

Abstract
In this work, the components of the protein electrostatic potentials in solution are analyzed with NMR paramagnetic relaxation enhancement experiments and compared with continuum solution theory, and multiscale simulations. To determine the contributions of the solution components, we analyze them at different ionic strengths from 0 to 745 mM. A theoretical approximation allows the determination of the electrostatic potential at a given proton without reference to the protein structure given the ratio of paramagnetic relaxation enhancements rates between a cationic and an anionic probe. The results derived from simulations show good agreement with experiment and simple continuum solvent theory for many of the residues. A discrepancy including a switch of sign of the electrostatic potential was observed for particular residues. By considering the components of the potential, we found the discrepancy is mainly caused by angular correlations of the probe molecules with these residues. The correction for the correlations allows a more accurate analysis of the experiments determining the electrostatic potential of proteins in solution.
Graphical Abstract
INTRODUCTION
Experimental determination of the electric potential and field around proteins is an important challenge. Recently, Yu et al.1 introduced an NMR-based method to measure near-surface electrostatic potentials for individual protein residues without chemically attaching probes or using structural information. In this method, paramagnetic relaxation enhancements (PRE) arising from analogous oppositely charged paramagnetic probes are determined for 1H nuclei of a protein. The electrostatic potentials near the molecular surface proximal to the observed 1H nuclei are then determined from the ratio of PRE rates reflecting different spatial distributions of cationic and anionic probes around the biomolecules. The measured potentials at ionic strengths of 30 and 130 mM agree reasonably well with the theoretical predictions of the Poisson−Boltzmann (PB) equation using a standard fixed charge model. The root-mean-square deviation (RMSD) between the experimental and theoretical values was about 6 mV for secondary structure regions.1 A few discrepancies, including a change of sign, at certain residues existed, which limits the possible uses of the analysis.
The approximations made to allow for a structure-independent analysis of the ratio of paramagnetic relaxation rates1 in principle can be relaxed. The components contributing to the electrostatic fields can be separated theoretically and considered in terms of both chemical components and how they are affected by various approximations. The PRE experiments can be analyzed with preexisting structures to aid in considering the origin of the discrepancies. Questions of whether protein conformational flexibility, the proximity approximation used in the analysis, or even the underlying simulation force field might cause differences between theory and experiment are considered here. The experimental determination of the protein electrostatic field has the potential to validate and ultimately improve theoretical model force fields which are in wide use.2–5
The electrostatic potential generated by a molecular system can not only be employed to describe many of the molecular properties but also used to interpret intermolecular interactions related to mechanism.6,7 Once average atomic charges are determined, the application of Coulomb’s law is a straightforward pairwise additive form; the long-ranged nature of the electrostatic field, however, can present a variety of challenges in implementation and interpretation for a given computational method. As a result, many approximations which are computationally convenient exist. One of the most widely used methods for modeling biomolecular electrostatics is the PB method which assumes that the mobile ions in a system follow the Boltzmann distribution in a continuum solvent as point charges.8,9 However, the PB method has well-known limitations when ion correlations exist and/or highly charged molecules are involved.
Simulation of a polar molecule in explicit water molecules and ions usually relies on approximate methods to ensure or force convergence of the electrostatic properties. Commonly, electrostatic potentials can be derived from reaction fields10–12 or the Ewald summation method13,14 using a periodic boundary condition. The Ewald sum decomposes the potential into two parts: a short-ranged real space part (computed in a unit cell) and a long-ranged Fourier space part (computed for image cells). The task of computing the electrostatic potential is still the most time-consuming part of any explicit all-atom simulation. Fortunately, many techniques (e.g., particle mesh Ewald method,15 the fast multipole method,16 fast Fourier transforms,17 etc.) have been applied to accelerate the calculations for particular conditions and size ranges.
Experimentally, the molecular potential can be derived, in principle, from X-ray or electron diffraction by fitting a multipolar density function to the data.18 The method requires availability of very high-resolution data (as is the possible case with some inorganic materials). As a result, it is not applicable to most chemical compounds and biomolecules. Another experimental method is vibrational spectroscopy,19 which utilizes the sensitivity of vibrational transitions to electric fields (the Stark effect) to provide a local probe of electrostatic fields in systems such as proteins. However, the approach provides only limited information and requires covalently attached probes, which may perturb the native conformation of some systems. Inferences can also be made from ion counting and similar experimental methods.20 Historically, pKa data have often been used to examine electrostatic models.21,22 In the past decade, there was significant progress in experimental pKa measurements for protein side chains.23,24 However, pKa data do not explicitly report electrostatic potentials or fields. Thus, the NMR PRE ratio method has advantages over other experimental methods for biomolecular electrostatics.
Here we apply the PRE ratio method to analyze the components of the electric potential of ubiquitin with and without prior knowledge of a protein structure. In the analysis of the experimental data, we include aspects of the known structure of ubiquitin in a multiscale computational approach and thus eliminate many of the approximations used in the previous analysis. Direct calculations of PREs for a protein are problematic due to the inverse sixth power dependence on the distance between the probe and a hydrogen on the protein. We adopt a multiscale computational strategy combining molecular dynamics (MD) simulations and Brownian dynamics (BD) simulations on the fields from MD calculated with a well-known potential model25 to evaluate terms in the theoretical analysis of the experimental data. We test the effectiveness and accuracy of the approximations and methods of analysis in determination of electrostatic potentials in the near-surface zone of ubiquitin in total ionic strengths up to 745 mM. We explore the discrepancy of the potentials between the experiment and simple model computations by studying possible effects on the interpreted potentials from sources including protein conformational flexibility and correlations with mobile ions, water, and charged probe orientations.
THEORY
The PRE rate arising from paramagnetic cosolutes for a transverse 1H magnetization (Γ2) previously derived is1,26
| (1) |
where ξ is a parameter involving 1H nuclei of a given protein and electron gyromagnetic ratios (see ref 1 for explicit expression), cP is the concentration of the paramagnetic probe, τc is the effective correlation time for the dipole–dipole interaction between the probe’s unpaired electron and the 1H nucleus, r is the distance between the paramagnetic center of the cosolute and the 1H nucleus, ⟨r−6⟩represents the ensemble average of r−6, W(r) is the angle-averaged potential of mean force between the probe cosolute and the 1H nucleus along r, kB is the Boltzmann constant, and T is the absolute temperature.
The integral in eq 1 may be discretized on a three-dimensional (3D) lattice grid surrounding the macromolecule:1
| (2) |
where i indexes the grid points around the solute to be probed and Δv is the volume of the voxels.
Since only the distance between the unpaired electron in the spin probe and the nucleus being excited is required, one could employ a simple spherical model of the probe tuned to obtain the distance dependence.1 The spin probe usually has nontrivial orientational correlations with the target molecule, which in part affects the potential of mean force or distribution function with respect to distance from the solute. Thus, in some strongly correlated cases, the PRE interpretation may require the potential of mean force, W, with all degrees of freedom between the probe molecules and the target.
A complete model would consider the full complement of intermolecular interactions including the electrostatic interactions, the combination of which causes the correlations between the PRE probe molecule, solvent, and the protein solute. In general, two rigid bodies have six degrees of freedom between them. The full pair intermolecular probability distributions, , are related to the potential of mean force W, which includes both the direct potential, U, and the indirect or solvent induced environmental influences, ΔW.
| (3) |
in which the subscript “1” denotes the target (here a protein), the subscript “2” denotes other solvent species such as the probe molecules, β is 1/kBT, and Ωi represents the orientation of the molecules. We note that while U may be a straightforward pairwise additive form as often found in molecular mechanics intermolecular potentials, the indirect contributions are generally not a simple sum over pairs of molecules.
From eq 1, we write
| (4) |
Integrating over the angles modifies the probability distribution with respect to the distance r versus a system with no angle dependence such as a sphere.
We may convert to a coordinate system which fixes the protein in a specific orientation at the origin. Probe orientations affect the distribution of distances, r, so we require the full six-dimensional distribution for probe molecules around the protein. We use a Cartesian grid around the protein (x, y, z), and at each spatial grid point, we have three Euler angles to fully describe the position and orientation of the molecules in the solvent (water, ions, and probes) with respect to the protein. Thus, eq 4 is reduced to
| (5) |
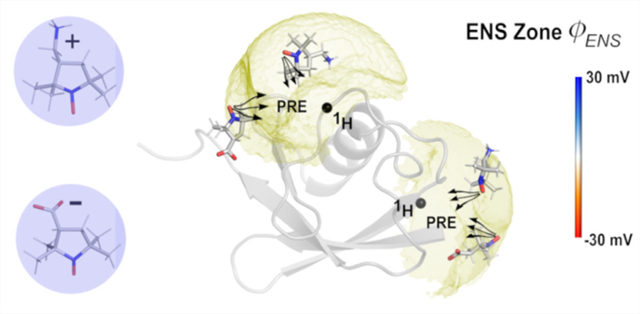
For a probe with nontrivial orientational correlations with the protein, we must sample the angular dependence at each grid point. The value of Γ2 is proportional to the integral of the inverse sixth power of distance between the probe and the target nucleus, so the Γ2 rate is dominated by terms in a zone close to the observed 1H nucleus. We have referred to this as the effective near-surface (ENS) zone.1 To consider the near-surface electrostatic potential, the experiment is run with two different probes of opposite charge. Then at any grid point surrounding the protein we can compute the ratio of the PRE rates of the cationic (+) and anionic (−) probes
| (6) |
In the case of assuming spherically shaped probes with a point charge,1 the excluded volume effects in ΔW between probes and protein cancel by using an identical radius of exclusion in the sum. We define an effective cation potential of mean force, , in the near-surface proximity zone voxels νENS as
| (7) |
where C is the normalization for the zone. If the potential of mean force is relatively constant within a given proximity zone (denoted with ′), which assumes that the effects of the averaged angular orientations are equal as well as the indirect potentials for both the cationic and anionic probe, then we obtain a simple approximation for the average electrostatic potential in the near-surface region
| (8) |
where is the effective Coulomb potential energy between the probe and the protein in the effective near-surface zone, e is the elementary charge, and ϕENS is the corresponding effective electrostatic potential. Only if the steric and indirect parts of the potential of mean force between the probes and the protein cancel does this approximation hold and allow a simple structure-free analysis of the underlying macromolecular electrostatics. This also assumes that the probe molecules, cation, or anion correlations with the protein hydrogens of interest are dominated by electrostatics. We can consider the dominant contributions of the indirect potential as the dielectric response of the solvent and ion screening.
