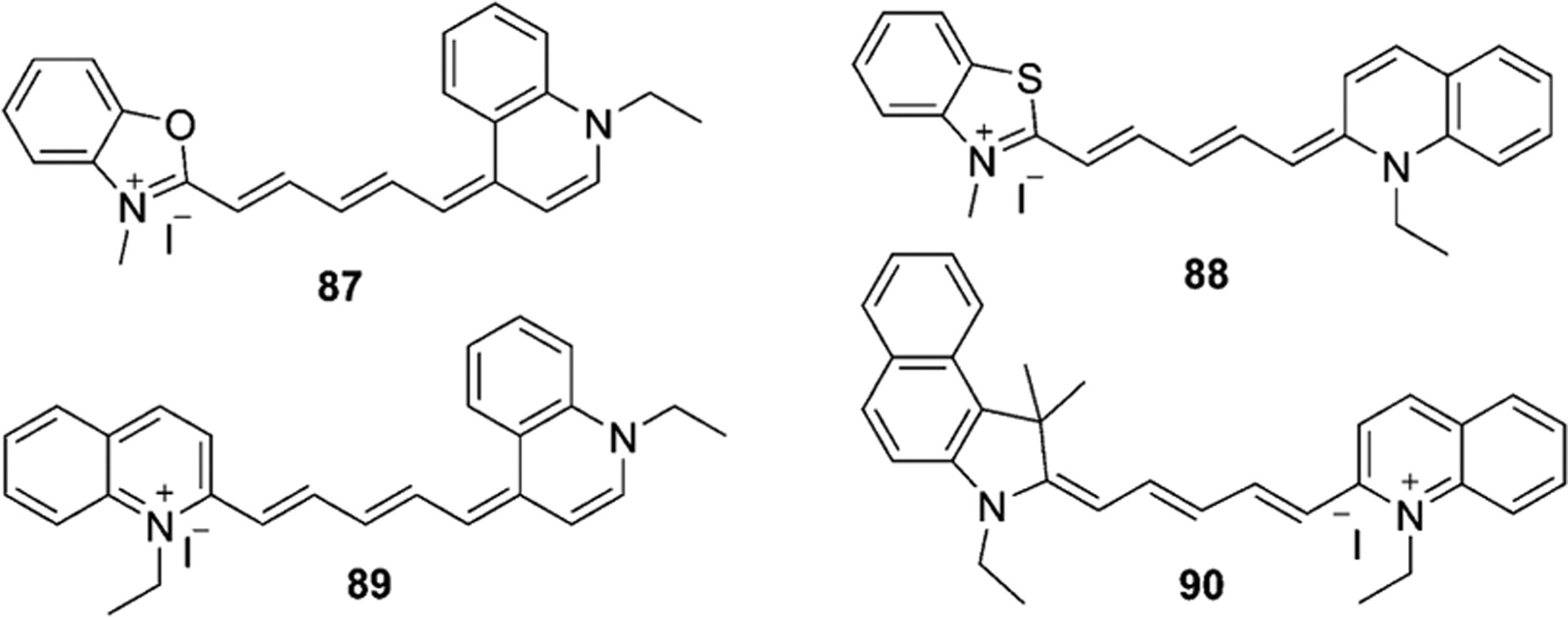
Pentamethine cyanines synthesized by solid-phase method.
Synthesis of Heptamethine Cyanine Dyes Containing Quinoline Moieties
Heptamethine cyanine dyes usually absorb in the near infrared (NIR) region (750–900 nm) and contain seven carbons in a chain. Heptamethine dyes usually have substituents at different positions of the chain, as they can help to improve the photophysical and photochemical properties of the dyes. Strekowski and co-workers found that the introduction of a chloro-cyclohexenyl ring in the middle of the polymethine chain may enhance the dye’s photostability.[69] That is why the majority of synthesized heptamethine cyanines have a six-membered ring in the chain.
Chloro heptamethine cyanine dyes have traditionally been synthesized by the condensation of the quaternary ammonium salt and bisaldehyde or its equivalent with the subsequent addition of the second molar equivalent of the salt. The process usually takes place in acetic anhydride or ethanol with sodium acetate, triethylamine, piperidine, or pyridine. But sometimes a benzene/butanol mixture may be also used as the solvent (Scheme 15).[70]
Scheme 15.
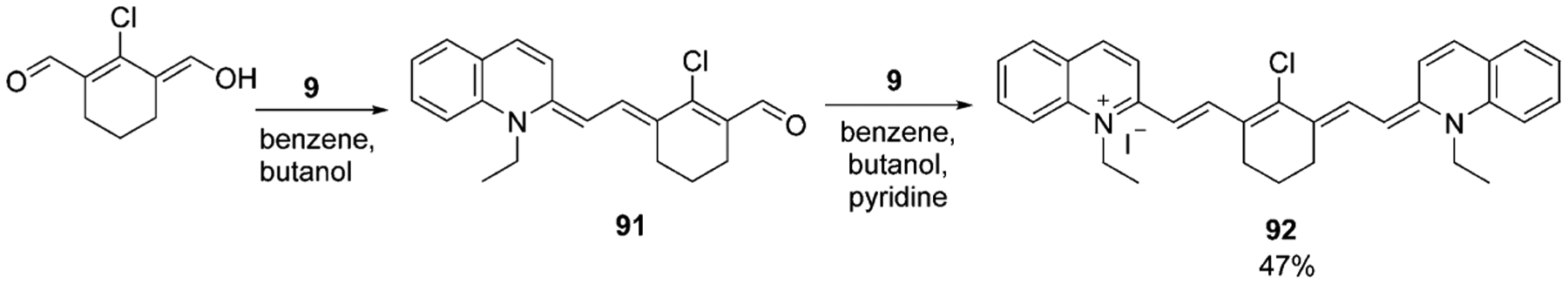
Synthesis of symmetrical heptamethine cyanine dye.[70]
The cyclohexene moiety in the polymethine core can be replaced or modified with substituents instead of the chlorine atom with phenyls, or at any other position of the chain, as in dyes 93–97 [Eq. (8), Scheme 16]. The synthesis of symmetrical heptamethine dyes 93–97 implies the synthesis of 2 moles of quinolinium salts and 1 mole of the linker, which allows formation of the dye within one step.[71–73] The resulting iodides 94 and 95 may be treated with sodium tetraphenylborate to form new fluorophores 96 and 97 in excellent yields (93–94 %).
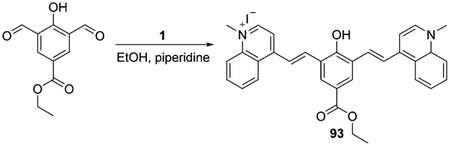 |
(8) |
Scheme 16.
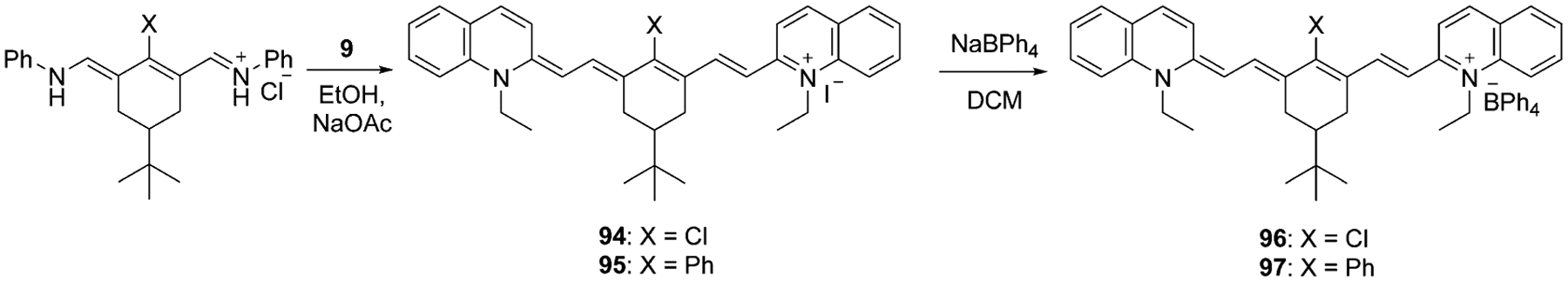
Synthesis of symmetrical heptamethine cyanine dyes with substitutions at the center of the bridge.
The same synthetic route is used to synthesize asymmetrical cyanine dyes, but instead of 2 moles of salts, one salt differs from another one as illustrated in Schemes 17 and 18.[17, 74] For the synthesis of fluorophores 99 and 101, the first step is the preparation of hemicyanines 98 and 100 accordingly, which are heated at reflux with other salts under basic conditions.
Scheme 17.
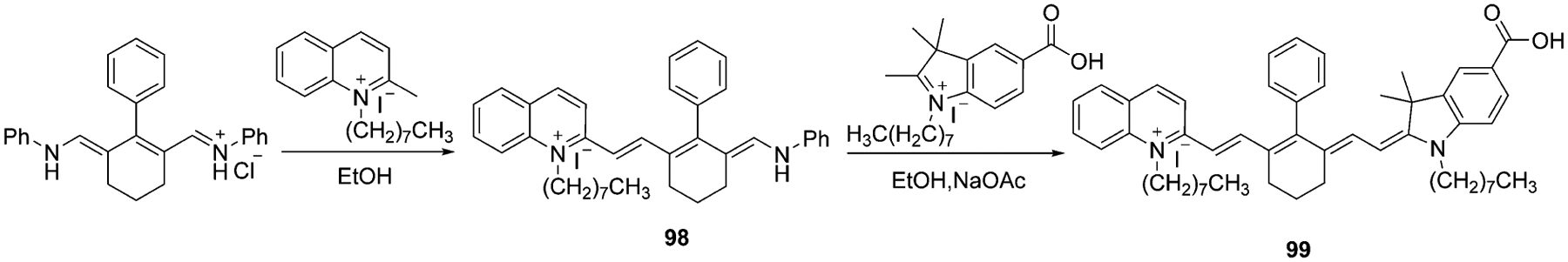
Synthesis of asymmetrical heptamethine cyanine dye 99.[17]
Scheme 18.

Synthesis of asymmetrical heptamethine cyanine dye 101.[74]
The methine bridge may be modified and have substituents at different positions in the chain. Štacková et al. offered a new approach for the generation of heptamethine cyanine dyes with the introduction of substituents at the C3 position of the chain. The synthesis includes the condensation of 3-phenylpyridine and 2,4-dinitrophenyl p-toluenesulfonate. The subsequent reaction of the resulting semi-product 102 with 3 equivalents of salt 9 in the presence of sodium acetate and 4-bromoaniline forms the final product 103 in 47 % yield (Scheme 19).[72]
Scheme 19.
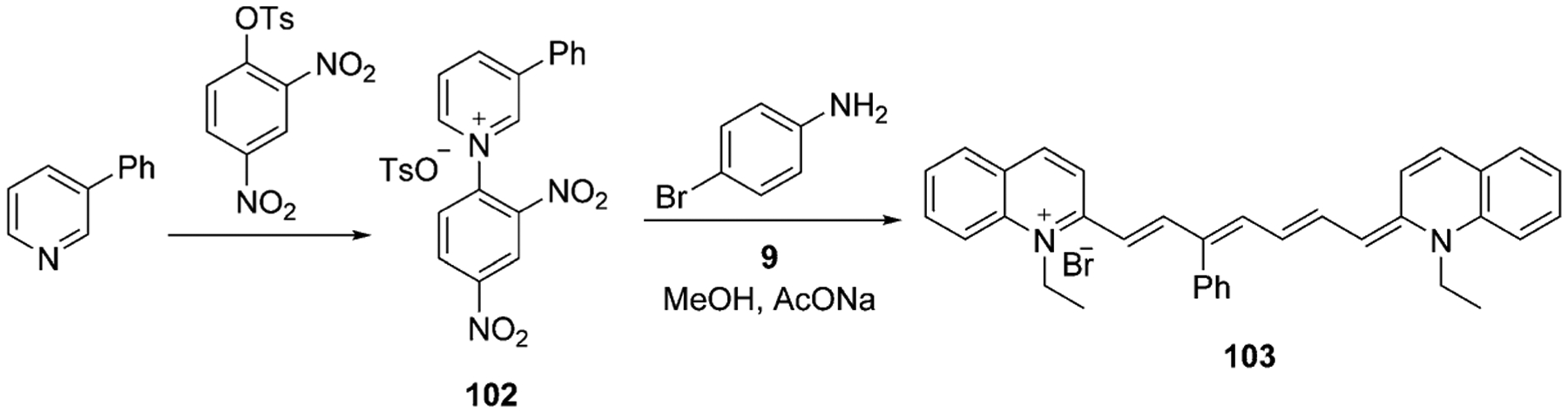
Synthesis of heptamethine cyanine dye 103 with the substituent at the C3 position.
The synthetic routes of cyanine dyes containing quinoline moieties that were published in the literature are summarized and presented in the schemes and equations outlined above. The optical properties and recent applications of some selected fluorophores are further discussed in the next section of this review article.
