

Abstract
The proximal tubule of the kidney is programmed to reabsorb all filtered glucose and fructose. Glucose is taken up by apical sodium-glucose cotransporters SGLT2 and SGLT1 whereas SGLT5 and potentially SGLT4 and GLUT5 have been implicated in apical fructose uptake. The glucose taken up by the proximal tubule is typically not metabolized but leaves via the basolateral facilitative glucose transporter GLUT2 and is returned to the systemic circulation or used as an energy source by distal tubular segments after basolateral uptake via GLUT1. The proximal tubule generates new glucose in metabolic acidosis and the postabsorptive phase, and fructose serves as an important substrate. In fact, under physiological conditions and intake, fructose taken up by proximal tubules is primarily utilized for gluconeogenesis. In the diabetic kidney, glucose is retained and gluconeogenesis enhanced, the latter in part driven by fructose. This is maladaptive as it sustains hyperglycemia. Moreover, renal glucose retention is coupled to sodium retention through SGLT2 and SGLT1, which induces secondary deleterious effects. SGLT2 inhibitors are new anti-hyperglycemic drugs that can protect the kidneys and heart from failing independent of kidney function and diabetes. Dietary excess of fructose also induces tubular injury. This can be magnified by kidney formation of fructose under pathological conditions. Fructose metabolism is linked to urate formation, which partially accounts for fructose-induced tubular injury, inflammation, and hemodynamic alterations. Fructose metabolism favors glycolysis over mitochondrial respiration as urate suppresses aconitase in the tricarboxylic acid cycle, and has been linked to potentially detrimental aerobic glycolysis (Warburg effect).
Introduction
The kidneys handle large amounts of d-glucose. d-glucose is found naturally in plants and vegetables, and can also be formed by animals. In comparison, l-glucose does not occur naturally in nature but can be synthesized in the laboratory. In the following, “glucose” refers to d-glucose. Glucose is a valuable energy substrate the body does not want to waste or lose. Considering a GFR of approximately 180 liter/day in a healthy adult and glucose concentrations in the plasma of 5 to 10mM (90–180 mg/dL), large amounts of glucose (≈180 g/day) are filtered by the glomeruli and subsequently reabsorbed by the tubular system, primarily in the proximal tubule (345, 388). Moreover, and like the liver, the kidneys generate new glucose through the process of gluconeogenesis. The latter process also resides in the proximal tubule and contributes to systemic glucose homeostasis but also to acid-base balance since it involves the generation of new bicarbonate (366). Proximal tubule cells, which reabsorb most of the filtered glucose and generate glucose, normally do not utilize glucose as a fuel, possibly to prevent a futile cycle. In comparison, distal tubular segments make use of glucose as a primary energy substrate (388). Proximal tubule handling of glucose is important in healthy individuals, with added implications during an acid load and fasting, and gains pathophysiological relevance in disease states. This particularly includes diabetic or hyperglycemic conditions but is also relevant in nondiabetic settings (388, 438). In diabetes, renal glucose retention becomes maladaptive and a contributor to hyperglycemia. Therefore, drugs have been developed that inhibit renal glucose reabsorption to cause urinary glucose loss and lower hyperglycemia. These drugs, however, can also protect the kidney and cardiovascular system, and the involved pleiotropic mechanisms go beyond glucose homeostasis (409, 410, 413).
Similar to glucose, fructose is a hexose and monosaccharide that is metabolized in the kidney. Fructose is a component of sucrose (table sugar), which is a disaccharide of fructose and glucose. Fructose is sweeter than glucose and is included in high fructose corn syrup (HFCS) that has been added for sweetness and taste in many foods. Only a small fraction of dietary fructose escapes the liver metabolism and reaches the systemic circulation, and plasma fructose concentrations are normally only at 0.1 to 0.8mM (227). As a consequence of lower plasma concentrations, healthy kidneys filter about 10 times less fructose than glucose. Filtered fructose is predominantly reabsorbed in the proximal tubule (6, 172), where, under physiological conditions, it is primarily utilized as a substrate for gluconeogenesis to help maintain systemic glucose concentrations (255). In addition, the kidney is capable of endogenously generating fructose from glucose through the activation of the polyol pathway (197, 255). Excessive dietary fructose intake or aberrant endogenous production of fructose as observed in pathological conditions, however, can induce deleterious effects on the kidney. In fact, excessive endogenous fructose generation in the kidney has been proposed to contribute to the development and progression of several renal diseases, including diabetic nephropathy and ischemia-reperfusion acute kidney injury, but also is found in the aging kidney (255). Deleterious effects of fructose have partially been related to urate, which is a byproduct of fructose metabolism and linked to several pathological processes, including insulin resistance, endothelial dysfunction, and renal tubular injury (175, 254, 260). As such, blocking specific aspects of fructose metabolism could be considered in the treatment of specific kidney diseases (112, 159, 161, 255).
This overview article discusses the renal handling of glucose and fructose. This includes the molecular players involved in their transport, generation, and metabolism, as well as the physiological integration in a healthy kidney. The pathophysiology implications of these two sugar molecules and their interaction are discussed together with therapeutical strategies impinging on them. The interested reader is referred to recent reviews on the topic by the authors, forming the basis for the current work (99, 159, 161, 255–257, 259, 268, 388, 389, 395, 409, 413) as well as by other authors (45, 82, 170, 345, 349, 366, 367, 438). For a broader discussion of glucose transport in other organs and species, see Ref. 186.
Physiology of Renal Glucose Transport
In many organisms including human, the cellular uptake and metabolism of d-glucose provide a significant energy source (191, 453). For normal function, the brain alone requires a continuous glucose supply and uptake of approximately 125 grams of glucose every day. To assure a constant delivery, blood glucose is highly controlled. This regulation comprises hormones like glucagon and insulin that control the glucose uptake into cells as well as its storage and endogenous production (191, 453).
Glucose is a small molecule with a molecular mass of 180 that is uncharged and filtered freely in the glomeruli of the kidneys. In healthy individuals, the kidneys daily filter 160 to 180 g of glucose assuming normal glomerular filtration rate (GFR, ≈180 liter/day) and blood glucose concentrations (approximately 100 mg/dL). This amount of filtered glucose equals about one-third of the daily energy expenditure that, if not recovered by the tubular system, would be lost with the urine. Instead and in euglycemic conditions, >99% of the filtered glucose is reabsorbed by the intact tubular system. This reabsorption of glucose primarily occurs in the proximal tubule as indicated by micropuncture studies in the dog, rat, and mouse (92, 314, 402, 429) (see Figure 1). Micropuncture studies in the rat indicated that the permeability of glucose across the proximal tubules is small, and, therefore, under normal free-flow conditions, the passive transport component plays no role in glucose reabsorption (220). As discussed in the following sections, glucose is reabsorbed from the glomerular filtrate by two Na+-coupled glucose cotransporters, SGLT2 and SGLT1, which are located in the brush border membrane of the early and later proximal tubule, respectively. The reabsorption of glucose through these transporters is a saturable process. The average maximum renal transport capacity (Tmax) for glucose has been reported at around 430 and 500 g/day in female and male healthy individuals, respectively (78, 247). These maximum transport rates equal about 3-fold the typical tubular glucose load derived from glomerular filtration, that is, the tubular machinery for glucose uptake is not saturated under normal conditions. Theoretically, at a normal GFR, the Tmax should be reached and glucose begins to appear in the urine at a plasma glucose threshold of 280 mg/dL. However, even in healthy adults, individual nephrons show variation in Tmax and, as a consequence, small rates of glucose can spill into the urine starting at modestly elevated levels of plasma glucose of about 180 to 200 mg/dL. When blood glucose concentrations surpass 270 to 290 mg/dL, a linear increase in glucose excretion is observed (see Figure 2). When GFR is elevated, like in diabetes or pregnancy, glucosuria may occur at lower plasma glucose levels. In kidney disease, when GFR is reduced, glucosuria may require higher blood glucose levels unless the tubular glucose transport capacity is likewise impaired.
Figure 1. Glucose reabsorption in the kidney.

(A) Under normoglycemia, SGLT2 in the early proximal tubule reabsorbs approximately 97% of filtered glucose. The remaining approximately 3% of glucose is reabsorbed by SGLT1 in the late proximal tubule, such that urine is nearly free of glucose. SGLT2 inhibition shifts glucose reabsorption downstream and unmasks the glucose reabsorption capacity of SGLT1 (≈40% of filtered glucose, depending on glucose load; see numbers in parentheses). (B) Cell model of glucose transport: The basolateral Na+-K+-ATPase lowers cytosolic Na+ generates a negative interior voltage, thereby providing the driving force for Na+-coupled glucose uptake through SGLT2 and SGLT1 across the apical membrane. The facilitative glucose transporter GLUT2 mediates glucose transport across the basolateral membrane down its chemical gradient. Basolateral GLUT1 may contribute to reabsorb glucose or take glucose up from peritubular space. Na+-glucose cotransport is electrogenic and accompanied by paracellular Cl− transcellular K+ secretion to stabilize membrane potential; K+ early and late proximal tubule, respectively. Modified, with permission, from Vallon V, 2011 (390).
Figure 2. Tubular glucose reabsorption can be saturated.

Tubular reabsorption of glucose increases linearly with the filtered glucose load up to the point when reabsorption reaches its maximum (Tmax glucose) and glucose starts to appear in urine. Theoretically in humans, a Tmax of approximately 350 mg/min and normal GFR would result in a plasma glucose threshold of approximately 280 mg/dL. The Tmax, however, varies between individual nephrons and, therefore, low-level spilling of glucose into the urine initiates at modestly elevated plasma glucose levels of approximately 180 to 200 mg/dL in a healthy adult (see “Splay”). Normoglycemia is defined as fasted plasma glucose levels < 100mg/dL (<5.5 mM). SGLT2 inhibition reduces the renal glucose reabsorption to the transport capacity of SGLT1, that is, it reduces the renal glucose threshold (≈55–65 mg/dL) and Tmax (≈70 mg/min). Modified, with permission, from Vallon V, 2020 (388).
SGLT2 in the early proximal tubule is largely responsible for renal glucose reabsorption
Studies in the early 1980s by Barfuss and Schafer, using isolated rabbit proximal tubule segments, indicated that early and late proximal tubule segments have different uptake rates and affinities for glucose (19). These differences were subsequently ascribed by Turner and Moran to the presence of two different glucose transporters located in the brush border membrane (383). These observations and follow-up studies identified the Na+-glucose cotransporters SGLT2 (SLC5A2) and SGLT1 (SLC5A1) as the key genes and transport pathways for glucose reabsorption in the kidney. These studies included mRNA expression analyses in rabbit and rat nephron segments as well as glucose transport studies in membrane vesicles, and finally the cloning of the responsible genes by the Wright and Hediger lab, to a large extent completed between 1981 and 1995 (19, 135, 166, 206, 302, 383, 428, 437, 439, 444). These studies established the concept that the bulk of tubular glucose reabsorption occurs in the early proximal tubule or S1/2 segment and is mediated across the apical membrane by the low-affinity and high-capacity SGLT2. In comparison, SGLT1, which has a higher affinity and lower transport capacity for glucose than SGLT2, was proposed to take up most of the remaining luminal glucose in the later parts of the proximal tubule, that is, the S2/S3 segment (see Figure 1). With the use of well-validated antibodies, the primary expression of SGLT2 and SGLT1 in the brush border membrane of the early and late proximal tubule, respectively, have been confirmed in rodent and human kidney (17, 319, 402, 418). Some species differences may exist with regard to the localization of the highest expression level of SGLT1 within the late proximal tubule: in mouse kidney, SGLT1 protein expression was higher in S2 segments than in S3 segments of the medullary rays and outer stripe (228), whereas in human kidney, the strongest expression of SGLT1 was located to the S3 segment (418). The mouse renal epithelial cell atlas (“Mouse-RNA” database) is based on RNA sequencing of micro-dissected tubular segments and confirmed the differential expression of Sglt2 (Slc5a2) versus Sglt1 (Slc5a1) in the S1 versus the S2/S3 segment, respectively (Table 1). The predominant expression of SGLT2 mRNA in the S1 segment was also documented in microdissected rat tubular segments in the rat renal epithelial cell atlas (“Rat-RNA” database), which for some reason did not detect a signal for SGLT1 in the proximal tubule ((205), Table 2). Quantitative proteomics analysis of micro-dissected rat kidney tubule segments (“Rat-Protein” database) likewise confirmed the predominant expression of SGLT2 versus SGLT1 in S1 versus S2/S3 segments, respectively ((213), Table 3). Consistent with the distinct expression pattern for SGLT2 and SGLT1 along the proximal tubule, free-flow renal micropuncture studies showed a lack of glucose reabsorption in the early proximal tubule in mice lacking SGLT2 (402) (see Figure 3A), whereas in mice lacking SGLT1 fractional glucose reabsorption up to the late proximal convoluted tubule accessible on the kidney surface (corresponding to S2 segments) was only reduced from 97% to 94% (120).
Table 1.
RNA-seq Analysis of Microdissected Mouse Kidney Tubule Segmemts
| Gene | PTS1 | PTS2 | PTS3 | DTL1 | DTL2 | DTL3 | ATL | mTAL | cTAL | DCT | CNT | CCD | OMCD | IMCD | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| SGLT1 | Slc5a1 | 0.3 | 93 | 38.5 | 6.4 | 1.3 | 0.7 | 0.8 | 1 15.3 | 391.6 | 2.6 | 0.1 | 0.3 | 0.5 | 3.6 |
| SGLT2 | Slc5a2 | 2721.8 | 0.7 | 0 | 13 | 2.6 | 1.9 | 0.2 | 0.1 | 0.9 | 12.7 | 1.1 | 0.4 | 0.4 | 0.2 |
| SGLT3 | Slc5a4a | 0 | 0 | 0 | 0.1 | 0 | 0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SGLT3 | Slc5a4b | 0.1 | 0.3 | 1.3 | 1.3 | 0.7 | 0.6 | 0.6 | 0 | 0 | 0 | 0 | 0 | 0.3 | 0 |
| SGLT4 | Slc5a9 | 37.8 | 44.8 | 35.7 | 2.1 | 0.7 | 1 | 0.2 | 0 | 0.1 | 0.1 | 0.1 | 0.4 | 1.4 | 0.4 |
| SGLT5 | Slc5a10 | 1 | 466.6 | 125 | 7.7 | 2 | 0.5 | 0.4 | 0 | 0.6 | 0.2 | 0.1 | 0.8 | 0.3 | 0.1 |
| GLUT1 | Slc2a1 | 8.4 | 14.8 | 7.2 | 69.1 | 308.1 | 185.8 | 50.2 | 164.8 | 131.7 | 84.2 | 255.7 | 410.3 | 404.6 | 32.2 |
| GLUT2 | Slc2a2 | 314.8 | 49.3 | 9.8 | 3.1 | 1 | 1.3 | 1 | 0.2 | 0.3 | 1.4 | 0.4 | 0.9 | 0.6 | 0.1 |
| GLUT4 | Slc2a4 | 0.1 | 0.2 | 0 | 0.5 | 4.5 | 0.4 | 0 | 125.1 | 282 | 219.2 | 147.4 | 94.5 | 63.7 | 0.4 |
| GLUT5 | Slc2a5 | 138.8 | 201.7 | 99.1 | 24.3 | 2.2 | 1.3 | 0.5 | 0.2 | 0.3 | 0.8 | 0.1 | 0.5 | 0.3 | 0.1 |
| GLUT9 | Slc2a9 | 2.5 | 2 | 1 | 10.3 | 4.5 | 3.9 | 5.2 | 0.2 | 0.2 | 25.3 | 26.7 | 3.8 | 2.6 | 0.9 |
| GLUT12 | Slc2a12 | 8.8 | 0.7 | 0.3 | 0.4 | 1 | 0.4 | 2.5 | 6.3 | 13.9 | 20.9 | 8.9 | 8.5 | 4.3 | 0.3 |
| Hexokinase 1 | Hk1 | 1.2 | 2.4 | 4.5 | 93.7 | 394.9 | 126.2 | 165 | 391.2 | 405.5 | 263.7 | 337.7 | 361.1 | 315.9 | 65.6 |
| PFK, liver type | Pfkl | 25.1 | 36.3 | 18.7 | 124.2 | 139.4 | 281 | 173.4 | 223.2 | 182.1 | 71.6 | 145.6 | 171.2 | 186.6 | 63.4 |
| PFK, muscle type | Pfkm | 20.6 | 11.1 | 3.8 | 29.9 | 52.5 | 21.1 | 8.8 | 56.2 | 116 | 125.1 | 105.2 | 80.1 | 49.2 | 7.9 |
| PFK, platelet type | Pfkp | 7.9 | 33.3 | 33.1 | 72.8 | 271.5 | 58.2 | 266.6 | 311.3 | 284.6 | 171.4 | 217.7 | 254.5 | 139.6 | 15 |
| Pyruvate kinase | Pkm | 2 | 38.9 | 50.1 | 366.9 | 846.4 | 894.5 | 330.5 | 1 153 | 1 246.8 | 1558.3 | 1365.1 | 1233.7 | 940.8 | 664.2 |
| Ketohexokinase | Khk | 530 | 1283.5 | 1335.2 | 91.3 | 28.6 | 25.1 | 23.8 | 41.6 | 43.5 | 36.3 | 43.3 | 48.7 | 48 | 15.6 |
| Aldolase B | Aldob | 11309 | 6615.7 | 5262.5 | 575.9 | 50.8 | 3.8 | 7.6 | 2.1 | 13.4 | 44.5 | 17 | 19.7 | 0.7 | 0.6 |
| Triokinase | Tkfc | 63.2 | 130.3 | 87 | 17.3 | 4.5 | 6.4 | 5.4 | 3.3 | 3.2 | 4.5 | 4.5 | 3.8 | 4.8 | 4 |
Kidneys from 6-week-old WT male and female mice (only female proximal tubules were included in this database) were perfused, sliced, and dissociated in perfusion buffer supplemented with collagenase B, followed by microdissection and RNA sequencing. Transcript abundances were calculated in the units of transcripts per million (TPM) using RSEM (https://github.com/deweylab/RSEM).
Terminology: PTS1 the initial segment of the proximal convoluted tubule; PTS2, proximal straight tubule in cortical medullary rays; PTS3, last segment of the proximal straight tubule in the outer stripe of outer medulla; DTL1, the short descending limb of the loop of Henle; DTL2, long descending limb of the loop of Henle in the outer medulla; DTL3, long descending limb of the loop of Henle in the inner medulla; ATL, thin ascending limb of the loop of Henle; MTAL, medullary thick ascending limb of the loop of Henle; CTAL, cortical thick ascending limb of the loop of Henle; DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct. Other abbreviations: PFK, phosphofructokinase. Website created by Lihe Chen and Mark A. Knepper https://esbl.nhlbi.nih.gov/MRECA/Nephron/
Table 2.
RNA-seq Analysis of Microdissected Rat Kidney Tubule Segmemts
| Gene | PTS1 | PTS2 | PTS3 | DTL1 | DTL2 | DTL3 | ATL | mTAL | cTAL | DCT | CNT | CCD | OMCD | IMCD | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| SGLT1 | Slc5a1a | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| SGLT2 | Slc5a2 | 90.9 | 8.2 | 0.5 | 0.3 | 0.1 | 0.2 | 0.4 | 0.0 | 2.4 | 5.1 | 4.0 | 4.9 | 0.0 | 1.0 |
| SGLT4 | Slc5a9 | 1.4 | 2.7 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| SGLT5 | Slc5a10 | 4.3 | 54.0 | 44.1 | 0.0 | 0.0 | 0.4 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.3 | 0.0 |
| GLUT1 | Slc2a1 | 0.1 | 0.3 | 1.3 | 71.8 | 20.5 | 96.0 | 73.3 | 17.9 | 25.5 | 14.2 | 43.5 | 46.2 | 65.3 | 20.1 |
| GLUT2 | Slc2a2 | 0.3 | 0.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| GLUT4 | Slc2a4 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.2 | 0.1 | 2.9 | 13.7 | 3.3 | 0.0 | 1.3 | 0.0 | 20.6 |
| GLUT5 | Slc2a5 | 0.1 | 2.0 | 4.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| GLUT9 | Slc2a9 | 0.0 | 1.6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| GLUT12 | Slc2a12 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.0 | 0.2 | 1.5 | 0.0 | 0.0 | 0.0 | 0.0 |
| Hexokinase 1 | Hk1 | 0.2 | 0.1 | 1.2 | 18.1 | 36.2 | 26.3 | 36.1 | 23.1 | 29.4 | 41.5 | 33.4 | 50.8 | 42.0 | 49.9 |
| PFK, liver type | Pfkl | 0.1 | 0.5 | 1.2 | 1.1 | 0.1 | 26.8 | 5.8 | 2.0 | 1.8 | 10.8 | 3.4 | 5.1 | 1.8 | 9.1 |
| PFK, muscle type | Pfkm | 0.6 | 0.8 | 0.4 | 6.7 | 5.6 | 5.7 | 7.4 | 0.3 | 10.5 | 21.2 | 5.3 | 10.7 | 39.2 | 10.9 |
| PFK, platelet type | Pfkp | 0.3 | 0.0 | 0.1 | 0.8 | 36.6 | 3.6 | 6.8 | 32.2 | 8.1 | 5.2 | 5.6 | 19.6 | 2.4 | 13.6 |
| Pyruvate kinase | Pkm | 0.2 | 0.2 | 2.2 | 6.3 | 14.7 | 11.4 | 2.9 | 9.5 | 12.8 | 17.0 | 7.8 | 24.5 | 41.0 | 51.6 |
| Ketohexokinase | Khk | 24.0 | 77.0 | 76.2 | 0.0 | 0.1 | 0.1 | 0.0 | 0.1 | 0.8 | 1.0 | 0.4 | 0.3 | 0.3 | 1.3 |
| Aldolase B | Aldob | 5101 | 2930 | 2388 | 0.4 | 2.3 | 2.0 | 9.0 | 133.8 | 145.7 | 240.5 | 8.9 | 18.0 | 7.5 | 0.6 |
| Triokinase | Tkfc | Not included in analysis | |||||||||||||
Adapted, with permission, from Lee JW, et al., 2015 (205).
This database provides median RPKM values for the transcriptome of 14 separate renal tubule segments in untreated male Sprague-Dawley rats (200–250g BW). Rat renal tubule segments manually dissected under microscope were lysed in cell lysis buffer containing reverse transcriptase and oligo-dT primers. After reverse transcription of poly(A)-tailed mRNA transcripts, cDNAs were amplified and sequenced using lllumina HiSeq 2000 sequencer. FASTQ sequences were mapped to rat reference genome (rn5).
Terminology for nephron segments: PTS1, first segment of the proximal tubule; PTS2, second segment of the proximal tubule; PTS3, third segment of the proximal tubule; DTL1, short descending limb of the loop of Henle; DTL2, long descending limb of the loop of Henle in the outer medulla; DTL3, long descending limb of the loop of Henle in the inner medulla; ATL, thin ascending limb of the loop of Henle; mTAL, medullary thick ascending limb of the loop of Henle; cTAL, cortical thick ascending limb of the loop of Henle; DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct. Other abbreviations: PFK, phosphofructokinase.
Unclear why no signal for Slc5al detected in proximal tubule. https://helixweb.nih.gov/ESBL/Database/NephronRNAseq/index.html.
Table 3.
Quantitative Proteomics of Microdissected Rat Kidney Tubule Segments
| Gene | PTSl | PTS2 | PTS3 | DTL1 | DTL2 | DTL3 | ATL | mTAL | cTAL | DCT | CNT | CCD | OMCD | IMCD | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| SGLT1 | Slc5a1 | 0 | 431,973 | 420,276 | 0 | 24 | 0 | 0 | 0 | 32,797 | 201 | 0 | 0 | 0 | 0 |
| SGLT2 | Slc5a2 | 2,452,145 | 53,473 | 544 | 17 | 11,453 | 0 | 0 | 1262 | 47,209 | 55,032 | 2959 | 884 | 16 | 0 |
| SGLT4 | Slc5a9 | 0 | 53,803 | 54,913 | 0 | 0 | 0 | 0 | 0 | 10 | 87 | 0 | 0 | 0 | 0 |
| SGLT5 | Slc5a10 | 0 | 288,423 | 640,243 | 0 | 15 | 0 | 0 | 0 | 226 | 15 | 0 | 6 | 0 | 0 |
| GLUT1 | Slc2a1 | 0 | 136,188 | 126,849 | 5703 | 174,015 | 5700 | 38 | 55,707 | 1120 | 0 | 98,715 | 504,420 | 189,573 | 103,794 |
| GLUT2 | Slc2a2 | 125,996 | 10,650 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 341 | 0 | 4 | 0 | 0 |
| GLUT4 | Slc2a4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 103 | 608 | 0 | 0 | 0 | 0 | 0 |
| GLUT5 | Slc2a5 | 0 | 66,641 | 224,326 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 |
| GLUT9 | Slc2a9 | 0 | 39,752 | 310 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| GLUT12 | Slc2a12 | No matchiing record | |||||||||||||
| Hexokinase-1 | Hk1 | 0 | 589 | 46,318 | 137,611 | 457,531 | 158,284 | 1 94,094 | 1,143,010 | 1,074,067 | 1,080,539 | 955,560 | 550,374 | 600,736 | 250,752 |
| PFK, liver type | Pfkl | 574 | 132,032 | 299,892 | 48,803 | 39,923 | 82,881 | 56,989 | 10,666 | 68,599 | 60,721 | 98,214 | 163,898 | 60,313 | 261,614 |
| PFK, muscle type | Pfkm | 17,612 | 845 | 34 | 15,721 | 44,548 | 26,029 | 17,981 | 79,784 | 222,277 | 345,686 | 313,507 | 426,853 | 104,681 | 109,668 |
| PFK, platelet type | Pfkp | 26,350 | 43,623 | 1 27,970 | 76,312 | 138,189 | 46,042 | 121,633 | 489,855 | 617,372 | 497,353 | 421,280 | 429,269 | 402,654 | 234,861 |
| Pyruvate kinase | Pkm | 0 | 501,760 | 2,053,341 | 1,096,344 | 1,679,900 ' | 1,235,711 | 1,589,524 | 1,903,743 | 2,290,476 | 3,497,352 | 5,640,768 | 9,272,883 | 5,821,671 | 20,774,678 |
| Ketohiexokinase | KFik | 1,382,043 | 5,708,196 | 7,999,941 | 87,262 | 81,724 | 4088 | 0 | 31,519 | 103,808 | 67,388 | 22,485 | 31,230 | 13,533 | 4237 |
| Aldolase B | Aldob | 136,537,124 | 71,700,294 | 62,371,936 | 2,468,314 | 1,930,093 | 34,271 | 121,673 | 1,861,171 | 4,476,370 | 3,816,445 | 703,081 | 821,589 | 157,310 | 0 |
| Triokinase | Tkfc | 762,319 | 6,383,718 | 9,036,537 | 185,625 | 211,109 | 19,693 | 15,443 | 69,707 | 179,425 | 159,381 | 81,538 | 85,098 | 38,048 | 36,871 |
Adapted, with permission, from Limbutara K, et al, 2020 (213).
Each renal tubule segment was microdissected from male Sprague Dawley rats and analyzed with Orbitrap Lumos mass spectrometry. Protein copy number per cell was estimated using ‘Proteomic Ruler’ approach. For each segment, average values from 3–4 biological replicates are reported.
Terminology for nephron segments: PTS1, proximal tubule directly attached to the glomerulus; PTS2, straight part of proximal tubule obtained from medullary ray; PTS3, final portion of proximal tubule from outer medulla before transitioning into thin limb; DTL1, descending thin limb of Henle’s loop of short-loop nephrons; DTL2, descending thin limb of Henle’s loop of long-loop nephrons in outer medulla; DTL3; descending thin limb of Henle’s loop of long-loop nephrons in inner medulla; ATL, ascending thin limb of Henle’s loop; mTAL, medullary thick ascending limb; cTAL, cortical thick ascending limb; DCT, distal convoluted tubule within around 0.5 mm from macula densa; CNT, connecting tubule; CCD; cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct. Other abbreviations: PFK, phosphofructokinase.
Figure 3. Defining the contribution of SGLT2 and SGLT1 to renal glucose reabsorption.

