

Abstract

Pharmacophore models are widely used as efficient virtual screening (VS) filters for the target-directed enrichment of large compound libraries. However, the generation of pharmacophore models that have the power to discriminate between active and inactive molecules traditionally requires structural information about ligand–target complexes or at the very least knowledge of one active ligand. The fact that the discovery of the first known active ligand of a newly investigated target represents a major hurdle at the beginning of every drug discovery project underscores the need for methods that are able to derive high-quality pharmacophore models even without the prior knowledge of any active ligand structures. In this work, we introduce a novel workflow, called apo2ph4, that enables the rapid derivation of pharmacophore models solely from the three-dimensional structure of the target receptor. The utility of this workflow is demonstrated retrospectively for the generation of a pharmacophore model for the M2 muscarinic acetylcholine receptor. Furthermore, in order to show the general applicability of apo2ph4, the workflow was employed for all 15 targets of the recently published LIT-PCBA dataset. Pharmacophore-based VS runs using the apo2ph4-derived models achieved a significant enrichment of actives for 13 targets. In the last presented example, a pharmacophore model derived from the etomidate site of the α1β2γ2 GABAA receptor was used in VS campaigns. Subsequent in vitro testing of selected hits revealed that 19 out of 20 (95%) tested compounds were able to significantly enhance GABA currents, which impressively demonstrates the applicability of apo2ph4 for real-world drug design projects.
Introduction
According to the official IUPAC definition, “a pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response.”1 With an appropriate pharmacophore model in hand, compound libraries containing millions of compounds may be subjected to virtual screening (VS) in order to identify compounds showing the same feature pattern as the query pharmacophore.2 If the query pharmacophore models the interactions of compounds showing activity toward a particular target, the obtained hit lists can be expected to contain a share of likewise active molecules that is significantly higher than the one in randomly selected subsets. Hit lists enriched with active compounds represent a considerable economic advantage because testing hundreds of promising VS hits rather than testing thousands of compounds is clearly more resource saving.3 Due to its simplicity, pharmacophore-based VS is also computationally quite efficient and millions of compounds can be screened in relatively short amounts of time—something that is out of reach for traditional docking-based approaches.4−6 The generation of pharmacophore models utilizing co-crystal structures in a so-called structure-based (SB) manner is relatively straightforward and done by identifying present non-covalent interactions between a bound ligand and residues at the binding pocket surface.7 Several advanced tools, for example, LigandScout,8 Phase,9 or Catalyst,9 are available for this purpose. In cases where structural information about a ligand–target complex is not available, it is still possible to generate reliable pharmacophore models, provided that at least one or more molecules that bind to the target receptor of interest are known. Within this so-called ligand-based approach, conformers of the active ligands are superimposed to identify common, spatially similarly arranged structural features whose sum then constitutes the corresponding 3D pharmacophore model. However, for many biological targets, apo-protein structures or homology models are available; this is evident as over 41,000 of 150,000 (27%) X-ray structures in the Protein Data Bank (PDB)10 do not contain a small-molecule ligand (excluding inorganic ions and water).11 Moreover, for many allosteric pockets, potent binders have not yet been discovered which then also do not permit the application of ligand-based modeling approaches. The most commonly used approach to enrich compound libraries for apo-sites is molecular docking.12 However, docking a large library of molecules is computationally quite intense and often requires simplifications to be feasible, for example, docking only pre-generated rigid molecule conformations.6,13,14 While it is possible to generate pharmacophore features solely from an apo-crystal structure or from a homology model, the derivation of high-quality apo-site pharmacophores is generally a challenging task because there may be a large number of potential interactions with the binding site residues—a criterion where no single molecule is likely to fulfil.15 Recently, our group has developed a grid-based method for molecular interaction analysis that enables the identification and visualization of potential binding site interaction hot spots.16 In addition, several other methods exist to predict pharmacophoric interactions; most of these similar grid-based approaches work by calculating interaction energies of distinct probes for certain feature types located at the points of a three-dimensional (3D) lattice superimposed with a biomolecule.14−17 Yet, due to the near infinite number of combinations of possible pharmacophore features, it is difficult to create a proper query pharmacophore for VS runs.15 Pharmacophoric features may be filtered by buriedness or predicted interaction energies but are still very reliant on subjective decisions to obtain a pharmacophore model that is reasonable for VS, especially when no other information apart from the 3D structure of the apo-site is available.15
In the last two decades, several methods for receptor-focused pharmacophore generation have been developed in an attempt to fill this gap in the list of pharmacophore modeling approaches.15,17−23 However, none of these methods were established as a gold standard (see a recent publication by Volkamer et al. for an excellent overview).17 This is mostly due to the qualitative or conceptional nature of those methods, or the lack of reliant scoring to limit the number of pharmacophore features. In need of a method to reliably generate apo-site pharmacophores for several projects, we sought to develop a streamlined workflow, called apo2ph4, for rapid and straightforward receptor-based pharmacophore generation. The approach presented herein utilizes fragment docking to generate an array of SB pharmacophore models, which are then clustered and scored based on feature density to output pharmacophore models suitable for VS. Despite being prototypical in nature, apo2ph4 has shown tremendous prospective value for in-house projects (unpublished work); hence, we have decided to make it publicly available. A short qualitative evaluation as well as a prospective use case for discovering novel GABAAR ligands demonstrating the workflow’s prospective value will be presented herein. In order to probe whether apo2ph4 can be successfully applied to a wider variety of systems, the workflow was employed to all 15 targets of the recently published LIT-PCBA dataset.24 Also here, apo2ph4 has shown excellent performance, and the obtained results will be presented and discussed in this work.
Methods
The apo2ph4 workflow comprises four steps: (A) selection of a (potential) binding site of a resolved protein structure or a homology model, (B) docking of a fragment library into that binding site, (C) generation of SB pharmacophore models for each selected docking pose, and (D) scoring, clustering, and filtering of features to output a single pharmacophore model ready for VS (Figure 1). The necessary scripts for the apo2ph4 workflow are available on GitHub.25
Figure 1.

Overview of the separate steps performed by the apo2ph4 workflow: (A) selection of a suitable binding site. (B) Docking of a diverse set of medium-sized fragments into this binding site. (C) Generation of a SB pharmacophore model for each binding pose of each docked fragment. (D) Scoring, clustering, and filtering of features to output a single pharmacophore model ready for VS.