Optical Properties and Applications of Cyanine Dyes Containing Quinoline Moieties
The first synthesized cyanine dyes were mostly used as photosensitizers in photography. In 1936, Scheibe and Jelley discovered pseudoisocyanine (PIC; Figure 7), which showed a new type of aggregation with two absorption maxima at 490 and 525 nm and the formation of a third peak in aqueous solutions with DNA at 580 nm (Figure 17). This type of dye was named J-aggregates, which may be formed through dispersion forces, induced by the high molecular polarizability. The J-aggregates have highly emissive properties, whereas a monomeric PIC shows no emission.[75] Pseudoisocyanine has become one of the most investigated organic compounds owing to its unique properties and has been used in photographic processes as a component of signal processing light-harvesting materials.[76] For many years, J-aggregates of cyanines have been used in silver halide photography and as photodetectors. The majority of known fluorophores, forming J-aggregates, have been designed for binding and staining the membranes of living cells.[77]
Figure 17.
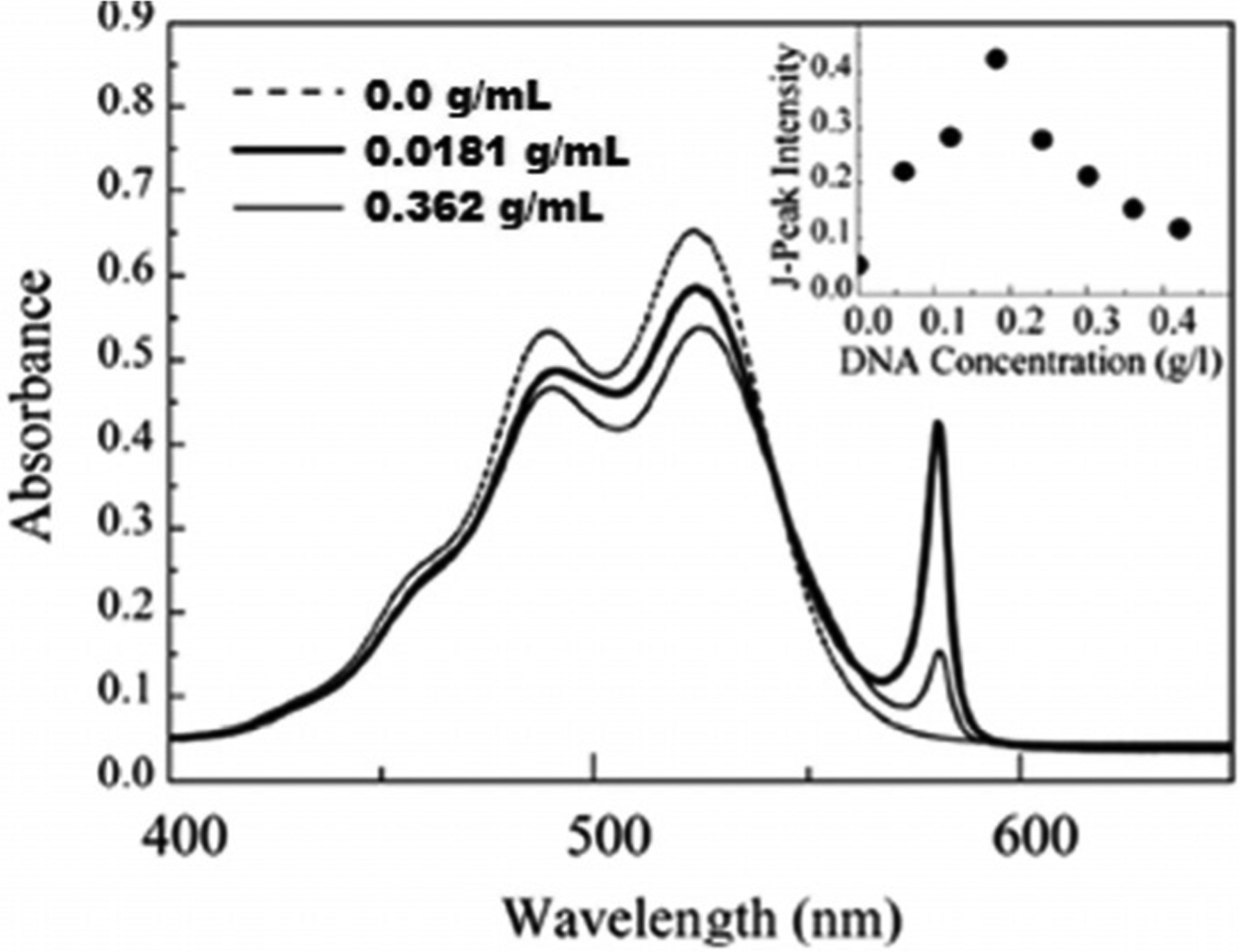
The absorption spectra of PIC in a mixture of water and methanol in the absence (dash) and the presence (solid) of DNA at different concentrations.[78]
The major application of monomethine cyanine dyes is as fluorescent labels for DNA and imaging probes for macromolecules, because of their fluorescence enhancement and outstanding photophysical properties. Thiazole Orange (TO) 15, Oxazole Yellow (YO) 17 [Eq. (3)], and their derivatives are used as fluorescent polymer base substitutions, imaging probes in biology, and DNA and RNA detection.[64, 79–80] TO is an example of a dye that has favorable physical, optical, and biological staining characteristics with absorption wavelength at 502–510 nm both in methanol and with the addition of DNA, RNA, and protein. In the absence of DNA or RNA, TO has quite low fluorescence because of the absence of a binding site. TO is very sensitive, whether it is free or bound to the nucleic acid. In general, if dyes are sensitive to the environment and have high quantum yields, they are useful for imaging of biological and other systems.[81] This property of the dyes has led to the development of “light-up probes” where TO is attached to the end of the probe strand.[82]
YO is like TO, having a high enhancement of quantum yield after binding to DNA. The molecular structures of TO and YO often undergo modifications to achieve higher sensitivity, photostability, or imaging of bioactivity in living cells.[83] Both TO and YO derivatives usually have quite low fluorescence, but become strongly fluorescent with high quantum yield after binding to biomolecules.[84]
TO-PRO-1 and YO-PRO-1 can form the dimers TOTO and YOYO (Figure 18), which excite at 490 and 460 nm and emit at 530 and 510 nm, respectively, bounded to double-strand (ds)DNA in solution.[85] The length of the linker between two TO monomers affects the quantum yield. As a result, TOTO exhibits a nucleic acid binding affinity 100 times greater than the TO monomeric form.[86] TOTO causes a single-strand cleavage approximately five times less efficiently than YOYO. The binding of YO and YOYO to Bacteriophage T5 has been analyzed and has proved the usefulness of dyes as spectroscopic probes for T5, despite the YOYO association to the T5 phages being much slower than to DNA, whereas YO association is quite similar. YO shows very fast association to T5 within just 1 min.[87]
Figure 18.
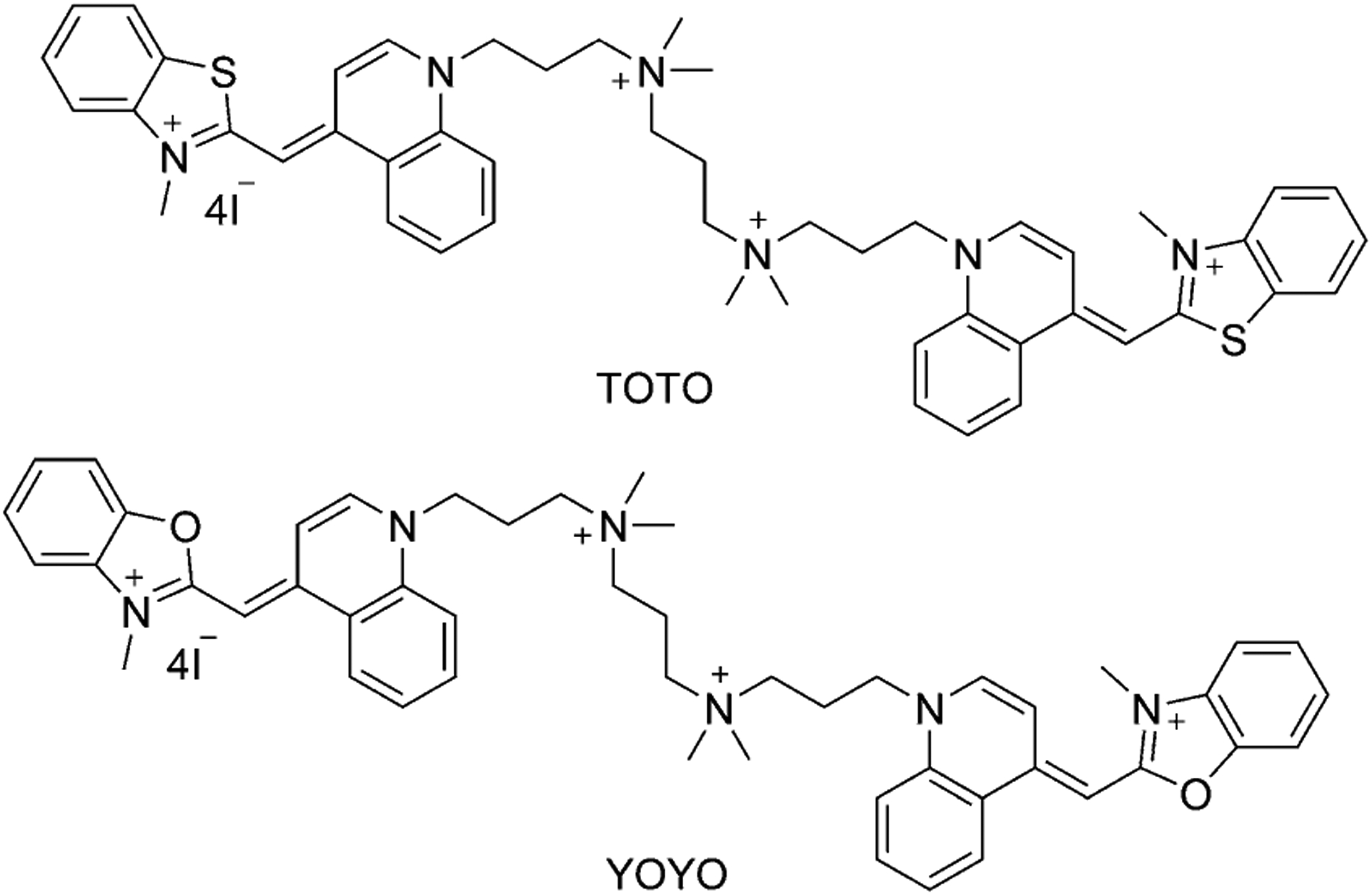
Structure of TOTO and YOYO dimers.
Table 1 presents the spectral properties of two similar dimethine cyanine dyes, 38 and 48 (Figure 11), differing only by the position the linker attaches to the quinoline moiety (4-position for 38 and 2-position for 48). According to the spectral data, both absorption and fluorescence wavelengths for dye 38 are higher than for dye 48 in four different solvents. Stokes shift values increase with the increase of solvent polarity, whereas the molar extinction coefficient of dye 38 is decreasing. The analysis of spectral characteristics of dyes shows that dye 38 has very low cytotoxicity, which is helpful for applications in biomedicine as fluorescent probes and in cellular imaging. The results of cell staining of dye 38 by incubation with A172 cells demonstrate that 38 may stain cytoplasm (Figure 19).
Table 1.
The spectral characteristics of dyes 38 and 48 in different solvents.[15]
| Solvents | Characteristics | Dye 38 | Dye 48 |
|---|---|---|---|
| H2O | λex [nm] | 473 | 458 |
| λem [nm] | 574 | 554 | |
| Stokes shift [nm] | 101 | 96 | |
| ε [× 10−4 Lmol−1 cm−1] | 1.91 | 3.52 | |
| DMSO | λex [nm] | 503 | 481 |
| λem [nm] | 596 | 565 | |
| Stokes shift [nm] | 93.6 | 84 | |
| ε [× 10−4 Lmol−1 cm−1] | 2.09 | 3.21 | |
| MeOH | λex [nm] | 503 | 479 |
| λem [nm] | 581 | 558 | |
| Stokes shift [nm] | 78.6 | 79.4 | |
| ε [× 10−4 Lmol−1 cm−1] | 2.28 | 4.13 | |
| CHCl3 | λex [nm] | 513 | 503 |
| λem [nm] | 570 | 568 | |
| Stokes shift [nm] | 57 | 65 | |
| ε [× 10−4 Lmol−1 cm−1] | 2.82 | 2.16 |
Figure 19.
