

Abstract
Background:
Previous research has demonstrated the utility of subanesthetic doses of ketamine in decreasing binge (Drinking-in-the-Dark, or DID) 20% alcohol intake in female inbred (C57BL/6J) mice when administered 12 hours prior to alcohol access (Crowley et al., 2019). In the current study, we assess the efficacy of a similar ketamine pretreatment using male and female selectively bred, crossed High Alcohol Preferring (cHAP) mice, which also drink to intoxication, but are not inbred. We hypothesized that ketamine would decrease binge alcohol intake without impacting locomotor activity.
Methods and Results:
Subjects were 28 adult cHAP mice. Mice first received a two-week DID drinking history using 2-hour/day alcohol access. On day 12, prior to ketamine treatment, the average blood ethanol concentration (BEC) was 130 mg/dL, confirming that mice reliably reached intoxicating BECs. On day 15, mice were given 0, 3 or 10 mg/kg of ketamine 12-hours prior to the DID session. Ketamine did not decrease total (2-hour) alcohol consumption or locomotion. Interestingly, the 10 mg/kg dose of ketamine did alter the drinking pattern in male mice, decreasing frontloading for a single day. We opted to then increase the doses to 32 or 100 mg/kg (i.e., an anesthetic dose) two days after the initial treatment, keeping the saline control. Mice of both sexes decreased total binge alcohol intake at the 100 mg/kg dose only, but again, the effect only lasted one day.
Conclusions:
The current study found that cHAP mice reached more than double the BECs observed in C57BL/6J mice during DID, but did not respond to subanesthetic ketamine. Modest efficacy was found for ketamine pretreatment at anesthetic doses. Differences in findings may be due to differential intake during DID, or genetic differences between C57Bl/6J mice and cHAP mice. Drug efficacy in multiple models is important for discovering reliable pharmacotherapies for alcoholism.
Keywords: Ketamine, alcohol, binge drinking, cHAP mice, selectively bred mice, drinking patterns, frontloading
Introduction
Binge drinking is a drinking pattern resulting in blood alcohol levels of 80 mg/dL or higher in a timespan of two hours or less (NIAAA, 2004). Binge drinking is common, with models estimating that one fifth of the global adult population have engaged in at least one episode of binge drinking within the past month (Peacock et al., 2018). The consequences of binge drinking are severe, given that longitudinal studies indicate that multiple binge drinking sessions predict the future development of alcohol use disorder (AUD) (Chassin, Pitts, & Prost, 2002; Dawson, Li, & Grant, 2008; Zucker et al., 2006). Despite the serious consequences of binge drinking and need for novel interventions, the validation of new pharmacological treatments for AUD is challenging due to the complexity of ethanol’s (EtOH) molecular targets, plasticity changes induced by EtOH, and circuitry involved (Abrahao, Salinas, & Lovinger, 2017). Notwithstanding these challenges, N-methyl-D-aspartate receptors (NMDARs) have been identified as a potential target for treatment (Chandrasekar, 2013; Holmes, Spanagel, & Krystal, 2013; Hopf, 2017; Ivan Ezquerra-Romano, Lawn, Krupitsky, & Morgan, 2018; Lovinger, 1996; C. E. Strong & Kabbaj, 2020).
Ketamine, an NMDAR antagonist, has been shown to reduce EtOH intake in rodent models of binge drinking (Crowley et al., 2019; Ruda-Kucerova, Babinska, Luptak, Getachew, & Tizabi, 2018) and has been shown to reduce EtOH intake in humans diagnosed with AUD when combined with therapy (Dakwar et al., 2019) or retrieval of maladaptive reward memories (Das et al., 2019). Further, and of particular interest in the current study, several recent studies indicate that individuals with a family history of AUD (FH+) have fewer adverse effects when treated with ketamine than those without a family history of AUD (FH−); please see Comstock et al. (2019) for a review. For example, in a study comparing reaction to ketamine in healthy individuals (FH+ vs. FH−) following an infusion of ketamine, FH+ participants displayed lower dysphoria, psychosis, and perceptual responses than those who were FH− (Petrakis et al., 2004). This decrease in undesirable side effects seen in FH+ individuals may play a beneficial role in FH+ AUD patients adhering to a pharmacological treatment plan. Past pre-clinical research has utilized alcohol-preferring rats, which are selectively bred for high alcohol intake and represent a rodent model of FH+ (Bell, Rodd, Lumeng, Murphy, & McBride, 2006), to determine the effect of ketamine on EtOH intake. One study indicated that alcohol-preferring rats administered 7.5 or 10 mg/kg of ketamine 15 minutes prior to two-bottle choice (10% EtOH vs. water) displayed a decrease in EtOH intake and preference 0–2 hours following treatment, where female rats decreased intake and preference more than males (Rezvani, Levin, Cauley, Getachew, & Tizabi, 2017). Similarly, another study indicated that alcohol-preferring rats (male only) administered 20 mg/kg of ketamine 30 minutes prior to 10% EtOH operant self-administration decreased EtOH intake (Sabino, Narayan, Zeric, Steardo, & Cottone, 2013). Despite this converging cross-species evidence indicating ketamine’s potential effectiveness in treating AUD, there remains a critical need to investigate ketamine’s efficacy in preclinical models of FH+ during binge drinking. It is of importance to assess the utility of pharmacological interventions within this genetically vulnerable population during this prevalent and risky form of alcohol consumption.
In the current study, we have opted to use a treatment timepoint (12-hours prior to DID) which has been shown to be successful in reducing binge EtOH intake in C57BL/6J mice (Crowley et al., 2019). Although previous studies utilizing P rats (a model of FH+) have used short pretreatment times, we did not want mice in the current study to be under the influence of ketamine during DID EtOH access. Effects on alcohol intake in short pretreatment designs are complicated by the fact that ketamine is a dissociative anesthetic, which could lead to non-specific changes in consummatory behavior (or in alcohol sensitivity) while intoxicated. Even if lower drinking persists after ketamine intoxication subsides, conditioned taste aversion to ethanol could be the cause of lower drinking when animals are drinking alcohol while experiencing ketamine intoxication, because the flavor of alcohol could be associated with ketamine’s effect (e.g., see Gill et al., 1986 for a similar situation). Further, we tested the 12-hour pretreatment timepoint as this gives time for ketamine to fully metabolize out of the system, which has a half-life of about 13 min following i.p. administration in mice (Maxwell et al., 2006). In any case, a treatment where individuals diagnosed with AUD must be persistently intoxicated with ketamine to avoid drinking alcohol is clearly untenable.
We utilized a common binge drinking paradigm (‘drinking-in-the-dark’, DID) to assess EtOH intake of cHAP mice, who are selectively bred for high alcohol intake/preference and represent a FH+ population (Oberlin, Best, Matson, Henderson, & Grahame, 2011). We hypothesized that a subanesthetic dose of ketamine would result in lower binge EtOH intake without impacting locomotion. Further, we hypothesized that female mice would decrease total intake more than male mice, as previous studies have indicated sex differences wherein ketamine is more efficacious in female rodents (Crowley et al., 2019; Rezvani et al., 2017). Lastly, we strived to characterize EtOH intake patterns to determine if/how ketamine impacts frontloading, wherein the amount or proportion of EtOH consumed is highly skewed toward the onset of EtOH access. Frontloading has been proposed to represent an increase in the motivation to experience the rewarding and/or the post-absorptive effects of EtOH, as frontloading generally increases over EtOH access days (Ardinger, Grahame, Lapish, & Linsenbardt, 2020; Darevsky et al., 2019; Linsenbardt & Boehm, 2014, 2015; Rhodes et al., 2007; Salling et al., 2018; Wilcox, Dekonenko, Mayer, Bogenschutz, & Turner, 2014). Therefore, determination of if/how frontloading is impacted by ketamine treatment administration provides additional evidence (when coupled with assessment of total binge intake) for the efficacy of the drug.
