

Abstract
Sepsis is a persistent systemic inflammatory condition involving multiple organ failures resulting from a dysregulated immune response to infection, and one of the hallmarks of sepsis is endothelial dysfunction. During its progression, neutrophils are the first line of innate immune defence against infection. Aside from traditional mechanisms, such as phagocytosis or the release of inflammatory cytokines, reactive oxygen species and other antibacterial substances, activated neutrophils also release web‐like structures composed of tangled decondensed DNA, histone, myeloperoxidase and other granules called neutrophil extracellular traps (NETs), which can efficiently ensnare bacteria in the circulation. In contrast, excessive neutrophil activation and NET release may induce endothelial cells to shift toward a pro‐inflammatory and pro‐coagulant phenotype. Furthermore, neutrophils and NETs can degrade glycocalyx on the endothelial cell surface and increase endothelium permeability. Consequently, the endothelial barrier collapses, contributing to impaired microcirculatory blood flow, tissue hypoperfusion and life‐threatening organ failure in the late phase of sepsis.
Keywords: endothelial cell dysfunction, neutrophil, neutrophil extracellular traps, sepsis
1. Neutrophils show increased lifespan and impaired migration, both of which promote organ damage.
2. Neutrophils and neutrophil extracellular traps (NETs) induce pro‐inflammatory and pro‐angiogenic responses in endothelial cells.
3. Neutrophils and NETs degrade glycocalyx and increase endothelial permeability.
4. Neutrophils and NETs induce a pro‐coagulant endothelial cell phenotype via degradation of the anti‐coagulation system and up‐regulation of tissue factors.
1. INTRODUCTION
Sepsis is defined as a lethal organ dysfunction resulting from a dysregulated immune response to infection. It still has a high rate of morbidity and mortality. 1 During sepsis, neutrophils play a critical role in the host's inflammatory response against invading pathogens. 2 , 3 They exert effector functions primarily through three approaches: phagocytosis, degranulation and releasing neutrophil extracellular traps (NETs). 4 , 5 , 6 NETs are extracellular, web‐like decondensed nuclear or mitochondrial DNA structures composed of histones, cytosolic and granule proteins with microbicidal activity. 7 , 8 , 9 NETs neutralize and kill bacteria, fungi and viruses, and can inhibit their dissemination. 10 However, if dysregulated, excessive NETs can further induce inflammation and organ injury contributing to the progression of sepsis. 11 , 12 Other immune cells, such as macrophages and eosinophils, can also release extracellular traps (ETs) that result in the killing of pathogens and participate in sepsis and other diseases, such as autoimmune processes. 13 , 14
During sepsis, neutrophils display increased lifespan and impaired migration. This confines them to blood vessels and results in overwhelming vascular inflammation via the release of cytokines, reactive oxygen species (ROS) and NETs. 15 , 16 Notably, neutrophils and NETs stimulate pro‐inflammatory and pro‐angiogenic responses in endothelial cells that cause further dysregulation of the immune system. 17 , 18 Activated endothelial cells display an increased glycolysis rate, further promoting inflammation and oxidative stress. 19 , 20 Direct destructive effects from NET components, accompanied by an inflammatory environment and oxidative stress, will degrade glycocalyx existing on the surface of the endothelial cells and increase endothelial permeability via junction cleavage, high expression of adhesion molecules and apoptosis. 21 Additionally, neutrophils and NETs induce a pro‐coagulant endothelial cell phenotype via degradation of the anti‐coagulation system and up‐regulation of tissue factor (TF). 22 , 23 Consequently, the collapse of the endothelial barrier enhances microvascular leakage that triggers vascular hypotension, tissue oedema and lethal organ failure in sepsis. 24
In this review, we will focus our discussion on the role of neutrophils, NET formation and sepsis progression, and then summarize their impacts on endothelial dysfunction. Lastly, we will provide evidence on potential targets for therapy.
2. NEUTROPHIL, NET FORMATION AND SEPSIS
Neutrophils show a critical role in innate immunity since they are the first line of defence against pathogens. 25 During infection or inflammation, neutrophils escape from the bone marrow in large numbers. 26 This process is accompanied by releasing proteolytic enzymes, ROS and reactive nitrogen species (RNS). 27 , 28 Under normal conditions, neutrophils display the shortest lifespan of all leukocytes and experience regulated cell death, which is crucial for preventing sustained inflammatory responses and tissue repair. 29 , 30 During sepsis, most immune cells tend to undergo apoptosis forming an immunosuppressive environment, while neutrophils display delayed apoptosis, resulting in prolonged inflammation. 15 Activation of several signal pathways explains neutrophil resistance to apoptosis. In the peripheral circulation, pro‐inflammatory factors like complement component 5a (C5a) and lipopolysaccharide (LPS) activate extracellular regulated protein kinases (ERK) 1/2 and phosphoinositide‐3 kinases (PI‐3K), which results in the phosphorylation of Akt and subsequent Bad phosphorylation. Finally, this prevents the formation of the apoptosome. 31 , 32 C5a also enhances the expression of anti‐apoptotic protein Bcl‐xL and reduces Bim expression, which further suppresses caspase, a terminal splicing enzyme essential for cell apoptosis. When activated by LPS, the myeloid nuclear differentiation antigen prevents proteasomal degradation of myeloid cell leukaemia‐1 (MCL‐1), an anti‐apoptotic factor of the Bcl‐2 family. 33 In rodents, single‐cell RNA sequencing confirmed that after LPS stimulation, multiple subclusters of neutrophils were differentiated and the programmed cell death ligand 1 (PD‐L1) was highly expressed in a particular cluster. 34 Mechanistically, sepsis can enhance PD‐L1 expression on neutrophils, triggering lymphocyte apoptosis via direct contact, and finally promoting sepsis‐induced immunosuppression. 35
During the early stages of sepsis, neutrophils expressing CXC receptor 2 (CXCR2) are recruited from the blood to the infection site, responding to CXC ligand 2 (CXCL2). 36 After migrating to the site of the infection, neutrophils release NETs, ROS and RNS to kill the pathogens. However, during severe sepsis, neutrophil migration is impaired.
One of the mechanisms involved in this process is the activation of toll‐like receptors (TLRs) expressed on neutrophils. Alves‐Filho et al. found that direct activation of TLR2 on neutrophils contributed to CXCR2 down‐regulation and chemotaxis impairment. 37 Consequently, neutrophils aggregate in blood vessels, facilitating the spread of pathogens. Studies have demonstrated that CC receptor 2 (CCR2), primarily absent in neutrophils under physiological conditions, is expressed in circulating neutrophils through TLRs activation. CCR2 drives inappropriate infiltration of neutrophils into remote organs that produce CC ligand 2 (CCL2), further eliciting tissue damage in organs such as the lung, liver and kidney. 38
It is well known that older septic patients have poor prognoses. 39 Inflammed aged tissues show a high frequency of neutrophil reverse transendothelial migration (rTEM) into the circulation, further contributing to remote organ damage. 17 Barkaway et al. demonstrated that aged mice showed high levels of rTEM driven by mast cell‐derived CXCL1. In this paradigm, the intensified endothelial atypical chemokine receptor 1‐CXCL1 promoted desensitization of CXCR2 on neutrophils and loss of their directional motility, which further contributed to vascular leakage and organ damage. 17 Moreover, an increased NET release was also found in the models of aged mice, which might exacerbate inflammation. 40
In 2004, Brinkmann et al. proved the ability of neutrophils to release NETs. The authors speculated that NET formation was an early event in cell death. 41 NET release occurs after stimulation with interleukin‐8 (IL‐8), LPS, or phorbol‐12‐myristate‐13‐acetate and involves activation of protein kinase C (PKC). Nicotinamide adenine dinucleotide phosphate (NADPH) helps neutrophil elastase (NE) translocate from cytosolic granules to the nucleus, where it causes chromatin breakdown by cleaving histone. 42 NADPH oxidase activated by PKC and Raf‐MEK‐ERK signalling pathways promotes ROS production, which leads to an increased calcium influx, peptidyl arginine deaminase 4 (PAD4) activation and histone citrullination. Myeloperoxidase (MPO) facilitates the breakdown of chromatin and nuclear envelope, and granular mixing in the NET vacuole. After intracellular NET formation (NETosis), mature NETs are extruded due to the rupture of the neutrophil outer membrane. 42 Remijsen et al. found that the formation of NETs involved both NADPH‐oxidase‐mediated superoxide production and autophagy, and both took place independently of each other. 43 However, the involvement of autophagy has been controversial. Simon et al. demonstrated that NET formation was not dependent on autophagy in both mouse and human cell lines. 44
After direct interaction between neutrophil and platelet through CD11a, gram‐negative bacteria induce NET formation via TLR4 activation in platelets. 45 Gram‐positive‐induced NETosis requires TLR2 and complement receptor 3. 46 Aside from suicidal NETosis as noted above, neutrophils are capable of releasing NETs without membrane rupture. During NET production, vesicles of DNA, like beads on a string, bud from the nuclear envelope, pass through the cytoplasm and fuse with the outer membrane. Finally, NETs are transported out of the cell. 42 This type of NETosis is ROS‐independent and requires a short period (30 min), while suicidal NETosis takes hours. 42 It has been suggested that neutrophils could release mitochondrial DNA (mtDNA) in a cell death‐independent process. 47 mtDNAs do not contain histones and are similar to plasmid DNA. After transporting nuclear DNA and mtDNA out of the cell, neutrophils are still equipped with effector functions, like chemotaxis, recruitment and phagocytosis (Figure 1).
FIGURE 1.