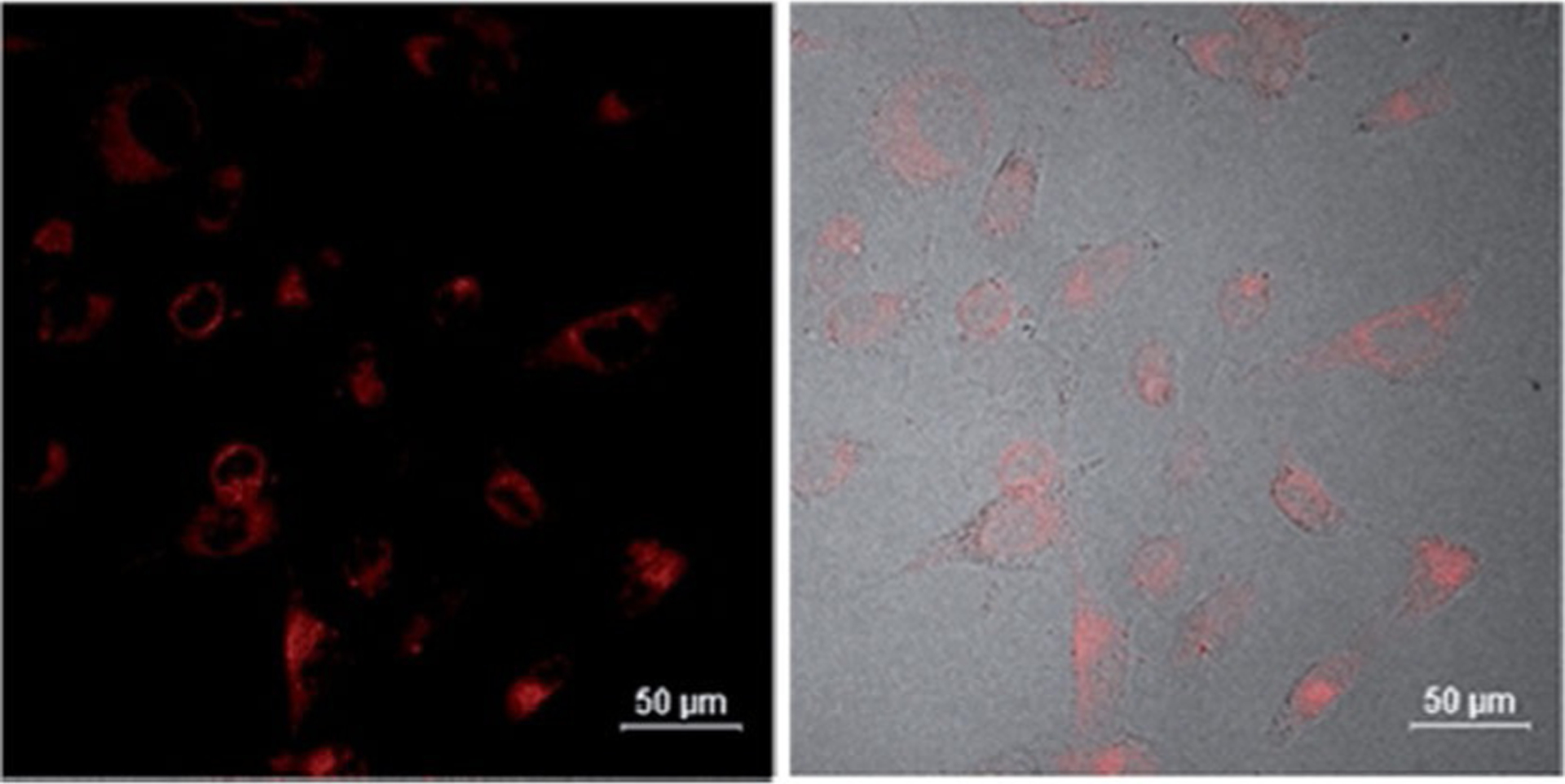
Confocal fluorescence microscopic images of A172 cells incubated with dye 38 at concentration 20 μmolL−1 at 37 °C for 10 min.[15]
Another dimethine cyanine SLM 40 (Figure 11) was investigated as an excellent theranostic agent for diagnosis and therapy of Alzheimer’s disease (AD). This compound has been successfully applied to perform near infrared in vivo imaging of Aβ species in transgenic (Tg) and wild-type (WT) mice in vivo (Figure 20). Dye 40 strongly binds to the Aβ species in the brain of Tg mice, and as a result, the mice can recover from Alzheimer’s disease. Moreover, SLM-treated mice have shown a considerable reduction in each of oligomeric Aβ content and τ proteins in their brain. These findings prove that dye 40 is a good theranostic agent with in vivo effectualness for diagnosing and treatment of AD in mouse models.[18] Dye SLOH 41 (Figure 11) also shows potential as a therapeutic agent for Alzheimer’s disease, inhibiting the aggregation of Aβ peptides and oligomers.[88]
Figure 20.
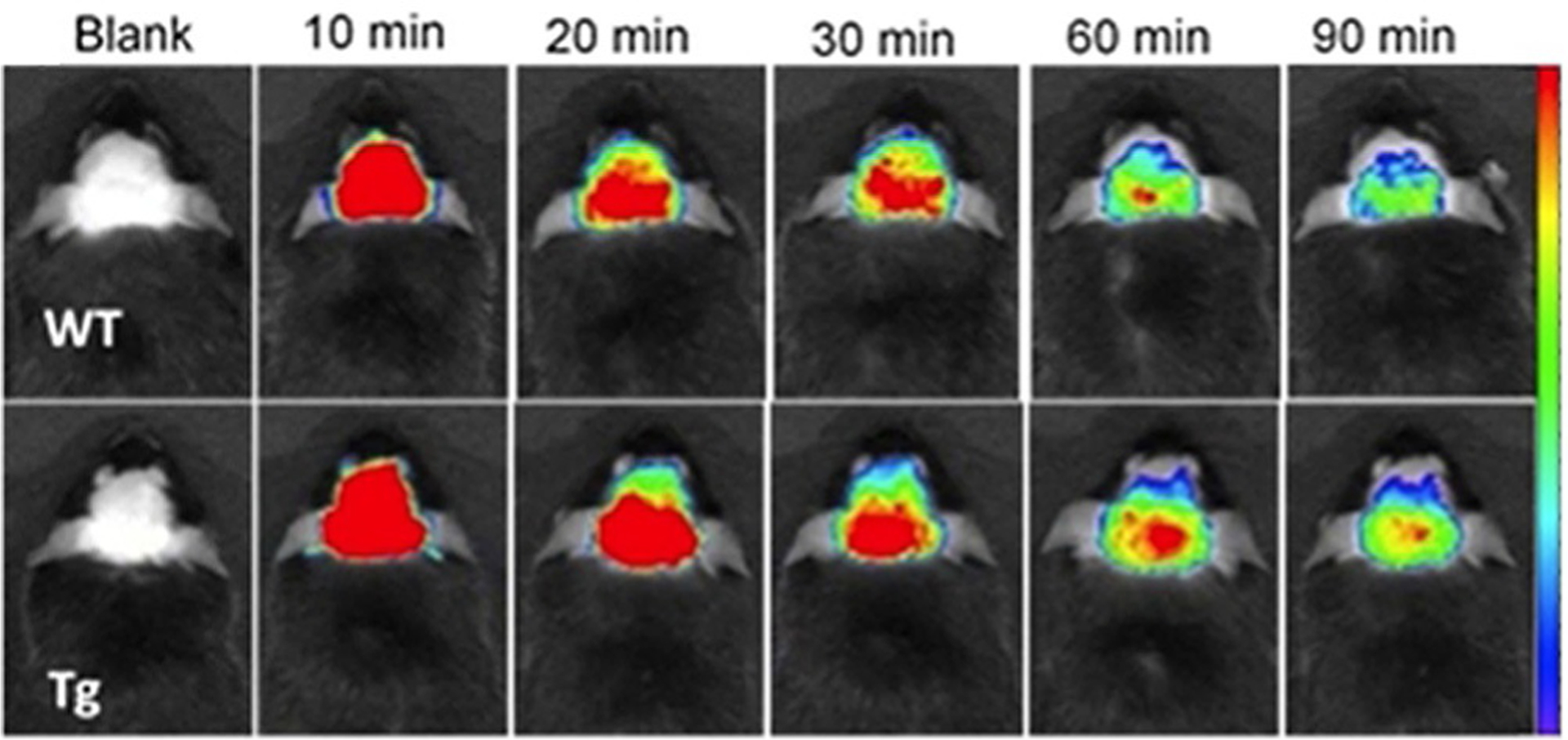
Fluorescence images of Tg (9 months old) and wild-type mice at different time points before (blank) and after intravenous injection of SLM 40 (dosage of 5 mgkg−1) with recordings at 10, 20, 30, 60, 90 min.[18]
Intensity properties for labeling living cells with low cell cytotoxicity and little photobleaching are shown by dyes 42 and 43 (Figure 11).[58] The report says that these dyes can be applied as fluorescent probes in flow cytometry and as double staining agents for measuring sperm cell viability.[58] Figure 21 illustrates the fluorescence intensity profile of the sperm cells stained with dyes 42 and 43.
Figure 21.
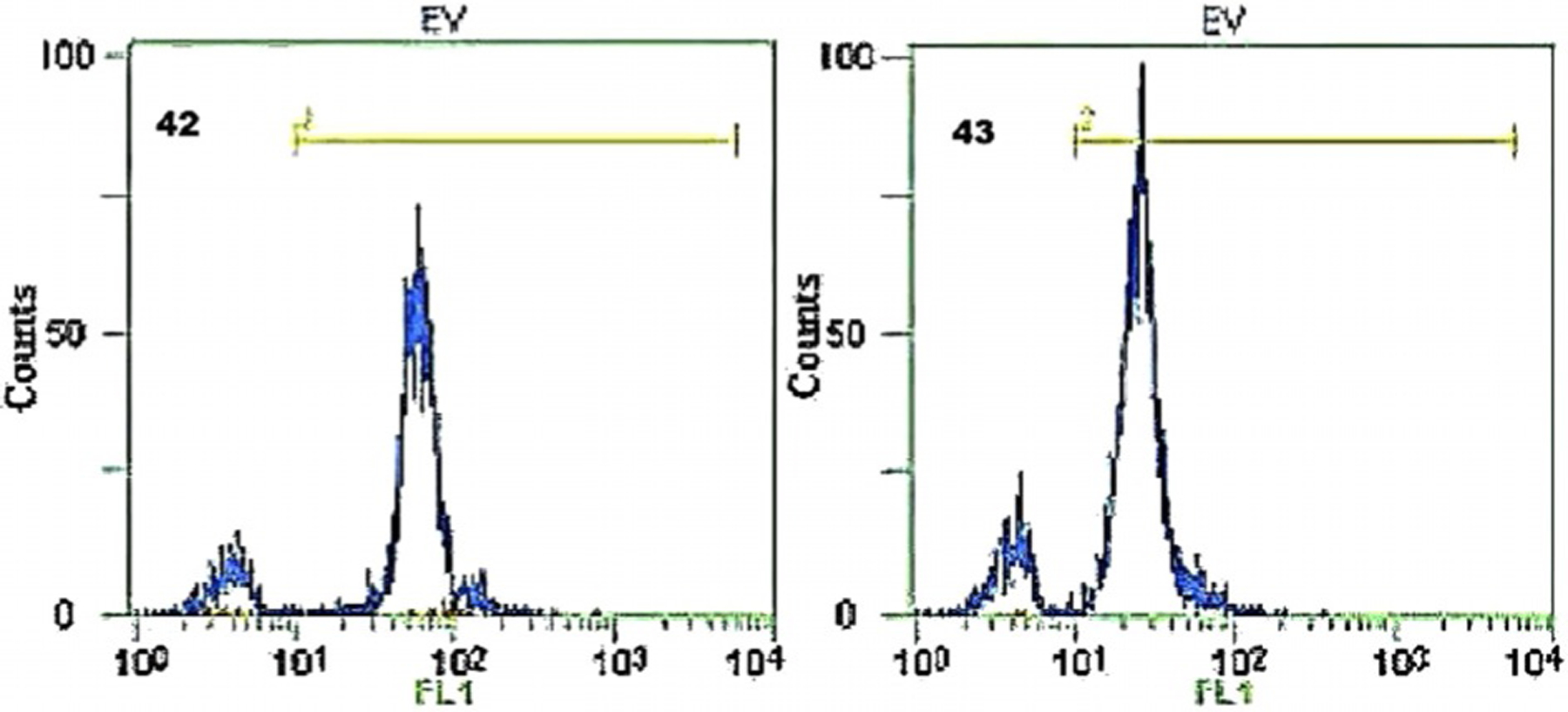
Flow cytometry histograms of dyes 42 and 43. The x-axis presents the fluorescence intensity of stained sperm cells, and the y-axis shows the number of the stained sperm cells.[58]
As previously mentioned, Sensitol Red (Figure 7), a symmetrical trimethine cyanine dye, was widely used as a photosensitizer in photography and against staphylococci and B. coli. Another symmetrical trimethine cyanine dye Kryptocyanine (Figure 12) after study in red blood cell (RBC) membranes and isolation of mitochondria, shows the ability to be useful for photochemotherapy in tumors, as a light-activated cytotoxic agent, and for photodestruction of leukemic cells.[89]
Dyes 55 (TO-3) and 57 (TO-PRO-3; Figure 13) are trimethine analogs of TO dyes 15 (TO) and 16 (TO-PRO-1; Equation (3)). Compared with TO-1, the TO-3 and TO-PRO-3 contain an extended carbomethine bridge that considerably shifts the absorption and emission to the red at 642 and 661 nm, respectively. Like TO, TO-PRO-3 strongly binds to nucleic acids with a high fluorescence enhancement. Moreover, the dye can bind to human cytoplasmic, and mitochondrial A-site RNAs.[90] TO-PRO-3 is also used in the pretreatment of samples for gel or capillary electrophoresis, determination of viability and resistance to staining in experiments with several labels.[90]
TO-PRO-3 (57, Figure 13), the same as TO-PRO-1 (16, Equation (3)), has a dimeric analog, TOTO-3 (Figure 22). TOTO-3 has the same absorption and emission wavelengths, strongly stains the cytoplasmic and nucleolar RNAs but weakly stains the nuclear DNA.[91] YO-PRO-3 (58, Figure 13) has a dimeric analog YOYO-3 (Figure 22), which has the same optical properties and applications for DNA staining.