(A) Left two panels: free-flow collections of tubular fluid were performed by micropuncture to establish a profile for fractional reabsorption of glucose versus fractional reabsorption of fluid along accessible proximal tubules at the kidney surface. Glucose reabsorption is prevented in the early proximal tubule in mice lacking SGLT2 (Sglt2−/−), but enhanced in the later proximal tubule, suggesting compensation by SGLT1. Right panel: In renal inulin clearance studies, the reduction in fractional renal glucose reabsorption in Sglt2−/− mice correlated with the amount of filtered glucose. (B) In metabolic cage studies, the SGLT2 inhibitor empagliflozin dose-dependently increased glucose excretion in WT mice. The response curve was shifted leftward and the maximum response doubled in Sglt1−/− mice. The difference between the 2 dose-response curves reflects glucose reabsorption via SGLT1 in WT mice. Glucosuria is initiated in WT mice when SGLT1-mediated glucose uptake is maximal (red arrow). The difference between curves was maintained for all higher doses (same length of vertical green lines), indicating selectivity of the drug for SGLT2 versus SGLT1 in this dose range. (C) Using genetic knockout models and pharmacologic tools in renal inulin clearance studies indicated that the glucose reabsorption preserved during SGLT2 knockout or inhibition (≈40%) is mediated by SGLT1. The SGLT2 inhibitor empagliflozin was applied at low and high doses to establish free plasma concentrations (similar to concentrations in glomerular filtrate) close to IC50 for mouse SGLT2 (≈1–2 nM) or 10-fold higher. Reused, with permission, from Rieg T, et al., 2014 (312); Vallon V, et al., 2011 (402).
Besides the expression of SGLT1 in the late proximal tubule, recent studies in mouse and human kidneys also established its expression in the luminal membrane of the thick ascending limb (TAL) and the macula densa (MD) (228, 418, 449). In accordance, the Mouse-RNA, Rat-RNA, and Rat-Protein database indicated additional SGLT1 expression in cTAL (Tables 1–3). The distinct role of SGLT1 in the macula densa is discussed in later sections. The Rat-Protein database also indicated SGLT2 protein signals in further distal segments, including cTAL and DCT, associated with some mRNA signal in Rat-RNA and Mouse-RNA database (Tables 1–3), but the relevance remains unclear.
Consistent with the above discussion, humans carrying mutations in the genes for SGLT1 (SLC5A1) and SGLT2 (SLC5A2) have very distinct renal phenotypes. Individuals with mutations in SGLT2 present with “Familial Renal Glucosuria” (OMIM 233100), that is, very prominent glucosuria ranging from 1 to up to 100 g/day, whereas intestinal glucose handling is normal as SGLT2 is not expressed in this tissue (326). No significant other problems (e.g., impaired kidney function or urinary tract infections) have been consistently observed or reported in individuals with SGLT2 loss-of-function mutations (326, 438). While these mutations are infrequent and thus the amount of information limited, these insights supported the rationale to target SGLT2 as a potentially safe pharmacological strategy to lower hyperglycemia (as discussed in detail below). In comparison to SGLT2, individuals with mutations in SGLT1 show little or no glucosuria. Due to the decisive importance of SGLT1 in active intestinal glucose reabsorption, however, these individuals suffer from intestinal Glucose Galactose Malabsorption” (Online Mendelian Inheritance in Man [OMIM] 182380) (185, 233, 438). For example, feeding of galactose or glucose to newborns with mutations in SGLT1 or gene-targeted mice lacking SGLT1 (120) can induce life-threatening diarrhea. Consistent with the human phenotypes, mouse models with genetic inactivation of SGLT2 presented with glucosuria (222, 298, 402). Moreover, genetic and pharmacologic inhibition in euglycemic mice revealed that approximately 97% of glucose is reabsorbed by SGLT2, whereas SGLT1 “cleans up” the remaining approximately 2% to 3% (120, 312, 402) (see Figures 1 and 3C).
Blocking of SGLT2 unmasks the substantial renal glucose transport capacity of SGLT1
As discussed above, SGLT2 reabsorbs almost all the filtered glucose in the normal kidney. Studies in humans and rodents, however, showed that fractional renal reabsorption of glucose is preserved at 40% to 50% in the presence of a selective SGLT2 inhibitor (138, 190, 342) (see Figures 1 and 3). Also in euglycemic mice lacking SGLT2, the fractional renal reabsorption of glucose varied between 10% and 60% and inversely with the amount of filtered glucose, with a mean value of about 40% (402) (see Figure 3A). This conundrum was resolved in subsequent studies showing that during inhibition of SGLT2 the substantial transport capacity of downstream SGLT1 is unmasked (see Figures 1 and 3). The first evidence came from micropuncture studies in SGLT2 knockout mice: while these mice lacked net glucose reabsorption in the early proximal tubule, net glucose reabsorption was prominent and enhanced versus wild-type mice in the later parts of the proximal convoluted tubule, consistent with high SGLT1 expression in mouse S2 segments (402) (see Figure 3A). Metabolic cages studies showed that the dose-response curve for urinary glucose excretion of a selective SGLT2 inhibitor was shifted leftward in SGLT1 knockout mice (312) (see Figure 3B). In other words, glucosuria appeared at lower doses of the SGLT2 inhibitor when SGLT1 was absent since it could no longer compensate. Moreover, the studies indicated that in wild-type mice the SGLT2 inhibitor was only glucosuric after the SGLT1-mediated glucose transport had been saturated, consistent with SGLT1 being located downstream of SGLT2 along the tubular system and compensating. Finally, the maximum glucosuric response induced by the SGLT2 inhibitor was doubled in the absence of SGLT1, consistent with the notion that SGLT1 can reabsorb approximately 50% of filtered glucose when SGLT2 is inhibited (312) (see Figure 3B). Associated renal clearance studies demonstrated that a high dose of a selective SGLT2 inhibitor reduced fractional renal glucose reabsorption to 44% in wild-type mice and prevented any net renal glucose reabsorption in SGLT1 knockout mice (see Figure 3C). Furthermore, absence of net renal glucose reabsorption was also demonstrated in both female and male mice carrying a SGLT2/SGLT1 double knockout (312) (see Figure 3C). These studies established that SGLT2 and SGLT1 together accounted for all net renal glucose reabsorption in nondiabetic and euglycemic mice (312). Thus, SGLT1 carries a sizable and relevant renal glucose transport capacity, consistent with high maximal glucose transport rates of human SGLT1 observed in in vitro studies (150). This transport capacity is unmasked and becomes engaged when more glucose is provided to SGLT1, for example, during inhibition of SGLT2. As a consequence, dual inhibition of SGLT1 and SGLT2 approximately doubles glucosuria versus sole SGLT2 inhibition, as shown in nondiabetic and diabetic mice (298, 312, 354), and indicated by studies using a dual SGLT2/SGLT1 inhibitor in mice, rats, and dogs (299). Furthermore, the results of the above studies were used to estimate a 3:1 to 5:1 ratio for the glucose reabsorption capacities of SGLT2 versus SGLT1 in the nondiabetic mouse kidney (99).
The proximal tubule takes up large amounts of glucose but, in contrast to further distal segments, has little glycolytic activity
The healthy kidney takes up most of the filtered glucose into the early proximal convoluted tubules by SGLT2 and the remaining small fraction into the late proximal convoluted tubule and the proximal straight tubule by SGLT1 (see above). However, these cells have no robust capacity for aerobic and anaerobic glycolysis, and glucose is not a substantial contributor to the cellular metabolism of these segments (124, 182, 384). In fact, glucose that is taken up across the luminal membrane or newly formed by proximal tubules (as discussed below) is primarily exiting the cells across the basolateral membrane into the tubular interstitium through facilitative glucose transport, primarily GLUT2 (see Figure 1; as discussed below). In the late 20th century, many studies investigated the activities of enzymes along the nephron and demonstrated that the glycolytic potential was rather low in proximal tubules compared with further distal parts of the nephron and collecting duct system. For example, hexokinase (HK), phosphofructokinase (PFK), and pyruvate kinase (PKM), all of which are key enzymes for glycolysis) (see Figure 4), are not highly expressed in proximal tubular epithelial cells (124, 335) (Tables 1–3), particularly in the S1 segment, where most of the glucose is reabsorbed through SGLT2.
Figure 4. Glucose and fructose metabolism.

Most of the filtered glucose is taken up by SGLT2 in the S1 segment of the proximal tubule and leaves via basolateral GLUT2 (not shown). This is because the glycolytic potential is rather low in proximal tubules compared with further distal parts of the nephron and collecting duct system. In accordance, hexokinase (HK), the gateway enzyme of glucose metabolism that phosphorylates glucose to glucose-6-phosphate (Glc6P), is less active in proximal tubules relative to the rest of nephron and collecting duct. Within the proximal tubule, the highest expression and activity of HK (I, II) is in S2/S3 segments. Other key enzymes of glycolysis include phosphofructokinase (PFK) and pyruvate kinase (PKM). In contrast to glucose, fructose, which is primarily taken up by SGLT5 in S2/S3 segments, is readily phosphorylated by fructokinase (known as ketohexokinase, KHK) to produce fructose 1-phosphate (Fru1P). Fru1P is subsequently cleaved by aldolase B (AldoB) into dihydroxyacetone phosphate (DHAP) and glyceraldehyde (GA). DHAP and GA feed into gluconeogenesis via fructose 1,6-biphosphate (Fru1,6BP) or into glycolysis via glyceraldehyde-3-phosphate (G3P). G3P enters the glycolytic pathway distal to PFK and the formation of Fru1,6BP. While PFK is the most heavily regulated enzyme and is considered as the gating step of glycolysis, fructose metabolism bypasses this enzyme and lacks a negative regulatory step. In parallel, fructokinase activation sequesters a phosphate, so that intracellular phosphate and ATP levels are transiently reduced. The rapid reduction of phosphate consequently activates AMP deaminase (AMPD), which cleaves AMP to IMP. The decline in phosphate levels is attenuated by the relatively slower metabolism of Fru1P by AldoB. The latter is slowed down further by the increase in IMP, which inhibits AldoB. IMP metabolism drives urate formation. Fructose metabolism is linked to the pentose phosphate pathway (PPP) to generate nucleotides and amino acids, but also to lipid generation like triglycerides and cholesterol, and to lactate formation as an alternative energy form. Urate suppresses aconitase in TCA cycle, thereby favoring glycolysis over mitochondrial oxidative phosphorylation (OXPHOS), similar to Warburg effect. For gluconeogenesis, DHAP, G3P, and Fru1,6BP are metabolized toward glucose (red arrows). TK, triose kinase.
In accordance, hexokinase, the gateway enzyme of glucose metabolism that phosphorylates glucose to glucose6-phosphate (Glc6P) (see Figure 4), is less active in proximal tubules (early proximal tubule < proximal straight tubule) relative to the entire nephron (124). The glucose reabsorption rate in proximal tubules is approximately 35pmoles/min/mm, whereas hexokinase activity is approximately 2pmoles/min/mm, suggesting only 5% of glucose entering the cell is phosphorylated toward glycolysis in proximal convoluted tubules (92). In comparison, further distal segments have much higher activity of glycolytic enzymes and protein and mRNA expression, including for hexokinase (Tables 1–3). Hexokinase activity in the thick ascending limb of Henle’s loop in rats was found to be 15 times higher than that of the proximal tubules (336). Likewise, pyruvate kinase, an enzyme catalyzing another irreversible step in the glycolytic pathway, had a 10 to 20 fold higher activity in collecting ducts than in proximal tubules (335). Based on studies in isolated rabbit tubule segments, Chamberlin et al. proposed that glycolysis can meet the energy needs of distal nephron segments including the medullary thick ascending limb (49). Notably, glucose enters distal tubular segments primarily across the basolateral membrane via GLUT1 (see below).
The molecular basis of renal glucose transport
Sodium-coupled glucose cotransport across the apical membrane
Na+ reabsorption in the tubular system of the kidney is driven by the Na+/K+ ATPase, which is located in the basolateral membrane and constitutes the primary active and ATP-consuming transport step. The Na+/K+ ATPase lowers cytosolic Na+ concentrations thereby establishing the concentration gradient that energizes the uptake of Na+ as well as the secondary uptake of other molecules co-transported with Na+ from the tubular lumen across the brush border into the tubular cells (see Figures 1 and 5). In 1960, the Na+ glucose cotransport hypothesis was proposed by Crane. The hypothesis stated that active glucose transport in the intestinal epithelium is energized by the Na+ gradient across the cell membrane (which expresses SGLT1) (see Ref. 438 for review). This concept was swiftly applied to other molecules and ions including the cotransport of Na+ with glucose in the kidney (438).
Figure 5. Coordination of glucose transport and gluconeogenesis in the proximal tubule.

(1) Insulin is a physiological stimulator of SGLT2, which may serve to maximize renal glucose reabsorption capacity in situations of increased blood glucose levels, for example, following a meal. (2) At the same time, enhanced Na+-glucose uptake and insulin suppress renal gluconeogenesis. (3) The latter, in contrast, is stimulated in the postabsorptive phase (fasting) by increased catecholamine and reduced insulin levels and involves primarily lactate as a precursor. (4) The newly formed glucose is delivered to the systemic circulation by basolateral GLUT2. (5) In metabolic acidosis, the increase in gluconeogenesis from glutamine is linked to the formation of (i) ammonium (NH4+), a renally excreted acid equivalent, and (ii) new bicarbonate, which is taken up into the circulation. The Na+-H+-exchanger NHE3 contributes to apical H+/NH4+ secretion and Na+/bicarbonate reabsorption. (6) The newly formed glucose can be used as fuel for proximal tubule H+ secretion or, after intercellular transfer, for intercalated cell H+ secretion. (7) SGLT2 and NHE3 are both stimulated by insulin to enhance Na+ and glucose reabsorption and their functions may be positively linked through the scaffolding protein MAP17. ?, indicates pathways that need further confirmation. Modified, with permission, from Vallon V, 2020 (388).
SGLT1 and SGLT2 belong to the human SLC5 solute carrier family, which includes 12 members, of which SGLT1 and SGLT2 have been the most intensively characterized. Six members are identified as SGLTs, and they have varying preferences for the binding of glucose, galactose, fructose, mannose, myoinositol, and choline, but also short-chain fatty acids and other anions (438). All SGLTs are characterized by 15 exons that code for proteins with 580 to 718 amino acids and molecular weights of 60 to 80kDa (438). Wright, Hediger, and their group have pioneered the studies on the molecular nature of SGLTs including the cloning of SGLT1 and SGLT2 (see Refs. 127, 438 for review of SLC5 family). Human SGLT1 and SGLT2 have an amino acid identity of 59% (439). To further define the molecular model of Na+ and sugar cotransport, Wright’s group used insights from the crystal structure of a sodium galactose bacterial isoform in Vibrio parahaemolyticus. According to the model the outside gate is opened by binding of Na+ to the outside of the transport protein. This permits sugar to bind from the outside and be trapped. This induces a confirmation change, such that the inward gate opens and Na+ and sugar are co-released into the cell cytoplasm. The transport cycle is completed by a conformation change from a ligand-free inward-facing state to a ligand-free outward-facing state (77, 438).
The sugar selectivity and transport kinetics of cloned SGLTs were studied by the application of electrophysiological techniques to various expression systems. Studies on SGLT1 revealed a similar affinity for glucose and galactose. In comparison, glucose but not galactose is a substrate for SGLT2, and neither transports fructose (438). Studies in transfected human embryonic kidney (HEK) 293T cells indicated that the apparent affinities (Km) for d-glucose for human SGLT2 and human SGLT1 are in the range of 5 and 2mM, respectively, that is, they are rather similar (150). Na+ is required for sugar binding to the transporters with Km values for Na+ transport of 25 and 70mM being reported for human SGLT2 and human SGLT1, respectively (150). The data indicate that in euglycemic conditions, glucose levels in the glomerular filtrate are in the range of the Km of SGLT2, whereas luminal Na+ concentration, which is close to the plasma concentration of approximately 140mM, is much higher than the Km of SGLT2 and thus not rate-limiting.
The Na+-glucose coupling ratio of SGLT2 and SGLT1 is 1:1 and 2:1, respectively (150). This enhances the glucose concentration power of SGLT1 and thereby its ability to reabsorb glucose in the late proximal tubule despite dropping luminal glucose concentrations as a consequence of upstream SGLT2 activity (see Figure 1). The electrogenic nature of Na+-glucose cotransport requires paracellular Cl− reabsorption and transcellular K+ secretion to help preserve the membrane potential and thereby the driving force. The K+ channel KCNE1/KCNQ1 has been located to the luminal membrane of the mid to late proximal tubule and implicated for the membrane potential stabilization during Na+-dependent glucose uptake (397, 398) (see Figure 1). KCNE1, but not KCNQ1, could also be detected in mouse early proximal tubule, indicating that the beta subunit KCNE1 may interact with another K+ channel alpha subunit in the early proximal tubule.
In addition to SGLT2 and SGLT1, three more members of the SLC5 family that have been linked to glucose handling have been detected in the kidney on the mRNA level, namely SGLT3, SGLT4, and SGLT5 (436). The presence of SGLT3 (SLC5A4) facilitates the depolarization of the plasma membrane in response to glucose application in a saturable and Na+-dependent manner, which can be blocked by the SGLT inhibitor phlorizin. It has been proposed that SGLT3 acts as a glucose sensor, however, its renal protein expression, location, and specific function remain to be determined (356). Sglt3 (Slc5a4) was not detected in Rat-protein and Rat-mRNA database base with a faint signal in Mouse-mRNA database of unclear relevance (Tables 1–3). In contrast, Sglt4 (Slc5a9) mRNA expression was detected in mouse S1 to S3 and rat S1/S2 segments and the protein in rat S2 and S3 of the proximal tubule, respectively (Tables 1–3). Studies in COS-7 cells revealed that SGLT4 can transport glucose but the transporter has a much higher affinity for mannose than for glucose (Ki 0.15 vs. 8mM), indicating that SGLT4 may play a primary role in mannose transport (369), and potentially fructose, as discussed below. SGLT5 (SLC5A10), the mRNA and protein expression of which was detected in S2/S3 segments (Tables 1–3), is a Na+-dependent sugar transporter with a relatively high affinity and capacity for mannose and fructose compared with glucose and galactose (107, 123). High levels of Sglt5 mRNA have been detected in kidney cortex (52, 123), and studies in knockout mice implicated SGLT5 as the major luminal transporter responsible for renal fructose reabsorption (95), as discussed in detail below. Thus, SGLT1 and SGLT2 are the primary pathways for apical glucose uptake in the tubular system of the kidney, consistent with the above-discussed functional studies.
Facilitative transport of glucose across the basolateral membrane
As mentioned above, glucose that is taken up across the luminal membrane or newly formed by proximal tubules (as discussed below) is typically not metabolized by these cells but primarily exiting the cells across the basolateral membrane, particularly in the early proximal tubule. This exit step occurs through facilitative glucose transporters, primarily GLUT2 (71), and is driven by the glucose concentration gradient (see Figure 1). Glucose is subsequently taken up through convection and fenestrated endothelial cells into peritubular capillaries. Within the kidney, the low glucose affinity, “liver-type” glucose transporter GLUT2 (SLC2A2) (Km of 15–20mM) is primarily expressed in the basolateral membrane of the S1 and S2 segments with lesser or no expression in S3 segments (54) (see also Tables 1–3). For a review of the SLC2 family, see Ref. 145. GLUT2 is the primary pathway for basolateral exit of glucose in proximal convoluted tubules (70, 376, 433). In comparison, in the rat kidney little or no expression was found for the higher affinity, “erythroid/brain-type” transporter GLUT1 (SLC2A1) (Km; 1–2mM) in the S1 segment whereas a robust signal was particularly detected in the S3 segment, and this transporter has been proposed to support transcellular glucose transport in the straight part of the proximal tubule (70, 376, 433) (Tables 2, 3). In mouse kidney, Glut1 (Slc2a1) mRNA expression appeared more evenly distributed along S1 to S3 segments (Table 1). However, in both species, the highest renal expression of GLUT1 has been reported in basolateral membranes of further distal tubule segments. As analyzed in rat kidney, this includes the medullary thin and thick ascending limbs with the highest levels of expression found in connecting segments and collecting ducts, including both principal and intercalated cells with a particular prominent expression in the latter (376), and a similar expression pattern was documented for mRNA expression in the mouse (Table 1–3). The expression of GLUT1 correlates well with the glycolytic activity of nephron and collecting duct segments, consistent with the concept that glucose enters in particular distal tubule segments for energy supply via basolateral GLUT1. Supporting a greater role of GLUT2 versus GLUT1 for the basolateral exit of glucose in proximal tubules (see Figure 1), application of positron emission tomography in mice indicated that GLUT2 gene knockout abolished the renal reabsorption of glucose (320). This is in accordance with the observation in humans that loss of function mutations in GLUT2 cause the Fanconi-Bickel syndrome, which is characterized by a tubulopathy that impairs proximal tubule function, including glycosuria, aminoaciduria, phosphaturia, hyperuricemia, and proteinuria (288, 327, 328). The resulting generalized impairment of proximal tubular function may reflect the glucotoxicity induced by the accumulation of intracellular glucose that results when glucose cannot leave the cell across the basolateral membrane. For comparison, mutations in GLUT1 primarily cause neurologic problems and no obvious renal phenotype (288, 341).
A few other members of the SLC2 gene family were reported to be expressed in the kidney with potential implication in glucose transport, however, the available information is limited with regard to their functional relevance (251). In this regard, GLUT4 has been detected in the TAL of rat kidney, both on mRNA and protein level (55) (see also Tables 2 and 3). Moreover, GLUT4 was co-expressed with insulin-like growth factor IGF-I, and vasopressin, a stimulator of Na+ transport in TAL, also increased GLUT4 expression. Thus, it has been speculated that GLUT4 may play a potential role in local fuel control in this segment (55). Glut4 (Slc2a4) mRNA was also detected in mouse TAL (Table 1). Glut5 (Slc2a5) is expressed in the apical membrane of rat and mouse S2 and S3 segments and proposed to primarily serve as a fructose transporter (55, 362) (see also Tables 1–3), as discussed below. GLUT12 (SLC2A12) is able to transport glucose and has been proposed to be expressed in the apical membrane of distal tubules and collecting ducts of the rat, but the functional relevance remains to be determined (215). Glut12 (Slc2a12) mRNA was detected in some mouse and rat distal segments (Tables 1 and 2).
Physiology of Renal Fructose Transport
Physiological role of fructose
Fructose is a simple sugar that is present in fruit (fruit sugar) and has an identical chemical composition as glucose (C6H12O6, also a hexose) but its biological effects are distinct. Fructose has several unique properties relevant for survival that differ from glucose (161). Fructose stimulates the storage of fat, thereby contributing to the storage of energy that can be used when a lack of food is encountered. For example, the freshwater Pacu fish actively feast on ripe fruits fallen into the river thereby becoming fat in the rainy season (163) (see Figure 6). Long-distance migrating birds eat fruits to increase their fat stores prior to their migration (15). Hibernating mammals, such as bears and ground squirrels eat fruits to accumulate fat and survive the subsequent winter season (47, 360). Moreover, fresh fruits were the main dietary staple for early primates during the evolution of mankind (86, 156). Another survival effect of accumulating fat is water conservation: fat can produce metabolic water as metabolizing 100g of fat results in 110mL of metabolic water (241). Furthermore, studies in mice indicate that fructose can indirectly promote body metabolic water storage in the form of fat by stimulating vasopressin, potentially via fructokinase activation in hypothalamus rather than an osmotic effect (5).
Figure 6. Fructose from either diet or endogenous production can be utilized for survival.

Fructose is natural fruits that stimulates fat accumulation, thereby contributing to the storage of energy that can be used when a lack of food is encountered. The freshwater Pacu fish actively feast on ripe fruits fallen into the river and become fat in the rainy season. Long-distance migrating birds eat fruits prior to their migration to increase their fat stores. Hibernating mammals, such as bears and the ground squirrel, actively eat natural fruits to accumulate fat as an internal storage of energy for next winter season. Fresh fruits were the main dietary staple for early primates during the evolution of mankind. Moreover, pregnant women and other female mammals have the ability to produce endogenous fructose in the placenta, where fructose is used for fetal organ development during pregnancy. Naked mole rats can produce fructose endogenously in several organs for their survival under hypoxic condition. Adapted, with permission, from Johnson RJ, et al., 2019 (161); Junk WJ, 1985 (163).
In addition, pregnant women and other female mammals, including ungulates and whales, have the ability to produce endogenous fructose in the placenta (13, 20, 118, 143), where fructose is used for fetal organ development during pregnancy (179, 340, 430). For example, fructose-labeling studies demonstrated that fructose is utilized by the fetus to generate nucleic acids, especially RNA (149, 430). This involves the stimulation of the pentose phosphate pathway (PPP) by fructose, leading to the production of ribose-5-phosphate, which is then used in the synthesis of nucleotides and nucleic acid, as well as the production of erythrose-4-phosphate, which is used in the synthesis of tyrosine, citrulline, and proline (154). The naked mole-rat endogenously generates fructose in several organs, which helps to develop tolerance to hypoxia and survive under hypoxic conditions (287). Dietary fructose can also help to retain sodium, the importance of which has been implicated when during evolution amphibians conquered the land (338). Moreover, fructose induces peripheral insulin resistance, which helps shift the utilization of glucose from peripheral tissues to the central nervous system (161). All these examples illustrate fructose as a survival factor for humans and other animals (see Figure 6).
Issues with fructose in the modern society
While fructose is a survival factor that helps during starvation and was appropriate for our evolution as hunter and gatherers, it became deleterious with the agricultural and industrial evolution when energy supply became more abundant and excessive, respectively. The easy access to unlimited food resources and calories in the Western world facilitates overeating and secondary health problems. Based on our genetic armamentarium and metabolic programming as hunter and gatherers, sugar, which is composed of glucose and fructose, is very attractive to us. However, an increase or excess of fructose consumption, for example, in the form of added sucrose or sweeteners in the form of corn syrup, can drive the features of metabolic syndrome, including obesity, elevated blood pressure, dyslipidemia, and kidney diseases in both humans and animals (7, 14, 254, 259). Proposed pathophysiological mechanisms include the ability of fructose to cause endothelial dysfunction, fatty liver, insulin resistance, proteinuria and kidney dysfunction, and hypertension (112, 158, 254, 260). A recent study in mice indicated that suppressing vasopressin with hydration both prevented and ameliorated fructose-induced metabolic syndrome (5). An improved understanding of both the physiological and pathological roles of fructose can help to better appreciate and unravel its complexity.
While fructose is metabolized by several organs, including the liver, intestine, and kidney, it has been assumed that the liver is the primary site for dietary fructose metabolism. However, recent studies demonstrated that the small intestine also plays a substantial role in dietary fructose metabolism (153). The epithelium likely utilizes dietary fructose as a substrate for intestinal gluconeogenesis, which contributes approximately 25% of systemic gluconeogenesis both after prolonged fasting and in diabetes (357). Intestinal fructose metabolism also determines sweet taste preference and sugar intake but seems not to contribute to the development of metabolic syndrome (7). In turn, excessive fructose intake saturates the intestinal metabolic capacity and induces a spillover to the colon and liver (153). In the colon, fructose is likely digested by microbiota, which use fructose carbons to generate TCA intermediates and essential amino acids, and short-chain fatty acids (153). Furthermore, fructose spill over to the liver is taken up by hepatocytes via GLUT2 and contributes to metabolic syndrome (7), in part by increasing hepatic fatty acid synthesis and malonyl-CoA levels, reducing fatty acid oxidation, and modifying the mitochondrial proteome (351).
Fructose transporters in the kidney
Fructose uptake across the apical membrane of the proximal tubule
Fructose is metabolized in several types of cells including hepatocyte in the liver, the intestinal epithelial cells, and proximal tubular epithelial cells in the kidney. In healthy individuals, the kidneys daily filter between 4 and 25g of fructose assuming a normal glomerular filtration rate (GFR, ≈180 liter/day), and pre- and postprandial blood fructose concentrations of approximately 2mg/dL (0.11mM) and 14mg/dL (0.77mM), respectively (227). This is about 10% of the amounts of glucose filtered in a healthy kidney under euglycemic conditions. Most of the filtered fructose is taken up by the proximal tubules (39). This fructose uptake is at least in part mediated by the sodium-glucose cotransporter SGLT5, which is a high affinity transporter for fructose (Km 0.62mM) and mannose (Km 0.45mM) in both humans and mice, respectively (95, 123) (Figure 7). Consistent with a role of SGLT5 in renal fructose reabsorption, SGLT5 is strongly expressed in the kidney and only slightly in testes, but has not been detected in other organs in humans and mice (95, 123). As shown in Tables 1–3, both Sglt4 (Slc5a9) and Sglt5 (Slc5a10) mRNA and protein expression reside predominantly in S2 and S3 segments. As shown in Figure 8, Fukuzawa et al. showed that deletion of Sglt5 in mice resulted in an increase in urinary fructose excretion despite unchanged plasma fructose concentration (95). This was observed when mice were given plain water and the difference was largely enhanced when the mice were given 30% fructose water, which significantly upregulated renal Sglt5 mRNA expression. The same study isolated brush border membrane vehicles (BBMVs) from proximal tubular epithelial cells and showed that [14C]-d-fructose was incorporated into BBMVs of wild type mice, but not those of Sglt5 knockout mice, indicating that SGLT5 is located at the brush border of proximal tubular cells, where it is responsible for fructose uptake (95). Recently, another group detected the potential fructose transporters SGLT5 and SGLT4 in the apical membrane of isolated rat proximal tubule S2 segments (115). Moreover, 86% of fructose uptake in these S2 segments was sensitive to extracellular Na+ removal, and pharmacological inhibition of SGLT with phlorizin reduced fructose reabsorption by 65%, consistent with the assumption that a large part of fructose reabsorption in the S2 segment is mediated by SGLT4 or SGLT5 (115). Moreover, the SGLT4 and SGLT5 expression and the fructose reabsorption were upregulated when the rats had been pretreated with a high fructose diet (115), indicating that the S2 segments adapt the fructose uptake machinery and activity to the tubular fructose load (see Figure 7).
Figure 7. Fructose transporters and metabolism in proximal tubular cells.

There are several fructose transporters located at the apical membrane of proximal tubular epithelial cells, primarily in S2/S3 segments, including SGLT4/5 and GLUT5 as observed in rodents and humans. The rat sodium-dependent glucose transporter-1 (rNaGLT1) has been localized to the convoluted and the straight portions of proximal tubules in rats. In turn, GLUT2 is a facilitative transporter for fructose and glucose exit across the basolateral membrane. The proximal tubular cells are equipped with several enzymes for fructose metabolism, including fructokinase/ketohexokinase (FK/KHK), aldolase B (AldoB), and fructose 1,6 bisphosphatase (FBPase). Under fasting condition, a low level of fructose 2,6 biphosphate (Fru2,6BP) favors metabolism by FBPase over phosphofructokinase-1 (PFK1) thereby metabolizing fructose 1,6 biphosphate (Fru1,6BP) toward gluconeogenesis. In turn, under satiation, a high level of Fru2,6BP activates PFK-1 and promotes glycolysis. DHAP, dihydroxyacetone phosphate; G3P; glyceraldehyde 3-phosphate; G6Pase, glucose 6-phosphatase; GA, glyceraldehyde; HK, hexokinase; TK, triose kinase. Adapted, with permission, from Grempler R, et al., 2012 (123); Fukuzawa T, et al., 2013.
Figure 8. Fructosuria in mice lacking Sglt5.