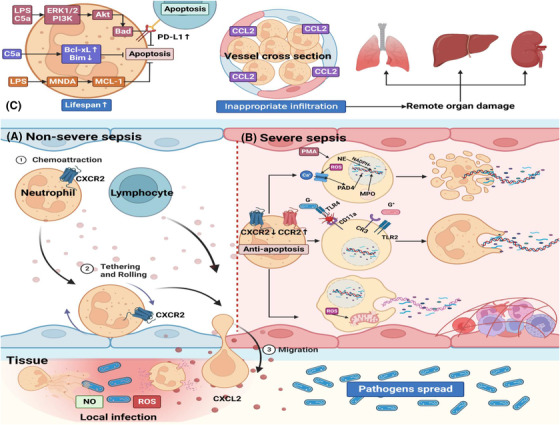
Neutrophil, NET formation and sepsis. (A) During non‐severe sepsis, neutrophils that express CXCR2 are recruited from blood to the infection site responding to CXCL2. Neutrophils migrate to the location of infection and kill pathogens through the release of antibacterial substances, such as ROS, NO and NETs. Other immune cells such as lymphocytes can also migrate to the infection site to prevent pathogens spread. (B) However, during severe sepsis, neutrophils show increased lifespan and impaired function due to the down‐regulation of CXCR2 and up‐regulation of CCR2. On the one hand, impaired migration results in pathogens' spread. On the other hand, many neutrophils are confined to vessels and release NETs, resulting in vascular inflammation, endothelial damage, and thrombosis. NET formation can be classified into three types. The first type is suicidal NETosis since NETs are released via cell lysis. The second type allows NET release and conventional live neutrophil functions, such as phagocytosis, to coexist. The third type is mtDNA NETosis. Viable neutrophils release mtDNA to form NETs, and this process does not depend on cell death but is dependent on ROS. (C) LPS and C5a in the peripheral circulation can induce neutrophils’ resistance to apoptosis via three signalling pathways. Moreover, neutrophils induce lymphocyte apoptosis via PD‐L1 up‐regulation. Additionally, CCR2, which is absent in neutrophils under normal conditions, is upregulated in neutrophils through TLR activation. CCR2 drives inappropriate infiltration of neutrophils into remote organs which produce CCL2 and further elicit tissue damage in remote organs such as lungs, liver, and kidneys. C5a, complement component 5a; CCL2, CC ligand 2; CCR2, CC receptor 2; CR3, complement receptor 3; CXCL2, CXC ligand 2; CXCR2, CXC receptor 2; ERK1/2, extracellular regulated protein kinases 1/2; G‐, gram‐negative bacteria; G+, gram‐positive bacteria; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; MCL‐1, myeloid cell leukemia‐1; MNDA, myeloid nuclear differentiation antigen; MPO, myeloperoxidase; mtDNA, mitochondrial DNA; NE, neutrophil elastase; NETs, neutrophil extracellular traps; PAD4, peptidyl arginine deaminase 4; PD‐L1, programmed cell death ligand 1; PI‐3K, phosphoinositide‐3 kinases; PI3Kγ, phosphoinositide‐3 kinase gamma; PMA, phorbol 12‐myristate 13‐acetate; ROS, reactive oxygen species; TLR, Toll‐like receptor
As previously indicated, NETs play a pivotal role in host immunity. LL‐37, an antibacterial protein externalized on NETs, improved septic mice survival, possibly by preventing the release of pro‐inflammatory cytokines. 48 However, the excessive release of NET promotes inflammation. In the extracellular space, host cell‐free DNA can serve as a damage‐associated molecular pattern (DAMP). 49 , 50 Also, mtDNA stimulates the secretion of tumour necrosis factor (TNF‐α) and IL‐1β from mouse splenocytes and macrophages, respectively. 51 Extracellular histones are also considered DAMPs since they activate immune cells via TLR and nod‐like receptor signalling pathways. Xu et al. demonstrated that histones contributed to high levels of IL‐6, IL‐10 and TNF‐α. 52 , 53 , 54
3. NEUTROPHIL, NETs AND IMMUNOTHROMBOSIS
Engelmann et al. reviewed the mechanism of NET‐mediated thrombosis, coining this process “immunothrombosis.” Although this process can ensnare pathogens and prevent dissemination, excessive thrombosis may further contribute to sepsis progression. 55 Within NETs, DNA fibre and histone networks provide a scaffold to recruit erythrocytes, platelets, leukocytes and plasma proteins, thus forming a positive feedback loop that augments thrombosis in vivo and ex vivo. 56 A recent study demonstrates that extracellular DNA in NETs can interact with von Willebrand Factor (vWF) and further promote thrombus formation and inflammation. 57 , 58 Moreover, the inflammation of damaged endothelial cells propagates the release of acute phase reactants, such as vWF and cell‐surface adhesion receptors into the circulation. 59 Von Brühl et al. showed that NETs promoted the interaction between factor XII (FXII) and neutrophils, and initiated the intrinsic coagulation pathway. 60 Data from intravital microscopy in septic mice indicated that the collaborative interaction between histone H4 in NETs induced intravascular coagulation. 61 Additionally, NETs contributed to thrombosis by restraining anticoagulants, like antithrombin (AT), activated protein C (APC) and TF pathway inhibitor. 62
A recent study demonstrated that neutrophils could also release NETs carrying active TF. 63 High levels of TF‐enriched NETs in sepsis patients might result in immunothrombosis and worse disease outcomes. 64 Thrombosis induced by NETs contributes to organ ischemic damage and disseminated intravascular coagulation (DIC). 65 The rate of DIC is high in the late stage of sepsis and is linked to multiple organ failure and refractory septic shock. 66
4. NEUTROPHILS AND NETs INDUCE A PRO‐INFLAMMATORY AND PRO‐ANGIOGENIC ENDOTHELIAL CELL PHENOTYPE
During sepsis, NETs trigger activation of the endothelium and may impair its structure and/or function. 18 Engagement of pathogen recognition receptors, such as TLRs, drives endothelial cells to reprogram toward a pro‐inflammatory and pro‐angiogenic phenotype. 21
Wojciak‐Stothard et al. found that markers of NETosis were increased in patients with pulmonary hypertension. The authors demonstrated that NETs could induce pro‐inflammatory and pro‐angiogenic responses in human pulmonary artery endothelial cells (HPAECs) via MPO/H2O2‐dependent activation of TLR4/NF‐κB signalling. They also showed that NETs markedly induced the expression levels of intercellular adhesion molecule‐1 (ICAM‐1), and several pro‐angiogenic factors, such as platelet‐derived growth factor (PDGF) and heparin‐binding EGF‐like growth factor in HPAECs. 18
Activation of NF‐κB signalling enhances pro‐inflammatory and pro‐angiogenic responses in endothelial cells via increased expression of vascular cell adhesion molecule (VCAM‐1), platelet endothelial cell adhesion molecular‐1 (PECAM‐1) and elevated secretion of IL‐6, IL8 and vascular endothelial growth factor (VEGF). 67 It has been reported that NETs induce neutrophil effector functions, such as exocytosis, ROS production and NET formation. These occur through several pathways related to the phosphorylation of Akt, ERK1/2 and p38. 10 Interestingly, the production of IL‐8 is strongly linked to NET formation via mitogen‐activated protein kinase (MAPK) pathway activation. 68
NETs induce an “M1‐like” macrophage phenotype characterized by the release of inflammatory cytokines like IL‐1, IL‐6, IL‐8 and TNF in vitro and in vivo. 69 M1 phenotype macrophages also secrete several pro‐angiogenic factors, including VEGF and fibroblast growth factor (FGF). 70 VEGF, as one of the most important growth factors, is capable of stimulating glycolysis via increased expression levels of glucose transporter 1 (GLUT1), fructose‐2,6‐bisphosphatase‐3 (PFKFB3) and lactate dehydrogenase‐A (LDH‐A). 71 , 72 , 73 , 74 In addition, activated VEGFR2 stimulates signalling via PI3K/Akt pathway. It thus suppresses the expression of growth‐inhibiting transcription factor FOXO1, which maintains endothelial cell quiescence through inhibiting glycolysis and mitochondrial respiration. 75 , 76 The growth‐enhancing transcription factor MYC resides in the nuclei of endothelial cells to drive the expression of genes that cooperatively facilitate proliferation and glycolysis. 75
A previous study demonstrated that NE, as one of the components of NET, participated in the proteolytic cleavage of some growth factors crucial for angiogenesis, such as VEGF and PDGF. 77 Ebos and Kerbel argued that the blockade of one growth factor could compensatorily upregulate the expression of others and stimulate angiogenesis again. 78 This could explain why the efficacy of anti‐VEGF treatments varied among patients and resulted in only partial vessel regression. 79 In animal models of inflammation, Mirabelli et al. reported that topical anti‐VEGF treatment reduced corneal neovascularization by only 14%. 80 Mechanistically, when VEGF is blocked, the expression of other growth factors like FGF may be elevated, thus promoting glycolysis again through the FGF‐MYC‐HK2 axis. 81 In the early phase of sepsis, various pro‐inflammatory cytokines and growth factors are overwhelmingly released; therefore, endothelial cells still retain a proliferative state characterized by high levels of glycolysis.
Notably, enhanced glycolysis promotes NF‐κB‐driven endothelial inflammation. Increased activity of PFKFB3 stimulates the expression of ICAM‐1 and VCAM‐1 in vitro and in models of acute lung injury. This appears to occur via NF‐κB activation, which further induces neutrophil and macrophage recruitment. 82 Proliferating endothelial cells display high levels of glycolysis even in the presence of oxygen, similar to the Warburg effect observed in tumour cells. 83 A similar metabolic shift in cancer vasculature can serve to explain changes in the metabolism of endothelial cells in sepsis. During tumour angiogenesis, lactate, the end‐product of glycolysis, activates NF‐κB by promoting the phosphorylation and degradation of the NF‐κB inhibitor IκB‐α. 19 Subsequently, activated NF‐κB enhances endothelial cell pro‐inflammatory responses, as noted above. 84 , 85 All of these changes aggravate inflammation and oxidase stress during sepsis (Figure 2).
FIGURE 2.
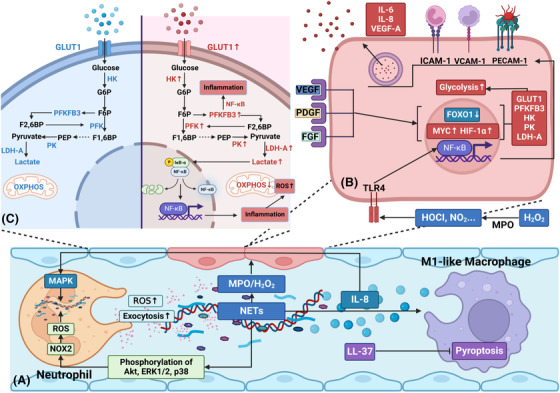
Neutrophils and NETs induce a pro‐inflammatory and pro‐angiogenic endothelial cell phenotype via activation of NF‐κB signalling. (A) NETs stimulate pro‐inflammatory and pro‐angiogenic responses in human endothelial cells via MPO/H2O2‐mediated NF‐κB activation. Moreover, NETs activate several neutrophil functions, such as exocytosis, ROS production, and NET formation, which may be related to the phosphorylation of Akt, ERK1/2 and p38. In addition to the impacts on neutrophils, NETs induce an “M1‐like” macrophage phenotype characterized by releasing inflammatory cytokines, such as IL‐8. Moreover, the concentration of IL‐8 is found to be strongly linked with NET formation via MAPK pathway activation. LL‐37, an antibacterial protein externalized on NETs, can suppress macrophage pyroptosis and then inhibit the release of pro‐inflammatory cytokines. (B) NF‐κB signalling enhances endothelial cell pro‐inflammatory and pro‐angiogenic responses via up‐regulation of ICAM‐1, VCAM‐1, PECAM‐1, and increased secretion of IL‐6, IL‐8, and VEGF‐A. Additionally, many pro‐angiogenic factors, such as VEGF, PDGE and FGF, are released during the process. These factors increase expression levels of glycolytic enzymes and transporters via up‐regulation of MYC and HIF‐1α and down‐regulation of FOXO1. (C) During inflammatory and angiogenic responses, proliferated endothelial cells show a higher rate of glycolysis than quiescent ones due to the up‐regulation of glycolytic enzymes and transporters, such as GLUT1, PFKFB3, HK, PK and LDH‐A. Notably, PFKFB3 and glycolytic product lactate promote NF‐κB‐driven vascular inflammation. F1,6BP, fructose‐1,6‐biphosphate; F2,6BP, fructose‐2,6‐biphosphate; F6P, fructose‐6‐phosphate; FGF, fibroblast growth factor; G6P, glucose‐6‐phosphate; GLUT1, glucose transporter 1; HIF‐1α, hypoxia‐inducible factor‐1α; HK, hexokinase; ICAM‐1, intercellular adhesion molecule‐1; LDH‐A, lactate dehydrogenase‐A; MAPK, mitogen‐activated protein kinase; NF‐κB, nuclear factor‐κB; NOX2, NADPH oxidase 2; OXPHOS, oxidative phosphorylation; PDGF, platelet‐derived growth factor; PECAM‐1, platelet endothelial cell adhesion molecule‐1; PEP, phosphoenolpyruvate; PFK‐1, phosphofructokinase‐1; PFKFB3, fructose‐2,6‐bisphosphatase‐3; PK, pyruvate kinase; VCAM‐1, vascular cell adhesion molecule‐1; VEGF, vascular endothelial growth factor
In short, neutrophils and NETs can induce pro‐inflammatory and pro‐angiogenic responses in endothelial cells via NF‐κB activation. Enhanced glycolysis in endothelial cells drives pro‐inflammatory programs and ROS accumulation. Consequently, this forms a vicious cycle that contributes to sustained vascular inflammation and overwhelming oxidative stress.