If a ligand occupying the protein structure is used as a center point for docking, its center of mass is calculated. If other molecules are present in the protein structure, they must be removed beforehand. For apo-proteins, or in an effort to generate a pharmacophore for a potential allosteric binding pocket, center coordinates may either be defined manually or by previously placing dummy atoms in the desired binding site which can be conveniently done by using PyMol.26 If structurally important water molecules or non-metal containing co-factors are contained within the protein structure file, a PDB file must be prepared manually for docking and subsequent steps.
Fragment Library
We have chosen a subset of Prestwick Chemical’s fragment set for use as a fragment library.27 This library contains a diverse set of 1456 lead-like fragments which have been derived from FDA approved drugs and was originally intended for fragment-based in vitro testing. To limit the time required for docking, a subset of 200 molecules was picked using the RDKit28 diversity KNIME29 node using a random seed. The molecular weight of the fragments ranges from 81 to 301 Da following a roughly Gaussian contribution. This is also the case for other critical descriptors such as c Log P and hydrogen donor and acceptor counts. As required for later steps, the number of potential ligand-based pharmacophore features is also calculated at this point using CDPKit.30
Docking
The protein and the fragments are then converted into the PDBQT format, which is required for docking with AutoDock Vina31 via a shell script employing MGLTools 1.5.6 (included in AutoDockTools432). This conversion may alternatively be performed by using OpenBabel.33 The PDBQT files are then submitted to docking using AutoDock Vina. A 20 × 20 × 20 Å grid box was chosen, and exhaustiveness was set to 20—it should be noted that using the default value of 8 is also acceptable, which may result in a slightly attenuated pharmacophore model quality, to significantly reduce the time required for docking if needed. Only the best two poses per fragment are kept if they are within an energy threshold of 2 kcal/mol. Additionally, AutoDock432 grid energies, using a 30 × 30 × 30 Å box with 0.3 Å spacing, of the protein complex were calculated using AutoGrid as they are needed for later steps.
Generation of SB Pharmacophore Models for Each Fragment
Using a python script employing the PyMOL API, mock co-crystal complexes of docking poses of each docked fragment with the protein structure were generated and saved as PDB files. Utilizing the batch version of KNIME, these files were then submitted to LigandScout’s SB pharmacophore-creator KNIME node and the generated pharmacophores were saved in a single LigandScout PML file.
SB Pharmacophore Generation for Fragments
By means
of a python script, pharmacophore features were then extracted from
the PML file using a functionality provided by CDPKit30 and got binned according to the feature type [hydrophobic
(H), aromatic (AR), positively ionizable (PI), negatively ionizable
(NI), hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), and
exclusion volume]. In the next step, distances between all features
of the same type are calculated and each feature is assigned a score
that depends on the density (number and distance) of neighboring features
(eq 1). For every neighboring
feature, a value is added to the total score, strongly rewarding features
within close proximity. For this, a relatively steep logistic scoring
function is applied: features that share the same coordinates get
assigned a value of nearly 1, while, for example, features with a
distance (eq 1: dist)
of 1 Å contribute a value of 0.5 and features with a distance
of 2 Å contribute a near-zero value (0.02). The list of features
is then sorted by the assigned score and pruned by discarding all
features that are within 3 Å of a higher-scored feature of the
same type. 3 Å was chosen as a threshold, as this is the default
minimum distance two LigandScout pharmacophore features can be apart
from each other without overlapping. To ensure a fairer comparison
of scores between feature types, each score is normalized dividing
by the number of maximum possible ligand-based pharmacophore features
(eq 1: nfeat.) calculated earlier to yield a final score (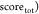 . While this approach
worked well for hydrophobic
features, additional considerations had to be made for other feature
types. During preliminary optimization, we discovered that PI and
NI group features were often poorly placed, resulting in the same
or less enrichment compared to random selection. This may be explained
as very polar moieties are simply placed in a position that results
in the least penalty by the docking algorithm’s scoring function.
This issue could rapidly be resolved by considering the electrostatic
potential map calculated via AutoGrid: PI features displaying a value
over −0.7 kcal/mol and NI features displaying a value under
0.9 kcal/mol were removed after the scoring but before the pruning
step.
. While this approach
worked well for hydrophobic
features, additional considerations had to be made for other feature
types. During preliminary optimization, we discovered that PI and
NI group features were often poorly placed, resulting in the same
or less enrichment compared to random selection. This may be explained
as very polar moieties are simply placed in a position that results
in the least penalty by the docking algorithm’s scoring function.
This issue could rapidly be resolved by considering the electrostatic
potential map calculated via AutoGrid: PI features displaying a value
over −0.7 kcal/mol and NI features displaying a value under
0.9 kcal/mol were removed after the scoring but before the pruning
step.
| 1 |
Unlike hydrophobic and ionizable features, aromatic features are modeled as planes because their interaction with side-chain aromatic groups must be within a certain angle to be of favorable energy. Hence, aromatic features with vastly different plane orientations should not be considered to be a similar feature for purposes of scoring. Employing DBSCAN implemented in scikit-learn,34 aromatic features were clustered by their plane angles and scored individually.
HBD and acceptor features may be presented as spherical features; however, as the correct angular geometry and distance between a donor and acceptor pair are critical for interactions to be energetically favorable,35 we opted to represent donor/acceptor features as vector features for VS.8 Hence, acceptor features were clustered by their initial point and donor features by their terminal point before scoring. In the default implementation, it is not possible for two vector features of the same type to be present at the same location as it is very difficult to satisfy two hydrogen bonds with the same functional group adequately. By default, if two or more vector features overlap, they are instead outputted as a spherical feature.
Exclusion volumes were treated differently as they are solely dependent on protein geometry; hence, all generated exclusion volumes were kept, with only duplicates being removed for screening efficiency. After overlapping features were removed and all scores are assigned, the defined number of features is selected based on the previously obtained score and written in LigandScout’s PML format. For most systems, it has proven to be beneficial to limit the number of features of a certain type; thus, hydrophobic features are limited to a maximum of four, PI and NI features to a maximum of one (each) and HBDs to a maximum of two, using default settings.
