

Abstract
Modification of polyketides with fluorine offers a promising approach to develop new pharmaceuticals. While synthetic chemical methods for site-selective incorporation of fluorine in complex molecules have improved in recent years, approaches for the biosynthetic incorporation of fluorine in natural compounds are still rare. Here, we report a strategy to introduce fluorine into complex polyketides during biosynthesis. We exchanged the native acyltransferase domain (AT) of a polyketide synthase (PKS), which acts as the gatekeeper for selection of extender units, with an evolutionarily related but substrate tolerant domain from metazoan type I fatty acid synthase (FAS). The resulting PKS/FAS hybrid can utilize fluoromalonyl coenzyme A and fluoromethylmalonyl coenzyme A for polyketide chain extension, introducing fluorine or fluoro-methyl units in polyketide scaffolds. We demonstrate the feasibility of our approach in the chemoenzymatic synthesis of fluorinated 12- and 14-membered macrolactones and fluorinated derivatives of the macrolide antibiotics YC-17 and methymycin.
The majority of new FDA-approved drugs are small organic molecules, making up over 90 percent of pharmaceuticals on the market today 1. Among those, natural products are highly represented 2, as their structures are presumed to undergo preselection during evolution to interact with cellular biomacromolecules 3. Fluorination has been widely used in medicinal chemistry for lead structure optimization, as its electronegativity and its small size can strongly impact molecular properties, thereby modulating protein–ligand interactions, bioavailability and metabolic stability 4. Reflecting the importance of fluorination, about a quarter of all small molecule drugs contain at least one fluorine atom, including the antidepressant Prozac, the cholesterol-lowering drug Lipitor and quinolone antibiotic Ciprobay 5. Applications of organofluorine chemistry toward natural product biosynthesis are rare due to the paucity of enzymes that catalyze addition of F atoms in secondary metabolism 6. Thus, new methods that enable fluorine derivatization are urgently needed to bridge the gap between the inherent bioactivity of a natural compound and its development as a therapeutic agent.
Polyketide natural products comprise over 10,000 molecules with a wide range of bioactivities and are among the most prominent classes of approved clinical agents 7,8. In nature, polyketides are assembled mainly from simple monomeric acetate and propionate units by polyketide synthases (PKSs). In the type I cis-AT subclass, PKSs occur as multi-functional protein mega-complexes comprising a series of catalytic domains organized in modules on one or few polypeptide chains. Typically, one PKS module requires a minimum of three domains for a two-carbon extension of a growing polyketide intermediate: an acyltransferase (AT) domain that selects an acyl-coenzyme A (CoA) extender unit and transfers the acyl moiety to the acyl carrier protein (ACP) domain, and a ketosynthase (KS) domain that accepts a growing chain from the ACP of the previous module and catalyzes a decarboxylative Claisen condensation to extend the polyketide chain. A canonical PKS module may further contain up to three additional domains, a ketoreductase (KR), a dehydratase (DH) and an enoylreductase (ER) that tailor the β-keto function prior to the next round of chain extension. The final module in the biosynthetic assembly line typically includes a thioesterase (TE) domain located at the C-terminus and is responsible for polyketide chain release as a linear chain or a macrocyclic product (Fig. 1a). Engineering of modular polyketide biosynthesis for the directed assembly of new-to-nature polyketides is a highly aspired aim 9 and offers an alternative or complementary approach to organic synthesis. Changing substrate specificity of a single enzymatic domain of a specific PKS module by protein engineering enables, for example, the regioselective modification of the product during biosynthesis.
