

Abstract
High‐throughput (HT) screening drug discovery, during which thousands or millions of compounds are screened, remains the key methodology for identifying active chemical matter in early drug discovery pipelines. Recent technological developments in mass spectrometry (MS) and automation have revolutionized the application of MS for use in HT screens. These methods allow the targeting of unlabelled biomolecules in HT assays, thereby expanding the breadth of targets for which HT assays can be developed compared to traditional approaches. Moreover, these label‐free MS assays are often cheaper, faster, and more physiologically relevant than competing assay technologies. In this review, we will describe current MS techniques used in drug discovery and explain their advantages and disadvantages. We will highlight the power of mass spectrometry in label‐free in vitro assays, and its application for setting up multiplexed cellular phenotypic assays, providing an exciting new tool for screening compounds in cell lines, and even primary cells. Finally, we will give an outlook on how technological advances will increase the future use and the capabilities of mass spectrometry in drug discovery.
Keywords: affinity selection, drug discovery, high‐throughput screening, MALDI‐TOF, mass spectrometry
Subject Categories: Methods & Resources, Pharmacology & Drug Discovery
This Review summarizes advantages and disadvantages of high‐throughput mass spectrometry techniques used in drug discovery and discusses how technological advances could increase the capabilities of mass spectrometry in drug discovery in the future.

Glossary
- BLAZE mode
The name of the RapidFire hardware modification that improves the speed of the system by enabling cycling times of 2.5 s per sample
- Chemoproteomics
A broad set of techniques used to identify and characterize the mode of action of a drug. This can include quantitative MS‐based proteomics
- Data‐independent acquisition MS
A recently developed global MS‐based proteomics strategy that first isolates precursor ions into pre‐defined isolation windows, which are then fragmented and analysed
- Fragment‐based drug discovery
Method used to develop potent small‐molecule compounds starting from fragments binding weakly to targets
- Limited proteolysis MS
Used to measure protein structural transitions directly in biological matrices and on a proteome‐wide scale
- Mechanism of action
Refers to the specific biochemical interaction through which a drug substance produces its pharmacological effect
- PhAbit
PhotoAffinity bits. A reversible ligand with a photoreactive warhead incorporated to facilitate covalent binding
- Phosphoproteomics
Proteomics analysis that seeks to determine the overall level of protein phosphorylation and the identity of proteins, which are phosphorylated, and amino acid residues, which hold the phosphate group
- RapidFire
Is a proprietary automated microfluidic sample collection and purification system that interfaces directly to standard ESI‐MS instruments. This system uses high‐speed robotics to directly aspirate fluidic samples from 96‐ or 384‐well screening plates, rapidly removes non‐volatile assay components such as salts, buffers and detergents in an online fractionation step, and delivers purified analytes to the mass spectrometer
- Size exclusion chromatography
A chromatographic separation technique that separates analytes by size, and, therefore, relative to molecular weight
- Thermal proteome profiling
A quantitative MS‐based proteomics tool used to monitor the melting profile of thousands of proteins simultaneously
- Warhead
A reactive group that is strategically incorporated onto a reversible ligand to facilitate the formation of a covalent bond to a target biomolecule
Introduction
The drug discovery and development pipeline is an interdisciplinary process that engages multiple phases of research to facilitate the generation of effective therapies (Mohs & Greig, 2017). The historical aspects of the traditional drug discovery pipeline have been extensively reviewed and demonstrate the advantages and challenges of drug discovery throughout R&D including productivity, attrition, and evolution of new technologies (Moffat et al, 2017; Vincent et al, 2022). The drug discovery phase contains the target identification and validation phase, as well as hit finding, typically through high‐throughput screening (HTS) campaigns employing large compound libraries of several hundred thousands of compounds. At the end of this phase, chemistry is performed to optimize the activity and physicochemical properties of the molecule, both of which influence its in vivo behavior as it relates to potency, clearance, and safety. Early adoption of new technologies can be critical to improving R&D as there are often lengthy cycle times and high failure rates of drug discovery projects prior to pre‐clinical development. There is, therefore, a focus across industry and academia on the development of more biologically relevant and diverse approaches to the discovery of chemical starting points, to address both the success rates and pace of research.
Mass spectrometry (MS) is a powerful, versatile technique with applications spanning the full spectrum of the drug discovery and development pipeline. For example, MS techniques such as proteomics, metabolomics and analysis of clinical tissue samples are an important part of target validation, as well as later in discovery where these techniques can be used to gain insight into a compound's cellular mechanism of action (MoA). During lead optimization, MS has for decades played the central role in determining both the structure and pharmacokinetic properties of compounds. MS is also increasingly important in the target identification step of the drug discovery pipeline. For example, limited proteolysis‐coupled MS (Schopper et al, 2017) is routinely used to determine proteome‐wide specificity and uncover small molecule binding sites, thermal proteome profiling (Franken et al, 2015) for small molecule target finding, and data‐independent acquisition MS for HT analysis of cell systems for global proteomics and phosphoproteomics (Kitata et al, 2021).
Despite MS being a powerful tool within the overall drug discovery process, its application to HT screening has lagged, often due to a lack of throughput and lack of associated automation. Current HTS assays are often performed using fluorescence and chemiluminescence‐based detection modalities that although HT, are susceptible to compound‐dependent screening artefacts leading to false positives or negatives (Winter et al, 2018). Here, MS presents itself as an attractive alternative technology as it is already an established, sensitive, and versatile technique in research for the analysis of small and large biomolecules. A key advantage of MS has been the potential to build label‐free assays that improve hit confirmation rates and ultimately accelerate the drug discovery process. HTS‐MS has been demonstrated to be an effective tool for removing potential detection‐based false positives and thus mitigating sources of assay interference (Adam et al, 2015). From orthogonal to traditional hit‐finding approaches, MS presents the opportunity to explore alternative hit‐identification strategies that focus on detecting protein‐target binders, or compounds that directly modulate cellular function to reverse or treat a disease phenotype.
The aim of this review is to provide an overview of the recent developments in HT‐MS for drug discovery. We outline how these advancements in MS have enabled the development of HTS‐MS platforms and their applications. Finally, we provide an outlook of how technological advances could further drive alternative capabilities of MS in drug discovery.
Basic principles of mass spectrometry instrumentation
MS is an analytical technique that measures both the mass‐to‐charge ratio (m/z) and abundance of ions to generate a mass spectrum that can in turn yield chemically relevant information such as empirical mass or structure about a particular analyte. In its simplest form, a mass spectrometer consists of an ionization source coupled to a mass analyzer and detector. The ion source transfers sample molecules into the gas phase as charged ions which then are transferred into a mass analyzer. Here, ions are separated based on their m/z and detected, thus generating a mass spectrum. As not only the m/z but also the number of detected ions is recorded, MS can be a highly quantitative technique with a linear range of up to ~105 (Collings et al, 2014).
HT‐MS‐based readouts in drug discovery have been largely dominated by instruments comprising of solid‐phase extraction (SPE) coupled to electrospray ionization (ESI), or surface‐based techniques such as matrix‐assisted laser/desorption ionization (MALDI). Self‐assembled monolayers (SAMs) coupled with desorption/ionization (SAMDI), as well as some more recent approaches such as acoustic mist ionization (AMI), and acoustic droplet ejection (ADE) open port interface (OPI) MS have been added to the toolbox. These principles are described in Fig 1 (surface‐based, Fig 1A and electrospray‐based Fig 1B). Each of these ionization techniques can be combined with different mass analyzers to access different levels of mass resolution, dynamic ranges, analysis time, and sample throughput. For a detailed review, please see Challen and Cramer (2022).
Figure 1. Schematic of main ionization techniques employed for HTS‐MS.
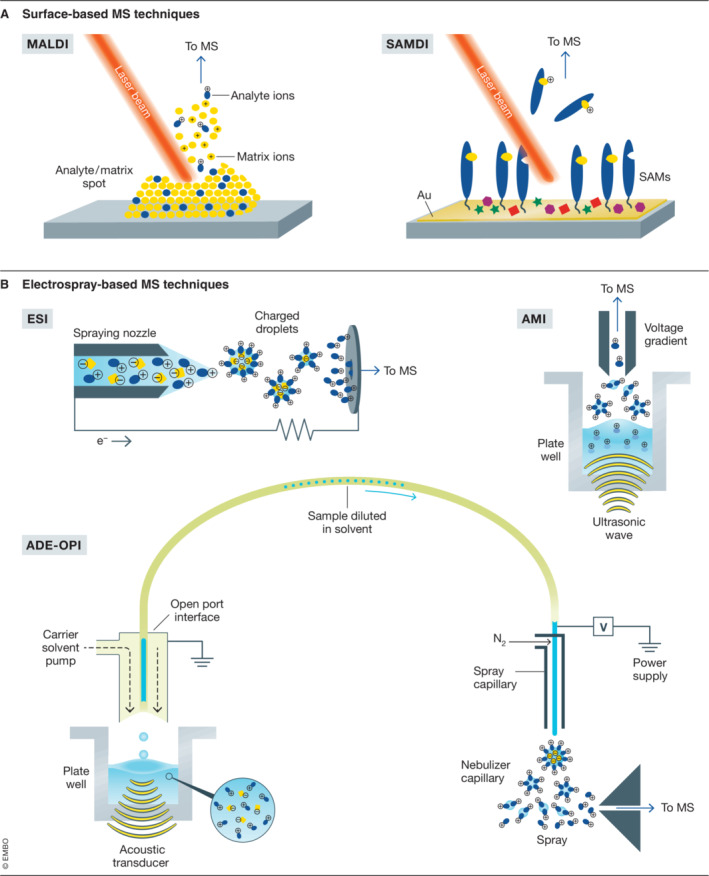
(A) Surface‐based: MALDI. Samples are co‐crystallized with a matrix on a conductive target plate. Laser shots are used to activate matrix molecules and evaporate analyte and matrix. In the reactive cloud, protons are transferred from the matrix to ionize the analyte molecules (Karas et al, 1985). SAMDI. Components of an enzymatic reaction (either enzymes or substrates) are immobilized onto self‐assembled monolayers (SAMs) in an array format, and upon irradiation with a laser, the monolayers are desorbed from the surface through cleavage of the thiolate‐gold bond and ionized (Gurard‐Levin et al, 2011). (B) Electrospray‐based: ESI. The analytes are dissolved in a liquid carrier phase, and a high voltage is applied to the tip of the metal capillary relative to the mass spectrometer's sampling cone. The electric field causes the dispersion of the sample solution resulting in nebulization. Charged droplets containing the analytes are generated at the exit of the electrospray tip. The solvent of the droplets is vaporized by a drying gas or heat and the charged analytes are guided by a potential gradient toward the analyzer region of the MS (Fenn et al, 1989; El‐Aneed et al, 2009). AMI. An acoustic transducer and charging cone are used to generate nanolitre‐sized charged droplets that are guided through an ion transfer line into a MS (Sinclair et al, 2015). ADE‐OPI. A pulse of acoustic energy ejects sample droplets upward into the inverted OPI, where a fluid pump delivers carrier solvent to a sample capture region. The sample is captured, diluted, and guided to MS by conventional ESI (Zhang et al, 2021).