Methods
Subjects
Subjects in this study were 13 adult (post-natal day 70–90 at the beginning of DID) female cHAP mice and 15 adult cHAP male mice from the 44th generation of selection. Please see Oberlin et al. (2011) for information regarding the creation and selective breeding of the cHAP line. All mice were bred in the AAALAC-approved School of Science Vivarium at Indiana University-Purdue University Indianapolis (IUPUI) and were single-housed in standard shoebox cages in a room with a 12-hour reverse light–dark cycle one week prior to the beginning of DID testing. All mice always had ad libitum access to standard laboratory rodent chow (LabDiet 5001), including during DID and pharmacological experiments. Mice also had ad libitum access to water via standard home cage water bottles, except during 2-hour DID sessions wherein normal water bottles were replaced with specialized sipper tubes containing 20% EtOH (see EtOH Solution below). Standard home cage water bottles did not utilize the specialized sippers but did contain sippers with identical-sized drinking orifices. All procedures were approved by the IUPUI School of Science Animal Care and Use Committee.
EtOH Solution
EtOH for drinking experiments was prepared by diluting 190 proof EtOH from Pharmco, Inc. (Brookfield, CT), to 20% v/v in tap water. Drinking solution was prepared at the beginning of the experiment and stored in sealed fluid reservoirs connected to the volumetric drinking monitor (VDM) system (Columbus Instruments Inc., Columbus, OH), equipped with specialized sipper tubes that monitor fluid volume consumed with high temporal resolution. EtOH within the reservoirs was topped-off halfway through the two-week drinking testing period.
Ketamine
Ketamine (100 mg/mL; Henry Schein) was diluted in sterile 0.9% saline to inject intraperitoneally (i.p.) at a volume of 10 mL/kg (i.e. for the groups receiving a dose of 3 or 10 mg/kg ketamine, the concentration of the drug was 0.3 and 1.0 mg/mL, respectively) to control for volume injected across different dose groups. Drug concentration, rather than injection volume, was increased correspondingly for assessment of the effects of 32 and 100 mL/kg ketamine doses.
Drinking-in-the-dark
DID procedures have been previously described (Ardinger et al., 2020; Linsenbardt & Boehm, 2014, 2015). Briefly, 3 hours into the dark cycle, each mouse’s home cage water bottle was replaced with a volumetric sipper tube (Columbus Instruments Inc) containing 20% v/v EtOH. Mice were given 2 hours of access to EtOH. All mice received 20% EtOH access for two hours on all DID testing days. On all DID days, the volume of consumed fluid was measured in 1-minute bins, allowing for within-session analyses of binge-drinking patterns. Mice were then pseudo-randomly assigned to a drug treatment group (where groups were counterbalanced by family and sex). Intakes on day 14 (prior to ketamine treatment) did not differ between treatment groups. Twelve hours prior to the start of DID on the 15th day, mice were injected with either saline (control), 3 mg/kg, or 10 mg/kg of ketamine. On day 16, mice did not receive additional drug treatment, but received 2-hour DID access to EtOH to determine if any changes in EtOH intake due to treatment prior to day 15 continued (or emerged) two days after drug administration. Twelve hours prior to day 17, mice received higher doses of ketamine. Mice who received 3 mg/kg prior to day 15 were given 32 mg/kg of ketamine, and mice who received 10 mg/kg group prior to day 15 were given 100 mg/kg of ketamine. Mice previously dosed with saline were given another dose of saline (control). The 32 and 100 mg/kg doses were selected as they are log steps above the initial doses, and separating dosing by log intervals is a common practice in pharmacology research (Lewandowski & Norman, 2015). Further, we wanted to assess response to a full anesthetic dose (100 mg/kg). Lastly, on days 18 and 19 mice again received 2-hours of DID access to EtOH without additional drug treatment. Please see Figure 1 for a description of the study timeline and Table 1 for a breakdown of sample size within each treatment group.
Figure 1.

Timeline of experiment.
Table 1.
Sample sizes for treatment groups.
| Day | 15 | 17 | ||||
| Saline | 3 mg/kg ketamine | 10 mg/kg ketamine | Saline | 32 mg/kg ketamine | 100 mg/kg ketamine | |
| Female | n = 4 | n = 5 | n = 4 | n = 4 | n = 5 | n = 4 |
| Total n = 13 | Total n = 13 | |||||
| Male | n = 3 | n = 6 | n = 6 | n = 3 | n = 5* | n = 6 |
| Total n = 15 | Total n = 14 | |||||
Note that one male mouse who was designated for the 32 mg/kg group accidentally received a dose of 100 mg/kg. This mouse’s data are not included in analyses for days 17, 18, and 19 to allow for the same treatment history (i.e. 3 mg/kg to a subsequent 32 mg/kg two days later) within this group.
Blood EtOH Concentrations (BECs)
Immediately following the end of the 2-hour drinking session on day 12, 50 μl of periorbital sinus blood was drawn from all mice. As periorbital sinus blood collection can interfere with drinking patterns, we chose day 12 to allow mice to restabilize their binge intake prior to ketamine or control treatment on day 15. Samples were centrifuged, and plasma was withdrawn and stored at −20°C. BECs were determined using an Analox EtOH Analyzer (Analox Instruments, Lunenburg, MA).
Home Cage Locomotion
Home cage locomotor activity was monitored using ANY-maze software (Stoelting Co., Wood Dale, IL) and Logitech C920 cameras. Distance traveled was recorded for each mouse in centimeters for the duration of the 2-hour DID session. Locomotor activity was primarily used to assess if there was sedation in mice receiving ketamine treatment.
Statistics: 2-Week Drinking History
Mean total (2-hour) EtOH intake, percentage of intake within the first 15 minutes (frontloading), and home cage locomotor activity on days 1 to 14 were analyzed using 2 (sex) × 14 (day) mixed-methods 2-way analysis of variance (ANOVA)s. Greenhouse–Geisser corrections were applied to these analyses when normality tests indicated non-normal distributions of intake or movement (see results). For analysis of frontloading during the 2-week drinking history, we note that 15-minutes accounts for 12.5% of the total 2-hour DID session, with mice needing to consume significantly more than 12.5% (dotted line, Figure 2C) of their total intake within the first 15 minutes of the DID session to be considered as having frontloaded on a given day. This was assessed using 1-sample t-tests where percentage of intake within the first 15 minutes was compared to the 12.5% threshold to determine whether front-loading was statistically significant, as we have done in our previous work assessing intake patterns in several HAP lines (Ardinger et al., 2020). Lastly, day 12 BEC was assessed using a simple linear regression wherein EtOH intake was the independent variable and BEC was the dependent variable.
Figure 2.
