

Abstract
Highly effective modulator therapies for cystic fibrosis (CF) make it a treatable condition for many people. However, although CF respiratory illness occurs after birth, other organ systems particularly in the digestive tract are damaged before birth. We use an ovine model of CF to investigate the in utero origins of CF disease since the sheep closely mirrors critical aspects of human development. Wildtype (WT) and CFTR ‐/‐ sheep tissues were collected at 50, 65, 80, 100, and 120 days of gestation and term (147 days) and used for histological, electrophysiological, and molecular analysis. Histological abnormalities are evident in CFTR‐/‐ ‐/‐ animals by 80 days of gestation, equivalent to 21 weeks in humans. Acinar and ductal dilation, mucus obstruction, and fibrosis are observed in the pancreas; biliary fibrosis, cholestasis, and gallbladder hypoplasia in the liver; and intestinal meconium obstruction, as seen at birth in all large animal models of CF. Concurrently, cystic fibrosis transmembrane conductance regulator (CFTR)‐dependent short circuit current is present in WT tracheal epithelium by 80 days gestation and is absent from CFTR ‐/‐ tissues. Transcriptomic profiles of tracheal tissues confirm the early expression of CFTR and suggest that its loss does not globally impair tracheal differentiation.
Keywords: CF development, CFTR, CF GI tract, CF liver, CF lung, CF pancreas, CF sheep model, CF trachea, early CF pathology
1. INTRODUCTION
The major focus of therapeutic approaches to the inherited disorder cystic fibrosis (CF) has been the correction or alleviation of lung disease, which is the primary cause of mortality. CF lung pathology is primarily a postnatal phenotype; however, profound CF disease is often evident prenatally in several organ systems, particularly within the digestive system.
Most notable is pancreatic pathology, which gave rise to the original description of the disease “cystic fibrosis of the pancreas” (reviewed in Ref. 1). Most people with CF (pwCF) have some evidence of pancreatic disease at birth with many developing pancreatic insufficiency. Additional gastrointestinal manifestations may include intestinal obstruction exhibiting as meconium ileus and less frequently early signs of liver disease. In the reproductive system, nearly all males with CF lack intact genital ducts, either through loss of the distal duct (vas deferens) and/or damage to the epididymis. Since the prenatal disease course in CF cannot be studied in humans, our goal was to use a highly relevant animal model to determine the cellular origin and time course of CF pathology before birth. Sheep are a well‐established surrogate for investigating aspects of early human development {Strang, 2 #5990;Barry, 3 #6004}.
The CF sheep model exhibits a profound phenotype at birth with obstructive intestinal disease, pancreatic hypoplasia and fibrosis, biliary fibrosis/cirrhosis and hepatic cholestasis, gallbladder hypoplasia, and absence or hypoplasia of the vas deferens. 4 , 5 Moreover, tracheal epithelial cells from CF sheep with human‐specific mutations (F508del and G542X) are responsive to human CFTR potentiator and corrector drugs. 5 The pathological and physiological similarities between CF sheep and pwCF, in addition to the extremely well‐characterized developmental biology of the sheep lung, 2 , 6 , 7 make the CF sheep model particularly well‐suited for studies of the early CF disease process. 8 We showed previously that the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which is the causative gene in CF, is expressed in the sheep lung by 52 days of gestation and is also an abundant transcript by 83 days in tissues from multiple sheep organs. 9 Here we established a time course of wildtype (WT) and CF sheep development through accurately monitored pregnancies and collected tissues from multiple organ systems through gestation. This enabled a detailed examination of the initiating events in CF pathology at the tissue, cellular and molecular levels. Furthermore, the availability of tracheal tissue facilitated electrophysiological experiments to investigate the onset of electrogenic ion transport and the differential contributions of sodium absorption and chloride secretion to epithelial function. Evidence for CF‐associated disease is apparent in the pancreas, intestine, and liver by 80 days gestation (147 days full term in sheep) suggesting that novel therapeutic approaches to prevent the destruction of the pancreas and other organs in CF may require intervention in the mid‐trimester of human gestation.
2. MATERIALS AND METHODS
2.1. Animals
American Romney breed of domestic sheep (Ovis aries) was used in this study. All animal studies were approved and monitored by the Institutional Animal Care and Use Committee (IACUC) at Utah State University (IACUC protocol # 10089) and conformed to the National Institute of Health guidelines. WT sheep were bred according to standard protocols. Briefly, ewes were synchronized at estrus using an intramuscular (IM) injection of 2 ml EstruMate (Merck Animal Health) and introduced to the ram on the same day. Ultrasonography around 40 days confirmed pregnancy status and day 1 of gestation was defined as ~48 h post‐injection. CFTR ‐/‐ sheep were generated by somatic cell nuclear transfer (SCNT) using genetically modified sheep fetal fibroblast (SFFs) (CF 60 male, exon 2 targeted) derived from American Romney fetuses as described previously. 4 SCNT recipient ewes were synchronized prior to embryo transfer. Fetuses were recovered at days 50, 65, 80, 100, and 120 of gestation and 147–150 days (term) for histological, molecular, and cellular analysis as described below. WT‐term animals were born naturally and time after birth until euthanasia was recorded.
2.2. Histopathologic analysis
A necropsy was performed on all fetuses collected, to examine for gross lesions and the findings were documented. The following tissue samples were collected and fixed in 10% neutral buffered formalin for histology: trachea, lung (proximal and distal separately), forestomach, small and large intestine, pancreas, liver, gallbladder, kidney, testis, male genital ducts, spleen, thyroid gland, adrenal gland, heart, and skeletal muscle. Formalin‐fixed tissue sections were processed and embedded in paraffin according to routine histologic techniques. Sections, 5‐μm thick, were stained with hematoxylin and eosin (H&E), alcian blue, or periodic acid–Schiff (PAS) stain according to standard methods and examined by light microscopy.
2.3. Tissue collection for RNA processing
Simultaneously with tissue collection for histology, tissue samples were snap‐frozen in liquid nitrogen and stored at −80°C for RNA purification.
Tissues were homogenized in TRIzol (Life Technologies) in a dry ice‐cooled MPBio FastPrep Classic homogenizer. RNA isolation was according to the TRIzol manufacturer's protocol. RNA quality was assessed by NanoDrop, formaldehyde gel electrophoresis, and Tapestation prior to choosing high‐quality samples for RNA‐seq.