Mass spectrometry screening assays for drug discovery
Biochemical and functional assays to identify inhibitors of enzymes
Once target proteins have been identified as a potential drug target in a specific disease, biochemical in vitro assays are often performed to identify molecules that modulate protein function. For protein targets that are enzymes, target inhibition or activation can be measured via the generation of a product, or the decrease of a substrate, in a biochemical reaction (Fig 2A). Unlike most traditional biochemical assays, MS allows the direct, label‐free quantitative measurement of both substrate and product in these in vitro assays, as long as a mass shift occurs; therefore, most enzyme targets are principally amenable for mass spectrometric analysis. In recent years, ion mobility separation has been integrated within new HTS capable mass spectrometers, thus enabling the separation of complex and isobaric compounds such as lipid classes (Djambazova et al, 2020). This will likely broaden the development of HTS‐compatible MS assays for challenging enzymes, such as isomerases, in future years.
Figure 2. Types of high‐throughput mass spectrometry drug discovery assays.
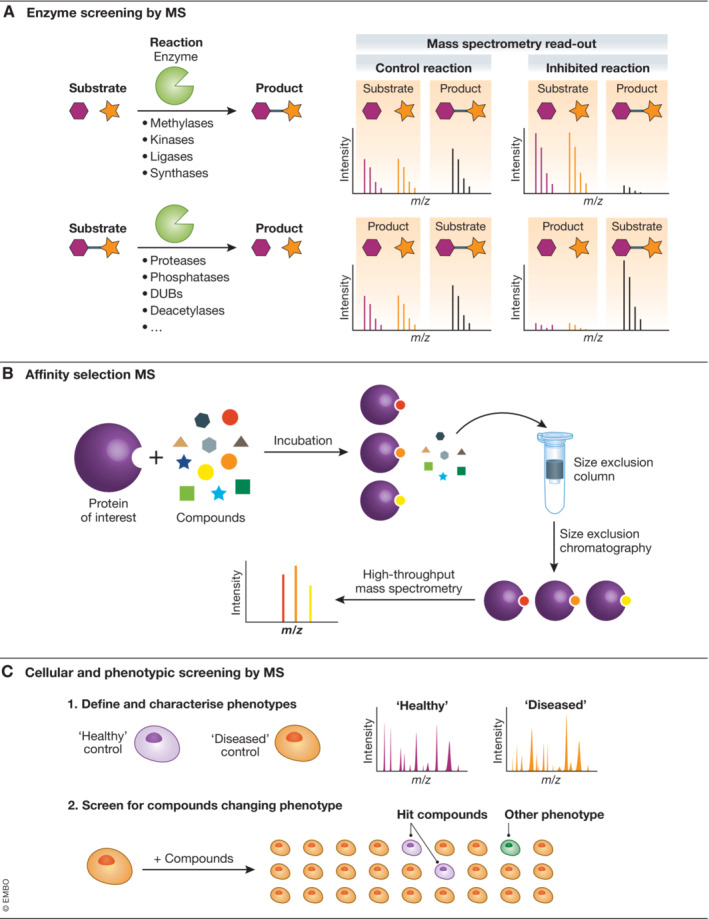
(A) Enzyme activity screening by mass spectrometry. In vitro reactions of enzymes with substrates are stopped at appropriate time points and the resulting mixture analysed by mass spectrometry to identify substrate to product conversion. Addition of chemical compounds that affect the reaction are identified by reduced product conversion. (B) Affinity Selection Mass Spectrometry. Compounds bind to a protein of interest and non‐binding compounds are removed by size‐exclusion chromatography. Binding compounds are identified by mass spectrometry. (C) Cellular and phenotypic screening by mass spectrometry. Cellular phenotypes of “healthy” and “diseased” controls are defined by a read‐out of a cellular “fingerprint” of specific biomolecules. Chemical compounds that shift the “diseased” phenotype to “healthy” are considered hits.
For ESI, different technologies such as the RapidFire system in BLAZE‐mode (Bretschneider et al, 2019) or ADE‐OPI MS approach have been described for enzymatic‐type assays (Häbe et al, 2020; Simon et al, 2021a). The versatility of the instrument setup allows the analysis of many different biomolecules including lipids (Highkin et al, 2011; Dittakavi et al, 2020), peptides (Hutchinson et al, 2011; Liddle et al, 2020), and metabolites (Soulard et al, 2008; Maxine et al, 2009), from a wide range of matrix systems including blood, plasma (Highkin et al, 2011; Bretschneider et al, 2017) and cell lysates (Gordon et al, 2016; Dittakavi et al, 2020). Ambient ionization, such as desorption electrospray ionization (DESI), which commonly does not require sample preparation, is a new and attractive alternative for HT analysis. DESI‐MS displays remarkably high salt tolerance, making this technique ideal for the analysis of complex samples without any sample preparation. Using DESI, samples are ionized outside of a mass spectrometer under native conditions. Due to its ability to rapidly scan a surface, DESI‐MS has been amenable to HT applications (Wleklinski et al, 2018), at rates approaching 10,000 reactions per hour, and for the analysis of enzymatic reactions directly from the bioassay matrix (Morato et al, 2020).
The development of instrumentation and improvements in sample preparation have enabled MALDI‐ time‐of‐flight (TOF) MS to rival the more conventional HTS assays with throughputs of 10–20 samples per second for conventional MALDI (Haslam et al, 2015) and liquid atmospheric pressure MALDI (Krenkel et al, 2022) reported. The first drug discovery studies using the high speed (1,536 spots in less than 8 min) of these new‐generation MALDI‐TOF mass spectrometers for drug discovery was the development of a HTS compatible assay to study the specificity and drugability of deubiquitylases (DUBs; Ritorto et al, 2014). In this work, individual DUBs were incubated with ubiquitin dimers of different linkage type and the quantitation of mono‐ubiquitin using an isotopically labelled internal standard enabled the determination of DUB specificity, and this was further applied to drug screening. This assay was unique to the field as it used native substrates, rather than the previously used rhodamine fluorescently labelled reagents (Hassiepen et al, 2007), and also had the potential to be expanded to a HT drug screening platform.
HT MALDI‐TOF MS assays targeting post‐translational modifications have grown rapidly in the past decade as the technique can be applied to potentially any reaction that involves a mass change. This importantly allows label‐free quantitation, a gold standard for assays in the drug discovery field with respect to simplicity and cost. Successful MALDI‐TOF MS assays now include the study of kinases (Beeman et al, 2017; Heap et al, 2017), methyltransferases (Guitot et al, 2017), and phosphatases (Winter et al, 2018).
Most of the MALDI‐TOF‐based HTS‐compatible approaches conducted so far have focused on in vitro assays with simple readouts (with often just a single substrate and product) and have been limited to peptide/protein‐centric activity assays (Ritorto et al, 2014; Guitot et al, 2017; Heap et al, 2017; De Cesare et al, 2018; Winter et al, 2018; Simon et al, 2020). Applying this technology for cellular assays and metabolomics‐based drug discovery remains a challenge mostly due to (i) interference from matrix peaks in the low‐mass range, (ii) matrix‐dependent analyte selectivity, and (iii) limited metabolite coverage due to low sensitivity of certain classes of metabolites. Although, recently, individual metabolites such as trimethylamine (Winter et al, 2019), acetylcholine (Chandler et al, 2016), 3‐methoxytyramine (Winter et al, 2022), and cyclic GMP‐AMP (at a throughput of ~60,000 samples per day; Simon et al, 2020) have been used in MALDI‐TOF HTS campaigns, new tools and methods need to be developed to meet the opportunities and challenges toward HT metabolic profiling for drug discovery.
The SAMDI technology is a promising strategy for HTS that uses the same MALDI‐TOF MS instrumentation but in a more targeted approach where immobilized proteins are used to capture substrates or products (Gurard‐Levin et al, 2011). Although generally not label‐free, as the protein needs to have a tag to be immobilized, this technology enables the specific capture of analytes and is well suited for measuring a broad range of enzyme activities as SAMs can be customized to use a variety of immobilization chemistries (Mrksich, 2008). An exemption to this statement is traceless‐SAMDI (Helal et al, 2018). This work introduced a truly label‐free approach for analysing HT reactions by using a photogenerated carbene to non‐selectively attach molecules to the SAMs, from which can then be analyzed by MS. SAMDI has also been used for in vitro recombinant enzyme/substrate screen on diverse enzyme classes, such as methyltransferases (Swalm Brooke et al, 2013), glycosyltransferases (Ban et al, 2012), and deacetylases (Gurard‐Levin et al, 2010). Selected publications describing HTS compatible MS assays can be found in Table 1.
Table 1.