(A) Plasma fructose concentrations measured under anesthesia after 3 h of fasting. Open circles represent individual mice. (B and C) Wild-type (WT) mice and Sglt5-deficient mice (Sglt5−/−) given plain water or 30% fructose water were maintained in metabolic cages and 24-h urine samples collected. Despite similar plasma fructose concentrations, absolute urinary fructose excretion is significantly greater in Sglt5−/− vs WT mice given plain water, and the difference is further enhanced when given fructose water. Data are presented as means ± S.E.M (n = 8–10/group). +++, P < 0.001 versus WT mice given plain water. ***, P < 0.001 versus WT mice given fructose water. ## and ###, P < 0.01 and P < 0.001 versus respective plain water controls. Adapted, with permission, from Fukuzawa T, et al., 2013 (95).
Theratsodium-dependentglucosetransporter-1(rNaGLT1) is also expressed at the apical membrane of the epithelial cells in both convoluted and straight proximal tubules in the rat and may also mediate fructose transport although the affinity is relatively low (Km of 4–5mM) (147) (see Figure 7). It is notable that the rNaGLT1 mRNA level in rat kidney is higher than for SGLT1 and SGLT2 (147). The function of rNaGLT1 (147) has been examined by in vitro experiments using both isolated BBMVs from proximal tubular epithelial cells and rNaGLT1-transfected HEK293 cells (147). These experiments demonstrated that [14C]-d-fructose was dose-dependently incorporated into BBMVs as well as the rNaGLT1-transfected HEK293 cells. The specificity of fructose transport was demonstrated by the fact that the transport of fructose was inhibited by nonradiolabeled fructose, phlorizin, and absence of sodium, but not by 2-deoxyglucose (147).
The kidneys of humans, mice, and rats also express GLUT5, a facilitative fructose transporter that has been localized most consistently to the apical membrane of the straight proximal tubule segments (8, 54, 173, 260, 362) (see Figure 7). Renal GLUT5 has a Km value of 12.6mM for fructose, a value similar to the small intestine in rats (236) and approximately 20-fold higher than for SGLT5. Besides the kidney and small intestine, GLUT5 is also expressed in the brain (307) while the liver appears to take up fructose primarily via GLUT2 (72). Renal GLUT5 has a molecular weight of 51kDa and is thus smaller than GLUT5 in the small intestine, which has a molecular weight of 58kDa (362), but the functional differences and implications remain poorly understood. The role of GLUT5 in the small intestine is well illustrated by the fact that Glut5 deficient mice exhibit a 75% decrease of fructose absorption in the jejunum associated with a 90% decrease in serum fructose concentration, suggesting that GLUT5 plays a major role in intestinal fructose absorption (22).
The expression of GLUT5 can be induced by several stimuli. During prenatal and suckling periods, Glut5 mRNA expression is very low in the small intestine in rats, rabbits, and humans (71), although GLUT5 is required for the reabsorption of the fructose contained in milk (119). However, Glut5 is dramatically up-regulated following the consumption of solid food, suggesting that exposure to food per se in the intestinal lumen directly induces Glut5 expression. Furthermore, fructose or sucrose in the diet increases intestinal GLUT5 levels in rats, while levels fall when fructose or sucrose are eliminated from the diet (239). These data support the notion that the induction of GLUT5 expression is a physiological adaptation to absorb available fructose, similar to the data discussed above on proximal tubule SGLT5.
In contrast to the intestine, relatively little is known about the quantitative role and expression regulation of GLUT5 in the kidney. Mouse and rat Glut5 (Slc2a5) mRNA and protein expression reside most consistently in the straight proximal tubule S2 and S3 segments, similar to Sglt4 and Sglt5 expression (Tables 1–3). Mice deficient in Glut5 have not been used to estimate the contribution of GLUT5 to renal fructose reabsorption. Glut5 mRNA is upregulated in glomerular mesangial cells and proximal tubules in response to streptozotocin (STZ)-induced diabetes in mice (54), which may be a response to elevated fructose generation via the polyol pathway from the high amounts of circulating glucose (197), but further studies are needed to establish the role of GLUT5 in renal fructose transport and function.
Fructose transport across the basolateral membrane of the proximal tubule
Fructose being taken up into the straight portion of the proximal tubules can be metabolized by these cells (39) (see below), but could also leave the cells into the interstitium through GLUT2 in the basolateral membrane (115, 232) (see Figure 7). However, to our knowledge and in contrast to glucose (see above), the role of GLUT2 in the proximal tubular handling of fructose has not yet been directly determined. Tables 1–3 indicate limited overlap between the expression of apical fructose transporters SGLT4, SGLT5, and GLUT5 and basolateral GLUT2, which may have consequences for transcellular fructose transport. Given the fact that GLUT2 is a facilitative transporter that is primarily expressed in the early proximal tubule and that is operating by passive diffusion, fructose could exit through GLUT2 in case the concentration of intracellular fructose is greater than the concentration in the basolateral interstitium. This may occur in the case of diabetes, where large amounts of glucose can be taken up by SGLT2 and converted into fructose by the proximal tubules (see below). On the other hand, a proposed targeting of GLUT2 to the apical membrane in STZ-induced diabetic rats (see discussion above) may suggest that excess fructose could also be excreted into urinary space.
GLUT9 (SLC2A9) is a member of the facilitative GLUT gene family but is now primarily described as a urate transporter, which can exchange both fructose and glucose for urate (68). GLUT9 has two splice variants, GLUT9a (full length) and GLUT9b (ΔN), both of which are present in the human kidney (11). GLUT9a (540 amino acids) is expressed in the basolateral membrane of the proximal tubular epithelial cells whereas GLUT9b (512 amino acids) is found apically in the proximal tubules (350) and the collecting ducts (11, 180) in humans. The GLUT9a located in the basolateral membrane of the proximal tubules favors urate transport back into the circulation from the tubular cells while the GLUT9b at the apical site likely takes up urate from tubular fluid into cells (238). The distinct functions between these two isoforms could be mediated by intra- and extracellular hexose concentrations. In fact, a recent study using oocytes expressing either transfected-GLUT9a or GLUT9b showed that GLUT9a-mediated urate efflux was significantly more accelerated by glucose than by fructose while GLUT9b-mediated urate influx was promoted more by fructose compared with glucose (432). The rat kidney tubules expressed GLUT9 mRNA and protein in S2 segments (Tables 2 and 3) whereas the mouse expressed small amounts of mRNA in proximal tubules and more in DCT and CNT (Table 1) but the analyses did not differentiate splice variants. This transporter may play a role under pathological conditions, including diabetes, when the tubular fluid in the collecting ducts could contain substantial amounts of sugar. The effect of SGLT2 inhibition on renal urate handling and the link of fructose metabolism to urate formation is discussed below.
General considerations on fructose metabolism
Fructose is firstly metabolized by fructokinase (known as ketohexokinase, KHK), which phosphorylates fructose to fructose 1-phosphate (Fru1P) (see Figure 4). KHK exists as two alternative spliced isoforms produced by mutual exclusion of the adjacent exons 3C and 3A within the KHK gene (134). The “A” isoform is ubiquitously expressed with a low activity for its substrate (Km 8 mM) (66). Expression of the “C” isoform is primarily restricted to metabolic tissues including the liver, kidney, and intestine, and this form has 10-fold higher affinity for fructose (Km 0.8 mM) (10, 66). In the kidney, KHK mRNA and protein expression are strongest in the proximal tubule segments (Tables 1–3). Recent studies have identified a primary role of the intestine for the metabolism of dietary fructose. Jang et al. demonstrated in mice that lower doses of dietary fructose were primarily cleared by the intestine while higher doses of fructose overcome the intestinal fructokinase capacity and reach the liver and systemic circulation (153). Interestingly, antibiotic treatment was capable of blocking fructose metabolism in murine small intestine (153). Along these lines, Zhao et al. showed that dietary fructose was converted to acetate by the gut microbiota in mice and that the deletion of the microbiota potently suppressed the hepatic generation of acetyl-CoA and fatty acid from dietary fructose (452), suggesting a key role of microbiota for fructose metabolism. Moreover, recent studies using mice with selective deletion of the fructokinase (Khk) gene in liver or intestinal epithelia concluded that intestinal epithelia fructokinase has an important role in the clearance and body uptake of dietary fructose while the liver metabolism of fructose is responsible for most of the features of metabolic syndrome (7).
Fru1P is cleaved by aldolase B into dihydroxyacetone phosphate and glyceraldehyde-3-phosphate, which then enters the glycolytic pathway distal to phosphofructokinase and the formation of F1,6P (see Figure 4). The tubular aldolase B expression pattern is similar to fructokinase with highest levels in the proximal tubule (Tables 1–3). Phosphofructokinase is the most heavily regulated enzyme and is considered as the gating step of glycolysis. The metabolism of fructose, however, bypasses this enzyme and lacks a negative regulatory step. In parallel, fructokinase activation sequesters a phosphate, so that intracellular phosphate and ATP levels are transiently reduced (229). The rapid reduction of phosphate levels activates AMP deaminase, which cleaves AMP to IMP. The phosphate levels recover in part due to the slowing down of the aldolase B reaction, in part by the increased IMP, which is an aldolase B inhibitor (434). These events can also be linked to urate production (200, 254, 259) (see Figure 4).
As described above and opposed to glucose, dietary fructose rather reduces ATP levels when it is metabolized under aerobic conditions (305) and is typically utilized, primarily by the liver, for the production of lipids (triglycerides) and glycogen, all of which are stored in the body as alternative sources of energy in case glucose concentration falls. Thus, dietary fructose can be used to store alternative backup energy in preparation for a lack of glucose or during food deficiency. The metabolism of fructose in the kidney is discussed in more detail below.
Physiology of Tubular Formation of Glucose and Fructose
Renal gluconeogenesis
The kidneys reabsorb the large amounts of glucose that are filtered by the glomeruli and, in addition, the kidneys generate new glucose through a process called gluconeogenesis. To our knowledge, renal gluconeogenesis was first documented in a study in 1937 by Benoy and Elliott showing that rat kidney slices produced glucose in response to pyruvate and lactate (24). A subsequent study in hepatectomized dogs reported that blood glucose declined more rapidly following removal of the kidneys (61). Moreover, in 1949, Reinecke and Hauser established in dogs that serum glucose concentration in the renal vein was higher than in the renal artery (310). Studies in the late seventies determined that the rat kidney produces approximately 26% of serum glucose in the normal fed state and that the glucose released by the kidneys rises by 46% in response to starvation (177). The contribution of renal gluconeogenesis to systemic glucose homeostasis is likewise substantial and physiologically relevant in humans. The healthy human kidneys generate 15 to 55 g of glucose every day, particularly during the periods of fasting. The human kidneys provide approximately 45% of blood glucose after prolonged starvation (281), and in the postabsorptive state (i.e., 12–16 h after the last meal) the gluconeogenesis by the human kidneys generates similar amounts of glucose as the liver (102). The second important stimulator of gluconeogenesis in the kidney, but not the liver, is metabolic acidosis (151, 335). This is because the process of renal gluconeogenesis from glutamine via the formation of alpha-ketoglutarate facilitates the new formation of bicarbonate (see below).
Like in the liver, which is the major gluconeogenic organ, the enzymes required and specific for gluconeogenesis are expressed in the kidney (60). Like glucose reabsorption, renal gluconeogenesis occurs along the entire proximal tubule (41, 63, 73, 124). In accordance, within the kidney, the enzymes specific to gluconeogenesis are found almost exclusively in the proximal tubule. This includes phosphoenolpyruvate carboxykinase (PEPCK), enzymes of the glucose 6-phosphatase (G6Pase) system, and fructose 1,6-phosphatase (FBPase). In particular, the activity and expression of PEPCK, which converts oxaloacetate to phosphoenolpyruvate (PEP), are restricted to proximal tubules (41, 125), are positively correlated with renal gluconeogenesis (4), and are markers of renal gluconeogenetic activity.
In response to starvation, gluconeogenesis is uniformly enhanced along the whole length of the proximal tubule; in comparison, metabolic acidosis stimulates gluconeogenesis primarily in the early proximal tubule S1 and S2 segments (41, 73, 124). Under starved conditions, low levels of fructose 2,6-biphosphate (Fru2,6BP) inhibit PFK-1 activity and glycolysis but activate fructose 1,6-phosphatase (FBPase) and promote glucose production, whereas high levels of Fru2,6BP activate PFK-1 and promote glycolysis in satiation (131, 139) (see Figure 7). Renal gluconeogenesis is inhibited by insulin and stimulated by epinephrine; thus, in the fasting state, the expected associated changes in insulin (down) and epinephrine (up) concentrations upregulate renal gluconeogenesis (102) (see Figure 5). In contrast to the liver, glucagon appears not to be a relevant regulator of renal gluconeogenesis (102).
Various precursors are used by the proximal tubule for the generation of glucose-6-phosphate. Renal gluconeogenesis in the postabsorptive state primarily makes use of lactate as substrate, followed by glutamine, glycerol, and alanine (103). In comparison, gluconeogenesis induced by metabolic acidosis primarily utilizes glutamine. Ascending vasa recta carry lactate-rich blood to the S3 segments in the outer medulla. The blood is lactate-rich due to anaerobic metabolism in the inner medulla, which consumes glucose and produces lactate (see discussion of potential intra-renal Cori cycle below). Anaerobic glucose usage by the medullary TAL constitutes another local lactate source. Under acidotic conditions, glutamine is converted to glutamate and alpha-ketoglutarate, which is associated with the generation of ammonium (NH4+). The latter is excreted as an acid equivalent into the urine. The generation of glucose from alpha-ketoglutarate is linked to the formation of new bicarbonate, which exits the cells across the basolateral membrane and acts as an acid buffer in the systemic circulation (see Figure 5) (102, 103). The glucose-6-phosphate formed during gluconeogenesis is dephosphorylated by glucose-6-phosphatase to generate free glucose, which can then exit the cell, usually via GLUT2 across the basolateral membrane.
Tubular glucose transport and formation are coordinated
To protect the epithelial cells of the early proximal tubules from glucose overload and its potential glucotoxicity, cellular uptake of glucose via SGLT2 and gluconeogenesis should be coordinated. In support of this notion, studies in HK2 cells and mice indicated that apical glucose uptake through SGLT2 or SGLT1 can inhibit gluconeogenic genes (see Figure 5) by glucose-induced and sirtuin 1-mediated deacetylation of peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) (330). Studying the consequences of tubular knockdown of the Na-H-exchanger NHE3, Onishi et al. provided first evidence that the reverse may also be true, that is, that an increase in renal gluconeogenesis suppresses SGLT2 (277). NHE3 mediates bicarbonate reabsorption and ammonium secretion in the proximal tubule (see Figure 5). Mice lacking NHE3 in the tubular system are able to preserve systemic acid-base balance at least in part by strongly upregulating the renal expression of PEPCK (277), the principal gluconeogenic enzyme (303). Notably, this upregulation of gluconeogenesis was associated with a 50% down-regulation of renal SGLT2 mRNA and protein expression, consistent with an intracellular negative feedback loop that regulates SGLT2 to limit excessive cellular accumulation of glucose (see Figures 5 and 9). In accordance with a negative control of SGLT2 expression by intracellular glucose concentrations, renal SGLT2 protein expression is increased in response to pharmacologic SGLT2 inhibition in nondiabetic mice (396). Moreover, tubular NHE3 knockdown enhances renal mRNA expression of SGLT1, possibly to compensate in part for the suppression of upstream SGLT2, suggesting a coordinated response to assure effective glucose retention. In fact, despite a 50% down-regulation of SGLT2, urinary glucose excretion was not elevated in nondiabetic mice with a tubular knockdown of NHE3; significantly enhanced glucosuria, however, was induced by superimposing Akita type 1 diabetes mellitus (T1DM) onto the absence of tubular NHE3, thereby overwhelming the compensation capacity of SGLT1 by increasing the filtered tubular glucose load (277). Surprisingly at first, the robust, 50% increase in glucosuria and thus the urinary loss of significant amounts of calories in diabetic mice with tubular NHE3 knockdown did not have an anti-hyperglycemic effect or induce compensatory increases in food intake, as observed in response to the glucosuric effect of an SGLT2 inhibitor (396, 405) (see below). Onishi et al. proposed that this may be the consequence of the upregulation of renal gluconeogenesis and thus a greater glucose delivery to the systemic circulation of these mice, consistent with the observed upregulation of not only renal mRNA of PEPCK but also of glucose-6-phosphatase and GLUT2 (277), which are responsible for the formation of free glucose and its basolateral exit, respectively. Finally, this study showed an upregulation in the renal mRNA expression of rate-limiting glycolytic enzymes in mice with tubular NHE3 knockdown. The authors speculated that newly formed glucose is in part metabolized by glycolysis to help drive ATP-consuming processes that facilitate renal bicarbonate reabsorption or ammonium secretion (277). Potential transporters include the basolateral Na+-K+-ATPase and the luminal H+-ATPase in proximal tubules as well as the luminal H+-ATPase in type A intercalated cells (see Figure 5). Glucose may reach the type A intercalated cells by intercellular transfer, that is, glucose is exiting the proximal tubular cell via basolateral GLUT2 and is taken up into intercalated cells via basolateral GLUT1 (376). The high glycolytic capacity of mitochondriadense type A intercalated cell has been confirmed using multiparametric imaging (106). Upregulation of distal tubule glycolytic markers in mice with tubular knockdown of NHE3 may also reflect the expected shift in the reabsorption of Na+ from proximal to more distal tubular segments (277). The study by Onishi proposes the coordination between the transport, generation, and usage of glucose in the kidney (see Figure 5).
Figure 9. Cellular processes in the early proximal tubule linked to SGLT2 and its inhibition.

Hyperglycemia enhances filtered glucose and, via SGLT2, the reabsorption of glucose and Na+ (1). Diabetes can increase SGLT2 expression (2); proposed mechanisms include tubular growth, Ang II, and HNF-1α, which may respond to basolateral hyperglycemia sensed by GLUT2. Hyperinsulinemia and tubular growth upregulate proximal tubular transport systems, including SGLT2, NHE3, URAT1, and Na-K-ATPase (3). The apical transporters may be functionally coupled via scaffolding proteins, such as MAP17 (4). The resulting proximal tubular Na+ retention enhances the GFR via tubuloglomerular feedback, which by increasing brush border torque can further increase transporter density in the luminal membrane. The increase in intracellular glucose may lower SGLT2 expression via negative feedback (5). Diabetes, in part due to the associated acidosis, can enhance gluconeogenesis (6). Gluconeogenesis can be inhibited by tubular injury, hyperinsulinemia, and enhanced glucose uptake via SGLT2 (6). HNF-1α and HNF-3β upregulate GLUT2 (7) and thereby the basolateral exit of glucose and maintains hyperglycemia (8). Excessive SGLT2-mediated glucose uptake may trigger apical translocation of GLUT2 (9). Hypoxia due to diabetes-induced hyperreabsorption or kidney injury may induce HIF1α, which enhances basolateral glucose uptake via GLUT1, induces a metabolic shift to glycolysis and inhibits apical transport (10). Induction of TGF-β1 and tubular growth may be particularly sensitive to basolateral glucose uptake via GLUT1 (11). Excessive intracellular glucose may also stimulate mTORC1 and attenuate autophagy (12). TGF-β1 enhances cyclin-dependent kinase inhibitors p21 and p27 and together with mTORC1 activation promotes tubular senescence, which is linked to inflammation and fibrosis. SGLT2 inhibition attenuates these deleterious effects linked to excessive intracellular glucose and hyperreabsorption. SGLT2 inhibition can also enhance gluconeogenesis, in part by lowering hyperinsulinemia. Gluconeogenesis enhances removal of intermediates from TCA cycle (cataplerosis) thereby facilitating the feeding of fatty acids and ketone bodies into the TCA cycle (anaplerosis), and enhancing oxidative phosphorylation (OxPhos) and ATP generation (13). This is associated with enhanced kidney delivery of fatty acids and ketone bodies in response to SGLT2 inhibition. Abbreviations: Ang II, angiotensin II; GFR, glomerular filtration rate; GLUT, facilitative glucose transporter; HIF-1α, hypoxia-inducible factor 1 alpha; HNF, hepatic nuclear factor; MAP17, 17-kDa membrane-associated protein; NHE3, Na-H-exchanger 3; OA, organic anion; TGF-β1, transforming growth factor β1; URAT1, urate transporter 1. ?, indicates pathways that need further confirmation. Adapted, with permission, from Vallon V, 2020 (388).
Proposed oscillations in TAL metabolism and a potential intrarenal Cori cycle?
The Cori cycle, which is named after its discoverers, Carl Ferdinand Cori and Gerty Cori, describes a cyclic metabolic pathway that involves and links glucose and lactate metabolism: lactate is produced as a consequence of anaerobic glycolysis in skeletal muscles, and the lactate is subsequently transported to the liver, where it is used for gluconeogenesis, and the newly formed glucose returns to the muscles and is metabolized again to lactate. Bankir and Yang proposed the hypothesis of an intrarenal Cori cycle (18). According to the hypothesis, the glucose, which is produced from lactate in the medullary S3 segment, enters the tubular lumen using reversed transport through SGLT1 (76). The tubular flow transfers glucose to the downstream tubular segments that take up and use the glucose as an energy resource for glycolysis or transfer it to local collecting duct or interstitial cells. Glucose is proposed to be delivered this way particularly to the inner medulla, including thin segments of long looped-nephrons, inner medullary collecting ducts, and the abundant medullary interstitial cells. The lactate formed by these cells in the medulla is returned by ascending vasa recta to mTAL segments as an energy substrate or to S3 segments for gluconeogenesis, which closes the cycle. In accordance with the hypothesis, it has been proposed that lactate is a better gluconeogenic precursor than glutamine in human proximal tubule S2 and S3 segments (63). The reverse transport via SGLT1 is important for the hypothesis formulated by Bankir and Yang. Along these lines, these authors also hypothesized that urea is secreted via SGLT1 in the S3 segment of the proximal tubule (18). Notably, mice lacking SGLT1 have increased plasma urea levels compared with wild-type mice despite similar GFR, when these mice are fed a high protein diet, which enhances hepatic urea formation and thereby the need for renal urea excretion (266). Also, studies in mice, rats, and humans recently indicated that the TAL, in addition to basolateral GLUT1, expresses SGLT1 in the luminal membrane (17, 228, 449). Additional studies are required to demonstrate quantitatively relevant tubular secretion of urea and glucose via SGLT1 in the late proximal tubule.
In euglycemic conditions, the most prominent glucose source for distal tubule glucose utilization, including TAL, may be provided by basolateral uptake of glucose, which is delivered by renal blood flow via peritubular capillaries. In addition, the possibility has been proposed that the glucose generated from lactate in S3 segments of the proximal tubule may in part reach the neighboring TAL by intercellular transport (see above) and that the lactate generated by anaerobic glycolysis in the medullary TAL is then transported back through the interstitium to the S3 segments for gluconeogenesis and closing of a Cori cycle (388).
Studies by Schurek and Johns found that placing a pO2 sensor upon glomeruli and tubuli at the kidney surface in halothane anesthetized rats exhibited pO2 oscillations with exactly the same frequency (mean of 30 mHz; ≈1.8/min) (339) as have been described for single nephron GFR (SNGFR), proximal tubular pressures or distal fluid conductivities (146, 404). The authors proposed that a limited oxygen supply to the nephron forces TAL segments to oscillate between aerobic and anaerobic energy production. A switch to glycolysis would reduce the transport efficiency of TAL, and the resulting rise in NaCl concentrations at the macula densa would lower SNGFR via tubuloglomerular feedback and thereby adapt the tubular workload to the oxygen supply of the individual nephron (339). This way the metabolism in the TAL would oscillate between aerobic (with lower transport load) and anaerobic (with higher transport load) metabolism with the above frequency. This may also protect proximal tubules from oxygen deficiency, which is essential due to their low glycolytic capacity (339), particularly the S1 segment.
Fructose is a substrate for gluconeogenesis in proximal tubules
Since the proximal tubular epithelial cells express fructose transporter(s) (see above), one could assume that fructose plays a physiological role in the kidney. In 1970, Bowman et al. examined isolated rat kidneys and found that fructose stimulated glucose production at the same rate as pyruvate production, and that fructose was more readily converted to glucose than was dihydroxyacetone (33). Likewise, Burch et al. showed that an intraperitoneal injection of fructose rapidly increased serum glucose levels in rats and that the rise in serum glucose was associated with a large accumulation of Fru1P and a marked depletion of ATP in the kidney (40) (see Figures 4 and 7 for illustration of fructose metabolism). Both studies also documented that fructose intermediates, including Fru1P, lactate, and pyruvate, were utilized for gluconeogenesis in the kidney. Moreover, there are several studies showing that the injection of fructose into the renal artery resulted in the release of glucose from the kidney (27, 322).
In 1980, Burch et al. aimed to localize fructose metabolism in the rat kidney following intraperitoneal infusion of fructose (39). The group utilized quantitative microchemical techniques to isolate convoluted and straight portions of the renal proximal tubules for biochemical assays. A key finding was that fructokinase was primarily expressed in proximal tubules, with the highest expression in the straight portion, suggesting that this portion could be the primary site for fructose metabolism in the kidney. However, further investigation illustrated that the convoluted portion of the proximal tubules was also likely to contribute to fructose metabolism as this part expressed both fructokinase and aldolase B (see also Tables 1–3). In addition, fructose injection increased the concentrations of Fru1P and glyceraldehyde 3-phosphate, and activated aldolase B, which is an enzyme that catalyzes Fru1P to dihydroxyacetone phosphate (DHAP) and glyceraldehyde (GA) (39) (see Figures 4 and 7). This notion was further supported by the finding that a lack of aldolase B in people with hereditary fructose intolerance causes Fru1P accumulation in proximal convoluted tubules (192). In micro-dissected mouse and rat tubular segments, the highest fructokinase (KHK) and aldolase B protein and mRNA expression were also detected in proximal tubule segments (Tables 1–3). Gluconeogenesis is stimulated under fasting conditions as low levels of fructose-2,6-biphosphate inhibit PFK-1 activity and glycolysis, and activate fructose-1,6-biphosphatase and glucose production (139) (see Figure 7).
Fructose appears to be the preferred substrate for renal gluconeogenesis based on the speed and efficiency of the reaction compared with classic substrates such as lactate, glutamine, alanine, and pyruvate (33, 193). By utilizing in situ perfusion in the rat and directly measuring the arteriovenous difference of fructose and glucose concentrations after bolus infusion of 25mg of fructose into the peripheral vessels, Salmon et al. observed that decreases in fructose concentration on the passage of blood through the kidney were closely associated with equivalent increases in renal venous glucose: the disappearance of fructose and appearance of glucose averaged approximately 19% (322). Moreover, following intravenous infusion of fructose at 2 mmol/min for 135 min into the peripheral vein of humans 20% of the infused fructose was taken up by the kidney (in the range of the glomerular filtration fraction), and this was associated with an increase in glucose concentration in the renal vein by approximately 0.17mmol/L, such that the net glucose release from the kidney could be derived from 55% of the net renal uptake of fructose (27).
Endogenous fructose production from glucose in the kidney
In addition to producing glucose, the kidney is capable of endogenously producing fructose under physiological condition as shown in mice, and this fructose production can be amplified under several pathological conditions (197). The machinery involved in fructose production is the polyol pathway, in which glucose is reduced to sorbitol by aldose reductase, followed by the oxidation of sorbitol into fructose by sorbitol dehydrogenase (see Figure 4). Fructose was detected in liver and kidney of wild-type mice (197, 198) and naked mole rats (287), and in serum of nondiabetic individuals (172), suggesting that glucose under physiological conditions might be converted into fructose in the polyol pathway. Nevertheless, this pathway can be further activated by several conditions, including hypoxia and osmotic stress due to high glucose and high salt (6, 198). Under physiological conditions, the renal medulla operates under a low oxygen availability as the partial oxygen pressure is in the range of 10 to 20 mmHg, contrasting with that in the cortex of about 50 mmHg (35). Oxygen supply is poor in the medulla because of oxygen shunting, which includes the medullary countercurrent system of vessels, in which oxygen diffuses from arterial to venous vasa recta. In addition, several active transporters are highly expressed in tubular and collecting duct segments in the outer medulla (e.g., NKCC2 in TAL), demanding a large amount of oxygen. In addition, some tubules are anatomically remote from vessels and thereby more exposed to hypoxia (35). Since those cells in the medulla seem constantly threatened by hypoxia under physiological conditions, kidneys need a protective machinery for the low oxygen tension under physiological conditions and, as discussed in later sections, under pathophysiological conditions. See also the above discussion on oscillating TAL metabolism and tubuloglomerular feedback.
Fructose is metabolized in the entire proximal tubule, and most strongly in the straight portion of the proximal tubule, and as discussed above, is physiologically utilized for gluconeogenesis. In addition, fructose may also contribute to protect the kidney from physiological hypoxia as the straight segment of the proximal tubule enters the outer medulla, a region where epithelial cells are physiologically exposed to borderline hypoxia. Studies in cultured cardiomyocytes found that fructose metabolism was further amplified by hypoxia together with evidence that hypoxia-inducible factor-1 (HIF-1) induced the mRNA expression of the liver fructokinase isoform (KHK-C), which has a high affinity for fructose and phosphorylates fructose as the first step of its metabolism (245). Studies in naked mole rats illustrated the role of endogenous fructose as a survival mechanism under low oxygen conditions: the experiments identified a potential mechanism by which several organs, including kidney and liver, were capable of producing fructose, which was then metabolized to generate glycolytic products (287). The precise mechanisms as to why fructose exerts protective effects under low oxygen conditions remain unclear, but it might be accounted for by the ability of fructose to provide not only energy but also serve the formation of other metabolites that are necessary for maintaining physiological functions. As shown in Figure 4, the increased glycolysis from Fru1P activation and the increased use of the pentose-phosphate pathway via transketolase activation can provide ATP, NADPH, and ribose for generation of lipids, hexosaminoglycans, and nucleic acid, which are all helpful for cell survival (154, 179). Basically, fructose metabolism, as opposed to glucose, is not associated with mitochondrial respiration so that oxygen levels are likely less important (see Figure 10). A potential mechanism is that urate, a by-product of fructose metabolism, suppresses aconitase, which converts citrate into iso-citrate in the TCA cycle thereby disconnecting it from mitochondrial oxidative phosphorylation (see Figures 4 and 10). Thus, fructose intermediates link to other metabolic pathways, including the PPP, hexosamine pathway, and lipid synthesis (see Figure 4). The PPP comprises two distinct phases, the oxidative pathway, and the nonoxidative pathway. Glucose 6-phosphate is metabolized to NADPH in the oxidative pathway while the nonoxidative pathway for nucleotide formation through ribose 5-phosphate is used for nucleotide formation, and erythrose 4-phosphate is metabolized into amino acids. In turn, the hexosamine pathway, which usually accounts for only 2% to 5% of total glucose metabolism, has been associated with posttranslational protein modification by glycosylation and the synthesis of glycolipids, proteoglycans, and glycosylphosphatidylinositol anchors (333). The hexosamine pathway is involved in both physiological and pathological processes. Examples include podocyte maturation under physiological condition (279) and TGF-β induction under diabetic condition (333).
Figure 10. Basic differences in glucose versus fructose metabolism.