5. NEUTROPHILS AND NETs DAMAGE ENDOTHELIAL CELLS’ GLYCOCALYX AND INCREASE ENDOTHELIAL PERMEABILITY
As previously mentioned, neutrophils and NETs damage endothelial cells’ glycocalyx and increase endothelial permeability. 21 This phenomenon may further contribute to dysregulated inflammation, impaired microcirculatory blood flow, tissue hypoperfusion and life‐threatening organ failure. 24
The matrix meshwork covering the endothelial cells is glycocalyx, which consists of glycosaminoglycans (GAG), proteoglycans and glycoproteins. The GAG chains contain heparan sulfate, chondroitin sulfate and hyaluronic acid that binds to CD44. Selectins and integrins are glycoproteins that participate in neutrophil adhesion and coagulation. 21 The endothelial glycocalyx also tethers the extracellular superoxide dismutase (SOD3), which prevents oxidative stress. 86 The glycocalyx is also found in the intercellular spaces and on the side of endothelial cells close to the basal membrane. Damage to glycocalyx will cause an impaired vascular barrier and elevated microvascular permeability. 87 As a result of glycocalyx disruption, endothelial cell adhesion molecules are exposed, which triggers further inflammation, rolling and adhesion of leukocytes and platelets. 88
A clinical study demonstrated that pediatric trauma patients with high levels of histone‐complexed DNA (hcDNA) showed elevation in plasma levels of syndecan‐1 (a proteoglycan), suggesting the degradation of the glycocalyx. 89 Enzymatic digestion by MPO, matrix metalloproteinase (MMP) and other NET‐containing proteinases can also induce glycocalyx degradation. 86 , 90 While the role of MMP‐9 activity is controversial, a clinical trial showed that the active form of MMP‐9 was only present in patients with severe sepsis. 91 Additionally, many inflammatory cytokines, such as TNF‐α, IL‐6 and IL‐8, induced by neutrophils and NETs can directly damage glycocalyx. 92 Mast cells are stimulated by TNF‐α, and they can release cytokines, proteases, histamine and heparinase, contributing to further glycocalyx degradation. 93 A recent study demonstrated that disintegrin and metalloproteinase 15 were upregulated during inflammation and they cleaved CD44 at the membrane‐proximal region, which disrupted the integrity of the endothelial glycocalyx. 94 In addition, oxidative stress‐induced histone deacetylase can upregulate MMP expression and inhibit tissue inhibitors of MMPs (TIMP1 and TIMP3), further activating MMP2 and MMP9, promoting syndecan‐1 and SOD3 shedding from the endothelial cell surface, and subsequently result in derangement of the endothelial glycocalyx. 86
Para‐endothelial permeability increases during sepsis mainly due to junction cleavage. There are two common types of junctions existing between endothelial cells: tight junctions (TJs) and adherens junctions. TJs contain occludins and claudins, which are anchored to the actin cytoskeleton via zonula occludins (ZO). 21 TNF‐α can disrupt claudin‐5 at cell‐cell junctions of endothelial cells by activating the NF‐κB pathway. 95 ROS causes rearrangement of occludin from intercellular junctions, thus promoting its dissociation from ZO‐1 and augmenting endothelial soluble permeability. 96
Adherens junctions mainly comprise vascular endothelial (VE)‐cadherins, which are linked to the actin cytoskeleton through catenins. 21 VE‐cadherin is easily cleaved by MMP because of its vulnerability to enzymatic degradation, and this causes further damage to the junction integrity. 97 , 98 During NETs‐induced inflammatory response, cAMP/Rac1 signalling is suppressed. This, in turn, stimulates Rho activity and kinases such as Src and Pyk2, 99 both of which lead to VE‐cadherin phosphorylation, dissociation from catenin and endocytosis. 100 Endocytosis of VE‐cadherin increases gaps between endothelial cells, exacerbating the permeability of the vascular barrier. 101
ICAM‐1 expression is significantly upregulated during inflammation and angiogenesis. Several studies have suggested that this adhesion molecule may assume a significant role in regulating barrier integrity. 102 , 103 On the one hand, emerging evidence demonstrates that ICAM‐1 promotes phosphorylation of VE‐cadherin, which in turn disrupts endothelial permeability in a neutrophil‐dependent fashion. 104 On the other hand, Sumagin et al. suggested that ICAM‐1 overexpression increased endothelial permeability via PKC‐dependent signalling in the absence of leukocytes. 105 Research also suggests that ICAM‐1 signalling can participate in transcellular permeability regulation, aside from its key role in regulating paracellular permeability. For example, caveolae are one of the main mechanisms of transporting albumin from the luminal side of the endothelium to the basement membrane. Importantly, Src phosphorylation of caveolin‐1 is facilitated by ICAM‐1 ligation. 21
Endothelial cell apoptosis results in hyperpermeability. 24 NETs promote endothelial apoptosis during sepsis. Saffarzadeh et al. found that NETs directly induced endothelial cell death and cytotoxicity partially mediated by histone and MPO. 106 Furthermore, under pro‐inflammatory conditions, neutrophils and NETs trigger the production of O2 − and NO by 1000‐fold, increasing the formation of peroxynitrite (ONOO−) and causing a reduction in NO availability. Peroxynitrite induces permeabilization of the mitochondrial outer membrane, allowing the efflux of various pro‐apoptotic signalling molecules. Peroxynitrite is also capable of inducing DNA damage and then activates PARP‐1, a DNA repair enzyme. Upon severe DNA injury, excessive activation of PARP‐1 depletes the cellular stores of NAD+, a pivotal cofactor of the glycolytic pathway. Consequently, the loss of NAD+ results in the reduction of adenosine triphosphate (ATP), causing further endothelial dysfunction 107 (Figure 3).
FIGURE 3.
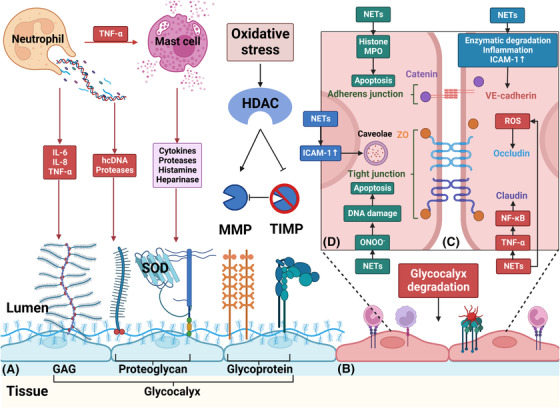
Neutrophils and NETs damage endothelial cells’ glycocalyx and increase endothelial permeability. (A) During sepsis, NETs can directly cause glycocalyx degradation through hcDNA and proteases. Additionally, a considerable number of inflammatory cytokines, such as IL‐6, IL‐8 and TNF‐α, induced by neutrophils and NETs can also damage glycocalyx. Moreover, mast cells are activated by TNF‐α and release cytokines, proteases, histamine and heparinase, further degrading glycocalyx. Furthermore, oxidative stress‐induced HDAC can upregulate MMP expression and inhibit tissue inhibitors of MMPs (TIMP1 and TIMP3), thereby activating MMPs to promote the shedding of the glycocalyx. (B) After degradation of the glycocalyx, endothelial cell adhesion molecules are exposed, triggering further inflammation, rolling, and adhesion of leukocytes and platelets. (C) Para‐endothelial permeability increases mainly due to junction cleavage. TNF‐α is shown to cause disruption of claudin through NF‐κB activation. ROS causes the redistribution of occludin, limiting its association with ZO‐1. VE‐cadherin is susceptible to enzymatic degradation. Additionally, inflammatory mediators promote VE‐cadherin phosphorylation and dissociation from p120 catenin and induce VE‐cadherin endocytosis via several signalling pathways. Upregulated ICAM‐1 can also promote VE‐cadherin phosphorylation. (D) ICAM‐1 can also increase trans‐endothelial permeability through caveolin‐1 phosphorylation, a major component of caveolae. However, endothelial cell apoptosis is the main cause of increased trans‐endothelial permeability. NETs contribute to apoptosis through MPO and histone. Moreover, the formation of ONOO− during oxidative stress also induces cell death via DNA damage. GAG, glycosaminoglycan; hcDNA, histone‐complexed DNA; HDAC, histone deacetylase; MMP, matrix metalloproteinase; ONOO‐, peroxynitrite; SOD, superoxide dismutase; VE‐cadherin, vascular endothelial‐cadherin; ZO, zonula occluding
Neutrophils may also play a partial protective role in sepsis by reducing vascular leakage in some forms of pathogen‐specific infections. 108 , 109 For instance, during meningococcemia caused by Neisseria meningitides (Nm), neutrophil recruitment allows efficient phagocytosis, partially reducing vascular leakage induced by bacteria. 109 Interestingly, Nm is a potent NET inducer. Lappann et al. found that NETs inhibited bacterial growth but could not exert killing activity. 110
6. NEUTROPHILS AND NETS INDUCE A PRO‐COAGULANT ENDOTHELIAL CELL PHENOTYPE VIA DEGRADATION OF THE ANTI‐COAGULATION SYSTEM AND UP‐REGULATION OF TISSUE FACTOR
The glycocalyx covering the luminal side of the endothelium is extremely important in maintaining anti‐thrombogenicity in the vascular lumen. Once the glycocalyx is damaged by NETs, ICAM‐1, E‐selectin and other adhesion molecules are exposed to the denuded endothelium, which accelerates the recruitment of neutrophils and platelets. 111 Additionally, GAGs prevent coagulation under physiological conditions. 112 In the vasculature, heparan sulfate is the major component of GAGs, representing 50%‐90% of the total content. 113 Heparan sulfate primarily binds to AT and dermatan sulfate to heparin cofactor II (HCII). Moreover, heparan sulfate can also interact with HCII. Antithrombin exerts its anticoagulant effects by inactivating both thrombin (factor IIa) and factor Xa, while HCII only inhibits thrombin. 114 , 115