2.4. RNA sequencing (RNA‐seq) and data analysis
RNA samples passing strict QC criteria were used as templates for cDNA library synthesis with oligo dT priming, amplified libraries were pooled and sequenced (100 bp paired ends) on a NovaSeq 6000 machine. Raw sequence reads were aligned to the Oar_v4.0 (oviAri4) genome of the Texel sheep breed with STAR version 2.7.0e_0320 (https://github.com/alexdobin/STAR). 10 Aligned reads were assigned to genomic features with featureCounts version 1.6.3 in the Subread package (http://subread.sourceforge.net/). 11 Gene expression analysis to identify differentially expressed genes (DEGs) was performed using edgeR version 3.34.1. 12 Genes were filtered for expression and normalized by tagwise dispersion, then comparisons were calculating the glmTreat function in sequential pairwise comparisons. DEGs were filtered for those with a >1.5 or <−1.5 fold change (log2 +/− 0.58) and an adjusted p‐value ≤0.001.
2.5. Tissue collection for electrophysiology
Tracheal tissue was excised during necropsy and placed in HEPES‐buffered Ringer solution on ice. The trachea was stripped of connective tissue, cut longitudinally and one or more segments mounted directly in Ussing chambers and maintained under short‐circuit conditions mounted as described previously. 13 The tissues were treated sequentially with: amiloride (100 μM, apical), forskolin (10 μM, apical), GlyH 101 (50 μM), and steady‐state short circuit current (Isc) was recorded for each condition.
3. RESULTS
3.1. Pancreatic pathology is evident by 80 days of gestation
Earlier data from human fetal tissues 14 , 15 , 16 showed abnormal pancreatic histology by 20/40 weeks of gestation, which is equivalent to ~75 days of sheep gestation. Here we observe the initiation of characteristic CF pancreatic disease by 80 days gestation in the CFTR ‐/‐ sheep fetus (Figure 1). No pancreatic lesions were observed at 50 and 65 days gestation. At 80 days gestation, in comparison to normal WT acinar tissue (Figure 1A), a small to moderate number of pancreatic acini and ducts in the CFTR ‐/‐ tissues are dilated, with variable acinar cell atrophy in the affected acini (Figure 1B,D, arrow). Accumulation of periodic acid Schiff (PAS)‐ and alcian blue‐staining material consistent with mucus is also present in the lumen of some acini and ducts (Figure 1D,F). Dilated acini and ducts are rarely observed in the WT pancreas and lack mucus accumulation (Figure 1C,E). The progression of histological abnormalities through gestation at 80, 100, 120 days, and term (147–150 days) in the CFTR ‐/‐ pancreas is shown in Figure S1. The amount and/or severity of acinar and ductular dilation increase over time and accumulation of cell debris and neutrophils are seen in some dilated acini by 120 days (Figure S1F, long arrowheads). Excessive loose stroma separating acini and ducts is evident by 120 days, and minimal interstitial neutrophilic inflammation is also present (Figure S1F,H, asterisk). In addition, abundant dense collagen surrounds some ducts. Histologic lesions vary in severity between fetuses and may involve upto 100% of pancreas sections at term, furthermore, nearly all animals showed pancreatic hypoplasia by 120 days of gestation. Hence, as in humans loss of functional CFTR has a profound impact on sheep pancreas histology by the middle of fetal life and the organ is progressively destroyed through gestation.
FIGURE 1.
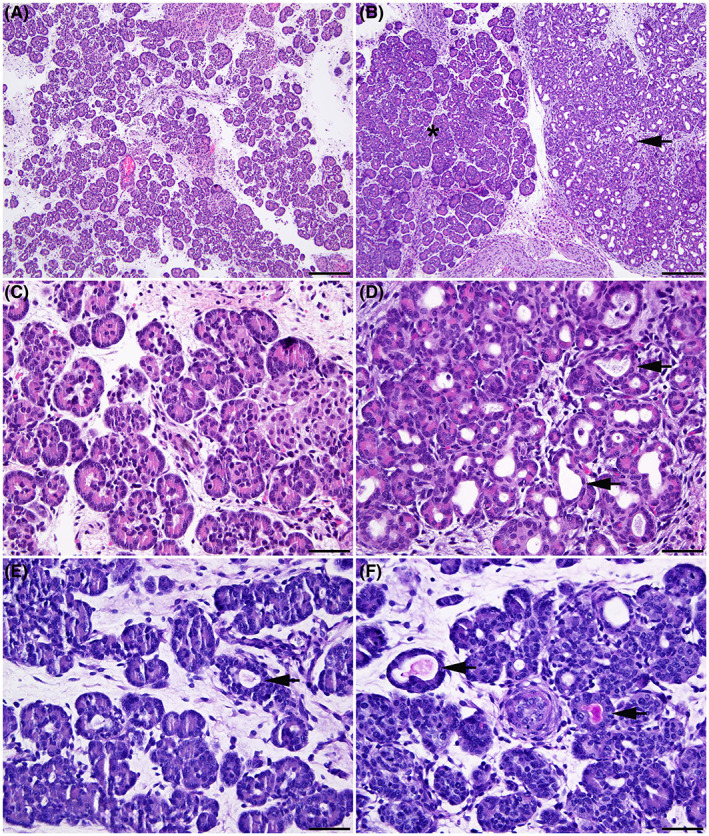
Pancreatic pathology is evident by 80 days gestation in CFTR −/− sheep. Wild‐type (WT) (A, C, E) and CFTR −/− (B, D, F) sheep fetal pancreas at 80 days gestation. Normal pancreatic histology in the WT fetus (A). In the CFTR −/− fetus (B) both normal acini (asterisk) and an area with substantial acinar dilation (arrow) are adjacent to each other. Higher magnification images show normal pancreatic tissue in a WT fetus (C), and dilatation of multiple pancreatic acini (arrow) in a CFTR −/− fetus (D). Periodic Acid Schiff (PAS) staining of a normal pancreas in a WT fetus (E) and CFTR −/− pancreas (F). Note that occasional dilated acini are present in the WT pancreas, but no PAS‐staining material is visible in the lumen (arrow). In the CFTR −/− pancreas some dilated acini contain PAS‐staining material stains consistent with mucus. Hematoxylin and eosin stain (A to D). 100× (A and B); bar = 200 μm. 400× (C and D); bar = 50 μm. Period acid‐Schiff (PAS) stain (E and F). 400×; bar = 50 μm.
3.2. Intestinal obstruction begins before 80 days of gestation
All animal models of CF show a more severe intestinal obstruction phenotype at birth than the majority of newborn babies with the disease. This is particularly severe in the large animal models, the sheep and the pig. 4 , 5 , 17 Intestinal lesions were not observed at 50 or 65 days gestation but substantial abnormalities were seen by 80 days gestation in CFTR −/− sheep (Figure 2), showing that this phenotype is also an early event during development. Although the WT intestinal tract shows a generally constant diameter along its length at 80 days (Figure 2A), the CFTR −/− aboral small intestine and colon have markedly smaller diameters (Figure 2B). At this gestational age, the CFTR −/− small intestine oral to the narrowing is filled with meconium (Figure 2D) and the colon contains excessive luminal mucus. Furthermore, the colonic mucosa has many mucus‐distended goblet cells (Figure 2F) that stain with alcian blue (Figure 2H). The severity of intestinal lesions in the CFTR −/− sheep increases through gestation from 80 days to term with progressive meconium/mucus accumulation in the lumen, and compression of the mucosa (Figure S2).