Previous analysis of ϕENS potentials showed good agreement for most residues of ubiquitin between the experiment and PB theory using simple spherical models for the probe molecule’s exclusion zone.1 In this study, we raise the resolution and details of the calculations of electrostatic potential field using a multiscale simulation method. We evaluate the electric fields with MD simulations of ubiquitin in explicit solvent at the specified ionic strengths of the experiment. Then, we improve the calculations of ϕENS potentials by considering the angular correlation between the probe molecule and ubiquitin, which can be accomplished by performing BD simulations on the fields produced in the MD simulations.
METHODS
Protein and Other Materials for NMR Experiments.
Chemical reagents were purchased from Sigma-Aldrich unless indicated otherwise. A pET-50b-derivative plasmid harboring a synthetic gene of human ubiquitin was purchased from GenScript. Escherichia coli BL21(DE3) cells were transformed with this plasmid and were cultured at 37 °C in minimal media containing 1 g/L 15NH4Cl (Cambridge Isotope Laboratories) as the sole nitrogen source in the presence of 30 μg/L kanamycin. When the optical density at 600 nm reached 0.8 for the culture, 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to induce expression of ubiquitin. The culture was continued at 18 °C for 16 h. Ubiquitin (15N-labeled) was purified through the procedures of Sundd et al.27 and additionally, through size-exclusion chromatography using a Sephacryl S-100 column (GE Healthcare) equilibrated by a buffer of 100 mM ammonium acetate at pH 7.0. The purified 15N ubiquitin was lyophilized and kept at −20 °C until use.
NMR Samples.
Solutions of 0.4 mM 15N ubiquitin in a buffer at pH 7.5 containing 20 mM acetate, 28 mM tris, 10 mM DMSO, 5% D2O, and 0, 100, 300, or 700 mM KCl were prepared for the NMR experiments. Paramagnetic samples contained 10 mM 2,2,5,5-tetramethylpyrrolidine-N-oxyl nitroxide (PROXYL) derivative (either carboxy-PROXYL or amino-methyl-PROXYL) for the 0 and 100 mM KCl samples and 25 mM PROXYL derivative for the 300 and 700 mM KCl samples. The higher PROXYL concentration was chosen for the high ionic strength samples because PRE rates are smaller when electrostatic attraction of charged PROXYL molecules to the protein is weaker (see below). The PROXYL derivatives in stock solutions were quantified as previously described.1 To ensure the expected concentration of the paramagnetic concentration, the PROXYL derivatives were added to the protein solutions at the final stage of sample preparation. For the NMR samples at 0 and 100 mM KCl, 0.5 mL of solution was sealed in standard 5 mm NMR tubes (Norell). To mitigate the adverse impact of high ionic strength on the sensitivity of NMR detection,28 thinner tubes with an outer diameter of 4 mm (Wilmad; part no. 435-PP-7) together with a 4 mm spinner turbine (Wilmad; part no. B-PEEK-4-NS) were used for the NMR samples at 300 and 700 mM KCl (0.35 mL each).
NMR PRE Measurements.
PRE rates Γ2 for 1H transverse magnetizations were measured for protein backbone 1HN nuclei of 15N-labeled ubiquitin. For each molecular system, the PRE experiments were conducted with three samples: one without any paramagnetic cosolute (diamagnetic) and the others with carboxy-(anionic) or aminomethyl-PROXYL (cationic) for each. The two time-point approach29 with a 10 ms difference was used to measure PRE Γ2 rates. The uncertainty in the Γ2 rate was estimated using the standard deviations of the noise σd and σp in the spectra for the diamagnetic and paramagnetic samples, as described.29 All NMR experiments were carried out at 25 °C using a Bruker Avance III spectrometer equipped with a QCI cryogenic probe operated at the 1H frequency of 600 MHz. The spectra were processed with the NMRPipe software.30 Spectra were analyzed using the NMRFAM-SPARKY software.31 PRE rates were calculated using the MATLAB software (Math-Works).
Determination of Effective Near-Surface Electrostatic Potentials.
Effective near-surface electrostatic potentials, ϕENS, for individual 1H nuclei were determined from Γ2,+ and Γ2,− data using eq 8. The uncertainties in ϕENS were estimated through error propagation 32 using
| (9) |
in which Γ2,+ and Γ2,− are the experimental PRE rates from aminomethyl-PROXYL and carboxy-PROXYL, respectively; σ+ and σ−, the uncertainties in Γ2,+ and Γ2,− respectively. PRE rates with statistical significance were selected using the following criteria: Γ2,+ > 3σ+, Γ2,− > 3σ−, and . Only PRE data satisfying these criteria were used for the determination of ϕENS potentials. The ϕENS data were analyzed using MATLAB scripts.
MD Simulations.
To calculate the electrostatic field around ubiquitin, four MD simulations of ubiquitin in 0, 130, 345, and 745 mM KCl salt solutions were carried out using NAMD 2.14 software33 with the CHARMM36 force field parameter set.2 The salt concentrations represent a total salt ionic strength including probes and excess salt in the experiment except for the 0 mM concentration used to check the theoretical limit of the field without salt screening. The probes were not explicitly simulated but simply counted as salt, because it would be computationally inconvenient to obtain sampling convergence of the full angularly dependent spatial correlations by direct MD simulation due to the small number of probe molecules and their slow translational and rotational diffusion combined with the inverse sixth power dependence on the distance.1 In each simulation, the initial structure of ubiquitin (Protein Data Bank (PDB) code: 1UBQ) was solvated in a box ~58.0 Å on a side containing ~6200 TIP3P34 water molecules. Because ubiquitin itself has no net charge, one simulation was set up in the absence of salt as a control. In the other three simulations, K+ and Cl− ions were randomly added to set the expected ionic strength. The resulting systems contain about ~20K atoms. The details of the systems are listed in Table S1.
Particle mesh Ewald15 was used to calculate the long-range electrostatic interactions, and van der Waals interactions were truncated at 12 Å. All bonds were constrained using the SETTLE algorithm35 with a time step of 2 fs. The temperature was controlled with Langevin dynamics with a damping coefficient of 5 ps−1. The Nosé–Hoover method36,37 with a Langevin piston was used to maintain a pressure of 1 atm. After energy minimization, the ubiquitin systems were first heated from 0 to 298.15 K, then equilibrated in the NPT (constant pressure) ensemble for 20 ns, from which one snapshot with a temperature close to 298.15 K, and a pressure close to 1 atm was chosen as the starting step for the production run in the NVT ensemble.
In the production runs, we removed the translation and rotation of the protein by introducing two collective variable restraints by means of harmonic potentials with the corresponding force constants for the positional restraint of ubiquitin as kr = 1000 kcal/(mol·Å2) and orientation of ubiquitin as kΩ = 2000 kcal/(mol·deg2). All the simulations were run for ~1.0 μs, and the trajectories were saved at an interval of 1 ps for analysis.
Electrostatic Potential Calculations.
To obtain the near-surface electrostatic potential for each residue of ubiquitin, we first calculated the electrostatic potentials on 3D grid points with a grid spacing 0.5 Å. From MD simulations, the electrostatic potential at any grid point (grid potential) was calculated using the standard Ewald summation13,14 (see the details in the Supporting Information). This avoids the problem of cutoffs in the electrostatic potential at the expense of a slight computational complexity. Sampling in the simulation reveals the time scales needed for convergence. Autocorrelation analysis of the time dependence of the backbone RMSDs of the protein from the MD simulations produces a decay time of ~50 ns. Potentials and the protein structure were calculated and averaged in several 50 ns blocks. The final grid potentials were averaged over the simulation production time. For comparisons, grid potentials from the PB theory were obtained by solving the full PB equation using APBS package9 on the MD structures on the same resolution grid.
Brownian Dynamic Simulations.