Validation of apo2ph4 by Pharmacophore Generation and Virtual Screening for Targets of the LIT-PCBA Dataset
All active and decoy datasets were used in the form provided by the LIT-PCBA website.24,36 By means of PyMol, the provided .mol2 files of ligands and targets were merged into a corresponding PDB file to apply apo2ph4. Conformations of active and inactive compounds were generated using CDPKit’s CONFORT conformer generator (confgen) using default settings, allowing up to 25 conformations per molecule to be generated.30,37 For VS, the generated multi-conformer SD files were then converted to LigandScout’s .ldb format via KNIME nodes provided by the LigandScout KNIME extension.38 To generate pharmacophore models for VS, apo2ph4 was run using all-default settings. The number of features to be matched was determined by screening the active dataset: the highest number of features was chosen that was able to retrieve at least one active compound. For enrichment calculations using the corresponding active and decoy datasets, VS runs were carried out on a HPC cluster using LigandScout Remote.39 All query pharmacophore models employed in the VS runs are provided as Supporting Information.
Pharmacophore Generation and Virtual Screening for the GABAA Receptor
Using a cryo-EM Structure of the GABAA receptor in complex with etomidate, we have employed the workflow presented herein, using etomidate enclosed by the C and D chains (PDB chain ID) as the center point for docking. We have generated a pharmacophore model containing seven features as a query for subsequent VS. We opted to use MolPort’s compound library (downloaded March 2020) as a VS database, which at the time of VS contained roughly 7 million commercially available compounds.40 Using Quacpac,41 compounds were set to their most favorable ionization state at the physiological pH of 7.4 and 25 conformations per compound were generated with Omega.42 Using LigandScout, Remote39 VS was performed, while allowing one feature to be omitted, yielding about 19,573 hits. As a first filtering step, only the hits with a pharmacophore fit score within the 70th percentile were kept (5872 hits remaining). Using LigandScout, the MMFF9443 energy of the molecules was then minimized in the protein structure. Standard properties and interacting feature counts were then calculated using LigandScout for the remaining molecules to allow a filtering according to Lipinski’s Ro5.44,45 Additionally, molecules having more than 10 rotatable bonds or a polar surface area greater than 140 Å2 were filtered out (Veber’s Rule).46 In order to comply with the known high lipophilicity of the pocket, only hits displaying a c Log P of greater than 2 were kept (4273 hits remaining). To filter out molecules with severely shifted poses due to minimization, their RMSD from the original poses was calculated and only molecules within an RMSD below 2 Å were kept (2925 hits remaining). Molecules which were strongly deformed as a result of minimalization (e.g., out of plane aromatic rings) were filtered out by only keeping molecules displaying a negative MMFF94 binding enthalpy, which was calculated within LigandScout (608 hits remaining). The remaining molecules were then redocked into the binding pocket using AutoDock Vina (default settings), and only molecules were kept if at least one docking pose resembled the pose minimized earlier within an rmsd of 1.5 Å. Following this rather harsh filtering procedure, 420 hits remained, which were sorted by their pharmacophore fit score. After visual inspection, comparing their pharmacophore fit with the query pharmacophore, consideration of actual commercial availability and diversity, a subset of 20 hits was chosen for subsequent in vitro assays.
Animals and Animal Welfare
All experiments involving animals were approved by the Austrian Animal Experimentation Ethics Board in compliance with the European convention for the protection of vertebrate animals used for experimental and other scientific purposes ETS no.: 123, which is in line with the EU Directive 2010/63/EU (GZ 66.011/0123-II/3b/2015 and 66.006/0029-WF/V/3b/2014). Female Xenopus laevis (X. laevis) frogs were purchased from NASCO (Fort Atkinson, USA) and kept in groups in temperature-controlled, continuous-flow water tanks (20 ± 1 °C), %; a 12 h light–dark cycle was in operation (lights on from 07.00 to 19.00).
Ion Channel Expression in X. laevis Oocytes and Two-Microelectrode Voltage Clamp Assay
Preparation of stage V–VI oocytes from X. laevis and expression of recombinant GABAA receptors (α1β2γ2s) in X. laevis oocytes by cRNA injection were performed as previously described.47 Female X. laevis frogs were anesthetized by 15 min incubation in a 0.2% MS-222 (methanesulfonate salt of 3-aminobenzoic acid ethyl ester; Sigma-Aldrich, Vienna, Austria) solution before removal of parts of the ovaries. Follicle membranes from isolated oocytes were enzymatically digested with 1 mg/mL collagenase (Type 1A; Sigma-Aldrich, Vienna, Austria). Selected oocytes were injected with 10–50 nL of DEPC-treated water (diethyl pyrocarbonate; Sigma, Vienna, Austria) containing different cRNAs at a concentration ranging between 200 and 3000 pg/nL/subunit. To ensure expression of the γ2S subunit, cRNAs were mixed in a ratio of 1:1:10.48 Oocytes were stored at +18 °C in ND96 solution (all from Sigma-Aldrich, Vienna, Austria). Two-microelectrode voltage clamp measurements were performed between days 1 and 5 after injection of cRNA of the respective subunits, using a TURBO TEC 03X amplifier (npi Electronic) at a holding potential of −70 mV and pCLAMP 10 data acquisition software (Molecular Devices). The bath solution contained 90 mM NaCl, 1 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM HEPES (pH 7.4). Electrode filling solution contained 3 M KCl. Test solutions (150 μL) were applied to the oocytes at a speed of 300 μL/s using the ScreeningTool (npi electronic) automated fast perfusion system. GABA EC3–5 was determined through a concentration–response experiment with GABA. Stock solution of compounds (30 mM) in DMSO was diluted with a bath solution containing GABA EC3–5 to obtain appropriate working solutions according to a validated protocol. Enhancement of the IGABA was defined as (I(GABA+Comp)/IGABA) – 1, where I(GABA+Comp) is the current response in the presence of a given compound and IGABA is the control of the GABA-induced chloride current. The data were analyzed using Origin Software (OriginLab Corporation, USA) and are given as mean ± SEM of 3 oocytes from ≥2 batches.
Results and Discussion
Parameter Selection
In order to cover a big part of the drug-like feature space within a very small number of fragments, we selected the Prestwick fragment library since it is directly derived from FDA approved drugs. To cut down computational cost, we selected a diverse subset of 200 molecules. It should be noted that a larger number of fragments appears to only very slightly improve results. Hence, when computational cost is negligible or only a small number of pharmacophore models are to be generated, a larger fragment library may be used. While a 20 × 20 × 20 Å space for docking usually far exceeds the area of a binding pocket, we opted for a rather lose box to allow ligands to dock outside the desired area instead of forcing fragments to “bind” within the desired binding pocket. In some cases, when two binding pockets are very close to each other, this area may be adjusted accordingly. For the same reason, to discard suboptimal binding poses, only the top 2 poses were kept and only if they were within calculated 2 kcal/mol of each other.