Fig. 1: Modular PKSs and hybrid design.
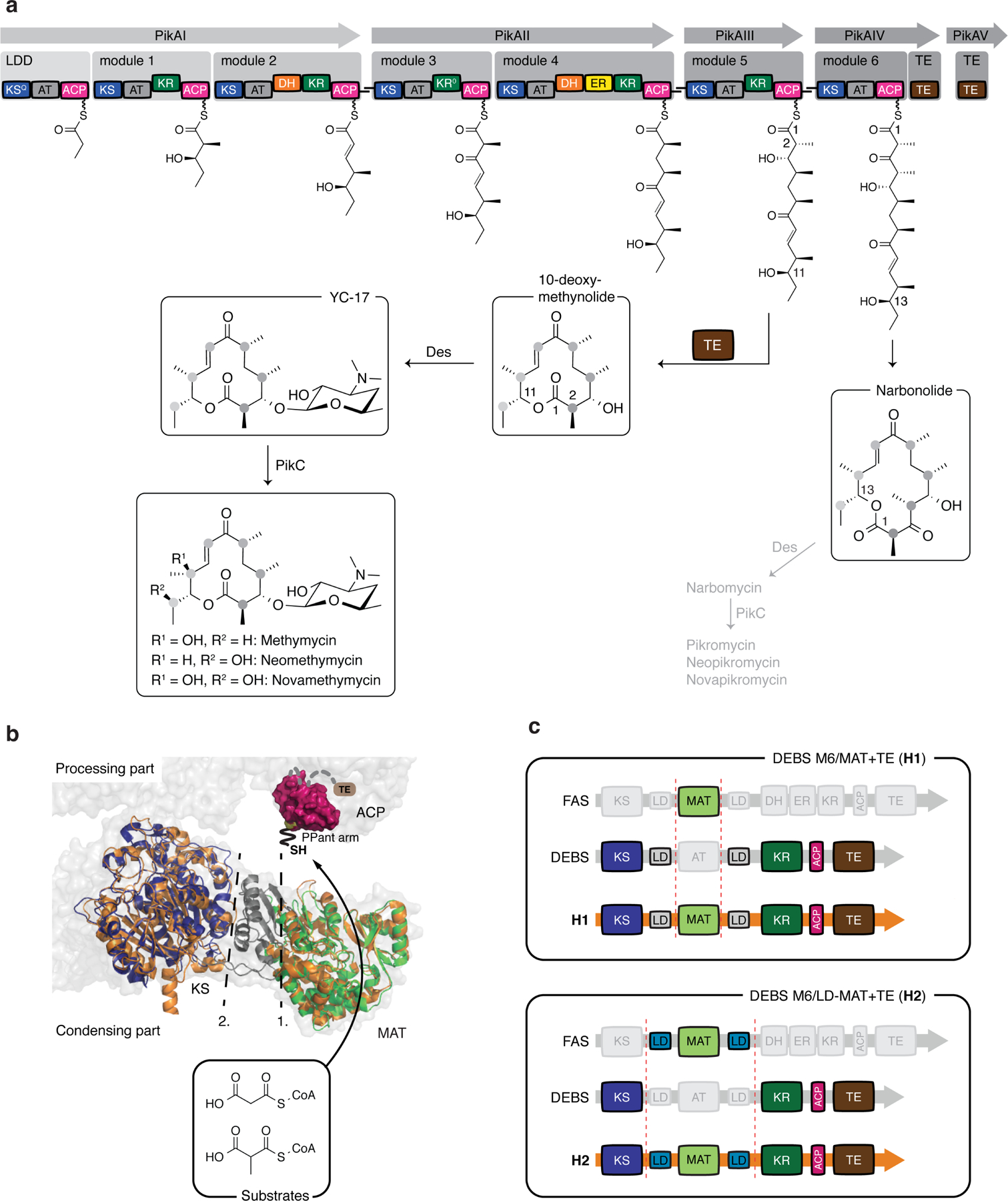
a, Assembly line like biosynthesis in the methymycin/pikromycin pathway. The modular pikromycin synthase can either produce a 12- or a 14-membered macrolactone. The polyketide products are subsequently glycosylated and oxidized by post-PKS enzymes. b, Function of the murine MAT domain and its insertion into the KS-MAT didomain (KS: blue; LD: grey; MAT: green; PDB code: 5my0). Atomic coordinates: porcine FAS (grey; PDB code: 2vz9), the ACP domain (purple; PDB code: 2png) and DEBS module 5 KS-AT didomain (orange; PDB code: 2hg4) 13,15,19,20. c, Design of DEBS/FAS hybrids H1 and H2.
Enzymes that catalyze direct fluorination of polyketides remain unknown. Previous efforts have shown that engineered PKS assembly lines can utilize non-canonical extender substrates 10 and, by using fluoromalonyl coenzyme A (F-Mal-CoA), allow fluorine to be introduced into triketide lactones 11,12. However, the application of this concept to the formation of a complete fluorinated macrolide structure had not been demonstrated. In canonical modular PKSs, extender subunits are selected by the AT domains, which act as the “gatekeepers” of polyketide biosynthesis and typically ensure the introduction of a defined acyl-CoA with high substrate specificity (Supplementary Fig. 1). We have recently demonstrated that the promiscuous AT domain from metazoan fatty acid synthase (FAS), termed malonyl-/acetyl transferase (MAT), is able to transfer various acyl-CoA moieties with high efficiencies 13, different to AT domains from PKSs 14. We hypothesized that this domain may also transfer fluorinated extender substrates and can be integrated in a PKS module, since FASs and PKSs are structurally and biochemically related (Fig. 1b) 15–17. To test the feasibility of this approach, we chose to work with module 6 of DEBS including its C-terminal TE domain (DEBS M6+TE) (Fig. 1c) 18.
Results and discussion
Initially, we analyzed whether the polyspecific MAT domain of murine FAS is generally suitable for divergent evolution for its perspective use in microbial polyketide production 21. In a preliminary screen on 42 MAT mutants, we were able to confirm important properties in this regard: mutations in the active site changed the substrate specificity significantly, while preserving the stability of the fold (Supplementary Fig. 2–4). Hence, we proceeded by investigating whether the MAT domain is also able to select F-Mal-CoA as a substrate, and whether it accepts the ACP domain of the DEBS M6 for substrate processing. F-Mal-CoA (1) was chemically synthesized following the four-step route to the fluoromalonic acid halfthioester by Saadi and Wennemers 22 with the subsequent transacylation of the F-Mal moiety to free CoA (Fig. 2a, Supplementary Fig. 5–6.). F-Mal-CoA was most likely received as a diastereomeric mixture of two compounds that are epimeric in the fluoromalonyl-moiety. In an enzyme-coupled fluorometric assay with the domains as individual proteins 13, we observed excellent transfer kinetics of MAT for F-Mal-CoA (Km/kcat = 6.9 × 106 M−1 s−1) as well as its ability to catalyze transacylation with DEBS ACP6 (Supplementary Fig. 7–10). The specificity constant of MAT for loading ACP6 with methylmalonyl moieties (Km/kcat = 6.9 × 106 M−1 s−1) was 2 – 3 orders of magnitude higher than DEBS AT6 (Supplementary Table 1), which can be explained by the inherently high transacylation rates of the MAT domain.
Fig. 2: Function of the hybrid DEBS/FAS modules.