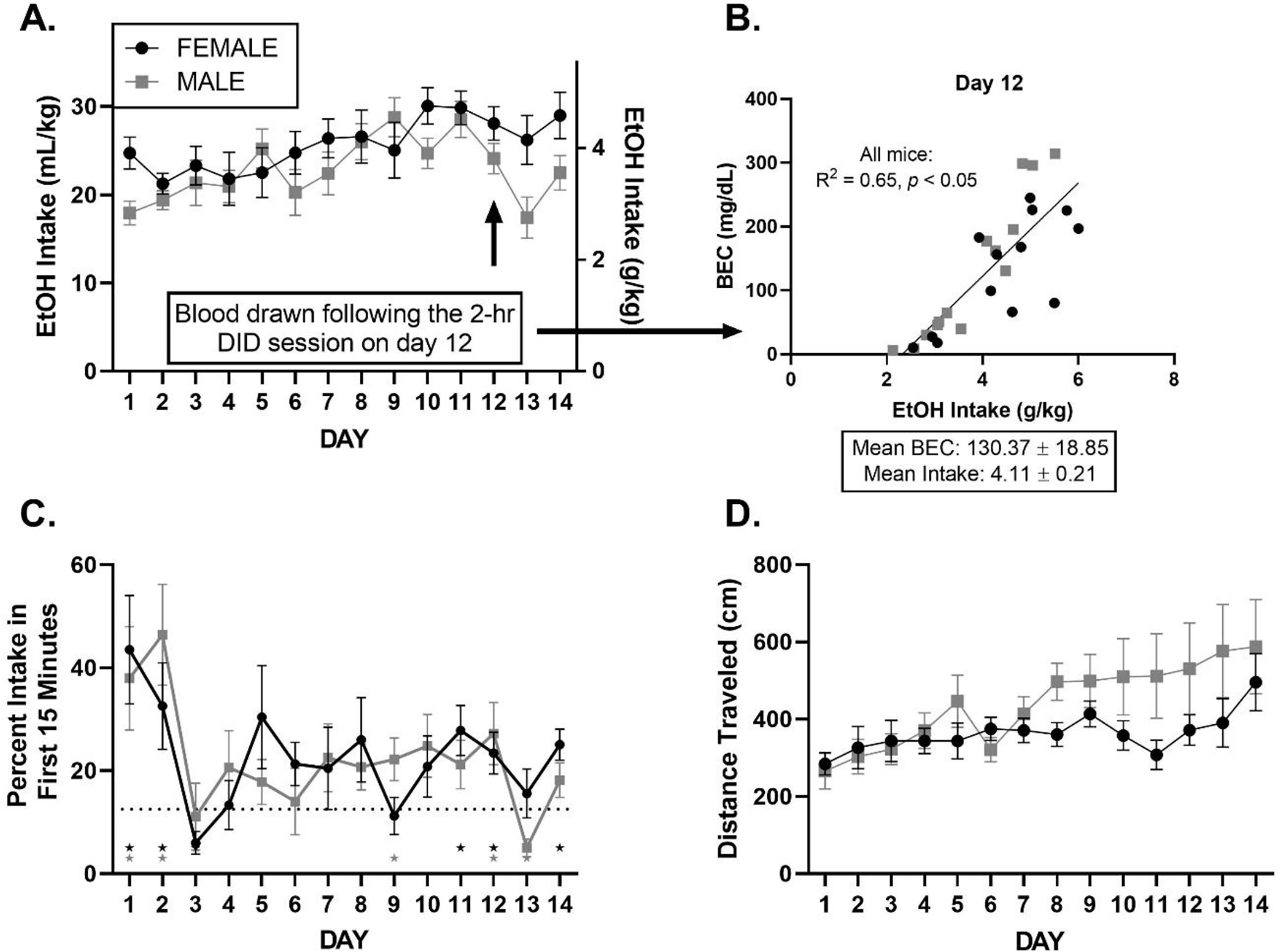
All graphs are displayed as mean ± SEM over days of 2-hr DID prior to ketamine or saline treatment. A: EtOH Drinking History. Total 20% EtOH intake varies over days. B. BEC. There is a relationship between EtOH intake and BEC during the 2-week drinking history. Intake (g/kg) and BEC (mg/dL) mean ± SEM are presented. C: Frontloading: Percent of EtOH intake within the first 15 minutes. Stars indicate that mice consumed significantly higher than 12.5% (dashed line) of their total intake within the first 15 minutes of the DID session on a given day (12.5% represents the frontloading threshold, please see further description in Methods: Statistics). D: Locomotion varies over days.
Statistics: Post-Ketamine Treatment
EtOH intake, percentage of intake within the first 15 minutes (frontloading), and home cage locomotor activity following ketamine treatment were analyzed using 2 (sex) × 3 (dose) 2-way ANOVAs calculated separately for each day post-treatment. To better characterize prominent within-session pattern alterations (of both EtOH intake and distance traveled), a central moving average was calculated on 15-minute bin increments as described previously (Ardinger et al., 2020). Briefly, data were binned into 15-minute averages and “moved” forward in time in 1-minute increments such that each subsequent bin included 1 additional minute into the future and excluded 1 minute furthest in time. Fifteen-minute increments were chosen because observations of intake data suggest that substantial changes occurred over DID sessions within the first 15 minutes of EtOH access (frontloading) and to allow for direct comparisons to previously published data assessing intake patterns during DID (Ardinger et al., 2020; Linsenbardt & Boehm, 2014, 2015).
Results
Days 1–14: Drinking history.
Total EtOH intake over days.
Analyses identified a main effect of day, F (6.612, 168.4) = 3.59, p < 0.05, where EtOH intake increased slightly over days, but there was no significant main effect of sex or interaction between sex and day, ps > 0.05; Figure 2A. Analysis of BECs from day 12 indicated a correlation between total EtOH intake and BEC, where regression lines for females vs. males did not differ, p > 0.05. Thus, we collapsed the regression line across sex and report R2 = 0.65, p < 0.05, Figure 2B.
Percent of intake in the first 15 minutes.
Like total intake, analyses revealed a main effect of day, F (13, 332) = 4.235, p < 0.05, where frontloading shifted over days, declining from its high levels on the first two days. However, significant frontloading was seen in 3 of the last 4 days of DID drinking. There was no significant main effect of sex or interaction between sex and day, ps > 0.05, Figure 2C.
Locomotor activity.
Analyses identified a main effect of day, F (2.099, 51.52) = 5.62, p < 0.05, where total distance travelled in 2-hours generally increased over DID testing days, and a sex × day interaction, F (13, 319) = 2.038, p < 0.05, where male mice surpassed female mice in total movement over the two-week drinking history period. There was no main effect of sex, p > 0.05, Figure 2D.
Days 15–16: Treatment with subanesthetic doses of ketamine.
Total EtOH intake following first ketamine treatment.
On day 15, there was a significant main effect of sex, F (1, 22) = 7.07, p < 0.05, where females drank more EtOH than males regardless of ketamine dose administered 12-hours prior to DID. However, there was no significant effect of dose, p > 0.05, or interaction between dose and sex, p > 0.05, Figure 3A. On day 16, when no additional ketamine treatment was administered prior to DID, there were no main effects of sex or dose, ps > 0.05, or interaction between sex and dose, p > 0.05, Figure 3D.
Figure 3.