Brownian dynamics simulation was used to sample the correlations of a fully atomistic paramagnetic probe molecule with the protein. With the protein fixed at the origin, we allowed the probe molecule to move relative to the protein in the electrostatic potential field generated from the MD simulation at a given ionic strength. The locations and orientations of the probe molecule were collected from BD trajectories. In this way, the probe molecule spatial and orientational sampling is performed in a preaveraged field of the solvent. This allowed us to calculate the Boltzmann-weighted effective potential energies between the probe molecule and the protein. We note that due to the multidimensional nature of the angular correlations many forms of enhanced sampling would not be as efficient as the multiscale route chosen here using a precomputed solvation field from MD simulations sampled by BD.
For the protein structure in the BD simulations, we used an averaged structure from the MD simulation. The model structures of amino-methyl-PROXYL and carboxy-PROXYL were built and optimized using Avogadro software38 by modifying the structures of reduced analogs (Zinc409214 and Zinc156924, whose chiral carbon atoms are in the S-enantiomeric form, from the Zinc database),39,40 respectively. The parameters pertaining to the nitroxide moiety were taken from a quantum chemical study of nitroxide systems41 and CGenFF3.0.1.42,43 The structures and partial charges of aminomethyl-PROXYL and carboxy-PROXYL are shown in Figure 1.
Figure 1.
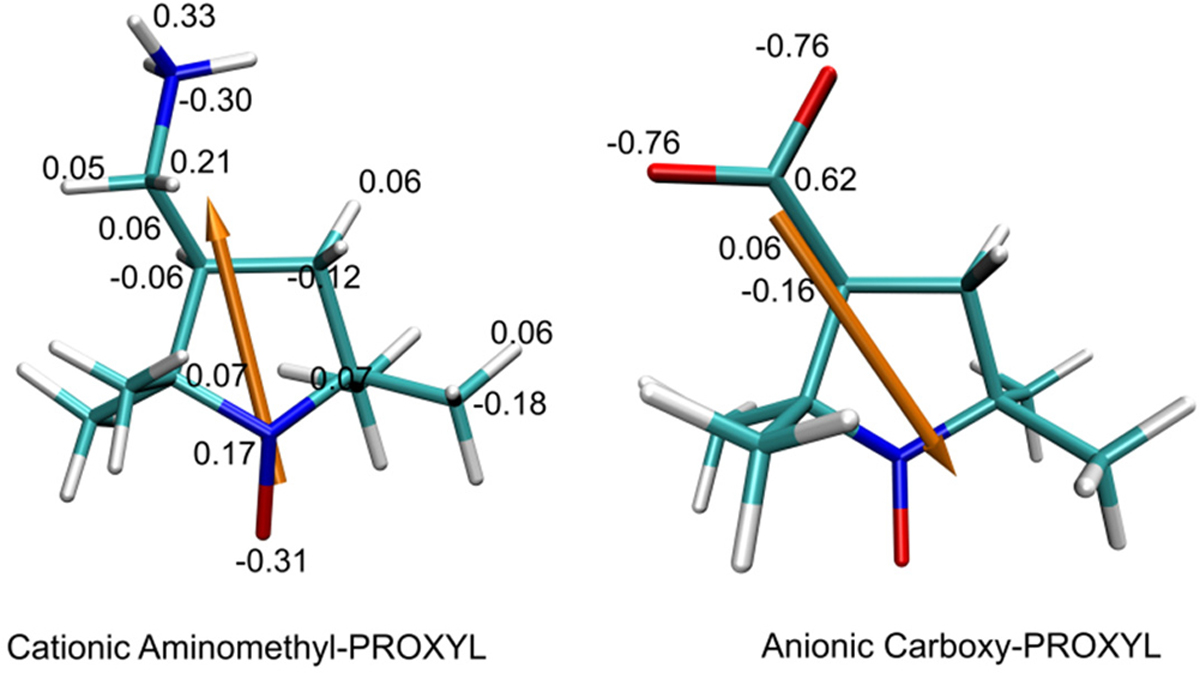
Structures and partial charges of amino-methyl-PROXYL and carboxy-PROXYL, respectively. The atoms are rendered in red for O, blue for N, cyan for C, and white for H. The orange arrow demonstrates the direction of the dipole of each probe molecule with respect to the center of charge.
We modified the software package SDA4.2344 to perform BD simulations. Only the electrostatic and repulsive exclusion forces were considered. The translational diffusion coefficient was 1.49 × 10−6 cm2/s for ubiquitin45 and 5.2 × 10−6 cm2/s for each probe.1 The rotational diffusion coefficients were estimated to be 4.344 × 10−5 radian/ps for ubiquitin and 1.850 × 10−3 radian/ps for each probe from the Stokes−Einstein relations. Each run of the BD simulations starts with the probe randomly placed and oriented at a center-to-center distance of 40 Å and is terminated when the probe moves outside of a center-to-center distance of 60 Å. The time step was 2.0 ps when the distance is <30 Å and then increased linearly with a slope of 0.45 ps/Å until the probe molecule reached 60 Å. Other details of the BD simulation have been described previously.25 We carried out 20 million such BD runs here to collect averages for our analysis.
RESULTS
ϕENS Potentials Determined from NMR Experiments.
According to eq 8, the ϕENS potentials for the backbone 1HN nuclei of ubiquitin at 0, 100, 300, and 700 mM KCl were determined by measuring the Γ2,+ and Γ2,− data from NMR experiments for 15N-labeled ubiquitin. Due to ionic components other than K+ and Cl− ions, the overall ionic strengths of the analyzed samples were 30, 130, 345, and 745 mM. The 1H−15N HSQC spectra recorded for ubiquitin at different KCl concentrations were similar, though 15N chemical shifts systematically increased upon an increase in the KCl concentration (Figure S1). In our previous study, PRE rates Γ2,+ and Γ2,− for ubiquitin at 0 and 100 mM KCl were measured using 10 mM PROXYL derivatives.1 In the current study, the Γ2,+ and Γ2,− rates at 300 and 700 mM KCl were measured using 25 mM PROXYL derivatives. The higher concentration of paramagnetic probes was chosen for those salt concentrations because the PREs arising from the charged PROXYL derivatives are smaller at higher ionic strength due to weaker electrostatic attraction. The Γ2,+ and Γ2,− data are shown in Figure 2A (see also Tables S2 and S3). Differences between Γ2,+ and Γ2,− rates (|Γ2,+ − Γ2,−|) were generally smaller for 700 mM KCl, suggesting that abundant mobile ions considerably attenuate the electrostatic bias in the spatial distribution of charged probes around the protein.
Figure 2.
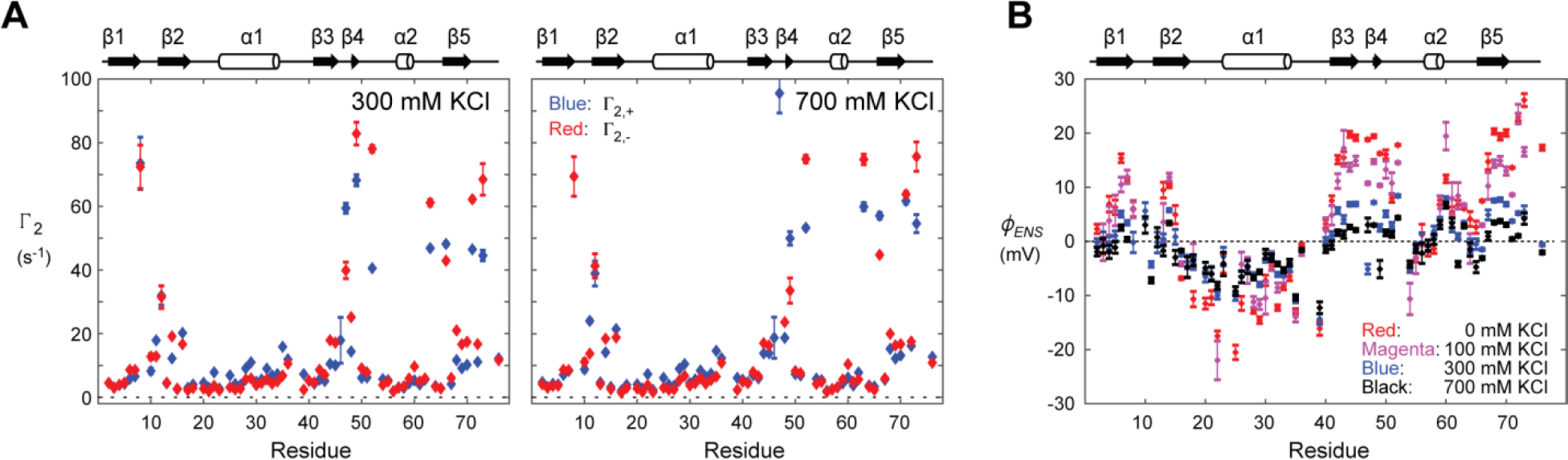
Experimental data of ϕENS potentials measured for ubiquitin at various ionic strengths. (A) PRE rates Γ2,+ and Γ2,− for ubiquitin at 300 and 700 mM KCl. (B) ϕENS potentials measured for backbone HN atoms of ubiquitin at 0, 100, 300, 700 mM KCl.