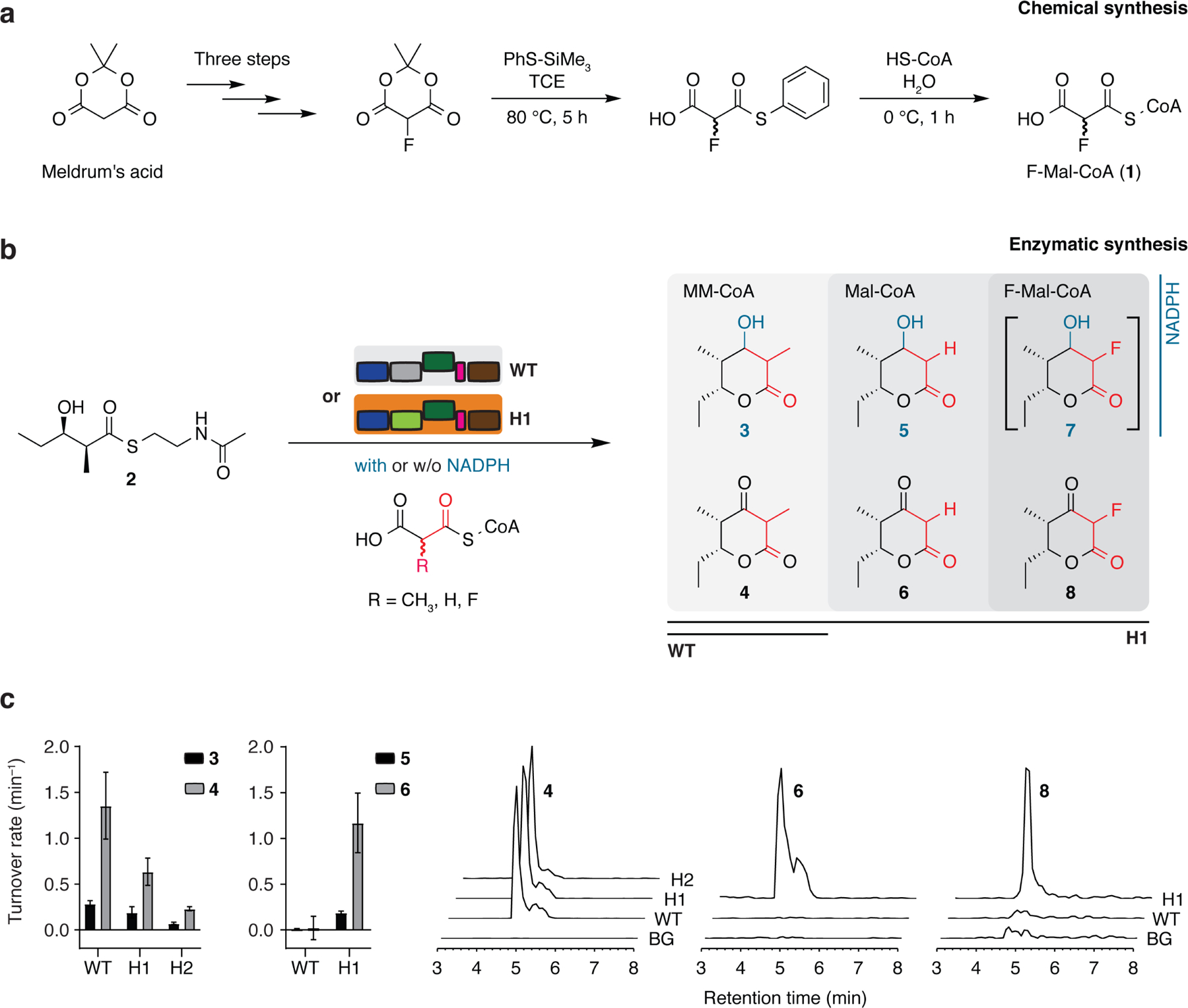
a, Synthetic route to F-Mal-CoA. Two diastereomers are obtained from chemical synthesis indicated with wavy lines at the epimeric center. b, Hybrid PKS-mediated synthesis of triketide lactones (TKLs) from 2 and MM-CoA, Mal-CoA or F-Mal-CoA. Compound 2 directly binds to the KS active site upon release of N-acetylcysteamine (not shown), and the KS catalyzes the decarboxylative Claisen condensation with the incoming ACP-bound malonyl or malonyl derivative (release of CO2 not shown) (for details, see Supplementary Fig. 14). Compound 7 was only produced in traces (not shown), presumably due to a substrate selective KR domain. c, Turnover rates for the WT-, H1- and H2-mediated formation of TKLs and detection by HPLC-MS (EIC: 4 [M-H]− m/z = 169.12; 6 [M-H]− m/z = 155.16; 8 [M-H]− m/z = 173.11). Data show mean and standard deviation of three independent experiments (biological replicates).
We constructed two hybrids of Saccharopolyspora erythraea DEBS and murine FAS, which differed in the DEBS/FAS interface by substituting DEBS AT6 with or without its adjacent linker domain, giving construct H1 (MAT hybrid) or H2 (LD-MAT hybrid), respectively (Fig. 1c, Supplementary Fig. 11). Domain boundaries for the AT exchanges were defined based on protein structures and the previous report by Yuzawa et al. (Supplementary Fig. 11–12) 23. The hybrids H1 and H2 were produced in Escherichia coli in yields similar to the wildtype protein (WT), but with different oligomeric stability (Supplementary Fig. 13). This provided overall yields of purified dimeric species of about 4 and 2 mg per liter of cell culture for H1 and H2, respectively (compared to 8 mg of DEBS M6+TE WT). Given that PKS modules occur as dimers in solution 24, deviations from a dimeric state were treated as indication for fold instabilities. Accordingly, the high fraction of oligomeric species observed in the SEC profile of H2 preparations, disfavored its further use. Native PAGE moreover indicated contamination with degraded or disassembled PKS proteins. On the basis of H1 showing significantly higher turnover rates in synthesizing triketide lactones (TKLs) from the N-acetylcysteamine-activated diketide, (2S,3R)-2-methyl-3-hydroxypentanoyl-N-acetylcysteamine thioester (2) and MM-CoA (see Supplementary Fig. 14 for details of TKL synthesis), we decided to pursue further efforts with H1 only (Fig. 2b–2c, Supplementary Fig. 15 and Supplementary Table 2, see Supplementary Table 3 for an overview of substrates used). In addition, H1 was able to produce C-2 derivatives of TKLs and the turnover rate using malonyl-CoA (Mal-CoA) to generate compound 6 was even twice as fast as using MM-CoA to produce compound 4. Also Yuzawa et al. observed faster kinetics with a non-native extender substrate for a hybrid module and we confirm that a PKS module is not necessarily optimized for the substrate loaded by the native AT 23. Given the production of compound 8 in the presence of F-Mal-CoA and H1, these data demonstrate that the substrate promiscuity of MAT enables substrate elongation with a fluorinated extender unit in a hybrid PKS module (see ref.11,23 for spectroscopic data on compound 6 and 8).