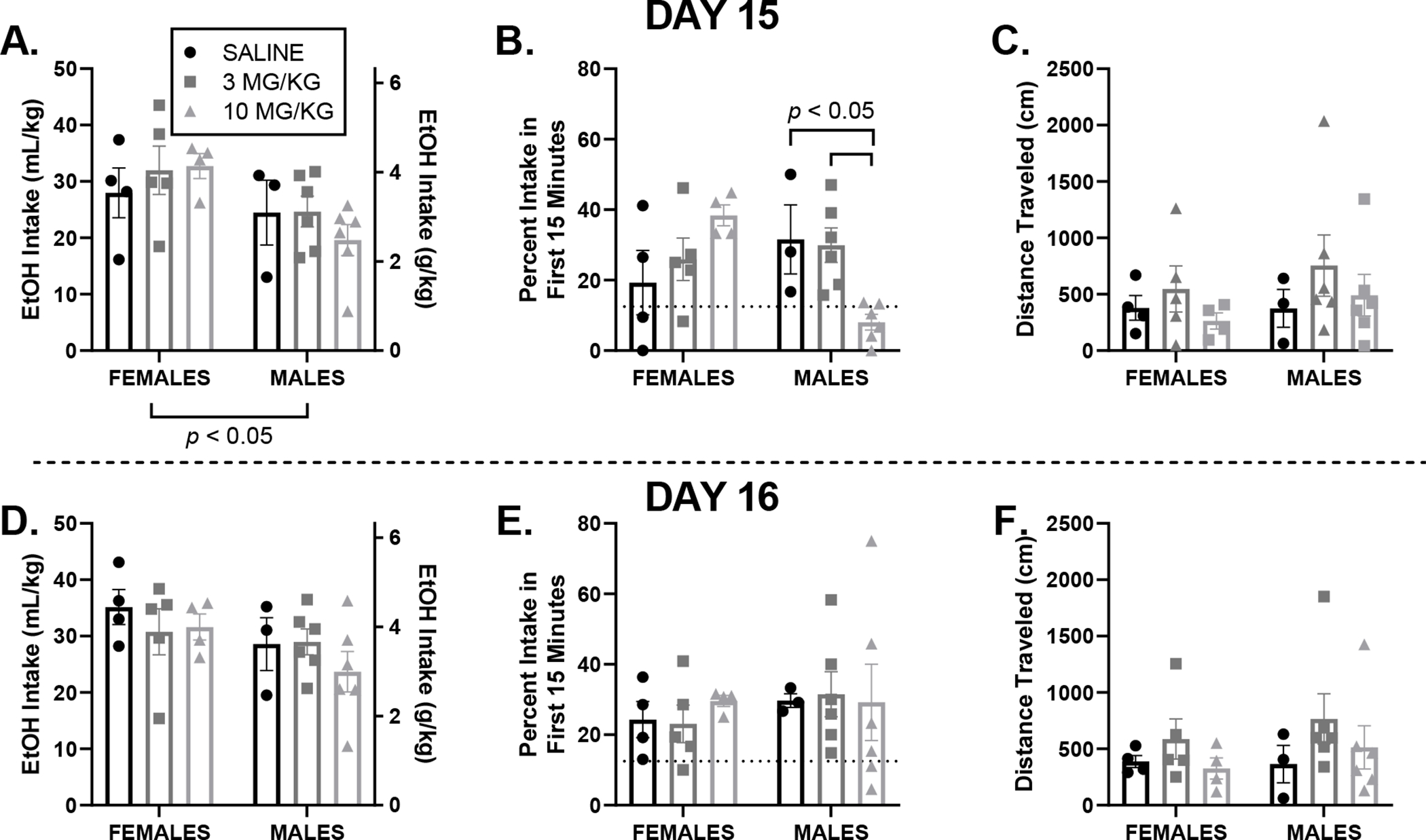
All graphs are displayed as mean ± SEM for day 15 (top) where drinking occurred 12-hours after an injection of saline, 3 or 10 mg/kg ketamine, and day 16 (bottom) one day following assessment of intake post-treatment. A, D: EtOH intake. Total 20% EtOH intake did not differ between sexes or dose groups. B, E: Frontloading: An interaction of sex and dose reveals that males in the 10 mg/kg ketamine group frontloaded significantly less than males in the saline or 3 mg/kg ketamine group on day 15, indicating that 10 mg/kg of ketamine transiently alters drinking patterns in a sex-dependent manner. C, F: Locomotor activity does not significantly differ between sexes or dose groups, indicating that differences observed in frontloading are not caused by sedation.
Percent of intake in the first 15 minutes following first ketamine treatment.
On day 15, there was no main effects of sex, p > 0.05, or of dose, p > 0.05. However, there was a significant interaction of sex and dose, F (2, 22) = 7.61, p < 0.05, where Tukey’s multiple comparison post-hoc tests indicated that males in the 10 mg/kg group had lower EtOH intake in the first 15-minutes than male mice in the 3 mg/kg and saline groups, p < 0.05, Figure 3B. This alteration in drinking pattern is further visualized in Figure 4. This decrease in frontloading in males who received 10 mg/kg of ketamine did not last. On day 16, when there was no additional ketamine treatment administered, there were no effects of sex, dose, or interaction of sex and dose, ps > 0.05, Figure 3E.
Figure 4.
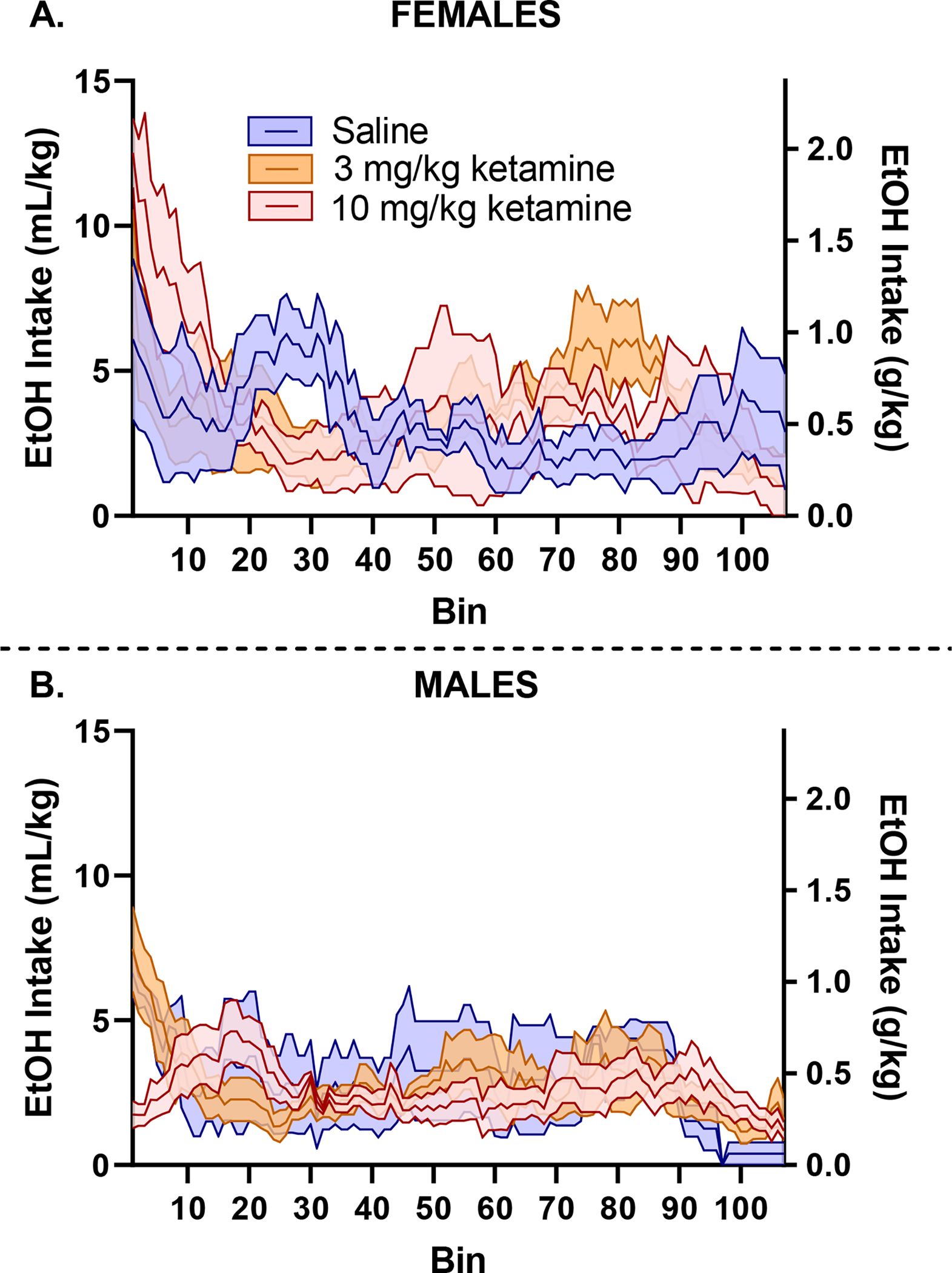
Day 15 intake patterns further demonstrate a decrease in frontloading in male mice who received 10 mg/kg of ketamine (B). No significant changes are observed in females (A).