We have set a default cutoff value of −0.4 kcal/mol to eliminate unfavorable hydrophobic features. However, for very lipophilic pockets, it may be beneficial to lower the default cutoff to −0.5 to −0.6 kcal/mol for improved placement of hydrophobic features. The employed empirically determined energy cutoffs for positively and negatively ionized features seemed to be generally very applicable for most systems. However, for many systems, these very polar features still might not result in energetically favorable interactions. Thus, it is generally advisable to also perform VS runs with PI/NI features marked as optional unless there is very strong indication that such features are beneficial [e.g., for muscarinic acetylcholine receptors (mAChRs)].49 Introducing energy cutoffs for HBDs and acceptors, as well as aromatic features, generally did not show any benefits. While HBDs are very often accurately placed, they may sometimes be generated too frequently compared to SB pharmacophore models of reference systems and during VS against known actives. Unfortunately, we were not able to find a suitable method to discard “unnecessary” HBDs. However, as many HBDs (e.g., phenols and peptides) are associated with metabolic instability and with decreased oral bioavailability,50−52 data may be skewed in reference datasets of known actives. We have also attempted to consider buriedness via implementing a crude 7-axis PSP score;53 however, this did not seem to improve the results, likely because buriedness is already indirectly taken into account by the docking algorithm. In case there is no information about known ligands available, we recommend VS with a higher number of features but with one omitted feature—which also ensures a broader diversity of the VS hits.
Comparison to SB Pharmacophores
Due to our experience in the development of orthosteric antagonists targeting mAChRs,54−56 we decided to qualitatively evaluate the predictive value of the apo2ph4 workflow using a crystal structure of M2 muscarinic acetylcholine receptor subtype (PDB-entry 3UON57). It should be noted that neither 3UON nor any other muscarinic acetylcholine receptor was used during internal optimization. After following the general procedure, an 8-feature pharmacophore model was obtained using default settings, comprising 4 hydrophobic, 2 HBA, 1 HBD, and 1 PI feature (Figure 2).
Figure 2.

Pharmacophore models derived from PDB-entry 3UON: (A) SB pharmacophore model obtained using LigandScout. (B) Pharmacophore model obtained by apo2ph4. (C) Superposition of the SB (light/translucent) and the apo2ph4 (dark) pharmacophore model. Legend: yellow sphere = hydrophobic feature; red vector = directed HBA feature; green vector = directed HBD feature; green sphere = undirected HBD feature; blue star = positive ionizable feature.
Using LigandScout, a SB pharmacophore model was created from 3UON to yield a 5-feature pharmacophore model consisting of 2 hydrophobic, 1 HBA, 1 HBD, and 1 PI feature (Figure 2). Both of the hydrophobic features from the SB model were closely matched by the ones generated without ligand information (within 0.62 and 0.66 Å). The same is true for the HBA interacting with Asn4046.52 and the PI feature interacting with Asp1033.32 (superscript numerals refer to the Ballesteros–Weinstein numbering scheme58 for GPCRs), which were found within 0.76 and 0.41 Å, respectively. The HBD feature of the SB pharmacophore interacting with Asn4046.52 was not present in the apo2ph4 pharmacophore. However, as evident by other published crystal structures or by recently developed ligands,55,56 this is not an essential feature (e.g., PDB-entry 4MQS59). Comparing the apo2ph4 pharmacophore with PDB-entry 5ZKB,60 a different crystal structure of the muscarinic receptor, the second HBA feature generated by the tool closely matches the respective HBA feature interacting with Tyr1043.33 from the SB model within 3.46 Å (Figure S1). The generated HBD feature interacting with Ser1073.36 from PDB-entry 5ZKB may also be matched with the HBD generated by the apo2ph4 workflow within a distance of 2.85 Å. While these observed distances appear to be high, superposition of 6UON and 5ZKB reveals that the oxygen atoms of side chains of Tyr1043.33 and Ser1073.36 are shifted by distances of 2.58 and 2.91 Å, respectively, in the same direction as the respective features. While there are no crystal structures of the M2 muscarinic acetylcholine receptor directly matching the two remaining hydrophobic features generated by apo2ph4, several known mAChRs ligands may possibly provide this feature. For example, the SB pharmacophore of the top scored docking pose generated for antagonist Tropicamide61 closely matches the hydrophobic feature near Asp1033.32 within 0.93 Å while also matching 4 other generated features within 2 Å (Figure S2). A SB pharmacophore of the top scored docking pose of muscarinic agonist Cevimeline62 reveals one hydrophobic feature placed within 1.69 Å of the last not yet assigned generated feature(Figure S2). The fact that six of the eight generated features could directly be confirmed by examining crystal structures and the remaining two features have a strong possibility to match interactions by known mAChRs ligands strongly emphasizes the quality of the obtained pharmacophoric features and their usefulness for VS.
Systematic Evaluation of apo2ph4’s Performance
To probe whether apo2ph4 can be successfully applied to a variety of systems, we decided to employ the recently published LIT-PCBA dataset.24 Older and more established datasets such as DUD-E63 are commonly employed for benchmarking VS techniques; however, they are known to contain significant biases and tend to overestimate the performance of VS techniques as elaborately discussed by Rognan and co-workers.24 While DUD-E only provides computer-generated decoys, LIT-PCBA’s decoy datasets contain true inactive compounds that were obtained from high-confidence high-throughput screening data. In addition, several measures were taken to de-bias the datasets. Hence, apo2ph4 was applied to PDB structures of all 15 targets of the dataset, using only default settings without any target-specific optimizations (Table 1). If only one active was retrieved during VS, possibly inflating enrichment, a second system is shown. Strikingly, VS runs using the apo2ph4-generated pharmacophore models led to significant enrichment for 13 out of 15 targets ranging from 1.7 to 24.2. Listed enrichment factors were calculated for the database subset retrieved as VS hits, that is, if a hit rate of 0.25% was achieved, the enrichment factor has been calculated for 0.25% of the total database (EF0.25%). This had to be done as most of the VS runs resulted in a hit rate of less than 1%, rendering it impossible to calculate standard metrics like EF1%. These results unequivocally show that apo2ph4 is applicable to a wide range of systems and VS runs using queries generated by apo2ph4 can be expected to lead to often significant enrichments of actives.