With the catalytically competent hybrid H1 in hand, we first aimed to produce 12-membered macrolactones from the pikromycin pathway that are diversified at the C-2 position (Fig. 1a, Supplementary Fig. 16). These are interesting compounds in drug discovery as exhibiting microbial activity against erythromycin-resistant Staphylococcus aureus strains and bind in a unique way to the 50S ribosomal subunit 25–27. To this end, we used a PIKS pentaketide primer in activated form, available via an eleven-step synthesis from Roche ester as previously reported 28. Several earlier studies have used the pentaketide for PKS-mediated conversion to 12-membered macrolactones 29–31. H1 mediated elongation of the pentaketide (9) with MM-CoA, Mal-CoA or F-Mal-CoA, respectively, produced 10-deoxymethynolide (10) as well as the desmethylated macrocycle (12) and the fluorinated analog (14). The absence of NADPH led to the C-3 oxidized species (11, 13 and 15) (compounds 10–15 were confirmed by HRMS; Fig. 3a and b, Supplementary Fig. 17 and Supplementary Table 2). The H1-mediated conversion rates were faster for Mal-CoA and slightly slower for F-Mal-CoA compared to the native substrate MM-CoA yielding compounds 12, 14 and 10, respectively (Fig. 3c, Supplementary Table 2).
Fig. 3: Enzymatic synthesis of 10-deoxymethynolide derivatives.
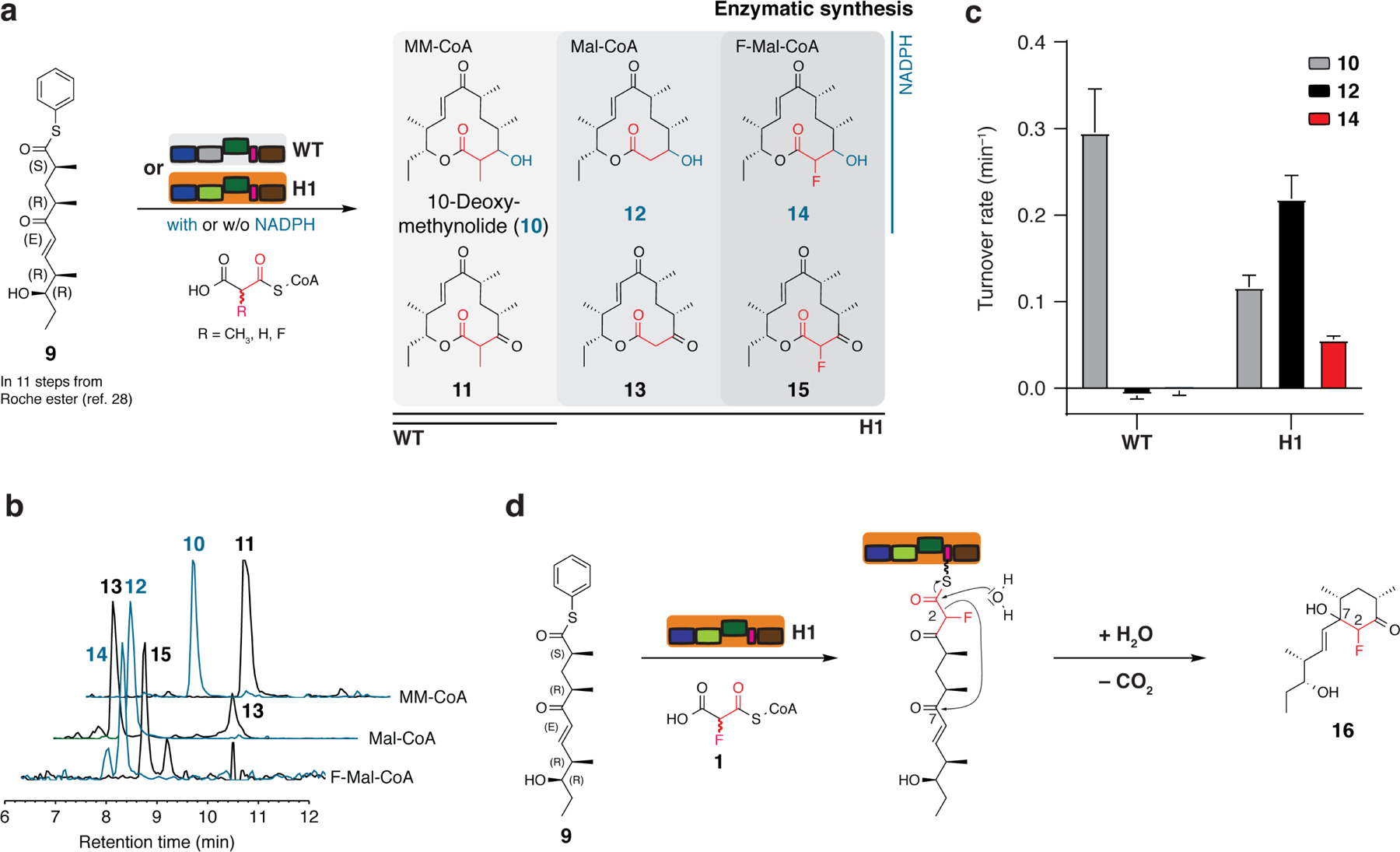
a, Reaction scheme for the H1-mediated conversion of pentaketide 9 to new derivatized keto- and macrolactones 10-15 (see also Supplementary Table 2). For details to the macrolactone synthesis, see Supplementary Fig. 14). b, Detection of macrolactones by HPLC-MS (EIC: 10 [M+Na]+ m/z = 319.11; 11 [M+Na]+ m/z = 317.09; 12 [M+H]+ m/z = 305.09; 13 [M+Na]+ m/z = 303.08; 14 [M+Na]+ m/z = 323.08 and 15 [M+Na]+ m/z = 321.07). c, Turnover rates for H1-mediated macrolactone formation in comparison with the WT turnover rate. Data show mean and standard deviation of three independent experiments (biological replicates). d, Formation of side product 16 during the synthesis of fluoro-compound 15. The mechanism involves hydrolysis, decarboxylation and cyclohexanone formation of the hexaketide intermediate, but the order of the steps is not known, as cyclohexanone formation can also occur first via aldol addition 29.
Intriguingly, when seeking to conduct scale-up to isolate milligram quantities, we faced challenges for the H1 mediated reactions (with F-Mal-CoA) to the fluoro-compounds 14 and 15. Here, very low amounts of products were obtained and reactions were contaminated with significant levels of side products. When working-up the reaction mixture to target compound 15, we identified compound 16 as the main product, presumably generated from the hexaketide intermediate via hydrolysis, decarboxylation and cyclohexanone formation (Fig. 3d). This side reaction has been reported previously as originating from the narrow substrate specificity of the DEBS TE domain, preventing macrolactonization to the 12-membered ring when a keto-group is present at C-3 29. We elected to pause further analysis of the origin for the low yields of compound 14, and reasoned that chemical instability relates to the C-2 HFC unit.