Locomotor activity following first ketamine treatment.
On day 15 and 16, there were no effects of sex, dose, or interaction between sex and dose, ps > 0.05, Figures 3C and F, suggesting that alterations in drinking patterns are not caused by sedation from ketamine.
Days 17–19: Treatment with higher doses of ketamine.
Total EtOH intake following second ketamine treatment.
On day 17, there was no main effect of sex, p > 0.05, but there was a main effect of dose, F (2, 21) = 5.35, p < 0.05, where mice receiving 100 mg/kg ketamine had significantly lower intake than mice who received saline. There was no interaction between dose and sex, p > 0.05, Figure 5A. On days 18 and 19, we observed a main effect of sex: F (1, 21) = 5.61, and F (1, 21) = 5.66, ps < 0.05, respectively, where female mice, regardless of treatment group, outdrank males, but no effect of dose or interaction between dose and sex, ps > 0.05, Figures 5D, 5G.
Figure 5.
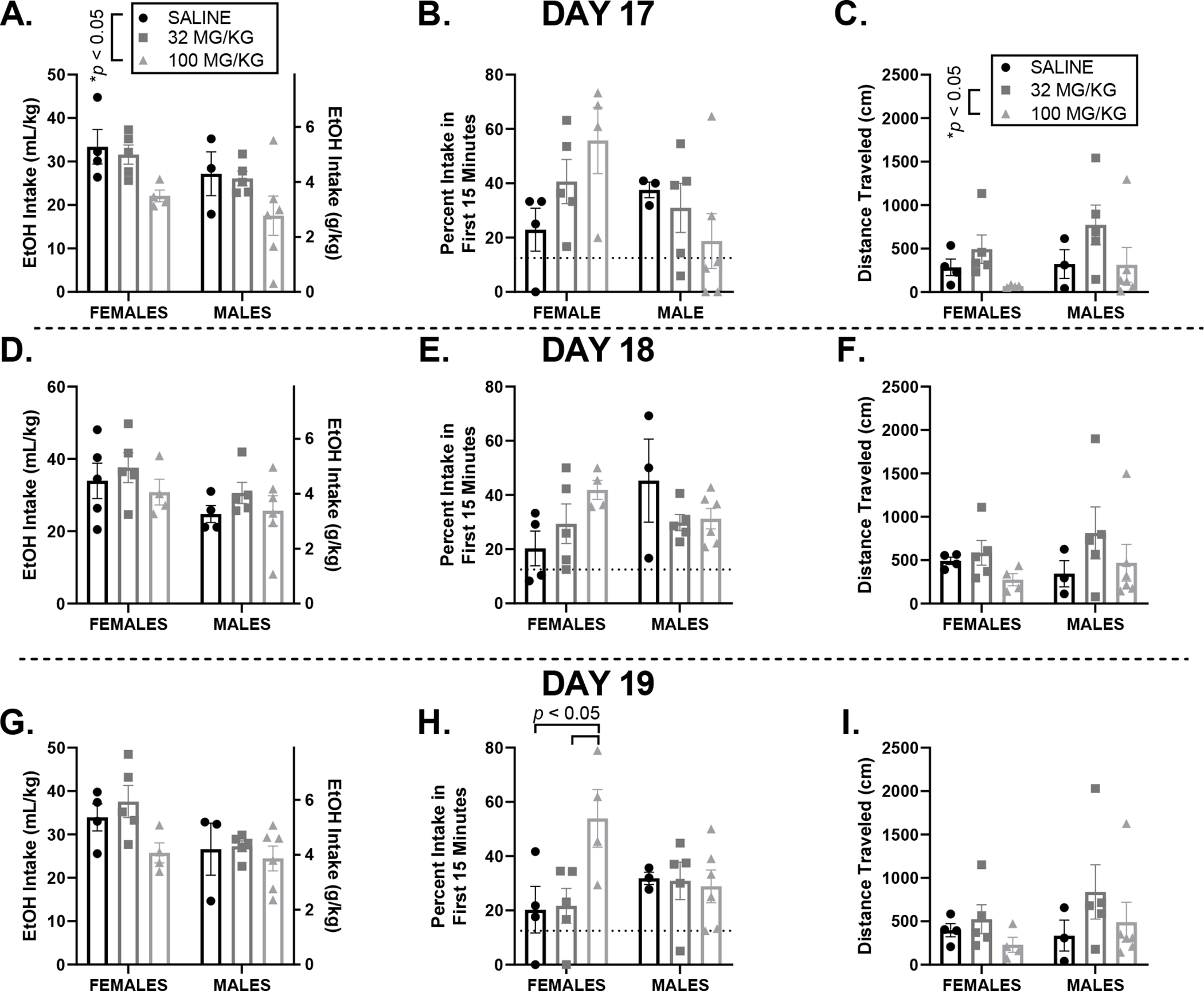
All graphs are displayed as mean ± SEM. Day 17 (top) where drinking occurred 12-hours after an injection of saline, 32 or 100 mg/kg ketamine, and day 18 (middle) one day post-treatment, and day 19 (bottom) two days following treatment. EtOH intake: Total 20% EtOH intake differed between dose groups 12-hours following treatment (Day 17; A), however this decrease in EtOH intake did not last into the next two days where no additional drug was given (days 18 and 19, D and G, respectively). Frontloading: there is no difference in frontloading between sex and dose groups on days 17 (B) and 18 (E), however an interaction of sex and dose on day 19 reveals that females in the 100 mg/kg ketamine group frontloaded significantly more than females in the saline or 32 mg/kg ketamine group (H). Locomotion: Mice in the 100 mg/kg group moved significantly less than mice in the 32 mg/kg group 12-hours following injection (day 17, C). Differences in movement between treatment groups did not persist one (day 18, F) or two (day 19, I) days later.
Percent of intake in the first 15 minutes following second ketamine treatment.
On day 17, there was no significant main effect of sex, main effect of dose, ps > 0.05, or interaction between dose and sex (although this finding was ‘trending towards significance’), F (2, 21) = 3.33, p = 0.0557, Figure 5D. To assess if any changes in frontloading persisted following a higher dose of ketamine treatment, percent of intake within the first 15 minutes on days 18 and 19 was also assessed. Day 18: no main effect of sex or main effect of dose, ps > 0.05. However, there was an interaction of sex and dose, F (2, 21) = 3.64, p < 0.05, Figure 5E. Like day 18, on day 19 there was no main effect of sex or main effect of dose, ps > 0.05. There was again an interaction of sex and dose, F (2, 21) = 3.88, p < 0.05, where post-hoc comparisons indicate that females in the 100 mg/kg group frontloaded significantly more than females in the saline and 32 mg/kg groups, p < 0.05, Figure 5F.
Locomotor activity following second ketamine treatment.
On day 17, there was no main effect of sex, p > 0.05. However, there was a main effect of dose, F (2, 21) = 3.53, p < 0.05, where mice in the 100 mg/kg group moved less than mice in the 32 mg/kg group. There was no interaction between dose and sex, p > 0.05, Figure 5G. To assess if any changes in locomotion persisted following a higher dose of ketamine treatment, total distance travelled on days 18 and 19 was also assessed. On both days, there were no effects of sex, dose, or their interaction, ps > 0.05, Figures 5H, 5I, respectively.
Intake and locomotor patterns following second ketamine treatment.