Table 1. Quantitative and Systematic Evaluation of apo2ph4 using the LIT-PCBA Dataset.
| entry | target | PDB template | actives | inactives | hits (actives) | hits (inactives) | no. of featuresc | hit rate (%) | EFhits |
|---|---|---|---|---|---|---|---|---|---|
| 1a | ADRB2 | 4LDE | 17 | 312,483 | 3 | 8243 | 3 | 2.64 | 6.7 |
| 2a | ALDH1 | 5AC2 | 7168 | 137,695 | 2 | 16 | 6 | 0.01 | 2.2 |
| 3a | ESR_ago | 2QR9 | 13 | 5583 | 2 | 64 | 4 | 1.18 | 13.0 |
| 4a | ESR_ant | 6B0F | 102 | 4948 | 1 | 2 | 4 | 0.06 | 16.5 |
| 4b | 2POG | 5 | 60 | 4 | 1.29 | 3.8 | |||
| 5a | FEN1 | 5FV7a | 369 | 355,402 | 3 | 881 | 5 | 0.25 | 3.3 |
| 6a | GBA | 3RIK | 166 | 296,052 | 3 | 384 | 5 | 0.13 | 13.8 |
| 7a | IDH1 | 4I3L | 39 | 362,049 | 1 | 382 | 5 | 0.11 | 24.2 |
| 7b | 5TQH | 5 | 8597 | 4 | 2.38 | 5.4 | |||
| 8a | KAT2A | 5MLJ | 194 | 348,548 | 1 | 205 | 5 | 0.06 | 8.7 |
| 8b | 5MLJ | 5 | 2741 | 4 | 0.79 | 3.3 | |||
| 9a | MAPK1 | 3W55 | 308 | 62,629 | 4 | 156 | 5 | 0.25 | 5.1 |
| 10a | MTORC1 | 4FAP | 97 | 32,972 | 32 | 8933 | 4 | 27.11 | 1.2 |
| 10b | 4FAPEF1%b | 4 | 333 | 4 | 1.00 | 4.0 | |||
| 11a | OPRK1 | 6B73 | 24 | 269,816 | 0 | 12 | 5 | 0.00 | 0.0 |
| 12a | PKM2 | 3ME3 | 546 | 245,523 | 4 | 585 | 6 | 0.24 | 3.1 |
| 13a | PPARG | 5Z5S | 27 | 5211 | 1 | 10 | 5 | 0.19 | 17.6 |
| 13b | 5Y2T | 3 | 340 | 4 | 6.55 | 1.7 | |||
| 14a | TP53 | 4AGQ | 79 | 4168 | 3 | 210 | 4 | 5.02 | 0.8 |
| 15a | VDR | 2A2I | 884 | 355,388 | 13 | 2917 | 5 | 0.82 | 1.8 |
A PI feature was removed from the pharmacophore model manually because a magnesium atom is known to occupy the location the feature was placed in. Currently, apo2ph4 cannot account for coordinating metal ions automatically.
Due to exceptionally high hit rate, only ranked hits representing top 1% of the entire dataset were used to calculate EF.
Number of features present in the pharmacophore model used for VS.
Prospective Value
We originally have developed the apo2ph4 workflow for internal purposes to find ligands acting as allosteric kinase activators, and it has led us to discover new binders for previously unknown binding pockets, which served as the start of a drug discovery campaign which is currently under development and not yet published. Similarly, we have applied the workflow to the GABAA receptor, which also served as a spark in the development of novel alpha-6 selective GABAAR positive allosteric modulators. This publication features our third VS campaign applying the apo2ph4 workflow. Recently, a new cryo-EM structure (PDB-entry 6 × 3 V64) of the human GABAA receptor in complex with etomidate was released. As this is the suspected binding site of β2/3 subunit-selective GABAAR modulators described earlier by our group as well as valerenic acid (VA) and loreclezole, we are interested in the development of novel ligands with improved pharmacodynamics.65
While in this case, a protein–ligand complex is available, upon generating a SB pharmacophore model with LigandScout, it quickly became apparent that this pharmacophore may not be suitable for VS: with three hydrophobic and one aromatic features, the number of features is not only very low but also, without any polar features, extremely generic and leads to an extremely high hit rate when used as a query pharmacophore (Figure 3). The generated seven-feature pharmacophore, in contrast, consists of 4 H, 1 AR, one HBA, and one HBD feature (Figure 3). In order to generate a more diverse set of hits, VS was performed with one omitted feature. After several filtering steps, 20 hits were purchased for in vitro testing (see Methods section for details). In vitro screening results on α1β2γ2S channels (GABA EC3–5) at a concentration of 30 μM revealed that 19 of 20 purchased compounds were able to significantly potentiate GABA activation. Thirteen compounds have shown a low potentiation of 30 to 100%, 5 of them were able to potentiate GABA current by 100–350%, and one ligand 19 was able to potentiate GABA current by almost 3000% (Figure 4). Ligand structures, with the exception of ligand 19, are disclosed in Supporting Information (Table S1). This extremely high hit rate again underscores the potential prospective value of the apo2ph4 workflow.
Figure 3.

Pharmacophore models derived from PDB-entry 6 × 3 V: (A) SB pharmacophore model obtained by LigandScout. (B) Pharmacophore model obtained by apo2ph4. Legend: yellow sphere = hydrophobic feature; red vector = directed HBA feature; green sphere = undirected HBD feature; blue disk = aromatic feature.
Figure 4.

Screening results (30 μM) showing IGABA enhancement through α1β2γ2S GABAA receptors of the generated compound library of hits compared to VA; error bars for compound 19 are not shown, for this see supplementary Figure S3. The data are presented as mean values ± SEM, n = 3.
As demonstrated by the previous examples, the apo2ph4 workflow often not only allows the derivation of features that may be matched by a known ligand but is also able to generate features found in other structures or find previously unknown features. While the workflow was primarily intended to be used on apo-protein structures, its potential usefulness to generate alternative pharmacophore models from existing co-crystal structures becomes apparent.