Figure 22.
Structure of the dimers TOTO-3 and YOYO-3.
According to the collected properties of TO and YO dicationic monomers and tetracationic dimers (Figure 23), monomers’ absorption and emission wavelengths correspond to the wavelengths of their dimers. TO derivatives’ wavelengths, differ from YO derivatives by 22–30 nm. Stokes shifts of all considered dyes (Table 2) lie in the range 16–19 nm. Molar extinction coefficients increase with the extension of chain length and number of cations. The dimers have bright fluorescence signals and may be used as nuclear and chromosome counterstains for multicolor fluorescence labeling experiments and for staining nucleic acids on solid supports, such as microarrays.[92]
Figure 23.
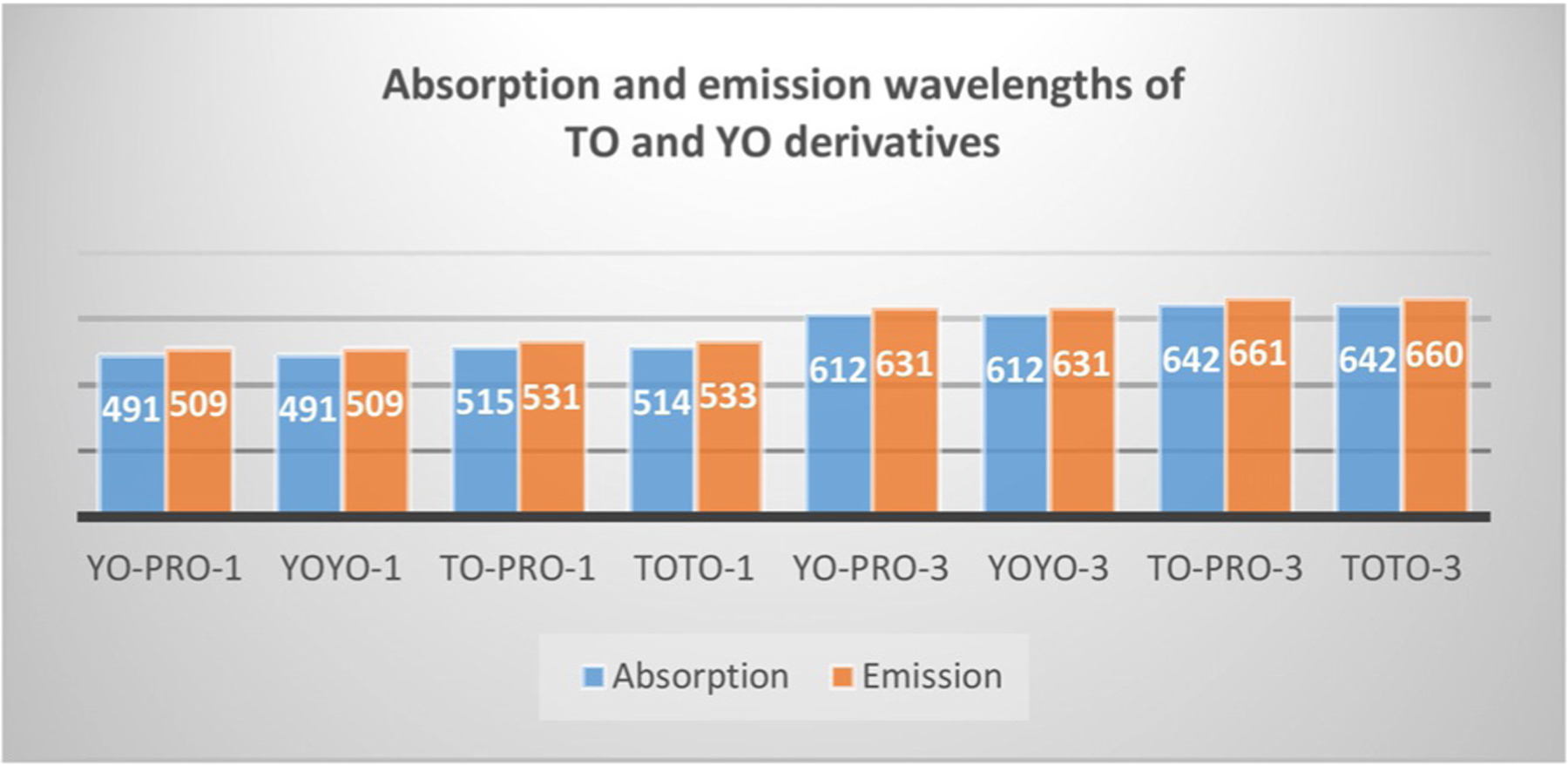
Absorption and emission wavelengths of TO and YO monomers and dimers.
Table 2.
The spectral characteristics of monomers YO-PRO-1 (18, Equation (3)), TO-PRO-1 (16, Equation (3)), YO-PRO-3 (58, Figure 13), TO-PRO-3 (57, Figure 13), and their dimers YOYO and TOTO (Figure 18), and YOYO-3 and TOTO-3 (Figure 22).[92]
| Dye | λabs [nm] | λem [nm] | Stokes shift [nm] | ε [× 10−4 Lmol−1 cm−1] |
|---|---|---|---|---|
| YO-PRO-1 | 491 | 509 | 18 | 5.2 |
| YOYO-1 | 491 | 509 | 18 | 9.9 |
| TO-PRO-1 | 515 | 531 | 16 | 6.3 |
| TOTO-1 | 514 | 533 | 19 | 11.7 |
| YO-PRO-3 | 612 | 631 | 19 | 10 |
| YOYO-3 | 612 | 631 | 19 | 16.7 |
| TO-PRO-3 | 642 | 661 | 19 | 10.2 |
| TOTO-3 | 642 | 660 | 18 | 15.4 |
Another trimethine dye 63 Dimethyl Indole Red (DIR; Scheme 11), exhibits a weak visible fluorescence when an excess quantity of calf thymus (CT) DNA and artificial dsDNA is present, thereby making it potential for specific light-up probes of G-quadruplexes emitting long wavelength.[59] The study of the conformational switch of G-quadruplex structure within the absence and presence of 63 has been explored by circular pleochroism (CD) spectroscopic analysis. The CD spectra of the parallel G-quadruplex exhibits a specific positive peak at 265 nm and a negative peak at 240 nm (Figure 24). Additionally, no elicited CD signal peaks within the wavelength longer than 350 nm can be detected. The probe DIR (63) has a negligible impact on the production of G-quadruplex structures. Such a G-quadruplex probe can disclose the distribution of intracellular shaped G-quadruplexes and avoid the controversy regardless of whether the detectable G-quadruplexes are present.
Figure 24.
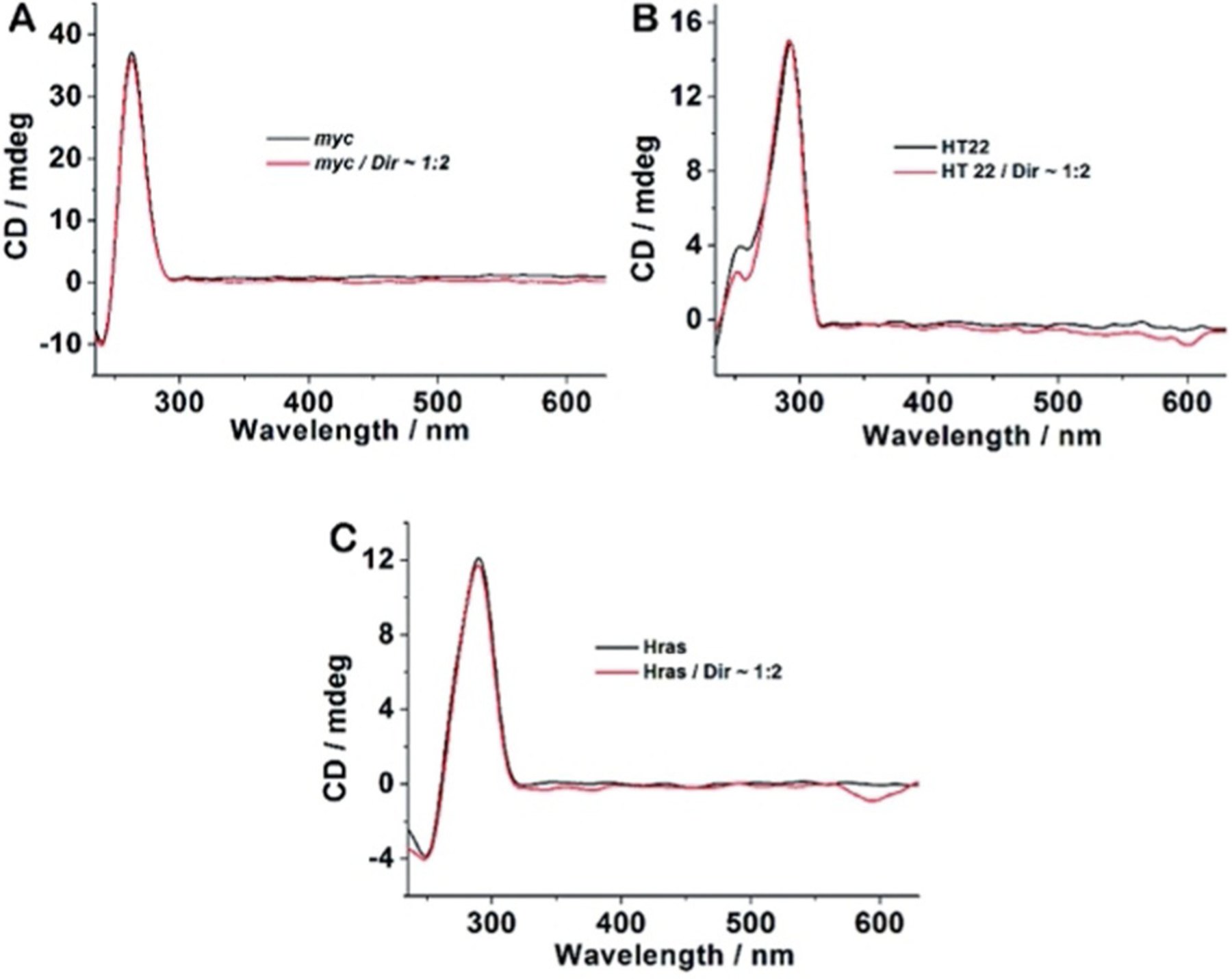
CD spectra of 4 μm G-quadruplex-forming oligonucleotides c-myc (A), HT 22 (B), and Hras (C) in the absence and presence of DIR 63 (8 μm) in 20 mm Tris-HCl buffer with 100 mm KCl, pH 7.4.[93]
For fluorophores 65–68 shown in Scheme 12, the effect of various substituent groups on the absorption and emission maxima can be seen in Figure 25. Absorption maxima of dyes 65–68 (Figure 25A) lies between 599 and 603 nm, whereas absorption of dye 68 with R=CH3 redshifts by 9–12 nm. In addition, intensities of Cl-substituted dyes 66 and 67 are higher than that of 65 (R=H) and 68 (R=CH3).
Figure 25.

UV/Vis (A) and fluorescence (B,C) spectra of dyes 65–68 excited at 480 nm (B) and 550 nm (C).[65]
According to Figure 25B, each curve has two peaks with the utmost emission wavelengths of indole quinoline dyes at 650 nm. The fluorescence intensity decreases in the order 67> 66>65>68. This means that the utmost peaks of dyes with electron-withdrawing groups redshift compared with those dyes with electron-donating groups. The second peaks were completely different, and the fluorescence intensity of 65 was stronger than the others.
