

Abstract
ERCC1/XPF is a heterodimeric DNA endonuclease critical for repair of certain chemotherapeutic agents. We recently identified that ERCC1- and p53-deficient lung cancer cells are tolerant to platinum-based chemotherapy. ATR inhibition synergistically re-stored platinum sensitivity to platinum tolerant ERCC1-deficient cells. Mechanistically we show this effect is reliant upon several functions of ATR including replication fork protection and altered cell cycle checkpoints. Utilizing an inhibitor of replication protein A (RPA), we further demonstrate that replication fork protection and RPA availability are critical for platinum-based drug tolerance. Dual treatment led to increased formation of DNA double strand breaks and was associated with chromosome pulverization. Combination treatment was also associated with increased micronuclei formation which were capable of being bound by the innate immunomodulatory factor, cGAS, suggesting that combination platinum and ATR inhibition may also enhance response to immunotherapy in ERCC1-deficient tumors. In vivo studies demonstrate a significant effect on tumor growth delay with combination therapy compared with single agent treatment. Results of this study have led to the identification of a feasible therapeutic strategy combining ATR inhibition with platinum and potentially immune checkpoint blockade inhibitors to overcome platinum tolerance in ERCC1-deficient, p53-mutant lung cancers.
Graphical Abstract
Graphical Abstract.
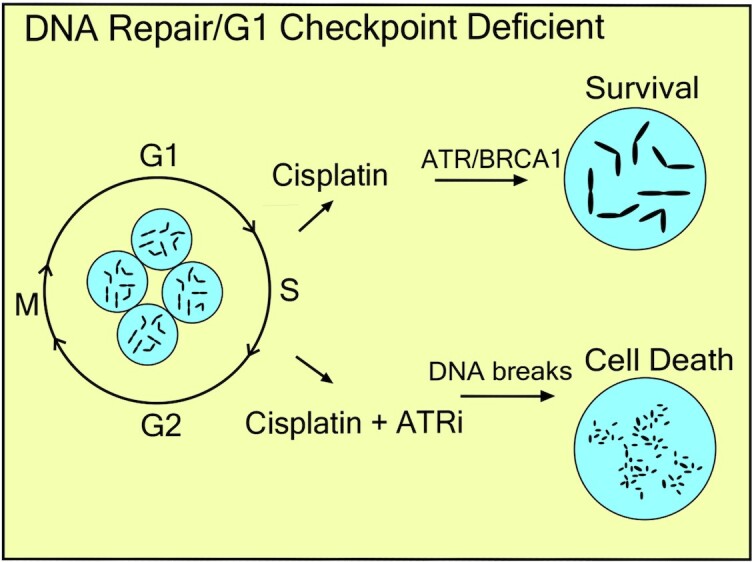
Tolerance to platinum-induced DNA damage in ERCC1 and p53 deficient lung cancers requires ATR signaling during S phase, which serves to limit global replication fork collapse and replication catastrophe.
INTRODUCTION
DNA crosslinking agents, including the platinum-based analogues remain mainstay treatments for a variety of neoplasms. These crosslinking agents function by covalently binding to guanines in the DNA thereby blocking DNA replication and inhibiting tumor cell growth. These agents form a variety of DNA lesions including monoadducts, intrastrand crosslinks and interstrand crosslinks which ultimately require different pathways for ultimate resolution of the DNA damage including Nucleotide Excision Repair, Homologous Recombination Repair and Interstrand Crosslink Repair.
Perhaps no DNA repair factor, excluding BRCA1 and BRCA2, has had more clinical interest in terms of biomarkers for response to cancer therapy than the nucleotide excision repair factor, ERCC1 (1–4). ERCC1 forms a heterodimer with the protein XPF, which constitutes a 5’-3’ structure-specific endonuclease. ERCC1/XPF has critical roles in multiple DNA repair pathways including nucleotide excision repair, interstrand crosslink repair, homologous recombination and single strand annealing. In general, it is thought that ERCC1/XPF nuclease activity is essential for repair of platinum-induced DNA damage. ERCC1 was first identified as a potential biomarker for predicting response to platinum-based chemotherapy in the late 1990s and early 2000s and up to 60% of lung adenocarcinomas and up to 30% of lung squamous cell carcinomas harbor low to undetectable ERCC1 expression (5–8). However, the clinical utility of ERCC1 expression has been hampered by problems with antibody specificity, splice variant expression and inconsistent results in retrospective clinical studies (4). Furthermore, an international Phase III clinical trial in non-small cell lung cancer (NSCLC) failed to show a survival benefit for patients with low ERCC1 who received a platinum agent (2, 9). Together, these observations suggest there remains an incomplete understanding of the biology of ERCC1 in human tumors.
Work from our lab identified a synthetic viable interaction between ERCC1 loss and p53 loss in a panel of ERCC1 knockout cell lines that was recapitulated in two separate patient data sets (10). We observed that ERCC1 deficient cell lines harboring a mutation in or that were null for p53 were tolerant to crosslinking agents including cisplatin and mitomycin C (MMC) and that tolerance was supported by DNA-PKcs and BRCA1 function as well as timely entry into S-phase following DNA damage (10). We hypothesized p53 was critical for sensing persistent, replication-associated DNA damage in the subsequent G1 phase in platinum-treated ERCC1 knockout cells and that this function of p53 is what accounted for differential phenotypes in response to DNA crosslinking agents with loss of ERCC1. However, when we deleted wildtype p53 from hypersensitive ERCC1 knockout cells, we could nearly completely rescue viability after platinum treatment, but only mildly increase clonogenicity (10). This led us to hypothesize that functional loss of p53 may be necessary but insufficient to completely account for the differences in sensitivity between ERCC1 knockout cell lines and that additional processes during replication (e.g. RPA bioavailability) may be critical for promoting clonogenicity after platinum treatment by suppressing the accumulation of replication-associated DNA damage (11).
In this study, we explored the possibility of using the ATR inhibitor M6620 to overcome platinum tolerance in ERCC1 deficient cells. Utilizing previously established ERCC1 knockout cell line models of hypersensitivity and tolerance to DNA crosslinking agents, we show that synthetic lethality between ERCC1 loss and ATR inhibition occurs in cells that are also hypersensitive to crosslinking agents. These data potentially link tolerance to DNA crosslinking agents in an ERCC1-deficient background to increased replication fork stability/protection. In support of this, we observe that tolerance to platinum and MMC with ERCC1 loss completely depends upon ATR function when p53 function is lost. Treating platinum tolerant, ERCC1 knockout cells with cisplatin did not dramatically lead to increases in DNA double strand breaks (DSBs) following treatment. However, the addition of an ATR inhibitor promoted substantial increases in DSBs following treatment and induced elevated rates of chromosome pulverization which has been associated with replication catastrophe (12–15). Furthermore, we show that M6620 treatment leads to enhanced cisplatin sensitization in platinum tolerant, ERCC1 knockout tumors in vivo. This work demonstrates the importance of ATR activity to promote tolerance to platinum-based chemotherapy in ERCC1 deficient cancers in vitro and in vivo and demonstrates that chemical inhibition of ATR kinase activity by M6620 is a potential strategy for overcoming platinum tolerance in ERCC1 deficient tumors harboring a mutation in p53.
MATERIALS AND METHODS
Cell Lines and Cell Culture
H460, H1299 and H1650 lung cancer cell lines were obtained from ATCC and were authenticated by the Karmanos Cancer Institute Biobanking and Correlative Sciences Core Facility. Cell lines were maintained in RPMI-1640 medium (Dharmacon) supplemented with 10% fetal bovine serum (Atlanta Biologicals) and 1% penicillin/streptomycin (Dharmacon) and cells were grown in a humidified incubator with 5% CO2. ERCC1 and TP53 knockout cell lines have been previously validated and published (10, 16).
Western Blot
100 μg protein was loaded onto 10% Mini-PROTEAN TGX precast gels (Bio-Rad; 456-1043) and run at 150 V for ∼40 min in Tris/glycine/SDS buffer (Bio-Rad; 1610732). Proteins were transferred onto PVDF membrane at 100 V for ∼35 min in transfer buffer (10 mmol/l CAPS, 10% methanol, pH 10.5). Membrane was blocked for 1 h with 5% non-fat milk in TBS–Tween. Proteins were probed overnight at 4°C with anti-ERCC1 (Abcam; ab76236; 1:1000), anti-XPF (Santa Cruz; sc-136153; 1:1000), or for 1 h at room temperature with anti-β-actin (Sigma-Aldrich; A5441; 1:100 000) or loading control α-tubulin (Sigma-Aldrich; T5168; 1:100 000) in antibody dilution buffer (3% w/v bovine serum albumin (BSA), 0.2% v/v sodium azide in PBS–Tween). For the ATR western blot, similar reactions were conducted with ATR antibody (Abcam; ab2905; 1:500). Excess antibody was removed by washing three times with PBS–Tween and the membrane was subsequently probed with goat anti-mouse or goat anti-rabbit secondary antibodies (Bio-Rad; 172-1011 and 172-1019; 1:2000) for 45 min at room temperature. Excess secondary antibody was removed by washing three times with PBS–Tween. PDX tumor tissue as well as resected lung tumor tissue for ERCC1 western blot assessment was obtained from the Karmanos Cancer Institute biorepository and PDX core with patient information de-identified and thus, IRB exempt.
