

Abstract
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) that poses a serious global public health threat. Due to the high incidence of adverse reactions associated with conventional treatment regimens, there is an urgent need for better alternative therapies. CpG oligodeoxynucleotides (CpG ODNs) are synthetic oligodeoxyribonucleotide sequences. They can induce a Th1-type immune response by stimulating Toll-like receptors (TLRs) in mammalian immune cells, thus killing Mtb. However, due to the negative charge and easy degradation of CpG ODNs, it is necessary to deliver them into cells using nanomaterials. PCN-224 (hereinafter referred to as PCN), as a metal–organic framework based on zirconium ions and porphyrin ligands, not only has the advantage of high drug loading capacity, but also the porphyrin molecule in it is a type of photosensitizer, which allows these nanocomposites to play a role in photodynamic therapy (PDT) while delivering CpG ODNs. In addition, since Mtb mainly exists in macrophages, targeting anti-TB agents to macrophages is helpful to improve the anti-TB effect. Phosphatidylserine (PS) is a biological membrane phospholipid that is normally found on the inner side of cell membranes in, for example, plant and mammalian cells. When apoptosis occurs, PS can flip from the inner side of the cell membrane to the surface of the cell membrane, displaying a specific “eat-me” signal that can be recognized by specific receptors on macrophages. Therefore, we can use this macrophage-targeting property of PS to construct bio-inspired targeted drug delivery systems. In this study, we constructed PCN-CpG@PS nanocomposites. PCN-CpG@PS, combining PDT and immunotherapy, is designed to target macrophages at the site of a lesion and kill latent Mtb. We physically characterized the nanocomposites and validated their bactericidal ability in vitro and their ability to stimulate the immune system in vivo. The results demonstrated that the targeted nanocomposites have certain in vitro antituberculosis efficacy with good safety.
A metal–organic framework-based nanotheranostic system was fabricated to achieve a macrophage-targeting ability and combined photodynamic and immunotherapy for Mtb.
Introduction
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) and is an epidemic that poses a serious global public health threat.1–3 At present, the World Health Organization recommends four first-line drugs, isoniazid (INH), rifampicin (RFP), pyrazinamide (PZA), and ethambutol (EMB), as the main drugs for the treatment of drug-susceptible TB with a 6 month course.4 However, this regimen has the disadvantages of a long course of treatment, high dose, and high incidence of adverse effects, which often lead to poor patient compliance, thus accelerating the development of drug-resistant tuberculosis (DR-TB), greatly increasing medical risks and the socioeconomic burden.5,6 Therefore, there is an urgent need for better alternative treatments.
CpG oligodeoxynucleotides (CpG ODNs) are synthetic single-stranded deoxyribonucleic acid (DNA) molecules containing unmethylated cytosine–phosphate–guanine (CpG) motifs, with hexamer sequence as the core.7 CpG ODNs can be used as an immune adjuvant or an immunotherapeutic agent alone.8,9 CpG ODNs can be easily recognized by the mammalian immune system and promote the production of T helper type 1(Th1) cytokines, such as IL-12, by stimulating Toll-like receptors (TLRs) in endolysosomes in cells such as macrophages, which in turn induce a strong Th1 immune response.9,10 This immunostimulatory activity makes the use of CpG ODNs in immunotherapy very attractive. It has been reported that CpG ODNs as an immune adjuvant can enhance the immune effect of the Bacillus Calmette-Guérin (BCG) vaccine.11,12 However, it is difficult for CpG ODNs to cross cell membranes because CpG ODNs is negatively charged and susceptible to degradation by nucleases.13 Therefore, how CpG ODNs is internalized into antigen-presenting cells is important to induce an immune response. The delivery of CpG ODN using nanomaterials can improve the cellular uptake of CpG ODNs and increase the therapeutic efficiency.14,15
Metal–organic frameworks (MOFs) are porous materials with specific structures formed by the self-assembly of metal clusters and organic ligands through coordination bonds, which have the advantages of easy synthesis, high drug loading and good biocompatibility.16,17 They have promising applications in drug delivery and disease treatment.18 As a kind of MOFs, the porphyrin-based Zr-MOFs PCN-224 (hereinafter referred to as PCN) not only has the above advantages but the porphyrin molecules in it can also be used directly as an efficient photosensitizer for photodynamic therapy (PDT).19,20 PDT is an oxygen-dependent therapy based on the interaction of photosensitizer (PS), light and molecular oxygen, i.e., photosensitizer produces reactive oxygen species (ROS), mainly singlet oxygen (1O2), under irradiation with a specific wavelength of light,21,22 which then disrupts cell membranes and DNA through ROS, leading to leakage of cellular components and membrane transport systems and breakage of single- and double-stranded DNA, respectively, thus providing bactericidal effects.23–26 In addition, PDT can also improve the bactericidal effect by activating the immune response of the body.27 The bactericidal effect of PDT on microorganisms was studied 100 years ago.28 However, the potential of PDT in inactivating pathogenic microorganisms is gradually being forgotten for various reasons, such as the poor response of some well-known pathogens, especially Gram-negative bacteria, to the most traditional photodynamic therapy and the discovery and successful application of antibiotics. Later, due to the emergence of drug-resistant bacterial pathogens such as methicillin-resistant Staphylococcus aureus (MRSA)29 and Vibrio traumaticus,30 PDT was reattempted for the treatment of bacterial infections. Studies have shown that PDT can inactivate Mycobacterium bovis (M. bovis),31Mycobacterium marinum (M. marinum)32 and Mtb33 in in vitro and animal infection models. This suggests that PDT can be used in the treatment of TB. However, since Mtb mainly exists in macrophages, nanomaterials used for PDT need to have macrophage-targeting properties for optimal therapeutic results.
Phosphatidylserine (PS) is abundant in the outer membrane of apoptotic cells and plays an important role in the recognition and phagocytosis of apoptotic cells by macrophages.34,35 Anti-tuberculosis therapy targeting macrophages can be achieved with PS-coated nanomaterials to increase focal drug concentrations and effectively reduce the systemic toxicity of the treatment. Studies have been reported on the use of PS for the functionalization of nanoparticles to target therapeutic sites and improve therapeutic efficacy, among other purposes.36,37
In this paper, nanocomposites targeting macrophages for photodynamic therapy combined with immunotherapy were constructed. PCN was used as a photosensitizer and a carrier loaded with CpG ODNs. Then, the CpG ODNs-loaded PCN was further encapsulated with PS to obtain the composite nanomaterial PCN-CpG@PS. Delivery of CpG ODNs with PCN can promote the cellular uptake of CpG ODNs on the one hand, and on the other hand, PCN can act as a photosensitizer for PDT. At the same time, PS achieved targeting of PCN-CpG@PS to macrophages. PDT combined with immunotherapy exerts anti-tuberculosis effects while reducing the incidence of adverse drug reactions and drug resistance (Scheme 1).