Usage of apo2ph4 for In Silico Neurotoxicity Prediction
Clozapine is an atypical antipsychotic primarily acting antagonistic on 5HT2A and dopamine receptors66,67 and is prone to induce seizures.68 This is attributed to Clozapine also binding to the GABAAR in an antagonistic fashion.69,70 Neurotoxicity, which is often caused by off-target effects, has become a huge concern for the development of new drugs.71 Hence, there is a big desire in being able to predict toxicity caused by off-target effects in silico. Thus, on behalf of the IM2 Project NeuroDerisk, we were developing methods to predict seizure causing off-target effects.72 Since the GABAAR is composed of five subunits of many different subtypes, a large variety of GABAARs are found in the human body, often associated with distinct physiological functions.73 In consequence, this means that there is a vast amount of known and possible binding sites, for many of which no ligand has been discovered yet or at least no cryo-EM structure of a ligand bound to a proposed binding site has been resolved. In addition to that, as demonstrated by the example above, SB models derived from structures of GABAAR in complex with a ligand often yield less than ideal pharmacophore models. Hence, we are currently applying apo2ph4 to derive pharmacophores which may be used to discover potential off-target interactions for newly developed compounds. While we plan to publish these pharmacophores in a separate work, one pharmacophore generated for the inhibitory pregnanolone sulfate binding site74 of the α1β2γ2 GABAAR, which was derived from derived from PDB-entry 6X3Z64 as apo-structure, is shown here (Figure 5). To discover GABAAR binders with a low false negative rate, we opted to generate pharmacophore models with a larger number of features but several optional features for subsequent VS runs.
Figure 5.

Pharmacophore model of the pregnanolone sulfate binding site of PDB-entry 6X3Z64 obtained by apo2ph4. Legend: yellow sphere = hydrophobic feature; red vector = directed HBA feature; green vector = directed HBD feature; blue disk = aromatic feature.
Conclusions
In the absence of reliable pharmacophore modeling methods for apo-pharmacophore models, apo2ph4 combines existing and commonly used tools in the field of computer-aided molecular design into a versatile workflow for generating receptor-based pharmacophore models. The combination of fragment docking and SB pharmacophore modeling seems to be a viable alternative to solely energy- or grid-based methods. This is likely as, in contrast to grid-based methods, steric and electrostatic properties of drug-like molecules are more strongly taken into account via docking rather than the use of simplified, often atomic, probes. Comparing generated pharmacophores with SB models derived from known systems, apo2ph4 appears to be largely successful on deriving important features devoid of any ligand information. By applying apo2ph4 to several targets of the LIT-PCBA dataset, we were able to show that apo2ph4 is applicable to a large number of diverse systems and that the obtained pharmacophore models, on average, lead to a significant enrichment of actives in VS experiments. In addition to the examples outlined in this publication, we verified the prospective value of apo2ph4 for two additional systems that resulted in hits currently undergoing further development. Lastly, apo2ph4 is currently being evaluated to generate toxicophores, which will be used to detect potential off-target effects of drug candidates in silico.
Data and Software Availability
The workflow published herein is available on GitHub.25 The fragment database is proprietary but may be requested from Prestwick Pharmaceuticals Inc.27 The MolPort compound library is proprietary and may be requested from MolPort.40 The 2D structures as well as the SMILES notations of the tested compounds are described in Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.2c00814.
Author Contributions
Conceptualization: J.H.; computational method development: J.H.; in silico validation: J.H. and J.K.; VS campaign and hit selection: J.H. and J.K.; in vitro experiments: A.G.; interpretation of in vitro results: A.G., S.H., J.H., and J.K.; visualization: J.H. and A.G.; supervision: T.S., T.L., and S.H.; original draft preparation: J.H., J.K., A.G., and T.S.; review and editing, all authors; resources: T.L., S.H., and T.S.; funding acquisition: T.L. All authors have read and agreed to the published version of the manuscript.
The authors J.H., T.L., and T.S. gratefully acknowledge the financial support by the NeuroDeRisk project, which has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement no 821528. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation program and EFPIA. The author A.G. has received funding from the Austrian Science Fund in the MolTag doctoral program FWF W1232. Open Access is funded by the Austrian Science Fund (FWF).
The authors declare no competing financial interest.
Supplementary Material
References
- Wermuth C. G.; Ganellin C. R.; Lindberg P.; Mitscher L. A. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. 10.1351/pac199870051129. [DOI] [Google Scholar]
- Seidel T.; Ibis G.; Bendix F.; Wolber G. Strategies for 3D pharmacophore-based virtual screening. Drug Discovery Today: Technol. 2010, 7, e221–e228. 10.1016/j.ddtec.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Good A.Virtual Screening. In Comprehensive Medicinal Chemistry II; Taylor J. B., Triggle D. J., Eds.; Elsevier: Oxford, 2007; pp 459–494. [Google Scholar]
- Chen Z.; Li H.-l.; Zhang Q.-j.; Bao X.-g.; Yu K.-q.; Luo X.-m.; Zhu W.-l.; Jiang H.-l. Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. 10.1038/aps.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas V.; Jain A.; Jain A.; Gupta A. Virtual Screening: A Fast Tool for Drug Design. Sci. Pharm. 2008, 76, 333. 10.3797/scipharm.0803-03. [DOI] [Google Scholar]