Colony survival assay
Clonogenic survival assays were performed as previously described (10). The day prior to treatment, 300–500 cells were seeded in complete medium in 60 mm plates. The day following seeding, cells were treated for varying times depending on the drug in serum-free medium. Cisplatin (Sigma-Aldrich; 479306) was prepared daily as a fresh 1 mmol/l stock in PBS. All cisplatin treatments in clonogenic assays were performed for two hours. M6620 (Selleckchem; S7102), VX-803 (Selleckchem; S9639), MK-1775 (Selleckchem; S1525), BMN 673 (Selleckchem; S7048), KU-55933 (Selleckchem; S1092) and CHIR-124 (Selleckchem; S2683) were prepared in DMSO, and treatments were performed for 4 h. Mitomycin C (Selleckchem; S8146) was prepared in DMSO and treatments were performed for 2 h. NERx329 (compound 43) was synthesized as described (17). Once colonies reached a size of at least 50 or more cells, plates were washed once with PBS, and crystal violet was added (20% ethanol, 1% w/v crystal violet). After counting, colony assay data were plotted and IC50s estimated using Sigma Plot version 10.0 or GraphPad Prism. For synergy studies, a constant cisplatin:M6620 or cisplatin:NERx329 ratio was used based upon the approximate IC50 value for each drug in H1299 ERCC1Δ cells. CI values for synergy studies were generated in CompuSyn and data plotted in GraphPad Prism (GraphPad Software, CA).
siRNA-mediated knockdown
siRNAs for ERCC1 (catalog #: L-006311-00-0010) and ATR (catalog #: L-003202-00-0010) were purchased from Dharmacon. The control siRNA was purchased from Invitrogen (catalog #: AM4635). One the day prior to transfection, ∼100 000 cells were seeded in six-well plates. Cells were transected with 100 nM siRNA using Dharmafect 2 (Horizon Discovery; T-2002). For ATR knockdown, a single transfection was performed while double transfection was used for ERCC1 knockdown. Clonogenic assays were performed ∼48 h post-transfection.
Immunofluorescence
Cells were treated with the indicated concentrations of cisplatin (2 h), M6620 (4 h) or combination. For γH2AX foci experiments in Figure 4A, 10 μmol/l CDK1 inhibitor (RO-3306; Selleckchem; S7747) was added and immunofluorescence performed ∼16 h after treatment. Cells were fixed with 4% paraformaldehyde for 20 min at room temperature, washed once with wash buffer (0.1% Triton-X 100 in PBS), followed by permeabilization with 0.3% Triton-X 100 in PBS for 15 min. Cells were washed twice with wash buffer in PBS and blocked for 1 h at room temperature using blocking buffer (0.02% Tween 20, 5% BSA in PBS). Cells were incubated with primary antibody (anti-cGAS; Cell Signaling; D1D3G; 1:500 and/or anti-H2AX-S139; EMD Millipore; JBW301; 1:1000) for 90 min at room temperature. Coverslips were washed with wash buffer and secondary antibody was added for one hour at room temperature in the dark (Alexa Fluor 488 goat anti-mouse IgG (H+L); Life Technologies; A11029; 1:2000 and Alexa Fluor 568 goat anti-rabbit IgG (H+L); Life Technologies; A11011; 1:1200). Coverslips were washed with wash buffer and with a final rinse with PBS. Cells were incubated with ProLong Gold antifade reagent with DAPI (Life Technologies; P36931) and coverslips were sealed with nail polish. Images were taken with a Nikon epifluorescent microscope using a 40x air objective. Micronuclei were quantified by visual inspection. Images were equally adjusted for presentation purposes.
Figure 4.

Effects of dual cisplatin and M6620 treatment on cell cycle dynamics, DSB formation and induction of chromosome pulverization. (A) Cell cycle profiles following cisplatin and ATR inhibitor treatment in H1299 isogenic cells. (B) Cell cycle profiles following cisplatin and ATR inhibitor treatment ±200 ng/ml nocodazole. (C) γH2AX staining by immunofluorescence ∼22 h after treatment in H1299 ERCC1 knockout cells. (D) Representative metaphase spreads prepared from H1299 wildtype and ERCC1 knockout cells ∼48 h following treatment with 100 nmol/l cisplatin, 100 nmol/l M6620 or combination. (E) Quantification of chromosome pulverization in H1299 wildtype and ERCC1 knockout cells following treatment with 500 nmol/l cisplatin for two hours and 750 nmol/l M6620 for 4 h. (F) Representative images showing colocalization of EdU with pulverized chromosomes in H1299 ERCC1 knockout cells treated with cisplatin and M6620. (G) Quantification of normal metaphases (NM) and chromosome pulverization (i.e. replication catastrophe (RC)) and colocalization with EdU staining in untreated and cisplatin + M6620 treated H1299 ERCC1 knockout cells. All experiments were performed three times. Error bars represent ± S.D. Statistical comparisons performed using t-test; * P< 0.05; ** P< 0.01.
Metaphase spreads
For experiments utilizing chronic exposure to low dose cisplatin and M6620, cells were treated with 100 nmol/l cisplatin, 100 nmol/l M6620 or 100 nmol/l cisplatin + 100 nmol/l M6620 in complete medium daily for 2 days. Experiments utilizing VX-803 were done in a similar manner, treating cells with 100 nmol/l cisplatin, 5 nmol/l VX-803 or 100 nmol/l cisplatin + 100 nmol/l 5 nmol/l in complete medium daily for 2 days. Additionally, for those experiments involving siRNA knockdown, 24 h after transfection, cells were treated with 100 nmol/l cisplatin for 48 h in complete medium. For experiments utilizing a single concentration of cisplatin and M6620, cells were treated with 500 nmol/l cisplatin for 2 h, followed by pulse labeling with 10 μmol/l EdU for 15 min, and subsequently treated with 750 nmol/l M6620 for 4 h. Forty-eight hours after chronic or single dose treatment, cells were incubated with Karyomax Colcemid (Life Technologies; 15212012) at 0.2 μg/ml for 90 min. Cells were subsequently suspended in ice cold 0.56% KCl for 15 min at room temperature. Next, cells were fixed on ice in 3:1 methanol:acetic acid for ∼1 h. ∼20 μl was added dropwise onto slides and allowed to air dry for 30 min followed by brief heat fixation. For EdU-incubated samples, slides were rehydrated with 3% BSA in PBS, followed by a 30-minute click chemistry reaction using an AF488 EdU Click It kit (Thermo Fisher; C10337). DNA was stained with anti-fade solution containing DAPI, a coverslip was added, and slides were sealed with nail polish. Spreads were counted on a Nikon epifluorescent microscope using a 40× oil objective. Images presented in this manuscript were taken on a Zeiss LSM 780 confocal microscope using a 63× oil objective. Images were equally sharpened or contrast adjusted for presentation purposes. For quantification, 50 or more spreads were counted for each condition to determine the percentage of spreads displaying pulverization. Experiments were performed three times.
Flow Cytometry
Cell cycle: 5 × 105 cells were seeded on 10 cm plates. The following day, cells were incubated with 2 mmol/l thymidine overnight. Thymidine was removed and cells were allowed to grow for 8 h followed by the addition of 2 mmol/l thymidine. For treatment, cells were treated with 1 μmol/l cisplatin in the presence of thymidine, and then released from the thymidine block into complete medium containing either no drug or 1 μmol/l M6620 for 4 h. Samples were collected for Flow cytometry at the 4-, 22-h and 46-h timepoints. Cells were fixed in 66% ethanol and stored at 4°C for no >4 days. Cells were prepared for detection of DNA content by flow cytometry using PI/RNase Staining Buffer (BD Biosciences; 550825) as per the manufacturer's instructions (with the exception that all spin steps were performed at 1000 rpm for 3 min). Flow cytometry was performed on a BD LSR II SORP Flow Cytometer (BD Biosciences). Data were analyzed using ModFit LT (Verity Software House) and FlowJo v10 (FlowJo, LLC). Apoptosis: On the day prior to treatment, cells were seeded onto 10 cm plates. Cells were treated with either 1 μmol/l cisplatin (2 h), 1 μmol/l M6620 (4 h) or combination. ∼48 h post-treatment, cells were processed for detection of PE and Annexin-V staining using the PE Annexin V Apoptosis Detection Kit I (BD Biosciences; 559763) as per the manufacturer's instructions. Flow cytometry was performed on a BD LSR II SORP Flow Cytometer (BD Biosciences). Data were analyzed using FlowJo v10 (FlowJo, LLC).