Scheme 1. (a) Schematic illustration of the in vivo application of the macrophage-targeted PCN-CpG@PS nanoparticles for combined photodynamic immunotherapy. (b) Schematic illustration of the specific process of PCN-CpG@PS exerting photodynamic combined immunotherapeutic effects in macrophages.

Experimental section
Materials
All reagents and solvents used in this work were analytical or higher grade. Phosphatidylserine (PS), sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC) and tris (2-chloroethyl) phosphate (TCEP) were purchased from Shanghai Macklin Biochemical Co. Ltd, Shanghai, China. CpG ODNs (2395) (5′-TCGTCGTTTTCGGCGCGCGCCG-3′), Cy3 labeled CpG ODNs (2395) (5′-TCGTCGTTTTCGGCGCGCGCCG-Cy3-3′) and Cy5 labeled and SH-modified CpG ODNs (2395) (5′-SH-TCGTCGTTTTCGGCGCGCGCCG-Cy5-3′) were synthesized by Sangon Biotechnology Co., Ltd., Shanghai, China. A Cell Counting Kit-8(CCK8) was purchased from Dōjindo Laboratories, Tokyo, Japan. High glucose Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin, and Dulbecco's phosphate buffered saline (PBS) were obtained from Wisent Biotechnology Co. Ltd, Nanjing, China. Opti-MEM I Reduced Serum Medium (referred to below as Opti-MEM) was purchased from Thermo Fisher Scientific. All of the other reagents were obtained from Beyotime Institute Of Biotechnology Co. Ltd, Shanghai, China. All reagents were used without further purification. Ultrapure water was used throughout all experiments and to prepare all solutions.
Synthesis of PCN-224
PCN-224 (hereinafter referred to as PCN) was synthesized according to the reported literature.38,39 In brief, 100 mg tetrakis (4carboxyphenyl) porphyrin (TCPP), 300 mg ZrOCl2·8H2O and 2.2 g benzoic acid were dissolved in 100 mL N, N-dimethylformamide (DMF) and stirred at 90 °C for 5 h. After this, the PCN nanoparticles were collected by centrifugation at 12 000 rpm for 30 min and washed with DMF three times. The obtained PCN nanoparticles were dispersed in DMF shielded from light for the following experiment.
Synthesis of PCN-CpG
The 10 µg mL−1 PCN solution was centrifuged at 10 000 rpm for 10 min. Then the supernatant was discarded, and the precipitate was washed with water three times, with centrifugation at 10 000 rpm for 10 min after each wash. Finally, the precipitate was resuspended in pure water. Then, 0.25 µM CpG 2395 solution was added to the resultant PCN solution. The mixed solution was vortexed, allowed to sit for 15 min at room temperature for adsorption, and then centrifuged to remove unadsorbed CpG 2395.
Synthesis of PCN-CpG@PS
300 mM TCEP solution, pH 5.5 PBS and 0.25 µM SH-modified and Cy5/Cy3 labeled or label-free CpG 2395 solution were mixed and stirred at RT for 1 h. The resultant solution was then transferred to a 3 kDa ultrafiltration tube, centrifuged at 8000 rpm for 6 min and washed with PBS three times and resuspended in PBS. Then, the solution was added to the PCN solution. The mixed solution was vortexed, allowed to sit for 15 min at room temperature for adsorption, and then centrifuged to remove unadsorbed CpG 2395. The precipitate was collected and resuspended in PBS, and the obtained solution was added to 10 mg mL−1 sulfo-SMCC and shaken overnight at RT. The solution was then transferred to a 3 kDa ultrafiltration tube, centrifuged at 8000 rpm for 10 min, washed with HEPES three times and resuspended in pH 8.0 HEPES. The resultant solution was added to 10 mg mL−1 PS. After vortexing for 10 s, the solution was kept for 4 h with continuous shaking. Finally, the mixed solution was transferred to a 3 kDa ultrafiltration tube, centrifuged at 8000 rpm for 10 min, washed with PBS three times and then resuspended in PBS for further experiments.
Characterization
X-ray diffraction (XRD) patterns were obtained with a D8 advance (Bruker). The sizes and morphologies of the samples were determined using scanning electron microscopy (SEM) (ZEISS). Zetasizer Nano ZS ZEN3600 (Malvern) was selected to measure the hydrodynamic sizes (DLS) and zeta potentials. The Ultraviolet-visible absorption (UV-vis) spectroscopy was recorded with a UV-Vis spectrophotometer (DS5, Edinburgh, UK). Fluorescence emission spectra were recorded with a fluorescence spectrofluorimeter (FS5C, Edinburgh, UK).
Mycobacterium tuberculosis strain and cell lines
The Mycobacterium tuberculosis (Mtb) strain H37Rv and Bacillus Calmette-Guérin (BCG) employed in this study came from Beijing Chest Hospital (Beijing, China). Each strain was suspended in a medium containing glycerol and stored at −80 °C. In preparation for use, each strain was removed from the freezer and placed at room temperature. Following thawing, the strain suspension was transferred to a sterile tube filled with 20 mL of 7H9 broth medium containing 10% (vol/vol) Middlebrook OADC (oleic acid, albumin, dextrose, catalase; Difco, Detroit, MI) and 0.05%(vol/vol) Tween 80 (Sigma, St. Louis, MO) and incubated at 37 °C. RAW264.7 cells (a mouse macrophage cell line) were gifted by the Medical Department of Peking University and were grown in high-glucose DMEM supplemented with 10% FBS and 1% penicillin-streptomycin at 37 °C in a 5% CO2 humidified incubator.
In vitro cytotoxicity of PCN, PCN-CpG and PCN-CpG@PS
RAW 264.7 cells were seeded at 1 × 104 cells per well in 96-well plates with 100 µL high glucose DMEM and cultured for 24 h. After removing the medium, RAW264.7 cells were treated with 100 µL Opti-MEM I Reduced Serum Medium (referred to below as Opti-MEM) containing PCN, PCN-CpG and PCN-CpG@PS (1–10 µg mL−1 PCN and/or 0.25 µM CpG 2395) for another 24 h. After that, 100 µL of DMEM containing 10% Cell Counting Kit-8 (CCK8) was added and the plates were incubated for 1 h under the same conditions. For the phototoxicity assay, after removing the medium, RAW264.7 cells were treated with 10 µg mL−1 PCN or PCN-CpG@PS for 4 h followed by light irradiation at a wavelength of 640 nm. 20 h after PDT, the cell viability was evaluated by the CCK-8. Finally, the absorbance was measured at 450 nm using an ELISA plate reader (Thermo Scientific). All experiments were performed in triplicate and the cell viability was calculated using the following formula:Cell viability = (absorbance of test wells/mean absorbance of untreated wells) × 100%.