As a next step, we postulated that direct installation of a C-2 fluoro-methyl (MeFC) unit might increase stability by abstracting the acidic proton while maintaining a similar size compared to the natural compound. Notably, this would give direct access to fluorination patterns of the erythromycin derivatives flurithromycin and solithromycin (Supplementary Fig. 18), two examples of semisynthetic next-generation macrolides, in which acidic protons have been replaced by fluorine 32. For solithromycin, the fluorine induces improved binding to the ribosome and modulates pharmacokinetics leading to superior antibiotic properties (23S rRNA binding) 33,34.
Natural polyketides with a MeFC unit are not known and just a handful of polyketides with a gem-dimethyl substitution (MeMeC unit) have been discovered to date, mainly ascribed to the methylation of the condensation product by C-methyltransferases. Recently, Keasling and coworkers demonstrated that modules of yersiniabactin and epothilone PKSs are capable of elongating the growing acyl-chain with dimethylmalonyl moieties 35, however disubstituted malonyl moieties have not yet been employed in directed biosynthesis. In order to incorporate the MeFC unit into macrolides, we established a route for chemical synthesis of fluoromethylmalonyl-CoA (17, F-MM-CoA, diastereomeric in the fluoromethylmalonyl-moiety) following a similar strategy as for F-Mal-CoA (Fig. 4a). Again, the MAT showed excellent transfer kinetics for F-MM-CoA (Km/kcat = 4.5 × 106 M−1 s−1) (Supplementary Fig. 19 and Supplementary Table 1). Finally, F-MM-CoA proved to be accepted by H1 for elongation of the pentaketide (9) in presence of NADPH to produce 2-fluoro-10-deoxymethynolide (18) (for mechanistic implications, see Supplementary Fig. 20). Conversion of the extender unit was verified by the NADPH consumption assay (turnover rate: 0.024 ± 0.02 min−1) as well as HRMS, and reaction scale-up provided full structural analysis by NMR (Fig. 4b). Macrolactone 18 features a (2S,3S)-configuration identical to solithromycin (see Supplementary Note). The stereoselectivity of this reaction indicates that the DEBS M6-derived construct H1 accommodates the F-MM moiety for substrate elongation with fluorine at the hydrogen position of the natively used methylmalonyl moiety (for formation of a cyclohexanone side product in absence of NADPH, see Supplementary Fig. 21). We note that the yields of compound 18 were improved significantly to 27 % when exchanging the DEBS TE with the PIKS TE, receiving hybrid H1.1, by following the design of Koch et al. (Supplementary Fig. 22) 36. The higher yield correlated also with higher turnover rates in the NADPH consumption assay (0.17 min−1) identifying the DEBS TE domain as a bottleneck of H1. 2-Fluoro-10-deoxymethynolide (18) was finally added to a culture of strain DHS316 or YJ112 for desosaminylation to isolate the 2-fluoro-YC-17 (19) and with YJ112 for desosaminylation with subsequent oxidation to isolate the 2-fluoro-methymycin (20) (Supplementary Fig. 23) 37. The generation of these macrolides demonstrates that post-PKS processing is generally not hindered by the introduced fluorine atom.
Fig. 4: Synthesis of new fluorinated macrolide antibiotics and 14-membered macrolactones.
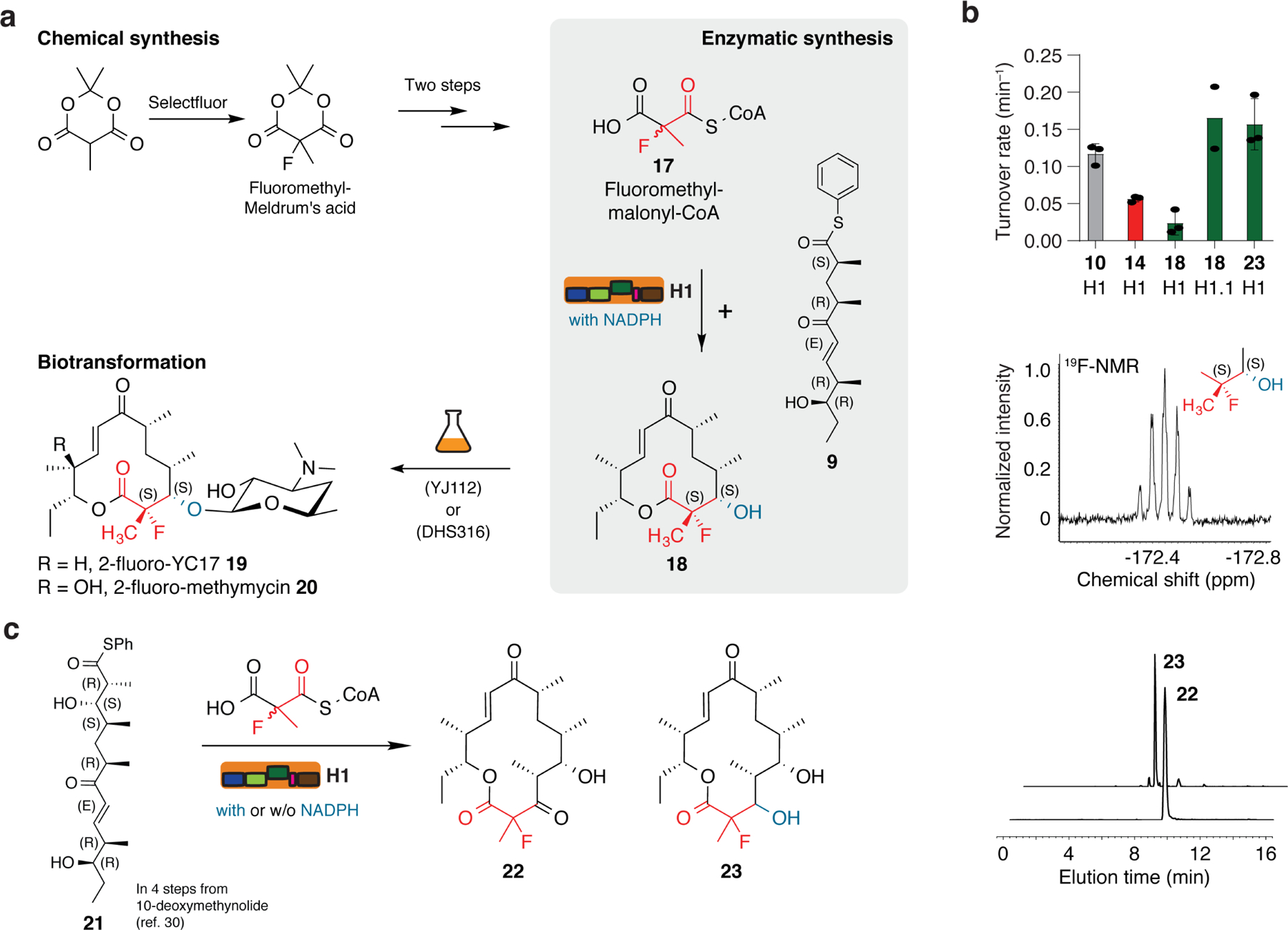
a, Chemoenzymatic approach to establish the MeFC unit at position C-2 in the macrolactone 10. Chemical synthesis was performed analogously to 1 from the respective Meldrum’s acid and the product was converted enzymatically with pentaketide 9 and NADPH to macrolactone 18. Compound 18 was transformed to the fluorinated derivatives of the antibiotic YC-17 (19) and methymycin (20) using the strain DHS316 or YJ112 37. b, Selected data on target compounds and enzymatic turnover. Turnover rates for the H1- and H1.1-mediated conversion of pentaketide 9 and hexaketide 21 with MM-CoA, F-Mal-CoA and F-MM-CoA yielding compounds 10, 14, 18 and 23, respectively. Each data point reflects an independent experiment (biological replicate); mean standard deviations are given, except for H1.1 data (left panel). Elongation using the substrate F-MM-CoA with subsequent reduction to compound 18 can be verified by the multiplicity in 19F-NMR as a quintet (middle panel). The production of compounds 22 and 23 was demonstrated by HPLC-HRMS (EIC: 22 [M+H]+ m/z = 371.2225; 23 [M+H]+ m/z = 373.2384 (right panel). c, Reaction scheme for the H1-mediated conversion of hexaketide 21 with F-MM-CoA to 2-fluoro-narbonolide (22) as well as the reduced analog (23) (for details of the synthesis, see Supplementary Fig. 14).