The fluorescence spectra of compounds excited at 550 nm are illustrated in Figure 25C. All fluorophores show similar fluorescent characteristics. There is only one peak in each line, and the fluorescence intensities of all compounds are higher than those excited at 480 nm. Most emission wavelengths are set between 647 nm and 657 nm. The Stokes shifts of dyes 65–68 are all around 100 nm. The longer wavelength signs and larger Stokes shifts indicate low UV interference when labeling the organism.[65]
Three identical symmetrical dyes, differing only by the number of carbons in the chain, such as monomethine Ethyl Red (Figure 7), trimethine Kryptocyanine (Figure 12), and pentamethine Dye 79 (Figure 15), have absorption wavelengths at around 600 nm, 700 nm, and 800 nm, respectively (Figure 26). Emission wavelengths also lie in the range 600–830 nm (Figure 26). Meanwhile, absorption and emission wavelengths for identical mono-, tri-, and pentamethine cyanines, PIC (Figure 7), Sensitol Red (Figure 12), and dye 83 (Figure 15), connected to the linker at the 2-position (instead of the 4-position) are 100 nm less for each one compared with the same ones at the 4-position and lie in the ranges 500–700 nm and 550–750 nm, respectively (Figure 27). This means that lepidine-containing cyanine dyes have higher excitation and emission wavelengths owing to the presence of an extra double bond between the nitrogen atoms of the heterocyclic rings.
Figure 26.
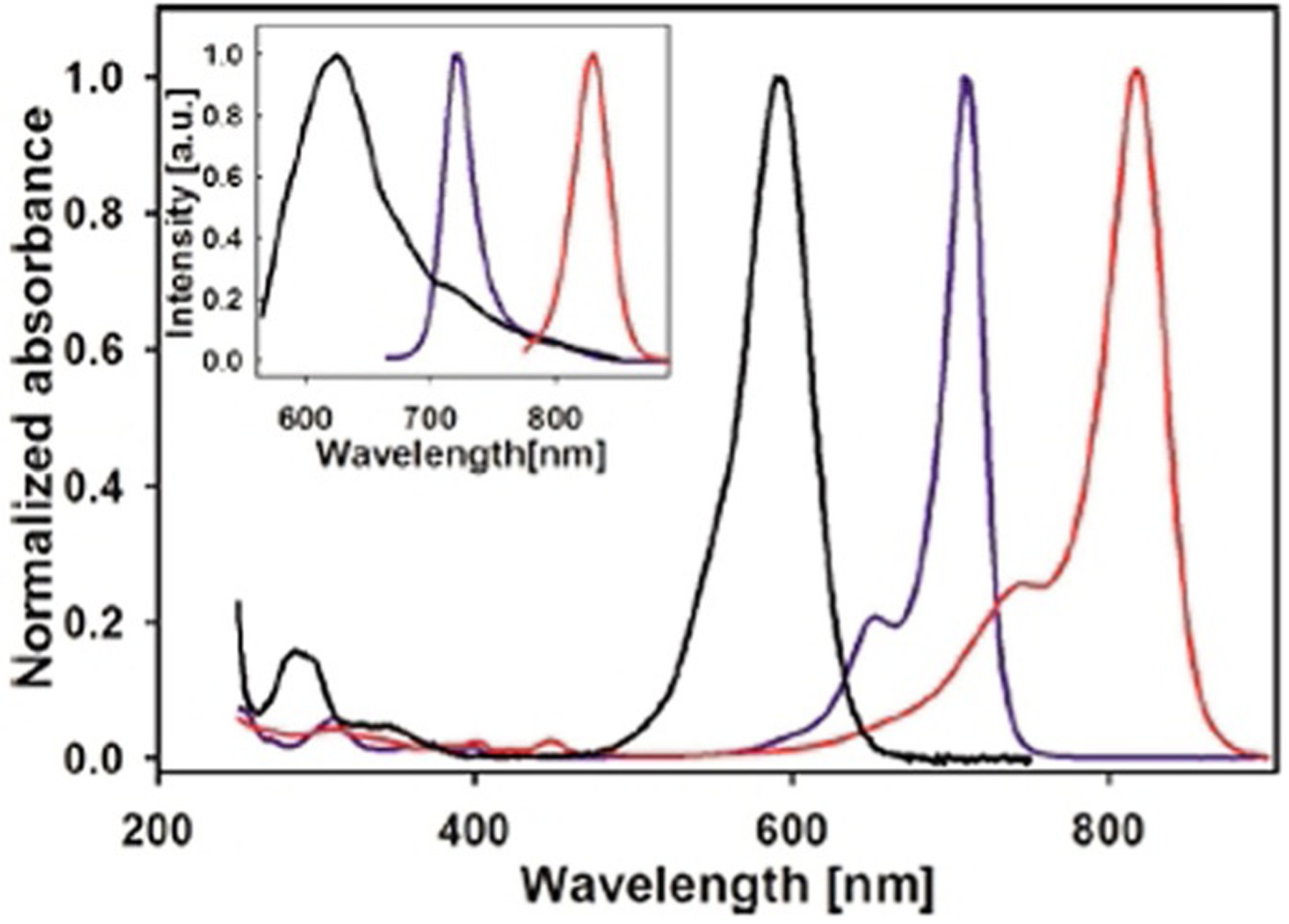
Absorption (main graph) and emission (inset) spectra of three symmetrical dyes: Ethyl Red (black), Kryptocyanine (blue), and 79 (red).[68]
Figure 27.
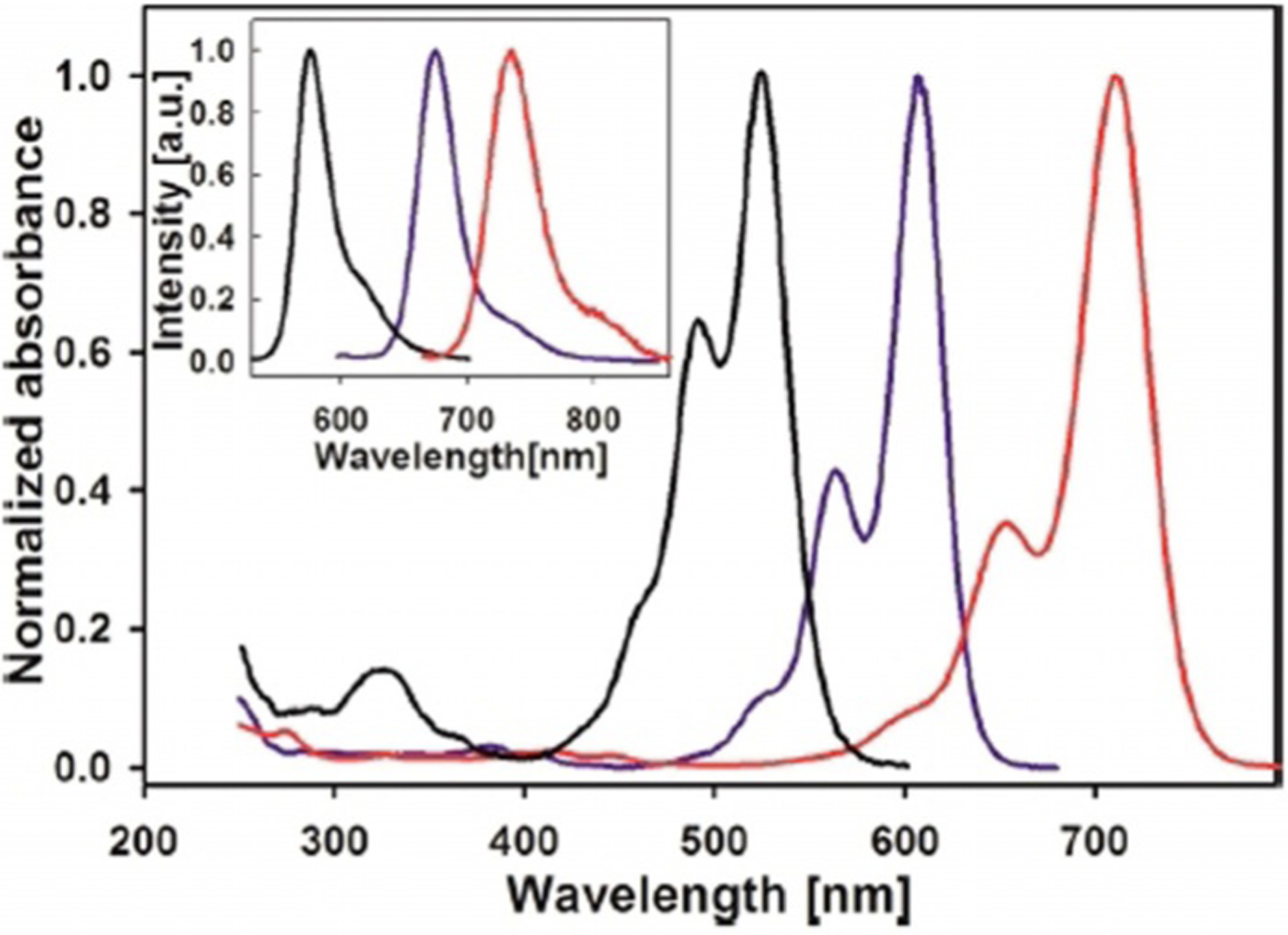
Absorption (main graph) and emission (inset) spectra of three symmetrical dyes: pseudoisocyanine (black), Sensitol Red (blue), and 83 (red).[68]
Four other dyes differ by the position of the chain connection to the moiety (at the 2- and 4-positions) and substitution at the meso carbon. Comparison of two symmetrical 4-methylquinoline based pentamethine dyes with meso-substitution of H (77) and Cl (78) in the polymethine bridge is illustrated in Figure 15, resulting in the 32–90 nm difference in the UV/Vis wavelength in DMSO, buffer, and buffer/DNA (Figure 28). In the presence of DNA, dye 78 shows high stabilization, in contrast to dye 77, owing to the presence of the electron-with-drawing chlorine atom. After the addition of CT DNA to dye 78, the absorption maximum of the dye increases to 805 nm. As a result, dye 78 shows excellent DNA photocleaving properties. Likewise, this dye was used to treat ES2 cancer cells and exposed to an 808 nm laser diode. The results showed the increase of intracellular reactive oxygen species (ROS) levels in the cells and the decrease of their viability (Figure 29).[16, 94]
Figure 28.

UV/Vis spectra of 10 μm of dyes 77 (left) and 78 (right): in DMSO, 10 mm sodium phosphate buffer, pH 7.0, and in buffer with 150 μmbp CT DNA.[16]
Figure 29.

(A) Superimposed fluorescence microscopy image revealing the intracellular localization of dye 78 (red) in ES2 cancer cells after 24 h incubation with the subsequent staining of nuclei Hoechst 33342 (blue). (B) ES2 cancer cell viability: with no treatment, under 808 nm laser exposure, after dye 78 incubation in the dark, after dye 78 incubation under 808 nm laser exposure.[16]
Fluorophores connected to the linker at the 2-position with meso-substitution of Br (81) and H (82) show similar results with stability in DMSO. With the addition of CT DNA, the absorbance of dye 81, the same as 78, stays constant, whereas dye 82 noticeably decreases even before DNA inclusion (Figure 30). The electron-withdrawing bromine atom allows the dye to stay stable with no autooxidation in aqueous solutions. After showing promising photocleaving properties, dye 81 was incubated with ES 2 ovarian carcinoma cells and shows ready uptake by cells and quick localization in the intracellular cytosolic and perinuclear regions (Figure 31).[94] Also, dye 81 shows high phototoxicity, decreasing ES 2 cells viability under 694 nm light irradiation.
Figure 30.
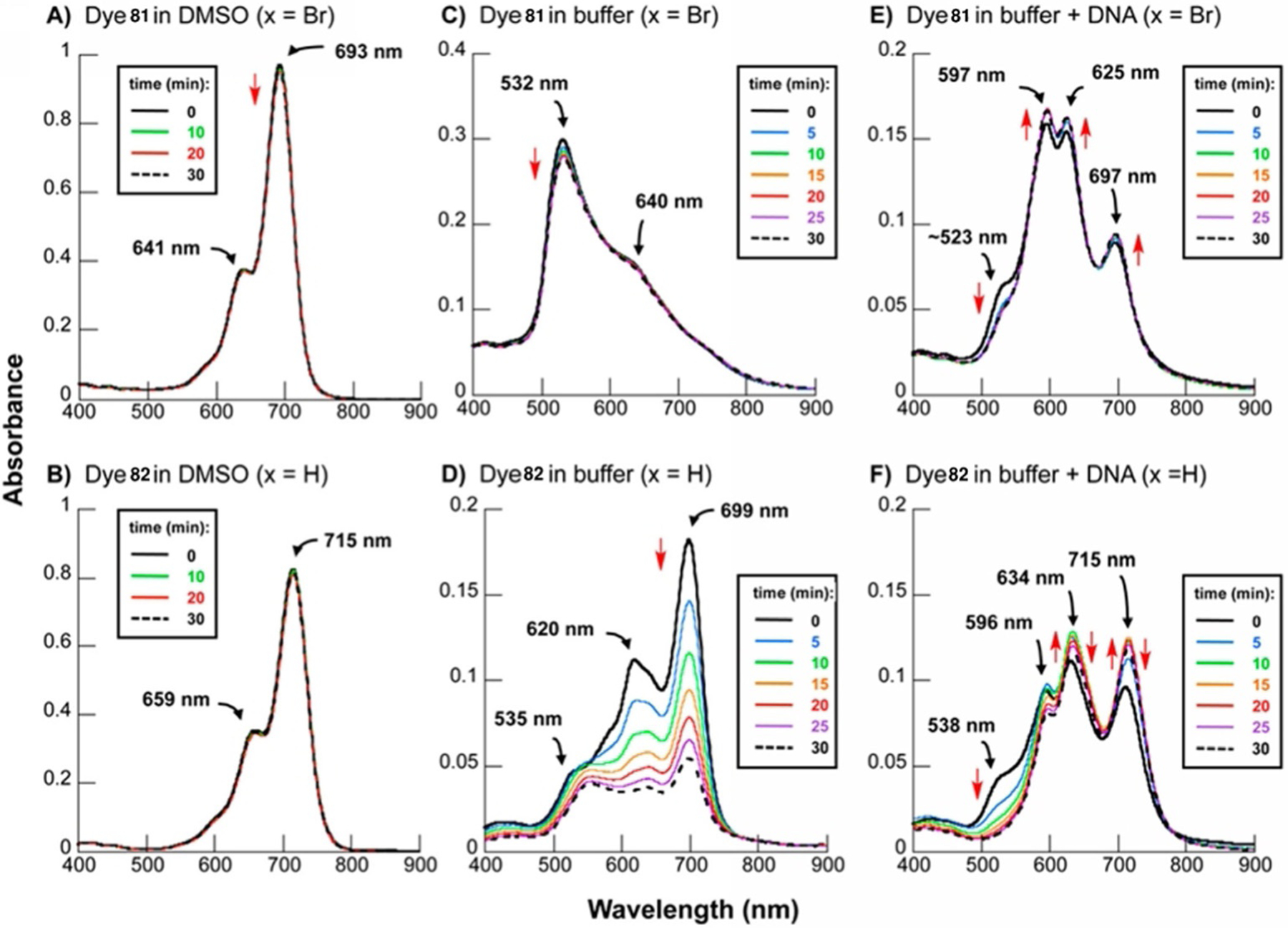
UV/Vis spectra of dyes 81 (top) and 82 (bottom) in DMSO (A, B), buffer pH 7.0 (C,D), and buffer pH 7.0 with 150 μm CT DNA (E,F).[94]
Figure 31.