Glucose metabolism in the kidney primarily occurs downstream of the proximal tubule and is determined by oxygen levels. (A) In the presence of sufficient oxygen (indicated by black arrow), glycolysis links to mitochondrial respiration/oxidative phosphorylation (OXPHOS) to efficiently produce high amounts of ATP. However, under low oxygen conditions (indicated by red arrow), mitochondrial respiration is disconnected from glycolysis, and lactate is produced. (B) In comparison, basic fructose metabolism is associated with glycolysis rather than mitochondrial respiration under either normoxia or hypoxia (indicated by blue arrow). A potential mechanism is that urate, a by product of fructose metabolism, suppresses aconitase (Aco), which converts citrate into iso-citrate as part of the TCA cycle, and disconnects fructose metabolism from mitochondrial respiration.
In contrast, glucose metabolism is more strongly linked to oxygen consumption. Under aerobic condition, glycolysis links to mitochondrial respiration to efficiently produce high amount of ATP whereas under anaerobic condition mitochondrial respiration is disconnected from glycolysis and lactate is produced. Alternatively, hypoxia activates aldose reductase in the polyol pathway to endogenously produce fructose from glucose, so that glucose metabolism could be operating more through the fructose pathway in hypoxia (see Figures 4 and 10). Further studies are needed to better understand the metabolic interactions between glucose and fructose in the kidney and other organs.
Glucose Handling in Renal Pathophysiology
Renal glucose transport in acute kidney injury
Acute kidney injury (AKI) is associated with high morbidity and mortality (3) and is a risk factor for chronic kidney disease (CKD) and end-stage kidney disease (ESKD) (87). Available treatments largely include supportive care rather than efficacious preventive or curative measures (51). As discussed above, the outer medulla of the kidney is particularly vulnerable (23, 140), due to high active solute reabsorption, including the S3 segment of the proximal tubule and the medullary TAL in combination with a relatively low blood and oxygen supply (132, 317).
The high transport rates in proximal tubules depend on high ATP turnover, which, in the healthy kidney, is primarily established by mitochondrial oxidative phosphorylation (124, 427). The proximal tubular metabolism can change in pathophysiological conditions that impair mitochondrial function. For example, proximal tubules that regenerate from AKI or proximal tubules that undergo atrophy show a shift to more glycolysis (196). This switch occurs during the early phase of proximal tubule regeneration and reverses during tubular recovery, but the shift persists and becomes even more apparent in the tubular cells that fail to re-differentiate and recover. In a murine model, tubular overexpression of HIF-1alpha increased the expression of GLUT1 mRNA in the kidney; this was associated with less oxygen consumption and increased glycolysis (79). Thus, hypoxia may enhance the basolateral facilitative uptake of glucose via GLUT1, and the glucose is then used as an energy source for anaerobic glycolysis. A hypoxia-induced upregulation of GLUT1 is likely to occur in distal tubule segments, but may also be relevant for the proximal tubule including medullary S3 segments (196), and may occur in response to hypoxic tubular injury and potentially in diabetic conditions (see Figure 9). In accordance, preliminary studies in a murine model of AKI (the renal ischemia-reperfusion model is described in more detail below) showed that while the renal mRNA expression of basically all other renal membrane transporters were reduced on day 1 after the injury, the renal expression of Glut1 mRNA was upregulated (80). A recent study confirmed that also the genes related to renal gluconeogenesis are downregulated in AKI, as observed in rodents and humans, and the authors associated impaired proximal tubule glucose metabolism/gluconeogenesis to mortality (208).
Renal ischemia-reperfusion (IR) injury is a clinically relevant cause of AKI that can occur in cardiac bypass surgery, shock, in kidney transplantation, or in response to decompensation of congestive heart failure (31, 231). Ischemic ATP depletion causes the rapid and reversible loss of proximal tubule brush border, a typical hallmark of early IR injury that is associated with a robust reduction in fluid and solute reabsorption. Studies in rats demonstrated that renal ischemia is associated with a reduced SGLT2 expression at the apical membrane and a transient reduction in SGLT-mediated glucose transport activity in brush border vesicles of proximal tubules (162, 248). This is consistent with preliminary RNA sequencing data performed on day 1 of reperfusion after 15 or 25 min of bilateral renal artery clamping in C57BL/6J mice that showed downregulation of renal Sglt2 and Sglt1 mRNA expression; in contrast, these studies found upregulation of Glut1 mRNA (80) as discussed above. Downregulation of SGLT2 expression and the related transport work may protect the early proximal tubule from further injury. According to a meta-analysis, SGLT2 inhibition is in fact associated with a reduction in AKI in people with type 2 diabetes mellitus (T2DM) (110, 270). In accordance, treatment of people with T2DM with the SGLT2 inhibitor dapagliflozin reduced urinary levels of markers of glomerular and tubular injury (65, 331) (effects of SGLT2 inhibition in diabetes are discussed in detail below). Moreover, the SGLT2 inhibitor luseogliflozin did not protect from initial AKI but ameliorated renal capillary rarefaction and reduced hypoxia and fibrosis in a nondiabetic murine model of renal IR (451). The study used combined renal artery and vein occlusion, luseogliflozin application was initiated 6 h after IR, and the mice were followed up for 1 to 4 weeks. The authors proposed that the beneficial effect of luseogliflozin was due to the lowering of intracellular glucose concentrations in early proximal tubules to an extent that the formation of vascular endothelial growth factor (VEGF) is stimulated (451). In another study in mice, the SGLT2 inhibitor dapagliflozin attenuated the increase in plasma creatinine at 24 h after IR when the drug was initiated 48 h earlier (50). For unclear reasons, SGLT2 gene-knockout in mice did not affect the extent of acute IR injury or the subsequent glomerular and tubular recovery when IR was induced by bilateral renal artery clamping in nondiabetic mice and follow-up for 23 days (267). The latter study confirmed IR-induced endothelial rarefaction in wild-type kidneys as shown by a decreased CD31-positive area, associated with a significant downregulation of renal Vegfa mRNA; the absence of SGLT2, however, did not prevent these changes (267). As discussed above, renal IR injury is associated with mitochondrial dysfunction and a metabolic shift of the recovering proximal tubules toward anaerobic glycolysis (9, 196). The studies in Sglt2 knockout mice indicated a similar upregulation of whole kidney mRNA expression of hexokinase Hk2, a key glycolytic enzyme, as well as downregulation of the transcription factor Ppargc1a, a master regulator of mitochondrial biogenesis and function (223), in both genotypes, suggesting that the renal transition to anaerobic glycolysis might not have been largely affected by the absence of SGLT2, at least on the whole kidney level. The authors note that the observed robust SGLT2 downregulation they have observed in their IR model may protect the kidney from further damage but would also have made it more difficult to detect beneficial effects of the SGLT2 knockout. All 3 studies used young adult male C57BL/6 mice; thus, differences in gender, age, and mouse strain cannot explain the observed differences. Further studies are needed to explain the different results and also compare genetic versus pharmacologic inhibition of SGLT2, including potential off-target effects of these drugs as recently proposed in the heart, including the ischemic heart (12, 50, 386, 387).
The downregulation of SGLT2 in response to IR can enhance the transport burden and oxygen consumption via SGLT1 in the downstream outer medullary S3 segment, which could worsen injury or recovery in the latter segment (343, 392). On the other hand, glycolysis has been proposed to increase in the injured and recovering proximal tubule (see above) and, thus, the glucose uptake via SGLT1 may provide a much-needed energy substrate. To address this issue, Nespoux et al. subjected mice lacking SGLT1 and their wild-type littermates to the bilateral renal artery clamping model of IR and monitored the mice over 16 days after IR (266). The study provided evidence for a sustained contribution of SGLT1 to tubular glucose reabsorption during the early injury and the subsequent recovery phase, associated with recovered renal mRNA expression of Sglt1 on day 16 after IR whereas Sglt2 expression was still strongly suppressed. While the absence of SGLT1 seemed not to affect early tubular injury and the associated impairment of kidney function after IR, the tubular and glomerular recovery was significantly improved. This included higher GFR, more completely restored urine and plasma osmolality, reduced tubular injury score in the cortex and outer medulla, better preserved renal mRNA expression of tubular transporters (Sglt2, Nkcc2), and a lesser rise in renal mRNA expression of markers of injury, inflammation, and fibrosis in Sglt1 knockout versus wild-type mice in response to IR. Whereas the upregulation of whole kidney mRNA expression of Hk2 on day 16 after IR occurred independent of SGLT1, the renal mRNA expression of Ppargc1a was higher in mice lacking SGLT1 suggesting a potential improvement in mitochondrial biogenesis and function. The study thus indicated a deleterious role of SGLT1 during recovery from renal IR (266) (see Figure 11), which is reminiscent of the deleterious role of cardiac SGLT1 proposed in heart IR injury (212).
Figure 11. A proposed deleterious role for SGLT1-mediated reabsorption during recovery from ischemia-reperfusion (IR)-induced acute kidney injury.

IR initially suppresses SGLT2 and SGLT1-mediated reabsorption in the early and later proximal tubule, respectively, which is associated with glucosuria. Early recovery of SGLT1 expression and SGLT1-mediated sodium reabsorption in late proximal tubule/outer medulla sustain IR-induced hypoxia. This sustains cell injury in the outer medulla and the inhibition of NKCC2-mediated NaCl reabsorption in the TAL, which impairs urine concentration and enhances Na-Cl-K delivery to macula densa ([NaCl-K]MD). The latter reduces renin expression and lowers GFR via tubuloglomerular feedback. The reduction in GFR enhances plasma creatinine and urea, the latter contributing to enhanced plasma osmolality. The sustained hypoxia and cell injury further enhance mitochondrial dysfunction, inflammation, and fibrosis, which can spread to the cortex and further suppress tubular function. Sustained suppression of SGLT2 maintains a high glucose load to downstream SGLT1, which may enhance the detrimental influence of SGLT1. Modified, with permission, from Nespoux J, et al., 2019 (266).
Diabetes increases renal glucose transport
Diabetes mellitus is associated with hyperglycemia and the glomerular filtration of larger amounts of glucose, as long as GFR is preserved. In the early stages of diabetes, GFR is often elevated (glomerular hyperfiltration; discussed below), which further increases the tubular glucose load. This is associated with an increase in the renal transport capacity for glucose by approximately 20% to 30% to approximately 500 to 600 g/day in people with T1DM (247) and T2DM (42, 78). This makes sense from the standpoint that glucose is a valuable energy substrate that the body wants to conserve. Thus, glomerular filtration and tubular reabsorption of glucose are typically increased in the early diabetic kidney. Despite increased blood glucose levels, diabetes may also stimulate renal gluconeogenesis (102, 177, 243). This can be due to diabetes-associated metabolic acidosis, activation of the sympathetic nervous system, or reduced insulin levels in T1DM (see Figures 5 and 9), as well as enhanced circulating fatty acids (102).
When the diabetic kidney makes more glucose, this can help to maintain acid-base balance or provide fuel for distal segments to reabsorb the enhanced tubular load due to diabetic glomerular hyperfiltration. However, renal glucose retention and enhanced glucose formation also sustain hyperglycemia, and, in this regard, are considered maladaptive (see Figure 9). While the genetic and biological programming of the kidneys to retain glucose evolved during times of scarce energy sources, these programs become maladaptive during excess calorie intake and hyperglycemia.
Upregulation of renal glucose transporter expression is likely to contribute to the increased glucose transport capacity observed in diabetes. Nevertheless, available preclinical and human studies provided inconsistent or conflicting results reporting reduced, unchanged, or increased renal glucose transporter expression and/or activity in response to hyperglycemia or diabetes (for review see Refs. 289, 399, 408). These differences may reflect dissociation between mRNA and protein expression, the use of nonspecific antibodies, variations in metabolic states and levels of kidney injury, or the use of dissimilar diabetes models. With regard to the latter, streptozotocin (STZ), which is often used in rodents to induce diabetes through pancreatic beta-cell apoptosis after being transported into these cells by GLUT2, may also enter and injure proximal tubule cells through the same pathway (122), which may contribute to contrasting effects in glucose transporter expression versus genetic diabetes models (405).
Increase in SGLT2 and GLUT2 expression in the diabetic kidney
Renal protein expression of SGLT2 has been reported to be increased by 40% to 80% in the early stages of genetic mouse models of T1DM (Akita) and T2DM (db/db) (396, 405). These studies used knockout mice as critical negative antibody controls. Consistent with a potential concerted regulation of luminal and basolateral glucose transport, up-regulation of GLUT2 expression has been reported in renal proximal tubules in multiple studies in diabetic rats (54, 69, 91, 165).
In STZ-diabetic rats and mice and in addition to its normal basolateral location, GLUT2 has also been detected in the brush border of proximal tubules (113, 142, 232) (see Figure 9). This has been linked to the activation of protein kinase C PKCβ1 (113, 295, 296) and may facilitate apical glucose diffusion into the proximal tubule cells if the luminal chemical potential of glucose were to rise above that in the cell and interstitial space. On the other hand, studies in mice indicated that SGLT2 and SGLT1 can account for all net renal glucose reabsorption in the nondiabetic and diabetic setting (354) inasmuch as net renal glucose reabsorption was eliminated when pharmacological SGLT2 inhibition was applied to genetic mouse models of T1DM (Akita) and T2DM (db/db) that lacked SGLT1. Notably, the small intestine senses an increases in luminal glucose concentrations via SGLT1, which is required for the co-insertion of GLUT2 into the brush border membrane (174). If SGLT2 plays a similar role in the early proximal tubule, then SGLT2 inhibition may lower renal glucose reabsorption in part by inhibiting apical GLUT2 translocation (see Figure 9). Alternatively, apical GLUT2 is leaking back glucose from the tubular cells into the lumen. This would facilitate apical recycling of glucose and strongly promote sodium reabsorption through SGLTs. Expression of GLUT2 in the brush border of the kidney needs to be confirmed in non-STZ models of diabetes.
The information available on the expression of glucose transporters in the kidneys of people with diabetes is sparse and variable. Primary cultures of human exfoliated proximal tubular epithelial cells that were harvested from fresh urine of people with T2DM showed increased protein expression of SGLT2 and GLUT2 associated with higher glucose uptake (304). Higher SGLT2 protein expression has also been reported in fresh kidney biopsies of people with T2DM that had advanced nephropathy (421). In comparison, in 19 people with T2DM and preserved kidney function being subjected to nephrectomy the renal mRNA expression of SGLT2 and GLUT2 were slightly reduced as compared with 20 nondiabetic individuals matched for age and estimated glomerular filtration rate (352). In another set of people with T2DM, lower levels were also reported for renal SGLT2 and GLUT2 mRNA expression but the results did not reach statistical significance (273).
An increase in SGLT2 expression in the diabetic kidney can be the result of the overall growth and hypertrophy of the proximal tubule and the associated increase in transport machinery (391, 409) (see Figure 9). Studies in HEK-293T cells showed that insulin can phosphorylate SGLT2 at Ser624 and that this was associated with higher Na+-glucose transport (109). Thus, the insulin release in the postprandial phase may activate SGLT2 in the early proximal tubule to reabsorb and conserve the increased amounts of filtered glucose (see Figure 5). Moreover, enhanced renal SGLT2 activity may result due to the hyperinsulinemia associated with insulin resistance in obesity and people with T2DM (378) (see Figure 9). Activation of Ang II AT1 receptors (280) and the transcription factor, hepatocyte nuclear factor HNF-1α (90) have been implicated in the up-regulation of SGLT2 expression in diabetic rats. Studies in proximal tubular cells proposed that high glucose can upregulate angiotensinogen expression, potentially through HNF-5 (420, 450), which through the local formation of Ang II may contribute to a local mechanism to enhance SGLT2 expression. On the other hand, basolateral hyperglycemia may be sensed through GLUT2 to trigger HNF-1α (385) (see Figure 9). HNF-1α and HNF-3β have also been implicated in the renal up-regulation of GLUT2 (91) (see Figure 9).
As intracellular glucose levels may induce a negative feedback regulation of SGLT2 expression (see above), the observation of subnormal SGLT2 expression in the diabetic kidney can reflect enhanced proximal tubular gluconeogenesis (e.g., due to metabolic acidosis; see Figures 5 and 9) or be the consequence of more severe tubular hypoxia and inflammation (334, 392, 445).
SGLT1 expression in the diabetic kidney
SGLT1 protein expression was enhanced in the kidney of leptin-deficient ob/ob mice (101), a model of T2DM. The serum and glucocorticoid-inducible kinase SGK1 has been proposed to stimulate SGLT1 activity and glucose reabsorption in proximal straight tubules in Akita diabetic mice, a model of T1DM (1). Moreover, SGK1 was upregulated in proximal tubules of people with diabetic nephropathy (201). In contrast, SGLT1 protein expression was lower in the kidneys of Akita mice in another study that used knockout mice to demonstrate the specificity of the antibody (405).
In contrast to SGLT2 (see above), insulin stimulation decreased SGLT1-mediated Na+-glucose transport in HEK-293T cells (109), suggesting that insulin regulates these two transporters differently. Notably, acute exogenous insulin infusion in hyperglycemic individuals enhanced glucosuria and natriuresis. People with T2DM were resistant to this effect, and the effect was not abolished by partial SGLT2 inhibition (85). These results would be consistent with an inhibitory effect of insulin on renal SGLT1. In contrast to the increase in SGLT2, SGLT1 protein expression determined in fresh kidney biopsies of people with T2DM and nephropathy was not significantly different versus nondiabetic controls (421).
Why should diabetes not increase renal SGLT1 expression to further enhance glucose reabsorption capacity but actually reduce SGLT1 expression? Other conditions that, like diabetes, enhance the glucose delivery to the late proximal tubule were also found to lessen renal SGLT1 protein expression: this included the response to SGLT2 knockout and pharmacological inhibition of SGLT2 inhibition in nondiabetic mice (396, 402). In vitro studies in proximal tubule cells showed that high glucose concentrations can lower both the expression of SGLT and the activity of Na+/glucose cotransport and that this response may be due to enhanced oxidative stress (130). Studies in a model of pig epithelial tubular cells (LLC-PK1) indicated that hypoxia-induced HIF-1alpha can suppress SGLT1 (and SGLT2) protein expression (445). Thus, an increase in the glucose load to the S3 segment can enhance the absolute Na+-glucose reabsorption via SGLT1 manifold (312); to limit the resulting hypoxia as well as the glucotoxicity in this segment, which has a high sensitivity to acute injury (392), however, SGLT1 is down-regulated by the associated increase in oxygen consumption (SGLT1 requires the reabsorption of 2Na+ per glucose) (see Figure 1). The role of SGLT1 in the macula densa is discussed below.
GLUT1 expression in the diabetic kidney
In STZ-induced diabetes in rats, GLUT1 protein expression was downregulated in isolated cortical proximal tubules at 2 and 4 weeks after STZ (69) but increased in whole kidneys at 30 weeks after STZ (215). In people with T2DM and preserved kidney function, renal GLUT1 mRNA expression was slightly less compared with nondiabetic individuals (352). The meaning and relevance of the available GLUT1 data in the diabetic kidney remain unclear. Also in diabetic kidneys, GLUT1 in distal tubule segments and potentially the proximal tubule may facilitate basolateral uptake of glucose. Basolateral exposure to 25mmol/L d-glucose of the proximal tubular cell line LLC-PK1, cultured and polarized on porous tissue culture inserts, enhanced glucose uptake via GLUT1; moreover, the intracellular metabolism of the absorbed glucose enhanced TGF-beta 1 synthesis and secretion, an effect not observed in response to apical glucose exposure (297). These in vitro studies indicate the possibility that it could be the persistent uptake of glucose via basolateral GLUT1 (or GLUT2?) facilitated by hyperglycemia, rather than the apical glucose uptake, that triggers the tubular synthesis of TGF-beta 1 and thereby the development of tubulointerstitial fibrosis and tubular growth (see Figure 9). More studies are required to better understand the role of basolateral renal glucose transport and its implications in health and disease.
Hyperglycemia is associated with tubular glycogen accumulation
In hyperglycemic conditions, cells of the TAL and, to a lesser extent, of the DCT and the CD show glycogen deposits, also known as glycogen nephrosis or Armanni-Ebstein lesions (26, 144, 308, 363). The glycogen deposits are largely confined to the kidney cortex and outer stripe of the outer medulla (308). With long duration of diabetes, glycogen accumulation has also been described in renal proximal tubules of diabetic individuals (313), and in preclinical rodent models (169, 262).
Glycogen accumulation in the diabetic kidney may reflect activation of glycogen synthase (GS). Normally, adiponectin, which is a regulator of glucose levels and fatty acid breakdown, is secreted by white adipocytes and filtered by glomeruli to bind to its receptor, the adiponectin receptor 1 (ADIPOR1) in the luminal membrane of the TAL to activate 5′ adenosine monophosphate-activated protein kinase (AMPK) and inhibit GS; this regulation was found to be impaired in STZ-diabetic rats (46). High glucose-induced dephosphorylate and inhibition of AMPK may also contribute to tubular GS stimulation and glycogen accumulation. Moreover, TALs from diabetic animals have higher concentrations of glucose-6-phosphate (46), an allosteric activator of GS, and in humans with diabetes, an increase in the renal expression of protein targeting to glycogen (PTG) (435) may enhance GS activity.
Most notable, whether the glycogen that abnormally accumulates in diabetic tubules is pathological, somehow protective or an inconsequential bystander is still unclear (363). Moreover, glycogen accumulation in the distal nephron of diabetic rats correlated with the delivery of glucose via the tubular fluid (144), but the role of apical versus basolateral glucose transporters in glycogen accumulation remains unclear.
Pleiotropic effects of SGLT2 inhibition in the diabetic and nondiabetic kidney
Therapies for T2DM usually target the small intestine, liver, pancreatic islets, adipose tissue, or skeletal muscle. Some of these therapies, including insulin, may not establish adequate glycemic control without relevant unwanted side effects, like hypoglycemia and weight gain, and may not reduce cardiovascular complications (105). The following sections describe the logic of SGLT2 inhibition in the diabetic kidney. Moreover, a better understanding of their mechanisms of action may teach us important lessons about the inner workings of the kidney.
Long-term access to abundant exogenous energy resources is not part of human evolution, and, therefore, the body’s responses can be maladaptive. In contrast, the body’s ability to adapt to environments with scarce energy resources has been tested and refined throughout evolution for the survival of the organism. Therefore, targeting the body’s metabolic “periphery” by inhibiting renal glucose reabsorption and spilling glucose as an energy resource and extra calories into the urine, which then activates metabolic counterregulatory mechanisms similar to fasting, may provide unique benefits as an anti-hyperglycemic approach (409).
The logic of inhibiting SGLT2 in the diabetic kidney as a therapeutic strategy starts with the role of tubular glucose reabsorption in maintaining hyperglycemia (see Figure 9). Multiple SGLT2 inhibitors have been approved as glucose-lowering agents for people with T2DM and their use is being explored in people with T1DM (81, 82, 409). SGLT2 inhibitors reach their target in the luminal cell membrane of the early proximal tubule from the extracellular surface (108), which is reached by glomerular filtration and, as shown for empagliflozin, also by tubular secretion via the organic anion transporter OAT3 (93) (HNF-1a upregulates both SGLT2 and OAT3 expression). SGLT2 inhibition lowers the renal capacity to reabsorb glucose to the amounts that can be reabsorbed by SGLT1, that is, approximately 80 g/day. Thus, SGLT2 inhibition causes the renal “glucose valve” to open at much lower and physiological blood glucose levels (see Figure 2). SGLT2 inhibitors dose-dependently excrete up to 40 to 80 g glucose per day, which in people with T2DM is associated with a decrease in Hb A1C levels of 0.5% to 0.7% (409). The higher the blood glucose level and GFR, the more glucose is filtered and reabsorbed by SGLT2 and, as a consequence, will be excreted in response to an SGLT2 inhibitor. The glucosuric effect of SGLT2 inhibitors increases the risk of genitourinary infections, which is the main side effect (100).
The observed small effect of SGLT2 inhibitors on blood glucose control alone cannot fully explain their protective effects on the kidney and heart that have been detected within a few months in large cardiovascular outcome trials (375). In contrast to SGLT2 inhibitors, other anti-hyperglycemic agents may have deleterious effects that offset the benefits of improving glycemic control, including a gain in body weight and an increase in the hypoglycemia risk. SGLT2 inhibitors do not increase the incidence of hypoglycemia (249, 263, 422, 454) because they become ineffective at lowering blood glucose once the filtered glucose load falls to approximately 80g/day, which can be handled by downstream SGLT1. Moreover, SGLT2 inhibitors leave the metabolic counterregulation intact and increase plasma glucagon concentrations and subsequently endogenous hepatic glucose production (gluconeogenesis) in people with T2DM (84, 242). The mechanism by which SGLT2 inhibitors increase glucagon secretion in pancreatic alpha cells remains controversial (194, 329, 361). Nevertheless, episodes of hypoglycemia impair the cardioprotective effects of anti-hyperglycemic therapy and have been linked to CKD and mortality (88, 105, 176, 246, 286), and thus, the intact metabolic counterregulation is potentially relevant for the improved cardiovascular outcome in response to SGLT2 inhibitors. SGLT2 inhibitors attenuate blood glucose highs and prevent blood glucose lows, which together has little effect on HbA1C values, but tightens 24 h blood glucose profiles within the desirable range with potential benefits for renal and cardiovascular outcome.
Body weight is reduced in response to SGLT2 inhibition due to the initial diuretic effect and the renal loss of calories. The latter shifts substrate utilization from carbohydrates to lipids and reduces body fat, including subcutaneous and visceral fat (409). The rise in free fatty acid availability drives the hepatic formation of ketone bodies, which are used as additional energy substrates, including in cardiac and kidney cells (83, 250, 301, 417). Tomita et al. proposed that the ketone bodies formed in response to SGLT2 inhibition protect from diabetic kidney disease, including podocytes and tubular structures, by inducing mTORC1 inhibition (381) (see Figure 9). On the other hand, SGLT2 inhibitors can increase the risk of diabetic ketoacidosis (301), particularly when the drugs are used off-label in people with T1DM (301). By lowering body weight and improving blood glucose control, SGLT2 inhibitors improve the sensitivity to insulin and beta-cell function as shown in people and rodent models with T2DM (84, 133, 164, 226, 242). By lowering hyperglycemia, SGLT2 inhibitors can attenuate the deleterious effects of glucotoxicity on the kidney and extrarenal organs (269).
In the following, we discuss direct and indirect kidney and heart-protecting effects of SGLT2 inhibition that are to a significant extent independent of blood glucose lowering and thus can help explain the proposed protective effects of these drugs on the kidney and heart in the absence of diabetes (137, 240, 283).
SGLT2 inhibition initially lowers GFR
Glomerular hyperfiltration, which is observed in a subset of people at the onset of T1DM and T2DM, not only increases the renal oxygen requirements but is also a risk factor for developing diabetic nephropathy (230, 399). Also in nondiabetic people with stage 1 hypertension, glomerular hyperfiltration predicted the development of microalbuminuria (284). Nearly all of the Na+ that is filtered in the glomeruli needs to be reabsorbed in the tubular system to match urinary excretion to dietary intake. Tubular transport, however, is the primary determinant of renal oxygen consumption. Therefore, an increase in GFR enhances renal transport work and oxygen consumption, while lowering GFR has the opposite effects (204).
The “tubular hypothesis” states that glomerular hyperfiltration in diabetes can be explained by a primary increase in tubular reabsorption and the physiology of the tubuloglomerular feedback (TGF) mechanism (for review see Refs. 393, 408, 410). Moderate levels of hyperglycemia increase proximal tubular reabsorption by providing more substrate for Na+-glucose cotransport via SGLT2 and SGLT1 and by causing the tubule to grow, which enhances the tubular transport machinery and transport capacity. The increased reabsorption in the proximal tubule reduces the NaCl and fluid delivery to the downstream macula densa (MD), which senses this reduction and subsequently increases GFR through the normal physiology of the TGF mechanism (see Figure 12). The TGF mechanism is mediated by basolateral release of ATP from MD cells, which occurs in proportion to the luminal NaCl concentration and delivery, and the subsequent extracellular conversion of ATP to adenosine; adenosine then adjusts the tone of the afferent arteriole (constriction via adenosine A1 receptor) and under some condition of the efferent arteriole (vasodilation via A2 receptors) (29, 311, 400, 412). This alters the GFR of the same nephron such that the change in NaCl delivery to the MD and the resulting change in GFR are inversely related (see Figure 12). The net effect is that the NaCl and fluid delivery downstream of the MD is stabilized by the TGF mechanism. The latter facilitates the fine regulation of NaCl and fluid balance in the more distal nephron by neurohumoral control. Thus, in the diabetic kidney, the physiology of TGF makes the GFR responsive to the primary increase in tubular reabsorption that occurs upstream of the macula densa, and as a consequence, the GFR increases (see Figure 13). Notably, also in nondiabetic individuals with hypertension, an index of proximal tubular hyperreabsorption (reduced lithium clearance) has been positively associated with larger kidneys, higher GFR and albuminuria, and total body exchangeable sodium, consistent with the tubular hypothesis of glomerular hyperfiltration (274). A primary increase in proximal reabsorption also lowers distal tubular flow rate, which lowers tubular back pressure, that is, the hydrostatic pressure in Bowman space, and thereby enhances the effective glomerular filtration pressure and GFR (see Figure 13). Mathematical modeling predicts that the TGF mechanism and the changes in tubular back pressure may contribute equally to the increase in GFR in diabetes (128).
Figure 12. Tubuloglomerular feedback, SGLT2 inhibition, and SGLT1 as a glucose sensor in the macula densa.