To further investigate the results described above, we calculated moving averages (please see Statistics: Post-Ketamine Treatment) for EtOH intake and distance travelled. Intake patterns (Figure 6A, C) further display the nearly significant differences in frontloading between treatment groups discussed above. Specifically, female mice in the 100 mg/kg group displayed high intake during the early portion of the 2-hr DID session, with no to low intake past the first hour. These data, coupled with further characterization of locomotor activity across the session (Figure 6B, D), suggest that the decrease in total distance travelled in the 100 mg/kg groups may not be indicative of a sedative effect of ketamine, but rather may be driven by alcohol sedation because of high frontloading.
Figure 6.
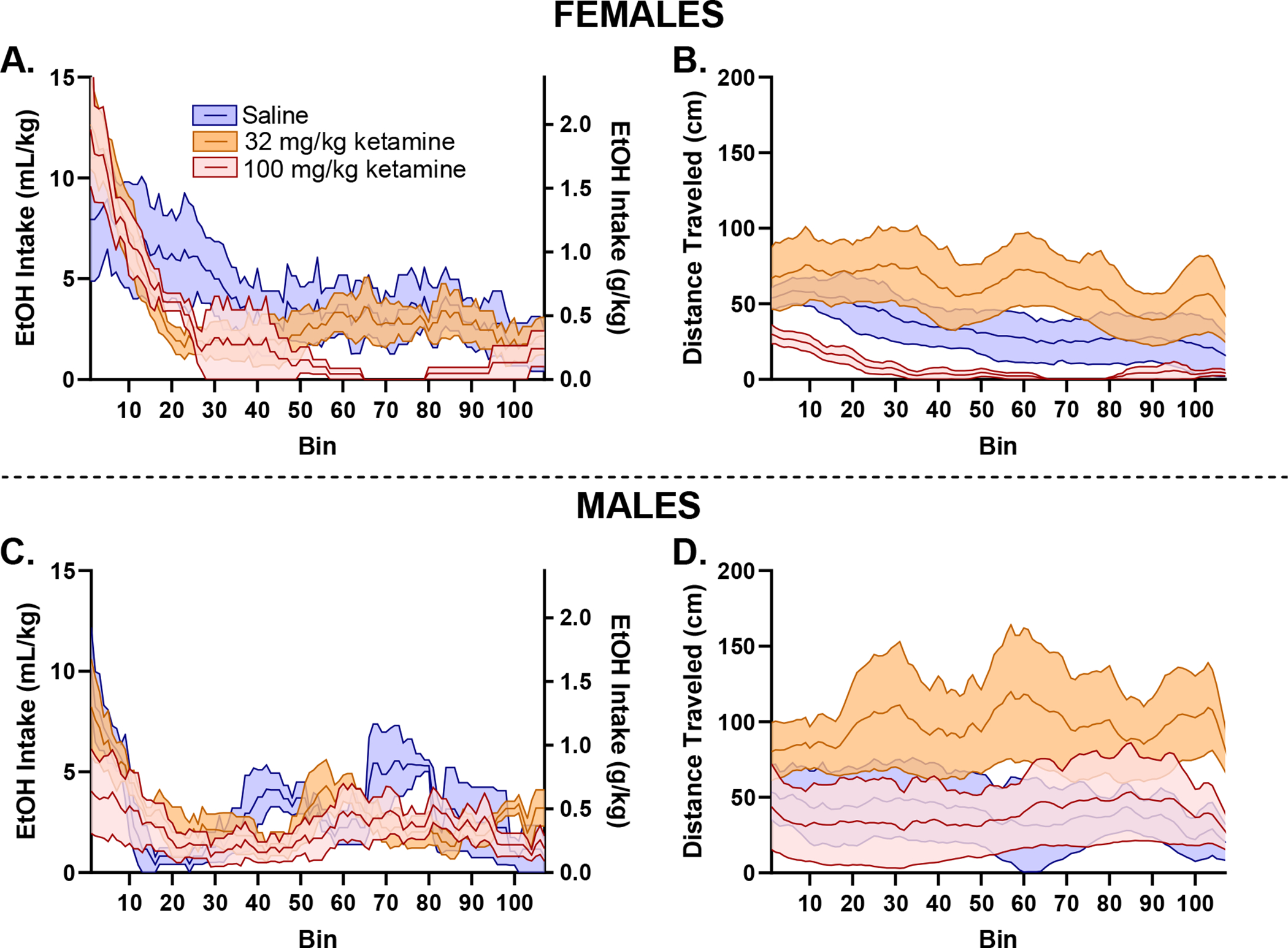
All figures are presented as mean (solid middle line) ± SEM (shaded area). To further investigate our finding that 100 mg/kg of ketamine decreased total EtOH intake and distance travelled, we calculated intake and movement patterns across the DID session on day 17 by sex: females (top), males (bottom). Intake patterns further demonstrate that female mice in the 100 mg/kg group (A) consume a disproportionately high amount in the early part of the 2-hr DID session, potentially resulting in sedation toward the middle of the session (B). Males shown for comparison (C, D). These results indicate that female mice were not sedated from the 100 mg/kg ketamine injection 12-hours prior, but rather the decrease in total distance travelled is driven by the mouse’s frontloading behavior.
Discussion
Our primary hypothesis, that a subanesthetic dose of ketamine would result in lower subsequent binge EtOH intake without impacting locomotion, was not supported. Locomotion was not impacted at these lower doses (Figure 3C); however, mice did not decrease binge EtOH intake following a dose of 3 or 10 mg/kg ketamine (Figure 3A). Previous studies using Wistar rats (Ruda-Kucerova et al., 2018) and C57BL/6J mice (Crowley et al., 2019), which are both strains who consume EtOH but do not represent a model of FH+, have demonstrated a decrease in binge EtOH intake following subanesthetic ketamine treatment. FH+ rodent models (alcohol-preferring rats) have shown similar results of a decrease in two-bottle choice EtOH intake (Rezvani et al., 2017) and operant self-administration (Sabino et al., 2013) following a subanesthetic dose of ketamine. The previous research utilizing FH+ rodents have administered treatment 15 (Rezvani et al., 2017) and 30 minutes (Sabino et al., 2013) prior to EtOH access. In the current study, we treated cHAP mice with ketamine 12 hours prior to DID EtOH access, a treatment timepoint which has shown efficacy in reducing binge intake in C57BL/6J mice (Crowley et al., 2019), and in turn more analogous to the human study (Dakwar et al., 2019) in that ketamine is no longer present when its effects on alcohol intake are observed. This earlier administration timepoint was appealing to both allow comparison to previous work in mice and ensure that alterations in EtOH intake are not due to acute ketamine intoxication. While the time and dose of ketamine administration relative to DID alcohol access was the same in both Crowley et al. (2019) and our study, the efficacy of ketamine seen previously in the Crowley lab’s C57BL/6J mice did not generalize to our selectively-bred high EtOH preference population. BECs that we observed during DID were at least double those observed in the Crowley study in C57BL/6J mice (averaging 130 mg/dl for cHAP and about 60 mg/dl for C57BL/6J). So, while subanesthetic ketamine was effective in female C57BL/6J mice but not cHAP mice, this may be driven by important differences in these drinking models, or may merely indicate that the findings in the inbred C57BL/6J mice may have limited generality to other high-drinking populations. Numerous authors have noted that extrapolating findings from a single inbred strain may be problematic (e.g., Voelkl et al., 2020). We would note that any development of a successful pharmacotherapy for human alcoholism should include multiple animal models to increase confidence in any preclinical findings.
Although we did not observe a decrease in total EtOH intake following administration of these lower ketamine doses (Figure 3A), 10 mg/kg of ketamine decreased frontloading in male cHAPs only (Figures 3B and 4B). However, we note that this decrease in male frontloading was not sustained, as determined by assessing EtOH intake and intake pattern one day later (day 16) when no additional treatment was administered (Figure 3E).