Senescence Assays
Cells were seeded in six-well plates on the day prior to treatment. Cells were subsequently left untreated, treated with 500 nmol/l cisplatin for 2 h, 500 nmol/l M6620 for 4 h or 500 nmol/l cisplatin + 500 nmol/l M6620. Cells were allowed to grow for 6 days after which cells were fixed and subsequently incubated with X-gal substrate overnight at 37°C as per the manufacturer's instructions using a β-galactosidase staining kit (Cell Signaling Technologies). Experiments were performed twice, and images taken on a Nikon epifluorescent microscope using a 20× air objective. Images were equally adjusted for presentation purposes.
Statistical analyses for cell line studies
All experiments were performed three times unless otherwise stated. IC50 values of drug sensitivity were estimated using Sigma Plot (v.10.0) or) or calculated in GraphPad Prism and values compared by Welch ANOVA. For comparisons of plating efficiency, normalized values were quantified and compared by t-test. For metaphase spread experiments, data were compared by two-sample t-test. For DNA Fiber analysis, data were compared by one-way ANOVA and multiple comparisons testing with Bonferroni test performed using GraphPad Prism.
TCGA analysis
Utilizing eight data sets (indicated in Supplemental Figure S1) from cancer patients routinely treated with platinum-based chemotherapy, mRNA expression (RNA Seq V2 RSEM) was compared between ERCC1 and ATR and ERCC1 and CHEK1 with Spearman correlation. Data were analyzed using tools at cBIOportal.org.
DNA fiber analysis
DNA fiber analysis was used to assess DNA replication fork stability. In brief, after treating H1299 ERCC1 knockout cells with cisplatin (50 μmol/l) for 1 h, cells were labeled with 20 μmol/l IdU (Sigma-Aldrich, I7125) for 20 min followed by labeling with 200 μmol/l CldU (Sigma, C6891) for 20 min. After washing off excess CldU with PBS, cells were left untreated or incubated with 2 μmol/l M6620 for 2 h ± 100 μmol/l mirin (Sigma-Aldrich, M9948). After treatment cells were trypsinized and resuspended in PBS at a concentration of ∼1–2 × 106 cells/ml. 2 μl of cell suspension was mixed with 8 μl lysis buffer (200 mmol/l Tris–HCl pH 7.5, 50 mmol/l EDTA, 0.5% SDS) on a positively charged glass slide and incubated for ∼6 min. DNA fibers were stretched by tilting slides to a ∼30–45° angle. Slides were air dried followed by fixation with 3:1 methanol:acetic acid. For immunostaining, DNA fibers were denatured with 2.5N HCl for one hour, washed with PBS, and blocked with 5% BSA in PBS-T (0.1% Tween 20 in PBS) for 1 h. Fibers were incubated with primary antibodies in a humidified chamber at 37°C for 1 h (rat anti-BrdU (1:50, Abcam, ab6326) and mouse anti-BrdU, BD Biosciences, 347580)). Excess antibody was removed by washing three times with PBS-T. Slides were incubated with secondary antibodies at 1:100 (Alexa Fluor 488 goat anti-mouse, Life Technologies, A11029) and Alexa Fluor 594 goat anti-rat, Invitrogen, A11007) for 45 min at room temperature. Slides were washed three times with PBS-T and a coverslip was mounted with Dako fluorescent mounting medium (Agilent, S3023). Images were acquired with a Nikon epifluorescence microscope using a 40× oil immersion objective. A total of 100 replication fibers were measured per experiment in ImageJ by using pixel length values to determine ratios of CldU/IdU. The experiments were repeated at least three times.
In vivo studies
For the study, 20 female SCID mice (five mice per group) were purchased from Envigo and were maintained in accordance with protocols approved by the Wayne State University Institutional Laboratory Animal Care and Use Committee. The mice were allowed to acclimate for 1 week. Established tumors were subcutaneously implanted into the right and left posterior flank of each mouse by trochar implant. Tumor volume was defined as (width2 × length/2). Weight of the mice was also measured regularly starting on the day of implantation and through the experimental endpoint. At 3 days post-tumor implantation, the mice were treated with vehicle, M6620 at 60 mg/kg by oral gavage or cisplatin (3 mg/kg) by intraperitoneal injection following the treatment scheme described in Figure 5A. The mice were sacrificed once the combined burden of the tumors within each flank reached ∼1500 mm3.
Figure 5.

M6620-potentiated platinum sensitization in H1299 ERCC1 knockout tumors in vivo. (A) Model depicting the treatment scheme for cisplatin and M6620 in H1299 ERCC1 knockout xenograft studies. (B) Plot depicting tumor growth for each individual SCID mouse in the study over the course of each treatment as measured by tumor volume, including vehicle control, M6620 at 60 mg/kg, cisplatin at 3 mg/kg, or combination (Cage #6 data not plotted). (C) Table depicting animal study design and efficacy analysis of each drug alone and in combination.
Efficacy analysis: endpoints for assessing antitumor activity for solid tumors
Standard experimental endpoints for SC tumor models include the quantitative measures: (i) tumor growth delay (T – C), where T is median days for the treatment group tumors to reach a pre-determined size (e.g. 1000 mg), and C is median time (in days) for the control group tumors to reach the same size (tumor-free survivors are excluded from these calculations and cures are tabulated separately); (ii) tumor cell kill, where log10 cell kill total (gross; GLK) = T – C value in days/(Td) (3.32), where T – C is tumor growth delay described above (i) and Td is tumor volume doubling time estimated from the best fit line from a log-linear growth plot of the control group tumors in exponential growth (100–800 mg range). Additional qualitative measures include: (iii) % T/C value (inverse of tumor growth inhibition, %TGI), where treated/control tumors were measured when control group tumors reach ∼700–1200 mg (logarithmic growth phase). The median for each group is determined as a measure of antitumor effectiveness. T/C < 42% is considered significant activity by the Drug Evaluation Branch of NCI; T/C < 10% is highly significant activity; (iv) activity rating, to facilitate direct comparisons of activity with standard agents or between tumor models, as reflected in relative log10 kill values (by this criterion, a gross log10 tumor cell line > 2.8 is considered highly active (++++); 2.0–2.8 (+++ activity); 1.3–1.9 (++ activity); 0.7–1.2 (+ activity) and a value <0.7 is considered inactive) (18–21).
RESULTS
Differential response of ERCC1Δ cell lines to cisplatin, mitomycin C and ATR inhibition
We previously established a panel of ERCC1 knockout lung cancer cell lines and observed a differential phenotype in response to cisplatin and mitomycin C (MMC) (10). We observed hypersensitivity to DNA crosslinks in ERCC1 deficient cells only when wildtype p53 was retained (H460, H522 and A549 cell lines) while cell lines that were null for or harbored a mutation in p53 (H1299, H358, H1650 and H1703 cell lines) were significantly more tolerant to DNA crosslinks despite loss of ERCC1 (Figure 1A). This led to the identification of two subsets of ERCC1-deficient tumors: platinum-hypersensitive and platinum-tolerant (Figure 1A). Since our previous work identified DNA-PKcs, BRCA1 and timely progression into S-phase as being critical regulators of platinum-tolerance with ERCC1 deficiency, we focused our efforts on understanding whether inhibition of the DNA damage kinase, ATR, by the selective small molecular inhibitors, M6620 and VX-803 could selectively sensitize platinum-tolerant, ERCC1-deficient cells to platinum-based chemotherapy. We used previously established lung cancer models of ERCC1-deficiency for our current studies, including the platinum hypersensitive model, H460 (p53 wildtype), and the platinum-tolerant, p53 null model, H1299 (Figure 1A and B). We confirmed our previously reported observations of a differential sensitivity to the DNA crosslinking agents, cisplatin and MMC, in our ERCC1-deficient cell line models. A clear hypersensitivity to cisplatin and MMC was observed in H460 ERCC1 knockout cells while the platinum-tolerant, ERCC1-deficient H1299 model was only moderately more sensitive to cisplatin or MMC compared to H460 and H1299 ERCC1-wildtype cells (Figure 1C and D). Together these data suggest that additional mechanisms exist which either partially compensate for loss of ERCC1 or lead to a reduction of platinum-induced DNA damage when p53 function is lost.
Figure 1.

Differential sensitivity of ERCC1-knockout cells to cisplatin. (A) Summary of previously established cell line models of ERCC1 deficiency. (B) Western blot depicting ERCC1 and XPF expression in H460 and H1299 ERCC1 knockout cell lines. Sensitivity of H460 (p53 wildtype) and H1299 (p53 null) isogenic cell lines to (C) cisplatin and (D) mitomycin (E), sensitivity of H460 and H1299 isogenic cell lines to the ATR inhibitor, M6620 and (F) the Chk1 inhibitor, CHIR-124. All clonogenic assays are presented as the average of three independent experiments ± standard deviation. Statistical comparisons were performed by comparing IC50 values by Welch ANOVA with Dunnett T3 test for multiple comparisons; **** P< 0.0001, *** P< 0.001, ** P< 0.01, * P< 0.05, n.s. P> 0.05. IC50 values are presented as mean ± SEM.