FIGURE 2.
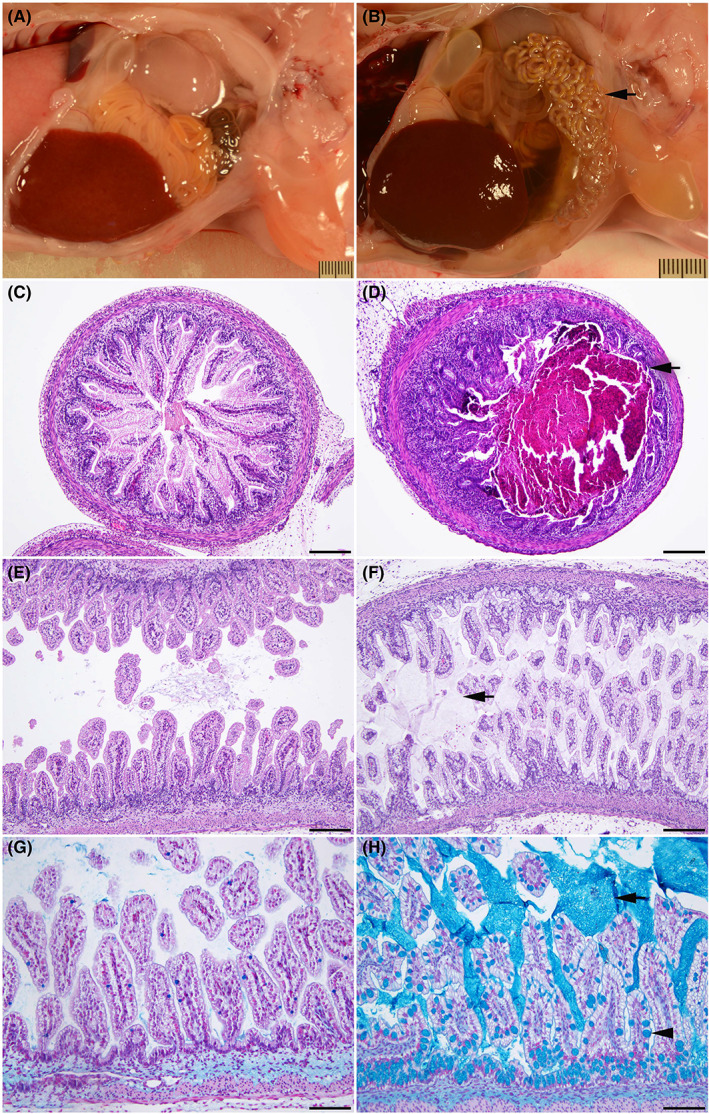
Intestinal obstruction is seen by 80 days gestation in the CFTR−/− sheep. WT (A, C, E, G) and CFTR −/− (B, D, F, H) sheep intestinal tracts at 80 days gestation. (A) Normal intestinal tract in a WT fetus. Note the constant diameter along the length of the intestine. (B) Early meconium ileus in a CFTR −/− fetus. Note the smaller diameter of a long segment of the small intestine and colon (arrow). (C) Normal small intestine in a wild‐type fetus. (D) Small intestine filled with meconium (arrow) in a CFTR −/− fetus. (E) Normal colon in a wild‐type fetus. (F) Colon with distended goblet cells in the mucosa and excessive luminal mucus (arrow) in a CFTR −/− sheep fetus. Note: hematoxylin and eosin (H and E) stain does not clearly stain the mucus. (G, H) Higher magnification images showing Alcian blue staining of mucus. A minimal amount of luminal mucus is seen in the WT colon (G). A large amount of luminal mucus (arrow) is evident in the CFTR −/− colon (H), where the goblet cells are also distended with mucus (arrowhead). H and E stain (A to F). 100× (C to F); bar = 20 μm. Alcian blue stain (G and H). 200× (G and H); bar = 100 μm.
3.3. Liver and gallbladder disease is evident by 80 days gestation
CF liver disease is of increasing clinical importance as pwCF achieve greater longevity. No hepatic lesions were observed at 50 and 65 days of gestation in the CFTR −/− sheep liver. However, early features of biliary fibrosis/cirrhosis were seen at 80 days, accompanied often by gallbladder hypoplasia. These lesions included portal tracts expanded by multiple poorly defined ducts, which often lacked a distinct lumen, and fibrosis (Figure 3D,F). Also, the onset of intrahepatic cholestasis is seen at the same time point (Figure 3F). These early features of liver disease progress through 100 days and 120 days and are pervasive in the liver at term (Figure S3). At term, these lesions resemble the uncommon early‐onset biliary cirrhosis in pwCF. 18 , 19 , 20
FIGURE 3.
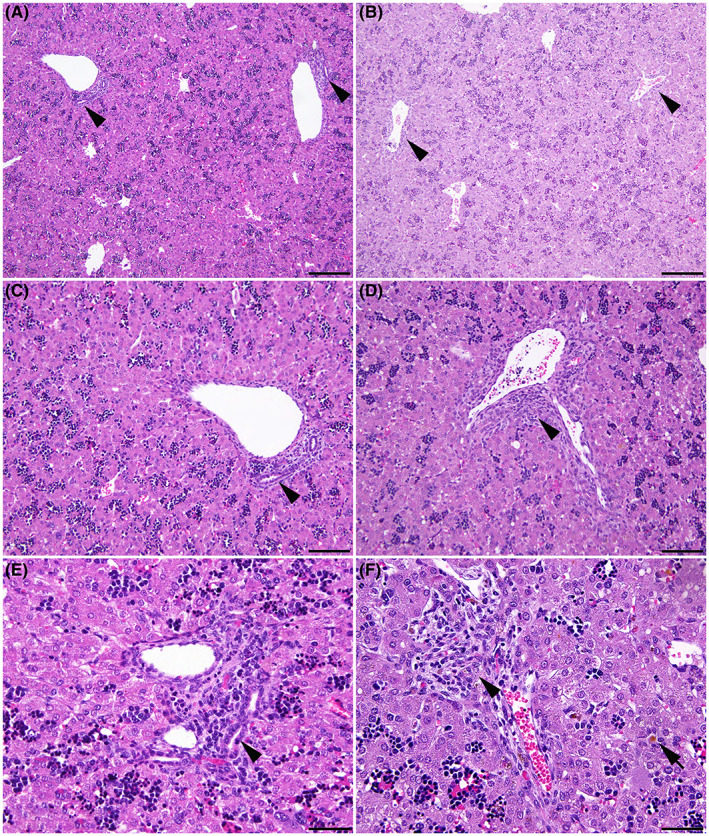
Biliary fibrosis and intrahepatic cholestasis are observed by 80 days gestation in the CFTR−/− sheep. WT (A, C, E) and CFTR −/− (B, D, F) fetal sheep liver at 80 days. (A) Normal liver in a WT fetus. Note the well‐defined bile ducts in portal tracts (arrowheads). (B) Liver of a CFTR −/− fetus. Higher magnifications (C–F) of (C and E) normal liver in a WT fetus showing the well‐defined bile duct in the portal tract (arrowhead), and (D and F) the portal tract with early biliary fibrosis (arrowhead) in a CFTR −/− fetus. (F) Multiple bile ducts with indistinct lumens expand the portal tract and intrahepatic cholestasis is seen (arrow). Hematoxylin and eosin stain (A to D). 100× (A and B); bar = 200 μm. 200× (C and D); bar = 100 μm. 400× (E and F); bar = 50 μm.