Cellular uptake
To evaluate the cellular uptake efficiencies of CpG 2395, PCN-CpG and PCN-CpG@PS, RAW264.7 cells were seeded into confocal culture dishes at a density of 2 × 105 per dish. After the cells adhered to the bottom, CpG 2395 (0.25 µM), PCN-CpG (10 µg mL−1 PCN and 0.25 µM CpG), and PCN-CpG@PS (10 µg mL−1 PCN, 83.2 µg mL−1 PS and 0.25 µM CpG) were added to the Petri dish and incubated with RAW264.7 cells. 4 hours later, the cells were washed 3 times with PBS buffer, fixed with paraformaldehyde, and stained with Hoechst 33258. Then, the cells were visualized via confocal laser scanning microscopy (CLSM) (Olympus, Japan) with a 40× objective. Laser 405 nm and Cy5 channel were activated for excitation of the nuclear stain Hoechst 33342 and Cy5 (Cy5-labeled CpG 2395), respectively. Flow cytometry was performed on a Guava easyCyte 6HT-2L Cytometer (Merck KGaA) and FCS files were analyzed using FlowJo software (version 10). Cells with PBS were used as controls for comparison.
Evaluation of in vitro ROS production
The intracellular reactive oxygen species (ROS) generation of PCN-CpG@PS with 640 nm laser irradiation was detected using 2′,7′-dichlorofluorescein diacetate (DCFH-DA) as a fluorescent probe. A continuous-wave laser at 640 nm wavelength was used as an illumination source. DCFH-DA itself has no fluorescence and can freely cross the cell membrane. After entering the cell, it can be hydrolyzed by intracellular enzymes to produce DCFH. However, DCFH cannot penetrate the cell membrane, so it can be easily loaded into the cell. In the presence of ROS, DCFH can be oxidized to produce the fluorescent substance DCF, and the green fluorescence intensity is proportional to the level of intracellular ROS. Therefore, the level of intracellular ROS can be determined by detecting the fluorescence of DCF. RAW264.7 cells were plated on confocal culture dishes at 2 × 105 cells per dish until cell attachment. Following incubation with 10 µg mL−1 PCN, PCN-CpG and PCN-CpG@PS for 4 h at 37 °C, the cells were washed three times with PBS and replaced with fresh medium. After a proper amount of DCFH-DA (5 µM) was added to the wells and incubated for another 0.5 h at 37 °C, the cells were incubated shielded from light or irradiated by a 640 nm laser for 10 min (0.15 W cm−2), including a circle of 5 irradiation fields, each for 2 minutes. The light source was mounted at a distance of 2 cm above the culture dish. Finally, the generated ROS were monitored by CLSM and flow cytometry. The generation of ROS in RAW264.7 cells treated with PCN without 640 nm laser irradiation was used as a control.
Mtb infection of macrophages in vitro
RAW264.7 cells were placed into six-well plates at a density of 8 × 105 cells per well with 2 mL high glucose DMEM for 24 h. Then the broth cultures of the H37Rv strain in log phage were centrifuged at 4000 × g for 5 min, and the pellets containing Mtb were resuspended in fresh DMEM. Subsequently, the confluent RAW264.7 cell monolayer was exposed for 4 h to Mtb at a multiplicity of infection (MOI) of 10 : 1. Three replicates of each sample were performed in the experiment.
Evaluation of PCN-CpG@PS killing intracellular bacteria
To evaluate the synergistic bactericidal effect of PCN-CpG@PS NPs as a PDT-immunotherapy agent, the Mtb-infected cells were washed three times with warm PBS and then incubated with PBS, CpG 2395, PCN, PCN-CpG, and PCN-CpG@PS(10 µg mL−1 PCN, 0.25 µM CpG 2395), respectively. After incubation for 4 h, they were washed three times with PBS to remove the excess material. In the NIR laser treatment group, a 640 nm laser with a power density of 0.15 W cm−2 was directly applied to cells for 10 min. RAW264.7 cells incubated with nanocomposites containing CpG 2395 without laser irradiation were considered the immunotherapy group. The PDT-immunotherapy group was treated with nanocomposites containing both PCN and CpG 2395 and irradiated under 640 nm irradiation for 10 min. Colony-forming units (CFUs) assays were performed on day 3 post-infection by lysing RAW264.7 cells in ice-cold sterile PBS containing 0.1% Triton-X-100, serially diluting them in PBS containing 0.01% Tween-80 and plating the lysate in triplicate on Middlebrook 7H10 agar. Mtb colonies that survived in macrophages were enumerated after incubation of the plates at 37 °C in humidified air for 2 weeks. To examine the effect of anti-tuberculosis treatments, CFUs recovered from cells incubated with PBS without laser irradiation were considered to be 100% growth.
Animal models
Female BALB/c mice aged 6–8 weeks (18–20 g) were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China) and bred under sterilized, specific pathogen-free (SPF) conditions. The mice were equally divided into cages with three mice per cage and given a week to acclimate to the environment before the experiment. For the Mtb-infected group, all 30 mice were aerosol infected with BCG via the inhalation route by using a Glas-Col Inhalation Exposure System (model 099C A4224). Three untreated mice were sacrificed 3 days after infection to determine if the infection was successful. All animal experimental protocols were conformed to the Guidelines for the Ethical Review of Laboratory Animal Welfare of People's Republic of China National Standard (GB/T 35892-2018) and were approved by the Experimental Animal Committee of Beijing Chest Hospital affiliated to Capital Medical University.
In vivo fluorescence imaging
9 healthy mice were intravenously injected with 100 µL of CpG 2395, PCN-CpG or PCN-CpG@PS and imaged by an IVIS imaging system at 6 h postinjection without laser irradiation. Then all groups of mice were sacrificed, and the major organs were imaged for the biodistribution study.
Histology
10 days after infection, BALB/c mice were treated differently(0.1 mL per mouse): (1) PBS; (2) PCN; (3) PCN-CpG; (4) PCN-CpG@PS; (5) CpG; (6) PBS exposed to 640 nm laser; (7) PCN exposed to 640 nm laser with the power intensity of 0.15 W cm−2 for 10 min (PDT); (8) PCN-CpG with 640 nm laser irradiation(0.15 W cm−2, 10 min); (9) PCN-CpG@PS irradiated by 640 nm laser (0.15 W cm−2, 10 min). Briefly, all solutions were injected via the tail vein. Two hours after administration, mice with their chest hair shaved in advance were anesthetized after inhalation of paraformaldehyde for a short period of time. The left and right chests were irradiated for a total of 10 min with 640 nm/0.15 W cm−2 laser optical fiber with its end in tight contact with the skin. All the treatments were performed at 1, 5, and 8 days after 10 days of infection. Five days post the final treatment, all the mice were sacrificed and the heart, liver, spleen, left lung and kidney were harvested and fixed in 4% paraformaldehyde for 24 h. Then, they were embedded in paraffin and cut into 4 µm tissue slices. Subsequently, the slices were stained with standard operating procedures. The images were observed and acquired under a fluorescence microscope (Olympus BX43, Tokyo, Japan).