There are mixed reports that ATR inhibition is synthetic lethal with loss of ERCC1, so we tested whether there were differences in response to the ATR inhibitor, M6620 and the Chk1 inhibitor, CHIR-124, between the platinum-hypersensitive and platinum-tolerant ERCC1 knockout cell line models (22–24). We observed that the H460 ERCC1 knockout cells were also sensitive to both ATR and Chk1 inhibition, however there was no synthetic lethality with ATR or Chk1 inhibition in the H1299 ERCC1 knockout cells, suggesting there are compensatory processes which not only render these cells tolerant to DNA crosslinking agents, but also to inhibitors of the ATR pathway (Figure 1E and F). While there was clear differential sensitivity between ERCC1 knockout cell lines in response to ATR inhibition, the sensitization of H460 ERCC1 knockout cells to Chk1 inhibition did not translate into a clear differential phenotype between H460 (p53 wildtype) and H1299 (p53 null) ERCC1 knockout cells (Figure 1E and F). Previous work identifying a synthetic lethal relationship between ATR inhibition and ERCC1-deficiency proposed in the absence of ERCC1 there was increased reliance upon ATR-mediated signaling to respond to damage associated with loss of ERCC1/XPF endonuclease activity (24). Interestingly, the cell line utilized for these studies was A549 lung cancer cells which is p53 wildtype and consistent with the data we observed in H460 p53 wildtype lung cancer cells. In line with the hypothesis that there is increased reliance upon ATR-mediated signaling for platinum tolerance in ERCC1 deficient cancers, we asked if there was a correlation between tumoral ERCC1 and ATR or Chk1 mRNA expression in tumors commonly treated with platinum-based chemotherapy. Utilizing eight TCGA patient data sets of tumor types commonly treated with platinum-based chemotherapy, we observed moderate inverse correlations between tumoral ERCC1 mRNA and ATR mRNA suggesting ATR activity may be generally important for compensating for loss of ERCC1 (Supplemental Figure 1). Interestingly, no notable correlations were observed between ERCC1 mRNA and Chk1 mRNA in the same data sets, suggesting either Chk1 expression may not be strongly controlled at the mRNA level in the absence of ERCC1 or that Chk1 activity may not be as critical as ATR in the absence of ERCC1 (Supplemental Figure 1). Together these data may indicate certain compensatory mechanisms exist in tolerant ERCC1 knockout cells to deal with endogenous damage that accumulates as a result ERCC1 loss.
ATR inhibition selectively sensitizes platinum-tolerant, ERCC1-deficient cells to cisplatin
Because our previous work suggested timely entry into S-phase was critical for platinum tolerance with loss of ERCC1, we tested whether ATR inhibition could sensitize platinum tolerant, ERCC1-deficient cells to cisplatin. Utilizing the ATR inhibitor, M6620, we observed a striking sensitization to cisplatin in H1299 ERCC1 knockout cells (1.50 μmol/l versus 0.19 μmol/l) (Figure 2A and C). Conversely, in cells already hypersensitive to cisplatin, the addition of an IC50 concentration of ATR inhibitor did not further sensitize to cisplatin (0.090 μmol/l versus 0.085 μmol/l) (Figure 2B and C). Consistent with this observation, we saw a similar pattern with MMC, where ERCC1-deficient cells tolerant to MMC could be sensitized by ATR inhibition without further enhancing MMC sensitivity in ERCC1 knockout cells already hypersensitive (Supplemental Figure 2A–D). We previously demonstrated that knockout of p53 (p53*) in hypersensitive H460 ERCC1 knockout cells could partially increase tolerance to cisplatin (10). Next, we asked whether H460 ERCC1 knockout/p53* cells could be re-sensitized to cisplatin by ATR inhibition. Indeed, we observed the increased platinum tolerance in H460 ERCC1 knockout/p53* cells could be overcome by ATR inhibition and H460 ERCC1 knockout/p53* cells were re-sensitized to the same level as H460 ERCC1 knockout/p53WT cells (Figure 2D and E).
Figure 2.

ATR inhibition overcomes platinum tolerance in a p53-null model of ERCC1 deficiency. Platinum tolerance with ERCC1 deficiency is overcome by inhibition of ATR. (A) Sensitization of H1299 (p53 null) ERCC1 knockout cells to cisplatin by M6620 treatment. IC50 values compared by t-test, **** P< 0.0001. (B) Lack of sensitization of H460 ERCC1 knockout cells by M6620 treatment. IC50 values were compared by t-test, n.s., not significant. (C) Effect on plating efficiency of H1299 and H460 ERCC1 knockout with the concentration of M6620 utilized in sensitization experiments. (D) Sensitization of H460 ERCC1 knockout/p53* cells to cisplatin by M6620 treatment representing two independent experiments (*P< 0.05). (E) Effect on plating efficiency of H460 ERCC1 knockout/p53* cells with the concentration of M6620 utilized in sensitization experiments. (F) Apoptotic cell death detected ∼48 h after treatment with 1 μmol/l cisplatin, 1 μmol/l M6620 or combination by 7AAD and PE-Annexin V staining and flow cytometry. Data is representative of two individual experiments. (G) b-Galactosidase staining in H1299 wildtype and knockout cells 6 days after treatment with 500 nmol/l cisplatin, 500 nmol/l M6620 or combination. Data is representative of two individual experiments.
ERCC1 has been shown to be nearly undetectable in 25–50% of lung cancer patient samples via IHC (8). ERCC1 has been shown to be variably expressed across lung tumor samples while being important for nucleotide excision repair (NER) in normal cells. Consistent with these observations, we demonstrate that ERCC1 is expressed at varying levels (e.g. very low/undetectable levels) in ∼25% of the tumor tissue derived from patient-derived xenografts (PDX) as well as resected lung cancer samples (Supplemental Figure 2E and F). These results highlight the clinical significance of utilizing ERCC1 knockout models which are a cleaner and more consistent model than transient knockdown models. We have previously shown that ERCC1 levels correlate with response to cisplatin treatment with higher levels of protein knockdown (e.g. >85%) (25). To confirm the synthetic lethal relationship between cisplatin and ATR inhibition we observe in ERCC1/p53-deficient cells is also applicable to cells with reduced ERCC1 expression, we performed combination treatment following transient ERCC1 knockdown (Supplemental Figure 2G and H). These results demonstrate that ERCC1 knockdown and knockout both yield significant hypersensitivity to combination treatment (Supplemental Figure 2G and H). Additionally, we confirmed this relationship in a second model of platinum tolerance with ERCC1 loss, H1650 ERCC1 knockout cells which harbor a X225_splice mutation in TP53. Once again, we observed ATR inhibition overcomes platinum tolerance in ERCC1-deficient cells (Supplemental Figure S3A). To ensure the effect is clearly due to targeting ATR, we utilized two additional approaches including a second ATR inhibitor, VX-803 (Supplemental Figure 3B) as well as siRNA-mediated ATR knockdown (Supplemental Figure 3C and D). In both instances, we observed enhanced sensitivity in combination with cisplatin treatment consistent with the results seen in H1299 ERCC1 knockout cells with cisplatin and M6620.
Furthermore, we detected increases in induction of apoptotic cell death in H1299 WT and ERCC1 knockout cells at 48 hours post-treatment by Annexin-V/7AAD staining, but apoptosis was not enhanced in knockout cells relative to wildtype cells (Figure 2F). This led us to hypothesize that in the absence of a robust ability to induce apoptosis with p53 loss, dual treatment promotes induction of cellular senescence. Consistent with this hypothesis, we observed increases in β-galactosidase staining in ERCC1 knockout cells following cisplatin and M6620 treatment 6 days after treatment (Figure 2G). These results are consistent with previous observations that ATR inhibition can selectively sensitize p53 mutant chronic lymphocytic leukemia cells to chemotherapy, although we are the first to report a specific benefit of ATR and platinum combination treatment in p53 deficient cells that are also deficient for ERCC1 (26). These data also suggest the relationship between ERCC1 loss and p53 in terms of platinum sensitivity may be related to levels of replication associated DNA damage which in turn leads to senescence.
Multiple protein targets have been studied in the context of sensitizing tumors to platinum-based chemotherapy, therefore, we tested whether the effects of ATR inhibition were independent of ATM inhibition. The addition of either 10 or 25 μmol/l KU-55933 did not enhance cisplatin sensitivity in H1299 ERCC1 knockout cells (Supplemental Figure S4A and B). These data confirm platinum-tolerance with ERCC1 relies specifically upon ATR function and is not related to generalized inhibition of DNA damage kinase activity. Since PARP inhibitors have also entered clinical trials in combination with platinum-based chemotherapy, we asked whether ATR inhibition was a stronger sensitizer of platinum-tolerant, ERCC1-deficient cells to cisplatin than the PARP inhibitor, BMN-673. BMN-673 weakly enhanced sensitivity to cisplatin in H1299 ERCC1 knockout cells and did not approach the level of sensitization induced by ATR inhibition (Supplemental Figure S4C and D). Finally, we asked whether inhibiting the ATR target Chk1 or inhibiting the G2/M checkpoint kinase Wee1 could also sensitize H1299 ERCC1 knockout cells to cisplatin. Somewhat surprisingly, we observed no increased sensitivity to cisplatin in H1299 ERCC1 knockout cells when Chk1 or Wee1 were chemically inhibited, suggesting that ATR-mediated platinum sensitization in this specific context may be independent of Chk1 or Wee1 kinase activity (Supplemental Figure S4E–H). These data would be consistent with previous observations described in the context of BRCA-mutant ovarian cancer cells where ATR inhibition did not disrupt Chk1 (auto)phosphorylation and Chk1 inhibition did not sensitize to cisplatin (27).