TF also plays a vital role in coagulation. This protein is normally expressed on endothelial cells and is not in direct contact with the blood. When neutrophils and NETs damage the endothelium, TF is exposed and initiates plasma‐mediated hemostasis (via the so‐called extrinsic pathway). In this process, TF acts as a cellular receptor for plasma factor VIIa. Then, the complex of TF/VIIa activates factor IX and factor X. In an important reaction that requires factor V, factor Xa proteolytically cleaves prothrombin to thrombin. Finally, FXIIIa (a transglutaminase activated by thrombin) induces the formation of an insoluble fibrin clot. 116 , 117
NETs can also upregulate TF expression in endothelial cells. Folco et al. found that treating human endothelial cells with NETs increased the expression of TF mRNA and enhanced endothelial cell TF activity. Mechanistically, NETs induced TF production through IL‐1α and cathepsin G. This serine protease is abundant in NET and cleaves pro‐IL‐1α precursor, which then activates IL‐1α to promote TF production. 23 Haubitz et al. showed that proteinase 3 (PR3) and elastase stimulated TF expression in human endothelial cells. While the elastase‐induced pro‐coagulant effect was blocked by α1‐antitrypsin (α1‐AT), suggesting that the effect depended on enzymatic activity. Interestingly, the pro‐coagulant effect of PR3 on endothelial cells was non‐enzymatic. 118
Histones, another NET component, trigger an endothelial pro‐coagulant state via up‐regulation of TF and down‐regulation of the anticoagulant thrombomodulin (TM) in a process regulated by TLR2 and TLR4. 119 Moreover, the cytotoxic effects of histone may trigger the exposure of phosphatidylserine (PS) on the endothelium. PS, normally existing on the inner layer of the plasma membrane, can stimulate the pro‐coagulant activity of TF by transferring it to the outer layer. 119 , 120 Hypochlorous acid, produced by NET component MPO, 121 increases the levels of TF mRNA expression in human saphenous vein endothelial cells (HSVECs) and stimulates endothelial TF activity. 122
A variety of anticoagulant mechanisms, especially the TM, protein C and endothelial protein C receptor (TM‐PC‐EPCR) anti‐coagulation pathway, are suppressed during sepsis or inflammation. The TM‐PC‐EPCR system is a crucial endogenous anti‐coagulation mechanism. 123 During coagulation, thrombin and PC bind to their receptors, TM and EPCR, respectively, which form a thrombin‐TM‐PC‐EPCR complex on the endothelium. This further contributes to the formation of APC, a proteinase generated by thrombin from the zymogen precursor protein C. 124 This proteinase has many anti‐coagulation properties, such as inactivating the procoagulant factors, FVa and FVIIIa and promoting the release of plasminogen activator. 125
As noted above, NETs can activate NF‐κB signalling in endothelial cells. This transcription factor has important functions in coagulation. 67 Song et al. developed a transgenic mouse overexpressing mutant I‐κBα, an inhibitor of NF‐κB, on endothelial cells. Transgenic mice had a decreased level of thrombin‐AT, indicating that NF‐κB signalling was likely to participate in septic‐induced coagulation. 126 Activation of endothelial‐specific NF‐κB signalling results in a decrease in cellular levels of TM and EPCR proteins in endothelial cells. Moreover, blocking endothelial NF‐κB signalling inhibits TNF‐α converting enzyme activity which is responsible for EPCR shedding, reducing plasma plasminogen activator inhibitor type‐1 level and restoring plasma level of APC 22 , 127 (Figure 4). Aside from its anticoagulant effects, APC also displays cytoprotective properties, such as blocking inflammatory cytokine release, 128 maintaining endothelial integrity and protecting cells from apoptosis. 129 , 130 So decreased plasma levels of APC are strongly linked with prognosis. Although controversial, APC has been used in severe sepsis. 131
FIGURE 4.
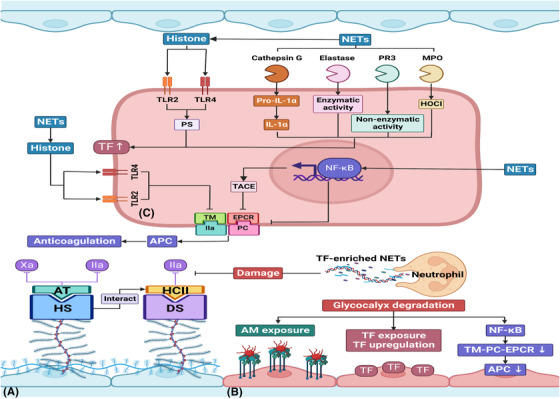
Neutrophils and NETs induce a pro‐coagulant endothelial cell phenotype via degradation of the anti‐coagulation system and up‐regulation of tissue factor. (A) GAGs play a key role in the anti‐coagulation system: HS primarily binds to AT to inhibit factor IIa and Xa, and DS mainly binds to HCII to inhibit factor IIa. Moreover, HS also interacts with HCII. So glycocalyx degradation induced by NETs contributes to the degradation of the anti‐coagulation system. (B) Furthermore, adhesion molecules and TF are exposed on the endothelium causing thrombus and fibrin formation. Aside from releasing TF‐enriched NETs, neutrophils increase expression levels of TF on endothelium through NET components. (C) Cathepsin G, elastase, PR3 and MPO can stimulate TF expression through different signalling pathways and further initiate the extrinsic coagulation pathway. Histone, another NET component, induces endothelial pro‐coagulant phenotype through up‐regulation of TF and down‐regulation of TM, and these effects are partly mediated by TLR2 and TLR4. Moreover, histone is capable of stimulating PS exposure, which enhances the pro‐coagulant activity of TF. Additionally, NETs can destroy the TM‐PC‐EPCR system, the most important natural anti‐coagulation mechanism, via NF‐κB activation. And NF‐κB signalling also promotes TACE activity, which is reportedly responsible for EPCR shedding. AM, adhesion molecule; APC, activated protein C; AT, antithrombin; DS, dermatan sulfate; HCII, heparin cofactor II; HOCI, hypochlorous acid; HS, heparan sulfate; PR3, proteinase 3; PS, phosphatidylserine; TACE, TNF‐α converting enzyme; TF, tissue factor; TM‐PC‐EPCR, thrombomodulin, protein C, and endothelial protein C receptor
7. POTENTIAL THERAPEUTIC TARGETS IN SEPSIS
7.1. Inhibition of NET formation in sepsis
PAD4 is an important enzyme involved in chromatin decondensation. By catalyzing histone citrullination, PAD4 weakens the combination of DNA and histones (FIGURE 5 and TABLE 1). 7 Overexpression of PAD4 contributes to severe vascular damage through the release of NETs and inducing expression of ICAM‐1 and VACM‐1 on endothelial cells. 23 Therefore, using PAD4 inhibitors can prevent the NET formation and avoid endothelial cell dysfunction. Martinod et al. found that PAD4−/− mice were partially protected from LPS‐induced shock. 132 The PAD4 inhibitor Cl‐amidine effectively prevented NET formation and improved overall survival in a murine sepsis model. 133 Inhibition of NET formation by the PAD4 inhibitor GSK484 led to an obvious reduction in thrombus formation in mouse lungs. 134 The neonatal NET‐inhibitory factor (nNIF), can inhibit PAD4 activity, nuclear histone citrullination and nuclear decondensation. Yost et al. demonstrated that nNIF and nNIF‐related peptides could block NET formation in mouse models of infection and systemic inflammation. 135 However, it is worth mentioning that PAD4 is not always required for NET release, since mtDNA is not equipped with histones, suggesting that its effects of inhibiting NET formation are limited.
FIGURE 5.
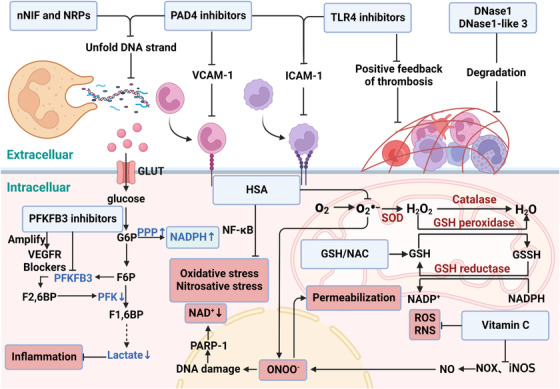
Potential therapeutic targets in sepsis. In blood vessels, nNIF, NRPs, and PAD4 inhibitors prevent NET formation by inhibiting chromatin decondensation. And PAD4 inhibitors may also reduce the expression of ICAM‐1 and VCAM‐1. TLR‐4−/− mice did not show an enhanced thrombotic response and exhibited markedly decreased circulating ICAM‐1 compared with wide‐type controls. DNase1 and DNase1‐like 3 blocks the positive feedback loop of thrombosis by degrading the scaffold of NET structures. PFKFB3 blockers may be used to control inflammation induced by excessive glycolysis during sepsis. Using PFKFB3 inhibitors can also promote NADPH production, which is likely to induce NET formation. Moreover, PFKFB3 blockade amplifies the anti‐angiogenic effect of VEGFR blockers. In a perfused endothelial cell model, GSH or NAC (the precursor of GSH) supplementation significantly decreased ROS production. HSA can suppress circulating and tissue O2 − production, and restrain activation of NF‐κB, thereby inhibiting oxidative stress and nitrosative stress. In addition to scavenging ROS and RNS directly, vitamin C reduces them by preventing NOX activation, decreasing iNOS expression, and enhancing NO bioavailability. Then decreased formation of ONOO− can efficiently prevent endothelial cell apoptosis. GSH, glutathione; HSA, human serum albumin; NAC, N‐acetylcysteine; nNIF, neonatal NET‐inhibitory factor; NRPs, nNIF‐related peptides; RNS, reactive nitrogen species
TABLE 1.