Selected publications describing HTS MS‐compatible assays in drug discovery.
| Enzyme | Substrate | Product | Platform | Citation |
|---|---|---|---|---|
| Phosphatidylserine decarboxylase | Phosphatidylserine | Phosphatidylethanolamine | RapidFire | Forbes et al (2007) |
| ERAP1 | Peptide | Peptide | RapidFire | Liddle et al (2020) |
| Acetyl‐coenzyme A carboxylase | Sphingosine in whole blood | Sphingosine‐1‐phosphate | RapidFire | Maxine et al (2009) |
| Autotaxin | Lysophosphatidyl choline | Lysophosphatidic acid | RapidFire | Soulard et al (2008) |
| Histone lysine demethylase | Trimethylated peptide | Demethylated peptide | RapidFire | Hutchinson et al (2011) |
| Histone deacetylase | Acetylated peptide | Peptide | AMI‐MS | Sinclair et al (2019) |
| Histone acetyltransferase | Peptide and acetyl‐CoA cofactor | Acetylated peptide | AMI‐MS | Belov et al (2020) |
| Diacylglycerol acyltransferase 2 | Diolein and oleoyl‐CoA | triolein | ADE‐OPI MS | Wen et al (2021) |
| Cyclic GMP‐AMP synthase | GTP + ATP | Cyclic GMP‐AMP | ADE‐OPI MS | Simon et al (2020) |
| Deubiquitylases | Diubiquitin | Ubiquitin | MALDI‐TOF | Ritorto et al (2014) |
| E3‐ligases | Diubiquitin | Ubiquitin | MALDI‐TOF | De Cesare et al (2018) and De Cesare et al (2020) |
| Kinases | Peptide | Phosphopeptide | MALDI‐TOF | Beeman et al (2017) and Heap et al (2017) |
| Methyltransferases | Peptide | Methylated peptide | MALDI‐TOF | Guitot et al (2017), Guitot et al (2014) and Haslam et al (2015) |
| Phosphatases | Phosphopeptide | Peptide | MALDI‐TOF | Winter et al (2018) |
| Acetylcholinesterase | Acetylcholine | Choline | MALDI‐TOF | Haslam et al (2015) |
| Cyclic GMP‐AMP synthase | GTP + ATP | Cyclic GMP‐AMP | MALDI‐TOF | Simon et al (2020) |
| Anthrax lethal factor | Peptide | Peptide | SAMDI | Min et al (2004) |
| Sirtuin 3 | Acetylated peptide | Peptide | SAMDI | Patel et al (2015) |
| Methyltransferases | Peptide | Methylated peptide | SAMDI | Swalm Brooke et al (2013) |
| Glycosyltransferases | Saccharides | Oligosaccharides | SAMDI | Ban et al (2012) |
| Deacetylases | Acetylated peptide | Peptide | SAMDI | Gurard‐Levin et al (2010) |
| Isocitrate dehydrogenase 1 | Isocitrate | α‐ketoglutarate | MALDI + ESI | Radosevich et al (2022) |
| Catechol‐O‐methyltransferase | Dopamine | 3‐methoxytyramine | MALDI‐TOF | Winter et al (2022) |
Affinity and binding assays
Affinity selection mass spectrometry (ASMS) is a HT and cost‐effective binding assay that enables rapid screening of a large number of compounds against a specific target biomolecule of interest (Prudent et al, 2021). In a traditional HT ASMS approach (Fig 2B), the biomolecular target is typically present in molar excess relative to the potential ligands that are then captured by the protein. Non‐bound ligands are separated from the protein using usually either an affinity enrichment or size exclusion chromatography (SEC). Bound ligands are then dissociated from the target protein and identified by their accurate mass with a suitable MS technique. Alternatively, ASMS can also be employed as an assay to further characterize ligand‐binding properties, such as to demonstrate proof of binding as well as performing competition experiments (Simon et al, 2021b). ASMS has emerged over the past two decades as a strategy complimentary to functional HTS assays (Annis et al, 2007). This approach leverages the label‐free and direct detection capability of MS and is most often coupled to SEC. In particular, it has been widely adopted in industry due to its scalability and led to the development of fully automated systems, such as the Automated Ligand Identification System (Annis et al, 2004), as well as the SpeedScreen system (Muckenschnabel et al, 2004; Zehender et al, 2004; Zehender & Mayr, 2007). Typically, a 1 million compound screen with a pooling strategy can take 5–7 days with follow‐up experiments ranging 1–3 weeks to re‐confirm and characterize compound binding depending on the strategy employed. The rationale behind the affinity selection approach is that binding must precede activity, therefore, the identification of small molecule binders can be a surrogate to reading out activity in a traditional HT biochemical assay during the first stages of a hit ID campaign. Advantageously, this can identify ligands that exhibit multiple MoA, potentially identifying agonists and antagonists in a single screen. An ASMS HTS can often be less complex to develop than a traditional biochemical HTS and can accommodate targets where very little knowledge of protein function or structure exists. By designing ASMS specific collections or mass encoded libraries, a broader screening of chemical space could be possible to reduce complex downstream deconvolution and redundancies. (Prudent et al, 2021).
MS has been instrumental in the development of ASMS strategies and HT screens of more than one million compounds have been achieved across in‐solution ASMS platforms. These include a diverse range of targets like beta‐secretase (Coburn et al, 2004), G‐protein coupled receptors (Whitehurst & Annis, 2008), RNA polymerase (Walker et al, 2017), CHK1 (Comess et al, 2006) and to probe druggable target space within the NF‐kβ pathway (Kutilek et al, 2016). These screens have historically been performed using pools of 100–2,000 compounds and analysis on high‐resolution MS instruments. This approach, although HT, does suffer a few analytical challenges. Typically, protein concentrations in the micromolar range are needed and good protein solubility over 12–24 h is critical, which can be problematic for some targets like membrane proteins. Furthermore, the use of large pools of compounds can increase overall DMSO concentration, reduce assay sensitivity, and could also denature the target protein structure. More recently, HTS capable MALDI‐TOF MS platforms that use faster instrument scanning speeds have been used to screen smaller pools of compounds by ASMS. This includes the SEC MALDI‐TOF MS platform proposed by Simon et al (2021b), as well as a SAMDI‐TOF MS approach, both of which use pools of tens of compounds rather than hundreds, yet can still reach the same sample throughput.
Covalent fragment assays in drug discovery
Fragment‐based drug discovery (FBDD) is an established, versatile strategy in drug discovery that aims to develop novel drugs from small, low molecular weight starting points. Sensitive technologies, including surface plasmon resonance, nuclear magnetic resonance, and MS, have been used to detect the binding or activity of these fragments. An excellent example of this approach is the discovery of vemurafenib, a selective inhibitor of the oncogenic target B‐RAF (Tsai et al, 2008). Advantages of FBDD often include reduced experimental costs, as well as novel strategies to developing new drugs that harness advances in HT chemistry.
One aspect of FBDD where MS technology has been instrumental is the development of reactive or covalent fragment screening strategies. This approach exploits the advances made in synthesis of small molecule libraries that can then be coupled to covalent warheads to accelerate screening efforts (Lu et al, 2021). Using small covalent fragments to probe biological systems and poorly characterized targets significantly enhances our ability to translate traditional biological research to the development of new medicines and understanding their MoA (Schreiber Stuart et al, 2015; Zhang et al, 2019). This has been particularly impactful where probes were used to explore the therapeutic effects of PKM2 activation in cancer (Anastasiou et al, 2012; Kung et al, 2012), and in the generation of a covalent inhibitor of KRAS that was previously thought to be undruggable (Naim et al, 2021). Novel chemotypes for anti‐malarial therapeutics have also been described, along with their MoA, through use of covalent probes (Heidebrecht et al, 2012); this in turn may infer on future paths of resistance (Lukens et al, 2015).
With this screening approach, an irreversible binding event of the fragment to a protein is observed. MS plays a key role in the screening of these compounds as the addition of the fragment molecule induces a shift in the protein molecular weight that can then be measured by MS. These MS techniques can then support the characterization of a compounds MoA as well as identify specific protein target engagement. The binding event itself is highly dependent on the synergistic relationship between structural biology and synthetic chemistry to enable binding to a relevant site of interest and often yields key mechanistic insights. In comparison to the use of a traditional screening collection, it has been shown that a fragment‐based screening approach can offer better coverage of chemical space, as well as identify novel chemical equity that interacts with a protein binding pocket (Hall et al, 2014). Furthermore, this approach has been particularly successful for studying targets where traditional compound collections have been unsuccessful (Coyne et al, 2010).
Perhaps the simplest reactive FBDD approach is the screening of fragment libraries against recombinant target proteins in vitro. Here, low molecular weight (typically < 300 Da) fragment libraries that typically contain electrophilic properties are synthesized and coupled to covalent warheads (Long & Aye, 2017). These libraries are then screened against a specific target protein and hits are distinguished by liquid chromatography (LC)‐MS (Fig 3). These libraries have historically been relatively small in the orders of 100–1000s of compounds and thus do not require the use of uHTS mass spectrometers. A LC–MS or RapidFire MS approach to covalent fragment screening is often in the order of 0.5–5 min per sample. Many studies have used this technology to screen fragments against various targets such as the E3 ligase HOIP (Johansson et al, 2019), BRD4 (Grant et al, 2019; Olp et al, 2020), GDP‐KRASG12C (Shin et al, 2019), and OTUB2 and NUDT7 (Resnick et al, 2019). Selected publications of fragment‐based MS assays can be found in Table 2.
Figure 3. Fragment‐based drug discovery assays.
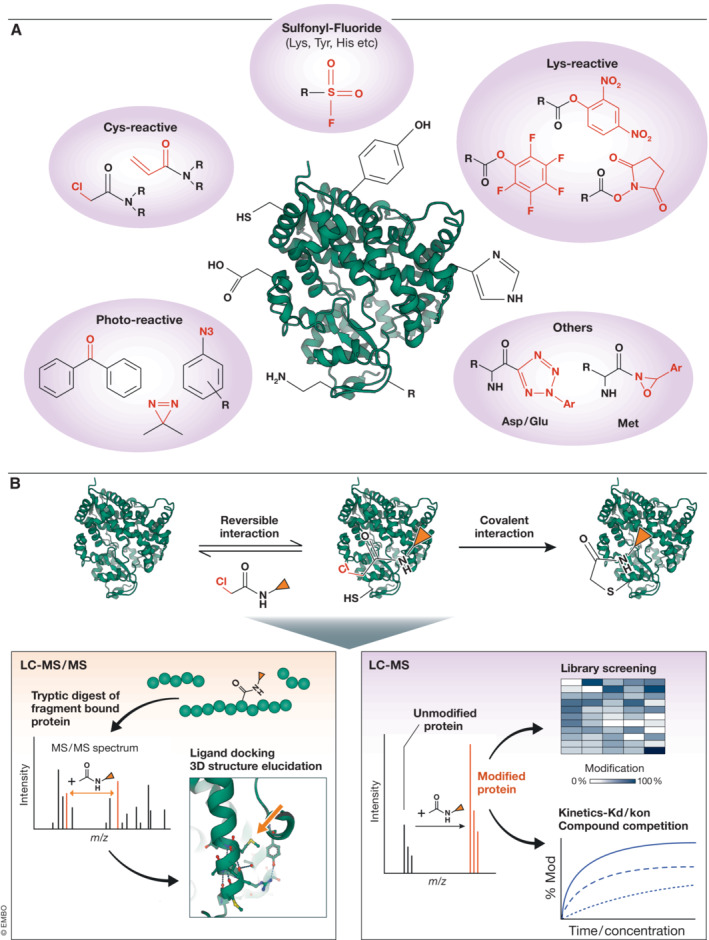
(A) Summary of amino acid targeting moieties with the reactive warheads highlighted in red. (B) Overview of a reactive fragment covalently binding to a protein target of interest and how LC–MS and LC–MS/MS contribute to the measurement of binding affinity, localization of covalently bound residue as well as kinetic and competition studies.