3.4. Lung disease is not grossly evident through gestation
The absence of overt lung disease in newborn humans with CF has always been somewhat of an enigma since CFTR is robustly expressed in the mid‐trimester lung in utero. 21 Similarly, histological analysis of the CFTR −/− sheep proximal and distal (Figure 4) lung from 80, 100, 120 day, and term fetuses did not reveal any abnormalities compared to WT age‐matched controls.
FIGURE 4.
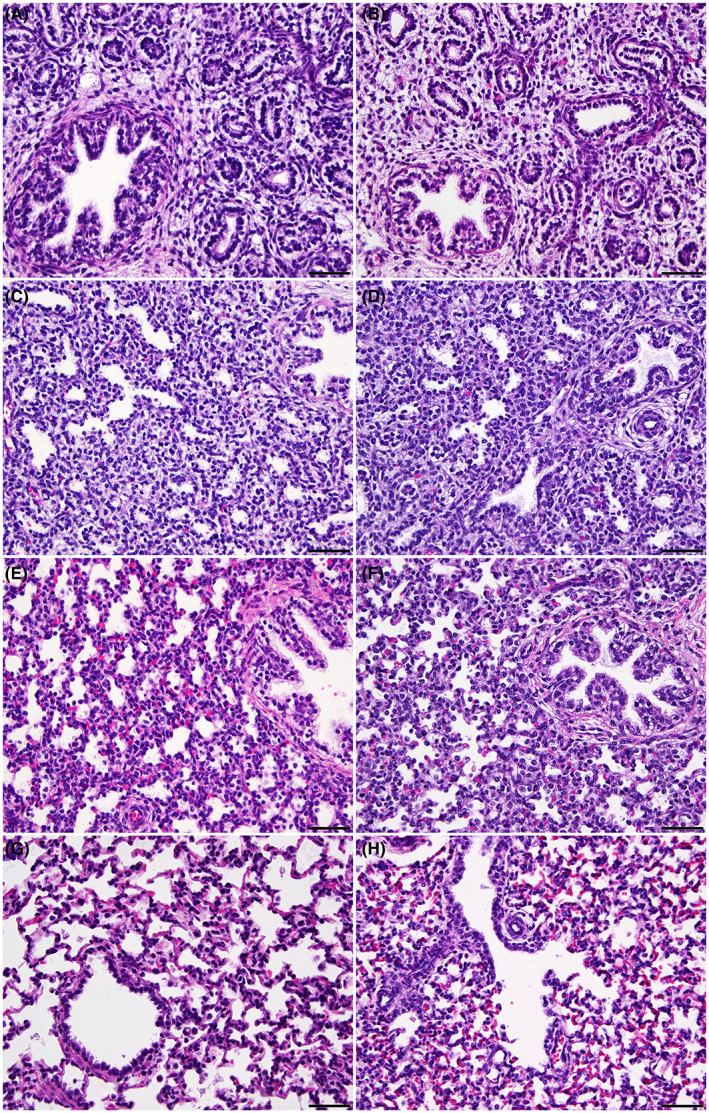
The CFTR ‐/‐ sheep distal lung shows no histological abnormalities through gestation. Comparison of WT (A, C, E, G) and CFTR −/− (B, D, F, H) sheep fetal lung at 80 (A, B), 100 (C, D), and 120 (E, F) days of gestation, and term (147 days) (G, H). The WT and CFTR −/− sheep lungs are identical histologically at each timepoint. Hematoxylin and eosin stain. 400×; bar = 50 μm.
A summary of histological data from the developmental time course, including the absence of the vas deferens, is shown in Table 1.
TABLE 1.