Potential targets for endothelial cell dysfunction therapy
| Clinical trials | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Potential target | Mechanism | Compound | Effects | In vitro | In vivo | I | II | III | IV | Ref |
| Inhibit NET formation | PAD4 inhibitor | Cl‐amidine | Effectively prevent the NET formation and improve overall survival in a murine sepsis model | [ 127 ] | ||||||
| GSK484 | Cause a dramatic reduction in thrombus deposition in mouse lungs | [ 128 ] | ||||||||
| TLR4 inhibitor | C34 | Reduce systemic inflammation in the mouse models of endotoxemia | [ 132 ] | |||||||
| TAK‐242 | Suppress inflammation by combining with cysteine 747 existing in the TIR domain of TLR4, but fail to reduce cytokine levels in patients with sepsis | [ 134 , 135 ] | ||||||||
| Eritoran | Inhibit LPS‐induced NF‐κB activation and inflammatory cytokine production in vitro and animal models, but do not reduce 28‐mortality among patients with severe sepsis | [ 136 , 137 , 138 ] | ||||||||
| Degrade DNA | DNase1 | Preferentially cleave protein‐free DNA and degrade the scaffold of NET structures | [ 143 ] | |||||||
| DNase1‐like 3 | Preferentially cleave DNA‐protein complexes and degrade the scaffold of NET structures | [ 143 ] | ||||||||
| NET‐inhibitory factor | nNIF and NRPs | Block NET formation in vitro and mouse models of infection and systemic inflammation, and show improved prognosis in these models | [ 144 ] | |||||||
| Inhibit excessive glycolysis | PFKFB3 blockade | 3PO | Protect mice from acute lung injury via suppression of NF‐κB‐mediated vascular inflammation | [ 77 ] | ||||||
| PFK158 | Suppress ATP production and proliferation in the mouse models of small cell lung cancer | [ 149 ] | ||||||||
| Phenoxyindole 44 | A novel inhibitor of PFKFB3 with higher selectivity | [ 150 ] | ||||||||
| Maintenance of the balance between ROS production and clearance | Antioxidant | GSH/NAC | Remarkably decrease ROS production when the cells were exposed to plasma from septic shock patients, impair NET formation in vitro and in vivo, contribute to shorter stays in ICU and improved severity scores | [ 153 , 154 , 155 , 156 ] | ||||||
| HSA | Reduce ROS and RNS production in vitro and in vivo, reverse sepsis‐induced hypotension in clinical trials | [ 157 , 158 ] | ||||||||
| Vitamin C | Scavenge ROS and RNS, prevent NOX activation, and decrease expression of iNOS, but do not obviously improve organ dysfunction scores or change markers of vascular damage and inflammation among patients with sepsis | [ 159 –162] | ||||||||
The green box denotes “Yes” and the red box denotes “No”.
TLR4 expressed on platelets is also responsible for NET induction in mice and humans. 45 Obi et al. reported that TLR4−/− mice did not show an enhanced thrombosis and exhibited conspicuously decreased circulating ICAM‐1 compared to wide‐type controls. 136 Therefore, it suggested that inhibition of TLR4 could improve clinical outcomes in patients with sepsis. 137 C34 is a novel inhibitor of TLR4 in vitro and in vivo. It reduces systemic inflammation in a mouse model of endotoxemia. 138 TAK‐242 is another inhibitor of the TLR4 signalling pathway. 139 This compound inhibits TLR4 signalling‐dependent inflammation by combining with cysteine 747 existing in the TIR domain of TLR4. 140 Despite its promising laboratory results, a randomized controlled trial showed that TAK‐242 failed to reduce cytokine levels in patients with sepsis. 141 Eritoran (E5564), a synthetic lipid A analogue of Rhodobacter aphaeroides, markedly inhibits LPS‐induced NF‐κB activation and inflammatory cytokine production in vitro and in vivo. 142 , 143 However, in a clinical study of severe sepsis, Eritoran did not reduce 28‐day mortality compared with a placebo. 144 It should be mentioned that TLR4 inhibitors show indirect functions on NET formation and TLR inhibition might have pleiotropic effects. 145 Claushuis et al. demonstrated that inhibition of TLR signalling in platelets did not alter the host immune response in murine sepsis models, arguing against the vital role of TLRs in sepsis progression. 146
DNases block the positive feedback loop of thrombosis by degrading the scaffold of NET structures. DNase1 is expressed in nonhematopoietic tissues and preferentially cleaves protein‐free DNA. 147 Moreover, immune cells secrete DNase1‐like3, which targets DNA‐protein complexes, such as nucleosomes. 148 DNase1 and DNase1‐like3 also protect the endothelium during sepsis in vitro and in vivo. 149
7.2. Inhibition of glycolysis in sepsis
Like tumour endothelial cells, activated endothelial cells are highly glycolytic during sepsis. 71 Cantelmo et al. reported that tumour endothelial cells were more glycolytic than normal endothelial cells, as indicated by a high expression of PFKFB3. Therefore, the therapeutic concept of using PFKFB3 inhibitor has been extended to patients with sepsis. Furthermore, targeting endothelial cell glycolysis prevents pathological angiogenesis during inflammation. 150 Wang et al. indicated that 3‐(3pyridinyl)‐1‐(4‐pyridinyl)‐2‐propen‐1‐one (3PO) treatment protected mice from acute lung injury via suppression of vascular inflammation. 82 A new class of 3PO derivatives has also been synthesized to improve their anti‐angiogenesis properties. For example, PFK158, a small molecule inhibitor of PFKFB3, can suppress ATP production and proliferation in mouse models of small‐cell lung cancer. 151 Additionally, phenoxyindole 44 shows a higher selectivity for PFKFB3 than the PFKFB1 and PFKFB2 isoforms. 152
7.3. Maintenance of the balance between ROS production and clearance
During sepsis, an imbalance between ROS production and clearance is one of the main mechanisms of endothelial cell dysfunction. Glutathione (GSH) or N‐acetylcysteine (NAC, the precursor of GSH) remarkably decreased endothelial cell death and ROS production when endothelial cells were exposed to plasma from septic shock patients. 153 Several clinical trials showed that patients treated with NAC had shorter stays in the ICU and improved sepsis severity scores. 154 , 155 Aside from clearing ROS, NAC was also shown to impair NET formation during bacterial infection, which could better explain the observed shorter stays and improved severity scores in ICU. 156
Albumin has antioxidant effects associated with its thiol group. Meziani et al. used human serum albumin (HSA) in a rodent model of septic shock. The authors proved that animals treated with HSA showed a decrease in ROS and RNS production. HSA reduces circulating and tissue O2 − production, further preventing endothelial cell dysfunction. Indeed, a decrease in O2 − results in a reduction in its interaction with NO and thus suppresses ONOO− production. Moreover, HSA inhibits the activation of NF‐κB signalling, thus reducing nitrosative stress and oxidative stress in the aorta. 157
Finally, vitamin C has been used in patients with sepsis because of its antioxidant effect. After scavenging ROS and RNS, vitamin C is oxidized to ascorbate‐free radicals. 158 Ascorbate further inhibits ROS and RNS production by preventing NOX activation, suppressing the expression of inducible nitric oxide synthase and enhancing NO bioavailability. 159 , 160 Unfortunately, a clinical trial showed that compared with a placebo, the infusion of vitamin C did not improve organ dysfunction scores or change markers of vascular damage and inflammation. 161
8. CONCLUSION AND PERSPECTIVE
In sepsis, neutrophils and NETs constitute a robust antimicrobial defence. At the same time, excessive neutrophil activation and NET release convert endothelial cells from an anti‐inflammatory, anti‐coagulant phenotype to a pro‐inflammatory, pro‐coagulant phenotype. Additionally, NETs degrade glycocalyx and increase endothelial permeability. Endothelial cell barrier destabilization further promotes sepsis progression.
Thus far, several potential targets for endothelial cell dysfunction therapy have been proposed. Although it is increasingly recognized that excessive NET release may be a vital therapeutic target, studies have been inconclusive. Most inhibitors of NET formation are still in preclinical status, and further clinical trials are needed to be conducted. Anti‐glycolysis treatments are primarily used to inhibit tumour growth, and the therapeutic dosage and the time of administration to sepsis patients need further research. Notably, key enzymes in glycolysis, such as hexokinase, phosphofructokinase‐1 and pyruvate kinase, are also likely to be novel and promising therapeutic targets.
FUNDING INFORMATION
This research was supported by the National Natural Science Foundation of China (NO. 82102253), Natural Science Foundation of Shanghai (NO. 21ZR1413400), Shanghai Sailing Program (NO. 21YF1406800) and the Shanghai Municipal 2021 “Science and Technology Innovation Action Plan” (NO. 21JC1401400).
CONFLICT OF INTERESTS
The authors declare that they have no conflict of interest
Zhang H, Wang Y, Qu M, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med. 2023;13:e1170. 10.1002/ctm2.1170
Hao Zhang and Yanghanzhao Wang Contributed equally to this work
Contributor Information
Hao Zhang, Email: eliteromes@126.com.
Juan P. Cata, Email: jcata@mdanderson.org.
Changhong Miao, Email: miaochangh@163.com.