(A) and (B) The tubuloglomerular feedback (TGF) establishes an inverse relationship between the delivery and concentration of Na-Cl-K at the macula densa and the single nephron GFR (SNGFR) of the same nephron. The operating point typically resides in the steepest part of the curve. In the diabetic kidney, a primary increase in proximal tubular reabsorption lowers Na-Cl-K delivery to the macula densa, which increases SNGFR through the physiology of TGF. SGLT2 inhibition attenuates proximal tubular reabsorption and increases Na-Cl-K delivery to the macula densa and lowers SNGFR through TGF. (C) (1 + 2) The macula densa senses an increase in luminal Na-Cl-K delivery by a NKCC2-dependent mechanism, which then enhances the basolateral release of ATP. (3) ATP is converted by endonucleotidases CD73/39 to adenosine (ADO). (4) ADO activates the adenosine A1 receptor in vascular smooth muscle cells (VSMC) of the afferent arteriole to increase cytosolic Ca2+ and induce vasoconstriction and lower GFR. (5) ADO can also activate adenosine A2 receptors on VSMC of the efferent arteriole to reduce cytosolic Ca2+ and induce vasodilation. (6) Both effects contribute to the TGF mechanism and lower glomerular capillary pressure (PGC). (7) Due to upstream tubular hyperreabsorption, diabetes lowers Na-Cl-K delivery to the macula densa. SGLT2 inhibition attenuates the hyperreabsorption, increase Na-Cl-K delivery to the macula densa, and lowers GFR and PGC. (8) An increased Na-Cl-K delivery also activates nitric oxide synthase NOS1 in the macula densa. (9) The formed nitric oxide (NO) diffuses across the interstitium and dilates the afferent arteriole, thereby partially offsetting the afferent arteriolar vasoconstrictor tone of TGF. (10) When glucose delivery to the macula densa is increased, SGLT1 in the luminal membrane takes up glucose, a process that is linked to the phosphorylation, activation, and increased expression of NOS1 in the macula densa. The resulting enhanced NO tone dilates the afferent arteriole and enhances GFR. This can contribute to diabetes-induced hyperfiltration, but also attenuate the reduction in GFR by SGLT2 inhibition. (11) On the other hand, SGLT2 inhibition can reduce macula densa NOS1/NO tone by inducing volume depletion. (12) ?, whether enhanced macula densa NO formation can also dilate the efferent arteriole remains to be determined. (C) Modified, with permission, from Vallon V, 2020 (388).
Figure 13. The tubular hypothesis of diabetic glomerular hyperfiltration.

(A) and (B) In vivo micropuncture studies in rats with superficial glomeruli were performed in nondiabetic and streptozotocin-diabetic rats (403). Small amounts of blue dye were injected into Bowman space to determine nephron configuration, including the first proximal tubular loop and the early distal tubule close to the macula densa. Tubular fluid was collected close to the macula densa to determine the tubuloglomerular feedback signal ([Na-Cl-K]MD) and single nephron glomerular filtration rate (SNGFR; by inulin clearance). Bowman space was punctured to determine the hydrostatic pressure (PBow). Measurements were performed under control conditions and following application of the SGLT2/SGLT1 inhibitor phlorizin into the early proximal tubule, that is, without changing systemic blood glucose levels. Basal measurements (con) revealed that glomerular hyperfiltration in diabetes was associated with reductions in [Na-Cl-K]MD and PBow. Adding phlorizin (P) had a small effect in nondiabetic rats, but normalized [Na-Cl-K]MD, PBow, and SNGFR in diabetes. (C) Kidneys are programmed to retain glucose. As a consequence, diabetes induces a primary hyperreabsorption in proximal tubules involving enhanced Na+-glucose cotransport and tubular growth. The concomitant enhanced reabsorption of sodium causes glomerular hyperfiltration through tubuloglomerular feedback ([Na-Cl-K]MD) and reducing tubular back pressure (PBow) thereby limiting sodium and volume retention. SGLT2 contributes to the tubular hyperreabsorption and as a consequence, SGLT2 inhibition mitigates these changes and lowers glomerular hyperfiltration. Modified, with permission, from Vallon V, and Thomson SC, 2017 (409).
In accordance with a prominent role of SGLT2 in the tubular hypothesis of diabetic glomerular hyperfiltration, SGLT2 inhibition attenuates proximal tubule hyperreabsorption in the diabetic kidney and thereby lowers diabetic glomerular hyperfiltration (see Figures 13 and 14). Micropuncture studies were performed in hyperfiltering STZ-diabetic rats with superficial glomeruli, which allowed to collect tubular fluid close to the MD (403) (see Figure 13). The study found that concentrations of Na+, Cl−, and K+ at the MD were lower by approximately 25% compared to nondiabetic controls, consistent with a primary increase in reabsorption upstream of the MD. When phlorizin, a potent inhibitor of SGLT2 with lesser affinity for SGLT1 (74), was added into Bowman’s space of the same nephrons, the electrolyte concentrations at the MD increased to normal and SNGFR declined to normal in diabetic rats. Phlorizin had similar effects in nondiabetic animals but the effects were less (see Figure 13). Similar findings were made in micropuncture studies in rats in response to acute or chronic systemic application of a selective SGLT2 inhibitor (373). Moreover, pharmacologic or genetic inhibition of SGLT2 reduced hyperfiltration in diabetic mice (354, 396, 405). The GFR lowering effect of SGLT inhibition was associated with an increase in the hydrostatic pressure in Bowman space (403) and was independent of effects on blood glucose (373, 403, 405). Studies in mice confirmed a role of A1R-mediated afferent arteriolar vasoconstriction in the GFR-lowering effect of empagliflozin (178). These results are consistent with diabetic hyperfiltration resulting at least in part from a primary increase in proximal tubular reabsorption due to enhanced sodium-glucose co-transport (see Figure 13).
Figure 14. Proposed mechanisms of kidney protection by SGLT2 inhibition.

SGLT2 inhibition counteracts the diabetes-induced hyperreabsorption of glucose and Na+ in the early proximal tubule and lowers blood glucose levels. This also increases the NaCl and K concentration ([Na-Cl-K]MD) and fluid delivery (V) to the macula densa, which lowers glomerular filtration rate (GFR) through the physiology of tubuloglomerular feedback (1) and by increasing hydrostatic pressure in Bowman’s space (PBow) (2). The GFR-lowering effect of tubuloglomerular feedback includes afferent arteriole constriction (via adenosine A1 receptor) and potentially efferent arteriole dilation (via adenosine A2 receptor), which both reduce glomerular capillary pressure (PGC). Lowering of GFR reduces tubular transport work (3), thereby lowering cortical oxygen demand (QO2) (4) and increasing cortical oxygen tension (PO2) (5). Lowering GFR (6) and hyperglycemia (7) attenuates filtration of tubulo-toxic compounds, including albumin, and reduces tubular growth and kidney inflammation. Tubular transport work is further reduced by lowering blood glucose and by cellular SGLT2 blockade itself, which reduces tubular glucotoxicity and has also been linked to inhibition of the Na-H-exchanger NHE3 (8). SGLT2 inhibition shifts glucose reabsorption downstream where SGLT1 compensates and reduces the risk of hypoglycemia (9). Shifting glucose and Na+ reabsorption downstream to S3 and mTAL segments increases QO2 (10) and lowers PO2 in the outer medulla (OM) (5). Furthermore, lower medullary PO2 may activate hypoxia-inducible factor (HIF) and enhance erythropoietin (EPO) release (11). The latter increases hematocrit (Hct) (12) and improves O2 delivery to kidney medulla and cortex (13) and the heart (14). Enhanced delivery of NaCl and fluid downstream of early proximal tubule may enhance responsiveness to atrial natriuretic peptide (ANP) and diuretics (15). The diuretic, natriuretic and kaliuretic effects of SGLT2 inhibition lower the risk of hyperkalemia and further increase Hct (16) and reduce extracellular (ECV) and interstitial (ISV) volume and blood pressure (17). These effects, which are also evident by compensatory upregulation of renin and vasopressin levels (18), can help protect the failing kidney and heart (19). The increased cortical oxygen availability together with lesser hyperglycemia, tubular glucotoxicity, filtered albumin, and tubulointerstitial inflammation improves the integrity of the tubular and endothelial system, thereby allowing to maintain a higher tubular transport capacity and GFR in the long term (20). The glucosuric effect lowers therapeutic and/or endogenous insulin levels and increases glucagon concentrations (21). This induces compensatory lipolysis and hepatic gluconeogenesis and ketogenesis. SGLT2 inhibitors are uricosuric, potentially involving urate transporter 1 (URAT1) inhibition and their glucosuric and insulin-lowering effect (22). These metabolic adaptations reduce urate levels, the hypoglycemia risk, and body and organ fat mass, which together with the resulting mild ketosis have the potential to further protect the kidney and heart (19, 23). The proposed effects are to a significant extend independent of hyperglycemia. Other abbreviations: NO, nitric oxide; UNaClV, urinary salt excretion; UV, urinary flow rate. Modified, with permission, from Vallon V, 2020 (388).
Studies in humans confirmed the GFR-lowering effect of short-term SGLT2 inhibition. In people with T1DM and baseline hyperfiltration, treatment with the SGLT2 inhibitor empagliflozin for 8 weeks decreased GFR and the response was independent of lowering blood glucose levels (53). Based on the estimation of glomerular hemodynamic responses, the latter study hypothesized a dominant effect on the afferent arteriole, whereas a study in people with T2DM hypothesized that the SGLT2 inhibitor dapagliflozin reduced GFR by reducing efferent arteriolar resistance (414). In accordance, in rats with STZ-induced T1DM, an agonist of adenosine A2a receptors dilated the efferent arteriole and lowered GFR (293). Constricting the afferent arteriole as well as dilating the efferent arteriole both lower glomerular capillary pressure (PGC), which has been proposed to underly at least in part the nephroprotective effects of SGLT2 inhibitors.
A micropuncture study in STZ-diabetic rats (374) indeed confirmed that acute SGLT2 inhibition reduced directly measured PGC by 5 to 8 mmHg and reduced GFR by approximately 25%. Moreover, the study showed that this effect required a functioning TGF system. The authors also inferred a contribution of postglomerular vasorelaxation to the TGF responses where decreases in PGC were large and decreases in GFR were small in response to the SGLT2 inhibitor (374). Previous studies have indicated that SNGFR is more sensitive over a lower range of macula densa inputs than was PGC (337, 371). In other words, the PGC curve of the TGF response is shifted to the right of the GFR curve (see Figure 15). This implied that typical TGF responses from the shoulder of the GFR curve through the TGF operating point involve parallel increases in resistances upstream and downstream of the glomerulus whereas responses towards the elbow of the GFR curve include decreased resistance in the efferent arteriole (374). TGF-induced adenosine formation can constrict the afferent arteriole and dilate the efferent arteriole, and thus could explain both effects of SGLT2 inhibitors (see Figures 12 and 15). Thus, the TGF response elicited by acute SGLT2 inhibition includes a variable mixture of preglomerular vasoconstriction and postglomerular vasorelaxation. The transition point to efferent dilation in such a system could be flexible and sensitive to manipulation by changing the differential expression of adenosine A1 and A2 receptors or by various modulators of the segmental responses (374). An enhanced efferent arteriolar tone may be more common in advanced CKD as a consequence of a higher sympathetic tone or impaired endothelial vasodilation, which both may sensitize the efferent arteriole to vasodilation by adenosine A2 receptor activation following SGLT2 inhibition. A prominent efferent dilation in response to SGLT2 inhibition facilitates stronger reductions in PGC versus GFR, which may particularly happen in advanced stages of CKD when the hyperfiltration in remaining nephrons pushes the TGF operating point to the elbow of the GFR curve (see Figure 15). This may explain a similar drop in albuminuria and kidney protection despite a lesser initial drop in GFR in response to SGLT2 inhibition observed in people with more advanced CKD, like CKD4 (16).
Figure 15. Glomerular hemodynamic effects of SGLT2 inhibitor superimposed on a model of tubuloglomerular feedback (TGF), which incorporates effects on pre- and postglomerular resistances.
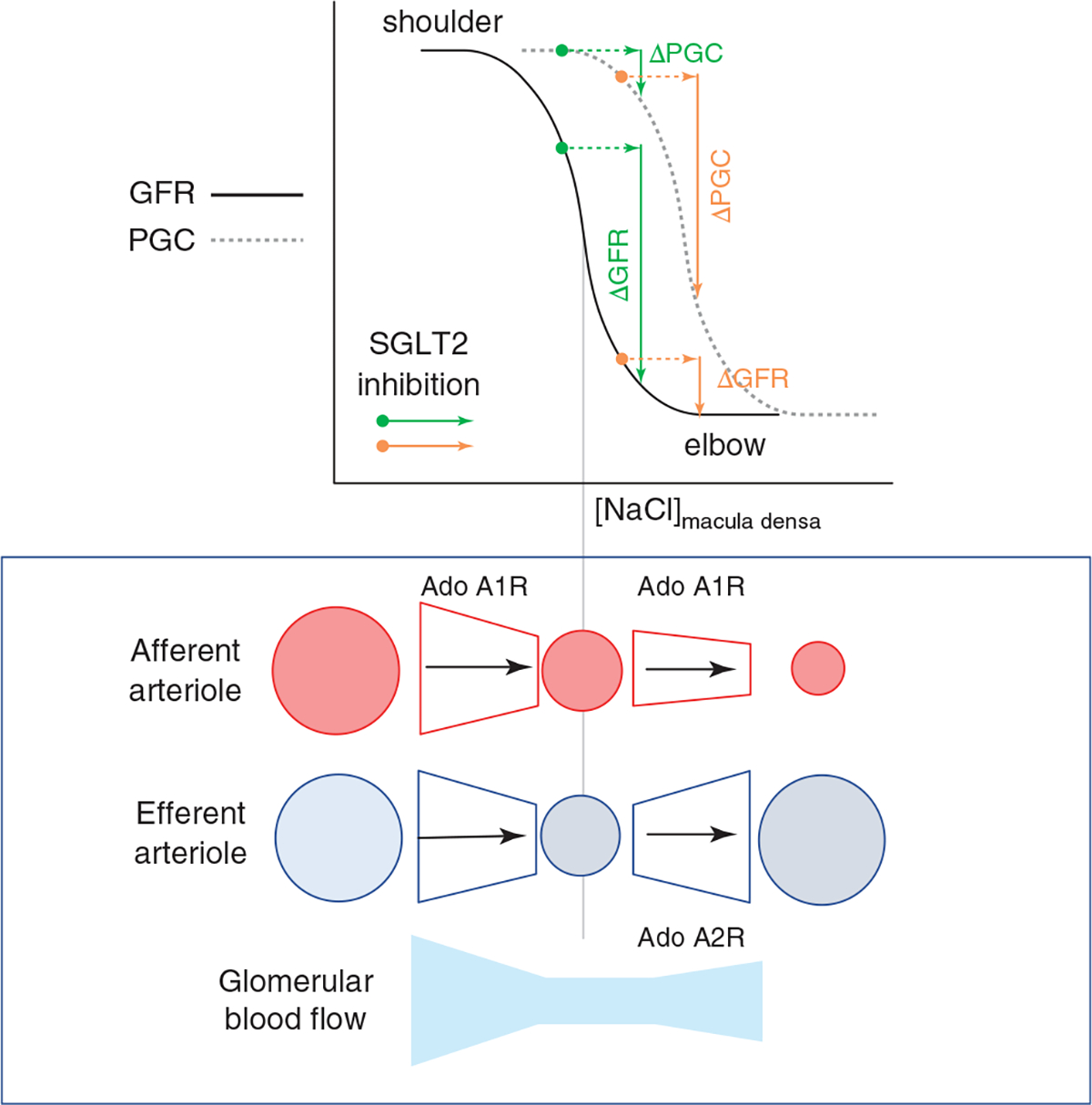
Idealized TGF curves are shown for glomerular filtration rate (GFR) and PGC. The PGC curve lies to the right of the GFR curve owing to differences in the way that pre- and postglomerular resistances react across the range of inputs, as shown in the bottom portion of the figure. Within a group of nephrons, the position of operating points along the respective TGF curves will form a distribution. The figure shows the effects on GFR and PGC when identical increases in macula densa delivery are imposed by SGLT2 inhibitor for two nephrons drawn from this distribution (nephrons A and B). For nephron A, the TGF response to SGLT2 blocker included a large decrease in GFR and small decrease in PGC. For nephron B, there was a smaller decline in GFR and a larger decline in PGC. The inverse relationship between decreases in whole kidney GFR and decreases in average PGC for that animal is expected if animals with the smaller decreases in GFR yet larger decreases in PGC had more nephrons operating near the elbow of their respective TGF curves and vice versa. In other words, the effect of SGLT2 inhibition on GFR can over or underestimate effects on PGC depending on the location of the operating point. Modified, with permission, from Thomson SC, and Vallon V, 2021 (374).
SGLT2 inhibition preserves GFR in long-term
Meta-analyses of clinical studies reported that SGLT2 inhibition can initially cause a small increase in serum creatinine (consistent with initial GFR lowering effect) but actually reduces the incidence of AKI (110, 270), consistent with kidney protective effects. In accordance, the SGLT2 inhibitor dapagliflozin decreased urinary levels of markers of glomerular and tubular injury in people with T2DM (65, 331). Moreover, in the long-term, and as shown in large clinical outcome trials, SGLT2 inhibition preserves eGFR and renal function in people with T2DM when compared with placebo treatment (263, 422, 446), including in individuals with kidney disease (292) (see Figure 14). See Ref. 375 for further review of the CANVAS, EMPA-REG, DELCARE as well as CREDENCE trial, respectively. The long-term studies typically show that SGLT2 inhibition induces a biphasic GFR profile, that is, an initial GFR reduction (see above) that is followed by long-term improvement in GFR preservation (136, 188, 291, 292, 422). Most importantly, after treatment discontinuation, eGFR increased to baseline in the SGLT2 inhibitor groups while eGFR remained unchanged at reduced levels in placebo groups (291, 422). Thus, SGLT2 inhibition induces an initial GFR reduction that is reversible, indicating a functional rather than structural cause, consistent with the tubular hypothesis of glomerular hyperfiltration. Due to lesser total glucose filtration, the blood glucose lowering effect of SGLT2 inhibition was attenuated in people with T2DM and CKD2 or CKD3—nevertheless, the short-term GFR lowering effect (21, 292, 441), the long-term GFR preservation (292), and the full reversibility of the GFR lowering effect after discontinuation of the SGLT2 inhibitor (21) remained, consistent with the outlined concept that the GFR responses are at least in part blood glucose independent.
How can an SGLT2 inhibitor by initially lowering GFR protect the kidney in the long term?
SGLT2 inhibition reduces PGC and GFR (374). This lowers the physical stress on the glomerular capillaries as well as the glomerular filtration of tubulo-toxic factors (e.g., glucose, albumin, advance glycation end products, growth hormones). The handling of these factors by the tubular system requires energy and promotes hypoxia, impairs autophagy, and triggers renal oxidative stress, inflammation, and fibrosis, which promotes the development and progression of diabetic kidney disease (410) (see Figures 9 and 14). A role for SGLT2 expression in podocytes has been proposed in protein overload conditions and remains to be confirmed (48).
Preserving renal cortical oxygenation appears decisive to preserve kidney function in people with CKD (300). GFR is the primary determinant of renal NaCl reabsorption and, thereby, of renal transport work and oxygen consumption. The proximal tubule reabsorbs most of the glomerular filtrate and is thus predicted to respond with the largest increase in oxygen consumption when GFR rises (203, 204). The tubular hypothesis of diabetic kidney disease proposes that lowering GFR and thereby the oxygen-consuming transport work helps to preserve the integrity of the remaining nephrons and overall kidney function in the long term (410) (see Figure 14). Mathematical modeling predicted that SGLT2 inhibition in the diabetic kidney reduces oxygen consumption in the proximal convoluted tubule and renal cortex, in part by lowering GFR (203, 204). The predicted increase in cortical O2 pressure has been confirmed in a diabetic rat model in response to phlorizin (264). Moreover, SGLT2 inhibition attenuated the cortical tubular expression of hypoxia-induced factor HIF-1α in db/db mice (25).
SGLT2 inhibition reduces renal glucotoxicity and alters renal metabolism
The molecular signature of tubular growth in the diabetic kidney involves early cell proliferation followed by cell cycle arrest and senescence and impaired autophagy. This process is characterized by upregulation of the mechanistic target of rapamycin complex 1 (mTORC1) and cyclin-dependent kinase inhibitors p21, p16, and p27, and, according to the tubular hypothesis of nephron filtration and diabetic kidney disease, links tubular growth to inflammation and fibrosis (for review see Refs. 391, 410). Consistent with a role of glucose uptake in this process, SGLT2 inhibition attenuated markers of renal fibrosis in a human kidney proximal tubule cell line (HK2) (285) and in db/db mice (98) and reduced proximal tubule mTORC1-driven fibrosis in T1DM Akita mice (187). Studies in T1DM Akita mice showed that SGLT2 inhibition reduced renal p21 and p27 upregulation (396), and knockdown of p21 or SGLT2 inhibited high glucose-induced senescence marker upregulation in human proximal tubular cells (181). Furthermore, empagliflozin reduced the renal accumulation of p62 in T1DM Akita mice, providing the first evidence that SGLT2 inhibition may improve autophagy in the diabetic kidney (396). In damaged proximal tubules of high-fat diet-fed ApoE-knockout mice, a model of nonproteinuric diabetic kidney disease, ATP production shifted from lipolysis to ketolysis due to hyperactivation of mTORC1; empagliflozin increased endogenous ketone body levels, and thereby prevented the decrease in renal ATP levels and organ damage (381). Empagliflozin also improved mitochondrial fragmentation and enhanced renal proximal tubule autophagic activity under high glucose conditions and in STZ-diabetic mice, potentially involving the AMPK and mTOR pathway, thereby leading to lesser apoptosis and tubulointerstitial fibrosis (207). The SGLT2 inhibitor ipragliflozin reversed the tubular and mitochondrial damage caused by high-fat diet in mice, independent of blood glucose levels (368).
Tubular hypoxia has been implicated in promoting fibrogenesis in vivo via HIF-1α stimulation (141, 364). Not only the tubular injury in AKI (see Discussion above), but also high glucose (217) can induce tubular HIF1α and is associated with aberrant glycolysis and fibrosis (Figure 9). In mice with STZ-induced diabetes, renal expression of the histone deacetylase sirtuin3 (SIRT3) was suppressed (359); this was associated with HIF1α accumulation and phosphorylation of signal transducers and activators of transcription-3 (STAT3), which was linked to abnormal proximal tubular glycolysis and fibrogenic programs and aspects of epithelial-mesenchymal transition (209); moreover, the SGLT2 inhibitors empagliflozin and canagliflozin attenuated these effects (209). In accordance with a prominent role of SIRT3 in the diabetic kidney, selective activation of SIRT3 enhanced tubular Sirt3 expression and attenuated albuminuria and glomerular damage in BTBR ob/ob mice (219). Notably, also the antifibrotic action of the angiotensin-converting enzyme inhibitor imidapril in mice with STZ-diabetes includes restoring SIRT3 protein and mitochondrial fatty acid oxidation and suppression of abnormal glucose metabolism (358).
Preliminary studies using urine metabolomics in T1DM Akita mice and people with T2DM also proposed a metabolic shift from mitochondrial oxidation to more glycolysis, an effect reversed by SGLT2 inhibition (64). Studies in kidney samples in people with diabetes, in mice with STZ-induced T1DM, and primarily cultured proximal tubule epithelial cells provided evidence that SGLT2 inhibition with dapagliflozin suppresses HIF1α-mediated metabolic switch from lipid oxidation to glycolysis (44). In accordance, in people with T2DM and albuminuria, dapagliflozin increased urinary metabolites linked to mitochondrial metabolism (252), an effect confirmed in T1DM Akita mice in response to empagliflozin (278). Onishi et al. further discovered that empagliflozin increases urinary azelaic acid in nondiabetic and diabetic mice (278). Azelaic acid is produced naturally by the peroxisomal ω-oxidation pathway from linoleic acid, a polyunsaturated essential fatty acid (216), and has been linked to mitochondrial biogenesis and autophagy (370), and oral administration of azelaic acid in mice reduces adiposity by rewiring fuel preference to fats (440). In this regard, empagliflozin reduced urinary stearate and palmitate in nondiabetic and diabetic mice, which may reflect this rewiring of the renal fuel preference to fats (278), which has been implicated in kidney health (168). Onishi et al. also found that empagliflozin upregulated renal gluconeogenesis in nondiabetic and diabetic mice and proposed a compensatory response for urinary glucose loss as well as for the partial NHE3 inhibition, as renal gluconeogenesis is linked to bicarbonate formation (Figure 5) (278). Remarkably, renal gluconeogenesis has been proposed as a marker of improved kidney health (208). In this regard, we speculate that SGLT2 inhibition promotes proximal tubule mitochondrial Citric Acid (TCA) cycle function, including fatty acid oxidation, by increasing gluconeogenesis (Figure 9): entry (anaplerosis) and removal (cataplerosis) of intermediates into and out of the TCA cycle are critical for its function, and gluconeogenesis supports cataplerosis (282). Thus, SGLT2 inhibitors may protect the kidney by reducing GFR and transport work, enhancing ketone bodies and renal gluconeogenesis, and lowering blood glucose levels and glucose toxicity in the early proximal tubule, thereby improving tubular energetics, attenuating senescence, and preserving mitochondrial function, including fatty acid oxidation, and autophagy (see Figures 9 and 14).
