

Abstract
Recent work has highlighted a lack of diversity in genomic studies. However, less attention has been given to epigenomics. Here, we show that epigenomic studies are lacking in diversity and propose several solutions to address this problem.
Research in diverse populations is critical for understanding disease etiology and risk. Several recent publications have highlighted the lack of racial or ethnic diversity in genetic studies and have called for more research in diverse populations1,2. However, less attention has been given to epigenomics. Over the past ten years, great progress has been made in the understanding of regulatory elements through the efforts of the International Human Epigenome Consortium (IHEC), which mapped regulatory elements in a wide range of tissues and cell types, and made many of these datasets freely available to the scientific community3. This comprehensive catalogue of cis-regulatory elements and chromatin datasets has proved useful for different areas such as genomic variant annotation4, fine-mapping of genetic loci5, genome editing approaches5, and design of pipelines for single cell-sequencing analyses6.
Current information regarding the race or ethnicity of IHEC samples is sparse. We queried publicly available IHEC datasets for different statistical metrics relating to race/ethnicity and country of origin, finding only 42.7% of experiments reporting any race or ethnicity information (Supplementary Table 1, downloaded from https://www.encodeproject.org/; we used US-based ENCODE data as it was the only publicly available dataset within IHEC). Of the 5,048 publicly available experiments with race or ethnicity information, 87.1% (n=4,397) were labelled as “European”, 9.3% (n=470) were reported as African, African American or Black, 1.7% (n=87) were of Asian ancestry, and the remainder (1.9%, n=94) were of other ancestries or a combination of racial/ethnic identities, showing considerable disparity in the samples utilized for analysis (Supplementary Table 1). From 2009 to 2021, the cumulative number of experiments on “European” samples increased, far outpacing experiments on samples from other races and ethnicities (Figure 1). Although a set of experiments based on specific African populations (e.g. Luhya, Maasai, Mende, Esan, and Gambia) was posted in 2021, increasing the diversity of data available, populations from other geographic regions (e.g., South Asia, Middle East) remain underrepresented.
Figure 1: Diversity in epigenetic data samples over time:
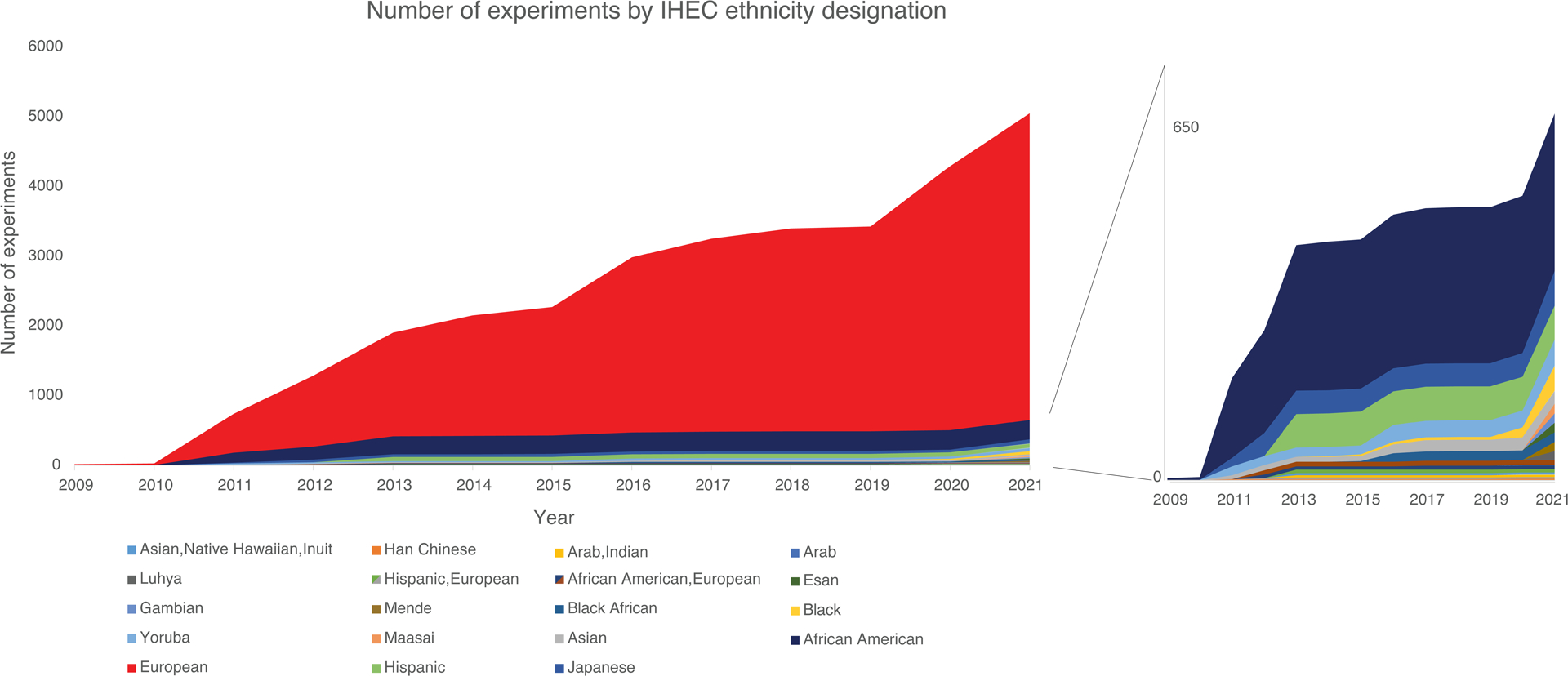
Shown is the cumulative number of samples per year in publicly-available IHEC data (2009–2020, left panel). Different segments of the chart are color-coded by ethnicity as found in IHEC. Ethnicity information was obtained from the relevant studies. Given the large proportion of samples in individuals of European ethnicity as found in IHEC (red), a zoom-in chart is provided (right panel) showing the different cumulative sample numbers in non-European populations across the same timeframe.