Immunofluorescence (IF) assay
After being dehydrated, washed, paraffin-embedded, sectioned, dewaxed and hydrated, the left lung tissue sections were antigenically repaired using EDTA antigen repair solution at pH = 8.0 and blocked with bovine serum albumin for 30 min. The sections were first incubated with rabbit anti-TNF-α and anti-IFN-γ antibodies overnight at 4 °C, and then incubated with secondary goat anti-rabbit antibody for 50 min at RT, followed by sealing with 4,6-diamidino-2-phenylindole (DAPI)-containing anti-fluorescence quenching blocking solution. Finally, images were photographed under a Nikon Eclipse Ci-L fluorescence microscope.
Evaluation of PCN-CpG@PS killing Mtb in vivo
The right lungs of mice were harvested after sacrifice and homogenized in PBS containing 0.01% Tween 80. The homogenate was inoculated on 7H10 agar plates after gradient dilution with PBS containing 0.01% Tween 80, and the live bacteria were counted by CFU assay after 4 weeks of incubation.
Statistical analysis
All data are based on at least 3 independent experiments. The statistical software SPSS23.0 for Windows (SPSS Inc., Chicago, IL, USA) was used. The data are presented as the means ± standard deviation (mean ± SD). Analysis of variance (ANOVA) was employed for multiple group comparisons, and statistical significance of the differences was indicated as P values * < 0.05, ** < 0.01, *** < 0.001, and **** < 0.0001.
Results and discussion
Structure and morphology
The illustration and preparation of PCN-CpG@PS are shown in Fig. S1† and 1a. First, PCN was synthesized as described previously. Next, PCN-CpG nanocomposites were synthesized by loading negatively charged CpG 2395 onto positively charged PCN by electrostatic interactions for CpG 2395 delivery and combining PDT and immunotherapy. Finally, PS was modified on the surface of PCN-CpG to target PCN-CpG@PS to macrophages.
Fig. 1. Preparation and characterization of PCN-CpG@PS. (a) A schematic illustration of the step-by-step synthesis of PCN-CpG@PS nanocomposites for targeted macrophages. (b) X-ray diffraction (XRD) pattern of PCN-224. (c–e) Scanning electron microscopy (SEM) images of (c) PCN-224, (d) PCN-CpG and (e) PCN-CpG@PS. (f) Hydrodynamic size distribution and (g) corresponding quantitative mean size analysis of PCN-224, PCN-CpG, PCN-CpG@PS. N = 3. (h) Zeta potential of PCN-224, PCN-CpG, PCN-CpG@PS. N = 3. Scale bar: 200 nm. Bar: mean; error bar: standard deviation.

As shown in Fig. 1b, the XRD of the synthesized PCN matched well with the stimulated patterns from the single-crystal X-ray data, indicating that a pure phase of PCN was obtained. As visualized by scanning electron microscopy (SEM), PCN showed a homogeneous sphere-like structure with an average diameter of ∼160 nm (Fig. 1c) because the physical natural form of PCN 224 is always as a nanoparticle. After CpG 2395 loading and PS modification, there was almost no change in morphology compared to PCN (Fig. 1d and e). Dynamic light scattering (DLS) analysis showed that the average diameter increased from 159 ± 2.27 nm for PCN to 224 ± 3.78 nm for PCN-CpG. However, PS modification reduced the size of PCN-CpG to 163 ± 5.19 nm (Fig. 1f and g). Fig. 1h shows the measured zeta potentials of PCN, PCN-CpG, and PCN-CpG@PS. It can be clearly seen that PCN possesses a positive charge (+26.7 mV). After adsorption with CpG 2395, the zeta potential of the obtained PCN-CpG changed from a positive charge to a negative charge (−24.4 mV), indicating the successful anchoring of CpG 2395 on the surface of PCN because of the negative charge of CpG 2395. Then, the modification of PS on PCN-CpG contributes to a decrease in the zeta potential to −29.6 mV, indicating that negatively charged PS successfully bound to PCN-CpG. The structure and purity of the synthesized PCN were confirmed by X-ray diffraction (XRD) Ultraviolet-visible (UV-vis) absorption spectroscopy was employed to confirm the existence of PCN and CpG 2395 in PCN-CpG@PS. As shown in Fig. S2,† PCN exhibited a strong absorption peak at 425 nm, which is the typical peak of PCN. CpG 2395 had an absorption peak at 260 nm. The spectrum of the PCN-CpG@PS clearly showed the characteristic absorption peak of PCN at 425 nm and the characteristic absorption peak of CpG 2395 at 250 nm, which may be the result of the interaction between the absorption peak of PCN at 240 nm and the absorption peak of CpG 2395 at 260 nm, which are too close to each other, indicating the successful formation of PCN-CpG@PS. The existence of CpG in PCN-CpG@PS was verified by excitation of CpG labeled with the Cy3 fluorescent group by fluorescence emission spectrum (Fig. S3†).
Cytotoxicity studies
Low toxicity or nontoxicity is required for biomedical uses of nanomaterials. Using a cell viability assay (CCK8), we investigated the dark toxicity of PCN, PCN-CpG and PCN-CpG@PS in RAW 264.7 cells. Three concentration gradients of nanomaterials, 1 µg mL−1, 5 µg mL−1 and 10 µg mL−1, were selected. Under dark conditions, as shown in Fig. 2, when the concentration of PCN solution was in the range of 1–10 µg mL−1, no significant cytotoxicity against RAW 264.7 cells was found (p > 0.05). All the results indicated that the concentrations of preparations (with 1–10 µg mL−1 for the PCN equivalent and 0.25 µM for the CpG 2395) used in the subsequent studies were safe for macrophages. We also tested the phototoxicity of PCN-CpG@PS (10 µg mL−1) to macrophages under 640 nm laser irradiation. As shown in Fig. S4,† the survival rate of macrophages was decreased compared to that of the PBS group. This may be due to the nonselective killing of the cellular components of macrophages by the ROS produced by PDT. However, the phototoxicity of the nanomaterials to macrophages would be reduced because the ROS produced by PDT were also consumed by Mtb in macrophages.
Fig. 2. The viabilities of RAW264.7 cells incubated with PCN, PCN-CpG and PCN-CpG@PS without laser irradiation. N = 5. Bar: mean; error bar: standard deviation. P1-way-ANOVA > 0.05.