As we did not observe a decrease in total EtOH intake following 3 or 10 mg/kg of ketamine, we opted to increase the dose of treatment where mice in the 3 mg/kg group subsequently received 32 mg/kg and mice in the 10 mg/kg group subsequently received 100 mg/kg (control mice received another dose of saline) 12-hours prior to DID EtOH access on day 17, venturing into anesthetic doses of ketamine. Mice of both sexes in the 100 mg/kg group drank significantly less EtOH than saline control mice 12-hours post-treatment (Figure 5A). We recognize that there may be concern regarding the small n within treatment groups. However, we do not believe that the failure to detect an effect at subanesthetic doses is due to insufficient power. We note that 12-hours after ketamine administration, we observed a 37% reduction in binge intake in the 100 mg/kg group as compared to the saline group. Previous research has observed a ~50% reduction in binge intake in female C57BL/6J mice administered 3 mg/kg of ketamine 12-hours prior to DID (Crowley et al., 2019). Therefore, the sample size in the current study provides the sensitivity to detect a similar reduction in alcohol intake as previous work, but we find that a higher dose is required in cHAP mice. We note that we would expect that an effective pharmacotherapy for AUD would decrease both total intake and avidity for alcohol consumption, as potentially measured by frontloading. This idea has been demonstrated in previous work assessing naltrexone’s efficacy in humans, where naltrexone has been demonstrated to not only decrease total intake, but also slow the progression of intake (Anton, Drobes, Voronin, Durazo-Avizu, & Moak, 2004). Thus, there is concern about the efficacy of ketamine as a treatment for AUD as an anesthetic dose decreased total intake but increased (although not significantly) frontloading in females. Further, we note that the decrease in alcohol intake in the 100 mg/kg group did not persist into days 18 or 19, where no additional drug was administered (Figures 5D and 5G, respectively). Further research using FH+ rodents should investigate if multiple treatments and/or earlier timepoints is more effective in reducing binge EtOH intake. However, this lack of persistence of ketamine’s effects is inconsistent with earlier findings in C57BL/6J mice (Crowley et al., 2019).
Previous research has highlighted robust sex differences wherein female mice have been observed to decrease EtOH intake more than males following ketamine administration (Crowley et al., 2019; Rezvani et al., 2017). EtOH inhibits glutamatergic action at NMDA receptors, with repeated EtOH drinking creating an upregulation in NMDA receptor-related binding (Hoffman et al., 1990). It is generally accepted that there is a consistent and replicable main effect of sex in EtOH intake when conducting drinking-in-the-dark (DID) studies, where females outdrink males (Sneddon, White, & Radke, 2019; M. N. Strong et al., 2010); and we have seen this same pattern in our laboratory’s two-bottle choice cHAP line selection data (Oberlin et al., 2011). A working theory is that the larger total amount of EtOH consumed by female mice over a drinking history period may lead to greater receptor sensitization in females over time. It has been proposed that administration of NMDAR antagonists may be working to reduce expression of EtOH tolerance (Krystal et al., 2003). This mechanism of action may work particularly well in rodents and individuals with problematic EtOH use which has led to impaired negative feedback signals to stop drinking (Krystal et al., 2003). This may explain why ketamine’s effect on alcohol intake has previously been more robust in female rodents as we note here that in the current study, we did not observe a main effect of sex during our DID drinking history (Figure 2A). Therefore, a lack of sex differences observed following ketamine treatment in the current study may not contradict the theory that higher female EtOH intake prior to ketamine treatment is necessary to facilitate a reduction in subsequent EtOH intake.
In conclusion, the current study suggests that although acute ketamine has previously been shown to decrease EtOH intake in FH+ rodents in two-bottle choice (Rezvani et al., 2017) and operant EtOH administration paradigms (Sabino et al., 2013), 12 hour pretreatment led to a similar and transient decrease in EtOH intake only at a high (100 mg/kg) dose in the current binge drinking paradigm. Future research should further consider the efficacy of ketamine treatment in various strains of rodents using a variety of alcohol intake paradigms to further assess the impact of ketamine on drinking patterns, total EtOH intake, and locomotor activity. This work will be crucial in determining if this pharmacological intervention shows promise for FH+ individuals who frequently engage in binge drinking, which represent one of the most vulnerable populations for the development of AUD.
Highlights.
cHAP mice reliably drink to intoxication during ‘drinking-in-the-dark’ (DID).
Ketamine (3 – 32 mg/kg, 12 h prior to DID) did not decrease alcohol intake.
100 mg/kg of ketamine 12 h prior to DID decreased alcohol intake in both sexes.
Future work is needed to assess ketamine’s efficacy in FH+ populations.
Acknowledgments
This work was supported in part by the Indiana Alcohol Research Center P60-AA007611.
Footnotes
Declarations of interest: none
References
- Abrahao KP, Salinas AG, & Lovinger DM (2017). Alcohol and the Brain: Neuronal Molecular Targets, Synapses, and Circuits. Neuron, 96(6), 1223–1238. doi: 10.1016/j.neuron.2017.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton RF, Drobes DJ, Voronin K, Durazo-Avizu R, & Moak D (2004). Naltrexone effects on alcohol consumption in a clinical laboratory paradigm: temporal effects of drinking. Psychopharmacology, 173(1), 32–40. doi: 10.1007/s00213-003-1720-7 [DOI] [PubMed] [Google Scholar]
- Ardinger CE, Grahame NJ, Lapish CC, & Linsenbardt DN (2020). High Alcohol–Preferring Mice Show Reaction to Loss of Ethanol Reward Following Repeated Binge Drinking. Alcoholism: Clinical and Experimental Research, 44(9), 1717–1727. doi: 10.1111/acer.14419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RL, Rodd ZA, Lumeng L, Murphy JM, & McBride WJ (2006). REVIEW: The alcohol-preferring P rat and animal models of excessive alcohol drinking. Addiction Biology, 11(3–4), 270–288. doi: 10.1111/j.1369-1600.2005.00029.x [DOI] [PubMed] [Google Scholar]
- Chandrasekar R (2013). Alcohol and NMDA receptor: current research and future direction. Front Mol Neurosci, 6, 14. doi: 10.3389/fnmol.2013.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassin L, Pitts SC, & Prost J (2002). Binge drinking trajectories from adolescence to emerging adulthood in a high-risk sample: predictors and substance abuse outcomes. J Consult Clin Psychol, 70(1), 67–78. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/11860058 [PubMed] [Google Scholar]
- Comstock SM, Vaidya JG, & Niciu MJ (2019). Neurophysiological Correlates and Differential Drug Response in Subjects With a Family History of an Alcohol Use Disorder. Chronic Stress (Thousand Oaks), 3. doi: 10.1177/2470547019865267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley NA, Magee SN, Feng M, Jefferson SJ, Morris CJ, Dao NC, . . . Luscher B (2019). Ketamine normalizes binge drinking-induced defects in glutamatergic synaptic transmission and ethanol drinking behavior in female but not male mice. Neuropharmacology, 149, 35–44. doi: 10.1016/j.neuropharm.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakwar E, Levin F, Hart CL, Basaraba C, Choi J, Pavlicova M, & Nunes EV (2019). A Single Ketamine Infusion Combined With Motivational Enhancement Therapy for Alcohol Use Disorder: A Randomized Midazolam-Controlled Pilot Trial. Am J Psychiatry, appiajp201919070684. doi: 10.1176/appi.ajp.2019.19070684 [DOI] [PubMed] [Google Scholar]