The breadth of epigenomic assays and tissues used is substantially more extensive for Europeans than for other races/ethnicities. Among assays, ATAC-seq, DNase-seq, ChIP-seq and DNA methylation arrays show the highest degree of diversity with data from more than 6 populations (Supplementary Table 1, Figure 2). Although Hispanics were represented in relatively few experiments (n=60), a more comprehensive set of annotations across main assay types, such as RNA-seq, DNase-seq and ChIP-seq (including ChIP-seq for CTCF and histone H3 modifications) is available for them compared to other non-European populations. We also noted that data from non-European populations largely come from cell lines. Although valuable, the immortalization and serial passage of cell lines can lead to epigenetic changes that are not present in the primary cells and tissues7. The experiments conducted in primary tissues are overwhelmingly from “Europeans”, with few primary tissue experiments in non-Europeans. Given limited non-European primary tissue samples, any differences in tissue-specific regulatory elements across populations will be hard to evaluate.
Figure 2: Diversity in epigenetic data samples by assay:

the doughnut chart (lower panel) shows the total number of samples by assay (outer ring) and by ethnicity (inner ring). Given the large proportion of samples in individuals of European ethnicity as found in IHEC, a zoom-in area chart is provided (top panel) showing the different sample number by assay in non-European populations.
An essential question in characterizing regulatory elements across populations is the role of DNA sequence variants. The extent to which ancestry-related DNA sequence variants affect epigenetic modifications is unknown. However, there is evidence for widespread epigenetic variation between populations, particularly with regards to DNA methylation8,9,10. While some sections of the epigenome are influenced by environmental exposures11,12,13, many epigenetic changes are driven by changes in the DNA sequence10,14,15,16. For example, twin studies have shown that the mean genetic heritability of DNA methylation is 19%, with some regions showing a heritability of over 90%15, suggesting that DNA methylation, particularly in those regions, is likely to be determined in large part by underlying genetic variants. Other studies have previously reported associations between individual ancestry-specific DNA sequence variants and DNA methylation differences between populations17,18. Given this evidence, we anticipate that more associations between genotype, DNA methylation and ancestry may be uncovered in the future, which could potentially help explain population disparities in disease risk. In short, the role of ancestry-related DNA sequence variants in driving epigenetic variation needs to be explored further, especially in regard to disease-associated regions.