Potential coupling of SGLT2 to other transporters in the early proximal tubule
Hyperinsulinemia co-stimulates SGLT2, NHE3, and URAT1 in the proximal tubule (388) (see Figure 9). A functional coupling of these transporters may facilitate glomerulotubular balance in response to a postprandial increase in GFR and insulin but also lead to renal glucose, NaCl, and urate retention in obesity and T2DM (388). Beneficial effects of SGLT2 inhibition include not only a glucosuric but also an uricosuric and plasma uric acid-lowering effect. The enhanced urate excretion in response to SGLT2 inhibition has been related to increased tubular delivery or urinary glucose loss (56, 225, 275), and studies in gene-targeted mouse models proposed a role for the luminal urate transporter URAT1 (275) (see Figure 14). SGLT2 inhibition may inhibit URAT1 through reduced insulin activity (382) or through other molecular interaction between SGLT2 and URAT1 in the early proximal tubule (see Figure 9). In addition, SGLT2 inhibition may upregulate ABCG2, which is involved in urate secretion in the proximal tubule (221).
SGLT2 is functionally coupled to the Na-H-exchanger NHE3, which is co-expressed with SGLT2 in the early proximal tubule, such that pharmacological blockade of SGLT2 partially inhibits NHE3 activity, potentially involving the scaffolding protein MAP17 (59, 94, 278, 294) (Figures 5 and 9). Onishi et al. provided the first evidence that the acute natriuretic and chronic volume and blood pressure effect of the SGLT2 inhibitor empagliflozin depends on intact tubular NHE3, particularly in nondiabetic mice (278). Moreover, the group discovered that SGLT2 inhibition in mice with T1DM and rats with T2DM enhanced the phosphorylation of NHE3 (235, 278), which is a marker for NHE3 redistribution to the coated pit region where NHE3 is inactive (62, 183, 184) or to the microvillar base (416) where its activity is depressed by local pH change caused by NHE3 clustering (34). Studies by Borges-Junior and colleagues subsequently confirmed an inhibitory effect of empagliflozin on NHE3 by showing that the upregulated renal NHE3 activity in a nondiabetic heart failure rat model was attenuated by the SGLT2 inhibitor, associated with enhanced NHE3 phosphorylation, preserved GFR, and restored euvolemia (32). Furthermore, Onishi and colleagues showed that tubular knockdown of NHE3 reduces SGLT2 expression (277), indicating bi-directional interactions. Inhibition of NHE3 in response to SGLT2 inhibition is expected to further reduce early proximal tubular transport activity and oxygen consumption and enhances the potential natriuretic and volume effect of these drugs and makes their effect more independent of glucose status. Whether SGLT2 or SGLT1 is also functionally coupled to apical fructose transporters is not known.
SGLT2 inhibition causes more equal distribution of transport work and may mimic systemic hypoxia at the renal oxygen sensor
The early proximal tubule has a high transport capacity and is predicted to show the largest absolute increase in oxygen consumption when GFR increases (203, 204). By inhibiting early proximal reabsorption of glucose, NaCl, bicarbonate, fluid, and potentially urate, SGLT2 inhibition shifts some of the reabsorption downstream and thereby more equally distributes the transport burden along the tubular and collecting duct system, which may help to preserve tubular integrity and function in the long-term.
Micropuncture studies measuring early distal chloride delivery in response to acute versus chronic application of a SGLT2 inhibitor in rats indicated upregulated chloride transport between the late proximal convoluted tubule and early distal tubule in the chronic setting (373). Moreover, glucose reabsorption via SGLT1 in the S3 segment uses double the energy per glucose compared with SGLT2. The increased transport in the outer medullary S3 segment and thick ascending limb in response to SGLT2 inhibition could further reduce the low O2 availability physiologically found in this region (203, 204, 264) (see Figure 14). The downregulation of renal SGLT1 protein expression observed in response to SGLT2 gene knockout or inhibition has been proposed to serve to protect the S3 segment from excessive transport burden (396, 402, 405). The observed increase in urinary adenosine excretion in people with T1DM (306) and T2DM (414) in response to SGLT2 inhibition likely reflects the increase in transport work in the downstream segments: the enhanced ATP consumption enhances the formation and release of adenosine, which inhibits transport activity in these segments according to its role in metabolic control of organ function (400). Furthermore, the concomitant reduction in blood glucose and GFR in response to SGLT2 inhibition attenuates the net increase in downstream transport activity (203, 204). Moreover, the reduction in oxygen pressure in the deep cortex and outer medulla may stimulate hypoxia-inducible factors HIF1 and HIF2. Gene knockout of SGLT2 increased the renal mRNA expression of hemoxygenase 1 (405), a tissue-protective gene induced by HIF1alpha. Furthermore, activation of HIF2 may enhance erythropoietin release from renal interstitial cells in response to SGLT2 inhibition. In accordance, empagliflozin was found to enhance renal mRNA expression of erythropoietin in nondiabetic mice (278). Together with the diuretic effect, the latter may contribute to the observed modest increase in hematocrit and hemoglobin observed in response to SGLT2 inhibition (152). This can improve the oxygenation of renal outer medulla and cortex and facilitate oxygen delivery to the heart and other organs (see Figure 14). According to mediation analyses, the strongest predictor of protective effects of SGLT2 inhibition on hard renal and cardiovascular outcomes was the increase in hematocrit and hemoglobin from baseline (152, 210, 211). Thus it has been proposed that, in addition to its volume effect, the transport shift induced by SGLT2 inhibition simulates systemic hypoxia at the oxygen sensor in the deep cortex and outer medulla of the kidney, and the induced response then helps the kidney and failing heart (202). These effects are likewise expected in the nondiabetic setting. Modeling studies indicate that this transport shift of SGLT2 inhibition can also be preserved in CKD (202). The latter study further indicated that some natriuretic and diuretic and kaliuretic effect of SGLT2 inhibition is preserved in CKD due to a high glucose load on the single nephron level in remnant nephrons (facilitated by lesser blood glucose lowering effect), which induced paracellular sodium secretion in the proximal tubule (202). This may contribute to the preserved effects of SGLT2 inhibitors on blood pressure and heart failure in individuals with CKD.
SGLT2 inhibition protects heart function
SGLT2 inhibition has been shown to reduce the incidence of hospitalization for heart failure and the composite of cardiovascular death and heart failure in people with normal heart function or preexisting heart failure, and these benefits were observed equally in those with and without diabetes, and across the entire range of baseline HbA1C (e.g., (240) and for review see Ref. 413). Mediation analyses indicated that similar factors were associated with protection of the heart and the kidney from failing: an increase in erythrocytes/hemoglobin, a reduction in albuminuria, and a lowering in plasma urate levels (210, 211). These effects are discussed above, and below only a few additional aspects are briefly mentioned.
SGLT2 inhibition causes an osmotic diuretic effect (100–470 mL/24 h) and a natriuretic effect and reduces body weight, thereby decreasing systolic blood pressure by 3 to 6 mmHg (409). The magnitude of this effect is expected to have cardiovascular protective consequences, particularly in high-risk individuals (89) (see Figure 14). In clinical studies, reduced proximal tubular sodium reabsorption in response to SGLT2 inhibition is indicated by enhanced fractional lithium excretion (75). SGLT2 inhibition enhances renin levels (75, 402) and vasopressin (or copeptin) levels (75, 224, 234, 235) and lowers renal free-water clearance (75, 234) in rodents and humans, associated with increased renal protein expression of vasopressin V2 receptors and phosphorylated aquaporin-2 in rats (234), indicating active compensation to counter the diuretic and natriuretic effects. Onishi et al. speculated based on studies in nondiabetic mice that the metabolic adaptations to empagliflozin include an enhanced proximal tubular formation of alpha-ketoglutarate, which may activate its luminal receptor in non-A non-B intercalated cells to stimulate NaCl reabsorption (278). Additional mechanisms to stabilize body fluid volume includes compensatory increases in fluid and food/carbohydrate intake (234, 235, 290, 396). The blood pressure-lowering effect of SGLT2 inhibition and modest reduction in plasma volume (195), and potentially interstitial volume (129) may quickly reduce cardiac pre- and afterload and thereby contribute to the rapid beneficial effects observed in larger outcome trials in people with heart failure (171, 263, 292, 454). The diuretic effect of SGLT2 inhibition facilitates renal K excretion indicated by similar plasma K levels despite suppressed aldosterone levels in Sglt2 ko mice (402) and consistent with a reduced incidence of hyperkalemia observed in clinical trials with empagliflozin, canagliflozin and when dapagliflozin was combined with an aldosterone antagonist (292, 344, 422). Most of the described effects of SGLT2 inhibition can occur independent of lowering blood glucose levels. For more detailed coverage of the heart protection by SGLT2 inhibition, including the reno-cardiac axis, effects on cardiac energetics and sympathetic nervous system, reduced inflammation and fibrosis, and improved vascular function and remodeling, and potential off-target effects of SGLT2 inhibitors, please see (413).
Macula densa SGLT1-NOS1 pathway: contributing to diabetic hyperfiltration and blood pressure regulation
The vasoconstrictor response of the TGF mechanism can be attenuated in diabetes (155, 394, 401). Modest hyperglycemia reduced the vasoconstrictor TGF activity in nondiabetic rats by effects of glucose in the tubular fluid beyond the late proximal tubule (28). Neuronal nitric oxide synthase in the macula densa (MD-NOS1) forms nitric oxide (NO) that affects afferent arteriolar tone and shifts the TGF curve rightward and makes it less steep, thereby increasing GFR and facilitating NaCl delivery downstream of the MD and renal sodium excretion (218, 407, 411, 431). MD-NOS1 is involved in the GFR increase in response to acute hyperglycemia and in STZ-induced diabetes in rats and mice (189, 372, 380, 449). The stimulus for MD-NOS1 activation in response to hyperglycemia and diabetes includes an enhanced glucose delivery that is sensed by SGLT1, which is expressed in the luminal membrane of the thick ascending limb and the MD in mice and humans (354, 449) (see Figure 16). In accordance, pharmacological inhibition of SGLT1 attenuated the high glucose-induced attenuation of TGF-induced afferent arteriolar constriction in the isolated perfused juxtaglomerular apparatus (449). Moreover, the absence of SGLT1 prevented the Akita diabetes-induced upregulation in MD-NOS1 expression and attenuated glomerular hyperfiltration in Akita mice and STZ-diabetic mice with minimal or no changes in blood glucose levels (354) (see Figures 12 and 16). Gene-knockout of SGLT1 not only lowered glomerular hyperfiltration, but also reduced kidney weight, glomerular size, and albuminuria in Akita mice (354). These findings indicated that SGLT1 can have implications for renal integrity beyond the reabsorption of glucose in the late proximal tubule. SGLT1 in the MD may orchestrate single nephron function and structure. For example, sensing of increased glucose delivery at the MD may indicate the need for more upstream transport capacity and trigger tubular growth during development but also in response to hyperglycemia (see Figure 16). The assessment of the therapeutic potential of SGLT1 inhibitions deserves further exploration (42, 273, 305, 309).
Figure 16. The integrated effects of SGLT1 in the diabetic kidney.
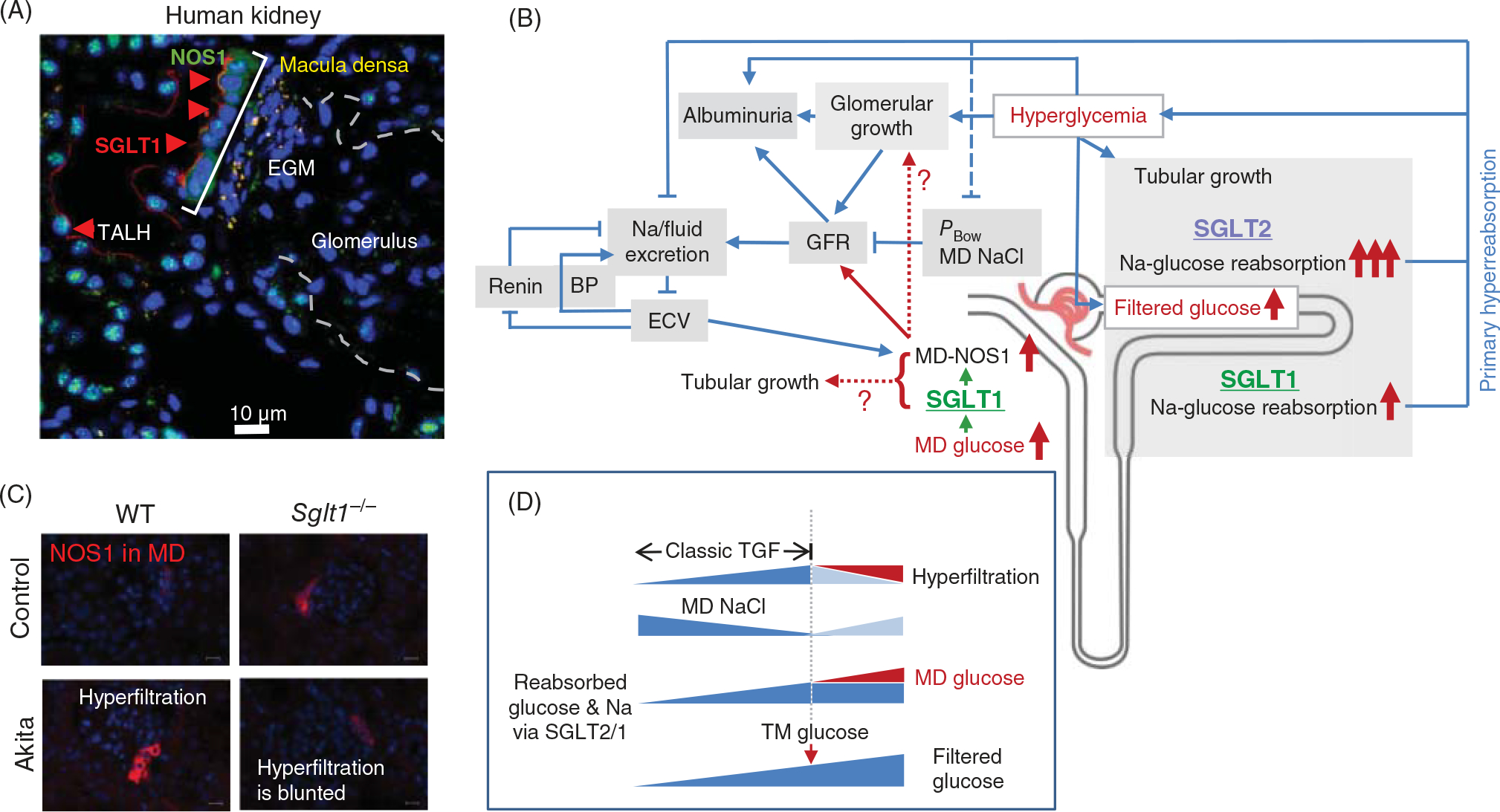
(A) SGLT1 is expressed in the luminal membrane of the macula densa (MD) in human kidney. (B) Blue arrows indicate positive interactions. Hyperglycemia enhances filtered glucose and induces tubular growth. This increases Na+-glucose cotransport, thereby maintaining hyperglycemia and reducing urinary Na+ and fluid excretion, with a larger contribution of SGLT2 versus SGLT1. Lesser urinary Na+ and fluid excretion increase effective circulating volume (ECV) and blood pressure (BP). Tubular hyperreabsorption lowers tubular backpressure in Bowman space (PBow) and the NaCl delivery and concentration at the MD, both increasing glomerular filtration rate (GFR) to restore urinary Na+ and fluid excretion. An increase in glucose delivery to the MD indicates saturated upstream Na+-glucose cotransport. This is sensed by SGLT1 in the MD and, by stimulating MD nitric oxide synthase 1 (NOS1), further increases GFR to compensate for maximized Na+-glucose cotransport. At the same time, SGLT1-mediated glucose sensing may trigger tubular growth to enhance tubular glucose transport capacity. SGLT1 inhibition has a relatively small effect on diabetic tubular hyperreabsorption and thus induces little natriuresis and diuresis. Through inhibition of MD-NOS1 upregulation and lowering of hyperfiltration, however, SGLT1 inhibition induces a relatively larger antinatriuretic and antidiuretic effect. As a consequence, SGLT1 inhibition can increase ECV with the resulting suppression in renin and increase in BP aim to restore renal Na+ and fluid excretion and ECV. (C) Diabetic Akita mice show enhanced MD NOS1 expression and higher GFR versus controls. Gene knockout of SGLT1 (Sglt1−/−) blunted both effects. (D) The classic TGF mechanism explains the increase in GFR when filtered glucose is increased up to the transport maximum for glucose (TM glucose). A further increase in filtered glucose can enhance MD NaCl and fluid delivery by the osmotic diuresis due to nonreabsorbed glucose, which would lower GFR. MD glucose sensing provides an additional stimulus to raise GFR and maintain hyperfiltration. EGM, extraglomerular mesangium, TALH, thick ascending limb. ?, indicate pathways that need further confirmation. Adapted, with permission, from Song P, et al., 2019 (354).
Why should an increase in macula densa glucose increase GFR?
GFR rises in the diabetic kidney, at least in part, to stabilize body fluid volume when tubular growth and enhanced sodium-glucose-cotransport cause a primary increase in upstream tubular reabsorption of sodium, glucose, and fluid (410) (see Figure 16). Glucose delivery to the MD indicates saturation of upstream SGLTs and thus hyperreabsorption of sodium, glucose, and fluid. The MD senses the increased luminal glucose via SGLT1, and the SGLT1-NOS1-GFR pathway enhances GFR to maintain urinary sodium and fluid excretion and volume balance (see Figure 16). Blunting this compensatory increase in GFR without significantly attenuating hyperreabsorption is expected to increase blood pressure, as a first-order mechanism for sodium homeostasis (126). In fact, blunted diabetes-induced glomerular hyperfiltration in SGLT1 knockout mice was associated with higher systolic blood pressure despite lower renal renin mRNA expression, indicating volume retention (see Figure 16). This response resembled the effect of a selective NOS1 inhibitor in diabetic rats (189)—in both cases, GFR was reduced and accompanied by an increase in blood pressure. Thus, it has been proposed that the MD-SGLT1-NOS1-GFR pathway complements the classic pathways of TGF and tubular back pressure in the compensatory adaptation of GFR to tubular hyperreabsorption in the diabetic kidney (354). While the two classic mechanisms primarily operate when tubular glucose reabsorption is below the maximum of glucose transport, the MD-SGLT1-NOS1-GFR pathway comes into play when the former is saturated (see Figure 16). It has been further proposed that deterioration of the MD-SGLT1-NOS1-GFR pathway could play a role in the transition from of an early hyperfiltering and normotensive diabetic patient to a later disease stage characterized by reduced GFR and hypertension (354, 449).
Interactions between MD-SGLT1-NOS1-GFR pathway and SGLT2 inhibition
Inhibition of SGLT1 and SGLT2 has additive effects on the early diabetic kidney as shown for T1DM Akita mice, including the improvement of blood glucose control and the lowering of GFR, renal glucose reabsorption, kidney weight, and glomerular size (354). SGLT2 inhibition lowers GFR by enhancing Na-Cl-K delivery to the MD (see Figures 12–14). This effect on GFR can be attenuated by the rise in glucose delivery to the MD and the engagement of the MD-SGLT1-NOS1-GFR pathway. SGLT2 inhibition enhances glucose delivery to the MD when it enhances glucosuria, which typically is observed in the clinical setting with euglycemia and moderate hyperglycemia. In accordance, SGLT2 inhibition increased MD-NOS1 expression in nondiabetic mice, and this effect was absent in SGLT1 knockout mice (354). SGLT2 inhibition in more severely hyperglycemic Akita mice did not increase glucosuria (due to the concomitant strong reduction in filtered glucose matching the inhibition of glucose reabsorption, as previously described (396, 405)), and actually reduced MD-NOS1 expression (354). The latter could reflect the inhibitory influence of volume loss on MD-NOS1, induced by the diuretic effect of SGLT2 inhibition (see Figures 12 and 16). Thus, the authors proposed that SGLT2 inhibitors may reduce hyperfiltration in part through inducing volume loss which impinges on the MD-NOS1 pathway (354). On the other hand, macula densa NO formation may also have protective effects on glomerular integrity and GFR (379), and it is possible that such protective effects may also be engaged by SGLT2 inhibition through the MD-SGLT1-NOS1 pathway, including in nondiabetic individuals. Further studies are required to define the nuances of MD glucose sensing, effects of MD-NOS1 derived NO on the afferent and efferent arterioles and the glomerular integrity, and their therapeutic potential.
Fructose and Renal Pathophysiology
Evidence that dietary fructose causes kidney disease in humans
The major added sugars are sucrose (table sugar), a disaccharide of fructose and glucose, while HFCS consists of a mixture of glucose and fructose with varying concentrations, but typically 55% fructose and 45% glucose. Fructose is also present in many foods, especially honey and fruits. Added sugars constitute approximately 15% of all calorie intake from the average western diet, with higher intakes in minority populations, young adults, adolescents, and toddlers (57, 318, 419).
A rise in fructose consumption over the last century is paralleled with an increased prevalence of metabolic syndrome, hypertension, obesity, fatty liver, and insulin resistance (259) (see Figure 17). Since these disorders are risk factors for renal injury, fructose could also drive CKD. Although several experimental studies showed the link of dietary fructose and kidney disease (104, 112, 260), as discussed in more detail below, there is, to our knowledge, only one study addressing this issue in clinical medicine. Brymora et al subjected twenty-eight people with Stages 2 and 3 CKD, who were switched from a regular diet containing 60 g fructose to a low fructose diet with 12 g fructose per day for 6 weeks, followed by a resumption of their regular diet for another 6 weeks (38). They found that although there were no effects on renal function, the low-fructose diet tended to improve blood pressure, significantly lowered serum uric acid, and reduced serum inflammatory markers, including high sensitivity C-reactive protein and soluble intercellular adhesion molecule (sICAM). Sugar-sweetened beverages (SSB) are a major source of added sugar and have been linked to obesity in children and adolescents (96). The dramatic increase in fructose consumption has stimulated a heated debate over the potential danger of SSB (160, 265). Given that table sugar is composed of one fructose and one glucose molecule, the findings of these studies might also tell us something about the effects of fructose on the kidney. In 2009, Bomback and colleagues performed a 5-year prospective cohort study in 447 persons with preexisting CKD (Multi-Ethnic Study of Atherosclerosis [MESA]) but found no association between sugar-sweetened soda and the incidence or prevalence of CKD on the basis of either eGFR or the urinary albumin to creatinine ratio (30). While the outcomes of the study should be appreciated, it examined the effect of up to just a single serving of SSB per day, which might not be sufficient to affect renal function (365). Similarly, Lin et al. subjected 3318 women and examined the effect of greater than one SSB per day, but again did not find any association between SSB and incidence of CKD (214). In these studies, only 3% of subjects consumed >one SSBs per day, which might be a reason for the negative results. In contrast, Saldana et al. examined the effect of higher doses of SSB (regular colas (6–7 g fructose/100 mL) and artificially sweetened colas (no fructose)) in a case-control study and found that greater than two SSBs per day was associated with an increased incidence of kidney disease (321). In this study, effects of other factors, including phosphoric acid, which was included in both regular colas and artificially sweetened colas, need to be considered. Likewise, Shoham et al. examined higher doses of SSB and found that the exposure of greater than or equal to two SSB per day was associated with a higher level of urinary albumin excretion compared to those who consumed less than two servings of SSBs per day (348). More recently, a 2019 study of the community-based cohort of black Americans also showed that a higher consumption of SSB was associated with significantly greater odds of incident CKD (309).
Figure 17. Mechanisms by which excessive fructose causes kidney diseases.