ATR or RPA inhibition in platinum-treated, ERCC1-deficient cells induces widespread DNA resection mediated by Mre11 and exacerbated by CDK2
Next, we hypothesized that ATR inhibition induces replication fork stalling at platinum-DNA adducts followed by subsequent DNA resection. To test this hypothesis, we used DNA fiber assays to monitor resection in the presence or absence of cisplatin (50 μmol/l) and M6620 (2 μmol/l) in H1299 ERCC1 knockout cells. The experimental design is pre-treatment with cisplatin, which induces DNA lesions, which will stall replication forks in the absence of ATR signaling, while the addition of the ATR inhibitor after the second nucleoside analog would enable us to directly monitor resection at stalled/broken replication forks. We found cisplatin treatment alone did not lead to noticeable fork degradation in these cells (Figure 3A). However, the addition of M6620 led to a significant increase in nascent strand degradation in cisplatin-treated cells (Figure 3A). We also identified Mre11 exonuclease activity as essential in promoting strand degradation in this context. Pharmacological inhibition of Mre11 by mirin (200 μmol/l) restored fork stability to the same level as observed in cells treated with cisplatin alone (Figure 3A). As excess origin firing is a common event occurring in cells treated with a DNA damaging agent plus an ATRi (28), we asked whether an increase in origin firing is associated with the strand degradation observed in platinum and ATRi-treated cells. After cells were treated with cisplatin, the CDK2 inhibitor, Roscovitine, was added prior to ATRi to limit new origin firing. Following all treatments, we observed a significant restoration of fork stability upon inhibition of new origin firing (Figure 3B). These data suggest in platinum-treated, ERCC1 deficient and p53 null cells, ATR activity is critical to support replication fork stability by limiting Mre11-dependent strand degradation and CDK2-mediated origin firing, possibly by limiting RPA depletion in these cells.
Figure 3.

Pharmacological inhibition of ATR or RPA induces fork instability after platinum treatment in p53-null, ERCC1-knockout cells. (A) Results from DNA fiber analysis depicting the effects of platinum, M6620 or combination treatment ± Mre11 inhibition on DNA end resection. Data presented are combined from three individual experiments (100 fibers analyzed per experiment; 300 fibers total) data analyzed by ANOVA with Bonferroni test for multiple comparisons (**** P< 0.0001). (B) Results from DNA fiber analysis depicting the effects of CDK2 inhibition on DNA end resection upon platinum, M6620 or combination-treatment (statistical analysis is the same as in A). (C) Chemical structure of NERx329, an inhibitor of RPA-ssDNA binding. (D) Clonogenic survival assay displaying the effects of RPA inhibition on sensitivity of H1299 wildtype and ERCC1 knockout cell lines to cisplatin. Data are presented as the average of three independent experiments, ± S.D. Statistical comparisons were performed by Welch ANOVA with Dunnett T3 test for multiple comparisons. **** P< 0.0001, ** P< 0.01. (E) Colorimetric plot depicting synergy or lack of synergy between cisplatin and M6620 or NERx329 in H1299 isogenic cell lines. Drug ratios listed on the x-axis indicate cisplatin:M6620 and cisplatin:NERx329 ratios used for synergy testing. (F) Results from DNA fiber analysis depicting the effects of platinum, NERx329 or combination treatment ± Mre11 inhibition on DNA end resection. Data presented are combined from three individual experiments (100 fibers analyzed per experiment; 300 fibers total) data analyzed by ANOVA with Bonferroni test for multiple comparisons (**** P< 0.0001).
Because RPA–ssDNA binding is essential for ATR activation at stalled replication forks, the data suggested RPA availability may be key to understanding how ATRi potently sensitized ERCC1/p53-deficient cells to cisplatin. We asked whether decreased RPA availability via chemical inhibition of RPA-DNA binding could also enhance platinum sensitivity of H1299 ERCC1 knockout cells to the same extent as ATR inhibition (Figure 3C) (17,29). Similar to ATRi, RPA inhibition (RPAi) by NERx329 sensitized H1299 ERCC1 knockout, but not wildtype cells, to cisplatin ∼10-fold (Figure 3D). Using a variety of ratios between RPAi and cisplatin, we found that RPAi-mediated sensitization of these cells to cisplatin was synergistic in H1299 ERCC1 knockout cells, similar to the interaction between ATRi and cisplatin (Figure 3E). Alternatively, in H1299 wildtype cells, RPAi was only additive or antagonistic, suggesting the observed effects are more specific to cells harboring low ERCC1 expression. Because RPA binding to ssDNA is key to ATR activation, we asked whether RPA inhibition also led to strand degradation in cisplatin treated, ERCC1 knockout cells. We observed a significant increase in strand degradation when RPA–ssDNA binding was inhibited with 50 μmol/l NERx329. Furthermore, this defect was rescued by concurrent treatment with an Mre11 inhibitor (Figure 3F). Together these data suggest in ERCC1-deficient cells, ATR activity is essential for regulating replication fork stability at platinum-DNA adducts via inhibition of excess origin firing in order to preserve RPA pools and protect single-stranded DNA. In ATRi treated cells, excess origin firing leads to widespread accumulation of stalled replication forks which require RPA for stabilization and to inhibit replication fork collapse. Thus, in the absence of ERCC1 and upon cisplatin and ATRi treatment, excess origin firing promotes widespread fork stalling leading to an accumulation of ssDNA which eventually gives way to RPA exhaustion and global replication fork collapse.
Dual treatment with cisplatin and M6620 enhances DSB formation and induces replication catastrophe
It is well-established that ERCC1 deficient cells strongly arrest in G2/M phase following treatment with DNA crosslinking agents. We next asked what effects M6620 had on cell cycle arrest and checkpoint activation in platinum-tolerant, ERCC1-deficient cells. As we suspected ATR inhibition in combination with cisplatin would lead to enhanced DNA damage compared to cisplatin alone, we hypothesized combination treatment would lead to increased cell cycle arrest in G2/M phase due to increased accumulation of DNA DSBs. To test this hypothesis, we treated H1299 wildtype and ERCC1 knockout cells with 1 μmol/l cisplatin, 1 μmol/l M6620, or combination and monitored cell cycle profiles by flow cytometry at ∼20 h post-treatment. In H1299 wildtype cells, we observed mild increases in G2/M arrest following treatment with cisplatin or combination treatment (Figure 4A). Consistent with previously reported observations, treatment of H1299 ERCC1 knockout cells with cisplatin led to G2/M arrest, but inconsistent with our initial hypothesis, combination treatment led to fewer cells arresting in G2/M (Figure 4A). While we observed fewer cells arresting in G2/M with combination treatment, we also observed the G1 peak broadened and thought it possible that either ATR inhibition was leading to arrest in S-phase or that ATR inhibition was leading to premature entry into mitosis which has been observed by other groups (30–32). To test which of these possibilities was the case, we performed the same treatments in ERCC1Δ cells and monitored cell cycle profiles in the presence or absence of 200 ng/ml nocodazole. In the presence of nocodazole, all treatment groups strongly arrested at G2/M which indicated that ATR inhibition was not leading to early S-phase arrest but to premature entry into mitosis (Figure 4B).
To further understand these events, we synchronized cells with a double thymidine block and monitored progression through the cell cycle following treatment at 4-, 22- and 46-h time points (Supplemental Figure S5A and B). In H1299 wildtype and ERCC1 knockout cells, we observed that treatment groups containing M6620 entered the subsequent G1 phase at a faster rate than untreated and cisplatin-treated groups (Supplemental Figure S5A and B). We also observed cells tended to accumulate and progress much more slowly through the subsequent S-phase indicating that cells were requiring more time for DNA replication possibly due to the persistence of replication-associated damage from the previous round of the cell cycle. In H1299 ERCC1 knockout cells, we observed cisplatin-treated cells arrested strongly at G2/M phase at the 4- and 22-h time points, but completely recovered from this G2/M arrest by 46 h post-treatment (Supplemental Figure S5B). Conversely, ATR-inhibited cells entered the subsequent G1 phase much faster than untreated control cells even in the presence of cisplatin. We also observed increases in the size of the S-phase peaks in both groups suggesting cells have difficulty progressing through the subsequent S-phase due to replication-related DNA damage persisting from the previous round of the cell cycle.