For the determination of ϕENS potentials, only PRE rates that are statistically significantly larger than zero (i.e., Γ2,+ > 3σ and Γ2,− > 3σ) were used. The ratio Γ2,+/Γ2,− should also be statistically significant and satisfy the condition indicated in the “Methods” section above. We found that these criteria are more difficult to satisfy at higher ionic strengths because of smaller PRE rates and lower NMR sensitivity at higher ionic strengths. The use of a higher concentration (25 mM) of the PROXYL derivatives was helpful to obtain a larger number of PRE data which satisfy the criteria for the analysis of ϕENS potentials at 300 and 700 mM KCl. As shown in Figure 2B, the NMR-derived data show that the overall magnitude of ϕENS potentials decreases upon an increase in ionic strength, clearly reflecting the electric-field screening effects by mobile ions.46
ϕENS Potentials from MD Simulations.
The ϕENS potentials for individual residues were determined for comparison with previous work1 using a spherical probe model with a radius of 3.5 Å. This optimal probe radius previously gave the least discrepancy between experiment and PB calculations using a crystal structure at low salt concentrations.1 Generally the structures of ubiquitin are stable and insensitive to the different ionic strengths used here (see details in the Supporting Information).
We wish to probe the electrostatic field of a protein in an aqueous saline solution. The electrostatic potential at a point in space is caused by all charges in the system. We decomposed the ϕENS potentials into two major components: one contributed from the protein itself and another contributed from the mobile ions, as shown in Figure 3A. The results show that the protein components are essentially the same within the statistical errors in different ionic strength solutions, reflecting the structural similarity and stability of the protein backbone structure over a range of solution conditions. In addition, this stability indicates that side-chain movements and low-populated conformational states have little influence on the ϕENS potential. As expected, the ionic components of the ϕENS potentials are anticorrelated with the protein component, reflecting the known screening effects of the ions on the field from the protein. We note that the protein electrostatic potential at 0 mM ionic strength, shown in Figure 3B, reflects the actual potential of our molecular mechanics model of ubiquitin without approximation. The salt screening effect becomes stronger as the ionic strength increases, consistent with the experimental observation. Including the solvent component, the contribution from all charges to the field is small and nearly featureless at this probe size, indicating that the major dielectric effects of water cancel in the ratio of eq 8 (Figure S3).
Figure 3.
MD-derived ϕENS potentials for individual residues using a spherical probe with a radius of 3.5 Å at different ionic strengths. (A) ϕENS potential components from the charges of the protein (solid line) and from the mobile ions (dash lines). (B) Total ϕENS potentials summed from the ϕENS potential components from the protein and the mobile ions.
Comparison of the MD-derived results with the NMR experimental and the continuum PB potentials at 130 mM ionic strength (Figure 4A) shows that all curves of the total ϕENS potentials follow a similar trend. We found that the MD potentials are somewhat smaller in magnitude by using the PB optimal probe size of 3.5 Å. Examination of the grid potentials around the protein (Figure S4) illustrates that the majority of the places having high magnitude potentials in the neighborhood of the protein atoms are excluded in the calculations of ϕENS potentials with such a large model probe. As a result, only positions having smaller magnitudes of potentials from the MD simulations are summed with the large probe size that best fits the experiment from the PB calculations.
Figure 4.
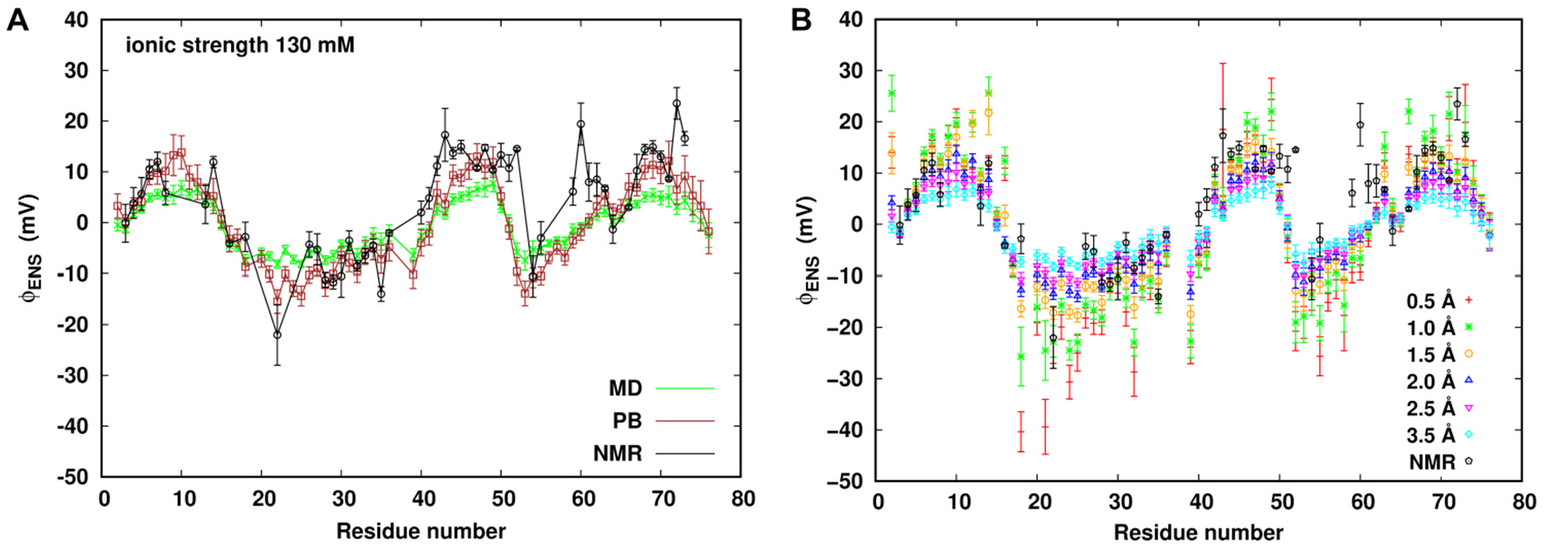
At 130 mM ionic strength, (A) a comparison of NMR-derived ϕENS potentials with MD- and PB-derived ϕENS potentials using the probe radius of 3.5 Å. (B) MD-derived ϕENS potentials dependence on the probe size compared with experiment at 130 mM ionic strength.
To further understand effects of spherical probe size on the calculated ϕENS potentials from explicit MD simulations, we calculated the ϕENS potentials using probes of radius from 0.5 to 3.5 Å, as shown in Figure 4B. We found that the unphysically smaller probe models sense stronger potential fields near the protein especially in crevices. However, in comparison with experiment the electrostatic correlations with the spherical probe model are apparently underestimated at the larger model probe radii for most residues. In addition, probe size adjustment over the range given does not improve the agreement between simulation and experiment with certain outlier residues.
Given the dependence of ϕENS potentials on probe size, we considered the fit of probe radius between simulation with experiment. We found that an effective probe radius between 1.5 and 2.0 Å produces MD potentials in better agreement with the experimental results than the larger probe sizes shown in Figure 4B. The difference in probe size required to fit the data is apparently due to the difference in the potential field produced by correlations in explicit solvent which are averaged or absent in the PB calculations.
Salt Dependence Comparison of ϕENS Potentials.
Figure 5 shows the comparison of the experimental ϕENS potentials with both MD and the PB values at 130 and 745 mM ionic strength. In this comparison, potentials derived from the MD simulation use a probe radius of 2.0 Å, and those from the PB theory use a probe radius of 3.5 Å. The comparison of ϕENS potentials in KCl-free solution and 345 mM ionic strength are displayed in Figure S5.
Figure 5.