In vitro cellular uptake
The cellular uptake efficiency of CpG 2395, PCN-CpG and PCN-CpG@PS was investigated by confocal laser scanning microscopy (CLSM) and flow cytometry after CpG 2395 was labeled with red-emitting Cy5. Therefore, confocal fluorescence imaging of Cy5 red emission demonstrated the colocalization of CpG nanocomposites. Fig. 3a showed the CLSM images of RAW264.7 cells incubated with different nanomaterials. The nuclei were stained blue, and the red spots throughout the RAW264.7 cells represent the Cy5-labeled CpG 2395 for easier observation. Both flow cytometry analysis and CLSM images revealed good cellular uptake of PCN-CpG@PS into RAW264.7 cells. Negative CpG 2395 may be degraded by nucleases and/or rarely ingested by RAW264.7 cells due to the electrostatic barrier of the cytoplasmic membrane. As shown in Fig. 3a, RAW264.7 cells treated with free CpG 2395 showed weak red fluorescence. However, bright fluorescence was observed in the cytoplasm of RAW264.7 cells after incubation with PCN-CpG, demonstrating that PCN-CpG showed better cellular uptake efficiency and crossed the cell membrane easily. Moreover, the red fluorescence intensity inside the cells treated with PCN-CpG@PS increased remarkably compared with PCN-CpG, indicating increased internalization efficiency of PCN-CpG@PS by RAW264.7 cells via PS-receptor mediated endocytosis compared to PCN-CpG. The gross medium fluorescence intensity (MFI) of treated cells was quantified for an accurate comparison. As shown in Fig. 3b and c, the MFI value of RAW264.7 cells treated with PCN-CpG was 11.2 times higher than that of RAW264.7 cells treated with free CpG 2395 (P < 0.0001). Similarly, the MFI value of RAW264.7 cells treated with PCN-CpG@PS was 2.3 times higher than that of RAW264.7 cells treated with PCN-CpG (P < 0.0001), which suggested that more CpG 2395 was taken up by RAW264.7 cells with the help of PCN and PS. Therefore, the well-designed PCN-CpG@PS shielded CpG 2395 against nuclease degradation through PCN while also overcoming the electrostatic barrier of the cell membrane through ligand–receptor interaction by PS, resulting in a considerable increase in cellular uptake.
Fig. 3. Endocytosis behavior of the nanocomposites. (a) Confocal laser scanning microscopy (CLSM) images of RAW264.7 cells treated with PBS, CpG 2395, PCN-CpG and PCN-CpG@PS for 4 h, (b) the corresponding flow cytometry analysis results and (c) corresponding quantitative mean fluorescence intensity (MFI) analysis. N = 3. Scale bar: 30 µm. Bar: mean; error bar: standard deviation. ****P1-way-ANOVA < 0.0001.

Optical properties and ROS detection
The photodynamic performance of nanocomposites (PCN-CpG@PS) was evaluated in vitro (Fig. 4a–c) by investigating the level of reactive oxygen species (ROS) in RAW264.7 cells induced by PCN, PCN-CpG, and PCN-CpG@PS under 640 nm laser irradiation by CLSM and flow cytometry. As expected, visible green fluorescence was observed in RAW264.7 cells of the PCN and PCN-CpG groups (Fig. 4a), which confirmed the PDT ability of PCN. In comparison to the PCN and PCN-CpG groups, stronger fluorescence was observed in the PCN-CpG@PS treated group, which could be attributed to the reaction of DCFH-DA with the higher levels of ROS produced, indicating that the light-triggered ROS generation capability of PCN-CpG@PS was markedly enhanced because of the PS modification. Simultaneously, flow cytometry was used to examine the intracellular fluorescence of ROS (Fig. 4b and c), which agreed with the CLSM findings, indicating that PCN-CpG@PS would provide an opportunity for enhanced PDT to achieve more effective antituberculosis treatment.
Fig. 4. In vitro ROS generation of the nanocomposites under 640 nm laser irradiation (0.15 W cm−2). (a) CLSM images of RAW264.7 cells treated with PCN without laser irradiation, PCN, PCN-CpG and PCN-CpG@PS with laser irradiation, (b) the corresponding flow cytometry analysis results and (c) corresponding quantitative mean fluorescence intensity (MFI) analysis. N = 3. Scale bar: 30 µm. Bar: mean; error bar: standard deviation. ****P1-way-ANOVA < 0.0001.

Verification of the bactericidal ability of targeted PDT combined with immunotherapy in vitro
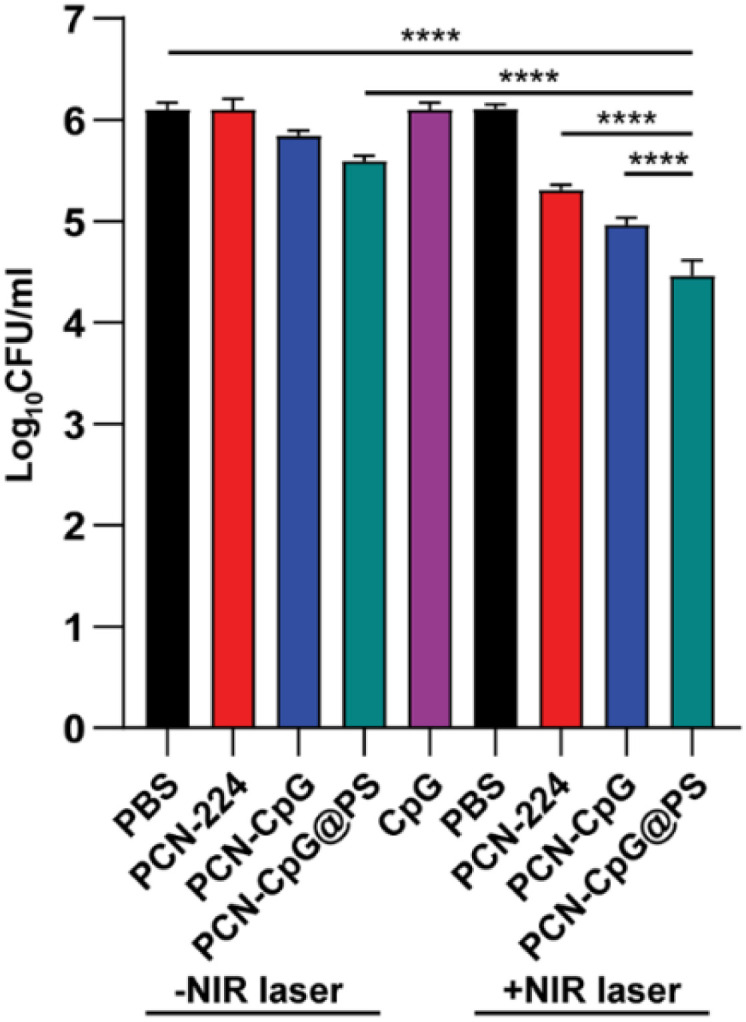
The antituberculosis efficacy of PCN-CpG@PS was estimated by a broth microdilution method. Following infection with Mtb H37Rv, RAW264.7 cells were treated with different nanocomposites. On the third postadministration day, the numbers of colony-forming units (CFUs) were counted. According to statistics, compared to the control group (6.097 ± 0.072), the groups treated with PBS + NIR (6.103 ± 0.047), PCN (6.100 ± 0.106) and CpG 2395 (6.097 ± 0.071) alone exhibited negligible changes in CFUs when treated for 72 h at a concentration of 10 µg mL−1 PCN and/or 0.25 µM CpG 2395 (Fig. 5). We investigated the ability of other nanocomposites to kill intracellular Mtb under the same conditions (72 h, 10 µg mL−1). The PCN + NIR (5.307 ± 0.050), PCN-CpG (5.843 ± 0.055) and PCN-CpG@PS (5.590 ± 0.056) groups showed some ability to kill Mtb in RAW264.7 cells compared to the PBS + NIR, PCN and CpG 2395 groups (a reduction of about 0.5 log units was observed). In the cells incubated with PCN-CpG + NIR (4.963 ± 0.071), the number of Mtb was smaller than that in the PCN + NIR and PCN-CpG@PS groups (a reduction of approximately 0.6 log units was observed), indicating that PDT combined with immunotherapy further inhibited the growth of intracellular Mtb compared with PDT or immunotherapy alone. In addition, the antibacterial efficacy of the PCN-CpG@PS + NIR group (4.460 ± 0.151) and PCN-CpG@PS group was better than that of the PCN-CpG + NIR group and PCN-CpG group, respectively, demonstrating that the targeting vector with PS can target nanocomposites to macrophages more effectively. The best intracellular Mtb killing ability was observed in the PCN-CpG@PS + NIR group, beyond those after treatment by either PDT or immunotherapy alone (a reduction of approximately 1 log units was observed), which was ascribed to the synergistic therapy of PS-targeted PDT-immunotherapy. The findings showed that PCN-CpG@PS has great potential for CpG 2395 delivery and targeted PDT combined with immunotherapy applications. We also counted the CFU immediately after PDT treatment. As shown in Fig. S5,† the Mtb survival was slightly decreased compared with that in the PBS group, which indicates the antimicrobial effect of PDT.