Next, we tested the effect of combination treatment on DSBs formation in platinum-tolerant ERCC1-deficient cells by monitoring γH2AX foci in cells arrested at the G2/M boundary post-treatment. Cisplatin-treated cells had very few γH2AX foci above untreated cells consistent with our previously published data (10). While the ATR inhibitor alone did not dramatically increase formation of DSBs, the combination treatment led to substantial increases in γH2AX foci (Figure 4C). Thus, ATR inhibition potentiates DSB formation or persistence after cisplatin treatment in platinum-tolerant, ERCC1-deficient cells. As ATR activity is critical for suppressing replication catastrophe after DNA damage by limiting depletion of available RPA pools, we asked whether combination treatment induced chromosome pulverization/fragmentation in metaphase spreads (13). Chromosome pulverization has been documented as a feature of replication catastrophe and refers to the dramatically fragmented appearance of chromosomes in metaphase spreads (12–15). We generated metaphase spreads following chronic treatment with cisplatin, M6620, or cisplatin and M6620 and observed that platinum-tolerant, ERCC1-deficient cells were more susceptible to chromosome pulverization than the parental ERCC1 wildtype cells (Figure 4D and E). In addition, we assessed induction of pulverized chromosomes in combination treatment with a second ATR inhibitor (VX-803) (Supplemental Figure 5C), in a second model of platinum tolerance (H1650 cells; p53 mutant and ERCC1 wildtype or knockout) (Supplemental Figure 5D), as well as upon ATR knockdown (Supplemental Figure 5E) to ensure these events are specifically related to ATR inhibition and are not specific to one cell line model. In all instances, we observed significant increases in chromosome pulverization with ATR targeting and cisplatin treatment in ERCC1 and p53 deficient cell lines. Next, we asked whether chromosome pulverization with combination treatment was specifically linked to events occurring during DNA replication. Therefore, we treated cells with cisplatin for two hours, pulse-labeled with EdU to mark actively replicating cells, followed by ATR inhibition for four hours. 48 hours post-treatment metaphase spreads were generated and stained for EdU to identify whether chromosome pulverization was enriched in cells undergoing DNA replication at the time of ATR inhibition. Compared to untreated cells, ERCC1 knockout cells positive for chromosome pulverization were significantly enriched for EdU positivity, presumably indicating that chromosome pulverization after combination treatment was specifically linked to inhibition of ATR during S-phase (Figure 4F and G). Together these data point to the importance of ATR activity during S-phase to limit replication-associated DNA damage after cisplatin treatment in ERCC1-deficient cells.
Combination treatment induces γH2AX-positive micronuclei formation
Platinum-based chemotherapy in combination with immune checkpoint inhibitors has become first-line treatment for most patients with advanced NSCLC. Next, we asked whether combination treatment with platinum and M6620 led to increased micronuclei formation in platinum-tolerant, ERCC1 knockout cells and if increased micronuclei were associated with DSBs and activation of the innate immune response. Activation of the innate immune response by cytosolic DNAs via cGAS-STING pathway has also been shown to influence response to immune checkpoint blockade inhibitors including anti-CTLA4 and anti-PD-L1 therapies (33, 34). We monitored formation of micronuclei in H1299 wildtype and ERCC1 knockout cells following treatment with cisplatin, ATRi, or combination. While we did not observe differences between wildtype and ERCC1 knockout cells in the number of cells positive for micronuclei formation, we did see a significant difference between ERCC1 wildtype and knockout cells when we quantified the number of cells harboring greater than two micronuclei (Supplemental Figure S6A & B). In the context of DNA damage, it was previously shown that micronuclei largely stain positive for γH2AX which we also observed (Supplemental Figure S6C) (33). Micronuclei were also capable of being bound by the innate immunomodulatory factor cGAS, which could indicate combination treatment with cisplatin and ATRi may have positive impacts in modulating responses to immunotherapy (Supplemental Figure S6D).
M6620 enhances response of ERCC1-deficient, p53-null tumors to cisplatin in vivo
From our in vitro data showing ATR activity is critical for supporting platinum tolerance in ERCC1- and p53-deficient cell line models, we asked if combined treatment with cisplatin and an ATR inhibitor could inhibit tumor growth in vivo. To this end, we implanted established tumors by subcutaneous (SC) trochar implantation into both posterior flanks of SCID mice. We included five mice in each treatment group including, vehicle control, M6620 (60 mg/kg; oral, PO), cisplatin (2 or 3 mg/kg; IV injection), or M6620 + cisplatin in combination. Treatment followed the outlined timeline and began three days post-trochar implant (Figure 5A). In this efficacy study against H1299 ERCC1Δ tumors, the combination of M6620 and cisplatin was found superior to either single agent regimen alone (Figure 5B and C). M6620 was completely ineffective (>100%/T/C; cage 2) against the ERCC1Δ tumors while standard-of-care agent cisplatin produced a moderate efficacious effect (21% T/C; 11–12 days of growth delay (T – C) resulting in 1.4–1.5 logs10 of gross cell kill; GLK; with an overall flat dose response; see cages 3–4) (Figure 5C). However, cisplatin efficacy was significantly enhanced when combined with M6620: Cg 5 utilizing the highest cisplatin dose level tested (3 mg/kg × 4) was toxic (LD20) to one mouse out of five (this mouse was euthanized due to signs of distress), but also produced significant efficacy: 0% T/C and a T – C of 37 days, resulting in 4.6 GLK and one tumor free survivor (day 134) (Figure 5B and C). To assess whether this tumor free ‘cured’ mouse was potentially a ‘leaky’ SCID mouse, this mouse was re-challenged with H1299 ERCC1Δ s.c. tumors and reached endpoint on day 157 (23 days to reach 1 g mass). The successful re-challenge indicated no immune influences were involved in generating the cure. Cg 6 was the highest non-toxic total dose level for the combination, also generating a significant efficacy response superior to that of either agent alone: 7% T/C, T – C of 17.5 days and 2.2 GLK (Figure 5C). The potentiating effect of M6620 with cisplatin warrants further refinements to dosing regimens to reduce toxicity encountered with this initial trial. This data provides proof-of-principle that a combination treatment strategy including cisplatin and M6620 could potentially benefit patients whose tumor is deficient for ERCC1 and p53.
DISCUSSION
Setbacks in the clinical implementation of ERCC1 expression as a first-in-class biomarker for predicting response to platinum-based chemotherapy suggest our current understanding of its predictive power remain unclear. Our recent work identified p53 status as a partial confounding variable in clinical evaluations of ERCC1 as a predictive biomarker of platinum-based chemotherapy (10). Patients with lung tumors harboring low ERCC1 and wildtype p53 had a 50% increase in overall survival compared to those with ERCC1 high/p53 wildtype tumors (10). Conversely, there was no overall survival benefit for patients whose tumors had low ERCC1 when p53 was mutated. Our previous work highlighted that p53 status plays a significant effect on cisplatin efficacy with loss of ERCC1 and more importantly, that likely the role of p53 in G1 cell cycle checkpoint is the critical function for mediating cisplatin hypersensitivity with loss of ERCC1 (10). We previously demonstrated that by inducing an artificial G1 arrest with CDK4/6 inhibitors or by blocking DNA replication initiation (Cdc7-Dbf4 inhibitor) maximum cisplatin sensitivity can be restored (10). Loss of the DNA repair factor ERCC1 with a functional G1 arrest drives cell sensitivity and senescence (10). NER is a critical cisplatin-DNA adduct repair pathway in G1 and thus, the inability to repair the platinum–DNA adducts triggers a G1 arrest and drives drug sensitivity. In cells with deficient G1 arrest, even with loss of ERCC1, cells enter S phase and rely on ATR and BRCA1 to mediate platinum tolerance. Our results suggest that following cisplatin/ATRi treatment, RPA exhaustion and nuclease degradation of the unprotected ssDNA regions following replication fork collapse drives replication catastrophe and cell killing. Blocking CDK2 reverses the DNA degradation observed in the DNA fiber assays with combination cisplatin/ATRi therapy. This is consistent with an important role of ATR in S phase and that blocking S phase entry reverses the DNA degradation which is similar to blocking Mre11 nuclease activity. This has significant clinical relevance as other important G1 cell cycle arrest factors like Rb, CDKN2A and ATM are commonly mutated in many cancer types which could alter response to platinum-based chemotherapy. Further studies are warranted to fully address the role of a functional G1 arrest in mediating maximum cisplatin efficacy with loss of DNA repair. In the current study, we show that ATR inhibition represents a potential therapeutic strategy for overcoming platinum tolerance in tumors harboring low ERCC1 and a functional deficiency in p53.