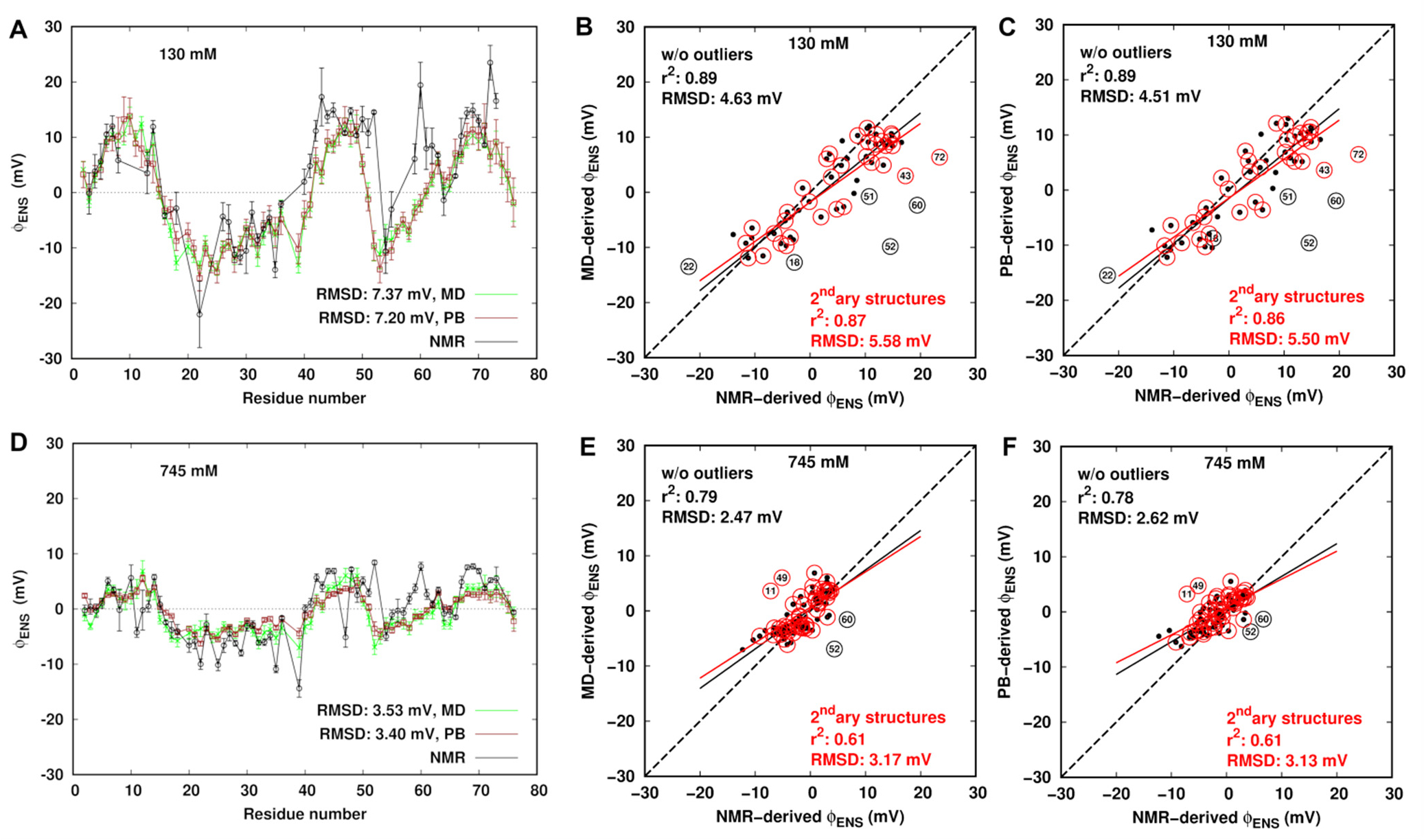
(A, D) Comparison of ϕENS potentials derived from NMR, MD simulation and PB theory at ionic strengths of 130 and 745 mM, respectively. (B, C, E, and F) Correlation plots comparing the NMR-derived ϕENS potentials and the MD- and PB-derived ones at ionic strengths of 130 and 745 mM. The outliers are labeled. The black dots represent ϕENS for the residues excluding the outliers. The red circles represent data for the residues in the secondary (secondary) structure region. The black and red solid lines represent linear regression on data points without outliers and the residues in the secondary structure, respectively. The dash line indicates predicted ϕENS = NMR-derived ϕENS.
Generally, the computed results show the same trends in all ionic strength solutions and are in good agreement (Figure 5A,D). The RMSD in ϕENS potentials between experiment and MD simulation is 7.37 mV with the correlation coefficient of r2 = 0.77 at 130 and 3.53 mV with r2 = 0.58 at 745 mM. As the ionic strength increases, the correlation diminishes slightly. The correlation plots comparing the NMR- and MD-derived ϕENS data show that the RMSD between them is 5.58 mV with r2 = 0.87 for secondary structure regions of ubiquitin at 130 mM (Figure 5B) and 3.17 mV with r2 = 0.61 at 745 mM (Figure 5E). Correlations between the NMR-derived ϕENS potentials and the computed ones are improved when only the residues in the secondary structure are considered resulting in smaller RMSD and higher r2 than using the entire original data set. Such improvement was also found in comparison between the experiment and PB theory (Figure 5C,F). Moreover, slightly better improvement is obtained if some outliers are removed from the original data points. Among these outliers, there exists a change of a sign between the calculated potentials and the experimental ones for several individual residues. The ϕENS potentials of Asp52 and Asn60 are positive in the experimental structure-free analysis, while they are negative in the MD and PB calculations in all four systems. In addition, the sign switch was also observed for Gln40 in the lowest salt solution and Lys11 at ionic strengths of 345 (Figure S5) and 745 mM.
Ubiquitin is known to have two stable conformations47–49 (see details in the Supporting Information): State1 is close to the crystal structure, and State2 has some residues shifting from the positions in State1. Such a conformational change is not large enough to give rise to a sign flip in the potentials (Figure S5A), nor did we find evidence for any changes in the pKa of the residues involved. We also found that trapping of water or mobile ions in cavities does not lead to the sign switch of ϕENS potentials for the affected residues. The protein itself generates a substantial dipolar potential field (Figure 6). Interestingly, we found that the residues having a sign switch are located close to the boundary of the positive and negative potential regions.
Figure 6.
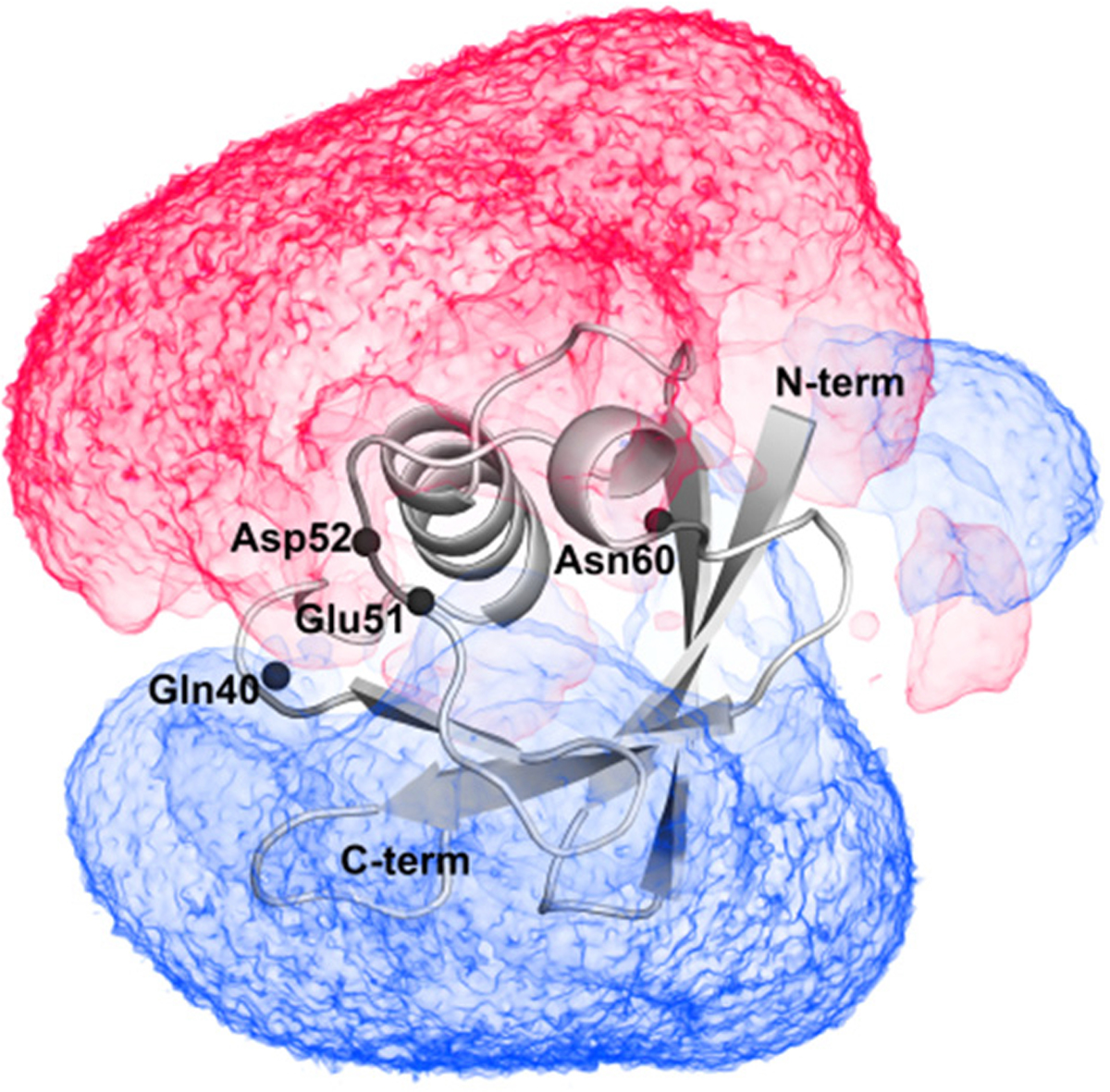
At 0 mM ionic strength, the volume map of electrostatic potentials in a range of −0.5 kBT/e (red color) and 0.5 kBT/e (blue color) around ubiquitin. Black spheres represent the backbone N atoms of labeled residues.