- Darevsky D, Gill TM, Vitale KR, Hu B, Wegner SA, & Hopf FW (2019). Drinking despite adversity: behavioral evidence for a head down and push strategy of conflict-resistant alcohol drinking in rats. Addict Biol, 24(3), 426–437. doi: 10.1111/adb.12608 [DOI] [PubMed] [Google Scholar]
- Das RK, Gale G, Walsh K, Hennessy VE, Iskandar G, Mordecai LA, . . . Kamboj SK (2019). Ketamine can reduce harmful drinking by pharmacologically rewriting drinking memories. Nat Commun, 10(1), 5187. doi: 10.1038/s41467-019-13162-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Li TK, & Grant BF (2008). A prospective study of risk drinking: at risk for what? Drug Alcohol Depend, 95(1–2), 62–72. doi: 10.1016/j.drugalcdep.2007.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill K, Shatz K, Amit Z, & Ogren SO (1986). Conditioned taste aversion to ethanol induced by zimeldine. Pharmacol Biochem Behav, 24(3), 463–468. doi: 10.1016/0091-3057(86)90542-3 [DOI] [PubMed] [Google Scholar]
- Hoffman PL, Rabe CS, Grant KA, Valverius P, Hudspith M, & Tabakoff B (1990). Ethanol and the NMDA receptor. Alcohol, 7(3), 229–231. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2158789 [DOI] [PubMed] [Google Scholar]
- Holmes A, Spanagel R, & Krystal JH (2013). Glutamatergic targets for new alcohol medications. Psychopharmacology (Berl), 229(3), 539–554. doi: 10.1007/s00213-013-3226-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW (2017). Do specific NMDA receptor subunits act as gateways for addictive behaviors? Genes Brain Behav, 16(1), 118–138. doi: 10.1111/gbb.12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan Ezquerra-Romano I, Lawn W, Krupitsky E, & Morgan CJA (2018). Ketamine for the treatment of addiction: Evidence and potential mechanisms. Neuropharmacology, 142, 72–82. doi: 10.1016/j.neuropharm.2018.01.017 [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Krupitsky E, Schutz C, Trevisan L, & D’Souza DC (2003). NMDA receptor antagonism and the ethanol intoxication signal: from alcoholism risk to pharmacotherapy. Ann N Y Acad Sci, 1003, 176–184. doi: 10.1196/annals.1300.010 [DOI] [PubMed] [Google Scholar]
- Lewandowski TA, & Norman J (2015). Dose-Response Assessment. In Torres JA & Bobst S (Eds.), Toxicological Risk Assessment for Beginners (pp. 43–66). Cham: Springer International Publishing. [Google Scholar]
- Linsenbardt DN, & Boehm SL 2nd. (2014). Alterations in the rate of binge ethanol consumption: implications for preclinical studies in mice. Addict Biol, 19(5), 812–825. doi: 10.1111/adb.12052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenbardt DN, & Boehm SL 2nd. (2015). Relative fluid novelty differentially alters the time course of limited-access ethanol and water intake in selectively bred high-alcohol-preferring mice. Alcohol Clin Exp Res, 39(4), 621–630. doi: 10.1111/acer.12679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM (1996). Interactions between ethanol and agents that act on the NMDA-type glutamate receptor. Alcohol Clin Exp Res, 20(8 Suppl), 187A–191A. doi: 10.1111/j.1530-0277.1996.tb01773.x [DOI] [PubMed] [Google Scholar]
- Maxwell CR, Ehrlichman RS, Liang Y, Trief D, Kanes SJ, Karp J, & Siegel SJ (2006). Ketamine produces lasting disruptions in encoding of sensory stimuli. J Pharmacol Exp Ther, 316(1), 315–324. doi: 10.1124/jpet.105.091199 [DOI] [PubMed] [Google Scholar]
- NIAAA. (2004). NIAAA Council Approves Definition of Binge Drinking. NIAAA Newsletter. [Google Scholar]
- Oberlin B, Best C, Matson L, Henderson A, & Grahame N (2011). Derivation and characterization of replicate high- and low-alcohol preferring lines of mice and a high-drinking crossed HAP line. Behav Genet, 41(2), 288–302. doi: 10.1007/s10519-010-9394-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock A, Leung J, Larney S, Colledge S, Hickman M, Rehm J, . . . Degenhardt L (2018). Global statistics on alcohol, tobacco and illicit drug use: 2017 status report. Addiction, 113, 1905–1926. doi: 10.1111/add.14234 [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Limoncelli D, Gueorguieva R, Jatlow P, Boutros NN, Trevisan L, . . . Krystal JH (2004). Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry, 161(10), 1776–1782. doi: 10.1176/ajp.161.10.1776 [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED, Cauley M, Getachew B, & Tizabi Y (2017). Ketamine Differentially Attenuates Alcohol Intake in Male Versus Female Alcohol Preferring (P) Rats. J Drug Alcohol Res, 6. doi: 10.4303/jdar/236030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JS, Ford MM, Yu CH, Brown LL, Finn DA, Garland T Jr., & Crabbe JC (2007). Mouse inbred strain differences in ethanol drinking to intoxication. Genes Brain Behav, 6(1), 1–18. doi: 10.1111/j.1601-183X.2006.00210.x [DOI] [PubMed] [Google Scholar]
- Ruda-Kucerova J, Babinska Z, Luptak M, Getachew B, & Tizabi Y (2018). Both ketamine and NBQX attenuate alcohol drinking in male Wistar rats. Neurosci Lett, 666, 175–180. doi: 10.1016/j.neulet.2017.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino V, Narayan AR, Zeric T, Steardo L, & Cottone P (2013). mTOR activation is required for the anti-alcohol effect of ketamine, but not memantine, in alcohol-preferring rats. Behav Brain Res, 247, 9–16. doi: 10.1016/j.bbr.2013.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salling MC, Skelly MJ, Avegno E, Regan S, Zeric T, Nichols E, & Harrison NL (2018). Alcohol Consumption during Adolescence in a Mouse Model of Binge Drinking Alters the Intrinsic Excitability and Function of the Prefrontal Cortex through a Reduction in the Hyperpolarization-Activated Cation Current. J Neurosci, 38(27), 6207–6222. doi: 10.1523/JNEUROSCI.0550-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon EA, White RD, & Radke AK (2019). Sex Differences in Binge-Like and Aversion-Resistant Alcohol Drinking in C57BL/6J Mice. Alcohol Clin Exp Res, 43(2), 243–249. doi: 10.1111/acer.13923 [DOI] [PubMed] [Google Scholar]
- Strong CE, & Kabbaj M (2020). Neural Mechanisms Underlying the Rewarding and Therapeutic Effects of Ketamine as a Treatment for Alcohol Use Disorder. Front Behav Neurosci, 14, 593860. doi: 10.3389/fnbeh.2020.593860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong MN, Yoneyama N, Fretwell AM, Snelling C, Tanchuck MA, & Finn DA (2010). “Binge” drinking experience in adolescent mice shows sex differences and elevated ethanol intake in adulthood. Horm Behav, 58(1), 82–90. doi: 10.1016/j.yhbeh.2009.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkl B, Altman NS, Forsman A, Forstmeier W, Gurevitch J, Jaric I, . . . Wurbel H (2020). Reproducibility of animal research in light of biological variation. Nat Rev Neurosci, 21(7), 384–393. doi: 10.1038/s41583-020-0313-3 [DOI] [PubMed] [Google Scholar]
- Wilcox CE, Dekonenko CJ, Mayer AR, Bogenschutz MP, & Turner JA (2014). Cognitive control in alcohol use disorder: deficits and clinical relevance. Rev Neurosci, 25(1), 1–24. doi: 10.1515/revneuro-2013-0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RA, Wong MM, Clark DB, Leonard KE, Schulenberg JE, Cornelius JR, . . . Puttler LI (2006). Predicting risky drinking outcomes longitudinally: what kind of advance notice can we get? Alcohol Clin Exp Res, 30(2), 243–252. doi: 10.1111/j.1530-0277.2006.00033.x [DOI] [PMC free article] [PubMed] [Google Scholar]