Summary of histological data from the developmental time course in WT and CFTR−/− animals
| Genotype | Age (days) | Sex | Pancreas | Intestinal tract | Lung | Liver | Vas deferens |
| WT | 50 | 2 males | No lesions | No lesions | No lesions | No lesions | Embryo too small to identify the vas deferens grossly |
| 65 |
2 males 1 female |
No lesions | No lesions | No lesions | No lesions |
Embryo too small to identify the vas deferens grossly N/A |
|
| 80 |
2 males 1 female |
No lesions | No lesions | No lesions | No lesions |
Embryo too small to identify the vas deferens grossly N/A |
|
| 100 | 2 females | No lesions | No lesions | No lesions | No lesions | N/A | |
| 120 |
2 males 2 females |
No lesions | No lesions | No lesions | No lesions |
Present N/A |
|
| TERM |
2 males 1 female |
No lesions | No lesions | No lesions | No lesions |
Present N/A |
|
| CF | 50 | 2 males | No lesions | No lesions | No lesions | No lesions | Embryo too small to identify the vas deferens grossly |
| 65 | 6 males | No lesions | No lesions | No lesions | No lesions | Embryo too small to identify the vas deferens grossly | |
| 80 | 3 males |
No lesions (1) Pancreatic acinar and duct dilation (2) |
Early meconium ileus (3) | No lesions |
No lesions (2), cholestasis (1), mild gallbladder hypoplasia (1) |
Not detected (3) but testes have not descended | |
| 100 | 2 males | Pancreatic acinar and duct dilation (2) | Meconium ileus (2) | No lesions |
Biliary fibrosis/cirrhosis (1) cholestasis (1), gallbladder hypoplasia (1) |
Presumptive vas deferens present (1) | |
| 120 | 3 males | Exocrine pancreatic hypoplasia (3) | Meconium ileus (3) | No lesions |
Biliary fibrosis/cirrhosis (3), cholestasis (3), gallbladder hypoplasia (3) |
No vas deferens (3) | |
| TERM | 4 males | Exocrine pancreatic hypoplasia (4) | Meconium ileus (4) | No lesions |
Biliary fibrosis/cirrhosis (4), cholestasis (3), gallbladder hypoplasia (2) |
No vas deferens (3), unliteral vas deferens (1) |
3.5. Development of ion transport mechanisms in the fetal trachea
The epithelial surface lining the trachea shares many functional properties with the lining of the large airways in the lung and contains cells that express CFTR. No histological difference was observed between WT and CFTR −/− sheep trachea through gestation (80‐day trachea is shown in Figure S4). The time course for the acquisition of fluid secretion in the fetal trachea and the ion channels involved are not well characterized, so we used sheep tracheal epithelium from the developmental time course to address this. Measurements of CFTR‐dependent short‐circuit current (Isc) in WT fetal tracheal tissue mounted in Ussing chambers showed activity by 80 days of gestation with increased levels at 100 days, 120 days, and in 2‐day neonates (Figure 5A). In contrast, only baseline Isc was seen in CFTR ‐/‐ animals (Figure 5B). Forskolin (FSK)‐stimulated Isc was minimal in WT fetal, neonatal, and adult tracheal tissues (Figure 5A,C), consistent with prior activation (in vivo and/or ex vivo). The higher (FSK)‐stimulated Isc at 80 days and in neonates warrants further investigation but may be an experimental artifact. While CFTR inhibitor I172 was completely ineffective in WT sheep tracheal tissues and primary cell cultures (data not shown), another CFTR inhibitor, GlyH 101 (50 μM) reduced the WT CFTR‐dependent current by ~50% (Figure 5A,D). Since CFTR‐dependent current appears to be fully activated prior to the addition of forskolin, the GlyH 101‐dependent inhibition of current is likely a better measure of CFTR channel function. The transepithelial resistance of WT and CFTR −/− tracheal tissues was similar and relatively constant in tissues from different gestational ages (Figure 5E), consistent with early (<80‐day gestation) formation of an intact epithelial barrier. The addition of ATP (100 μM, apical) elicited a small, transient (1–2 min) increase in current that was similar throughout gestation and not different between WT (10.3 ± 1.3 μAmp/cm2) and CFTR −/− (9.4 ± 2.1 μAmp/cm2) tracheal tissues (data not shown). Finally, consistent with previous reports we observed amiloride‐sensitive short‐circuit current (reflecting activity of the epithelial sodium channel, ENaC) only in tracheal tissues from postnatal animals (2‐day neonate and adult) (Figure 5A,F), and this was absent from CFTR−/− animals.
FIGURE 5.
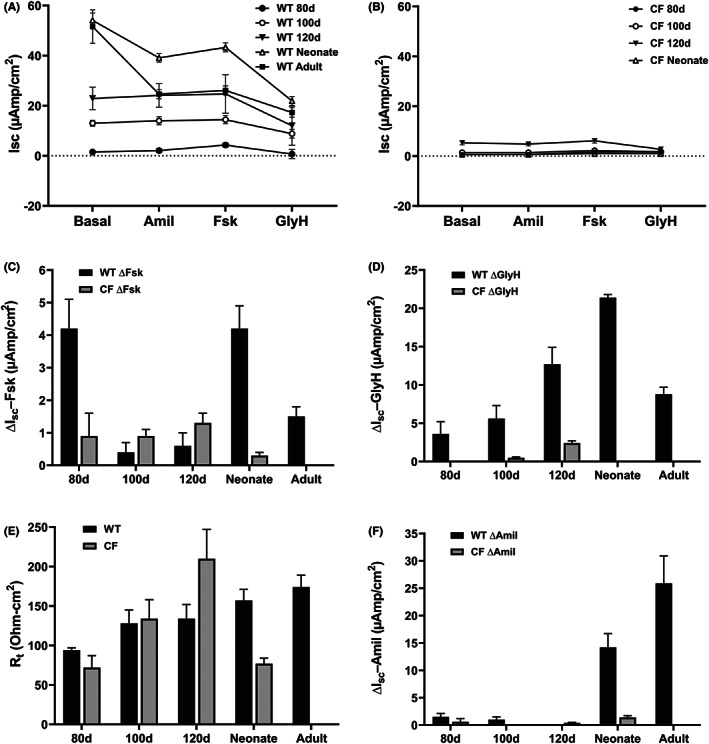
Electrophysiological characterization of tracheal epithelium in WT and CFTR ‐/‐ sheep through gestation. Excised tracheal tissue segments from fetal, neonatal, and adult WT sheep were prepared as described, mounted in Ussing chambers and maintained under short‐circuit current conditions. The tissues were treated sequentially with: amiloride (100 μM, apical) (A, B, F), forskolin (10 μM, apical) (A, B, C), and GlyH 101 (50 μM) (A, B, D) and steady‐state Isc was recorded for each condition. (C) Values represent delta Isc immediately before and 10 min after the addition of forskolin as in (A, B). (D) Values represent delta Isc immediately before and 10 min after the addition of GlyH 101 as in (A, B). 0 values for 80d and neonatal CF are due to no change in Isc upon the addition of GlyH101. (E) transepithelial resistance values for WT and CFTR ‐/‐ sheep tracheal tissues in (A, B). (F) Values represent delta Isc immediately before and 5 min after the addition of amiloride as in (A, B). 0 values for 100d CF and 120d WT, are due to no change in Isc upon the addition of amiloride. The values are mean +/ SEM for #animals/#tissues: 80d, 2/2; 100d, 2/4; 120d, 2/6; neonate, 1/6; adult, 9/19.
3.6. Gene expression profiles through gestation in the fetal trachea
Next, to examine the identity of ion channels and transporters that might contribute to the ion transport properties of the tracheal epithelium in vivo, as measured by Ussing chambers ex vivo we defined the tracheal transcriptome from 50 days gestation through to term by RNA‐seq. Tissues were collected in parallel from two animals at the same time points used for electrophysiology (80, 100, 120 days, and term) together with 50 and 65 days. The full list of differentially expressed genes through gestation is shown at GEO: GSE202024, but here we focus on the expression of specific ion channels, secreted glycoproteins, and transcription factors (Figure 6 and Figure S5).
FIGURE 6.