Fig. 5. The number of Mtb in RAW264.7 cells after different treatments. N = 3. Bar: mean; error bar: standard deviation. ****P1-way-ANOVA < 0.0001.
Fluorescence imaging of PCN-CpG@PS
The preferential aggregation of NPs in the lung facilitates better anti-tuberculosis efficacy. Therefore, we further evaluated the ability of PCN-CpG@PS to target lung tissue in vivo. Healthy mice were used for in vivo fluorescence imaging, and the change in in vivo fluorescence distribution was recorded with an IVIS imaging system. As shown in Fig. S6,† without laser irradiation, the lungs of mice injected with PCN-CpG@PS exhibited stronger fluorescence than those injected with PCN-CpG and free CpG 2395, demonstrating that PCN-CpG@PS has better lung biodistribution than PCN-CpG and free CpG 2395. Then the mice were sacrificed and the major organs (heart, liver, spleen, lung, kidney) were harvested for imaging (Fig. 6). Apparently, the organs with great abundance of macrophages such as lung, kidney and liver, isolated in the PCN-CpG@PS group showed stronger fluorescence compared to the PCN-CpG and free CpG 2395 groups, further suggesting that PS coating confers macrophage targeting ability to NPs due to receptor–ligand interactions.
Fig. 6. Fluorescence images of excised major organs at 6 h after tail vein injection of CpG 2395, PCN-CpG and PCN-CpG@PS, respectively.

In vivo safety evaluation of PCN-CpG@PS
The HE staining results showed that there were no significant pathological changes in the heart, liver or kidney of infected mice after administration of PCN-CpG@PS with laser irradiation when compared with the control group (Fig. S7†). These results revealed that PCN-CpG@PS nanoparticles in this study caused no obvious damage to the major organs with laser irradiation. In addition, the degree of lung and spleen tissue damage in the PCN-CpG@PS group was less severe than that in the control group. Therefore, PCN-CpG@PS showed very promising application potential due to its good biocompatibility.
Immunological responses after combined PDT immune treatment
The in vivo immunostimulatory activity of PCN-CpG@PS was assessed by immunofluorescence staining of the Th1-polarizing cytokines, TNF-α and IFN-γ in the lung tissue of mice. As shown in Fig. S8, S9† and 7, PBS, PCN, CpG or PBS + NIR did not secrete any visible levels of TNF-α or IFN-γ, indicating that there was no obvious effect of laser irradiation alone and the addition of PCN without laser irradiation or CpG alone on the expression levels of TNF-α and IFN-γ. For the PCN-CpG group, TNF-α and IFN-γ production could be increased to some level, but compared to the PCN-CpG@PS group, the production capacity was lower, probably due to the increased intracellular uptake of CpG in PCN-CpG@PS. There was also a difference in the TNF-α production between the PBS group and PCN-CpG@PS group without irradiation, indicating the immunological actions of CpG. In addition, incubation of RAW264.7 cells with PCN-CpG + NIR dramatically increased the expression levels of TNF-α and IFN-γ compared to the PCN-CpG group, indicating that PDT promoted TNF-α and IFN-γ secretion in the PCN-CpG + NIR group to some extent. Notably, the secreted TNF-α and IFN-γ levels stimulated by PCN-CpG@PS + NIR were considerably more robust than those of the PCN-CpG + NIR group and other groups. In conclusion, PS-targeted CpG-loaded PCN promoted the activation and production of inflammatory cytokines. Noteworthy, since IFN-γ is produced by different immune cells, including innate and adaptive cells, which may respond differently to infection. And IFN-γ-producing cells preferentially accumulate at the site of infection.40 TNF-α is mainly secreted by macrophages and monocytes. It is generally believed that Mtb proliferates in alveolar macrophages and DCs.41 In this study, the TB model was established in an aerosol manner, so IFN-γ and TNF-α may be mainly concentrated in the lungs, while other parts are not as significantly highly expressed. Therefore, there would be no risk of immunopathology or septic shock.
Fig. 7. Immunofluorescence staining of the left lungs of differently treated BALB/c mice under laser irradiation. Composite image of TNF-α and IFN-γ (a & c) staining and the corresponding intensity value (b & d) of lung sections of differently treated BALB/c mice. Nuclei are visualized with Hoechst. Scale bar = 50 µm. N = 3. Bar: mean; error bar: standard deviation. *P1-way ANOVA < 0.05, ***P1-way ANOVA < 0.001, ****P1-way ANOVA < 0.0001.

Antimycobacterial activity of targeted PDT combined with immunotherapy in vivo
In vivo, all groups except CpG, PBS + NIR, and PCN-224 had anti-tuberculosis capacity compared to the untreated control group to some extent (Table 1). Among them, PCN-CpG@PS + NIR had the best anti-tuberculosis effect, which was similar to the experimental results obtained in vitro. In contrast to the results of in vitro experiments, in vivo, the anti-TB effect of the PCN-224 + NIR group was not significantly different from that of the PCN-CPG-NIR group, which may be the result of the involvement of multiple cells in the immune response in vivo. Even with some benefits, the therapeutic effects of external laser light irradiation were not particularly ideal.
Pulmonary CFU in the BCG-infected mouse infection model after different treatments (mean ± SD, N = 3).
| Laser | log10CFU ml−1 (mean ± SD) of different treatments | ||||
|---|---|---|---|---|---|
| PBS | PCN | PCN-CpG | PCN-CpG@PS | CpG | |
| −NIR | 4.99 ± 0.13a | 5.01 ± 0.08 | 4.57 ± 0.04 | 4.17 ± 0.07b | 5.15 ± 0.13 |
| +NIR | 5.02 ± 0.15 | 4.58 ± 0.11c | 4.17 ± 0.20d | 3.77 ± 0.17a,b,c,d | |
P 1-way ANOVA < 0.0001.