- Gaurav A.; Gautam V. Structure-based three-dimensional pharmacophores as an alternative to traditional methodologies. J. Recept., Ligand Channel Res. 2014, 7, 27–38. 10.2147/jrlcr.s46845. [DOI] [Google Scholar]
- Greenidge P. A.; Carlsson B.; Bladh L.-G.; Gillner M. Pharmacophores Incorporating Numerous Excluded Volumes Defined by X-ray Crystallographic Structure in Three-Dimensional Database Searching: Application to the Thyroid Hormone Receptor. J. Med. Chem. 1998, 41, 2503–2512. 10.1021/jm9708691. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Doman T. N.; Thorner D. A.; Bodkin M. J. 3D QSAR Methods: Phase and Catalyst Compared. J. Chem. Inf. Model. 2007, 47, 1248–1257. 10.1021/ci7000082. [DOI] [PubMed] [Google Scholar]
- Berman H.; Henrick K.; Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Shao C.; Westbrook J. D.; Lu C.; Bhikadiya C.; Peisach E.; Young J. Y.; Duarte J. M.; Lowe R.; Wang S.; Rose Y.; Feng Z.; Burley S. K. Simplified quality assessment for small-molecule ligands in the Protein Data Bank. Structure 2022, 30, 252–262. 10.1016/j.str.2021.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira N. M. F. S. A.; Gesto D.; Oliveira E. F.; Santos-Martins D.; Brás N. F.; Sousa S. F.; Fernandes P. A.; Ramos M. J. Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys. 2015, 582, 56–67. 10.1016/j.abb.2015.05.011. [DOI] [PubMed] [Google Scholar]
- Palacio-Rodríguez K.; Lans I.; Cavasotto C. N.; Cossio P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 5142. 10.1038/s41598-019-41594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E.; Walters W. P.; Charifson P. S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004, 56, 235–249. 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- Tran-Nguyen V.-K.; Da Silva F.; Bret G.; Rognan D. All in One: Cavity Detection, Druggability Estimate, Cavity-Based Pharmacophore Perception, and Virtual Screening. J. Chem. Inf. Model. 2019, 59, 573–585. 10.1021/acs.jcim.8b00684. [DOI] [PubMed] [Google Scholar]
- Schuetz D. A.; Seidel T.; Garon A.; Martini R.; Körbel M.; Ecker G. F.; Langer T. GRAIL: GRids of phArmacophore Interaction fieLds. J. Chem. Theory Comput. 2018, 14, 4958–4970. 10.1021/acs.jctc.8b00495. [DOI] [PubMed] [Google Scholar]
- Mortier J.; Dhakal P.; Volkamer A. Truly Target-Focused Pharmacophore Modeling: A Novel Tool for Mapping Intermolecular Surfaces. Molecules 2018, 23, 1959. 10.3390/molecules23081959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G.; Gong B.; Li J.; Song Y.; Li S.; Lu X. An Improved Receptor-Based Pharmacophore Generation Algorithm Guided by Atomic Chemical Characteristics and Hybridization Types. Front. Pharmacol. 2018, 9, 1463. 10.3389/fphar.2018.01463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Lill M. A. Exploring the Potential of Protein-Based Pharmacophore Models in Ligand Pose Prediction and Ranking. J. Chem. Inf. Model. 2013, 53, 1179–1190. 10.1021/ci400143r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Lill M. A. Protein Pharmacophore Selection Using Hydration-Site Analysis. J. Chem. Inf. Model. 2012, 52, 1046–1060. 10.1021/ci200620h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintori C.; Corradi V.; Magnani M.; Manetti F.; Botta M. Targets Looking for Drugs: A Multistep Computational Protocol for the Development of Structure-Based Pharmacophores and Their Applications for Hit Discovery. J. Chem. Inf. Model. 2008, 48, 2166–2179. 10.1021/ci800105p. [DOI] [PubMed] [Google Scholar]
- Chen J.; Lai L. Pocket v.2: Further Developments on Receptor-Based Pharmacophore Modeling. J. Chem. Inf. Model. 2006, 46, 2684–2691. 10.1021/ci600246s. [DOI] [PubMed] [Google Scholar]
- Goto J.; Kataoka R.; Hirayama N. Ph4Dock: Pharmacophore-Based Protein–Ligand Docking. J. Med. Chem. 2004, 47, 6804–6811. 10.1021/jm0493818. [DOI] [PubMed] [Google Scholar]
- Tran-Nguyen V.-K.; Jacquemard C.; Rognan D. LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4263–4273. 10.1021/acs.jcim.0c00155. [DOI] [PubMed] [Google Scholar]
- Heider J.apo2ph4 github repository. https://github.com/aglanger/apo2ph4/.
- Schrödinger LLC The PyMOL Molecular Graphics System, version 2.0.
- Prestwick Drug-Fragment Library . Fragments arising from smart fragmentation of approved drugs, 1456. http://prestwickchemical.com/screening-libraries/prestwick-drug-fragment-library/.(accessed May 15 2022).
- RDKit . Open-source cheminformatics. http://www.rdkit.org (accessed May 15 2022).
- Berthold M. R.; Cebron N.; Dill F.; Gabriel T. R.; Kötter T.; Meinl T.; Ohl P.; Sieb C.; Thiel K.; Wiswedel B.. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications; Preisach C., Burkhardt H., Schmidt-Thieme L., Decker R., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2008; pp 319–326. [Google Scholar]
- Seidel T.The Chemical Data Processing Toolkit. https://github.com/aglanger/CDPKit (accessed May 15 2022).
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIT-PCBA A dataset for virtual screening and machine learning. https://drugdesign.unistra.fr/LIT-PCBA/ (accessed Dec 05 2022).
- Seidel T.; Permann C.; Wieder O.; Kohlbacher S.; Langer T.. High-Quality Conformer Generation with CDPKit/CONFORT: Algorithm and Performance Assessment., Preprint; Research Square, 2022. https://europepmc.org/article/ppr/ppr491854. [DOI] [PMC free article] [PubMed]
- LigandScout Expert Knime Extensions. http://www.inteligand.com/download/LigandScout_Knime_Nodes.pdf (accessed Dec 05 2022).
- Kainrad T.; Hunold S.; Seidel T.; Langer T. LigandScout Remote: A New User-Friendly Interface for HPC and Cloud Resources. J. Chem. Inf. Model. 2019, 59, 31–37. 10.1021/acs.jcim.8b00716. [DOI] [PubMed] [Google Scholar]
- The MolPort database. https://www.molport.com/shop/database-download (accessed May 15 2022).
- QUACPAC 2.1.3.0; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com (accessed May 15 2022).
- OMEGA 4.1.2.0; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com (accessed May 15 2022).