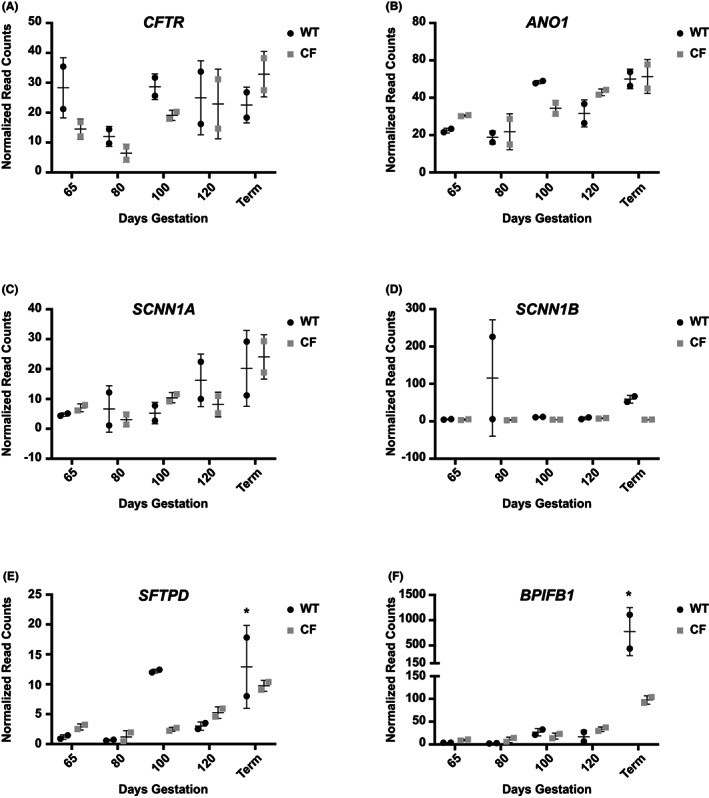
Differential expression of key marker genes in WT and CFTR ‐/‐ fetal trachea through gestation. Data from RNA seq analysis of tracheal tissue samples at 65, 80, 100, 120 days gestation and term (147 days). (n = 2 at each time point). Individual graphs show normalized read counts for WT, black circles, and CF (CFTR ‐/‐), gray squares, of A) CFTR, B) ANO1, C) SCNN1A, D) SCNN1B, E) SFTPD, F) BPIFB1. * denotes term samples from ~6 h (low) and ~15 h (high) after birth.
As noted above, CFTR‐mediated Isc was evident by 80 days gestation in WT sheep fetal tracheal epithelium, and activity increased through 100 and 120 days gestation and remained high in the neonate. In the CFTR ‐/‐ trachea only baseline activity was seen. At the transcriptional level, no consistent differences were seen in CFTR transcript abundance in the WT and CFTR ‐/‐ tracheal tissues through gestation (Figure 6A). Moreover, transcript abundance did not change substantially with gestational age in either WT or CFTR ‐/‐ tissues. It is possible that a small decrease in CFTR mRNA at 80 days gestation compared to 65 days, with levels restored at 100 days may reflect the cellular composition of individual processed tissue samples. Another CF‐relevant anion channel in the trachea is the calcium‐activated chloride channel (CaCC/TMEM16A) encoded by the anoctamin 1 (ANO1) gene. ANO1 transcript abundance increased gradually in both WT and CFTR ‐/‐ tracheal tissue between 80 days gestation and term (Figure 6B). Expression of the alpha subunit of the epithelial sodium channel (SCNN1A) showed a marked increase toward the end of gestation in both WT and CFTR ‐/‐ tracheal tissue (Figure 6C), while SCNN1B transcripts only increased slightly in the WT animals at term (Figure 6D), consistent with amiloride‐sensitive short circuit current only being present in post‐natal WT animals. The SCNN1G gene that encodes the third subunit of ENaC showed a very similar expression pattern to SCNN1B (not shown).
Other important proteins in tracheal epithelial function are secreted glycoproteins with surfactant and/or host defense functions. These include surfactant proteins encoded by the SFTP gene family 22 and BPI fold containing family members (BPIF). 23 Transcript levels of SFTPA1 and SFTPB in the sheep fetal trachea are either very low or inconsistent. Expression of SFTPB mRNA is noticeably absent from the CFTR ‐/‐ tracheal tissue though is higher at 100 days in the WT tissues (not shown). In contrast transcripts of SFTPD, which are also more abundant in 100 days WT tissues, generally increase through gestation in both WT and CFTR ‐/‐ animals (Figure 6E). Another key secreted protein encoded by the BPIFB1 gene is not seen during gestation but is greatly upregulated at term in WT animals, consistent with its key role in mucosal defense, and also to a lesser extent in CFTR ‐/‐ animals (Figure 6F). Of note, the large difference in expression of SFTPD and BPIFB1 between the two WT term animals is likely due to a difference in time between birth and tissue collection (euthanasia, ~6 h for the lower value and ~15 h for the higher value).
Next, turning to stem cell markers in the tracheal epithelium through gestation, it is of interest that the transcription of the basal cell marker KRT5, which increases between 80‐ and 100‐ days gestation in both WT and CFTR −/− animals subsequently falls in WT tissues but remains constant in CFTR −/− tissues (Figure S5A). Furthermore, the subset of KRT5+ basal cells that also express TP63 (the stem cell compartment) substantially decreases between 65 and 80 days gestation, is restored to the 65‐day levels by 100 days, and then gradually decreases to term in WT tissues while increasing in CFTR −/− tissues (Figure S5B).
Finally, we looked at the expression of cell‐type‐specific and lineage‐specific forkhead box (FOX) transcription factors through tracheal development. Sample‐to‐sample variation in levels of FOXJ1, a marker of ciliated cells, prevented consistent interpretation of the data, while expression of the pulmonary ionocyte marker FOXI1 was extremely low, reflecting a paucity of these cells in the intact tissue (data not shown). The lineage marker FOXA1 24 , 25 shows an identical profile of expression in tracheal tissue from WT and CFTR ‐/‐ animals, with a drop between 65 and 80 days gestation, followed by an increase at 100 days that is maintained through to term (Figure S5C). This bimodal pattern of expression, possibly indicative of different roles early and late in gestation is also seen for Ets Homologous Factor (EHF). EHF, which has a pivotal role in human lung epithelial function, 26 , 27 is an abundant transcript in the trachea through sheep gestation in both WT and CFTR ‐/‐ animals.
We also performed gene ontology process enrichment analysis on the differentially expressed genes across the developmental time course in WT and CFTR ‐/‐ tracheal samples. We interpret these data with caution due to the relatively small numbers of animals analyzed at each time point but note that the tracheal tissue shows relatively few processes that change significantly with time. In the WT animals, structural changes involving fibrillar collagens (COL1A1, COL6A1, and COL14A1) are greatly downregulated between 65 and 100 days and then upregulated between 100 days and 120 days gestation (Figure S6). Notable in the CF trachea is the large number of transcription factors that are upregulated between 120 days and term (GEO: GSE202024).