While a model of cisplatin hypersensitivity could not be further sensitized to cisplatin by ATR inhibition, a model of cisplatin tolerance with ERCC1 deficiency could be sensitized to cisplatin by ATR inhibition in a synergistic manner (approximately ten-fold in vitro) (Figures 2A, B and 3E). These data suggest a model where at least one reason for platinum-tolerance is likely increased replication fork protection or a general response to replication-associated damage (Figure 6). This hypothesis is supported by the observation that pharmacological inhibition of RPA binding to ssDNA or ATR kinase activity in the presence of cisplatin exacerbates resection at stalled replication forks as shown by DNA fiber assays in vitro (Figures 3A, F and 6). Assessing cell cycle profiles after combination treatment revealed that ATR inhibition by M6620 leads to abrogation of the G2/M cell cycle checkpoint after cisplatin treatment (Figure 4A and B). Similar observations were recently made in BRCA mutant tumors when treated with the ATR inhibitor AZD6738 in combination with olaparib (35). Additionally, via synchronization studies, we observed ATR-inhibited cells enter the subsequent G1 phase much faster than untreated control cells which could be related to premature entry into mitosis which has been observed by other groups in the context of inhibiting ATR (Supplemental Figure S5A and B) (30–32). After this bypass of the G2/M checkpoint, we detected accumulation of cells in the subsequent S-phase, likely indicating the presence of persistent DNA damage from the previous round of DNA replication and a defect in the G1 cell-cycle checkpoint due to loss of p53 function. In terms of sensitization of platinum-tolerant ERCC1 knockout cells to cisplatin by ATR inhibition, we hypothesize that ATR inhibition during S-phase leads to enhanced replication-associated DNA damage with cisplatin and it is these effects in combination with altered cell-cycle checkpoints that is critical for promoting cisplatin sensitivity in the absence of ERCC1 and functional p53 (Figure 6). Considering that platinum largely induces intrastrand adducts, our data is reminiscent of previous observations described in the context of ultraviolet light (UV) (36). In the context of UV, BRCA1 was demonstrated to act in an NER-independent manner to promote excision of the lesions as well as post-replicative repair (36). Furthermore, recruitment of RPA to ssDNA and ATRIP, an activator of ATR signaling, was suppressed in the absence of BRCA1. Our observations that platinum tolerance in ERCC1 and p53 deficient cells is abolished by depletion of BRCA1 or inhibition of ATR make it tempting to speculate that the phenotypes we observe are reporting on a synthetic lethal relationship between loss of NER and Post-replicative repair (10).
Figure 6.

Proposed model describing how platinum-tolerant, ERCC1-deficient tumors can be sensitized to platinum-based chemotherapy by ATR inhibition.
In platinum tolerant ERCC1-deficient cells, dual treatment coincided with substantial increases in DNA DSBs as shown by γH2AX staining (Figure 4C). Subsequent analysis revealed that dual treatment led to increased rates of pulverized chromosomes (Figures 4D, E and 6). Increased levels of DSBs were also linked to increased micronuclei formation which were more numerous in ERCC1 knockout cells and capable of being bound by the innate immuno-modulatory factor, cGAS (Supplemental Figure S6A–D). Micronuclei formation has become an established marker for activation of the innate immune response that is associated with increased PD-L1 expression and may positively impact response of tumors to immunotherapy (33, 37, 38). Similar observations with micronuclei formation in the context of ERCC1 deficiency and PARP inhibition have also been observed (39). In that context, increased micronuclei formation was associated with increased membranous PD-L1 expression, activation of interferon signaling mediated by cGAS-STING, and secretion of CCL5 (39). These observations have important implications for lung cancer therapy as first-line treatment for ∼70% of advanced NSCLC patients includes a platinum-based agent in combination with immune checkpoint inhibitor therapy.
In vivo studies utilizing platinum-tolerant H1299 ERCC1 knockout tumors confirmed that these tumors were not particularly sensitive to platinum-based chemotherapy which is striking considering ERCC1/XPF activity is thought to be essential for the repair of DNA intrastrand and interstrand crosslinks (Figure 5B). However, combination treatment lead to greatly enhanced responses in H1299 ERCC1 knockout tumors supporting our hypotheses that 1. ATR activity supports tolerance to platinum-based chemotherapy in this context and that 2. ATR kinase inhibitors may represent a therapeutic strategy for overcoming platinum tolerance in ERCC1 deficient tumors harboring a functional deficiency in p53 (Figure 6).
M6620 is currently in multiple clinical trials in combination with a variety of agents. This highly selective inhibitor of ATR kinase activity has shown promising activity in two Phase I studies. Results from a Phase I study combining topotecan with M6620 in 21 patients with advanced solid tumors who had failed at least one prior line of therapy showed two partial responses and eight patients with stable disease (40). Strikingly, three of five small cell lung cancer patients with platinum-refractory disease had durable clinical benefit from M6620 and topotecan combination therapy. Additionally, preliminary results from a Phase I study combining cisplatin and M6620 in triple negative breast cancers showed an objective response rate of nearly 39% and the disease control rate was approximately 72% (41). Clinical trial data suggests promising activity of M6620 in combination with cytotoxic chemotherapy in a subset of patients, particularly those with deficiencies in DNA repair associated genes. The RPA-DNA binding requirement for ATR activation opens up additional opportunities to target this pathway. Our data with the RPAi, NERx329, suggests that impinging on this pathway via targeting the sensor activity of RPA can also impact the DDR and sensitivity to cisplatin in ERCC1-deficient cancers.
In summary, we recently identified p53 status as a confounding variable in clinical assessments of ERCC1 status as a first-in-class biomarker for predicting response to platinum-based chemotherapy. Building upon this work, we have identified ATR kinase activity as essential for tolerance to platinum-based chemotherapy in ERCC1/p53-deficient tumors and propose that these specific patients would benefit from combination treatment with M6620, a platinum analogue and potentially immune checkpoint inhibitor therapy.
DATA AVAILABILITY
The data underlying this article are available in FlowRepository at http://flowrepository.org/, and can be accessed with Repository IDs FR-FCM-Z48H and FR-FCM-Z4B4.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Karmanos Cancer Institute for internal support for this project.
Notes
Present address: Joshua R. Heyza, Division of Synthetic Biology, Institute for Quantitative Health Science and Engineering, Michigan State University, East Lansing, MI, USA.
Contributor Information
Joshua R Heyza, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Elmira Ekinci, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Jacob Lindquist, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Wen Lei, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Christopher Yunker, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Vilvanathan Vinothkumar, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Rachelle Rowbotham, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Lisa Polin, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Natalie G Snider, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Eric Van Buren, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Donovan Watza, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Jessica B Back, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Wei Chen, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Hirva Mamdani, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Ann G Schwartz, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
John J Turchi, Departments of Medicine and Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, USA; NERx Biosciences, Indianapolis, IN, USA.
Gerold Bepler, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
Steve M Patrick, Department of Oncology, Wayne State University and Karmanos Cancer Institute, Detroit, MI, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
T32CA009531 (to J.R.H. and D.W.); NIH [R01CA229535 to S.M.P., R01CA180710 to J.J.T.]; NIH [R01CA141769 to A.G.S.]; Microscopy, Imaging and Cytometry Resources, Biostatistics, Animal Model and Therapeutic Evaluation, and Biobanking and Correlative Sciences Core Facilities are supported, in part, by NIH Cancer Center Support Grant [P30CA022453 to the Karmanos Cancer Institute at Wayne State University and R50CA251068-01 to Kamiar Moin, Wayne State University].
Conflict of interest statement. J.J.T. is President and Chief Scientific Officer of NERx Biosciences. All other authors declare no conflict of interest.