- Halgren T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. . [DOI] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1. Adv. Drug Deliv. Rev. 2001, 46, 3–26. 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Khom S.; Baburin I.; Timin E. N.; Hohaus A.; Sieghart W.; Hering S. Pharmacological Properties of GABAA Receptors Containing γ1 Subunits. Mol. Pharmacol. 2006, 69, 640–649. 10.1124/mol.105.017236. [DOI] [PubMed] [Google Scholar]
- Boileau A. J.; Baur R.; Sharkey L. M.; Sigel E.; Czajkowski C. The relative amount of cRNA coding for γ2 subunits affects stimulation by benzodiazepines in GABAA receptors expressed in Xenopus oocytes. Neuropharmacology 2002, 43, 695–700. 10.1016/s0028-3908(02)00036-9. [DOI] [PubMed] [Google Scholar]
- Kruse A. C.; Hu J.; Kobilka B. K.; Wess J. Muscarinic acetylcholine receptor X-ray structures: potential implications for drug development. Curr. Opin. Pharmacol. 2014, 16, 24–30. 10.1016/j.coph.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrap P.; Zheng G.; Wan Y.; Li T.; Hu W.; Li W.; Zhang H.; Zhang Z.; Hu J. Discovery of a widespread metabolic pathway within and among phenolic xenobiotics. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 6062–6067. 10.1073/pnas.1700558114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J.-F.; Yang H.; Zhao Y.-Z.; Xue M. Metabolism of Peptide Drugs and Strategies to Improve their Metabolic Stability. Curr. Drug Metab. 2018, 19, 892–901. 10.2174/1389200219666180628171531. [DOI] [PubMed] [Google Scholar]
- Han Y.; Gao Z.; Chen L.; Kang L.; Huang W.; Jin M.; Wang Q.; Bae Y. H. Multifunctional oral delivery systems for enhanced bioavailability of therapeutic peptides/proteins. Acta Pharm. Sin. B 2019, 9, 902–922. 10.1016/j.apsb.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendlich M.; Rippmann F.; Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graphics 1997, 15, 359–363. 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- Kilian J.; Millard M.; Ozenil M.; Krause D.; Ghaderi K.; Holzer W.; Urban E.; Spreitzer H.; Wadsak W.; Hacker M.; Langer T.; Pichler V. Synthesis, Biological Evaluation, and Docking Studies of Antagonistic Hydroxylated Arecaidine Esters Targeting mAChRs. Molecules 2022, 27, 3173. 10.3390/molecules27103173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian J.; Ozenil M.; Millard M.; Fürtös D.; Maisetschläger V.; Holzer W.; Wadsak W.; Hacker M.; Langer T.; Pichler V. Design, Synthesis, and Biological Evaluation of 4,4’-Difluorobenzhydrol Carbamates as Selective M1 Antagonists. Pharmaceuticals 2022, 15, 248. 10.3390/ph15020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozenil M.; Aronow J.; Piljak D.; Vraka C.; Holzer W.; Spreitzer H.; Wadsak W.; Hacker M.; Pichler V. Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting mAChR M1. Pharmaceuticals 2020, 13, 437. 10.3390/ph13120437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K.; Kruse A. C.; Asada H.; Yurugi-Kobayashi T.; Shiroishi M.; Zhang C.; Weis W. I.; Okada T.; Kobilka B. K.; Haga T.; Kobayashi T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H.. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon S. C., Ed.; Academic Press, 1995; Vol. 25; pp 366–428. [Google Scholar]
- Kruse A. C.; Ring A. M.; Manglik A.; Hu J.; Hu K.; Eitel K.; Hübner H.; Pardon E.; Valant C.; Sexton P. M.; Christopoulos A.; Felder C. C.; Gmeiner P.; Steyaert J.; Weis W. I.; Garcia K. C.; Wess J.; Kobilka B. K. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suno R.; Lee S.; Maeda S.; Yasuda S.; Yamashita K.; Hirata K.; Horita S.; Tawaramoto M. S.; Tsujimoto H.; Murata T.; Kinoshita M.; Yamamoto M.; Kobilka B. K.; Vaidehi N.; Iwata S.; Kobayashi T. Structural insights into the subtype-selective antagonist binding to the M2 muscarinic receptor. Nat. Chem. Biol. 2018, 14, 1150–1158. 10.1038/s41589-018-0152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dei S.; Bellucci C.; Ghelardini C.; Romanelli M. N.; Spampinato S. Synthesis, characterization and pharmacological profile of tropicamide enantiomers. Life Sci. 1996, 58, 2147–2153. 10.1016/0024-3205(96)00208-1. [DOI] [PubMed] [Google Scholar]
- Ono M.; Takamura E.; Shinozaki K.; Tsumura T.; Hamano T.; Yagi Y.; Tsubota K. Therapeutic effect of cevimeline on dry eye in patients with Sjögren’s syndrome: a randomized, double-blind clinical study. Am. J. Ophthalmol. 2004, 138, 6–17. 10.1016/j.ajo.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. J.; Gharpure A.; Teng J.; Zhuang Y.; Howard R. J.; Zhu S.; Noviello C. M.; Walsh R. M.; Lindahl E.; Hibbs R. E. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature 2020, 585, 303–308. 10.1038/s41586-020-2654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M.; Monticelli S.; Seidel T.; Luger D.; Salzer I.; Boehm S.; Holzer W.; Schwarzer C.; Urban E.; Khom S.; Langer T.; Pace V.; Hering S. Design, Synthesis, and Pharmacological Evaluation of Novel β2/3 Subunit-Selective γ-Aminobutyric Acid Type A (GABAA) Receptor Modulators. J. Med. Chem. 2019, 62, 317–341. 10.1021/acs.jmedchem.8b00859. [DOI] [PubMed] [Google Scholar]
- Meltzer H. Y. The Role of Serotonin in Antipsychotic Drug Action. Neuropsychopharmacology 1999, 21, 106S–115S. 10.1016/s0893-133x(99)00046-9. [DOI] [PubMed] [Google Scholar]
- Naheed M.; Green B. Focus on Clozapine. Curr. Med. Res. Opin. 2001, 17, 223–229. 10.1185/03007990152673864. [DOI] [PubMed] [Google Scholar]
- Khoury R.; Ghossoub E. Antipsychotics and seizures: What are the risks?. Curr. Psychiatry 2019, 18, 26–33. [Google Scholar]
- Yokota K.; Tatebayashi H.; Matsuo T.; Shoge T.; Motomura H.; Matsuno T.; Fukuda A.; Tashiro N. The effects of neuroleptics on the GABA-induced Cl- current in rat dorsal root ganglion neurons: differences between some neuroleptics. Br. J. Pharmacol. 2002, 135, 1547–1555. 10.1038/sj.bjp.0704608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bampali K.; Koniuszewski F.; Silva L. L.; Rehman S.; Vogel F. D.; Seidel T.; Scholze P.; Zirpel F.; Garon A.; Langer T.; Willeit M.; Ernst M. Tricyclic antipsychotics and antidepressants can inhibit α5-containing GABAA receptors by two distinct mechanisms. Br. J. Pharmacol. 2022, 179, 3675. 10.1111/bph.15807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. L.; Imam S. Z.; Roberts R. A. Drug discovery and development: Biomarkers of neurotoxicity and neurodegeneration. Exp. Biol. Med. 2018, 243, 1037–1045. 10.1177/1535370218801309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurotoxicity De-Risking in Preclinical Drug Discovery. https://cordis.europa.eu/project/id/821528 (accessed May 15 2022).
- Rudolph U.; Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discovery 2011, 10, 685–697. 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverty D.; Thomas P.; Field M.; Andersen O. J.; Gold M. G.; Biggin P. C.; Gielen M.; Smart T. G. Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nat. Struct. Mol. Biol. 2017, 24, 977–985. 10.1038/nsmb.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The workflow published herein is available on GitHub.25 The fragment database is proprietary but may be requested from Prestwick Pharmaceuticals Inc.27 The MolPort compound library is proprietary and may be requested from MolPort.40 The 2D structures as well as the SMILES notations of the tested compounds are described in Supporting Information.