Encouraged by these findings, we further tested the ability of H1 to produce 14-membered macrolactones with the activated PIKS hexaketide 21 that is accessible within four steps from 10-deoxymethynolide as reported before 30. When F-MM-CoA was supplied to the reaction solution, 2-fluoro-narbonolide (22) as well as the reduced analog (23) in presence of NADPH were produced and verified by HPLC-HRMS (Fig. 4b & c and Supplementary Fig. 24). We observed similar turnover rates in the NADPH consumption assay (0.16 ± 0.03 min−1) for compound 23 as compared to H1.1 producing 18 from the pentaketide substrate. Both macrolactones, compound 18 and 22, are valuable direct precursor molecules for medicinal chemistry, as a plethora of fluorinated 12- and 14-membered macrolides can now be generated by attaching different sugar moieties or by varying oxidation patterns (Supplementary Fig. 16) to screen for the macrolide with the highest potency against resistant strains 25.
In conclusion, we report the regioselective derivatization of polyketides with fluorine by utilizing the promiscuous MAT domain of metazoan FAS, integrated as a domain in a bacterial modular PKS. Our approach extends previous findings of Chang and coworkers, who introduced F-Mal-CoA metabolism in the cell 11,12. The AT exchange strategy maintains the overall protein architecture and integrity of the vectorial synthesis of type I PKSs, which paves the way for precursor-mediated site-selective incorporation of non-canonical chemical functions at multiple positions in a diversity of polyketides during biosynthesis. Specifically, we demonstrate the relevance of this method by producing regioselectively fluorinated 12- and 14-membered macrolactones and the new macrolide antibiotics 2-fluoro-YC-17 and 2-fluoro-methymycin with the MeFC unit selectively introduced in the S-configuration (stereochemistry as in solithromycin). This approach could be extended into in vivo applications with specialized MAT variants and in combination with further achievements in the field, mainly to a microbial host engineered for precursor supply, and for handling the toxicity and reactivity of fluorinated substrates and products.
Methods
Methods, additional references and spectra are available in the supplementary information.
Synthesis of priming substrates
(i) Diketide SNAC (2) was synthesized in three steps. First, Evans auxiliary was elongated with propanal by aldol addition 38. Then, LiOH/H2O2 was used for cleavage of the Evans oxazolidinone, and the generated acid was esterified with N-acetylcysteamine using ethyl chloroformate to receive the thioester product (2) 39. (ii) The activated PIKS pentaketide was produced in 11 steps from Roche ester as described before 28. Key steps are the alkylation of an iodinated intermediate derived from Roche ester with (S,S)-pseudoephedrine propionamide, and cross metathesis of an α,β-unsaturated ketone intermediate, , with a silyl ether,. (iii) The hexaketide was produced in four steps from 10-deoxymethynolide beginning with 3-OH protection followed by reductive ring opening, TEMPO-catalyzed selective oxidation and finally thioester formation 30.
General synthesis of fluorinated CoA-Ester
Fluoro-Meldrum’s and fluoromethyl-Meldrum’s acid were synthesized from Meldrum’s acid and methyl-Meldrum’s acid, respectively, in three steps using a previously described method utilizing Selectfluor® 22. The Meldrum’s acids were treated with trimethyl(phenylthio)silane to produce the respective malonic acid halfthioesters. Fluorinated CoA-esters were eventually synthesized by transacylation from malonic acid halfthioesters to free coenzyme A (CoASH) 40.