Under physiological conditions, fructose links to gluconeogenesis, and may protect from hypoxia and maintain systemic glucose concentrations, and preserves renal function. In turn, endogenous fructose formation is enhanced under several pathological conditions, including diabetes, ischemia, dehydration, senescence, and pressure overload. Either excessive dietary fructose or aberrant endogenous fructose generation stimulates the pathological pathway to cause tubulointerstitial damage and endothelial dysfunction in association with urate production and inflammation. Fructose metabolism links to the Warburg effect, which favors an unbalanced increase of glycolysis with suppressed mitochondrial respiration and stimulation of lactate production under aerobic conditions. These processes could be involved in the development of acute kidney injury (AKI), diabetic nephropathy, chronic kidney disease (CKD), senescence, or deleterious consequences of dehydration. Recurrent dehydration has been proposed to induce renal injury via a fructokinase-dependent mechanism, likely from the generation of endogenous fructose via the polyol pathway. Adapted, with permission, from Roncal Jimenez CA, et al., 2014 (316).
Experimental evidence for a causal role of fructose in kidney disease
Multiple experimental studies support deleterious effects of high fructose intake on kidney outcome. Based on kidney morphological analyses, feeding normal rats with high fructose diet for 8 weeks caused mild tubulointerstitial injury, and the damage was particularly confined to proximal tubular epithelial cells (260). Cells of the proximal straight tubules exhibited cellular damage with cell proliferation and aberrant expression of vimentin while the glomerulus, vessels, and other tubular cell compartments appeared unaffected (260). Along with tubular injury, the development of interstitial fibrosis was indicated based on an increase in interstitial collagen III deposition around the damaged tubular cells. These data suggest that excessive fructose consumption in normal rats can have detrimental effects on proximal tubular cells, in particular the straight segment, and the surrounding interstitium (see Figure 17). Similar to rats, mice have also been reported to develop tubulointerstitial injury in response to a high-fructose diet (8).
In addition to initiating tubular injury in the normal rodent kidney, there is evidence that fructose also accelerates the progression of renal diseases in CKD. When a high fructose diet was administered to a rat CKD model, in which 5/6th of total renal mass had been surgically removed, the preexisting renal injury was further deteriorated compared to an isocaloric glucose diet (104). In particular, the tubular damage was further accelerated to manifest ballooning tubules with higher degree of tubular epithelial cell proliferation and severe Interstitial immune-cell infiltration. The preexisting glomerular sclerosis was likewise further deteriorated with high fructose diet in the same model (104). These morphological changes were associated with deterioration of renal impairment, including a significant decline in renal creatinine clearance and an increase in urinary protein excretion. Since the proximal tubular epithelial cells express multiple fructose transporters in the luminal membrane (see above), excessive fructose derived from glomerular filtration may directly affect the proximal tubular cells.
In terms of inflammation, fructose can cause inflammation by acting on renal tubular epithelial cells and endothelial cells (see Figure 17). An in vitro study showed that cultured proximal tubular cells exposed to fructose released inflammatory cytokines, showed signs of enhanced oxidative stress, and produced uric acid (58). Tubular cells exposed to fructose expressed enhanced monocyte chemoattractant protein-1 (MCP-1) (58), while the exposure of endothelial cells to fructose induced intercellular adhesion molecule-1 (ICAM-1) (112). Feeding rats with high fructose diet for 20 weeks increased soluble ICAM-1 concentrations in serum and predominantly enhanced ICAM-1 expression in the endothelial cells of glomerular and peritubular capillaries (112). A proposed underlying mechanism is the ability of fructose to reduce nitric oxide (NO) availability in endothelial cells as NO donors mitigated the fructose-induced ICAM-1 expression. In this regard, fructose was found to uncouple the endothelial NO synthase (eNOS), thereby reducing NO availability and increasing oxidative stress (253, 258, 346). Recently, it was shown that blocking fructokinase activated eNOS and protected endothelial cells from senescence-associated cellular dysfunction (315). Therefore, fructose-induced endothelial dysfunction is likely a key mechanism for the progression of renal disease. Since uric acid directly impairs endothelial function (175, 254, 324), uric acid formation from fructose could be also involved in this process.
Excess fructose intake is associated with hypertension in humans (271). Experimental studies showed that rats fed with high fructose diet showed an increase in blood pressure in response to additional salt intake, suggesting that fructose increases the salt sensitivity of blood pressure (116). The majority of Na+ filtered by the glomeruli is reabsorbed by the proximal tubular epithelial cells via Na+/H+ exchangers (NHEs), primarily NHE3, located in the apical membrane (43, 116, 406). Fructose stimulates both the expression and activity of NHE/NHE3 and increases Na+ reabsorption in proximal tubules (43, 116). While NHEs are regulated by angiotensin II, the fructose-induced upregulation of NHE/NHE3 expression results in sensitizing the proximal tubules to angiotensin II (43, 115, 116). In addition, urate also plays a key role in the development of salt-sensitive hypertension in response to fructose as it causes several insults including arteriolopathy, tubulointerstitial injury, and reduced NO formation in endothelial cells (112, 175, 254, 325, 425). A sustained elevation in plasma renin despite of salt loading, increased free radicals, and increased renal nerve stimulation have also been implicated in fructose-induced salt-sensitivity of blood pressure (121, 353, 447). The major feature of the fructose transcriptome signature in rat renal cortex is a change in metabolic programs of the renal proximal tubule consistent with gluconeogenesis and de-novo lipogenesis (117).
Role of endogenous fructose in diabetic kidney disease
While endogenous fructose production may serve protective roles under physiological condition, it could turn to be deleterious in cases when fructose is unphysiologically produced (see Figure 17). An example is diabetic nephropathy, in which an excessive amount of glucose is available and converted to fructose. This reaction is a consequence of the activation of the polyol pathway and is considered a key mechanism for developing diabetic complication, including diabetic nephropathy (37). A pathological role of the polyol pathway was indicated in studies using mice with overexpression of human aldose reductase cDNA, which developed thrombosis in renal vessels with collagen deposits in Bowman’s capsule, resembling the histology of human diabetic nephropathy (442). Since this mouse model also developed some features of human diabetic retinopathy, including cataracts and occlusion of the retinochoroidal vessels, aldose reductase appears to play a key role for the development of diabetic complication (442). We found that the kidneys of streptozotocin-induced diabetic mice contained higher levels of fructose and uric acid associated with higher aldose reductase expression, compared with nondiabetic mice (197). Moreover, streptozotocin-induced diabetic mice lacking fructokinase were found to be protected from diabetic nephropathy and manifested less albuminuria, better renal function, and less tubular injury (197), suggesting that fructose metabolism, as a consequence of the activation of the polyol pathway contributes to diabetic kidney disease.
Is human diabetic nephropathy also mediated by endogenous fructose? To our knowledge, there is currently no study to examine intrarenal fructose content in people with diabetes. However, endogenous fructose is likely produced in the kidneys of people with diabetes. In fact, Kawasaki et al. demonstrated that diabetic subjects have higher levels of fructose in the serum and urine (172). Since these individuals were admitted and received healthy hospital diet, elevated serum fructose concentrations were unlikely due to excessive dietary fructose, but potentially due to endogenous production of fructose from glucose in these people with diabetes.
Nonetheless, the pathological role of the polyol pathway remains elusive in clinical settings. One practical way to address this issue would be to use aldose reductase inhibitors in people with diabetes. The Aldose Reductase Inhibitor-Diabetes Complications Trial Study Group (148) reported that inhibition of aldose reductase with epalrestat slowed the progression of diabetic nephropathy, and another study found that the compound reduced lipid hydroperoxides in erythrocytes of people with T2DM (276). Further trials are warranted to determine if aldose reductase inhibitors might protect the kidney in people with diabetes mellitus.
Endogenous fructose in other types of kidney diseases
Endogenous fructose has been proposed to also play a role in AKI in both mice and humans. In a clinical study subjecting twelve pediatric individuals to undergo cardiac bypass surgery, seven out of twelve individuals developed AKI after surgery, and they had significantly higher levels of fructose in their urines compared with those without AKI (6). Given that all 12 individuals were admitted and given the hospital diet, urinary fructose in people with AKI may not be of dietary origin but may reflect endogenously produced fructose, perhaps in the kidney, although an impairment of tubular fructose reabsorption was not excluded. The precise mechanism for an increase in urinary fructose was investigated using mice, in which AKI was induced by renal ischemia-reperfusion (IR) (5). Wild-type mice with IR-AKI developed severe tubular injury, which was associated with the activation of the polyol pathway, as evidenced by high levels of aldose reductase, sorbitol, and fructose in the injured kidney. The same study examined the role of endogenous fructose by using mice with deletion of fructokinase. The kidneys in fructokinase knockout mice were partially protected from IR injury and exhibited less tubular injury and less renal dysfunction, suggesting that fructose contributes to IR-mediated kidney injury in mice (see Figure 17). A deleterious role for endogenous fructose was also confirmed in radiocontrast-induced AKI (6).
There are several pathological conditions stimulating endogenous fructose production (see Figure 17). Recurrent heat stress and dehydration can cause CKD in mice (316), and this may be relevant to the epidemics of CKD that are occurring in Central America, Sri Lanka, and India (111). Chronic recurrent dehydration resulted in aldose reductase activation in both the kidney and hypothalamus, leading to fructoneogenesis at both sites in mice (316, 355). Again, the kidney in mice lacking fructokinase was largely protected from repeated heat stress and dehydration (316). Finally, senescence (aging) changes in the mouse kidney were found to be mediated by fructokinase activation: aging wild-type mice exhibited renal dysfunction characterized by an increase in urinary albumin excretion, glomerular collagen IV deposition, and tubulointerstitial injury. However, these changes were ameliorated in mice with a systemic deletion of the fructokinase gene (315).
Nonkidney disease: fructose can cause cardiac hypertrophy
In addition to the kidney, there are several organs, which are capable of producing fructose, including the liver, brain, and muscle (287, 355). Likewise, the placenta and fetus are also likely to convert glucose to fructose during pregnancy as fructose is required for fetal organ development (179, 340, 430). Recently, Mirtschink et al. showed that cardiac myocytes were also able to endogenously produce fructose and the fructose generated was involved in pathological processes. Specifically, HIF-1α induced the shift in the expression of fructokinase mRNA splice variants A to C (the latter having a higher affinity for fructose) under low oxygen conditions and this process was critical for the development of heart hypertrophy in response to hypertension induced either by the 1-kidney-1-clip model or by transverse aortic constriction (245). Moreover, the heart in fructokinase knockout mice was also protected from pressure-overload cardiac remodeling, and in humans, biopsy samples of individuals with hypertrophic cardiomyopathy showed upregulated fructokinase mRNA in cardiomyocytes (245).
Fructose accelerates renal injury under hypoxia
There are several causes for the development of kidney diseases. However, as kidney disease progresses, there is a common pathway toward end stage renal disease, and hypoxia contributes to this process (35, 261). For example, glomerular sclerosis causes tubular hypoxia as the loss of glomerular capillaries decreases the blood flow to the following peritubular capillaries and reduces oxygen supply to the tubules. Tubular hypoxia is also induced by alternative factors, including anemia, an increase in metabolic demand of tubules, and the constriction of efferent arterioles due to vasoconstrictive factors (35).
Importantly, hypoxic conditions activate aldose reductase, leading to endogenous fructose production. The fructose produced is preferentially metabolized under hypoxic condition because fructose does not require high amounts of oxygen, suggesting that hypoxic conditions could unphysiologically amplify fructose production and metabolism. Since fructose can cause tubulointerstitial injury (see above), however, the endogenous fructose produced and metabolized under hypoxic conditions could be involved in a common pathway toward progressive and end stage renal disease.
The observation that fructose is often beneficial, but occasionally turns to be deleterious for the kidney and other organs is a conundrum. A clue for understanding the complexity might be hidden in the availability of fructose under either physiological or pathological conditions (see Figure 17). An excessive amount of fructose usually induces deleterious effects and can cause metabolic syndrome (254, 259) while a physiological dose of fructose can act as a survival factor under starvation or dehydration in wild animals and humans (161). For example, during pregnancy, physiologically produced fructose in the placenta seems a beneficial factor as it is utilized for the development of the fetus; however, an excessive amount of fructose, due to maternal consumption of a diet high in fructose or aberrantly produced endogenous fructose, can cause organ damages in fetus and maternal preeclampsia (179, 340, 430). Fructose metabolism and its metabolites are physiologically beneficial on adequate timing under the right condition. However, if these factors are provided when tissues do not require them for maintaining function or developing tissues, then unphysiological fructose and its metabolites can link to pathological consequences. Therefore, in the kidney, a physiological dose of fructose plays a protective role under physiological levels of hypoxia (e.g., in the outer medulla), while excessive amounts of fructose, due to the aberrantly amplified production of fructose under pathological hypoxia, would go awry to cause kidney injury (255).
Fructose metabolism: links to urate generation and the Warburg effect
The gateway enzyme for fructose metabolism is fructokinase (known as ketohexokinase), which phosphorylates fructose to produce Fru1P. During this reaction, fructokinase activation requires a phosphate, so that intracellular phosphate levels are reduced and ATP is depleted (229). The rapid reduction of phosphate activates AMP deaminase, which in turn drives adenine nucleotide turnover and stimulates urate production (200, 254, 259). As such, an excess amount of fructose either from diet or from endogenous production could result in high amounts of intracellular uric acid (259). Uric acid likely plays a pivotal role in fructose metabolism (see Figure 4). A key function of uric acid is to stimulate mitochondrial oxidative stress (324). The potential mechanism is attributed to the ability of uric acid to inhibit aconitase activity, which is an enzyme of the TCA cycle converting citrate to iso-citrate, so that blocking aconitase disconnects fructose metabolism from mitochondrial respiration, leading to the aberrant production of mitochondrial oxidative stress, at least in liver cells (199).
The pathological roles of uric acid have been demonstrated either in cell culture systems or in animal models. Indeed, uric acid causes endothelial dysfunction, vascular injury, and inflammation, leading to glomerular hypertension, tubulointerstitial injury, and an elevation in systemic blood pressure in rodent models (112, 167, 175, 254, 259, 260). Uric acid exerts biological effects, including a reduction in endothelial nitric oxide formation, an increase in oxidative stress, and renin-angiotensin activation that may all predispose to hypertension (425). In addition, uric acid also modulates fructose metabolism and increases the risk for animals to develop insulin resistance and metabolic syndrome (254). Uric acid can also feedback to amplify both aldose reductase and fructokinase expression (323), further increasing fructose production and metabolism. While there is robust evidence that uric acid can contribute to the development of hypertension, insulin resistance, and potentially renal and cardiac diseases (157, 332), three recent trials, using urate-lowering xanthine oxidase inhibitors febuxostat or allopurinol did not improve kidney outcomes despite large and sustained reductions in serum urate levels (377).
Triose phosphates, which are metabolites from the reaction of Fru1P by aldolase B, can enter the glycolytic pathway distal to phosphofructokinase and drive aerobic glycolysis with the pathological activation of gluconeogenesis and lipogenesis (see Figure 4). These reactions result in the production of glucose, glycogen, lactate, and triglycerides, resembling the “Warburg effect” (257). In 1924, Otto Heinrich Warburg described that cancer cells, as opposed to normal cells, exhibit a unique property to ferment glucose into lactate even in the presence of sufficient oxygen (423, 424). Recently it has been proposed that the Warburg effect could contribute to the progression of renal diseases in which aerobic glycolysis contributes to renal fibrosis (448). To block the Warburg effect, two pharmaceutical ways are currently available. One is to use 2-deoxyglycose to block glycolysis, and the other is to apply dichloroacetate (DCA), which switches aerobic phosphorylation back to oxidative phosphorylation. DCA activates the pyruvate dehydrogenase and increases delivery of pyruvate into the mitochondria. As predicted, DCA depolarizes the mitochondria, returning the membrane potential towards the levels of noncancer cells (244). It was shown that these compounds inhibited renal fibrosis development in the obstructed kidney (426). Although the clinical relevance of the obstructed kidney model is limited, the Warburg effect could be a novel mechanism for renal fibrosis (67, 443) that could be driven by fructose and uric acid (255), but more studies are needed to further establish this hypothesis.
Interactions of Renal Glucose and Fructose Handling in Disease
While healthy proximal tubular cells undergo little glycolysis, but rather release glucose into the systemic circulation, the damaged tubular cells turn to glucose fermentation in the pathological setting. Lan et al. demonstrated that the proximal tubular cells undergoing atrophy display mitochondrial alterations, evidenced by a reduction in mitochondrial number with large autophagolysosomes, leading to a metabolic switch from mitochondrial oxidative phosphorylation to glycolysis associated with amplified expression of glycolytic enzymes (196). Thus, both fructose, as discussed above, and glucose are metabolized in injured proximal tubular cells. In other words, proximal tubular metabolism of both fructose and glucose need to be considered under pathophysiological conditions (see Figures 4, 7, and 9).
In the liver, the combination of fructose with glucose influences glucokinase, the first enzyme for glycolysis. Van Schaftingen et al. (415) and Agius and Peak (2) demonstrated in hepatocyte that glucokinase was positively regulated by Fru1P whereas it was inhibited by Fru6P. The mechanism by which Fru1P activates glucokinase involves promoting the release of glucokinase from glucokinase regulatory protein (GKRP), which sequesters glucokinase in the nucleus (36, 272). Even at small concentrations, intracellular fructose is rapidly metabolized to Fru1P. Therefore, Fru1P-induced glucokinase activation could explain why fructose facilitates glucose utilization. Moreover, Shiota et al. showed that the effect of small amounts of fructose enhanced hepatic glucose uptake in the dog (347). Furthermore, fructose metabolism also increases fructokinase activity, which depletes intracellular ATP. Since ATP negatively regulates the glycolytic pathway by inhibiting phosphofructokinase and pyruvate kinase, the ATP depletion due to fructokinase activation would enhance glycolysis. In fact, this phenomenon has been recently demonstrated in a model of colon cancer in mice (114). However, glucokinase (Hexokinase IV) is basically only expressed in hepatocyte and pancreatic β cells (237) while the proximal tubular cells possess hexokinase I and II (97, 196). Studies are needed to explore the effect of Fru1P on hexokinase activity in the gateway step of glucose metabolism in the proximal tubules.
Conclusion and Perspectives
The glucose transporters SGLT2, SGLT1, and GLUT2 play key roles for transcellular glucose reabsorption in the proximal tubule. The proximal tubule, in addition, has the ability to generate glucose, which is important for glucose homeostasis, especially during fasting, and during metabolic acidosis. In contrast to the proximal tubule, the more distal renal segments typically make use of glucose as an energy source, and basolateral GLUT1 has been proposed as an entry path. We are only beginning to learn about the coordination between renal glucose transport, glucose formation, and the intrarenal glucose usage. Is there a quantitative role for intercellular glucose transfer within the kidney or an intra-renal Cori cycle? Is cellular glucose uptake good or bad for an injured epithelial cell? Is this different for proximal versus more distal segments? Does it matter whether glucose enters the cell across the apical or basolateral membrane? Is there a primary role for basolateral glucose uptake through GLUT1 or GLUT2 in the deleterious effects of hyperglycemia on cellular metabolism and integrity of renal epithelia? Accumulating evidence also suggests a role for glucose uptake via apical SGLT2 in kidney pathophysiology, including in nondiabetic settings. Moreover, pharmacological inhibition of SGLT2 may indirectly inhibit other apical transporters in the early proximal tubule, including NHE3, with consequences on volume status. Much has been learned about the role of apical SGLT2 and SGLT1 in diabetic glomerular hyperfiltration. SGLT1 has been discovered as a glucose sensor in the macula densa that affects local NO formation in the juxtaglomerular apparatus and GFR, which may contribute to the larger role of these cells in the orchestration of kidney function, structure, and integrity. The tubular hypothesis of diabetic glomerular hyperfiltration and nephropathy illustrates the pathophysiological potential of SGLT2, which couples the retention of glucose to conserve an energy source, to the reabsorption of sodium, which affects volume status. These concepts contribute to the unexpected logic of SGLT2 inhibition in the diabetic kidney. New insights relate to a proposed efferent dilation, in addition to afferent constriction, in the lowering of glomerular capillary pressure by SGLT2 inhibition. New hypotheses include a proposed transport shift to the outer medulla, such that SGLT2 inhibition simulates systemic hypoxia at the oxygen sensors in this kidney region, with the resulting erythropoietin release and increase in erythrocyte numbers facilitating oxygen support for the kidney and other organs. Better understanding the protective effects of SGLT2 inhibitors provides an opportunity to better understand the needs of a failing kidney and heart.
Fructose is a natural sugar present in fruits and honey and metabolized in the kidney. Under physiological condition, fructose is freely filtered by the glomeruli and subsequently reabsorbed via several types of fructose transporters in the apical membrane of the proximal tubular epithelial cells. In the cytosol, fructose is metabolized by fructokinase and utilized as a substrate for gluconeogenesis. However, when provided in excess, fructose metabolism can stimulate pathological processes and promote tubular injury. A unique characteristic of fructose, as opposed to glucose, is to produce urate as a by-product in the metabolic process. While urate mediates several pathological reactions, including endothelial dysfunction, inflammation, tubular injury, and insulin resistance, it also suppresses aconitase in the TCA cycle and reduces mitochondrial oxidation. As a result, fructose favors glycolysis over mitochondrial respiration, which is similar to the process of the Warburg effect in cancer. Since aberrant glycolysis links PPP, hexosamine pathway, and lipid synthesis to provide a pool of biosynthetic precursors including nucleic acids, amino acids, fatty acids, and ATP, this pathway could provide fuel supply for several pathological reactions, such as inflammation and fibrosis. Another important issue is that fructose is endogenously produced under several pathological conditions, including hypoxia, diabetes, osmotic pressure, senescence, and pressure overload, all of which are risk factors for CKD. Given the fact that several types of kidney diseases share the Warburg effect as a potential mechanism, we hypothesize that endogenous fructose is a common contributor to CKD through the Warburg effect. Compared to glucose, the mechanism by which fructose causes CKD has yet to be rigorously studied as are the interactions between fructose and glucose metabolism in kidney tubules in disease state.
Didactic Synopsis.
Major teaching points
The kidneys filter 160 to 180 g/day of glucose (≈30% of daily energy expenditure), which is primarily reabsorbed by the sodium-glucose cotransporter SGLT2 (≈97%) in the early proximal tubule.
Diabetes raises the glomerular filtration of glucose; this increases proximal tubule reabsorption of glucose and sodium and thereby enhances kidney transport work and oxygen consumption, and sustains hyperglycemia.
SGLT2 inhibitors induce blood glucose-dependent and -independent pleiotropic effects that reduce the risk of renal and heart failure independent of kidney function and T2DM.
The kidneys filter 4 to 25 g/day of fructose, which is taken up by fructose transporters in the proximal tubules.
Fructose is metabolized in proximal tubules and under physiological conditions primarily utilized as a substrate for gluconeogenesis.
Excess amounts of fructose cause tubulointerstitial injury.
Urate (a by-product of fructose metabolism) and the Warburg effect may contribute to fructose-associated chronic kidney disease.
Acknowledgments
The author was supported by NIH grants R01DK112042, R01DK106102, R01HL142814, RF1AG061296, the UAB/ UCSD O’Brien Center of Acute Kidney Injury NIHP30DK079337, and the Department of Veterans Affairs. TN has an equity with XORTX therapeutics which is developing novel xanthine oxidase inhibitors.
Footnotes
Competing Interests
Over the past 36 months, VV has served as a consultant and received honoraria from Astra-Zeneca, Boehringer Ingelheim, and Retrophin, and received grant support for investigator-initiated research from Astra-Zeneca, Boehringer Ingelheim, Gilead, Janssen, Kyowa-Kirin, Merck, and Novo-Nordisk.
References
- 1.Ackermann TF, Boini KM, Volkl H, Bhandaru M, Bareiss PM, Just L, Vallon V, Amann K, Kuhl D, Feng Y, Hammes HP, Lang F. SGK1-sensitive renal tubular glucose reabsorption in diabetes. Am J Physiol Renal Physiol 296: F859–F866, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agius L, Peak M. Intracellular binding of glucokinase in hepatocytes and translocation by glucose, fructose and insulin. Biochem J 296 (Pt 3): 785–796, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali T, Khan I, Simpson W, Prescott G, Townend J, Smith W, Macleod A. Incidence and outcomes in acute kidney injury: A comprehensive population-based study. J Am Soc Nephrol 18: 1292–1298, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Alleyne GA, Scullard GH. Renal metabolic response to acid base changes. I. Enzymatic control of ammoniagenesis in the rat. J Clin Invest 48: 364–370, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andres-Hernando A, Jensen TJ, Kuwabara M, Orlicky DJ, Cicerchi C, Li N, Roncal-Jimenez CA,Garcia GE, Ishimoto T, Maclean PS, Bjornstad P, Sanchez-Lozada LG, Kanbay M, Nakagawa T, Johnson RJ, Lanaspa MA. Vasopressin mediates fructose-induced metabolic syndrome by activating the V1b receptor. JCI Insight 6: e140848, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andres-Hernando A, Li N, Cicerchi C, Inaba S, Chen W, Roncal-Jimenez C, Le MT, Wempe MF, Milagres T, Ishimoto T, Fini M, Nakagawa T, Johnson RJ, Lanaspa MA. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat Commun 8: 14181, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, Lanaspa MA. Deletion of fructokinase in the liver or in the intestine reveals differential effects on sugar-induced metabolic dysfunction. Cell Metab 32 (1): 117–127, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aoyama M, Isshiki K, Kume S, Chin-Kanasaki M, Araki H, Araki S, Koya D, Haneda M, Kashiwagi A, Maegawa H, Uzu T. Fructose induces tubulointerstitial injury in the kidney of mice. Biochem Biophys Res Commun 419: 244–249, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Ash SR, Cuppage FE. Shift toward anaerobic glycolysis in the regenerating rat kidney. Am J Pathol 60: 385–402, 1970. [PMC free article] [PubMed] [Google Scholar]
- 10.Asipu A, Hayward BE, O’Reilly J, Bonthron DT. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes 52: 2426–2432, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Augustin R, Carayannopoulos MO, Dowd LO, Phay JE, Moley JF, Moley KH. Identification and characterization of human glucose transporter-like protein-9 (GLUT9): Alternative splicing alters trafficking. J Biol Chem 279: 16229–16236, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia 60: 568–573, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacon JS, Bell DJ. The identification of fructose as a constituent of the foetal blood of the sheep. Biochem J 40: xlii, 1946. [PubMed] [Google Scholar]
- 14.Baena M, Sanguesa G, Davalos A, Latasa MJ, Sala-Vila A, Sanchez RM, Roglans N, Laguna JC, Alegret M. Fructose, but not glucose, impairs insulin signaling in the three major insulin-sensitive tissues. Sci Rep 6: 26149, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bairlein F. How to get fat: Nutritional mechanisms of seasonal fat accumulation in migratory songbirds. Naturwissenschaften 89: 1–10, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Bakris G, Oshima M, Mahaffey KW, Agarwal R, Cannon CP, Capuano G, Charytan DM, de Zeeuw D, Edwards R, Greene T, Heerspink HJL, Levin A, Neal B, Oh R, Pollock C, Rosenthal N, Wheeler DC, Zhang H, Zinman B, Jardine MJ, Perkovic V. Effects of canagliflozin in patients with baseline eGFR <30 ml/min per 1.73 m(2): Subgroup analysis of the randomized CREDENCE trial. Clin J Am Soc Nephrol 15: 1705–1714, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balen D, Ljubojevic M, Breljak D, Brzica H, Zlender V, Koepsell H, Sabolic I. Revised immunolocalization of the Na+-D-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 295: C475–C489, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Bankir L, Yang B. New insights into urea and glucose handling by the kidney, and the urine concentrating mechanism. Kidney Int 81: 1179–1198, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Barfuss DW, Schafer JA. Differences in active and passive glucose transport along the proximal nephron. Am J Physiol 241: F322–F332, 1981. [DOI] [PubMed] [Google Scholar]
- 20.Barklay H, Haas P, Huggett ASG, King G, Rowley D. The sugar of the foetal blood, the amniotic and allantoic fluids. J Physiol 109: 98–102, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnett AH, Mithal A, Manassie J, Jones R, Rattunde H, Woerle HJ, Broedl UC. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2: 369–384, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, Wu X, Yu Y, Amlal H, Seidler U, Zuo J, Soleimani M. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem 284: 5056–5066, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benoy MP, Elliott KA. The metabolism of lactic and pyruvic acids in normal and tumour tissues: Synthesis of carbohydrate. Biochem J 31: 1268–1275, 1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bessho R, Takiyama Y, Takiyama T, Kitsunai H, Takeda Y, Sakagami H, Ota T. Hypoxia-inducible factor-1alpha is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci Rep 9: 14754, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biava C, Grossman A, West M. Ultrastructural observations on renal glycogen in normal and pathologic human kidneys. Lab Invest 15: 330–356, 1966. [PubMed] [Google Scholar]
- 27.Bjorkman O, Felig P. Role of the kidney in the metabolism of fructose in 60-hour fasted humans. Diabetes 31: 516–520, 1982. [DOI] [PubMed] [Google Scholar]
- 28.Blantz RC, Peterson OW, Gushwa L, Tucker BJ. Effect of modest hyperglycemia on tubuloglomerular feedback activity. Kidney Int Suppl 12: S206–S212, 1982. [PubMed] [Google Scholar]
- 29.Blantz RC, Vallon V. Tubuloglomerular feedback responses of the downstream efferent resistance: Unmasking a role for adenosine? Kidney Int 71: 837–839, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Bomback AS, Katz R, He K, Shoham DA, Burke GL, Klemmer PJ. Sugar-sweetened beverage consumption and the progression of chronic kidney disease in the multi-ethnic study of atherosclerosis (MESA). Am J Clin Nutr 90: 1172–1178, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borges-Junior FA, Silva Dos Santos D, Benetti A, Polidoro JZ, Wisnivesky ACT, Crajoinas RO, Antonio EL, Jensen L, Caramelli B, Malnic G, Tucci PJ, ACC G. Empagliflozin inhibits proximal tubule NHE3 activity, preserves GFR, and restores euvolemia in nondiabetic rats with induced heart failure. J Am Soc Nephrol Apr 12:ASN.2020071029, 2021. DOI: 10.2020071681/ASN.2020071029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowman RH. Gluconeogenesis in the isolated perfused rat kidney. J Biol Chem 245: 1604–1612, 1970. [PubMed] [Google Scholar]
- 34.Brasen JC, Burford JL, McDonough AA, Holstein-Rathlou NH, Peti-Peterdi J. Local pH domains regulate NHE3-mediated Na (+) reabsorption in the renal proximal tubule. Am J Physiol Renal Physiol 307: F1249–F1262, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brezis M, Rosen S. Hypoxia of the renal medulla–its implications for disease. N Engl J Med 332: 647–655, 1995. [DOI] [PubMed] [Google Scholar]
- 36.Brown KS, Kalinowski SS, Megill JR, Durham SK, Mookhtiar KA. Glucokinase regulatory protein may interact with glucokinase in the hepatocyte nucleus. Diabetes 46: 179–186, 1997. [DOI] [PubMed] [Google Scholar]
- 37.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 414: 813–820, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Brymora A, Flisinski M, Johnson RJ, Goszka G, Stefanska A, Manitius J. Low-fructose diet lowers blood pressure and inflammation in patients with chronic kidney disease. Nephrol Dial Transplant 27: 608–612, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burch HB, Choi S, Dence CN, Alvey TR, Cole BR, Lowry OH. Metabolic effects of large fructose loads in different parts of the rat nephron. J Biol Chem 255: 8239–8244, 1980. [PubMed] [Google Scholar]