P 1-way ANOVA < 0.05.
P 1-way ANOVA < 0.0001.
P 1-way ANOVA < 0.05.
Discussion
In our study, we have successfully developed versatile nanocomposites based on PCN nanoparticles (PCN-CpG@PS) loaded with CpG and modified with PS on the surface, in which the agents of photodynamic, immuno- and targeted therapy were well combined for their complementary advantages to achieve a synergistic anti-tuberculosis effect. In this study, first, cytotoxicity tests suggested the safety of the nanocomposites. Then, the ROS experimental results showed that the nanocomposites could effectively release a large amount of ROS under laser irradiation, and the lung immunofluorescence results demonstrated that the nanocomposites could enhance the Th1-type immune response.42 In addition, the uptake experiments indicated that PS could target nanocomposites to macrophages at Mtb infection site.43 Finally, the results of in vitro and in vivo bactericidal experiments both revealed the good anti-tuberculosis effect of the PCN-CpG@PS.
The increase in microbial resistance to conventional antibiotics is a major worldwide health problem that requires the development of new bactericidal strategies. Antimicrobial photodynamic therapy (PDT) that generates ROS acting on multiple targets is unlikely to induce bacterial resistance and thus has attracted much attention.44 In recent years, an increasing number of studies have concentrated on developing new strategies based on PDT. For example, single PDT45 or PDT combined with other therapies.46 These methods have shown excellent effects on both Gram-positive and Gram-negative bacteria, but many factors need to be considered. For example, the penetration depth of irradiation light, the rate of drug delivery, clearance of use, the nonselective killing effect of ROS etc.47 The study used external laser light irradiation for convenience and safety. However, since the lungs are tridimensional and of heterogeneous shape, there is a limited geometry of the irradiation spot (0.5 cm in diameter). Perhaps the results of groups with laser irradiation would be better if internal laser light irradiation was adopted. Also, there are still no standards for the clinical use of PDT. Targeted PDT has attracted increasing attention due to its ability to precisely target lesions, increase drug delivery, and reduce side effects. Phosphatidylserine has been widely used for macrophage targeting in tumors,35 hepatic fibrosis48 and other diseases treatment. There is also a study that explored phosphatidylserine decorated nanoplatform for the treatment of tuberculous meningitis,37 which demonstrated that phosphatidylserine is suitable for macrophage targeting. In the future, to overcome the antimicrobial resistance to conventional antibiotics, nanoparticles provide smart strategies for combining different elements together and show significant potential in clinical applications.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China (82001946), Beijing Municipal Natural Science Foundation (7214300, KZ202110025034), Beijing Municipal Science & Technology Commission (Z211100002921002), and Beijing Municipal Administration of Hospitals Incubating Program (PX2021061).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06334d
References
- Harding E. Lancet Respir. Med. 2020;8:19. doi: 10.1016/S2213-2600(19)30418-7. [DOI] [PubMed] [Google Scholar]
- Natarajan A. Beena P. M. Devnikar A. V. Mali S. Indian J. Tubercul. 2020;67:295–311. doi: 10.1016/j.ijtb.2020.02.005. [DOI] [PubMed] [Google Scholar]
- Miggiano R. Rizzi M. Ferraris D. M. Pathogens. 2020;9 doi: 10.3390/pathogens9050385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO operational handbook on tuberculosis: module 4: treatment: drug-susceptible tuberculosis treatment, World Health Organization © World Health Organization 2022, Geneva, 2022, https://www.who.int/publications/i/item/9789240050761 [Google Scholar]
- Dahanayake M. H. Jayasundera A. C. A. J. Microbiol. Methods. 2021;181:106127. doi: 10.1016/j.mimet.2020.106127. [DOI] [PubMed] [Google Scholar]
- Prasad R. Singh A. Gupta N. Indian J. Tubercul. 2019;66:520–532. doi: 10.1016/j.ijtb.2019.11.005. [DOI] [PubMed] [Google Scholar]
- Fehér K. Curr. Protein Pept. Sci. 2019;20:1060–1068. doi: 10.2174/1389203720666190830162149. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Kuo J. C. Yao S. Zhang C. Khan H. Lee R. J. Pharmaceutics. 2021:14. doi: 10.3390/pharmaceutics14010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayraklioglu N. Horuluoglu B. Klinman D. M. Methods Mol. Biol. 2021;2197:51–85. doi: 10.1007/978-1-0716-0872-2_4. [DOI] [PubMed] [Google Scholar]
- Chen W. Jiang M. Yu W. Xu Z. Liu X. Jia Q. Guan X. Zhang W. Int. J. Nanomed. 2021;16:5281–5299. doi: 10.2147/ijn.S317626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freidag B. L. Melton G. B. Collins F. Klinman D. M. Cheever A. Stobie L. Suen W. Seder R. A. Infect. Immun. 2000;68:2948–2953. doi: 10.1128/iai.68.5.2948-2953.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeyama J. I. Iho S. Suzuki F. Hayashi D. Yamamoto T. Yamazaki T. Goto Y. Ozeki Y. Matsumoto S. Yamamoto S. Tuberculosis. 2021;128:102067. doi: 10.1016/j.tube.2021.102067. [DOI] [PubMed] [Google Scholar]
- Zhang H. Cheng T. Lai L. Deng S. Yu R. Qiu L. Zhou J. Lu G. Zhi C. Chen J. Nanoscale. 2018;10:14516–14524. doi: 10.1039/c8nr03820a. [DOI] [PubMed] [Google Scholar]
- Shirai S. Shibuya M. Kawai A. Tamiya S. Munakata L. Omata D. Suzuki R. Aoshi T. Yoshioka Y. Front. Immunol. 2019;10:3018. doi: 10.3389/fimmu.2019.03018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens B. E. Geary S. M. Salem A. K. Immunotherapy. 2018;10:595–604. doi: 10.2217/imt-2017-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J. Li X. Tian H. Curr. Med. Chem. 2020;27:5949–5969. doi: 10.2174/0929867326666190618152518. [DOI] [PubMed] [Google Scholar]