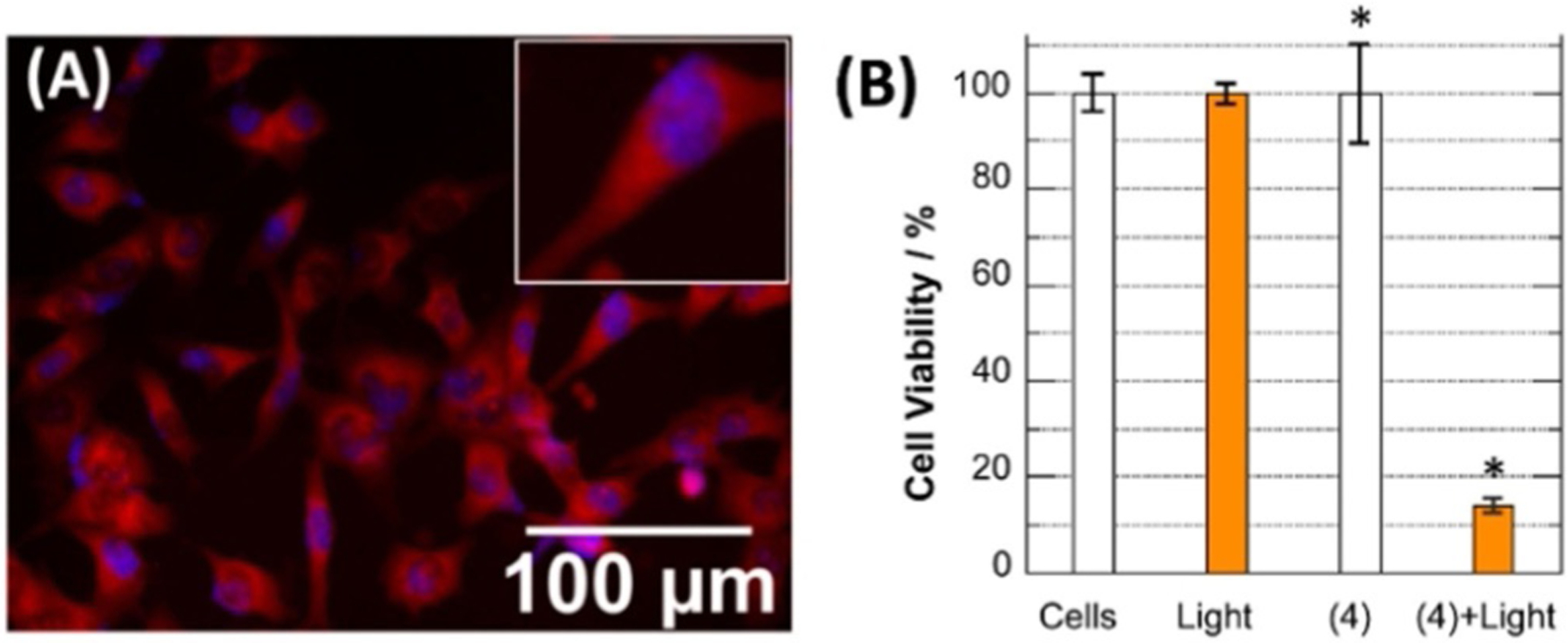
(A) Superimposed fluorescence microscopy image revealing intracellular localization of dye 81 (red) in ES2 ovarian cells after 24 h incubation with the subsequent staining of nuclei Hoechst 33342 (blue). (B) ES2 cancer cell viability: with no treatment, under 694 nm laser exposure, after dye 81 incubation in the dark, after dye 81 incubation under 694 nm laser exposure.[16]
As was mentioned earlier, heptamethine cyanine dyes are near-infrared (NIR) absorbing and emitting. This allows them to be applicable in photovoltaics and nonlinear optics and in vivo cell imaging. Heptamethine dyes 96 and 97 (Scheme 16) were synthesized with a chlorine atom and phenyl at the meso carbon, respectively. The neat thin-film absorption spectra of dyes and in CH2Cl2 solutions are presented in Figure 32. Interestingly, that phenyl substitution leads to the blueshift in CH2Cl2 compared with chloride substitution, owing to the inductive effect, whereas the neat thin film leads to a redshift.
Figure 32.
The absorption spectra for dyes 96 (black) and 97 (red) in CH2Cl2 (left) and as neat thin film (right).[71]
Heptamethine cyanine dye 99 (Scheme 17) has been investigated for application in dye-sensitized solar cells (DSCs). Figure 33 presents the absorption spectra of dye 99 and AN50 electrolyte as well as the transmission spectra of the DSC. Transmittance in the visible range 400–700 nm is achievable. Dye 99 has the high potential for application in DSCs sensitization.[74]
Figure 33.
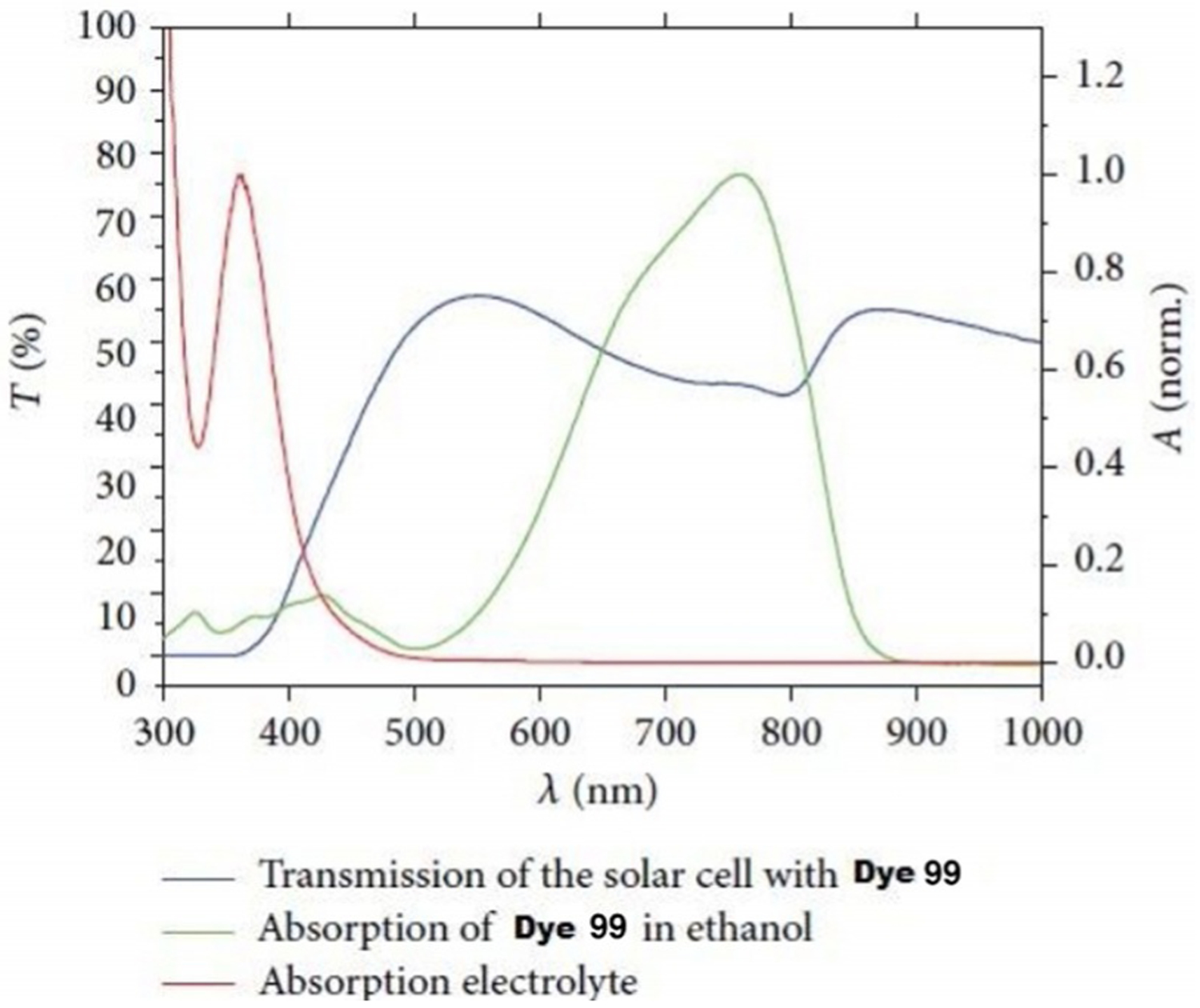
Transmission of DSC with dye 99.[74]
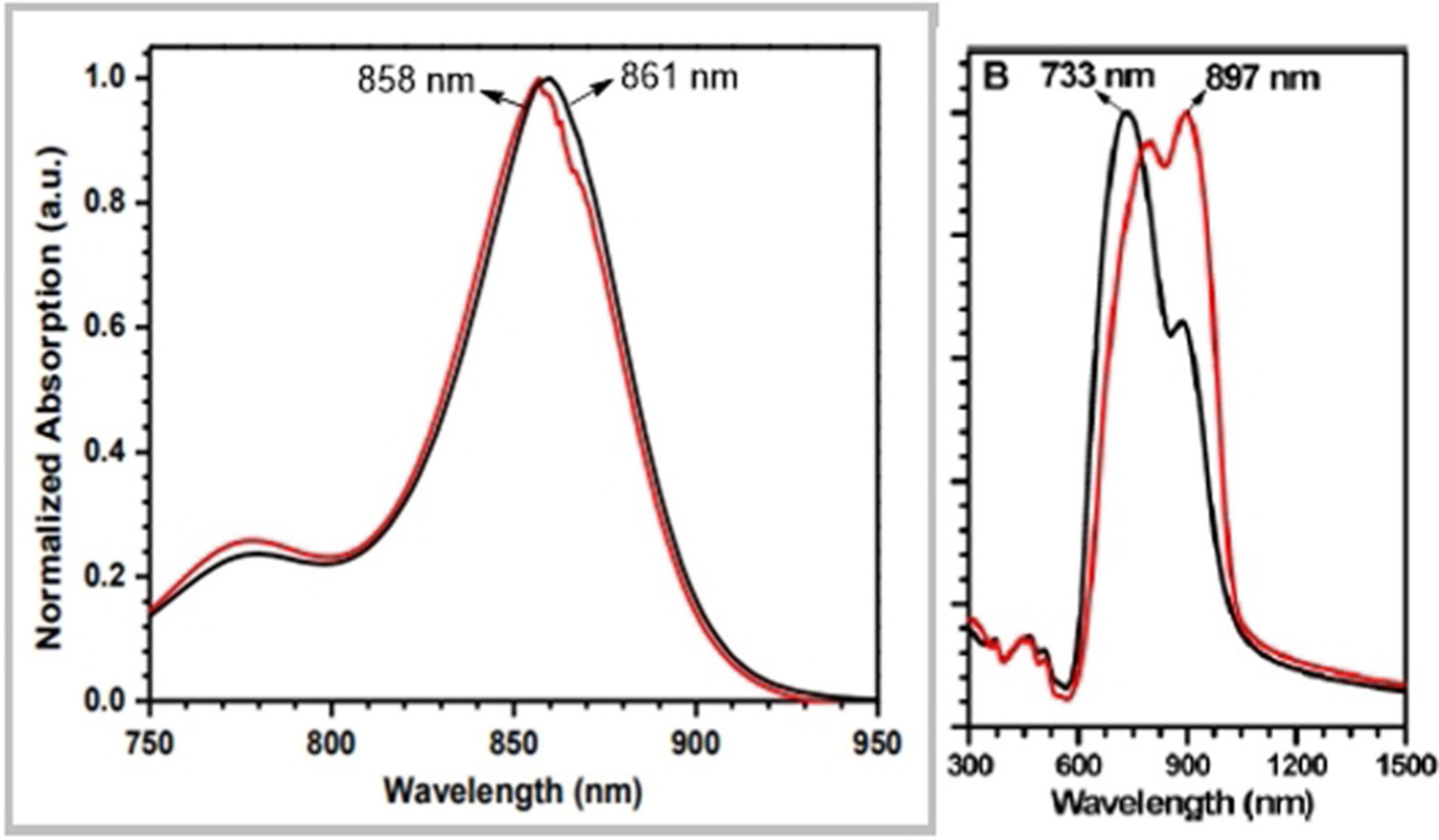
Fluorophore 101 (Scheme 18) was analyzed by Shimizu and co-workers after encapsulation in a lactosome and evaluated as a NIR fluorescent probe for in vivo tumor imaging.[17] The optical properties of dye 101 and its lactosome are shown in Table 3. The dye has a maximum emission wavelength in methanol at 932 nm, whereas its lactosome shows an emission maximum at 905 nm. Interestingly, the absorption maximum of dye lactosome in water is higher than of the dye itself in organic solvents. The excitation and emission wavelengths are higher for dye 101 in methanol. As for photostability, the absorbance of lactosome is stable within 1 h under tungsten lamp irradiation.