Table 2.
Selected publications of fragment‐based MS screening assays.
| Reactive fragment | Warhead | Target protein | MS platform | Citation |
|---|---|---|---|---|
| Cys‐reactive | Acrylate | Pappain | LC‐TOF | Kathman et al (2014) |
| Cys‐reactive | α,β‐unsaturated methyl ester | HOIP | LC‐TOF | Johansson et al (2019) |
| Cys‐reactive | Chloroacetamide | Pin1 | LC‐TOF | Dubiella et al (2021) |
| Cys‐reactive | Acrylate | BRD domains | LC‐Ion Trap | Olp et al (2020) |
| Cys‐reactive | Acrylamide | KRas/KRas(G12C) | LC‐TOF | Ostrem et al (2013) |
| Lys‐reactive | Acrylate/alpha‐beta unsaturated esters | HSP72 | LC‐qTOF and LC–MS/MS | Pettinger et al (2017) |
| Lys‐reactive | Aryl boronic acid carbonyl | Mcl‐1 | LC‐TOF | Akçay et al (2016) |
| PhABit | Alkyl diazirine/alkyne tag | BRD4/KRas | RapidFire‐TOF | Grant et al (2020) |
| PhABit | 5× photoreactive groups | CDK(2/7/9) | LC‐TOF | Grant et al (2019) |
| PhABit | Diazirine | CA(ii) | RapidFire‐TOF | Thomas et al (2021) |
| Tyr‐reactive | Sulfonyl‐fluoride | GST's | LC–MS/MS | Shishido et al (2017) |
| Ser‐reactive | Aryl fluorosulfate | DcpS | LC‐TOF/LC–MS/MS | Fadeyi et al (2017) |
As covalent fragment screening is still a relatively new approach, a number of challenges do hinder its potential to come to the forefront of drug discovery. For example, although warhead design is expanding to target a range of amino acids in active site pockets (Fig 3A), their selectivity can be limited and sometimes the reactivity of the warhead rather than fragment affinity can drive binding. Furthermore, some of the more reactive warheads currently used in screening, such as chloroacetamides, are not always suitable for translating into the clinic due to toxicity. Finally, current LC–MS approaches used for covalent fragment screening (Fig 3B) lack the throughput of other technologies discussed in this review such as MALDI and ADE. Advances in future MS instrumentation, such as ion mobility, may support the implementation of covalent FBDD in routine drug discovery efforts.
With MS underpinning the majority of chemical biology approaches to covalent drug discovery and chemoproteomics, many of the advances made in this field have been made hand‐in‐hand with the development of newer, faster, and higher resolution MS instruments. It is expected that chemical biology will continue to be a major driver of modern drug discovery approaches and understanding of the illusive, yet potentially therapeutic human proteome.
Moving toward cellular and phenotypic MS drug discovery assays
Phenotypic screening is common for target identification as a result of genetic perturbation of a biological system such as CRISPR edits and chemogenomic screening (Jones & Bunnage, 2017; Jost & Weissman, 2018). In some cases, phenotypic screening can also be used orthogonally to identify and validate molecules that alter a specific in vitro cellular phenotype in a mechanistically agnostic manner (Fig 2C). This approach can identify compounds that are active against multiple targets and unknown pathways in physiologically relevant disease models. Until recently, microscopy‐based approaches have been the most common way to read out parameters of the cellular phenotype and the potential shift toward a desired phenotype. Recently, there has been interest to explore MS approaches to unbiasedly screen the phenotype by monitoring specific metabolites, protein substrates and activation of biological pathways.
While MS‐based in vitro HTS assays are achievable on current instrumentation, there is a renewed interest in the pharmaceutical industry toward phenotypic cellular assays as these allow the identification of novel pathways that lead to the wanted outcome; most of these assays are currently performed using fluorescence microscopy or flow cytometry. Cellular assays for evaluating compound efficacy at moderating or reversing a cellular phenotype presents an interesting challenge for MS platforms as the system becomes inherently more complex.
A well‐established application for whole cell phenotyping is the classification of micro‐organisms by MALDI‐TOF MS, also known as biotyping (Claydon et al, 1996). Throughout the past decades, this has led to sensitive, robust, and inexpensive phenotyping of microorganisms (Mutters et al, 2014; Patel, 2015). Application of whole cell MALDI‐TOF MS methodologies to mammalian cells has not yet reached the heights of typical microbial biotyping methods but is rising as a promising technology for phenotypic screening and development of drug discovery assays. Compared to microbial cells, eukaryotic cells have a much higher complexity, with intricate cellular networks and cell cycle states influenced by their spatial anatomy (Munteanu & Hopf, 2013), making them an excellent target for MALDI‐TOF MS.
For mammalian cells, both the low‐molecular mass range (100–1,000 Da), which is mostly populated by lipids and high‐abundant cellular metabolites, and the high molecular range (2,000–20,000 Da), which contains peptides and small proteins, can be used for fingerprinting. Fingerprinting of mammalian cell protein biomarkers has been successfully applied to phenotype different cancer cell lines (Serafim et al, 2017), classify immune cells (Ouedraogo et al, 2012), iPSC embryonic stem cell differentiation (Heap et al, 2019), as well as monitor early stress or apoptosis signals in cell lines (Schwamb et al, 2013). The classification of cell lines from primary tissues can be complicated by cell heterogeneity, but MALDI‐TOF MS has proven to be sensitive and robust at distinguishing tissue‐derived cell mixtures (Petukhova et al, 2019), as well as classifying differentiated cells from primary blood monocytes (Portevin et al, 2015). Typically, these strategies use multivariate analysis and identification of unique features for classification that, when combined with flow cytometry, microscopy or known biomarker analysis, result in robust MALDI‐TOF MS methodologies. A proposed strategy to perform an intact cell HT assay using MALDI‐TOF MS is depicted in Fig 4. Cells grown with a “diseased” state and a “healthy” control state are cultured with relevant control compounds and a library of compounds to be tested. The metabolite profiling of these cells using MALDI‐TOF MS will provide a number of biomolecules (i.e., biomarkers) representing the “diseased” and “healthy” cellular states. Relevant biomarkers can be used as a read‐out for HTS using their ratio in the whole data set. Alternatively, unsupervised approaches or machine learning strategies can provide multidimensional insights if compounds return the “diseased” state back to “healthy.” If additional, known inhibitors of relevant pathways or inducers of cell toxicity are added, the assay can be multiplexed to obtain additional information on the compounds tested.
Figure 4. Cellular phenotypic assays by MALDI‐TOF mass spectrometry.
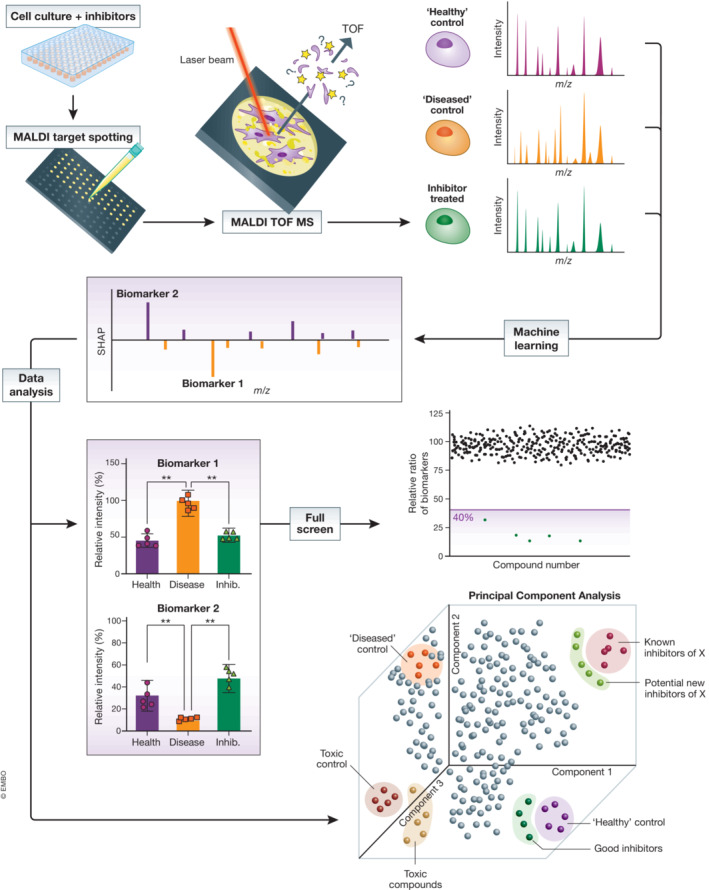
Cells or extracts of cells are spotted by liquid handling robots onto a MALDI target. MALDI‐TOF MS analysis of these samples defines “fingerprints” of “healthy” and “diseased” controls. Characterization of these complex fingerprints, potentially through machine learning or dimensionality reduction analysis (such as principal component analysis), allows the identification of biomarkers specific for the phenotypes. Changes of these biomarkers can be used as a read‐out for a drug discovery screen against many chemical moieties. Since the readout produces information‐rich multi‐dimensional data, the use of known inhibitors and cytotoxic compounds can be used to multiplex and identify novel compounds in these pathways.