We have assumed a spherically averaged probe model for the exclusion zone such that we need only consider the position of the probe with respect to the HN of any individual residue of the protein. However, the interaction of direct importance to the PRE experiments is that of the unpaired probe electron spin with a given protein 1H nucleus. As shown in Figure 1, the paramagnetic probe molecule (i.e., aminomethyl-PROXYL and carboxy-PROXYL) is a nonspherical molecule, which have the unpaired electron on a nitroxide group and either a cationic or anionic group tethered to the opposite end of the molecule. The shape and charge distribution of the probe in interaction with the protein electrostatic potential field can result in significant orientational correlations. According to eq 3, the potential energy, potential of mean force, and therefore the pair distribution are six-dimensional properties dependent on not only the probe’s relative position to an HN atom but also its angular orientation in the electrostatic field. To obtain the required potential of mean force depending only on the distance between the unpaired electron center and a given protein hydrogen, the Boltzmann-weighted angular correlations must be considered. At a position close to the boundary of the positive and negative protein potentials, it can happen that the nitroxide oxygen bearing an unpaired electron and the ionic group of the probe molecule can be located in potential fields having opposite signs.
ϕENS Potentials Using Atomistic Probe Models.
We carried out BD simulations at the ionic strength of 130 mM to calculate the Boltzmann-weighted potential energies of a fully atomistic probe model with the protein in the electrostatic potential field from the MD simulation and thus determined the ϕENS potentials for individual residues. Considering the effective interactions are weak between the probe molecules and ubiquitin, 20 million runs for one BD simulation were carried out to ensure sufficient sampling in the near-surface zone. Ten independent sets of simulations were carried out for statistical error estimates.
The value of ϕENS potential depends on the ratio , and a relaxation ratio greater than 1 indicates a negative near-surface electrostatic potential. The experimental ratio is measured directly from NMR experiments without further assumptions. A predicted ratio is calculated from eq 6 with the assumption that indirect potential ΔW cancels or is sufficiently small in a voxel volume for both the probes. As shown in Figure 7A, compared to the experiment, the RMSD in the ratio by using the atomistic model is 0.74 which is smaller than 0.90 by using a spherical model. The atomistic model, without being a fit to the experimental data, thus provides better agreement in the ratio with the experimental results than the spherical model for many residues. The ratio discrepancy observed in the spherical model is corrected by consideration of orientational correlations of the probe in the atomistic model. However, for residues 16, 22, and 53, large uncertainties were found, which could be because aspects of ΔW+(r, Ω+) and/or ΔW−(r, Ω−) cannot be neglected.
Figure 7.
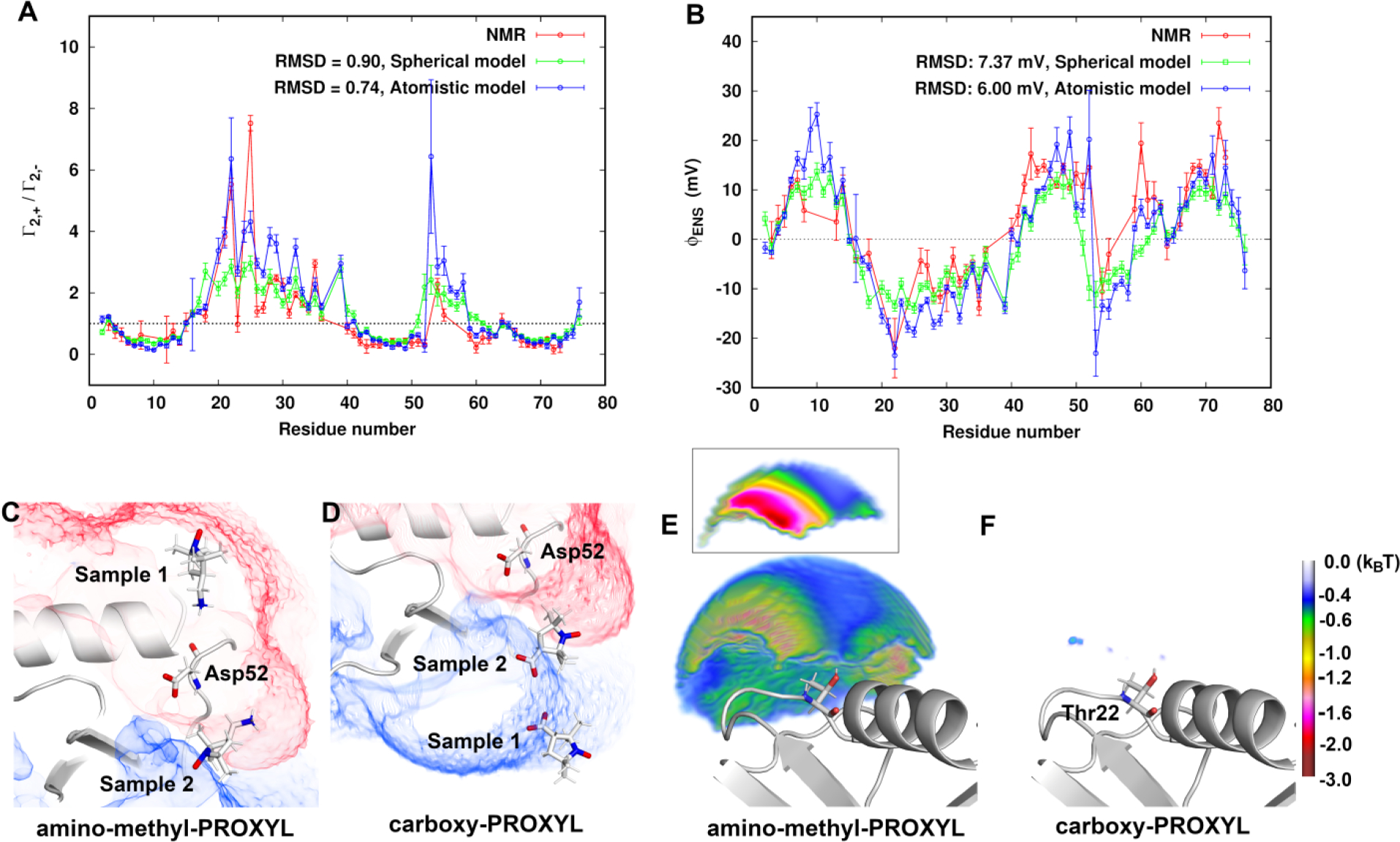
Comparison of (A) the ratio and (B) ϕENS potentials for backbone 1HN nuclei of ubiquitin using atomistic model and spherical model with the experimental results at 130 mM ionic strength. (C) and (D) display two orientational samples of amino-methyl-PROXYL and carboxy-PROXYL, respectively, at the boundary of the positive (+0.3 kBT/e in blue) and negative (−0.3 kBT/e in red) potential field around Asp52. (E) and (F) Favorable potential energies of amino-methyl-PROXYL and carboxy-PROXYL, respectively, in the near-surface zone within 10 Å from the HN atom of Thr22. The inset in (E) is a cross section showing the energy differences on the same grid points.
Figure 7B compares the predicted ϕENS potentials using the spherical and atomistic model with the NMR-derived ones. The atomistic model improves the agreement with the experiment with a smaller RMSD of 6.00 mV. In particular, for outliers such as Asp52 and Asn60, the calculated ϕENS potentials have the same sign as the experimental ones. The Boltzmann-weighted potential energies for the cationic probe and the anionic probe, show respectively, as expected, that the cationic probe is energetically favorably distributed in the negative potential field, and the anionic probe in the positive potential field (Figure S6C,D). Here we take Asp52 as an example. Far from the boundary between the positive and negative protein potential field, the whole probe is immersed in opposite field as the ionic moiety (Sample 1 around Asp52 in Figure 7C,D). However, for example, when the cationic probe is in proximity to the protein dipolar boundary, it is able to take an orientation in which the ionic end of the molecule stays in the negative potential field and the unpaired electron can report on a proton in the positive field (Sample 2 in Figure 7C). As a result, a sign switch can be observed in the estimated ϕENS potential of Asp52 from a negative value using a spherical probe to a positive value using an atomistic model having the chemical shape of the probe molecule.
Large uncertainties in ϕENS potentials were observed for residues 16, 22, 52, and 53. Similarly, in the NMR experiment, the ϕENS potentials are difficult to obtain for residues 15, 17, 20−25, 53, and 56−58 due to large uncertainties. residues 15−21, 53, and 56−58 are located near a potential boundary (Figures S7A and 6). In these boundary regions the effects of orientational correlations on the potential of mean force for both the probes cannot be exactly canceled, as discussed above. Residues 22−25 are in the deeply negative potential field (Figure S7A). Investigation of the potential energies for the cationic probe molecules in the near-surface zone within 10 Å from the HN atoms of the residues shows that the minimum potential energies for the above-mentioned residues are about −3 kBT, at least about 1 kBT lower than the other residues (Figure S7B). This indicates that the near-surface proximity zone of some residues (Thr22 as an example in Figure 7E) is not well-represented as an electrostatic mean field for the cationic probe. In addition, due to the low population of the anionic probe in the near-surface zone (Figure 7F), for both the probes cannot exactly be canceled. Therefore, the assumptions held in eq 8 cannot be properly satisfied in the assessment of ϕENS potentials from both the experiment and the calculations in the case of some residues.