Table 3.
Optical properties of dye 101 and its lactosome.[17]
| Entry | Solvent | Abs max | Ex max | Em max | ε [× 10−4 Lmol−1 cm−1] |
|---|---|---|---|---|---|
| Dye 101 | chloroform | 781 | 805 | 897 | 9.2 |
| methanol | 790 | 883 | 932 | 2.6 | |
| Dye 101 lactosome | water | 830 | 844 | 905 | 6.0 |
After injection of dye 101 lactosome into a mouse, the fluorescence intensity in the tumor region becomes visible and gradually grows 6 and 24 h after injection (Figure 34). The developed dye 101 lactosome probes, possessing low toxicity, good photostability, high fluorescence in the tumor, and low background signals in liver and muscle tissue, have been suggested for application in noninvasive in vivo imaging techniques to detect tumors.[17]
Figure 34.
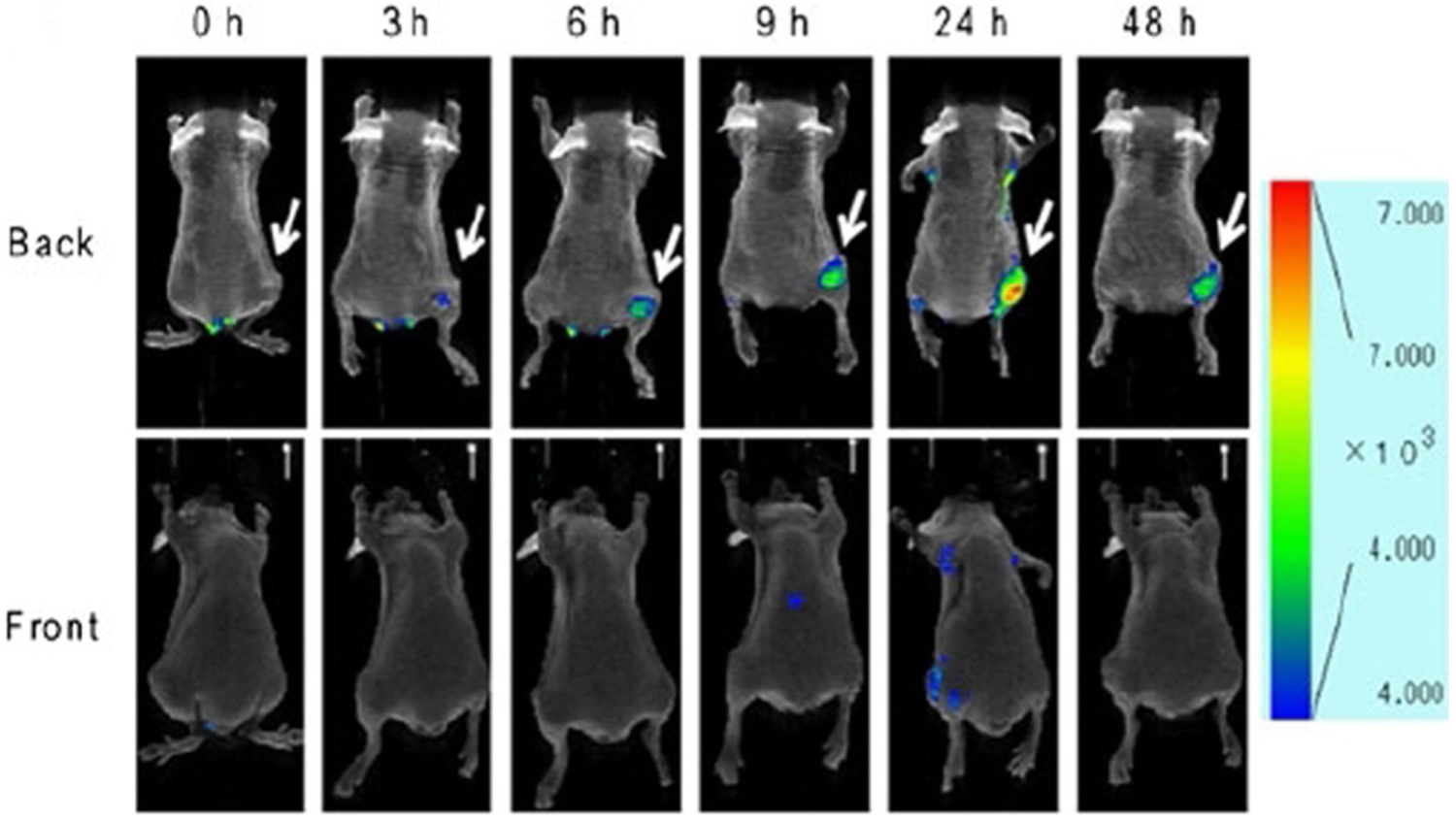
Back (top) and front (bottom) sides of mouse fluorescence images of FM3A cell xenografted mouse after injection of dye 101 lactosome, recorded at 0, 3, 6, 9, 24, and 48 h.[17]
Summary and Outlook
The cyanine fluorophores containing quinoline moieties are worthy of attention because of their optical properties such as stability, high molar extinction coefficients, high sensitivity, and low aggregation. Therefore, they are useful for biomedicine, as they possess antiseptic and photocleaving properties.
They have always had widespread applications in various fields such as pigmenting and photosensitizing, biomedical and pharmaceutical, and as probes in biomolecule labeling, detection of nucleic acids and tumors, and potential imaging agents.
This review summarizes the synthetic procedures with different initial compounds with no solvent, using solid-phase resin support, or microwave irradiation. Even though the preparation of these dyes follows traditional methodologies, the synthetic routes are very efficient to produce these dyes in high yields.
Based on the valuable optical properties and various applications of these fluorophores, as presented and discussed in this review, it is evident that these dyes have great potential for many useful applications. With further development of cyanines containing quinoline moieties, the forthcoming improvements and refinements of their structures, it would be possible to widen the practical applications of these dyes based on their excellent properties.
Acknowledgments
The authors would like to thank the Department of Chemistry at Georgia State University. This study was supported by grants from the Georgia State University Brains, Behavior Seed Grant, The Atlanta Clinical and Translational Science Institute Healthcare Innovation Seed Grant, The Georgia Research Alliance Ventures Phase 1 Grant, and The US NIH NIBIB # R01-EB022230.
Biographies

Kristina Ilina received her M.Sc. in chemical engineering from Volgograd State Technical University in 2016. She then moved to Atlanta, USA, and in 2020, she obtained her M.Sc. in organic chemistry from Georgia State University under the direction of Professor Maged Henary. Kristina worked on synthesis of symmetrical pentamethine cyanine dyes containing quinoline moieties for their potential use in photodynamic therapy.

Maged Henary is an Associate Professor in the Chemistry Department at Georgia State University (GSU). Dr. Henary earned his B.Sc. in chemistry from Alexandria University, Egypt, his M.Sc. in organic chemistry from Cairo University and his Ph.D. in medicinal chemistry from GSU. His research focuses on the synthesis of heterocyclic compounds and NIR dyes as anticancer and imaging agents. The NIH, the Health Innovation, and the Venture Laboratory Grants support Henary’s research.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/chem.202003697.
References
- [1].Ullah S, Ahmad I, Alias Y, Yusoff I, Ashraf MA, J. Chem 2013, 136908. [Google Scholar]
- [2].Ma X, Laramie M, Henary M, Bioorg. Med. Chem. Lett 2018, 28, 509–514. [DOI] [PubMed] [Google Scholar]
- [3].Henary M, Paranjpe S, Owens EA, Heterocycl. Commun 2013, 19, 1–11. [Google Scholar]
- [4].Henary M, Levitz A, Dyes Pigm. 2013, 99, 1107–1116. [Google Scholar]
- [5].Visible/Infrared Spectroscopy (VIRS) as a Research Tool in Economic Geology: Background and Pilot Studies from Newfoundland and Labrador (Eds.: Kerr A, Rafuse H, Sparkes GW, Hinchey J, Sandeman H), Current Research Newfoundland and Labrador Department of Natural Resources Geological Survey, Report 11–1, 2011, pp. 145–166. [Google Scholar]
- [6].Mojzych M, Henary M, Synthesis of Polymethine Dyes, Topics in Heterocyclic Chemistry, Springer, Heidelberg, 2008, Vol. 14, pp. 1–9. [Google Scholar]
- [7].Johnson EW, Chem. Inf. Bull 1984, 36, 13. [Google Scholar]
- [8].Shindy HA, Dyes Pigm 2017, 145, 505–513. [Google Scholar]
- [9].Hamer FM, The Chemistry of Heterocyclic Compounds, Wiley, Hoboken, 2009. [Google Scholar]
- [10].Brooker LGS, J. Am. Chem. Soc 1965, 87, 937–938. [Google Scholar]
- [11].Vogel HW, Ber. Dt. Chem. Ges 1873, p. 1320. [Google Scholar]
- [12].Shelkovnikov VV, Plekhanov AI, Optical and Resonant Non-Linear Optical Properties of J-Aggregates of Pseudoisocyanine Derivatives in Thin Solid Films in Macro To Nano Spectroscopy (Ed.: Uddin J), IntechOpen, London, 2012. [Google Scholar]
- [13].Clark M, Handbook of Textile and Industrial Dyeing, Woodhead, Cambridge, 2011. [Google Scholar]
- [14].Wainwright M, Kristiansen JE, Int. J. Antimicrob. Agents 2003, 22, 479–486. [DOI] [PubMed] [Google Scholar]
- [15].Jia H, Lv Y, Wang S, Sun D, Wang L, RSC Adv. 2015, 5, 4681–4692. [Google Scholar]
- [16].Basnet K, Fatemipouya T, St. Lorenz A, Nguyen M, Taratula O, Henary M, Grant KB, Chem. Commun 2019, 55, 12667–12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shimizu Y, Temma T, Hara I, Yamahara R, Ozeki E.-i., Ono M, Saji H, J. Fluoresc 2012, 22, 719–727. [DOI] [PubMed] [Google Scholar]
- [18].Li Y, Chen C, Xu D, Poon C-Y, Ho S-L, Zheng R, Liu Q, Song G, Li H-W, Wong MS, ACS Omega 2018, 3, 6812–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Czaplinska B, Spaczynska E, Musiol R, Med. Chem 2018, 14, 19–33. [DOI] [PubMed] [Google Scholar]
- [20].Collin GH, Quinoline and Isoquinoline in Ullmann’s Encyclopedia of Industrial Chemistry, Vol. 31, 2000, Wiley, New York, pp. 1–4. [Google Scholar]
- [21].Denmark SE, Venkatraman S, J. Org. Chem 2006, 71, 1668–1676. [DOI] [PubMed] [Google Scholar]
- [22].Jones G, Quinolines, Vol. 32, Wiley, New York, 1977. [Google Scholar]
- [23].Al-Rehaily AJ, Ahmad MS, Muhammad I, Al-Thukair AA, Perzanowski HP, Phytochemistry 2003, 64, 1405–1411. [DOI] [PubMed] [Google Scholar]
- [24].Chen I-S, Tsai I-L, Wu S-J, Sheen W-S, Ishikawa T, Ishii H, Phytochemistry 1993, 34, 1449–1451. [Google Scholar]
- [25].Achan J, Talisuna AO, Erhart A, Yeka A, Tibenderana JK, Baliraine FN, Rosenthal PJ, D’Alessandro U, Malar. J 2011, 10, l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gorka AP, Dios A. de, Roepe PD, J. Med. Chem 2013, 56, 5231–5246. [DOI] [PubMed] [Google Scholar]
- [27].Ji Ram V, Sethi A, Nath M, Pratap R, Six-Membered Heterocycles in The Chemistry of Heterocycles, Elsevier, Amsterdam, 2019, Chapter 2, pp. 3–391. [Google Scholar]
- [28].Sivaprasad G, Rajesh R, Perumal PT, Tetrahedron Lett. 2006, 47, 1783–1785. [Google Scholar]
- [29].Li J, Zhang J, Yang H, Jiang G, J. Org. Chem 2017, 82, 3284–3290. [DOI] [PubMed] [Google Scholar]
- [30].Ushakov EN, Vedernikov AI, Lobova NA, Dmitrieva SN, Kuz’ mina LG, Moiseeva AA, Howard JA, Alfimov MV, Gromov SP, J. Phys. Chem. A 2015, 119, 13025–13037. [DOI] [PubMed] [Google Scholar]
- [31].Dolgosheina EV, Jeng SC, Panchapakesan SS, Cojocaru R, Chen PS, Wilson PD, Hawkins N, Wiggins PA, Unrau PJ, ACS Chem. Biol 2014, 9, 2412–2420. [DOI] [PubMed] [Google Scholar]
- [32].Zhang X-H, Wang L-Y, Nan Z-X, Tan S-H, Zhang Z-X, Dyes Pigm. 2008, 79, 205–209. [Google Scholar]