Design and recombinant production of FAS/PKS hybrid proteins
Vectors (pET22b derived plasmids) encoding hybrid DEBS/FAS proteins (pMJD076 (H2) and pMJD077 (H1)) were produced by sequence and ligation independent cloning using the In-Fusion HD Cloning Kit (Takara Bio, USA). Domain borders were determined from structural and sequence information. Vector pMSR001 (H1.1) was also generated by sequence and ligation independent cloning following the design of Koch et al. 36. All constructs were expressed in E. coli BL21 Gold (DE3) cells in 1 L TB medium cultures at 20 °C after induction with 0.25 mM IPTG. After harvesting cells by centrifugation and lysis with French Press, the cleared cytosolic fraction was subjected to Ni-NTA affinity chromatography. After concentration to 10–20 mg mL−1, the proteins were frozen in liquid nitrogen and stored at −80 °C. Samples were thawed and further purified by SEC before analysis or product synthesis.
General procedure for the biosyntheses
Small scale biosynthesis of TKLs and macrolactones were carried out with 4 µM enzyme, 5 mM 2 or 1 mM 9 or 1 mM 21, 200 µM X-CoA and 0 or 60 µM NADPH in the assay buffer (400 mM phosphate buffer, 20 % (v/v) glycerol, 1 mM EDTA, 0.8 % DMSO, pH 7.2) at 25 °C. The reactions were followed fluorometrically by monitoring the consumption of NADPH. Products were extracted with EtOAc and confirmed by HPLC-MS.
General procedure for the up-scaled syntheses of macrolactones
In order to receive larger amounts of macrolactones, reactions were carried out in 10–50 mL scale with the final concentrations of 5–10 µM H1, 300–600 µM 9, 400–4000 µM X-CoA and 500–1000 µM NADPH in the reaction buffer at 25 °C. After at least 4 h of incubation, products were extracted with EtOAc and purified on a silica column. Compound 16 was additionally purified by HPLC on a C18 column. Biotransformation of 2-fluoro-10-deoxymethynolide (18) to obtain macrolides 2-fluoro-YC-17 (19) and 2-fluoro-methymycin (20) was conducted according to the published procedures with minor modifications 41,42.
Supplementary Material
Acknowledgments:
This work was supported by a Lichtenberg grant of the Volkswagen Foundation to M.G. (grant number 85701). Further support was received from the LOEWE program (Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz) of the state of Hesse conducted within the framework of the MegaSyn Research Cluster. We would like to thank Khanh Vu Huu and Kudratullah Karimi for MS-analysis of acyl carrier proteins and Karthik S. Paithankar for proofreading the manuscript. Further, we are grateful to the Bode group for the extensive support in HPLC-MS analysis and Julia Wirmer-Bartoschek and Gabriele Sentis for support in NMR analysis. D.H.S. is grateful to NIH grant R35 GM118101 and the Hans W. Vahlteich Professorship for support.
Funding:
LOEWE program (Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz) of the state of Hesse (MG)
Lichtenberg grant of the Volkswagen Foundation (MG)
NIH grant R35 GM118101 (DHS)
Footnotes
Competing interests: A.R. declares a financial interest as co-founder of kez.biosolutions GmbH (Potsdam, Germany). All other authors declare no competing interests.
Data and materials availability:
All data supporting the main findings of the article, including material and methods, are described in the Article or Supplementary Information. Alternatively, the data is available from the corresponding author on request.
References and Notes:
- 1.de la Torre BG & Albericio F The Pharmaceutical Industry in 2018. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 24, 809–820, doi; 10.3390/molecules24040809 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newman DJ & Cragg GM Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod 75, 311–335, doi: 10.1021/np200906s (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Nussbaum F, Brands M, Hinzen B, Weigand S & Häbich D Antibacterial Natural Products in Medicinal Chemistry—Exodus or Revival? Angew. Chem. Int. Ed 45, 5072–5129, doi: 10.1002/anie.200600350 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 51, 4359–4369, doi: 10.1021/jm800219f (2008). [DOI] [PubMed] [Google Scholar]
- 5.Müller K, Faeh C & Diederich F Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 317, 1881–1886, doi: 10.1126/science.1131943 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Carvalho MF & Oliveira RS Natural production of fluorinated compounds and biotechnological prospects of the fluorinase enzyme. Critical Reviews in Biotechnology 37, 1–18, doi: 10.1080/07388551.2016.1267109 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Hertweck C The Biosynthetic Logic of Polyketide Diversity. Angew. Chem. Int. Ed 48, 4688–4716, doi: 10.1002/anie.200806121 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Staunton J & Weissman KJ Polyketide biosynthesis: a millennium review. Nat. Prod. Rep 18, 380–416, doi: 10.1039/A909079G (2001). [DOI] [PubMed] [Google Scholar]
- 9.Klaus M & Grininger M Engineering strategies for rational polyketide synthase design. Natural Product Reports 35, 1070–1081, doi: 10.1039/C8NP00030A (2018). [DOI] [PubMed] [Google Scholar]
- 10.Kalkreuter E, CroweTipton JM, Lowell AN, Sherman DH & Williams GJ Engineering the Substrate Specificity of a Modular Polyketide Synthase for Installation of Consecutive Non-Natural Extender Units. J. Am. Chem. Soc 141, 1961–1969, doi: 10.1021/jacs.8b10521 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker MC et al. Expanding the Fluorine Chemistry of Living Systems Using Engineered Polyketide Synthase Pathways. Science 341, 1089–1094, doi: 10.1126/science.1242345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thuronyi BW, Privalsky TM & Chang MCY Engineered Fluorine Metabolism and Fluoropolymer Production in Living Cells. Angew. Chem. Int. Ed 56, 13637–13640, doi: 10.1002/ange.201706696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rittner A, Paithankar KS, Huu KV & Grininger M Characterization of the Polyspecific Transferase of Murine Type I Fatty Acid Synthase (FAS) and Implications for Polyketide Synthase (PKS) Engineering. ACS Chem. Biol 13, 723–732, doi: 10.1021/acschembio.7b00718 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Stegemann F & Grininger M Transacylation Kinetics in Fatty Acid and Polyketide Synthases and its Sensitivity to Point Mutations**. ChemCatChem 13, 2771–2782, doi: 10.1002/cctc.202002077 (2021). [DOI] [Google Scholar]