DISCUSSION
In this work, we combined NMR PRE experiments and multiresolution simulations to determine the near-surface electrostatic potentials for the residues of ubiquitin in different salt solutions. Salt dependence of ϕENS potentials was studied at ionic strengths from 0 to 745 mM. The ϕENS potential results from theory and simulation are in good agreement with those from experiment for most of residues using a simple spherical model of the probe and allowing for different effective probe sizes when considering the surface accessible volumes. A discrepancy of the sign of ϕENS potentials was observed for a few particular residues in both continuum and explicit solvent calculations. We found the discrepancy is mainly caused by angular correlations of the probe which are particularly damaging to an interpretation of the electrostatic potential field for residues near the dipolar boundary of the protein. More problematic situations may arise when the proton of interest is surrounded by even more complicated multipolar fields.
While in principle the probe molecules can be included in the MD simulations, the low concentrations used would require considerable computational effort to obtain converged results. The use of Brownian trajectories of an atomistic probe model in a precomputed MD solvent potential field overcomes this limitation and produces the required angular correlation dependence.
Probe Size Dependence in Calculations Using Spherical Model.
In the calculations of ϕENS potentials from experiment, two major assumptions of the analysis are considered. The first assumption is that the correlation times of the dipole−dipole interaction between the macromolecular 1H nucleus and the unpaired electron of the probe are identical for the cationic and anionic probes. This is confirmed by the nearly identical diffusion constants for both probes.1 The second assumption is that orientation correlations and the indirect part of the potential of mean force, ΔW, approximately cancel in the near-surface proximity zone of a residue. This allows the probe exclusion volume to be modeled as a sphere with a point charge. We used a best fit radius of 3.5 Å for the probe for predictions with PB theory and a smaller radius of 2.0 Å for the MD simulations which points out differences in modeling the effects of the aqueous solution. The PB predictions used an ionic probe radius of 2.0 Å to determine the ion accessibility function and used a dielectric continuum to represent the solution. Additionally, a spherical exclusion radius of 1.4 Å was used to define the boundary between the low dielectric protein and the high dielectric solvent. All-atom MD simulations with explicit solvent molecules provide access to the detailed correlations in a heterogeneous environment, where correlations produce the charge−charge response. Moreover, accessibility of ions and solvent to the surface of the protein is controlled by nonbonded interactions. In the radial distributions of the K+ ions, Cl− ions, and water molecules from the protein vdW surface (Figure S8), the first peak of the distribution is found at 2.6, 3.2, and 2.7 Å for the K+ ions, Cl− ions, and water oxygen atoms, respectively, indicating that the mobile ions and solvent have more accessibility to the vdW surface of the protein than with the PB model. Thus, the region near the vdW surface of the protein is influenced more deeply in the MD simulation than in the PB theory. This contributes to the smaller size of the spherical probe needed to best fit the experiment in the MD simulation analyses.
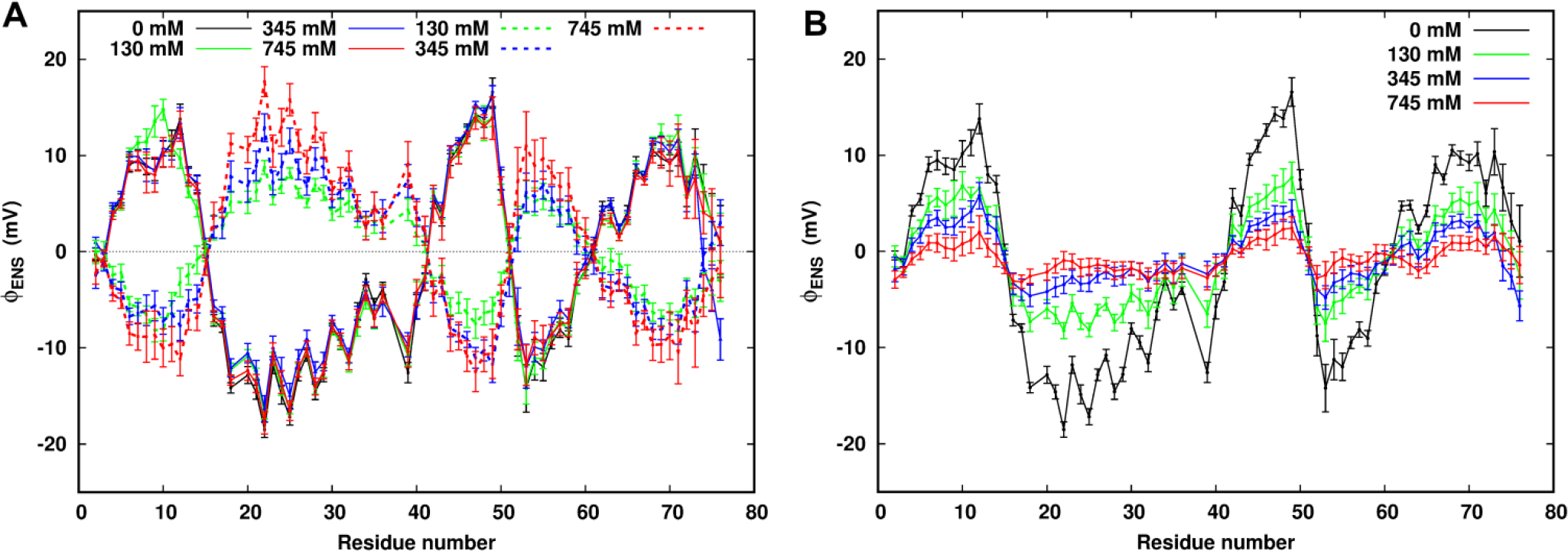
Salt Dependence of ϕENS Potentials.
Experiment (Figure S9A−C) and PB and MD simulations (Figure S9D–F) demonstrate the strong ionic strength dependence of ϕENS. As shown in Figure S9, the NMR-derived ϕENS potentials at 345 and 745 mM ionic strength were on average only 40 and 24%, respectively, of those at 30 mM ionic strength. The decrease in the magnitude of ϕENS potentials upon an increase in ionic strength is also demonstrated in Figure S9C, which shows the ionic strength dependence of ϕENS potentials for selected residues.
Compared to experiment, the PB-calculated ϕENS potentials demonstrate slightly smaller RMSDs with experiment than the MD-derived ones at 130 and 345 mM ionic strengths, but larger RMSDs at 745 mM. Due to the small number of mobile ions in the low-salt solutions in the MD simulations, more sampling is required for convergence of ion distribution, while in the high-salt solution the MD simulations should be converged and demonstrate nontrivial ion−ion correlations. At 745 mM, the ions contribute to a favorable amount of interaction energy of about −1.0 kBT in the region close to Glu16, Glu18, Asp21, and Lys29 (Figure S10). The ions not only play a role in screening the protein charges but also correlate to form an explicit interaction cluster among the residues. Nevertheless, in this case of ubiquitin, the PB equation with the large spherical probe model still provides a reasonable prediction of most residues near-surface zone potentials in solutions with an ionic strength as high as 745 mM.
Implication of ϕENS Potentials Calculated with BD from Atomistic Models.
Assessment of electrostatic potentials near a residue with this experimental technique relies on the distribution of probe molecules around the protein. The probes are nonspherical molecules each with a full charge and other multipoles dependent on origin. The atomistic models’ dipole moment with respect to the center of the molecule is 19.7 D for amino-methyl-PROXYL and 13.1 D for carboxy-PROXYL. The charge distribution controls the orientation of the probe near a dipolar boundary. Thus, the potential of mean force on the probe is not only a function of the distance of the probe with respect to the HN atoms but also a function of angular orientation. The probe’s orientational correlation determines whether the nitroxide oxygen stays in a potential field similar or opposite to the ionic moiety of the probe. As a result, far from the dipolar potential boundary the spherical probe model can report a reasonably true field. However, for some residues near the boundary (e.g., amino acids 15−21, 40, and 50−60), the neglect of orientational correlations can lead to the discrepancy of ϕENS potentials between the NMR experiments and the computations with the spherical probe model. The atomistic probe model demonstrates that the unpaired electrons on the probes are able to be in a potential field opposite to the electrostatic field sensed by the other end of the charged probe molecule. Locations of 1H nuclei that may exhibit such a discrepancy for ubiquitin are predictable to some extent through a relatively simple inspection of dipolar boundaries, as demonstrated above. Such an analysis extends to other protein hydrogens, in particular to those on aliphatic groups.50
CONCLUSIONS
In this work, we presented the determination of the ratios of PRE rates and the relation to near-surface electrostatic potentials of ubiquitin by NMR experiment and simulations in different ionic strength solutions. Decomposing contributions to the potential of mean force and the electrostatic potentials in an atomistic detail, we obtained structure-dependent yet more accurate comparisons with the experiment. We found that the angular correlation of the probe molecule with the protein cannot be neglected for 1H nuclei near the dipolar potential boundary of the protein. If the indirect effects of solvent and the effects of angular correlations are the same for the cationic and anionic probe in the near-surface zone of a residue, then we can accurately obtain a measure of the near-surface electrostatic potential with simple spherical exclusion models. The electrostatic potential is central to many applications like protein folding and molecular recognition. The advantage of the PRE rate experiment is that the method simultaneously provides the near-surface electrostatic potentials for many residues with no prior knowledge of structural information on the protein when spherical probe exclusion models may be used. This PRE rate method may help tune such model potentials in difficult cases, such as pKa shift due to change of chemical environment and electrostatic properties of intrinsically disordered protein.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Sealy Center for Structural Biology staff for help, Drs. Ka-yiu Wong and Gillian Lynch for helpful discussions, and Dr. Tianzhi Wang for maintenance of the NMR equipment. A portion of the computational research was carried out through Stampede2 and Frontera at the Texas Advanced Computing Center (TACC) at The University of Texas at Austin. Frontera is made possible by National Science Foundation award OAC-1818253. The molecular images were rendered by PYMOL.51 B.M.P. thanks NSF (CHE-1709310), NIH (R01-GM037657), and the Robert A. Welch foundation (H-013) for support. J.I. acknowledges support for the experimental components of this work supported by NIH (R35-GM130326).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.2c01611.