In the low‐molecular range, mammalian cells exhibit dynamic lipid profiles that are often indicative of cell phenotype or disease state (Reddy & Sambasiva Rao, 2006; Fuchs & Schiller, 2008; Goldberg Ira et al, 2012). Imaging mass spectrometry (IMS) has already demonstrated that MALDI‐MS is well suited for lipid analysis of cells, and therefore MS imaging methods for lipids have been expanded into cellular classification (Holčapek et al, 2015). These assays are exceptionally sensitive, requiring small numbers of cells or even allowed the profiling of single cells such as the classification of astrocytes and neurons by Neumann et al (2019a, 2019b), who were also able to show that this was robust across 30,000 individual rodent cerebellar cells. In another study, a proof‐of‐concept assay demonstrated that inhibitors of fatty acid synthase (FASN), which are key for cancer proliferation can be identified by MALDI‐MS (Weigt et al, 2019). Combined with automated liquid handling and sample preparation, this study demonstrates that lipid analysis of whole mammalian cells is suitable for development of drug discovery assays to identify inhibitors of lipid metabolism (Weigt et al, 2018).
More recently, an automated, label‐free MALDI‐TOF MS cell assay was developed to measure compound uptake and inhibition of that uptake through the transporter OATP2B1 (Unger et al, 2020, 2021), providing a proof of principle for the application of MALDI MS cellular assay for rapid direct assessment of drug transporter function. Here, a 384‐well plate was prepared in less 2 min and analyzed in 10 min. In another study, a label‐free cellular phenotypic drug discovery assay was developed to identify anti‐inflammatory drugs in human monocytes derived from acute myeloid leukaemia. The screen identified that the inhibitor nilotinib blocked LPS‐induced inflammatory responses (preprint: Marín‐Rubio et al, 2021).
Even though IMS is mainly used to visualize the localization of compounds in tissue sections, the instrumentation also lends itself well to microarray formats, which can increase sample throughput. These microarray formats can be used for tissue or organ sections as these grid‐like applications generates defined coordinates for systematic sampling across a surface (Groseclose et al, 2008). In a recent attempt to incorporate cell‐based assays using MALDI‐IMS, Guevara et al (2021) integrated microarrays and MALDI‐IMS to demonstrate the potential for miniaturized (down to 40 nl and 10 cells per spot) HT cell screening with the biochemical analysis capabilities of MS in a single platform.
Limitations, future outlook, and conclusions
Advancement in the instrumentation and methods have led to the increasingly widespread acceptance and utilization of MS‐based HTS platforms in drug discovery R&D. This has been particularly impactful in the HTS field where MS assays have typically lacked the throughput to compete with conventional fluorescence or luminescence assays. An ideal MS‐based HTS platform can now meet the following criteria: speed of analysis, robustness, low sample volume, high sensitivity, ease of use, wide mass range coverage, accurate and precise quantification without the need for compromises in assay design, and direct detection of native biological analytes. In the past decade, there has been a surge in the development of these assays covering a wide variety of MS techniques and biological targets covered in this review. MS‐based HTS approaches are continuously improving in terms of throughput and sensitivity; this is offset by certain limitations discussed herein. MS is often not compatible with biochemical assay reactions that contain high concentration of salts, detergents and common buffering agents as these can induce ion suppression and poor assay robustness and reproducibility (Chandler et al, 2016; Belov et al, 2020). This is particularly inherent to MALDI where poor spot‐to‐spot reproducibility can occur, and a highly variable response is observed as a consequence. This is also common in AMI/ADE approaches that require careful consideration of assay volumes, buffers, and solvents to ensure uniform ejection and ionization. The most appropriate MS technique for tackling non‐suitable assay compositions is often RapidFire‐MS. However, this is one of the lowest throughput technologies available for developing HTS MS assays. These problems can be mitigated through a more judicious selection of assay matrix components, by applying an appropriate internal standard, performing an on‐target washing step, or by conducting relative quantification by measuring substrate‐to‐product ratio. Moreover, MS‐based readouts are susceptible to isobaric interference, which can be a source of false results for analytes within the mass range of the test compounds. To alleviate this issue, Winter et al (2022) suggest using counter assays (tandem MS or orthogonal readout technologies) to rule out false positives.
Further method validation and multi‐site standardization of sample preparation, data acquisition and data processing strategies will be needed to define best practices and reporting guidelines. There have been considerable efforts in the last two decades to address the crisis of reproducibility for drug discovery by incorporating the best practise in assay methodologies. The recommendation put forth in the National Center for Advancing Translational Sciences Assay Guidance Manual, a guide originally developed by Eli Lilly, is a great resource that offers step‐by‐step guidance for drug developers for planning and creating projects for HTS, as well as other steps in the drug discovery pipeline (Markossian et al, 2004, 2021). This resource is updated quarterly with more than 100 authors' contributions to date.
Alternative hit‐identification strategies such as ASMS, covalent FBDD and phenotypic assays have also benefited greatly from the recent advancements in MS technologies. For ASMS and covalent fragment screening in particular, MS underpins the screening concept and is critical for identification and characterization of positive binders. The throughput of LC–MS ASMS now rivals that of traditional HTS for hit‐ID and in more recent years novel strategies and platforms such as MALDI/SAMDI‐TOF‐MS have also demonstrated promise. For covalent fragment drug discovery, MS plays an important role in both initial screening, subsequent characterization of binding, as well as chemoproteomics to understand MoA and target engagement in a biological system. These approaches do not necessarily fit HT criteria as of yet, with sample preparation and instrument limitations often being a bottleneck. The further development of novel, state‐of‐the‐art MS platforms will likely factor as to whether these alternative drug discovery approaches to classical hit ID campaign can be incorporated into routine discovery screening strategies.
Author contributions
Maria Emilia Dueñas: Conceptualization; investigation; writing – original draft; writing – review and editing. Rachel E Peltier‐Heap: Conceptualization; investigation; writing – original draft; writing – review and editing. Melanie Leveridge: Conceptualization; writing – original draft; writing – review and editing. Roland S Annan: Conceptualization; supervision; writing – original draft; project administration; writing – review and editing. Frank H Büttner: Conceptualization; writing – original draft; writing – review and editing. Matthias Trost: Conceptualization; supervision; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
REP‐H, ML, and RSA are employees of GSK. FHB is an employee of Boehringer Ingelheim. The other authors declare that they have no conflict of interest.
Acknowledgements
MED is a Marie Sklodowska Curie Fellow within the European Union's H2020 Marie Skodowska‐Curie Actions research and innovation programme (grant agreement No. 890296).
EMBO Mol Med (2023) 15: e14850
See the Glossary for abbreviations used in this article.
Contributor Information
Maria Emilia Dueñas, Email: maria.duenas@ncl.ac.uk.
Matthias Trost, Email: matthias.trost@ncl.ac.uk.
References
- Adam GC, Meng J, Rizzo JM, Amoss A, Lusen JW, Patel A, Riley D, Hunt R, Zuck P, Johnson EN et al (2015) Use of high‐throughput mass spectrometry to reduce false positives in protease uHTS screens. J Biomol Screen 20: 212–222 [DOI] [PubMed] [Google Scholar]
- Akçay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP et al (2016) Inhibition of Mcl‐1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol 12: 931–936 [DOI] [PubMed] [Google Scholar]
- Anastasiou D, Yu Y, Israelsen WJ, Jiang J‐K, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A et al (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8: 839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annis DA, Athanasopoulos J, Curran PJ, Felsch JS, Kalghatgi K, Lee WH, Nash HM, Orminati J-PA, Rosner KE, Shipps GW et al (2004) An affinity selection–mass spectrometry method for the identification of small molecule ligands from self-encoded combinatorial libraries. Int J Mass Spectrom 238: 77–83 [Google Scholar]
- Annis DA, Nickbarg E, Yang X, Ziebell MR, Whitehurst CE (2007) Affinity selection‐mass spectrometry screening techniques for small molecule drug discovery. Curr Opin Chem Biol 11: 518–526 [DOI] [PubMed] [Google Scholar]