REFERENCES
- 1. Bepler G., Kusmartseva I., Sharma S., Gautam A., Cantor A., Sharma A., Simon G. RRM1 modulated in vitro and in vivo efficacy of gemcitabine and platinum in non-small-cell lung cancer. J. Clin. Oncol. 2006; 24:4731–4737. [DOI] [PubMed] [Google Scholar]
- 2. Bepler G., Williams C., Schell M.J., Chen W., Zheng Z., Simon G., Gadgeel S., Zhao X., Schreiber F., Brahmer J. et al. Randomized international phase III trial of ERCC1 and RRM1 expression-based chemotherapy versus gemcitabine/carboplatin in advanced non-small-cell lung cancer. J. Clin. Oncol. 2013; 31:2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olaussen K.A., Dunant A., Fouret P., Brambilla E., André F., Haddad V., Taranchon E., Filipits M., Pirker R., Popper H.H. et al. DNA repair by ERCC1 in non–small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006; 355:983–991. [DOI] [PubMed] [Google Scholar]
- 4. Friboulet L., Olaussen K.A., Pignon J.-P., Shepherd F.A., Tsao M.-S., Graziano S., Kratzke R., Douillard J.-Y., Seymour L., Pirker R. et al. ERCC1 isoform expression and DNA repair in non–small-cell lung cancer. N. Engl. J. Med. 2013; 368:1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dabholkar M., Vionnet J., Bostick-Bruton F., Yu J.J., Reed E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J. Clin. Invest. 1994; 94:703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Metzger R., Leichman C.G., Danenberg K.D., Danenberg P.v., Lenz H.J., Hayashi K., Groshen S., Salonga D., Cohen H., Laine L. et al. ERCC1 mRNA levels complement thymidylate synthase mRNA levels in predicting response and survival for gastric cancer patients receiving combination cisplatin and fluorouracil chemotherapy. J. Clin. Oncol. 1998; 16:309–316. [DOI] [PubMed] [Google Scholar]
- 7. Lord R.V.N., Brabender J., Gandara D., Alberola V., Camps C., Domine M., Cardenal F., Sánchez J.M., Gumerlock P.H., Tarón M. et al. Low ERRC1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin. Cancer Res. 2002; 8:2286–2291. [PubMed] [Google Scholar]
- 8. Friboulet L., Barrios-Gonzales D., Commo F., Olaussen K.A., Vagner S., Adam J., Goubar A., Dorvault N., Lazar V., Job B. et al. Molecular characteristics of ERCC1-negative versus ERCC1-positive tumors in resected NSCLC. Clin. Cancer Res. 2011; 17:5562–5572. [DOI] [PubMed] [Google Scholar]
- 9. Lee S.M., Falzon M., Blackhall F., Spicer J., Nicolson M., Chaudhuri A., Middleton G., Ahmed S., Hicks J., Crosse B. et al. Randomized prospective biomarker trial of ERCC1 for comparing platinum and nonplatinum therapy in advanced non-small-cell lung cancer: ERCC1 Trial (ET). J. Clin. Oncol. 2017; 35:402–411. [DOI] [PubMed] [Google Scholar]
- 10. Heyza J.R., Lei W., Watza D., Zhang H., Chen W., Back J.B., Schwartz A.G., Bepler G., Patrick S.M. Identification and characterization of synthetic viability with ERCC1 deficiency in response to interstrand crosslinks in lung cancer. Clin. Cancer Res. 2019; 25:2523–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Belanger F., Fortier E., Dube M., Lemay J.F., Buisson R., Masson J.Y., Elsherbiny A., Costantino S., Carmona E., Mes-Masson A.M. et al. Replication protein A availability during DNA replication stress is a major determinant of cisplatin resistance in ovarian cancer cells. Cancer Res. 2018; 78:5561–5573. [DOI] [PubMed] [Google Scholar]
- 12. Duda H., Arter M., Gloggnitzer J., Teloni F., Wild P., Blanco M.G., Altmeyer M., Matos J. A mechanism for controlled breakage of under-replicated chromosomes during mitosis. Dev. Cell. 2016; 39:740–755. [DOI] [PubMed] [Google Scholar]
- 13. Toledo L.I., Altmeyer M., Rask M.B., Lukas C., Larsen D.H., Povlsen L.K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2014; 156:1088–1103. [DOI] [PubMed] [Google Scholar]
- 14. King C., Diaz H.B., McNeely S., Barnard D., Dempsey J., Blosser W., Beckmann R., Barda D., Marshall M.S. LY2606368 causes replication catastrophe and antitumor effects through CHK1-dependent mechanisms. Mol. Cancer Ther. 2015; 14:2004–2013. [DOI] [PubMed] [Google Scholar]
- 15. Stevens J.B., Abdallah B.Y., Liu G., Ye C.J., Horne S.D., Wang G., Savasan S., Shekhar M., Krawetz S.A., Hüttemann M. et al. Diverse system stresses: Common mechanisms of chromosome fragmentation. Cell Death. Dis. 2011; 2:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arora S., Heyza J., Zhang H., Kalman-Maltese V., Tillison K., Floyd A.M., Chalfin E.M., Bepler G., Patrick S.M. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget. 2016; 7:75104–75117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gavande N.S., Vandervere-Carozza P.S., Pawelczak K.S., Vernon T.L., Jordan M.R., Turchi J.J. Structure-guided optimization of replication protein A (RPA)-DNA interaction inhibitors. ACS Med. Chem. Lett. 2020; 11:1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. LoRusso P.M., Aukerman S.L., Polin L., Redman B.G., Valdivieso M., Biernat L., Corbett T.H. Antitumor efficacy of interleukin-2 alone and in combination with adriamycin and dacarbazine in murine solid tumor systems. Cancer Res. 1990; 50:5876–5882. [PubMed] [Google Scholar]
- 19. Polin L., Corbett T.H., Roberts B.J., Lawson A.J., Leopold W.R., White K., Kushner J., Hazeldine S., Moore R., Rake J. et al. Transplantable syngeneic rodent tumors: solid tumors in mice. Tumor Models Cancer Res. 2011; 43–78. [Google Scholar]
- 20. Polin L., White K., Kushner J., Paluch J., Simpson C., Pugh S., Edelstein M.K., Hazeldine S., Fontana J., LoRusso P. et al. Preclinical efficacy evaluations of XK-469: Dose schedule, route and cross-resistance behavior in tumor bearing mice. Invest. New Drugs. 2002; 20:13–22. [DOI] [PubMed] [Google Scholar]
- 21. Polin L., Valeriote F., White K., Panchapor C., Pugh S., Knight J., LoRusso P., Hussain M., Liversidge E., Peltier N. et al. Treatment of human prostate tumors PC-3 and TSU-PR1 with standard and investigational agents in SCID mice. Invest. New Drugs. 1997; 15:99–108. [DOI] [PubMed] [Google Scholar]
- 22. Hustedt N., Álvarez-Quilón A., McEwan A., Yuan J.Y., Cho T., Koob L., Hart T., Durocher D. A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol. 2019; 9:190156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mohni K.N., Thompson P.S., Luzwick J.W., Glick G.G., Pendleton C.S., Lehmann B.D., Pietenpol J.A., Cortez D. A synthetic lethal screen identifies DNA repair pathways that sensitize cancer cells to combined ATR inhibition and cisplatin treatments. PLoS One. 2015; 10:e0125482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mohni K.N., Kavanaugh G.M., Cortez D. ATR pathway inhibition is synthetically lethal in cancer cells with ercc1 deficiency. Cancer Res. 2014; 74:2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arora S., Kothandapani A., Tillison K., Kalman-Maltese V., Patrick S.M. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair (Amst.). 2010; 9:745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwok M., Davies N., Agathanggelou A., Smith E., Oldreive C., Petermann E., Stewart G., Brown J., Lau A., Pratt G. et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53-or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016; 127:582–595. [DOI] [PubMed] [Google Scholar]
- 27. Huntoon C.J., Flatten K.S., Wahner Hendrickson A.E., Huehls A.M., Sutor S.L., Kaufmann S.H., Karnitz L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013; 73:3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moiseeva T., Hood B., Schamus S., O’Connor M.J., Conrads T.P., Bakkenist C.J. ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nat. Commun. 2017; 8:1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. VanderVere-Carozza P.S., Gavande N.S., Jalal S.I., Pollok K.E., Ekinci E., Heyza J., Patrick S.M., Masters A., Turchi J.J., Pawelczak K.S. In vivo targeting replication protein A for cancer therapy. Front. Oncol. 2022; 12:826655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schoonen P.M., Kok Y.P., Wierenga E., Bakker B., Foijer F., Spierings D.C.J., van Vugt M.A.T.M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019; 13:2422–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nghiem P., Park P.K., Kim Y.S., Vaziri C., Schreiber S.L. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saldivar J.C., Hamperl S., Bocek M.J., Chung M., Bass T.E., Cisneros-Soberanis F., Samejima K., Xie L., Paulson J.R., Earnshaw W.C. et al. An intrinsic S/G2 checkpoint enforced by ATR. Science. 2018; 361:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harding S.M., Benci J.L., Irianto J., Discher D.E., Minn A.J., Greenberg R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017; 548:466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H., Hu S., Chen X., Shi H., Chen C., Sun L., Chen Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim H., George E., Ragland R.L., Rafail S., Zhang R., Krepler C., Morgan M.A., Herlyn M., Brown E.J., Simpkins F. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 2017; 23:3097–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pathania S., Nguyen J., Hill S.J., Scully R., Adelmant G.O., Marto J.A., Feunteun J., Livingston D.M. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol. Cell. 2011; 44:235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sen T., Rodriguez B.L., Chen L., della Corte C.M., Morikawa N., Fujimoto J., Cristea S., Nguyen T., Diao L., Li L. et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019; 9:646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. MacKenzie K.J., Carroll P., Martin C.A., Murina O., Fluteau A., Simpson D.J., Olova N., Sutcliffe H., Rainger J.K., Leitch A. et al. CGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017; 548:461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chabanon R.M., Muirhead G., Krastev D.B., Adam J., Morel D., Garrido M., Lamb A., Hénon C., Dorvault N., Rouanne M. et al. PARP inhibition enhances tumor cell–intrinsic immunity in ERCC1-deficient non–small cell lung cancer. J. Clin. Invest. 2019; 129:1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thomas A., Redon C.E., Sciuto L., Padiernos E., Ji J., Lee M.J., Yuno A., Lee S., Zhang Y., Tran L. et al. Phase i study of ATR inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J. Clin. Oncol. 2018; 36:1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Telli M.L., Lord S., Dean E., Abramson V., Arkenau H.-T., Becerra C., Tolaney S.M., Tang R., Penney M.S., Pollard J. et al. Initial results of a phase 1 dose expansion cohort of M6620 (formerly VX-970), an ATR inhibitor, in combination with cisplatin in patients with advanced triple-negative breast cancer NCT02157792). Ann. Oncol. 2017; 28:v76. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in FlowRepository at http://flowrepository.org/, and can be accessed with Repository IDs FR-FCM-Z48H and FR-FCM-Z4B4.