4. DISCUSSION
The increasing availability of highly effective modulator therapies (HEMT) for many pwCF is changing the focus of the investigation into CF disease mechanisms. Formerly the lung was the primary target since the pulmonary disease was the main determinant of longevity. More recently interest in other CF‐affected organs has undergone somewhat of a renaissance. Notably, understanding the etiology of pancreatic and liver disease, and male infertility are all high priorities. Though current HEMTs have a systemic effect, 28 they may have limited capacity to restore completely normal function to an organ that is profoundly damaged in utero such as the pancreas. However, early data suggest that in pediatric patients with CF, HEMTs can convert pancreatic insufficiency to sufficiency, based on clinical and biochemical parameters. 29 In this context, it is critical to determine the molecular basis of disease in those organs with profound CF‐associated pathology in utero and identify the initiating events. Clearly, these studies necessitate the use of suitable animal models of CF that closely model human development and the CF sheep model is particularly useful {Strang,2 #5990;Barry,3 #6004}. Here, we pursued a detailed analysis of the main CF‐affected organ systems through gestation in the CFTR ‐/‐ sheep model.
Pancreatic, liver, and intestinal disease initiate between 65 and 80 days of CFTR ‐/‐ sheep gestation, corresponding to about 21 weeks gestation in humans. The pancreatic pathology is consistent with an obstructive phenotype, rather than the failure of normal pancreatic duct branching and development. Acinar lumens become distended with mucoid material and later also contain cell debris and neutrophils. Interstitial fibrosis surrounding the affected pancreatic acini likely results mainly from stromal condensation following acinar loss; this lesion is present early in gestation and is progressive. Some pancreatic ducts are surrounded by dense collagenous stroma in late gestation also supporting excessive collagen production. During gestation and at term, the cystic acini and ducts, and fibrosis seen in CFTR ‐/‐ sheep pancreas closely mirror severe disease in the human CF pancreas. Biliary fibrosis/cirrhosis and cholestasis that start around 80 days in the CFTR ‐/‐ sheep liver have many common features with CF‐related early onset liver cirrhosis, which is relatively uncommon in newborns with CF but is a disorder mainly seen in children and adolescents. Gallbladder hypoplasia is a common phenotype in the CFTR ‐/‐ sheep by 80 days gestation consistent with observations in the neonatal CF pig. 30 Hence, the CFTR ‐/‐ sheep provides an excellent model for investigating the initiating events in CF pathology of the pancreas and liver. The intestinal phenotype of the CFTR ‐/‐ sheep is so profound already at 80 days gestation that, similar to the CF pig, 31 , 32 it may not be a good model for investigating the meconium ileus seen in a subset of newborns with CF. In contrast, unlike in the CF pig where structural lesions including smaller airway lumens were detectable in the trachea and lung by the pseudoglandular stage, 33 no histological lesions were seen in the CFTR ‐/‐ sheep lung at any time through gestation. This observation is consistent with apparently normal lung development in the majority of newborns and infants with CF. Hence, the CF sheep provides an excellent opportunity to investigate more subtle molecular deficits in CF lung development that do not manifest as microscopic features.
The electrophysiological characterization of the developing CFTR ‐/‐ sheep tracheal epithelium we describe here provides new information on the initiation of the CF phenotype. Previous studies of freshly isolated adult and late‐term fetal airway tissues from non‐CF sheep suggested that cAMP‐activated Cl secretion was present in the late‐term fetal trachea as well as adult bronchus and trachea. 13 Furthermore, amiloride‐sensitive sodium absorption was present in adult but absent in fetal airways. These data are consistent with in utero lung liquid secretion studies demonstrating net fluid secretion by the airway epithelium prior to birth and transition to net fluid absorption at or near birth. Our earlier studies with primary cultures of neonatal WT and CF sheep tracheal airway epithelial cells showed that WT tracheal air/liquid interface (ALI) cultures exhibited amiloride‐sensitive short‐circuit current and CFTR‐dependent short‐circuit current. 4 In contrast, primary ALI cultures from neonatal CF trachea exhibited amiloride‐sensitive short‐circuit current, but no cAMP‐activated (CFTR) current. Here we show that CFTR‐dependent short‐circuit current is present in the tracheal epithelium of WT animals by 80 days gestation, with increasing levels at 100 and 120 days, while no CFTR‐dependent Isc was seen in CFTR ‐/‐ animals through gestation and at term. The role of alternative, calcium‐activated Cl channels in lung liquid secretion is not known, though we provide molecular evidence for ANO1 expression in the tracheal epithelium that increases through gestation, and electrophysiological evidence of ATP‐stimulated current in both WT and CF tracheae. Since the loss of CFTR apparently has no impact on gross lung development it remains possible that the protein has a different role prior to birth in the respiratory epithelium, or that other channels in the fluid‐filled lung compensate for its absence.
Finally, it is important to focus on the clinical utility of the data reported here. Many women with CF, who are responsive to HEMT and thus have greatly improved health, decide to become pregnant. They are also choosing to continue modulator therapies through gestation without the usual safety studies having been performed. The CFTR ‐/‐ and F508del sheep models provide an excellent opportunity to investigate the potential prevention of in utero pathology by modulator therapies and address safety concerns.
AUTHOR CONTRIBUTIONS
Ann Harris and Irina A. Polejaeva conceived and managed the study. Arnaud J. Van Wettere performed the necropsy and histopathological analysis of the sheep fetuses and Calvin U. Cotton electrophysiological analysis of tracheal epithelium. Misha Regouski managed the project and the tissue collection, assisted by Iuri Viotti Perisse and Shih‐Hsing Leir. Zhiqiang Fan generated, maintained, and prepared CFTR ‐/‐ cells for cloning. Zhiqiang Fan and Ying Liu performed SCNT embryo production and evaluation. Jenny L. Kerschner isolated RNA from tissues for RNA‐seq. Alekh Paranjapye performed bioinformatic analysis of RNA‐seq data. Shih‐Hsing Leir, Jenny L. Kerschner, and Makayla Schacht analyzed data and prepared figures. Ann Harris, Irina A. Polejaeva, Shih‐Hsing Leir, and Kenneth L. White obtained funding. Ann Harris and Arnaud J. Van Wettere wrote the manuscript. All authors reviewed the manuscript.
FUNDING INFORMATION
This work was supported by the Cystic Fibrosis Foundation (POLEJA18G0, LEIR21G0, HARRIS21G0) and also by the US Department of Agriculture Multistate Project W‐4171 (IAP) and the Utah Agricultural Experiment Station (Project 1343).
CONFLICT OF INTEREST
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of the data, in the writing of the manuscript, or in the decision to publish the results.