- Ban L, Pettit N, Li L, Stuparu AD, Cai L, Chen W, Guan W, Han W, Wang PG, Mrksich M (2012) Discovery of glycosyltransferases using carbohydrate arrays and mass spectrometry. Nat Chem Biol 8: 769–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeman K, Baumgärtner J, Laubenheimer M, Hergesell K, Hoffmann M, Pehl U, Fischer F, Pieck J‐C (2017) Integration of an in situ MALDI‐based high‐throughput screening process: a case study with receptor tyrosine kinase c‐MET. SLAS Discov 22: 1203–1210 [DOI] [PubMed] [Google Scholar]
- Belov AM, Kozole J, Bean MF, Machutta CA, Zhang G, Gao EN, Ghislain L, Datwani SS, Leveridge M, Annan RS (2020) Acoustic mist ionization‐mass spectrometry: a comparison to conventional high‐throughput screening and compound profiling platforms. Anal Chem 92: 13847–13854 [DOI] [PubMed] [Google Scholar]
- Bretschneider T, Luippold AH, Romig H, Bischoff D, Klinder K, Nicklin P, Rist W (2017) Ultrafast and predictive mass spectrometry–based autotaxin assays for label‐free potency screening. SLAS Discov 22: 425–432 [DOI] [PubMed] [Google Scholar]
- Bretschneider T, Ozbal C, Holstein M, Winter M, Buettner FH, Thamm S, Bischoff D, Luippold AH (2019) RapidFire BLAZE‐mode is boosting ESI‐MS toward high‐throughput‐screening. SLAS Technol 24: 386–393 [DOI] [PubMed] [Google Scholar]
- Challen B, Cramer R (2022) Advances in ionisation techniques for mass spectrometry‐based omics research. Proteomics 22: e2100394 [DOI] [PubMed] [Google Scholar]
- Chandler J, Haslam C, Hardy N, Leveridge M, Marshall P (2016) A systematic investigation of the best buffers for use in screening by MALDI–mass spectrometry. SLAS Discov 22: 1262–1269 [DOI] [PubMed] [Google Scholar]
- Claydon MA, Davey SN, Edwards‐Jones V, Gordon DB (1996) The rapid identification of intact microorganisms using mass spectrometry. Nat Biotechnol 14: 1584–1586 [DOI] [PubMed] [Google Scholar]
- Coburn CA, Stachel SJ, Li Y‐M, Rush DM, Steele TG, Chen‐Dodson E, Holloway MK, Xu M, Huang Q, Lai M‐T et al (2004) Identification of a small molecule nonpeptide active site β‐secretase inhibitor that displays a nontraditional binding mode for aspartyl proteases. J Med Chem 47: 6117–6119 [DOI] [PubMed] [Google Scholar]
- Collings BA, Dima MD, Ivosev G, Zhong F (2014) A high dynamic range pulse counting detection system for mass spectrometry. Rapid Commun Mass Spectrom 28: 209–216 [DOI] [PubMed] [Google Scholar]
- Comess KM, Trumbull JD, Park C, Chen Z, Judge RA, Voorbach MJ, Coen M, Gao L, Tang H, Kovar P et al (2006) Kinase drug discovery by affinity selection/mass spectrometry (ASMS): application to DNA damage checkpoint kinase Chk1. SLAS Discov 11: 755–764 [DOI] [PubMed] [Google Scholar]
- Coyne AG, Scott DE, Abell C (2010) Drugging challenging targets using fragment‐based approaches. Curr Opin Chem Biol 14: 299–307 [DOI] [PubMed] [Google Scholar]
- De Cesare V, Johnson C, Barlow V, Hastie J, Knebel A, Trost M (2018) The MALDI‐TOF E2/E3 ligase assay as universal tool for drug discovery in the ubiquitin pathway. Cell Chem Biol 25: 1117–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare V, Moran J, Traynor R, Knebel A, Ritorto MS, Trost M, McLauchlan H, Hastie CJ, Davies P (2020) High‐throughput matrix‐assisted laser desorption/ionization time‐of‐flight (MALDI‐TOF) mass spectrometry–based deubiquitylating enzyme assay for drug discovery. Nat Protoc 15: 4034–4057 [DOI] [PubMed] [Google Scholar]
- Dittakavi S, Mahadevan L, Chandrashekar DV, Bhamidipati RK, Suresh J, Dhakshinamoorthy S, Li Z, Baerenz F, Tennagels N, Mullangi R (2020) High‐throughput screening assay for the quantification of Cer d18:1/16:0, d18:1/24:0, d18:1/24:1, d18:1/18:0, d18:1/14:0, d18:1/20:0, and d18:1/22:0 in HepG2 cells using RapidFire mass spectrometry. Biomed Chromatogr 34: e4790 [DOI] [PubMed] [Google Scholar]
- Djambazova KV, Klein DR, Migas LG, Neumann EK, Rivera ES, Van de Plas R, Caprioli RM, Spraggins JM (2020) Resolving the complexity of spatial lipidomics using MALDI TIMS imaging mass spectrometry. Anal Chem 92: 13290–13297 [DOI] [PubMed] [Google Scholar]
- Dubiella C, Pinch BJ, Koikawa K, Zaidman D, Poon E, Manz TD, Nabet B, He S, Resnick E, Rogel A et al (2021) Sulfopin is a covalent inhibitor of Pin1 that blocks Myc‐driven tumors in vivo. Nat Chem Biol 17: 954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Aneed A, Cohen A, Banoub J (2009) Mass spectrometry, review of the basics: electrospray, MALDI, and commonly used mass analyzers. Appl Spectrosc Rev 44: 210–230 [Google Scholar]
- Fadeyi OO, Hoth LR, Choi C, Feng X, Gopalsamy A, Hett EC, Kyne RE, Robinson RP, Jones LH (2017) Covalent enzyme inhibition through fluorosulfate modification of a noncatalytic serine residue. ACS Chem Biol 12: 2015–2020 [DOI] [PubMed] [Google Scholar]
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM (1989) Electrospray ionization for mass spectrometry of large biomolecules. Science 246: 64–71 [DOI] [PubMed] [Google Scholar]
- Forbes CD, Toth JG, Özbal CC, LaMarr WA, Pendleton JA, Rocks S, Gedrich RW, Osterman DG, Landro JA, Lumb KJ (2007) High‐throughput mass spectrometry screening for inhibitors of phosphatidylserine decarboxylase. J Biomol Screen 12: 628–634 [DOI] [PubMed] [Google Scholar]
- Franken H, Mathieson T, Childs D, Sweetman GMA, Werner T, Tögel I, Doce C, Gade S, Bantscheff M, Drewes G et al (2015) Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat Protoc 10: 1567–1593 [DOI] [PubMed] [Google Scholar]
- Fuchs B, Schiller J (2008) MALDI‐TOF MS analysis of lipids from cells, tissues and body fluids. In Lipids in Health and Disease, pp 541–565. Dordrecht: Springer Netherlands; [DOI] [PubMed] [Google Scholar]
- Goldberg Ira J, Trent Chad M, Schulze PC (2012) Lipid metabolism and toxicity in the heart. Cell Metab 15: 805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LJ, Allen M, Artursson P, Hann MM, Leavens BJ, Mateus A, Readshaw S, Valko K, Wayne GJ, West A (2016) Direct measurement of intracellular compound concentration by rapidfire mass spectrometry offers insights into cell permeability. J Biomol Screen 21: 156–164 [DOI] [PubMed] [Google Scholar]
- Grant EK, Fallon DJ, Eberl HC, Fantom KGM, Zappacosta F, Messenger C, Tomkinson NCO, Bush JT (2019) A photoaffinity displacement assay and probes to study the cyclin‐dependent kinase family. Angew Chem Int Ed 58: 17322–17327 [DOI] [PubMed] [Google Scholar]
- Grant EK, Fallon DJ, Hann MM, Fantom KGM, Quinn C, Zappacosta F, Annan RS, Chung C‐w, Bamborough P, Dixon DP et al (2020) A photoaffinity‐based fragment‐screening platform for efficient identification of protein ligands. Angew Chem Int Ed 59: 21096–21105 [DOI] [PubMed] [Google Scholar]
- Groseclose MR, Massion PP, Chaurand P, Caprioli RM (2008) High‐throughput proteomic analysis of formalin‐fixed paraffin‐embedded tissue microarrays using MALDI imaging mass spectrometry. Proteomics 8: 3715–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara C, Paulssen D, Popova AA, Hopf C, Levkin PA (2021) Fast nanoliter‐scale cell assays using droplet microarray–mass spectrometry imaging. Adv Biol 5: 2000279 [DOI] [PubMed] [Google Scholar]
- Guitot K, Scarabelli S, Drujon T, Bolbach G, Amoura M, Burlina F, Jeltsch A, Sagan S, Guianvarc'h D (2014) Label‐free measurement of histone lysine methyltransferases activity by matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry. Anal Biochem 456: 25–31 [DOI] [PubMed] [Google Scholar]
- Guitot K, Drujon T, Burlina F, Sagan S, Beaupierre S, Pamlard O, Dodd RH, Guillou C, Bolbach G, Sachon E et al (2017) A direct label‐free MALDI‐TOF mass spectrometry based assay for the characterization of inhibitors of protein lysine methyltransferases. Anal Bioanal Chem 409: 3767–3777 [DOI] [PubMed] [Google Scholar]
- Gurard‐Levin ZA, Kilian KA, Kim J, Bähr K, Mrksich M (2010) Peptide arrays identify isoform‐selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem Biol 5: 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard‐Levin ZA, Scholle MD, Eisenberg AH, Mrksich M (2011) High‐throughput screening of small molecule libraries using SAMDI mass spectrometry. ACS Comb Sci 13: 347–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häbe TT, Liu C, Covey TR, Simon RP, Reindl W, Büttner FH, Winter M, Bischoff D, Luippold AH, Runge F (2020) Ultrahigh‐throughput ESI‐MS: sampling pushed to six samples per second by acoustic ejection mass spectrometry. Anal Chem 92: 12242–12249 [DOI] [PubMed] [Google Scholar]
- Hall RJ, Mortenson PN, Murray CW (2014) Efficient exploration of chemical space by fragment‐based screening. Prog Biophys Mol Biol 116: 82–91 [DOI] [PubMed] [Google Scholar]
- Haslam C, Hellicar J, Dunn A, Fuetterer A, Hardy N, Marshall P, Paape R, Pemberton M, Resemannand A, Leveridge M (2015) The evolution of MALDI‐TOF mass spectrometry toward ultra‐high‐throughput screening: 1536‐well format and beyond. J Biomol Screen 21: 176–186 [DOI] [PubMed] [Google Scholar]
- Hassiepen U, Eidhoff U, Meder G, Bulber J‐F, Hein A, Bodendorf U, Lorthiois E, Martoglio B (2007) A sensitive fluorescence intensity assay for deubiquitinating proteases using ubiquitin‐rhodamine110‐glycine as substrate. Anal Biochem 371: 201–207 [DOI] [PubMed] [Google Scholar]
- Heap RE, Hope AG, Pearson L‐A, Reyskens KMSE, McElroy SP, Hastie CJ, Porter DW, Arthur JSC, Gray DW, Trost M (2017) Identifying inhibitors of inflammation: a novel high‐throughput MALDI‐TOF screening assay for salt‐inducible kinases (SIKs). SLAS Discov 22: 1193–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap RE, Segarra‐Fas A, Blain AP, Findlay GM, Trost M (2019) Profiling embryonic stem cell differentiation by MALDI TOF mass spectrometry: development of a reproducible and robust sample preparation workflow. Analyst 144: 6371–6381 [DOI] [PubMed] [Google Scholar]