- Li R. Chen T. Pan X. ACS Nano. 2021;15:3808–3848. doi: 10.1021/acsnano.0c09617. [DOI] [PubMed] [Google Scholar]
- Osterrieth J. W. M. Fairen-Jimenez D. Biotechnol. J. 2021;16:e2000005. doi: 10.1002/biot.202000005. [DOI] [PubMed] [Google Scholar]
- Li S. Y. Cheng H. Qiu W. X. Zhang L. Wan S. S. Zeng J. Y. Zhang X. Z. Biomaterials. 2017;142:149–161. doi: 10.1016/j.biomaterials.2017.07.026. [DOI] [PubMed] [Google Scholar]
- Nie X. Wu S. Hussain T. Wei Q. Nanomaterials. 2021;11 doi: 10.3390/nano11123162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan M. Zhao S. Liu W. Lee C. S. Zhang W. Wang P. Adv. Healthc. Mater. 2019;8:e1900132. doi: 10.1002/adhm.201900132. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski S. Knap B. Przystupski D. Saczko J. Kędzierska E. Knap-Czop K. Kotlińska J. Michel O. Kotowski K. Kulbacka J. Biomed. Pharmacother. 2018;106:1098–1107. doi: 10.1016/j.biopha.2018.07.049. [DOI] [PubMed] [Google Scholar]
- Idris N. M. Gnanasammandhan M. K. Zhang J. Ho P. C. Mahendran R. Zhang Y. Nat. Med. 2012;18:1580–1585. doi: 10.1038/nm.2933. [DOI] [PubMed] [Google Scholar]
- Han H. Jin Q. Wang H. Teng W. Wu J. Tong H. Chen T. Ji J. Small. 2016;12:3870–3878. doi: 10.1002/smll.201600950. [DOI] [PubMed] [Google Scholar]
- Cieplik F. Deng D. Crielaard W. Buchalla W. Hellwig E. Al-Ahmad A. Maisch T. Crit. Rev. Microbiol. 2018;44:571–589. doi: 10.1080/1040841x.2018.1467876. [DOI] [PubMed] [Google Scholar]
- Pérez-Laguna V. García-Malinis A. J. Aspiroz C. Rezusta A. Gilaberte Y. Ital G. Dermatol. Venereol. 2018;153:833–846. doi: 10.23736/s0392-0488.18.06007-8. [DOI] [PubMed] [Google Scholar]
- Valenzuela-Valderrama M. González I. A. Palavecino C. E. Photodiagnosis Photodyn. Ther. 2019;28:256–264. doi: 10.1016/j.pdpdt.2019.08.012. [DOI] [PubMed] [Google Scholar]
- Raab O. Biol Z. Uber die wirkung fluoriziender stoffe auf infusorien. Z. Biol. 1900;39:524–546. [Google Scholar]; . https://www.scienceopen.com/document?-1.ILinkListener-header-action∼bar-public∼button∼container-bookmark∼button-bookmark∼button&vid=a8c526fe-776b-4b6c-9f30-292b3ae293fd
- Maisch T. Mini-Rev. Med. Chem. 2009;9:974–983. doi: 10.2174/138955709788681582. [DOI] [PubMed] [Google Scholar]
- Wong T. W. Wang Y. Y. Sheu H. M. Chuang Y. C. Antimicrob. Agents Chemother. 2005;49:895–902. doi: 10.1128/aac.49.3.895-902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Riordan K. Sharlin D. S. Gross J. Chang S. Errabelli D. Akilov O. E. Kosaka S. Nau G. J. Hasan T. Antimicrob. Agents Chemother. 2006;50:1828–1834. doi: 10.1128/AAC.50.5.1828-1834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegell S. R. Kongshoj B. Wulf H. C. Arch. Dermatol. 2006;142:1241–1242. doi: 10.1001/archderm.142.9.1241. [DOI] [PubMed] [Google Scholar]
- Sung N. Back S. Jung J. Kim K. H. Kim J. K. Lee J. H. Ra Y. Yang H. C. Lim C. Cho S. Kim K. Jheon S. Photodiagn. Photodyn. Ther. 2013;10:694–702. doi: 10.1016/j.pdpdt.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Lemke G. Nat. Rev. Immunol. 2019;19:539–549. doi: 10.1038/s41577-019-0167-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M. Kang K. W. Arch. Pharmacal Res. 2019;42:617–628. doi: 10.1007/s12272-019-01167-4. [DOI] [PubMed] [Google Scholar]
- Konduru N. V. Tyurina Y. Y. Feng W. Basova L. V. Belikova N. A. Bayir H. Clark K. Rubin M. Stolz D. Vallhov H. Scheynius A. Witasp E. Fadeel B. Kichambare P. D. Star A. Kisin E. R. Murray A. R. Shvedova A. A. Kagan V. E. PLoS One. 2009;4:e4398. doi: 10.1371/journal.pone.0004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W. Peng H. Liu K. Wang Y. Wang W. Qu S. Li Y. Bi L. Zhang X. Zhang L. J. Biomed. Nanotechnol. 2021;17:2034–2042. doi: 10.1166/jbn.2021.3169. [DOI] [PubMed] [Google Scholar]
- Zeng J. Y. Zou M. Z. Zhang M. Wang X. S. Zeng X. Cong H. Zhang X. Z. ACS Nano. 2018;12:4630–4640. doi: 10.1021/acsnano.8b01186. [DOI] [PubMed] [Google Scholar]
- Li S. Y. Cheng H. Xie B. R. Qiu W. X. Zeng J. Y. Li C. X. Wan S. S. Zhang L. Liu W. L. Zhang X. Z. ACS Nano. 2017;11:7006–7018. doi: 10.1021/acsnano.7b02533. [DOI] [PubMed] [Google Scholar]
- Lyadova I. V. Panteleev A. V. Mediat. Inflamm. 2015;2015:854507. doi: 10.1155/2015/854507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matucci A. Maggi E. Vultaggio A. J. Rheumatol. Suppl. 2014;91:17–23. doi: 10.3899/jrheum.140098. [DOI] [PubMed] [Google Scholar]
- Troy A. Esparza-Gonzalez S. C. Bartek A. Creissen E. Izzo L. Izzo A. A. Tuberculosis. 2020;123:101949. doi: 10.1016/j.tube.2020.101949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. A. Jabeen R. Nasti T. H. Mohammad O. J. Antimicrob. Chemother. 2005;55:223–228. doi: 10.1093/jac/dkh522. [DOI] [PubMed] [Google Scholar]
- Suvorov N. Pogorilyy V. Diachkova E. Vasil'ev Y. Mironov A. Grin M. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22126392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchat M. Bertolini G. Giacomini P. Villanueva A. Jori G. J. Photochem. Photobiol. B Biol. 1996;32:153–157. doi: 10.1016/1011-1344(95)07147-4. [DOI] [PubMed] [Google Scholar]
- Gourlot C. Gosset A. Glattard E. Aisenbrey C. Rangasamy S. Rabineau M. Ouk T. S. Sol V. Lavalle P. Gourlaouen C. Ventura B. Bechinger B. Heitz V. ACS Infect. Dis. 2022;8:1509–1520. doi: 10.1021/acsinfecdis.2c00131. [DOI] [PubMed] [Google Scholar]
- Amos-Tautua B. M. Songca S. P. Oluwafemi O. S. Molecules. 2019;24 doi: 10.3390/molecules24132456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Pan W. Wang Y. Lei W. Feng B. Du C. Wang X. J. Drug Delivery. 2018;25:1–11. doi: 10.1080/10717544.2017.1399301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.