Epigenomic resources in diverse populations could contribute to annotating and interpreting disease-associated genomic regions. Genome-wide association studies (GWAS) have identified thousands of loci for various diseases and traits19,20,21. However, many of these variants are located in non-coding regions of the genome with unclear functional consequences4,22. Mapping these variants to the regulatory elements, including promoters, enhancers, and repressors, through epigenomic markers can provide important insights into possible functional mechanisms across a variety of tissues and cell types4,23. The extent to which current epigenomic mapping resources, which are mostly European-centric, facilitate interpretation of GWAS loci in diverse populations is unknown. However, expanded epigenomic mapping data in diverse populations may improve the interpretation of disease-associated loci across populations9,24 and offer additional insights. Expanded population-specific epigenomic maps may be particularly useful for annotating and fine-mapping variants in diseases with a higher burden in non-European populations, such as prostate cancer25, hypertension26, and chronic kidney disease27.
In conclusion, additional research is warranted to evaluate the diversity in the epigenome across populations and determine the extent of population variability. Current efforts to increase representation in genomic research in diverse populations should be paired with similar efforts in epigenomics, which have, thus far, received less attention and scientific scrutiny. The posting of ancestry information, which could be inferred from sequencing or genotype array data, with existing epigenomic data could be beneficial in helping researchers understand the potential limitations for annotating and interpreting GWAS loci from different populations. Regarding IHEC, we recommend that participating consortia post genetic ancestry assignment inferred using reference genomes. While consortia may include self-reported race/ethnicity (for example in the US-based consortium reported here), we recommend analyses at the international scale first focus on genetic ancestry given the substantial challenges in standardizing race/ethnicity reporting across different countries. In addition, efforts to diversify IHEC participating countries should be promoted. Future studies should concentrate on generating high-quality data across diverse populations, using ancestry-specific reference genomes for aligning or mapping chromatin peaks from diverse populations, and developing DNA methylation arrays that adequately capture epigenomic diversity across populations. Improvement of the diversity of epigenomic resources will likely accelerate research addressing disease risk and health disparities across populations.
Supplementary Material
Acknowledgments
This project was supported in part by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH (CEB, SIB).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Popejoy AB & Fullerton SM Genomics is failing on diversity. Nature 538, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirugo G, Williams SM & Tishkoff SA The Missing Diversity in Human Genetic Studies. Cell 177, 26–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claussnitzer M et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med 373, 895–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bravo González-Blas Carmen, M. L cisTopic: cis-Regulatory topic modelling on single-cell ATAC-seq data. Nature methods 16, 397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grafodatskaya D, Chung B, Szatmari P & Weksberg R Autism spectrum disorders and epigenetics. J Am Acad Child Adolesc Psychiatry 49, 794–809 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Husquin LT et al. Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation. Genome Biology 19, 222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breeze CE et al. Epigenome-wide association study of kidney function identifies trans-ethnic and ethnic-specific loci. Genome Medicine 13, 74 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraser HB, Lam LL, Neumann SM & Kobor MS Population-specificity of human DNA methylation. Genome Biology 13, R8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai P-C et al. Smoking induces coordinated DNA methylation and gene expression changes in adipose tissue with consequences for metabolic health. Clinical Epigenetics 10, 126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Philibert R et al. A quantitative epigenetic approach for the assessment of cigarette consumption. Front. Psychol 656 (2015) doi: 10.3389/fpsyg.2015.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu AT et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 11, 303–327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birney E, Smith GD & Greally JM Epigenome-wide Association Studies and the Interpretation of Disease -Omics. PLOS Genet 12, e1006105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Dongen J et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun 7, 11115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min JL et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat Genet 53, 1311–1321 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bell JT et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biology 12, R10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heyn H et al. DNA methylation contributes to natural human variation. Genome Research 23, 1363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visscher PM et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet 101, 5–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visscher PM, Brown MA, McCarthy MI & Yang J Five Years of GWAS Discovery. Am J Hum Genet 90, 7–24 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welter D et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42, D1001–D1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards SL, Beesley J, French JD & Dunning AM Beyond GWASs: Illuminating the Dark Road from Association to Function. Am J Hum Genet 93, 779–797 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trynka G et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet 45, 124–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tehranchi A et al. Fine-mapping cis-regulatory variants in diverse human populations. eLife 8, e39595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taitt HE Global Trends and Prostate Cancer: A Review of Incidence, Detection, and Mortality as Influenced by Race, Ethnicity, and Geographic Location. American Journal of Men’s Health 12, 1807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills KT et al. Global Disparities of Hypertension Prevalence and Control: A Systematic Analysis of Population-based Studies from 90 Countries. Circulation 134, 441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bikbov B et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet 395, 709–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.