Supporting information
Figures S1‐S6
Supplementary Figure Legends
ACKNOWLEDGMENTS
We thank Drs. Rusty Stott, Alexis Sweat, and Holly Clement for performing embryo transfer procedures, DJ Anderson and Taylor Martin for excellent assistance with animal care, and Jacob Keim and Tayler Patrick for technical assistance with SCNT. We also acknowledge Amanda Wilheim for the preparation of the histology slides.
Van Wettere AJ, Leir S‐H, Cotton CU, et al. Early developmental phenotypes in the cystic fibrosis sheep model. FASEB BioAdvances. 2023;5:13‐26. doi: 10.1096/fba.2022-00085
Contributor Information
Irina A. Polejaeva, Email: irina.polejaeva@usu.edu.
Ann Harris, Email: ann.harris@case.edu.
REFERENCES
- 1. Andersen DH. Cystic fibrosis of the pancreas. J Chronic Dis. 1958;7(1):58‐90. [DOI] [PubMed] [Google Scholar]
- 2. Strang LB. Growth and development of the lung: fetal and postnatal. Annu Rev Physiol. 1977;39:253‐276. [DOI] [PubMed] [Google Scholar]
- 3. Barry JS, Anthony RV. The pregnant sheep as a model for human pregnancy. Theriogenology. 2008;69(1):55‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fan Z, Perisse IV, Cotton CU, et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight. 2018;3(19):e123529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Viotti Perisse I, Fan Z, Van Wettere A, et al. Sheep models of F508del and G542X cystic fibrosis mutations show cellular responses to human therapeutics. FASEB Bioadv. 2021;3(10):841‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dawes GS, Mott JC, Widdicombe JG, Wyatt DG. Changes in the lungs of the new‐born lamb. J Physiol. 1953;121(1):141‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dawes GS, Mott JC. The vascular tone of the foetal lung. J Physiol. 1962;164:465‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harris A. Towards an ovine model of cystic fibrosis. Hum Mol Genet. 1997;6(13):2191‐2194. [DOI] [PubMed] [Google Scholar]
- 9. Broackes‐Carter FC, Mouchel N, Gill D, Hyde S, Bassett J, Harris A. Temporal regulation of CFTR expression during ovine lung development: implications for CF gene therapy. Hum Mol Genet. 2002;11(2):125‐131. [DOI] [PubMed] [Google Scholar]
- 10. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29(1):15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923‐930. [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, Lun AT, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA‐seq experiments using Rsubread and the edgeR quasi‐likelihood pipeline. F1000Res. 2016;5:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cotton CU, Lawson EE, Boucher RC, Gatzy JT. Bioelectric properties and ion transport of airways excised from adult and fetal sheep. J Appl Physiol. 1983;55(5):1542‐1549. [DOI] [PubMed] [Google Scholar]
- 14. Boue A, Muller F, Nezelof C, et al. Prenatal diagnosis in 200 pregnancies with a 1‐in‐4 risk of cystic fibrosis. Hum Genet. 1986;74(3):288‐297. [DOI] [PubMed] [Google Scholar]
- 15. Hyde K, Reid CJ, Tebbutt SJ, Weide L, Hollingsworth MA, Harris A. The cystic fibrosis transmembrane conductance regulator as a marker of human pancreatic duct development. Gastroenterology. 1997;113(3):914‐919. [DOI] [PubMed] [Google Scholar]
- 16. Reid CJ, Hyde K, Ho SB, Harris A. Cystic fibrosis of the pancreas: involvement of MUC6 mucin in obstruction of pancreatic ducts. Mol Med. 1997;3(6):403‐411. [PMC free article] [PubMed] [Google Scholar]
- 17. Rogers CS, Hao Y, Rokhlina T, et al. Production of CFTR‐null and CFTR‐DeltaF508 heterozygous pigs by adeno‐associated virus‐mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118(4):1571‐1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colombo C. Liver disease in cystic fibrosis. Curr Opin Pulm Med. 2007;13(6):529‐536. [DOI] [PubMed] [Google Scholar]
- 19. Debray D, Narkewicz MR, Bodewes F, et al. Cystic fibrosis‐related liver disease: research challenges and future perspectives. J Pediatr Gastroenterol Nutr. 2017;65(4):443‐448. [DOI] [PubMed] [Google Scholar]
- 20. Leung DH, Narkewicz MR. Cystic fibrosis‐related cirrhosis. J Cyst Fibros. 2017;16(Suppl 2):S50‐S61. [DOI] [PubMed] [Google Scholar]
- 21. Trezise AE, Chambers JA, Wardle CJ, Gould S, Harris A. Expression of the cystic fibrosis gene in human foetal tissues. Hum Mol Genet. 1993;2(3):213‐218. [DOI] [PubMed] [Google Scholar]
- 22. Haagsman HP, Diemel RV. Surfactant‐associated proteins: functions and structural variation. Comp Biochem Physiol A Mol Integr Physiol. 2001;129(1):91‐108. [DOI] [PubMed] [Google Scholar]
- 23. Bingle CD, Craven CJ. PLUNC: a novel family of candidate host defence proteins expressed in the upper airways and nasopharynx. Hum Mol Genet. 2002;11(8):937‐943. [DOI] [PubMed] [Google Scholar]
- 24. Wan H, Dingle S, Xu Y, et al. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J Biol Chem. 2005;280(14):13809‐13816. [DOI] [PubMed] [Google Scholar]
- 25. Paranjapye A, Mutolo MJ, Ebron JS, Leir SH, Harris A. The FOXA1 transcriptional network coordinates key functions of primary human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2020;319(1):L126‐L136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fossum SL, Mutolo MJ, Tugores A, et al. Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J Biol Chem. 2017;292(26):10938‐10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fossum SL, Mutolo MJ, Yang R, et al. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 2014;42(22):13588‐13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dave K, Dobra R, Scott S, et al. Entering the era of highly effective modulator therapies. Pediatr Pulmonol. 2021;56(Suppl 1):S79‐S89. [DOI] [PubMed] [Google Scholar]
- 29. Munce D, Lim M, Akong K. Persistent recovery of pancreatic function in patients with cystic fibrosis after ivacaftor. Pediatr Pulmonol. 2020;55(12):3381‐3383. [DOI] [PubMed] [Google Scholar]
- 30. Zarei K, Stroik MR, Gansemer ND, et al. Early pathogenesis of cystic fibrosis gallbladder disease in a porcine model. Lab Invest. 2020;100(11):1388‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rogers CS, Stoltz DA, Meyerholz DK, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321(5897):1837‐1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stoltz DA, Rokhlina T, Ernst SE, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123(6):2685‐2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meyerholz DK, Stoltz DA, Gansemer ND, et al. Lack of cystic fibrosis transmembrane conductance regulator disrupts fetal airway development in pigs. Lab Invest. 2018;98(6):825‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S6
Supplementary Figure Legends