- Heidebrecht RW, Mulrooney C, Austin CP, Barker RH, Beaudoin JA, Cheng KC‐C, Comer E, Dandapani S, Dick J, Duvall JR et al (2012) Diversity‐oriented synthesis yields a novel lead for the treatment of malaria. ACS Med Chem Lett 3: 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helal KY, Alamgir A, Berns EJ, Mrksich M (2018) Traceless immobilization of analytes for high‐throughput experiments with SAMDI mass spectrometry. J Am Chem Soc 140: 8060–8063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highkin MK, Yates MP, Nemirovskiy OV, Lamarr WA, Munie GE, Rains JW, Masferrer JL, Nagiec MM (2011) High‐throughput screening assay for sphingosine kinase inhibitors in whole blood using RapidFire® mass spectrometry. J Biomol Screen 16: 272–277 [DOI] [PubMed] [Google Scholar]
- Holčapek M, Červená B, Cífková E, Lísa M, Chagovets V, Vostálová J, Bancířová M, Galuszka J, Hill M (2015) Lipidomic analysis of plasma, erythrocytes and lipoprotein fractions of cardiovascular disease patients using UHPLC/MS, MALDI‐MS and multivariate data analysis. J Chromatogr B 990: 52–63 [DOI] [PubMed] [Google Scholar]
- Hutchinson SE, Leveridge MV, Heathcote ML, Francis P, Williams L, Gee M, Munoz‐Muriedas J, Leavens B, Shillings A, Jones E et al (2011) Enabling lead discovery for histone lysine demethylases by high‐throughput RapidFire mass spectrometry. J Biomol Screen 17: 39–48 [DOI] [PubMed] [Google Scholar]
- Johansson H, Isabella Tsai Y‐C, Fantom K, Chung C‐W, Kümper S, Martino L, Thomas DA, Eberl HC, Muelbaier M, House D et al (2019) Fragment‐based covalent ligand screening enables rapid discovery of inhibitors for the RBR E3 ubiquitin ligase HOIP. J Am Chem Soc 141: 2703–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LH, Bunnage ME (2017) Applications of chemogenomic library screening in drug discovery. Nat Rev Drug Discov 16: 285–296 [DOI] [PubMed] [Google Scholar]
- Jost M, Weissman JS (2018) CRISPR approaches to small molecule target identification. ACS Chem Biol 13: 366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas M, Bachmann D, Hillenkamp F (1985) Influence of the wavelength in high‐irradiance ultraviolet laser desorption mass spectrometry of organic molecules. Anal Chem 57: 2935–2939 [Google Scholar]
- Kathman SG, Xu Z, Statsyuk AV (2014) A fragment‐based method to discover irreversible covalent inhibitors of cysteine proteases. J Med Chem 57: 4969–4974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitata RB, Choong W‐K, Tsai C‐F, Lin P‐Y, Chen B‐S, Chang Y‐C, Nesvizhskii AI, Sung T‐Y, Chen Y‐J (2021) A data‐independent acquisition‐based global phosphoproteomics system enables deep profiling. Nat Commun 12: 2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkel H, Brown J, Richardson K, Hoyes E, Morris M, Cramer R (2022) Ultrahigh‐throughput sample analysis using liquid atmospheric pressure matrix‐assisted laser desorption/ionization mass spectrometry. Anal Chem 94: 4141–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung C, Hixon J, Choe S, Marks K, Gross S, Murphy E, DeLaBarre B, Cianchetta G, Sethumadhavan S, Wang X et al (2012) Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem Biol 19: 1187–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutilek VD, Andrews CL, Richards MP, Xu Z, Sun T, Chen Y, Hashke A, Smotrov N, Fernandez R, Nickbarg EB et al (2016) Integration of affinity selection–mass spectrometry and functional cell‐based assays to rapidly triage druggable target space within the NF‐κB pathway. J Biomol Screen 21: 608–619 [DOI] [PubMed] [Google Scholar]
- Liddle J, Hutchinson JP, Kitchen S, Rowland P, Neu M, Cecconie T, Holmes DS, Jones E, Korczynska J, Koumantou D et al (2020) Targeting the regulatory site of ER aminopeptidase 1 leads to the discovery of a natural product modulator of antigen presentation. J Med Chem 63: 3348–3358 [DOI] [PubMed] [Google Scholar]
- Long MJC, Aye Y (2017) Privileged electrophile sensors: a resource for covalent drug development. Cell Chem Biol 24: 787–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Kostic M, Zhang T, Che J, Patricelli MP, Jones LH, Chouchani ET, Gray NS (2021) Fragment‐based covalent ligand discovery. RSC Chem Biol 2: 354–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens AK, Heidebrecht RW Jr, Mulrooney C, Beaudoin JA, Comer E, Duvall JR, Fitzgerald ME, Masi D, Galinsky K, Scherer CA et al (2015) Diversity‐oriented synthesis probe targets Plasmodium falciparum cytochrome b ubiquinone reduction site and synergizes with oxidation site inhibitors. J Infect Dis 211: 1097–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín‐Rubio JL, Heap RE, Heunis T, Dueñas ME, Inns J, Scott J, Simpson AJ, Blair H, Heidenreich O, Allan JM et al (2021) A MALDI‐TOF assay identifies nilotinib as an inhibitor of inflammation in acute myeloid leukaemia. bioRxiv 10.1101/2021.03.29.437557 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markossian S, Grossman A, Brimacombe K, Arkin M, Auld D, Austin CP, Baell J, Chung TD, Coussens NP, Dahlin JL (2004) Assay guidance manual [Internet]
- Markossian S, Coussens NP, Dahlin JL, Sittampalam GS (2021) Assay guidance manual for drug discovery: robust or go bust. SLAS Discov 26: 1241–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxine J, LaMarr WA, Ozbal C (2009) Mass spectrometry in high throughput screening: a case study on acetyl‐coenzyme A carboxylase using RapidFire® – mass spectrometry (RF‐MS). Comb Chem High Throughput Screen 12: 752–759 [DOI] [PubMed] [Google Scholar]
- Min D‐H, Tang W‐J, Mrksich M (2004) Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nat Biotechnol 22: 717–723 [DOI] [PubMed] [Google Scholar]
- Moffat JG, Vincent F, Lee JA, Eder J, Prunotto M (2017) Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat Rev Drug Discov 16: 531–543 [DOI] [PubMed] [Google Scholar]
- Mohs RC, Greig NH (2017) Drug discovery and development: role of basic biological research. Alzheimers Dement (N Y) 3: 651–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morato NM, Holden DT, Cooks RG (2020) High‐throughput label‐free enzymatic assays using desorption electrospray‐ionization mass spectrometry. Angew Chem Int Ed 59: 20459–20464 [DOI] [PubMed] [Google Scholar]
- Mrksich M (2008) Mass spectrometry of self‐assembled monolayers: a new tool for molecular surface science. ACS Nano 2: 7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muckenschnabel I, Falchetto R, Mayr LM, Filipuzzi I (2004) SpeedScreen: label-free liquid chromatography–mass spectrometry-based high-throughput screening for the discovery of orphan protein ligands. Anal Biochem 324: 241–249 [DOI] [PubMed] [Google Scholar]
- Munteanu B, Hopf C (2013) Emergence of whole‐cell MALDI‐MS biotyping for high‐throughput bioanalysis of mammalian cells? Bioanalysis 5: 885–893 [DOI] [PubMed] [Google Scholar]
- Mutters NT, Hodiamont CJ, de Jong MD, HPJ O, van den Boogaard M, Visser CE (2014) Performance of kiestra total laboratory automation combined with MS in clinical microbiology practice. Ann Lab Med 34: 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim N, Moukheiber S, Daou S, Kourie HR (2021) KRAS‐G12C covalent inhibitors: a game changer in the scene of cancer therapies. Crit Rev Oncol Hematol 168: 103524 [DOI] [PubMed] [Google Scholar]
- Neumann EK, Comi TJ, Rubakhin SS, Sweedler JV (2019a) Lipid heterogeneity between astrocytes and neurons revealed by single‐cell MALDI‐MS combined with immunocytochemical classification. Angew Chem Int Ed 58: 5910–5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann EK, Ellis JF, Triplett AE, Rubakhin SS, Sweedler JV (2019b) Lipid analysis of 30 000 individual rodent cerebellar cells using high‐resolution mass spectrometry. Anal Chem 91: 7871–7878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olp MD, Sprague DJ, Goetz CJ, Kathman SG, Wynia‐Smith SL, Shishodia S, Summers SB, Xu Z, Statsyuk AV, Smith BC (2020) Covalent‐fragment screening of BRD4 identifies a ligandable site orthogonal to the acetyl‐lysine binding sites. ACS Chem Biol 15: 1036–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM (2013) K‐Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503: 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouedraogo R, Daumas A, Ghigo E, Capo C, Mege J‐L, Textoris J (2012) Whole‐cell MALDI‐TOF MS: a new tool to assess the multifaceted activation of macrophages. J Proteomics 75: 5523–5532 [DOI] [PubMed] [Google Scholar]
- Patel R (2015) MALDI‐TOF MS for the diagnosis of infectious diseases. Clin Chem 61: 100–111 [DOI] [PubMed] [Google Scholar]
- Patel K, Sherrill J, Mrksich M, Scholle MD (2015) Discovery of SIRT3 inhibitors using SAMDI mass spectrometry. J Biomol Screen 20: 842–848 [DOI] [PubMed] [Google Scholar]
- Pettinger J, Le Bihan Y‐V, Widya M, van Montfort RLM, Jones K, Cheeseman MD (2017) An irreversible inhibitor of HSP72 that unexpectedly targets lysine‐56. Angew Chem Int Ed 56: 3536–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova VZ, Young AN, Wang J, Wang M, Ladanyi A, Kothari R, Burdette JE, Sanchez LM (2019) Whole cell MALDI fingerprinting is a robust tool for differential profiling of two‐component mammalian cell mixtures. J Am Soc Mass Spectrom 30: 344–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portevin D, Pflüger V, Otieno P, Brunisholz R, Vogel G, Daubenberger C (2015) Quantitative whole‐cell MALDI‐TOF MS fingerprints distinguishes human monocyte sub‐populations activated by distinct microbial ligands. BMC Biotechnol 15: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent R, Annis DA, Dandliker PJ, Ortholand J‐Y, Roche D (2021) Exploring new targets and chemical space with affinity selection‐mass spectrometry. Nat Rev Chem 5: 62–71 [DOI] [PubMed] [Google Scholar]