- [33].Patrick MJ, Ernst LA, Waggoner AS, Thai D, Tai D, Salama G, Org. Biomol. Chem 2007, 5, 3347–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Martins CT, Lima MS, El Seoud OA, J. Phys. Org. Chem 2005, 18, 1072–1085. [Google Scholar]
- [35].Karthigha S, Kalainathan S, Hamada F, Yamada M, Kondo Y, RSC Adv. 2016, 6, 33159–33169. [Google Scholar]
- [36].Cong Z, Li Y, Xia G, Shen S, Sun J, Xu K, Jiang Z, Jiang L, Chen Y, Yu Q, Wang H, Dyes Pigm. 2019, 162, 654–661. [Google Scholar]
- [37].Alganzory HH, Arief MMH, Amine MS, Ebeid E-Z, J. Chem. Pharm. Res 2014, 6, 143–161. [Google Scholar]
- [38].Wang HC, Cai F, Zhou L, Li D, Feng D, Wei Y, Feng Z, Gu X, Li X, Wu Y, Polyhedron 2019, 170, 440–446. [Google Scholar]
- [39].Tung C-H, Pept. Sci 2004, 76, 391–403. [DOI] [PubMed] [Google Scholar]
- [40].Gonçalves MST, Chem. Rev 2009, 109, 190–212. [DOI] [PubMed] [Google Scholar]
- [41].Stadler AL, Delos Santos JO, Stensrud ES, Dembska A, Silva GL, Liu S, Shank NI, Kunttas-Tatli E, Sobers CJ, Gramlich PME, Carell T, Peteanu LA, McCartney BM, Armitage BA, Bioconjugate Chem. 2011, 22, 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Winstead AJ, Nyambura G, Matthews R, Toney D, Oyaghire S, Molecules 2013, 18, 14306–14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang M, Du W, Tian X, Zhang R, Zhao M, Zhou H, Ding Y, Li L, Wu J, Tian Y, J. Mater. Chem. B 2018, 6, 4417–4421. [DOI] [PubMed] [Google Scholar]
- [44].Sun J, Tian M, Lin W, Analyst 2019, 144, 2387–2392. [DOI] [PubMed] [Google Scholar]
- [45].Jeong J-H, Kim J-S, Campo J, Lee S-H, Jeon W-Y, Wenseleers W, Jazbinsek M, Yun H, Kwon OP, Dyes Pigm 2015, 113, 8–17. [Google Scholar]
- [46].Neelakantan H, Wang HY, Vance V, Hommel JD, McHardy SF, Watowich SJ, J. Med. Chem 2017, 60, 5015–5028. [DOI] [PubMed] [Google Scholar]
- [47].Long L, Yuan X, Cao S, Han Y, Liu W, Chen Q, Gong A, Wang K, Chem. Commun 2019, 55, 8462–8465. [DOI] [PubMed] [Google Scholar]
- [48].Feng Y, Chi S, Zhao Y, Zhang Y, Wu Y, Analyst 2019, 144, 1245–1252. [DOI] [PubMed] [Google Scholar]
- [49].Daehne S, Resch-Genger U, Wolfbeis OS, Near-Infrared Dyes for High Technology Applications, Springer, Amsterdam, 1998. [Google Scholar]
- [50].Mahmood T, Paul A, Ladame S, J. Org. Chem 2010, 75, 204–207. [DOI] [PubMed] [Google Scholar]
- [51].Deligeorgiev T, Kaloyanova S, Vasilev A, Dyes Pigm. 2011, 90, 170–176. [Google Scholar]
- [52].Deligeorgiev TG, Gadjev NI, Drexhage K-H, Sabnis RW, Dyes Pigm. 1995, 29, 315–322. [Google Scholar]
- [53].Fei X, Yang S, Zhang B, Liu Z, Gu Y, J. Comb. Chem 2007, 9, 943–950. [DOI] [PubMed] [Google Scholar]
- [54].Fu YL, Zhang BR, Wang S, Gao XX, Wang LY, Bull. Korean Chem. Soc 2013, 34, 489–494. [Google Scholar]
- [55].Lartia R, Asseline U, Chem. Eur. J 2006, 12, 2270–2281. [DOI] [PubMed] [Google Scholar]
- [56].Kuz’mina LG, Vedernikov AI, Lobova NA, Howard JAK, Strelenko YA, Fedin VP, Alfimov MV, Gromov SP, New J Chem. 2006, 30, 458–466. [Google Scholar]
- [57].Kuz’mina LG, Churakov AV, Howard JAK, Vedernikov AI, Lobova NA, Botsmanova AA, Alfimov MV, Gromov SP, Crystallogr. Rep 2005, 50, 234–253. [Google Scholar]
- [58].Zhang XH, Liu Q, Shi HJ, Wang LY, Fu YL, Wei XC, Yang LF, Dyes Pigm. 2014, 100, 232–240. [Google Scholar]
- [59].Abd El-Aal RM, Saber NM, Mina SM, Essam ZM, Arab. J. Chem 2017, 10, 82–90. [Google Scholar]
- [60].Schwechheimer C, Rönicke F, Schepers U, Wagenknecht H-A, Chem. Sci 2018, 9, 6557–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Arndt S, Walter H-K, Wagenknecht H-A, J. Vis. Exp 2016, 113, 54121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Matsui M, Yamamoto T, Kubota Y, Funabiki K, New J Chem. 2016, 40, 10187–10196. [Google Scholar]
- [63].Peng X, Wu T, Fan J, Wang J, Zhang S, Song F, Sun S, Angew. Chem. Int. Ed 2011, 50, 4180–4183; Angew. Chem. 2011, 123, 4266–4269. [DOI] [PubMed] [Google Scholar]
- [64].Constantin TP, Silva GL, Robertson KL, Hamilton TP, Fague K, Waggoner AS, Armitage BA, Org. Lett 2008, 10, 1561–1564. [DOI] [PubMed] [Google Scholar]
- [65].Gu Y, Fei X, Liu Y, Wang Y, Yang X, J. Lumin 2013, 134, 184–190. [Google Scholar]
- [66].Mason SJ, Hake JL, Nairne J, Cummins WJ, Balasubramanian S, J. Org. Chem 2005, 70, 2939–2949. [DOI] [PubMed] [Google Scholar]
- [67].Lee H, Berezin MY, Henary M, Strekowski L, Achilefu S, J. Photochem. Photobiol. A 2008, 200, 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Guarin CA, Villabona-Monsalve JP, Lopez-Arteaga R, Peon J, J. Phys. Chem. B 2013, 117, 7352–7362. [DOI] [PubMed] [Google Scholar]
- [69].Tarazi L, George A, Patonay G, Strekowski L, Talanta 1998, 46, 1413–1424. [DOI] [PubMed] [Google Scholar]
- [70].Ramos SS, Santos PF, Reis LV, Almeida P, Dyes Pigm. 2002, 53, 143–152. [Google Scholar]
- [71].Li ZA, Mukhopadhyay S, Jang S-H, Brédas J-L, Jen AKY, J. Am. Chem. Soc 2015, 137, 11920–11923. [DOI] [PubMed] [Google Scholar]
- [72].Štacková L, Štacko P, Klán P, J. Am. Chem. Soc 2019, 141, 7155–7162. [DOI] [PubMed] [Google Scholar]
- [73].Karton-Lifshin N, Albertazzi L, Bendikov M, Baran PS, Shabat D, J. Am. Chem. Soc 2012, 134, 20412–20420. [DOI] [PubMed] [Google Scholar]
- [74].Geiger T, Schoger I, Rentsch D, Véron AC, Oswald F, Meyer T, Nüesch F, Int. J. Photoenergy 2014, 258984. [Google Scholar]
- [75].Yao H, Morita Y, Kimura K, J. Colloid Interface Sci 2008, 318, 116–123. [DOI] [PubMed] [Google Scholar]
- [76].Bujdák J, Iyi N, Hrobáriková J, Fujita T, J. Colloid Interface Sci 2002, 247, 494–503. [DOI] [PubMed] [Google Scholar]
- [77].Bricks JL, Slominskii YL, Panas ID, Demchenko AP, Meth. Appl. Fluoresc 2017, 6, 012001. [DOI] [PubMed] [Google Scholar]
- [78].Kato S, Kawabe Y, Mol. Cryst. Liq. Cryst 2010, 15, 165/[441]–171/[447]. [Google Scholar]
- [79].Zhytniakivska O, Girych M, Trusova V, Gorbenko G, Vasilev A, Kandinska M, Kurutos A, Baluschev SB, Dyes Pigm. 2020, 180, 108446. [Google Scholar]
- [80].Zhang L, Liu X, Lu S, Liu J, Zhong S, Wei Y, Bing T, Zhang N, Shangguan D, ACS Appl. Bio. Mater 2020, 3, 2643–2650. [DOI] [PubMed] [Google Scholar]
- [81].Silva GL, Ediz V, Yaron D, Armitage BA, J. Am. Chem. Soc 2007, 129, 5710–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Waring MJ, Chaires JB, Armitage BA, DNA Binders and Related Subjects, Springer, Berlin, 2005. [Google Scholar]
- [83].Long W, Lu Y-J, Zhang K, Huang X-H, Hou J-Q, Cai S-Y, Li Y, Du X, Luyt LG, Wong W-L, Chow C-F, Dyes Pigm. 2018, 159, 449–456. [Google Scholar]
- [84].Vasilev AA, Kandinska MI, Stoyanov SS, Yordanova SB, Sucunza D, Vaquero JJ, Castaño OD, Baluschev S, Angelova SE, Beilstein J Org. Chem 2017, 13, 2902–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hirons GT, Fawcett JJ, Crissman HA, Cytometry 1994, 15, 129–140. [DOI] [PubMed] [Google Scholar]
- [86].Tajc SG, Miller BL, J. Am. Chem. Soc 2006, 128, 2532–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Eriksson M, Härdelin M, Larsson A, Bergenholtz J, Åakerman B, J. Phys. Chem. A 2007, 111, 1139–1148. [DOI] [PubMed] [Google Scholar]
- [88].Yang W, Wong Y, Ng OTW, Bai L-P, Kwong DWJ, Ke Y, Jiang Z-H, Li H-W, Yung KKL, Wong MS, Angew. Chem. Int. Ed 2012, 51, 1804–1810; Angew. Chem. 2012, 124, 1840–1846. [DOI] [PubMed] [Google Scholar]
- [89].Harriman A, Luengo G, Gulliya K, Photochem. Photobiol 1990, 52, 735–740. [DOI] [PubMed] [Google Scholar]
- [90].Sato Y, Yajima S, Taguchi A, Baba K, Nakagomi M, Aiba Y, Nishizawa S, Chem. Commun 2019, 55, 3183–3186. [DOI] [PubMed] [Google Scholar]
- [91].Suzuki T, Fujikura K, Higashiyama T, Takata K, J. Histochem. Cytochem 1997, 45, 49–53. [DOI] [PubMed] [Google Scholar]
- [92].Stefansson S, Adams DL, Tang C-M, BioTechniques 2012, 52, 10.2144/000113873. [DOI] [PubMed] [Google Scholar]
- [93].Chen X, Wang J, Jiang G, Zu G, Liu M, Zhou L, Pei R, RSC Adv. 2016, 6, 70117–70123. [Google Scholar]
- [94].Ahoulou EO, Drinkard KK, Basnet K, St. Lorenz A, Taratula O, Henary M, Grant KB, Molecules 2020, 25, 2926. [DOI] [PMC free article] [PubMed] [Google Scholar]