- 15.Maier T, Leibundgut M & Ban N The Crystal Structure of a Mammalian Fatty Acid Synthase. Science 321, 1315–1322, doi: DOI: 10.1126/science.1161269 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Dutta S et al. Structure of a modular polyketide synthase. Nature 510, 512–517, doi: 10.1038/nature13423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weissman KJ The structural biology of biosynthetic megaenzymes. Nature Chemical Biology 11, 660–670, doi: 10.1038/nchembio.1883 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Wu N, Kudo F, Cane DE & Khosla C Analysis of the Molecular Recognition Features of Individual Modules Derived from the Erythromycin Polyketide Synthase. J. Am. Chem. Soc 122, 4847–4852, doi 10.1021/ja000023d (2000). [DOI] [Google Scholar]
- 19.Tang Y, Kim CY, Mathews II, Cane DE & Khosla C The 2.7-Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U.S.A 103, 11124–11129, doi: 10.1073/pnas.0601924103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ploskoń E et al. A Mammalian Type I Fatty Acid Synthase Acyl Carrier Protein Domain Does Not Sequester Acyl Chains. J. Biol. Chem 283, 518–528, doi: 10.1074/jbc.M703454200 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Yoshikuni Y, Ferrin TE & Keasling JD Designed divergent evolution of enzyme function. Nature 440, 1078–1082, doi: 10.1038/nature04607 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Saadi J & Wennemers H Enantioselective aldol reactions with masked fluoroacetates. Nature Chemistry 8, 276–280, doi: 10.1038/nchem.2437 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Yuzawa S et al. Comprehensive in Vitro Analysis of Acyltransferase Domain Exchanges in Modular Polyketide Synthases and Its Application for Short-Chain Ketone Production. ACS Synth. Biol 6, 139–147, doi: 10.1021/acssynbio.6b00176 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Klaus M et al. Solution Structure and Conformational Flexibility of a Polyketide Synthase Module. JACS Au, doi: 10.1021/jacsau.1c00043 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinde PB et al. Combinatorial biosynthesis and antibacterial evaluation of glycosylated derivatives of 12-membered macrolide antibiotic YC-17. Journal of Biotechnology 168, 142–148, doi: 10.1016/j.jbiotec.2013.05.014 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Auerbach T et al. Structural basis for the antibacterial activity of the 12-membered-ring mono-sugar macrolide methymycin. Biotechnologia 84 (2009). [Google Scholar]
- 27.Almutairi MM et al. Co-produced natural ketolides methymycin and pikromycin inhibit bacterial growth by preventing synthesis of a limited number of proteins. Nucleic Acids Research 45, 9573–9582, doi: 10.1093/nar/gkx673 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen DA et al. Biocatalytic Synthesis of Pikromycin, Methymycin, Neomethymycin, Novamethymycin, and Ketomethymycin. J. Am. Chem. Soc 135, 11232–11238, doi: 10.1021/ja404134f (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen DA, Koch AA & Sherman DH Identification of a Thioesterase Bottleneck in the Pikromycin Pathway through Full-Module Processing of Unnatural Pentaketides. J. Am. Chem. Soc 139, 13450–13455, doi: 10.1021/jacs.7b06432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen DA, Koch AA & Sherman DH Substrate Controlled Divergence in Polyketide Synthase Catalysis. J. Am. Chem. Soc 137, 3735–3738, doi: 10.1021/ja511743n (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalkreuter E et al. Computationally-guided exchange of substrate selectivity motifs in a modular polyketide synthase acyltransferase. Nature Communications 12, 2193, doi: 10.1038/s41467-021-22497-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandes P, Martens E, Bertrand D & Pereira D The solithromycin journey—It is all in the chemistry. Bioorganic & Medicinal Chemistry 24, 6420–6428, doi: 10.1016/j.bmc.2016.08.035 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Donald BJ, Surani S, Deol HS, Mbadugha UJ & Udeani G Spotlight on solithromycin in the treatment of community-acquired bacterial pneumonia: design, development, and potential place in therapy. DDDT Volume 11, 3559–3566, doi: 10.2147/DDDT.S119545 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhanel GG et al. Solithromycin: A Novel Fluoroketolide for the Treatment of Community-Acquired Bacterial Pneumonia. Drugs 76, 1737–1757, doi: 10.1007/s40265-016-0667-z (2016). [DOI] [PubMed] [Google Scholar]
- 35.Poust S et al. Divergent Mechanistic Routes for the Formation of gem-Dimethyl Groups in the Biosynthesis of Complex Polyketides. Angew. Chem. Int. Ed 54, 2370–2373, doi: 10.1002/anie.201410124 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Koch AA et al. Probing Selectivity and Creating Structural Diversity Through Hybrid Polyketide Synthases. Angew. Chem. Int. Ed 59, 13575–13580 doi: 10.1002/ange.202004991 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung WS et al. Enhanced heterologous production of desosaminyl macrolides and their hydroxylated derivatives by overexpression of the pikD regulatory gene in Streptomyces venezuelae. Appl. Environ. Microbiol 74, 1972–1979, doi: 10.1128/AEM.02296-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma KK & Boddy CN The thioesterase domain from the pimaricin and erythromycin biosynthetic pathways can catalyze hydrolysis of simple thioester substrates. Bioorganic & Medicinal Chemistry Letters 17, 3034–3037, doi: 10.1016/j.bmcl.2007.03.060 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Peter DM et al. Screening and Engineering the Synthetic Potential of Carboxylating Reductases from Central Metabolism and Polyketide Biosynthesis. Angew. Chem. Int. Ed 54, 13457–13461, doi: 10.1002/anie.201505282 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Dunn BJ, Watts KR, Robbins T, Cane DE & Khosla C Comparative Analysis of the Substrate Specificity of trans- versus cis-Acyltransferases of Assembly Line Polyketide Synthases. Biochemistry 53, 3796–3806, doi: 10.1021/bi5004316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowell AN et al. Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics through Late-Stage Polyketide Assembly, Tailoring, and C—H Functionalization. J. Am. Chem. Soc 139, 7913–7920, doi: 10.1021/jacs.7b02875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeMars MD et al. Biochemical and Structural Characterization of MycCI, a Versatile P450 Biocatalyst from the Mycinamicin Biosynthetic Pathway. ACS Chem. Biol 11, 2642–2654, doi: 10.1021/acschembio.6b00479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.