- Radosevich AJ, Pu F, Chang‐Yen D, Sawicki JW, Talaty NN, Elsen NL, Williams JD, Pan JY (2022) Ultra‐high‐throughput ambient MS: direct analysis at 22 samples per second by infrared matrix‐assisted laser desorption electrospray ionization mass spectrometry. Anal Chem 94: 4913–4918 [DOI] [PubMed] [Google Scholar]
- Reddy JK, Sambasiva Rao M (2006) Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Phys Gastrointest Liver Phys Ther 290: G852–G858 [DOI] [PubMed] [Google Scholar]
- Resnick E, Bradley A, Gan J, Douangamath A, Krojer T, Sethi R, Geurink PP, Aimon A, Amitai G, Bellini D et al (2019) Rapid covalent‐probe discovery by electrophile‐fragment screening. J Am Chem Soc 141: 8951–8968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritorto MS, Ewan R, Perez‐Oliva AB, Knebel A, Buhrlage SJ, Wightman M, Kelly SM, Wood NT, Virdee S, Gray NS et al (2014) Screening of DUB activity and specificity by MALDI‐TOF mass spectrometry. Nat Commun 5: 4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopper S, Kahraman A, Leuenberger P, Feng Y, Piazza I, Müller O, Boersema PJ, Picotti P (2017) Measuring protein structural changes on a proteome‐wide scale using limited proteolysis‐coupled mass spectrometry. Nat Protoc 12: 2391–2410 [DOI] [PubMed] [Google Scholar]
- Schreiber Stuart L, Kotz Joanne D, Li M, Aubé J, Austin Christopher P, Reed John C, Rosen H, White EL, Sklar Larry A, Lindsley Craig W et al (2015) Advancing biological understanding and therapeutics discovery with small‐molecule probes. Cell 161: 1252–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamb S, Munteanu B, Meyer B, Hopf C, Hafner M, Wiedemann P (2013) Monitoring CHO cell cultures: cell stress and early apoptosis assessment by mass spectrometry. J Biotechnol 168: 452–461 [DOI] [PubMed] [Google Scholar]
- Serafim V, Shah A, Puiu M, Andreescu N, Coricovac D, Nosyrev A, Spandidos DA, Tsatsakis AM, Dehelean C, Pinzaru I (2017) Classification of cancer cell lines using matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry and statistical analysis. Int J Mol Med 40: 1096–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Jeong JW, Wurz RP, Achanta P, Arvedson T, Bartberger MD, Campuzano IDG, Fucini R, Hansen SK, Ingersoll J et al (2019) Discovery of N‐(1‐Acryloylazetidin‐3‐yl)‐2‐(1H‐indol‐1‐yl)acetamides as covalent inhibitors of KRASG12C. ACS Med Chem Lett 10: 1302–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido Y, Tomoike F, Kimura Y, Kuwata K, Yano T, Fukui K, Fujikawa H, Sekido Y, Murakami‐Tonami Y, Kameda T et al (2017) A covalent G‐site inhibitor for glutathione S‐transferase Pi (GSTP1‐1). Chem Commun 53: 11138–11141 [DOI] [PubMed] [Google Scholar]
- Simon RP, Winter M, Kleiner C, Ries R, Schnapp G, Heimann A, Li J, Zuvela‐Jelaska L, Bretschneider T, Luippold AH et al (2020) MALDI‐TOF mass spectrometry‐based high‐throughput screening for inhibitors of the cytosolic DNA sensor cGAS. SLAS Discov 25: 372–383 [DOI] [PubMed] [Google Scholar]
- Simon RP, Häbe TT, Ries R, Winter M, Wang Y, Fernández‐Montalván A, Bischoff D, Runge F, Reindl W, Luippold AH et al (2021a) Acoustic ejection mass spectrometry: a fully automatable technology for high‐throughput screening in drug discovery. SLAS Discov 26: 961–973 [DOI] [PubMed] [Google Scholar]
- Simon RP, Winter M, Kleiner C, Wehrle L, Karnath M, Ries R, Zeeb M, Schnapp G, Fiegen D, Häbe TT et al (2021b) MALDI‐TOF‐based affinity selection mass spectrometry for automated screening of protein–ligand interactions at high throughput. SLAS Discov 26: 44–57 [DOI] [PubMed] [Google Scholar]
- Sinclair I, Stearns R, Pringle S, Wingfield J, Datwani S, Hall E, Ghislain L, Majlof L, Bachman M (2015) Novel acoustic loading of a mass spectrometer: toward next‐generation high‐throughput MS screening. J Lab Autom 21: 19–26 [DOI] [PubMed] [Google Scholar]
- Sinclair I, Bachman M, Addison D, Rohman M, Murray DC, Davies G, Mouchet E, Tonge ME, Stearns RG, Ghislain L et al (2019) Acoustic mist ionization platform for direct and contactless ultrahigh‐throughput mass spectrometry analysis of liquid samples. Anal Chem 91: 3790–3794 [DOI] [PubMed] [Google Scholar]
- Soulard P, McLaughlin M, Stevens J, Connolly B, Coli R, Wang L, Moore J, Kuo M‐ST, LaMarr WA, Ozbal CC et al (2008) Development of a high‐throughput screening assay for stearoyl‐CoA desaturase using rat liver microsomes, deuterium labeled stearoyl‐CoA and mass spectrometry. Anal Chim Acta 627: 105–111 [DOI] [PubMed] [Google Scholar]
- Swalm Brooke M, Hallenbeck Kenneth K, Majer Christina R, Jin L, Scott Margaret P, Moyer Mikel P, Copeland Robert A, Wigle Tim J (2013) Convergent evolution of chromatin modification by structurally distinct enzymes: comparative enzymology of histone H3 Lys27 methylation by human polycomb repressive complex 2 and vSET. Biochem J 453: 241–247 [DOI] [PubMed] [Google Scholar]
- Thomas RP, Heap RE, Zappacosta F, Grant EK, Pogány P, Besley S, Fallon DJ, Hann MM, House D, Tomkinson NCO et al (2021) A direct‐to‐biology high‐throughput chemistry approach to reactive fragment screening. Chem Sci 12: 12098–12106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK et al (2008) Discovery of a selective inhibitor of oncogenic B‐Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA 105: 3041–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger MS, Schumacher L, Enzlein T, Weigt D, Zamek‐Gliszczynski MJ, Schwab M, Nies AT, Drewes G, Schulz S, Reinhard FBM et al (2020) Direct automated MALDI mass spectrometry analysis of cellular transporter function: inhibition of OATP2B1 uptake by 294 drugs. Anal Chem 92: 11851–11859 [DOI] [PubMed] [Google Scholar]
- Unger MS, Blank M, Enzlein T, Hopf C (2021) Label‐free cell assays to determine compound uptake or drug action using MALDI‐TOF mass spectrometry. Nat Protoc 16: 5533–5558 [DOI] [PubMed] [Google Scholar]
- Vincent F, Nueda A, Lee J, Schenone M, Prunotto M, Mercola M (2022) Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat Rev Drug Discov 21: 899–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SS, Degen D, Nickbarg E, Carr D, Soriano A, Mandal M, Painter RE, Sheth P, Xiao L, Sher X et al (2017) Affinity selection–mass spectrometry identifies a novel antibacterial RNA polymerase inhibitor. ACS Chem Biol 12: 1346–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigt D, Sammour DA, Ulrich T, Munteanu B, Hopf C (2018) Automated analysis of lipid drug‐response markers by combined fast and high‐resolution whole cell MALDI mass spectrometry biotyping. Sci Rep 8: 11260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigt D, Parrish CA, Krueger JA, Oleykowski CA, Rendina AR, Hopf C (2019) Mechanistic MALDI‐TOF cell‐based assay for the discovery of potent and specific fatty acid synthase inhibitors. Cell Chem Biol 26: 1322–1331 [DOI] [PubMed] [Google Scholar]
- Wen X, Liu C, Ghislain L, Tovar K, Shah V, Stout SJ, Cifelli S, Satapati S, O'Donnell G, Sheth PR et al (2021) Direct analysis from phase‐separated liquid samples using ADE‐OPI‐MS: applicability to high‐throughput screening for inhibitors of diacylglycerol acyltransferase 2. Anal Chem 93: 6071–6079 [DOI] [PubMed] [Google Scholar]
- Whitehurst EC, Annis AD (2008) Affinity selection‐mass spectrometry and its emerging application to the high throughput screening of G protein‐coupled receptors. Comb Chem High Throughput Screen 11: 427–438 [DOI] [PubMed] [Google Scholar]
- Winter M, Bretschneider T, Kleiner C, Ries R, Hehn JP, Redemann N, Luippold AH, Bischoff D, Büttner FH (2018) Establishing MALDI‐TOF as versatile drug discovery readout to dissect the PTP1B enzymatic reaction. SLAS Discov 23: 561–573 [DOI] [PubMed] [Google Scholar]
- Winter M, Bretschneider T, Thamm S, Kleiner C, Grabowski D, Chandler S, Ries R, Kley JT, Fowler D, Bartlett C et al (2019) Chemical derivatization enables MALDI‐TOF‐based high‐throughput screening for microbial trimethylamine (TMA)‐lyase inhibitors. SLAS Discov 24: 766–777 [DOI] [PubMed] [Google Scholar]
- Winter M, Simon RP, Wang Y, Bretschneider T, Bauer M, Magarkar A, Reindl W, Fernández‐Montalván A, Montel F, Büttner FH (2022) Differential analyte derivatization enables unbiased MALDI‐TOF‐based high‐throughput screening: a proof‐of‐concept study for the discovery of catechol‐O‐methyltransferase inhibitors. SLAS Discov 27: 287–297 [DOI] [PubMed] [Google Scholar]
- Wleklinski M, Loren BP, Ferreira CR, Jaman Z, Avramova L, Sobreira TJP, Thompson DH, Cooks RG (2018) High throughput reaction screening using desorption electrospray ionization mass spectrometry. Chem Sci 9: 1647–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehender H, Le Goff F, Lehmann N, Filipuzzi I, Mayr LM (2004) SpeedScreen: the “Missing Link” between genomics and lead discovery. SLAS Discov 9: 498–505 [DOI] [PubMed] [Google Scholar]
- Zehender H, Mayr LM (2007) Application of high-throughput affinity-selection mass spectrometry for screening of chemical compound libraries in lead discovery. Expert Opin Drug Discov 2: 285–294 [DOI] [PubMed] [Google Scholar]
- Zhang T, Hatcher JM, Teng M, Gray NS, Kostic M (2019) Recent advances in selective and irreversible covalent ligand development and validation. Cell Chem Biol 26: 1486–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu C, Hua W, Ghislain LP, Liu J, Aschenbrenner L, Noell S, Dirico KJ, Lanyon LF, Steppan CM et al (2021) Acoustic ejection mass spectrometry for high‐throughput analysis. Anal Chem 93: 10850–10861 [DOI] [PubMed] [Google Scholar]