Details of the simulations, NMR data, and other features of the electrostatic potential fields (PDF)
Contributor Information
Chuanying Chen, Department of Biochemistry and Molecular Biology, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Binhan Yu, Department of Biochemistry and Molecular Biology, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Razie Yousefi, Department of Biochemistry and Molecular Biology, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Junji Iwahara, Department of Biochemistry and Molecular Biology, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston, Texas 77555, United States.
B. Montgomery Pettitt, Department of Biochemistry and Molecular Biology, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston, Texas 77555, United States.
REFERENCES
- (1).Yu B; Pletka CC; Pettitt BM; Iwahara J De novo determination of near-surface electrostatic potentials by NMR. Proc. Natl. Acad. Sci. U. S. A 2021, 118, No. e2104020118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Best RB; Zhu X; Shim J; Lopes PE; Mittal J; Feig M; Mackerell AD Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory. Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tian C; Kasavajhala K; Belfon KAA; Raguette L; Huang H; Migues AN; Bickel J; Wang Y; Pincay J; Wu Q; Simmerling C ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem.Theory Comput 2020, 16, 528–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jorgensen WL; Tirado-Rives J The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- (5).Oostenbrink C; Villa A; Mark AE; van Gunsteren WF A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [DOI] [PubMed] [Google Scholar]
- (6).Sharp KA; Honig B Electrostatic interactions in macromolecules: Theory and applications. Annu. Rev. Biophys. Biophys. Chem. 1990, 19, 301–332. [DOI] [PubMed] [Google Scholar]
- (7).Fogolari F; Brigo A; Molinari H The Poisson-Boltzmann equation for biomolecular electrostatics: A tool for structural biology. J. Mol. Recognit 2002, 15, 377–392. [DOI] [PubMed] [Google Scholar]
- (8).Fogolari F; Zuccato P; Esposito G; Viglino P Biomolecular electrostatics with the linearized Poisson-Boltzmann equation. Biophys. J. 1999, 76, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Baker NA; Sept D; Joseph S; Holst MJ; McCammon JA Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Onsager L Electric moments of molecules in liquids. J. Am. Chem. Soc. 1936, 58, 1486–1493. [Google Scholar]
- (11).Hünenberger PH; van Gunsteren WF Alternative schemes for the inclusion of a reaction-field correction into molecular dynamics simulations: Influence on the simulated energetic, structural, and dielectric properties of liquid water. J. Chem. Phys. 1998, 108, 6117–6134. [Google Scholar]
- (12).Hummer G; Pratt LR; Garcia AE Free energy of ionic hydration. J. Phys. Chem. 1996, 100, 1206–1215. [Google Scholar]
- (13).Ewald PP The calculation of optical and electrostatic grid potential. Ann. Phys. 1921, 369, 253–287. [Google Scholar]
- (14).de Leeuw SW; Perram JW; Smith ER Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. London A 1980, 373, 27–56. [Google Scholar]
- (15).Darden T; York D; Pedersen L Particle mesh ewald: An N· log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- (16).Greengard L; Rokhlin V A fast algorithm for particle simulations. J. Comput. Phys. 1987, 73, 325–348. [Google Scholar]
- (17).York D; Yang W The fast Fourier Poisson method for calculating Ewald sums. J. Chem. Phys. 1994, 101, 3298–3300. [Google Scholar]
- (18).Tsirelson VG; Avilov AS; Lepeshov GG; Kulygin AK; Stahn J; Pietsch U; Spence JCH Quantitative analysis of the electrostatic potential in rock-salt crystals using accurate electron diffraction data. J. Phys. Chem. B 2001, 105, 5068–5074. [Google Scholar]
- (19).Chattopadhyay A; Boxer SG Vibrational stark effect spectroscopy. J. Am. Chem. Soc. 1995, 117, 1449–1450. [Google Scholar]
- (20).Bai Y; Greenfeld M; Travers KJ; Chu VB; Lipfert J; Doniach S; Herschlag D Quantitative and comprehensive decomposition of the ion atmosphere around nucleic acids. J. Am. Chem. Soc. 2007, 129, 14981–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schutz CN; Warshel A What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins 2001, 44, 400–417. [DOI] [PubMed] [Google Scholar]
- (22).Nielsen JE; Gunner MR; Garcia-Moreno E, The pKa cooperative B: A collaborative effort to advance structure-based calculations of pKa values and electrostatic effects in proteins. Proteins 2011, 79, 3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hass MA; Mulder FA Contemporary NMR studies of protein electrostatics. Annu. Rev. Biophys 2015, 44, 53–75. [DOI] [PubMed] [Google Scholar]
- (24).Kougentakis CM; Grasso EM; Robinson AC; Caro JA; Schlessman JL; Majumdar A; Garcia-Moreno EB Anomalous properties of Lys residues buried in the hydrophobic interior of a protein revealed with (15)N-detect NMR spectroscopy. J. Phys. Chem. Lett. 2018, 9, 383–387. [DOI] [PubMed] [Google Scholar]
- (25).Chen C; Pettitt BM The binding process of a nonspecific enzyme with DNA. Biophys. J. 2011, 101, 1139–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Okuno Y; Szabo A; Clore GM Quantitative interpretation of solvent paramagnetic relaxation for probing protein-cosolute interactions. J. Am. Chem. Soc. 2020, 142, 8281–8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sundd M; Iverson N; Ibarra-Molero B; Sanchez-Ruiz JM; Robertson AD Electrostatic interactions in ubiquitin: Stabilization of carboxylates by lysine amino groups. Biochemistry 2002, 41, 7586–7596. [DOI] [PubMed] [Google Scholar]
- (28).Voehler MW; Collier G; Young JK; Stone MP; Germann MW Performance of cryogenic probes as a function of ionic strength and sample tube geometry. J. Magn. Reson. 2006, 183, 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Iwahara J; Tang C; Clore GM Practical aspects of 1H transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 2007, 184, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (31).Lee W; Tonelli M; Markley JL NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bevington PR; Robinson DK Data Reduction and Error Analysis for the Physical Sciences, 3rd ed.; McGraw-Hill: New York, 2003; pp 75–94. [Google Scholar]
- (33).Phillips JC; Hardy DJ; Maia JDC; Stone JE; Ribeiro JV; Bernardi RC; Buch R; Fiorin G; Henin J; Jiang W; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Neria E; Fischer S; Karplus MJ Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar]
- (35).Miyamoto S; Kollman PA SETTLE: An analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar]
- (36).Nosé S A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar]
- (37).Hoover WG Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- (38).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Irwin JJ; Shoichet BK ZINC - a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sterling T; Irwin JJ ZINC 15 - ligand discovery for everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Barone V; Bencini A; Cossi M; Di Matteo A; Mattesini M; Totti F Assessment of a combined qm/mm approach for the study of large nitroxide systems in vacuo and in condensed phases. J. Am. Chem. Soc. 1998, 120, 7069–7078. [Google Scholar]
- (42).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; MacKerell AD Jr. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM allatom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yu W; He X; Vanommeslaeghe K; MacKerell AD Jr. Extension of the CHARMM General Force Field to sulfonylcontaining compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gabdoulline RR; Wade RC Brownian dynamics simulation of protein-protein diffusional encounter. Methods 1998, 14, 329–341. [DOI] [PubMed] [Google Scholar]
- (45).Altieri A; Hinton D; Byrd R Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J. Am. Chem. Soc. 1995, 117, 7566–7567. [Google Scholar]
- (46).Chazalviel JN Coulomb Screening by Mobile Charges: Application to Materials Science, Chemistry, and Biology; Springer, 1999. [Google Scholar]
- (47).Piana S; Lindorff-Larsen K; Shaw DE Atomic-level description of ubiquitin folding. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 5915–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lindorff-Larsen K; Maragakis P; Piana S; Shaw DE Picosecond to millisecond structural dynamics in human ubiquitin. J. Phys. Chem. B 2016, 120, 8313–8320. [DOI] [PubMed] [Google Scholar]
- (49).Kitahara R; Yokoyama S; Akasaka K NMR snapshots of a fluctuating protein structure: ubiquitin at 30 bar-3 kbar. J. Mol. Biol. 2005, 347, 277–285. [DOI] [PubMed] [Google Scholar]
- (50).Yu B; Pletka CC; Iwahara J Protein electrostatics investigated through paramagnetic NMR for nonpolar groups. J. Phys. Chem. B 2022, 126, 2196–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).The PyMOL Molecular Graphics System, version 2.0; Schrödinger, LLC, 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.