REFERENCES
- 1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA. 2016;315(8):801‐810. doi: 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kienle K, Glaser KM, Eickhoff S, et al. Neutrophils self‐limit swarming to contain bacterial growth in vivo. Science. 2021;372(6548):eabe7729. doi: 10.1126/science.abe7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Giovanni M, Tam H, Valet C, Xu Y, Looney MR, Cyster JG. GPR35 promotes neutrophil recruitment in response to serotonin metabolite 5‐HIAA. Cell. 2022;185(5):815‐830.e19. doi: 10.1016/j.cell.2022.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sekheri M, El Kebir D, Edner N, Filep JG. 15‐Epi‐LXA4 and 17‐epi‐RvD1 restore TLR9‐mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc Natl Acad Sci U S A. 2020;117(14):7971‐7980. doi: 10.1073/pnas.1920193117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosa BA, Ahmed M, Singh DK, et al. IFN signalling and neutrophil degranulation transcriptional signatures are induced during SARS‐CoV‐2 infection. Commun Biol. 2021;4(1):290. doi: 10.1038/s42003-021-01829-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adrover JM, Aroca‐Crevillén A, Crainiciuc G, et al. Programmed “disarming” of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol. 2020;21(2):135‐144. doi: 10.1038/s41590-019-0571-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thiam HR, Wong SL, Qiu R, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4‐mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117(13):7326‐7337. doi: 10.1073/pnas.1909546117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hidalgo A, Libby P, Soehnlein O, Aramburu IV, Papayannopoulos V, Silvestre‐Roig C. Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res. 2021:cvab329. doi: 10.1093/cvr/cvab329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Demkow U. Neutrophil extracellular traps (NETs) in cancer invasion, evasion and metastasis. Cancers. 2021;13(17):4495. doi: 10.3390/cancers13174495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dömer D, Walther T, Möller S, Behnen M, Laskay T. Neutrophil extracellular traps activate proinflammatory functions of human neutrophils. Front Immunol. 2021;12:636954. doi: 10.3389/fimmu.2021.636954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alsabani M, Abrams ST, Cheng Z, et al. Reduction of NETosis by targeting CXCR1/2 reduces thrombosis, lung injury, and mortality in experimental human and murine sepsis. Br J Anaesth. 2022;128(2):283‐293. doi: 10.1016/j.bja.2021.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun S, Duan Z, Wang X, et al. Neutrophil extracellular traps impair intestinal barrier functions in sepsis by regulating TLR9‐mediated endoplasmic reticulum stress pathway. Cell Death Dis. 2021;12(6):606. doi: 10.1038/s41419-021-03896-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doster RS, Rogers LM, Gaddy JA, Aronoff DM. Macrophage extracellular traps: a scoping review. J Innate Immun. 2018;10(1):3‐13. doi: 10.1159/000480373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim HJ, Sim MS, Lee DH, et al. Lysophosphatidylserine induces eosinophil extracellular trap formation and degranulation: implications in severe asthma. Allergy. 2020;75(12):3159‐3170. doi: 10.1111/all.14450 [DOI] [PubMed] [Google Scholar]
- 15. Sônego F, Castanheira FVES, Ferreira RG, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. 2016;7:155. doi: 10.3389/fimmu.2016.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Milot E, Fotouhi‐Ardakani N, Filep JG. Myeloid nuclear differentiation antigen, neutrophil apoptosis and sepsis. Front Immunol. 2012;3:397. doi: 10.3389/fimmu.2012.00397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barkaway A, Rolas L, Joulia R, et al. Age‐related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity. 2021;54(7):1494‐1510.e7. doi: 10.1016/j.immuni.2021.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aldabbous L, Abdul‐Salam V, McKinnon T, et al. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2016;36(10):2078‐2087. doi: 10.1161/ATVBAHA.116.307634 [DOI] [PubMed] [Google Scholar]
- 19. Polet F, Feron O. Endothelial cell metabolism and tumour angiogenesis: glucose and glutamine as essential fuels and lactate as the driving force. J Intern Med. 2013;273(2):156‐165. doi: 10.1111/joim.12016 [DOI] [PubMed] [Google Scholar]
- 20. Herrero‐Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C‐Cdh1. Nat Cell Biol. 2009;11(6):747‐752. doi: 10.1038/ncb1881 [DOI] [PubMed] [Google Scholar]
- 21. Ma Y, Yang X, Chatterjee V, Meegan JE, Beard RS Jr, Yuan SY. Role of neutrophil extracellular traps and vesicles in regulating vascular endothelial permeability. Front Immunol. 2019;10:1037. doi: 10.3389/fimmu.2019.01037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song D, Ye X, Xu H, Liu SF. Activation of endothelial intrinsic NF‐κB pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood. 2009;114(12):2521‐2529. doi: 10.1182/blood-2009-02-205914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Folco EJ, Mawson TL, Vromman A, et al. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin‐1α and cathepsin G. Arterioscler Thromb Vasc Biol. 2018;38(8):1901‐1912. doi: 10.1161/ATVBAHA.118.311150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joffre J, Hellman J, Ince C, Ait‐Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med. 2020;202(3):361‐370. doi: 10.1164/rccm.201910-1911TR [DOI] [PubMed] [Google Scholar]
- 25. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6(3):173‐182. doi: 10.1038/nri1785 [DOI] [PubMed] [Google Scholar]
- 26. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125(3):281‐288. doi: 10.1111/j.1365-2567.2008.02950.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677‐691. doi: 10.1083/jcb.201006052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Metzler KD, Fuchs TA, Nauseef WM, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2011;117(3):953‐959. doi: 10.1182/blood-2010-06-290171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2(12):965‐975. doi: 10.1038/nri957 [DOI] [PubMed] [Google Scholar]
- 30. Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3(5):401‐416. doi: 10.1038/nrd1383 [DOI] [PubMed] [Google Scholar]
- 31. Guo RF, Sun L, Gao H, et al. In vivo regulation of neutrophil apoptosis by C5a during sepsis. J Leukoc Biol. 2006;80(6):1575‐1583. doi: 10.1189/jlb.0106065 [DOI] [PubMed] [Google Scholar]
- 32. Perianayagam MC, Balakrishnan VS, Pereira BJG, Jaber BL. C5a delays apoptosis of human neutrophils via an extracellular signal‐regulated kinase and bad‐mediated signalling pathway. Eur J Clin Invest. 2004;34(1):50‐56. doi: 10.1111/j.1365-2362.2004.01273.x [DOI] [PubMed] [Google Scholar]
- 33. Shen XF, Cao K, Jiang JP, Guan WX, Du JF. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med. 2017;21(9):1687‐1697. doi: 10.1111/jcmm.13112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qi X, Yu Y, Sun R, et al. Identification and characterization of neutrophil heterogeneity in sepsis. Crit Care. 2021;25(1):50. doi: 10.1186/s13054-021-03481-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang JF, Li JB, Zhao YJ, et al. Up‐regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis‐induced immunosuppression: an animal study and a prospective case‐control study. Anesthesiology. 2015;122(4):852‐863. doi: 10.1097/ALN.0000000000000525 [DOI] [PubMed] [Google Scholar]
- 36. Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. 2006;6(12):907‐918. doi: 10.1038/nri1964 [DOI] [PubMed] [Google Scholar]
- 37. Alves‐Filho JC, Freitas A, Souto FO, et al. Regulation of chemokine receptor by Toll‐like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A. 2009;106(10):4018‐4023. doi: 10.1073/pnas.0900196106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Essential role of CCR2 in neutrophil tissue infiltration and multiple organ dysfunction in sepsis. Accessed November 30, 2022 https://pubmed.ncbi.nlm.nih.gov/20732989/ [DOI] [PubMed]
- 39. Barter J, Kumar A, Stortz JA, et al. Age and sex influence the hippocampal response and recovery following sepsis. Mol Neurobiol. 2019;56(12):8557‐8572. doi: 10.1007/s12035-019-01681-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong SL, Wagner DD. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018;32(12):6258‐6370. doi: 10.1096/fj.201800691R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532‐1535. doi: 10.1126/science.1092385 [DOI] [PubMed] [Google Scholar]
- 42. Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784‐2794. doi: 10.1182/blood-2013-04-457671 [DOI] [PubMed] [Google Scholar]
- 43. Remijsen Q, Berghe TV, Wirawan E, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21(2):290‐304. doi: 10.1038/cr.2010.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU. Neither eosinophils nor neutrophils require ATG5‐dependent autophagy for extracellular DNA trap formation. Immunology. 2017;152(3):517‐525. doi: 10.1111/imm.12790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463‐469. doi: 10.1038/nm1565 [DOI] [PubMed] [Google Scholar]
- 46. Yipp BG, Petri B, Salina D, et al. Infection‐induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18(9):1386‐1393. doi: 10.1038/nm.2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16(11):1438‐1444. doi: 10.1038/cdd.2009.96 [DOI] [PubMed] [Google Scholar]
- 48. Nagaoka I, Tamura H, Reich J. Therapeutic potential of cathelicidin peptide LL‐37, an antimicrobial agent, in a murine sepsis model. Int J Mol Sci. 2020;21(17):E5973. doi: 10.3390/ijms21175973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Magna M, Pisetsky DS. The alarmin properties of DNA and DNA‐associated nuclear proteins. Clin Ther. 2016;38(5):1029‐1041. doi: 10.1016/j.clinthera.2016.02.029 [DOI] [PubMed] [Google Scholar]
- 50. Li Y, Berke IC, Modis Y. DNA binding to proteolytically activated TLR9 is sequence‐independent and enhanced by DNA curvature. EMBO J. 2012;31(4):919‐931. doi: 10.1038/emboj.2011.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75(6):995‐1000. doi: 10.1189/jlb.0703328 [DOI] [PubMed] [Google Scholar]
- 52. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol Baltim Md 1950. 2011;187(5):2626‐2631. doi: 10.4049/jimmunol.1003930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ackermann M, Anders HJ, Bilyy R, et al. Patients with COVID‐19: in the dark‐NETs of neutrophils. Cell Death Differ. 2021;28(11):3125‐3139. doi: 10.1038/s41418-021-00805-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang YY, Ning BT. Signaling pathways and intervention therapies in sepsis. Signal Transduct Target Ther. 2021;6(1):407. doi: 10.1038/s41392-021-00816-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34‐45. doi: 10.1038/nri3345 [DOI] [PubMed] [Google Scholar]
- 56. Li T, Peng R, Wang F, et al. Lysophosphatidic acid promotes thrombus stability by inducing rapid formation of neutrophil extracellular traps: a new mechanism of thrombosis. J Thromb Haemost JTH. 2020;18(8):1952‐1964. doi: 10.1111/jth.14839 [DOI] [PubMed] [Google Scholar]
- 57. Sandoval‐Pérez A, Berger RML, Garaizar A, et al. DNA binds to a specific site of the adhesive blood‐protein von Willebrand factor guided by electrostatic interactions. Nucleic Acids Res. 2020;48(13):7333‐7344. doi: 10.1093/nar/gkaa466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grässle S, Huck V, Pappelbaum KI, et al. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler Thromb Vasc Biol. 2014;34(7):1382‐1389. doi: 10.1161/ATVBAHA.113.303016 [DOI] [PubMed] [Google Scholar]
- 59. Fernández S, Moreno‐Castaño AB, Palomo M, et al. Distinctive biomarker features in the endotheliopathy of COVID‐19 and septic syndromes. Shock Augusta Ga. 2022;57(1):95‐105. doi: 10.1097/SHK.0000000000001823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. von Brühl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819‐835. doi: 10.1084/jem.20112322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129(10):1357‐1367. doi: 10.1182/blood-2016-09-741298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16(8):887‐896. doi: 10.1038/nm.2184 [DOI] [PubMed] [Google Scholar]
- 63. Skendros P, Mitsios A, Chrysanthopoulou A, et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID‐19 immunothrombosis. J Clin Invest. 2020;130(11):6151‐6157. doi: 10.1172/JCI141374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang H, Zhou Y, Qu M, et al. Tissue factor‐enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis‐induced lung injury. Front Cell Infect Microbiol. 2021;11:677902. doi: 10.3389/fcimb.2021.677902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen Z, Zhang H, Qu M, et al. Review: the emerging role of neutrophil extracellular traps in sepsis and sepsis‐associated thrombosis. Front Cell Infect Microbiol. 2021;11:653228. doi: 10.3389/fcimb.2021.653228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gando S, Saitoh D, Ishikura H, et al. A randomized, controlled, multicenter trial of the effects of antithrombin on disseminated intravascular coagulation in patients with sepsis. Published online 2013:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mussbacher M, Salzmann M, Brostjan C, et al. Cell type‐specific roles of NF‐κB linking inflammation and thrombosis. Front Immunol. 2019;10:85. doi: 10.3389/fimmu.2019.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Abrams ST, Morton B, Alhamdi Y, et al. A novel assay for neutrophil extracellular trap formation independently predicts disseminated intravascular coagulation and mortality in critically ill patients. Am J Respir Crit Care Med. 2019;200(7):869‐880. doi: 10.1164/rccm.201811-2111OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Song C, Li H, Li Y, et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp Cell Res. 2019;382(2):111486. doi: 10.1016/j.yexcr.2019.06.031 [DOI] [PubMed] [Google Scholar]
- 70. Spiller KL, Anfang RR, Spiller KJ, et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014;35(15):4477‐4488. doi: 10.1016/j.biomaterials.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. De Bock K, Georgiadou M, Schoors S, et al. Role of PFKFB3‐driven glycolysis in vessel sprouting. Cell. 2013;154(3):651‐663. doi: 10.1016/j.cell.2013.06.037 [DOI] [PubMed] [Google Scholar]
- 72. Parra‐Bonilla G, Alvarez DF, Al‐Mehdi AB, Alexeyev M, Stevens T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am J Physiol‐Lung Cell Mol Physiol. 2010;299(4):L513‐L522. doi: 10.1152/ajplung.00274.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peters K, Kamp G, Berz A, et al. Changes in human endothelial cell energy metabolic capacities during in vitro cultivation. The role of “aerobic glycolysis” and proliferation. Cell Physiol Biochem. 2009;24(5‐6):483‐492. doi: 10.1159/000257490 [DOI] [PubMed] [Google Scholar]
- 74. Yeh WL, Lin CJ, Fu WM. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol. 2008;73(1):170‐177. doi: 10.1124/mol.107.038851 [DOI] [PubMed] [Google Scholar]
- 75. Potente M, Carmeliet P. The link between angiogenesis and endothelial metabolism. Annu Rev Physiol. 2017;79(1):43‐66. doi: 10.1146/annurev-physiol-021115-105134 [DOI] [PubMed] [Google Scholar]
- 76. Wilhelm K, Happel K, Eelen G, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529(7585):216‐220. doi: 10.1038/nature16498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Eustache JH, Tohme S, Milette S, Rayes RF, Tsung A, Spicer JD. Casting a wide net on surgery: the central role of neutrophil extracellular traps. Ann Surg. 2020;272(2):277‐283. doi: 10.1097/SLA.0000000000003586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ebos JML, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210‐221. doi: 10.1038/nrclinonc.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Keating AM, Jacobs DS. Anti‐VEGF treatment of corneal neovascularization. Ocul Surf. 2011;9(4):227‐238. doi: 10.1016/S1542-0124(11)70035-0 [DOI] [PubMed] [Google Scholar]
- 80. Mirabelli P, Peebo BB, Xeroudaki M, Koulikovska M, Lagali N. Early effects of dexamethasone and anti‐VEGF therapy in an inflammatory corneal neovascularization model. Exp Eye Res. 2014;125:118‐127. doi: 10.1016/j.exer.2014.06.006 [DOI] [PubMed] [Google Scholar]
- 81. Yu P, Wilhelm K, Dubrac A, et al. FGF‐dependent metabolic control of vascular development. Nature. 2017;545(7653):224‐228. doi: 10.1038/nature22322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang L, Cao Y, Gorshkov B, et al. Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS‐induced endotoxemia. Pharmacol Res. 2019;146:104292. doi: 10.1016/j.phrs.2019.104292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Appiah MG, Park EJ, Akama Y, et al. Cellular and exosomal regulations of sepsis‐induced metabolic alterations. Int J Mol Sci. 2021;22(15):8295. doi: 10.3390/ijms22158295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang R, Li R, Liu Y, Li L, Tang Y. The glycolytic enzyme PFKFB3 controls TNF‐α‐induced endothelial proinflammatory responses. Inflammation. 2019;42(1):146‐155. doi: 10.1007/s10753-018-0880-x [DOI] [PubMed] [Google Scholar]
- 85. Panieri E, Santoro MM. ROS signalling and redox biology in endothelial cells. Cell Mol Life Sci. 2015;72(17):3281‐3303. doi: 10.1007/s00018-015-1928-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ali MM, Mahmoud AM, Le Master E, Levitan I, Phillips SA. Role of matrix metalloproteinases and histone deacetylase in oxidative stress‐induced degradation of the endothelial glycocalyx. Am J Physiol‐Heart Circ Physiol. 2019;316(3):H647‐H663. doi: 10.1152/ajpheart.00090.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis‐induced alterations in vascular permeability. Crit Care. 2015;19(1):26. doi: 10.1186/s13054-015-0741-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res. 2010;87(2):300‐310. doi: 10.1093/cvr/cvq137 [DOI] [PubMed] [Google Scholar]
- 89. Russell RT, Christiaans SC, Nice TR, et al. Histone‐complexed DNA fragments levels are associated with coagulopathy, endothelial cell damage, and increased mortality after severe pediatric trauma. Shock. 2018;49(1):44‐52. doi: 10.1097/SHK.0000000000000902 [DOI] [PubMed] [Google Scholar]
- 90. Manchanda K, Kolarova H, Kerkenpaß C, et al. MPO (Myeloperoxidase) reduces endothelial glycocalyx thickness dependent on its cationic charge. Arterioscler Thromb Vasc Biol. 2018;38(8):1859‐1867. doi: 10.1161/ATVBAHA.118.311143 [DOI] [PubMed] [Google Scholar]
- 91. Gäddnäs FP, Sutinen MM, Koskela M, et al. Matrix‐metalloproteinase‐2, ‐8 and ‐9 in serum and skin blister fluid in patients with severe sepsis. Crit Care Lond Engl. 2010;14(2):R49. doi: 10.1186/cc8938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217‐1223. doi: 10.1038/nm.2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Alphonsus CS, Rodseth RN. The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia. 2014;69(7):777‐784. doi: 10.1111/anae.12661 [DOI] [PubMed] [Google Scholar]
- 94. Yang X, Meegan JE, Jannaway M, Coleman DC, Yuan SY. A disintegrin and metalloproteinase 15‐mediated glycocalyx shedding contributes to vascular leakage during inflammation. Cardiovasc Res. 2018;114(13):1752‐1763. doi: 10.1093/cvr/cvy167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Clark PR, Kim RK, Pober JS, Kluger MS. Tumor necrosis factor disrupts claudin‐5 endothelial tight junction barriers in two distinct NF‐κB‐dependent phases. PLOS ONE. 2015;10(3):e0120075. doi: 10.1371/journal.pone.0120075. Koval M, ed.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H 2 O 2 ‐mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol. 2000;279(1):C21‐C30. doi: 10.1152/ajpcell.2000.279.1.C21 [DOI] [PubMed] [Google Scholar]
- 97. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE‐cadherin in the control of vascular permeability. J Cell Sci. 2008;121(13):2115‐2122. doi: 10.1242/jcs.017897 [DOI] [PubMed] [Google Scholar]
- 98. Luplerdlop N, Missé D, Bray D, et al. Dengue‐virus‐infected dendritic cells trigger vascular leakage through metalloproteinase overproduction. EMBO Rep. 2006;7(11):1176‐1181. doi: 10.1038/sj.embor.7400814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Allingham MJ, van Buul JD, Burridge K. ICAM‐1‐Mediated, Src‐ and Pyk2‐dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179(6):4053‐4064. doi: 10.4049/jimmunol.179.6.4053 [DOI] [PubMed] [Google Scholar]
- 100. Radeva MY, Waschke J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol. 2018;222(1):e12860. doi: 10.1111/apha.12860 [DOI] [PubMed] [Google Scholar]
- 101. Lee WL. Sepsis and endothelial permeability. N Engl J Med. 2010;363(7):689‐691. [DOI] [PubMed] [Google Scholar]
- 102. Wautier JL, Wautier MP. Vascular permeability in diseases. Int J Mol Sci. 2022;23(7):3645. doi: 10.3390/ijms23073645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Molema G, Zijlstra JG, van Meurs M, Kamps JAAM. Renal microvascular endothelial cell responses in sepsis‐induced acute kidney injury. Nat Rev Nephrol. 2022;18(2):95‐112. doi: 10.1038/s41581-021-00489-1 [DOI] [PubMed] [Google Scholar]
- 104. DiStasi MR, Ley K. Opening the flood‐gates: how neutrophil‐endothelial interactions regulate permeability. Trends Immunol. 2009;30(11):547‐556. doi: 10.1016/j.it.2009.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sumagin R, Kuebel JM, Sarelius IH. Leukocyte rolling and adhesion both contribute to regulation of microvascular permeability to albumin via ligation of ICAM‐1. Am J Physiol Cell Physiol. 2011;301(4):C804‐813. doi: 10.1152/ajpcell.00135.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PloS One. 2012;7(2):e32366. doi: 10.1371/journal.pone.0032366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Radi R, Cassina A, Hodara R. Nitric oxide and peroxynitrite interactions with mitochondria. Biol Chem. 2002;383(3‐4). doi: 10.1515/BC.2002.044 [DOI] [PubMed] [Google Scholar]
- 108. Melican K, Michea Veloso P, Martin T, Bruneval P, Duménil G. Adhesion of Neisseria meningitidis to dermal vessels leads to local vascular damage and purpura in a humanized mouse model. PLoS Pathog. 2013;9(1):e1003139. doi: 10.1371/journal.ppat.1003139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Manriquez V, Nivoit P, Urbina T, et al. Colonization of dermal arterioles by Neisseria meningitidis provides a safe haven from neutrophils. Nat Commun. 2021;12(1):4547. doi: 10.1038/s41467-021-24797-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lappann M, Danhof S, Guenther F, Olivares‐Florez S, Mordhorst IL, Vogel U. In vitro resistance mechanisms of Neisseria meningitidis against neutrophil extracellular traps. Mol Microbiol. 2013;89(3):433‐449. doi: 10.1111/mmi.12288 [DOI] [PubMed] [Google Scholar]
- 111. Iba T, Levi M, Levy JH. Sepsis‐induced coagulopathy and disseminated intravascular coagulation. Semin Thromb Hemost. 2020;46(01):089‐095. doi: 10.1055/s-0039-1694995 [DOI] [PubMed] [Google Scholar]
- 112. Sobczak AIS, Pitt SJ, Stewart AJ. Glycosaminoglycan neutralization in coagulation control. Arterioscler Thromb Vasc Biol. 2018;38(6):1258‐1270. doi: 10.1161/ATVBAHA.118.311102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, oude Egbrink MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflüg Arch Eur J Physiol. 2007;454(3):345‐359. doi: 10.1007/s00424-007-0212-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tollefsen DM. Vascular dermatan sulfate and heparin cofactor II. Progress in Molecular Biology and Translational Science. Elsevier; 2010:351‐372. doi: 10.1016/S1877-1173(10)93015-9 [DOI] [PubMed] [Google Scholar]
- 115. Li W, Johnson DJD, Esmon CT, Huntington JA. Structure of the antithrombin–thrombin–heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol. 2004;11(9):857‐862. doi: 10.1038/nsmb811 [DOI] [PubMed] [Google Scholar]
- 116. de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16(1):19‐27. doi: 10.1038/s41423-018-0024-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Owens AP, Mackman N. Tissue factor and thrombosis: the clot starts here. Thromb Haemost. 2010;104(09):432‐439. doi: 10.1160/TH09-11-0771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Haubitz M, Gerlach M, Kruse HJ, Brunkhorst R. Endothelial tissue factor stimulation by proteinase 3 and elastase. Clin Exp Immunol. 2002;126(3):584‐588. doi: 10.1046/j.1365-2249.2001.01587.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kim JE, Yoo HJ, Gu JY, Kim HK. Histones induce the procoagulant phenotype of endothelial cells through tissue factor up‐regulation and thrombomodulin down‐regulation. Kanthou C, ed. PLOS ONE. 2016;11(6):e0156763. doi: 10.1371/journal.pone.0156763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rao LVM, Pendurthi UR. Regulation of tissue factor coagulant activity on cell surfaces: tissue factor . J Thromb Haemost. 2012;10(11):2242‐2253. doi: 10.1111/jth.12003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320(6):365‐376. doi: 10.1056/NEJM198902093200606 [DOI] [PubMed] [Google Scholar]
- 122. Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H, Libby P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase‐mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24(7):1309‐1314. doi: 10.1161/01.ATV.0000131784.50633.4f [DOI] [PubMed] [Google Scholar]
- 123. Salvaris EJ, Moran CJ, Roussel JC, Fisicaro N, Robson SC, Cowan PJ. Pig endothelial protein C receptor is functionally compatible with the human protein C pathway. Xenotransplantation. 2020;27(2):e12557. doi: 10.1111/xen.12557 [DOI] [PubMed] [Google Scholar]
- 124. Stojanovski BM, Pelc LA, Zuo X, Di Cera E. Zymogen and activated protein C have similar structural architecture. J Biol Chem. 2020;295(45):15236‐15244. doi: 10.1074/jbc.RA120.014789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Griffin JH, Zlokovic BV, Mosnier LO. Protein C anticoagulant and cytoprotective pathways. Int J Hematol. 2012;95(4):333‐345. doi: 10.1007/s12185-012-1059-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF‐kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med. 2008;205(6):1303‐1315. doi: 10.1084/jem.20071393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Qu D, Wang Y, Esmon NL, Esmon CT. Regulated endothelial protein C receptor shedding is mediated by tumour necrosis factor‐alpha converting enzyme/ADAM17. J Thromb Haemost JTH. 2007;5(2):395‐402. doi: 10.1111/j.1538-7836.2007.02347.x [DOI] [PubMed] [Google Scholar]
- 128. Kant R, Halder SK, Fernández JA, Griffin JH, Milner R. Activated protein C attenuates experimental autoimmune encephalomyelitis progression by enhancing vascular integrity and suppressing microglial activation. Front Neurosci. 2020;14:333. doi: 10.3389/fnins.2020.00333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Livnat T, Weinberger Y, Fernández JA, et al. Activated protein C (APC) and 3K3A‐APC‐induced regression of choroidal neovascularization (CNV) is accompanied by vascular endothelial growth factor (VEGF) reduction. Biomolecules. 2021;11(3):358. doi: 10.3390/biom11030358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Nazir S, Gadi I, Al‐Dabet MM, et al. Cytoprotective activated protein C averts Nlrp3 inflammasome‐induced ischemia‐reperfusion injury via mTORC1 inhibition. Blood. 2017;130(24):2664‐2677. doi: 10.1182/blood-2017-05-782102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Oto J, Fernández‐Pardo Á, Miralles M, et al. Activated protein C assays: a review. Clin Chim Acta Int J Clin Chem. 2020;502:227‐232. doi: 10.1016/j.cca.2019.11.005 [DOI] [PubMed] [Google Scholar]
- 132. Martinod K, Fuchs TA, Zitomersky NL, et al. PAD4‐deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood. 2015;125(12):1948‐1956. doi: 10.1182/blood-2014-07-587709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Biron BM, Chung CS, O'Brien XM, Chen Y, Reichner JS, Ayala A. Cl‐amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. 2017;9(1):22‐32. doi: 10.1159/000448808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Perdomo J, Leung HHL, Ahmadi Z, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin‐induced thrombocytopenia. Nat Commun. 2019;10(1):1322. doi: 10.1038/s41467-019-09160-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yost CC, Schwertz H, Cody MJ, et al. Neonatal NET‐inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J Clin Invest. 2016;126(10):3783‐3798. doi: 10.1172/JCI83873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Obi AT, Andraska E, Kanthi Y, et al. Endotoxaemia‐augmented murine venous thrombosis is dependent on TLR‐4 and ICAM‐1, and potentiated by neutropenia. Thromb Haemost. 2017;117(2):339‐348. doi: 10.1160/TH16-03-0218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Schattner M. Platelet TLR4 at the crossroads of thrombosis and the innate immune response. J Leukoc Biol. 2019;105(5):873‐880. doi: 10.1002/JLB.MR0618-213R [DOI] [PubMed] [Google Scholar]
- 138. Neal MD, Jia H, Eyer B, et al. Discovery and validation of a new class of small molecule Toll‐like receptor 4 (TLR4) inhibitors. PloS One. 2013;8(6):e65779. doi: 10.1371/journal.pone.0065779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK‐242 (resatorvid), a small‐molecule inhibitor of Toll‐like receptor (TLR) 4 signalling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. 2011;79(1):34‐41. doi: 10.1124/mol.110.068064 [DOI] [PubMed] [Google Scholar]
- 140. Gao W, Xiong Y, Li Q, Yang H. Inhibition of toll‐like receptor signalling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol. 2017;8:508. doi: 10.3389/fphys.2017.00508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Rice TW, Wheeler AP, Bernard GR, et al. A randomized, double‐blind, placebo‐controlled trial of TAK‐242 for the treatment of severe sepsis. Crit Care Med. 2010;38(8):1685‐1694. doi: 10.1097/CCM.0b013e3181e7c5c9 [DOI] [PubMed] [Google Scholar]
- 142. Mullarkey M, Rose JR, Bristol J, et al. Inhibition of endotoxin response by e5564, a novel toll‐like receptor 4‐directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304(3):1093‐1102. doi: 10.1124/jpet.102.044487 [DOI] [PubMed] [Google Scholar]
- 143. Savov JD, Brass DM, Lawson BL, McElvania‐Tekippe E, Walker JKL, Schwartz DA. Toll‐like receptor 4 antagonist (E5564) prevents the chronic airway response to inhaled lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 2005;289(2):L329‐L337. doi: 10.1152/ajplung.00014.2005 [DOI] [PubMed] [Google Scholar]
- 144. Opal SM, Laterre PF, Francois B, et al. Effect of eritoran, an antagonist of MD2‐TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309(11):1154‐1162. doi: 10.1001/jama.2013.2194 [DOI] [PubMed] [Google Scholar]
- 145. Wang Y, Zhang S, Li H, et al. Small‐molecule modulators of toll‐like receptors. Acc Chem Res. 2020;53(5):1046‐1055. doi: 10.1021/acs.accounts.9b00631 [DOI] [PubMed] [Google Scholar]
- 146. Claushuis TAM, Van Der Veen AIP, Horn J, et al. Platelet Toll‐like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets. 2019;30(3):296‐304. doi: 10.1080/09537104.2018.1445841 [DOI] [PubMed] [Google Scholar]
- 147. Napirei M, Ricken A, Eulitz D, Knoop H, Mannherz HG. Expression pattern of the deoxyribonuclease 1 gene: lessons from the DNase1 knockout mouse. Biochem J. 2004;380(Pt 3):929‐937. doi: 10.1042/BJ20040046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Sisirak V, Sally B, D'Agati V, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. 2016;166(1):88‐101. doi: 10.1016/j.cell.2016.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Jiménez‐Alcázar M, Rangaswamy C, Panda R, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358(6367):1202‐1206. doi: 10.1126/science.aam8897 [DOI] [PubMed] [Google Scholar]
- 150. Schoors S, De Bock K, Cantelmo AR, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19(1):37‐48. doi: 10.1016/j.cmet.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 151. Cargill KR, Stewart CA, Park EM, et al. Targeting MYC‐enhanced glycolysis for the treatment of small cell lung cancer. Cancer Metab. 2021;9(1):33. doi: 10.1186/s40170-021-00270-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Boyd S, Brookfield JL, Critchlow SE, et al. Structure‐based design of potent and selective inhibitors of the metabolic kinase PFKFB3. J Med Chem. 2015;58(8):3611‐3625. doi: 10.1021/acs.jmedchem.5b00352 [DOI] [PubMed] [Google Scholar]
- 153. Huet O, Cherreau C, Nicco C, et al. Pivotal role of glutathione depletion in plasma‐induced endothelial oxidative stress during sepsis. Crit Care Med. 2008;36(8):2328‐2334. doi: 10.1097/CCM.0b013e3181800387 [DOI] [PubMed] [Google Scholar]
- 154. Spapen H, Zhang H, Demanet C, Vleminckx W, Vincent JL, Huyghens L. Does N‐acetyl‐L‐cysteine influence cytokine response during early human septic shock? Chest. 1998;113(6):1616‐1624. doi: 10.1378/chest.113.6.1616 [DOI] [PubMed] [Google Scholar]
- 155. Ortolani O, Conti A, De Gaudio AR, Moraldi E, Cantini Q, Novelli G. The effect of glutathione and N‐acetylcysteine on lipoperoxidative damage in patients with early septic shock. Am J Respir Crit Care Med. 2000;161(6):1907‐1911. doi: 10.1164/ajrccm.161.6.9903043 [DOI] [PubMed] [Google Scholar]
- 156. Pylaeva E, Bordbari S, Spyra I, et al. Detrimental effect of type I IFNs during acute lung infection with pseudomonas aeruginosa is mediated through the stimulation of neutrophil NETosis. Front Immunol. 2019;10:2190. doi: 10.3389/fimmu.2019.02190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Meziani F, Kremer H, Tesse A, et al. Human serum albumin improves arterial dysfunction during early resuscitation in mouse endotoxic model via reduced oxidative and nitrosative stresses. Am J Pathol. 2007;171(6):1753‐1761. doi: 10.2353/ajpath.2007.070316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wilson JX. Mechanism of action of vitamin C in sepsis: ascorbate modulates redox signalling in endothelium. BioFactors Oxf Engl. 2009;35(1):5‐13. doi: 10.1002/biof.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Mortensen A, Lykkesfeldt J. Does vitamin C enhance nitric oxide bioavailability in a tetrahydrobiopterin‐dependent manner? In vitro, in vivo and clinical studies. Nitric Oxide Biol Chem. 2014;36:51‐57. doi: 10.1016/j.niox.2013.12.001 [DOI] [PubMed] [Google Scholar]
- 160. Wu F, Tyml K, Wilson JX. Ascorbate inhibits iNOS expression in endotoxin‐ and IFN gamma‐stimulated rat skeletal muscle endothelial cells. FEBS Lett. 2002;520(1‐3):122‐126. doi: 10.1016/s0014-5793(02)02804-1 [DOI] [PubMed] [Google Scholar]
- 161. Fowler AA, Truwit JD, Hite RD, et al. Effect of Vitamin C infusion on organ failure and biomarkers of inflammation and vascular injury in patients with sepsis and severe acute respiratory failure: the CITRIS‐ALI randomized clinical trial. JAMA. 2019;322(13):1261‐1270. doi: 10.1001/jama.2019.11825 [DOI] [PMC free article] [PubMed] [Google Scholar]