- 40.Burch HB, Lowry OH, Meinhardt L, Max P Jr, Chyu K. Effect of fructose, dihydroxyacetone, glycerol, and glucose on metabolites and related compounds in liver and kidney. J Biol Chem 245: 2092–2102, 1970. [PubMed] [Google Scholar]
- 41.Burch HB, Narins RG, Chu C, Fagioli S, Choi S, McCarthy W, Lowry OH. Distribution along the rat nephron of three enzymes of gluconeogenesis in acidosis and starvation. Am J Physiol 235: F246–F253,1978. [DOI] [PubMed] [Google Scholar]
- 42.Burckhardt BC, Burckhardt G. Transport of organic anions across the basolateral membrane of proximal tubule cells. Rev Physiol Biochem Pharmacol 146: 95–158, 2003. [DOI] [PubMed] [Google Scholar]
- 43.Cabral PD, Hong NJ, Hye Khan MA, Ortiz PA, Beierwaltes WH, Imig JD, Garvin JL. Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to angiotensin II. Hypertension 63: e68–e73, 2014. [DOI] [PubMed] [Google Scholar]
- 44.Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, Luo J, Zhang Y, Sun Q, Lv Y, Zen K, Jiang L, Zhou Y, Yang J. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1alpha-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis 11: 390, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caliceti C, Calabria D, Roda A, Cicero AFG. Fructose intake, serum uric acid, and cardiometabolic disorders: A critical review. Nutrients 9: 395, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cammisotto PG, Londono I, Gingras D, Bendayan M. Control of glycogen synthase through ADIPOR1-AMPK pathway in renal distal tubules of normal and diabetic rats. Am J Physiol Renal Physiol 294: F881–F889, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Carey HV, Andrews MT, Martin SL. Mammalian hibernation:Cellular and molecular responses to depressed metabolism and low temperature. Physiol Rev 83: 1153–1181, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Cassis P, Locatelli M, Cerullo D, Corna D, Buelli S, Zanchi C, Villa S, Morigi M, Remuzzi G, Benigni A, Zoja C. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 3, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chamberlin ME, LeFurgey A, Mandel LJ. Suspension of medullary thick ascending limb tubules from the rabbit kidney. Am J Physiol 247: F955–F964, 1984. [DOI] [PubMed] [Google Scholar]
- 50.Chang YK, Choi H, Jeong JY, Na KR, Lee KW, Lim BJ, Choi DE. Dapagliflozin, SGLT2 inhibitor, attenuates renal ischemia-reperfusion injury. PLoS One 11: e0158810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Busse LW. Novel therapies for acute kidney injury. Kidney Int Rep 2: 785–799, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen J, Williams S, Ho S, Loraine H, Hagan D, Whaley JM, Feder JN. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther 1: 57–92, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, Fagan NM, Woerle HJ, Johansen OE, Broedl UC, Von Eynatten M. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129: 587–597, 2014. [DOI] [PubMed] [Google Scholar]
- 54.Chin E, Zamah AM, Landau D, Gronbcek H, Flyvbjerg A, LeRoith D, Bondy CA. Changes in facilitative glucose transporter messenger ribonucleic acid levels in the diabetic rat kidney. Endocrinology 138: 1267–1275, 1997. [DOI] [PubMed] [Google Scholar]
- 55.Chin E, Zhou J, Bondy C. Anatomical and developmental patterns of facilitative glucose transporter gene expression in the rat kidney. J Clin Invest 91: 1810–1815, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chino Y, Samukawa Y, Sakai S, Nakai Y, Yamaguchi JI, Nakanishi T, Tamai I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos 35: 391–404, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi YJ, Yoon Y, Lee KY, Hien TT, Kang KW, Kim KC, Lee J, Lee MY, Lee SM, Kang DH, Lee BH. Uric acid induces endothelial dysfunction by vascular insulin resistance associated with the impairment of nitric oxide synthesis. FASEB J 28: 3197–3204, 2014. [DOI] [PubMed] [Google Scholar]
- 58.Cirillo P, Gersch MS, Mu W, Scherer PM, Kim KM, Gesualdo L, Henderson GN, Johnson RJ, Sautin YY. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J Am Soc Nephrol 20: 545–553, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coady MJ, El TA, Santer R, Bissonnette P, Sasseville LJ, Calado J, Lussier Y, Dumayne C, Bichet DG, Lapointe JY. MAP17 is a necessary activator of renal Na+/Glucose cotransporter SGLT2. J Am Soc Nephrol 28: 85–93, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coffee EM, Tolan DR. Gluconeogenesis. In: LEE B, Scaglia F, editors. Inborn Errors in Metabolism: From Neonatal Screening to Metabolic Pathways. New York: Oxford University Press, 2014, p. 68–91. [Google Scholar]
- 61.Cohn C, Kolinsky M. Effect of blood sugar levels and insulin lack on gluconeogenesis by the kidney of the dog. Am J Physiol 156: 345–348, 1949. [DOI] [PubMed] [Google Scholar]
- 62.Collazo R, Fan L, Hu MC, Zhao H, Wiederkehr MR, Moe OW. Acute regulation of Na+/H+ exchanger NHE3 by parathyroid hormone via NHE3 phosphorylation and dynamin-dependent endocytosis. J Biol Chem 275: 31601–31608, 2000. [DOI] [PubMed] [Google Scholar]
- 63.Conjard A, Martin M, Guitton J, Baverel G, Ferrier B. Gluconeogenesis from glutamine and lactate in the isolated human renal proximal tubule: Longitudinal heterogeneity and lack of response to adrenaline. Biochem J 360: 371–377, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Darshi M, Onishi A, Kim JJ, Pham J, Van Espen BF, Murphy A, Heerspink HJl, Vallon V, Sharma K. Metabolic reprogramming in diabetic kidney disease can be restored via SGLT2 inhibition. J Am Soc Nephrol 439: 439 (Abstract), 2017. [Google Scholar]
- 65.Dekkers CCJ, Petrykiv S, Laverman GD, Cherney DZ, Gansevoort RT, Heerspink HJL. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes Metab 20:1988–1993, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, Hayward BE, Asipu A, Bonthron DT. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem 57: 763–774, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, Luo J, Zen K, Yang J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol 313: F561–F575, 2017. [DOI] [PubMed] [Google Scholar]
- 68.Doblado M, Moley KH. Facilitative glucose transporter 9, a unique hexose and urate transporter. Am J Physiol Endocrinol Metab 297: E831–E835, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dominguez JH, Camp K, Maianu L, Feister H, Garvey WT. Molecular adaptations of GLUT1 and GLUT2 in renal proximal tubules of diabetic rats. Am J Physiol 266: F283–F290, 1994. [DOI] [PubMed] [Google Scholar]
- 70.Dominguez JH, Camp K, Maianu L, Garvey WT. Glucose transporters of rat proximal tubule: Differential expression and subcellular distribution. Am J Physiol 262: F807–F812, 1992. [DOI] [PubMed] [Google Scholar]
- 71.Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab 295: E227–E237, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Douard V, Ferraris RP. The role of fructose transporters in diseases linked to excessive fructose intake. J Physiol 591: 401–414, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Drewnowsk KD, Craig MR, Digiovanni SR, McCarty JM, Moorman AF, Lamers WH, Schoolwerth AC. PEPCK mRNA localization in proximal tubule and gene regulation during metabolic acidosis. J Physiol Pharmacol 53: 3–20, 2002. [PubMed] [Google Scholar]
- 74.Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: A review. Diabetes Metab Res Rev 21: 31–38, 2005. [DOI] [PubMed] [Google Scholar]
- 75.Eickhoff MK, Dekkers CCJ, Kramers BJ, Laverman GD, Frimodt-Moller M, Jorgensen NR, Faber J, Danser AHJ, Gansevoort RT, Rossing P, Persson F, Heerspink HJL. Effects of dapagliflozin on volume status when added to renin-angiotensin system inhibitors. J Clin Med 8: 799, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eskandari S, Wright EM, Loo DD. Kinetics of the reverse mode of the Na+/glucose cotransporter. J Membr Biol 204: 23–32, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, Wright EM, Abramson J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321: 810–814, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farber SJ, Berger EY, Earle DP. Effect of diabetes and insulin on the maximum capacity of the renal tubules to reabsorb glucose. J Clin Invest 30: 125–129, 1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Farsijani NM, Liu Q, Kobayashi H, Davidoff O, Sha F, Fandrey J, Ikizler TA, O’Connor PM, Haase VH. Renal epithelium regulates erythropoiesis via HIF-dependent suppression of erythropoietin. J Clin Invest 126: 1425–1437, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fattah H, Shigeoka A, Huang W, Patel R, Kasimsetty S, Singh P, McKay DB, Vallon V. Diverse gene expression patterns of renal transporters in AKI. J Am Soc Nephrol 29: 1045 (Abstract), 2018. [Google Scholar]
- 81.Fattah H, Vallon V. The potential role of SGLT2 inhibitors in the treatment of type 1 diabetes mellitus. Drugs 78: 717–726, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferrannini E Sodium-glucose co-transporters and their inhibition: Clinical physiology. Cell Metab 26: 27–38, 2017. [DOI] [PubMed] [Google Scholar]
- 83.Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 39: 1108–1114, 2016. [DOI] [PubMed] [Google Scholar]
- 84.Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, Broedl UC, Woerle HJ. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124: 499–508, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ferrannini E, Pereira-Moreira R, Seghieri M, Rebelos E, Souza AL, Chueire VB, Arvia C, Muscelli E. Insulin enhances renal glucose excretion: Relation to insulin sensitivity and sodium-glucose cotransport. BMJ Open Diabetes Res Care 8: e001178, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Finch CE, Stanford CB. Meat-adaptive genes and the evolution of slower aging in humans. Q Rev Biol 79: 3–50, 2004. [DOI] [PubMed] [Google Scholar]
- 87.Fiorentino M, Grandaliano G, Gesualdo L, Castellano G. Acute kidney injury to chronic kidney disease transition. Contrib Nephrol 193: 45–54, 2018. [DOI] [PubMed] [Google Scholar]
- 88.Fischer KF, Lees JA, Newman JH. Hypoglycemia in hospitalized patients. Causes and outcomes. N Engl J Med 315: 1245–1250, 1986. [DOI] [PubMed] [Google Scholar]
- 89.Foote C, Perkovic V, Neal B. Effects of SGLT2 inhibitors on cardiovascular outcomes. Diab Vasc Dis Res 9: 117–123, 2012. [DOI] [PubMed] [Google Scholar]
- 90.Freitas HS, Anhe GF, Melo KF, Okamoto MM, Oliveira-Souza M, Bordin S, Machado UF. Na(+) -glucose transporter-2 messenger ribonucleic acid expression in kidney of diabetic rats correlates with glycemic levels: Involvement of hepatocyte nuclear factor-1alpha expression and activity. Endocrinology 149: 717–724, 2008. [DOI] [PubMed] [Google Scholar]
- 91.Freitas HS, Schaan BD, David-Silva A, Sabino-Silva R, Okamoto MM, Alves-Wagner AB, Mori RC, Machado UF. SLC2A2 gene expression in kidney of diabetic rats is regulated by HNF-1alpha and HNF-3beta. Mol Cell Endocrinol 305: 63–70, 2009. [DOI] [PubMed] [Google Scholar]
- 92.Frohnert PP, Hohmann B, Zwiebel R, Baumann K. Free flow micropuncture studies of glucose transport in the rat nephron. Pflugers Arch 315: 66–85, 1970. [DOI] [PubMed] [Google Scholar]
- 93.Fu Y, Breljak D,Onishi A, Batz F, Patel R, Huang W, Song P, Freeman B, Mayoux E, Koepsell H, Anzai N, Nigam SK, Sabolic I, Vallon V. The organic anion transporter OAT3 enhances the glucosuric effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol 315: F386–F394, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fu Y, Gerasimova M, Mayoux E, Masuda T, Vallon V. SGLT2 inhibitor empagliflozin increases renal NHE3 phosphorylation in diabetic Akita mice: Possible implications for the prevention of glomerular hyperfiltration. Diabetes 63 (supplement 1): A132, 2014. [Google Scholar]
- 95.Fukuzawa T, Fukazawa M, Ueda O, Shimada H, Kito A, Kakefuda M, Kawase Y, Wada NA, Goto C, Fukushima N, Jishage K, Honda K, King GL, Kawabe Y. SGLT5 reabsorbs fructose in the kidney but its deficiency paradoxically exacerbates hepatic steatosis induced by fructose. PLoS One 8: e56681, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gaby AR. Adverse effects of dietary fructose. Altern Med Rev 10: 294–306, 2005. [PubMed] [Google Scholar]
- 97.Gall JM, Wong V, Pimental DR, Havasi A, Wang Z, Pastorino JG, Bonegio RG, Schwartz JH, Borkan SC. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int 79: 1207–1216, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gallo LA, Ward MS, Fotheringham AK, Zhuang A, Borg DJ, Flemming NB, Harvie BM, Kinneally TL, Yeh SM, McCarthy DA, Koepsell H, Vallon V, Pollock C, Panchapakesan U, Forbes JM. Once daily administration of the SGLT2 inhibitor, empagliflozin, attenuates markers of renal fibrosis without improving albuminuria in diabetic db/db mice. Sci Rep 6: 26428, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diab Vasc Dis Res 12: 78–89, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Geerlings S, Fonseca V, Castro-Diaz D, List J, Parikh S. Genital and urinary tract infections in diabetes: Impact of pharmacologicallyinduced glucosuria. Diabetes Res Clin Pract 103: 373–381, 2014. [DOI] [PubMed] [Google Scholar]
- 101.Gembardt F, Bartaun C, Jarzebska N, Mayoux E, Todorov VT, Hohenstein B, Hugo C. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am J Physiol Renal Physiol 307: F317–F325, 2014. [DOI] [PubMed] [Google Scholar]
- 102.Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: Therapeutic implications. Diabet Med 27: 136–142, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care 24: 382–391, 2001. [DOI] [PubMed] [Google Scholar]
- 104.Gersch MS, Mu W, Cirillo P, Reungjui S, Zhang L, Roncal C, Sautin YY, Johnson RJ, Nakagawa T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am J Physiol Renal Physiol 293: F1256–F1261, 2007. [DOI] [PubMed] [Google Scholar]
- 105.Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH Jr, Probstfield JL, Simons-Morton DG, Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 358: 2545–2559, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ghazi S, Bourgeois S, Gomariz A, Bugarski M, Haenni D, Martins JR, Nombela-Arrieta C, Unwin RJ, Wagner CA, Hall AM, Craigie E. Multiparametric imaging reveals that mitochondria-rich intercalated cells in the kidney collecting duct have a very high glycolytic capacity. FASEB J 34: 8510–8525, 2020. [DOI] [PubMed] [Google Scholar]
- 107.Ghezzi C, Gorraitz E, Hirayama BA, Loo DD, Grempler R, Mayoux E, Wright EM. Fingerprints of hSGLT5 sugar and cation selectivity. Am J Physiol Cell Physiol 306: C864–C870, 2014. [DOI] [PubMed] [Google Scholar]
- 108.Ghezzi C, Hirayama BA, Gorraitz E, Loo DD, Liang Y, Wright EM. SGLT2 inhibitors act from the extracellular surface of the cell membrane. Physiol Rep 2: pii: e12058, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ghezzi C, Wright EM. Regulation of the human Na+ dependent glucose cotransporter hSGLT2. Am J Physiol Cell Physiol 303: C348–C354, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gilbert RE, Thorpe KE. Acute kidney injury with sodium-glucose cotransporter-2 inhibitors: A meta-analysis of cardiovascular outcome trials. Diabetes Obes Metab 21: 1996–2000, 2019. [DOI] [PubMed] [Google Scholar]
- 111.Glaser J, Lemery J, Rajagopalan B, Diaz HF, Garcia-Trabanino R, Taduri G, Madero M, Amarasinghe M, Abraham G, Anutrakulchai S, Jha V, Stenvinkel P, Roncal-Jimenez C, Lanaspa MA, Correa-Rotter R, Sheikh-Hamad D, Burdmann EA, Andres-Hernando A, Milagres T, Weiss I, Kanbay M, Wesseling C, Sanchez-Lozada LG, Johnson RJ. Climate change and the emergent epidemic of CKD from heat stress in rural communities: The case for heat stress nephropathy. Clin J Am Soc Nephrol 11: 1472–1483, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Glushakova O, Kosugi T, Roncal C, Mu W, Heinig M, Cirillo P, Sanchez-Lozada LG, Johnson RJ, Nakagawa T. Fructose induces the inflammatory molecule ICAM-1 in endothelial cells. J Am Soc Nephrol 19: 1712–1720, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goestemeyer AK, Marks J, Srai SK, Debnam ES, Unwin RJ. GLUT2 protein at the rat proximal tubule brush border membrane correlates with protein kinase C (PKC)-betal and plasma glucose concentration. Diabetologia 50: 2209–2217, 2007. [DOI] [PubMed] [Google Scholar]
- 114.Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, Pauli C, Morris R, Taylor S, Bosch K, Yang S, Wang Y, Van Riper J, Lekaye HC, Roper J, Kim Y, Chen Q, Gross SS, Rhee KY, Cantley LC, Yun J. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363: 1345–1349, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gonzalez-Vicente A, Cabral PD, Hong NJ, Asirwatham J, Saez F, Garvin JL. Fructose reabsorption by rat proximal tubules: Role of Na(+)-linked cotransporters and the effect of dietary fructose. Am J Physiol Renal Physiol 316: F473–F480, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gonzalez-Vicente A, Cabral PD, Hong NJ, Asirwatham J, Yang N, Berthiaume JM, Dominici FP, Garvin JL. Dietary fructose enhances the ability of low concentrations of angiotens in II to stimulate proximal tubule Na(+) reabsorption. Nutrients 9: 885, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gonzalez-Vicente A, Garvin JL, Hopfer U. Transcriptome signature for dietary fructose-specific changes in rat renal cortex: A quantitative approach to physiological relevance. PLoS One 13: e0201293, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goodwin RF. Division of the common mammals into two groups according to the concentration of fructose in the blood of the foetus. J Physiol 132: 146–156, 1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goran MI, Martin AA, Alderete TL, Fujiwara H, Fields DA. Fructose in breast milk is positively associated with infant body composition at 6 months of age. Nutrients 9, 146, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gorboulev V, Schurmann A, Vallon V, Kipp H, Jaschke A, Klessen D, Friedrich A, Scherneck S, Rieg T, Cunard R, Veyhl-Wichmann M, Srinivasan A, Balen D, Breljak D, Rexhepaj R, Parker HE, Gribble FM, Reimann F, Lang F, Wiese S, Sabolic I, Sendtner M, Koepsell H. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61: 187–196, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gordish KL, Kassem KM, Ortiz PA, Beierwaltes WH. Moderate (20%) fructose-enriched diet stimulates salt-sensitive hypertension with increased salt retention and decreased renal nitric oxide. Physiol Rep 5: e13162, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goyal SN, Reddy NM, Patil KR, Nakhate KT, Ojha S, Patil CR, Agrawal YO. Challenges and issues with streptozotocin-induced diabetes - A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem Biol Interact 244: 49–63, 2016. [DOI] [PubMed] [Google Scholar]
- 123.Grempler R, Augustin R, Froehner S, Hildebrandt T, Simon E, Mark M, Eickelmann P. Functional characterisation of human SGLT-5 as a novel kidney-specific sodium-dependent sugar transporter. FEBS Lett 586: 248–253, 2012. [DOI] [PubMed] [Google Scholar]
- 124.Guder WG, Ross BD. Enzyme distribution along the nephron. Kidney Int 26: 101–111, 1984. [DOI] [PubMed] [Google Scholar]
- 125.Guder WG, Schmidt U. The localization of gluconeogenesis in rat nephron. Determination of phosphoenolpyruvate carboxykinase in microdissected tubules. Hoppe Seylers Z Physiol Chem 355: 273–278, 1974. [DOI] [PubMed] [Google Scholar]
- 126.Guyton AC. Blood pressure control–special role of the kidneys and body fluids. Science 252: 1813–1816, 1991. [DOI] [PubMed] [Google Scholar]
- 127.Gyimesi G, Pujol-Gimenez J, Kanai Y, Hediger MA. Sodium-coupled glucose transport, the SLC5 family, and therapeutically relevant inhibitors: From molecular discovery to clinical application. Pflugers Arch 472: 1177–1206, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hallow KM, Gebremichael Y, Helmlinger G, Vallon V. Primary proximal tubule hyperreabsorption and impaired tubular transport counterregulation determine glomerular hyperfiltration in diabetes: A modeling analysis. Am J Physiol Renal Physiol 312: F819–F835, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hallow KM, Helmlinger G, Greasley PJ, McMurray JJV, Boulton DW. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes Metab 20: 479–487, 2018. [DOI] [PubMed] [Google Scholar]
- 130.Han HJ, Lee YJ, Park SH, Lee JH, Taub M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am J Physiol Renal Physiol 288: F988–F996, 2005. [DOI] [PubMed] [Google Scholar]
- 131.Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest 128: 545–555, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol 40: 123–137, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hansen HH, Jelsing J, Hansen CF, Hansen G, Vrang N, Mark M, Klein T, Mayoux E. The sodium glucose cotransporter type 2 inhibitor empagliflozin preserves beta-cell mass and restores glucose homeostasis in the male zucker diabetic fatty rat. J Pharmacol Exp Ther 350: 657–664, 2014. [DOI] [PubMed] [Google Scholar]
- 134.Hayward BE, Bonthron DT. Structure and alternative splicing of the ketohexokinase gene. Eur J Biochem 257: 85–91, 1998. [DOI] [PubMed] [Google Scholar]
- 135.Hediger MA, Coady MJ, Ikeda TS, Wright EM. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature 330: 379–381, 1987. [DOI] [PubMed] [Google Scholar]
- 136.Heerspink HJ, Desai M, Jardine M, Balis D, Meininger G, Perkovic V. Canagliflozin slows progression of renal function decline independently of glycemic effects. J Am Soc Nephrol 28: 368–375, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, Mann JFE, McMurray JJV, Lindberg M, Rossing P, Sjostrom CD, Toto RD, Langkilde AM, Wheeler DC, Committees D-CT, and Investigators. Dapagliflozin in patients with chronic kidney disease. N Engl J Med 383: 1436–1446, 2020. [DOI] [PubMed] [Google Scholar]
- 138.Heise T, Seewaldt-Becker E, Macha S, Hantel S, Pinnetti S, Seman L, Woerle HJ. Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks’ treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab 15: 613–621, 2013. [DOI] [PubMed] [Google Scholar]
- 139.Hers HG, Van Schaftingen E. Fructose 2,6-bisphosphate 2 years after its discovery. Biochem J 206: 1–12, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heyman SN, Rosen S, Brezis M. The renal medulla: Life at the edge of anoxia. Blood Purif 15: 232–242, 1997. [DOI] [PubMed] [Google Scholar]
- 141.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 117: 3810–3820, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hinden L, Udi S, Drori A, Gammal A, Nemirovski A, Hadar R, Baraghithy S, Permyakova A, Geron M, Cohen M, Tsytkin-Kirschenzweig S, Riahi Y, Leibowitz G, Nahmias Y, Priel A, Tam J. Modulation of renal GLUT2 by the cannabinoid-1 receptor: Implications for the treatment of diabetic nephropathy. J Am Soc Nephrol 29: 434–448, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hitchcock MW. Fructose in the sheep foetus. J Physiol 108: 117–126, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Holck P, Rasch R. Structure and segmental localization of glycogen in the diabetic rat kidney. Diabetes 42: 891–900, 1993. [DOI] [PubMed] [Google Scholar]
- 145.Holman GD. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflugers Arch 472: 1155–1175, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Holstein-Rathlou NH, Marsh DJ. Oscillations of tubular pressure, flow, and distal chloride concentration in rats. Am J Physiol 256: F1007–F1014, 1989. [DOI] [PubMed] [Google Scholar]
- 147.Horiba N, Masuda S, Ohnishi C, Takeuchi D, Okuda M, Inui K. Na(+)-dependent fructose transport via rNaGLT1 in rat kidney. FEBS Lett 546: 276–280, 2003. [DOI] [PubMed] [Google Scholar]
- 148.Hotta N, Kawamori R, Fukuda M, Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: Multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabet Med 29: 1529–1533, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huggett AS. Carbohydrate metabolism in the placenta and foetus. Br Med Bull 17: 122–126, 1961. [DOI] [PubMed] [Google Scholar]
- 150.Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM. Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol 300: C14–C21, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Iles RA, Cohen RD, Rist AH, Baron PG. The mechanism of inhibition by acidosis of gluconeogenesis from lactate in rat liver. Biochem J 164: 185–191, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Inzucchi SE, Zinman B, Fitchett D, Wanner C, Ferrannini E, Schumacher M, Schmoor C, Ohneberg K, Johansen OE, George JT, Hantel S, Bluhmki E, Lachin JM. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care 41: 356–363, 2018. [DOI] [PubMed] [Google Scholar]
- 153.Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, Rabinowitz JD. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab 27: 351–361 e353, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jauniaux E, Hempstock J, Teng C, Battaglia FC, Burton GJ. Polyol concentrations in the fluid compartments of the human conceptus during the first trimester of pregnancy: Maintenance of redox potential in a low oxygen environment. J Clin Endocrinol Metab 90: 1171–1175, 2005. [DOI] [PubMed] [Google Scholar]
- 155.Jensen PK, Kristensen KS, Rasch R, Persson AEG. Decreased sensitivity of the tubuloglomerular feedback mechanism in experimental diabetic rats. In: Persson AEG, Boberg U, editors. The Juxtaglomerular Apparatus. Amsterdam New York: Elsevier, 1988, p. 333–338. [Google Scholar]
- 156.Johnson RJ, Andrews P, Benner SA, Oliver W. Theodore E. Woodward award. The evolution of obesity: Insights from the mid-Miocene. Trans Am Clin Climatol Assoc 121: 295–305; discussion 305–298, 2010. [PMC free article] [PubMed] [Google Scholar]
- 157.Johnson RJ, Bakris GL, Borghi C, Chonchol MB, Feldman D, Lanaspa MA, Merriman TR, Moe OW, Mount DB, Sanchez Lozada LG, Stahl E, Weiner DE, Chertow GM. Hyperuricemia, acute and chronic kidney disease, hypertension, and cardiovascular disease: Report of a scientific workshop organized by the National Kidney Foundation. Am J Kidney Dis, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY, Lanaspa MA. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 62: 3307–3315, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Johnson RJ, Sanchez-Lozada LG, Nakagawa T. The effect of fructose on renal biology and disease. J Am Soc Nephrol 21: 2036–2039, 2010. [DOI] [PubMed] [Google Scholar]
- 160.Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang DH, Gersch MS, Benner S, Sanchez-Lozada LG. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr 86: 899–906, 2007. [DOI] [PubMed] [Google Scholar]
- 161.Johnson RJ, Stenvinke lP, Andrews P, Sanchez-Lozada LG, Nakagawa T, Gaucher E, Andres-Hernando A, Rodriguez-Iturbe B, Jimenez CR, Garcia G, Kang DH, Tolan DR, Lanaspa MA. Fructose metabolism as a common evolutionary pathway of survival associated with climate change, food shortage and droughts. J Intern Med 287: 252–262, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Johnston PA, Rennke H, Levinsky NG. Recovery of proximal tubular function from ischemic injury. Am J Physiol 246: F159–F166, 1984. [DOI] [PubMed] [Google Scholar]
- 163.Junk WJ. Temporary fat storage, an adaptation of some fish species to the waterlevel fluctuations and related environmental changes of the Amazon river. Amazoniana 9: 315–351, 1985. [Google Scholar]
- 164.Jurczak MJ, Lee HY, Birkenfeld AL, Jornayvaz FR, Frederick DW, Pongratz RL, Zhao X, Moeckel GW, Samuel VT, Whaley JM, Shulman GI, Kibbey RG. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes 60: 890–898, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kamran M, Peterson RG, Dominguez JH. Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. J Am Soc Nephrol 8: 943–948, 1997. [DOI] [PubMed] [Google Scholar]
- 166.Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest 93: 397–404, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kang DH, Nakagawa T, Feng L, Watanabe S, Han L, Mazzali M, Truong L, Harris R, Johnson RJ. A role for uric acid in the progression of renal disease. J Am Soc Nephrol 13: 2888–2897, 2002. [DOI] [PubMed] [Google Scholar]
- 168.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kang J, Dai XS, Yu TB, Wen B, Yang ZW. Glycogen accumulation in renal tubules, a key morphological change in the diabetic rat kidney. Acta Diabetol 42: 110–116, 2005. [DOI] [PubMed] [Google Scholar]
- 170.Karalius VP, Shoham DA. Dietary sugar and artificial sweetener intake and chronic kidney disease: A review. Adv Chronic Kidney Dis 20: 157–164, 2013. [DOI] [PubMed] [Google Scholar]
- 171.Kato ET, Silverman MG, Mosenzon O, Zelniker TA, Cahn A, Furtado RHM, Kuder J, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Bonaca MP, Ruff CT, Desai AS, Goto S, Johansson PA, Gause-Nilsson I, Johanson P, Langkilde AM, Raz I, Sabatine MS, Wiviott SD. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation 139: 2528–2536, 2019. [DOI] [PubMed] [Google Scholar]
- 172.Kawasaki T, Akanuma H, Yamanouchi T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care 25: 353–357, 2002. [DOI] [PubMed] [Google Scholar]
- 173.Kayano T, Burant CF, Fukumoto H, Gould GW, Fan YS, Eddy RL, Byers MG, Shows TB, Seino S, Bell GI. Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6). J Biol Chem 265: 13276–13282, 1990. [PubMed] [Google Scholar]
- 174.Kellett GL, Brot-Laroche E, Mace OJ, Leturque A. Sugar absorption in the intestine: The role of GLUT2. Annu Rev Nutr 28: 35–54, 2008. [DOI] [PubMed] [Google Scholar]
- 175.Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, Krotova K, Block ER, Prabhakar S, Johnson RJ. Hyperuricemia induces endothelial dysfunction. Kidney Int 67: 1739–1742, 2005. [DOI] [PubMed] [Google Scholar]
- 176.Khunti K, Davies M, Majeed A, Thorsted BL, Wolden ML, Paul SK. Hypoglycemia and risk of cardiovascular disease and all-cause mortality in insulin-treated people with type 1 and type 2 diabetes: A cohort study. Diabetes Care 38: 316–322, 2015. [DOI] [PubMed] [Google Scholar]
- 177.Kida K, Nakajo S, Kamiya F, Toyama Y, Nishio T, Nakagawa H. Renal net glucose release in vivo and its contribution to blood glucose in rats. J Clin Invest 62: 721–726, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kidokoro K, Cherney DZI, Bozovic A, Nagasu H, Satoh M, Kanda E, Sasaki T, Kashihara N. Evaluation of glomerular hemodynamic function by empagliflozin in diabetic mice using in vivo imaging. Circulation 140: 303–315, 2019. [DOI] [PubMed] [Google Scholar]
- 179.Kim J, Song G, Wu G, Bazer FW. Functional roles of fructose. Proc Natl Acad Sci U S A 109: E1619–E1628, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kimura T, Takahashi M, Yan K, Sakurai H. Expression of SLC2A9 isoforms in the kidney and their localization in polarized epithelial cells. PLoS One 9: e84996, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kitada K, Nakano D, Ohsaki H, Hitomi H, Minamino T, Yatabe J, Felder RA, Mori H, Masaki T, Kobori H, Nishiyama A. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J Diabetes Complications 28: 604–611, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Klein KL, Wang MS, Torikai S, Davidson WD, Kurokawa K. Substrate oxidation by isolated single nephron segments of the rat. Kidney Int 20: 29–35, 1981. [DOI] [PubMed] [Google Scholar]
- 183.Kocinsky HS, Dynia DW, Wang T, Aronson PS. NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am J Physiol Renal Physiol 293: F212–F218, 2007. [DOI] [PubMed] [Google Scholar]
- 184.Kocinsky HS, Girardi AC, Biemesderfer D, Nguyen T, Mentone S, Orlowski J, Aronson PS. Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am J Physiol Renal Physiol 289: F249–F258, 2005. [DOI] [PubMed] [Google Scholar]
- 185.Koepsell H. Glucose transporters in the small intestine in health and disease. Pflugers Arch 472: 1207–1248, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Koepsell H, Vallon V. A special issue on glucose transporters in health and disease. Pflugers Arch 472: 1107–1109, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kogot-Levin A, Hinden L, Riahi Y, Israeli T, Tirosh B, Cerasi E, Mizrachi EB, Tam J, Mosenzon O, Leibowitz G. Proximal tubule mTORC1 is a central player in the pathophysiology of diabetic nephropathy and its correction by SGLT2 inhibitors. Cell Rep 32: 107954, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kohan DE, Fioretto P, Johnsson K, Parikh S, Ptaszynska A, Ying L. The effect of dapagliflozin on renal function in patients with type 2 diabetes. J Nephrol 29: 391–400, 2016. [DOI] [PubMed] [Google Scholar]
- 189.Komers R, Lindsley JN, Oyama TT, Allison KM, Anderson S. Role of neuronal nitric oxide synthase (NOS1) in the pathogenesis of renal hemodynamic changes in diabetes. Am J Physiol Renal Physiol 279: F573–F583, 2000. [DOI] [PubMed] [Google Scholar]
- 190.Komoroski B, Vachharajani N, Boulton D, Kornhauser D, Geraldes M, Li L, Pfister M. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther 85: 520–526, 2009. [DOI] [PubMed] [Google Scholar]
- 191.Kowalski GM, Bruce CR. The regulation of glucose metabolism: Implications and considerations for the assessment of glucose homeostasis in rodents. Am J Physiol Endocrinol Metab 307: E859–E871, 2014. [DOI] [PubMed] [Google Scholar]
- 192.Kranhold JF, Loh D, Morris RC Jr. Renal fructose-metabolizing enzymes: Significance in hereditary fructose intolerance. Science 165: 402–403, 1969. [DOI] [PubMed] [Google Scholar]
- 193.Krebs HA, Lund P. Formation of glucose from hexoses, pentoses, polyols and related substances in kidney cortex. Biochem J 98: 210–214, 1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kuhre RE, Ghiasi SM, Adriaenssens AE, Wewer Albrechtsen NJ, Andersen DB, Aivazidis A, Chen L, Mandrup-Poulsen T, Orskov C, Gribble FM, Reimann F, Wierup N, Tyrberg B, Holst JJ. No direct effect of SGLT2 activity on glucagon secretion. Diabetologia 62: 1011–1023, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab 15: 853–862, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, Venkatachalam MA. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27: 3356–3367, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, Tanabe K, Andres-Hernando A, Orlicky DJ, Finol E, Inaba S, Li N, Rivard CJ, Kosugi T, Sanchez-Lozada LG, Petrash JM, Sautin YY, Ejaz AA, Kitagawa W, Garcia GE, Bonthron DT, Asipu A, Diggle CP, Rodriguez-Iturbe B, Nakagawa T, Johnson RJ. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J Am Soc Nephrol 25: 2526–2538, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lanaspa MA, Kuwabara M, Andres-Hernando A, Li N, Cicerchi C, Jensen T, Orlicky DJ, Roncal-Jimenez CA, Ishimoto T, Nakagawa T, Rodriguez-Iturbe B, MacLean PS, Johnson RJ. High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci U S A 115: 3138–3143, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, Ishimoto T, Li N, Marek G, Duranay M, Schreiner G, Rodriguez-Iturbe B, Nakagawa T, Kang DH, Sautin YY, Johnson RJ. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J Biol Chem 287: 40732–40744, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, Li N, Roncal-Jimenez CA, Ishimoto T, Le M, Garcia GE, Thomas JB, Rivard CJ, Andres-Hernando A, Hunter B, Schreiner G, Rodriguez-Iturbe B, Sautin YY, Johnson RJ. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One 7: e47948, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lang F, Gorlach A, Vallon V. Targeting SGK1 in diabetes. Expert Opin Ther Targets 13: 1303–1311, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Layton AT, Vallon V. SGLT2 inhibition in a kidney with reduced nephron number: Modeling and analysis of solute transport and metabolism. Am J Physiol Renal Physiol 314: F969–F984, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Layton AT, Vallon V, Edwards A. Modeling oxygen consumption in the proximal tubule: Effects of NHE and SGLT2 inhibition. Am J Physiol Renal Physiol 308: F1343–F1357, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Layton AT, Vallon V, Edwards A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am J Physiol Renal Physiol 310: F1269–F1283, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lee WS, Kanai Y, Wells RG, Hediger MA. The high affinity Na+/glucose cotransporter. Re-evaluation of function and distribution of expression. J Biol Chem 269: 12032–12039, 1994. [PubMed] [Google Scholar]
- 207.Lee YH, Kim SH, Kang JM, Heo JH, Kim DJ, Park SH, Sung M, Kim J, Oh J, Yang DH, Lee SH, Lee SY. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am J Physiol Renal Physiol 317: F767–F780, 2019. [DOI] [PubMed] [Google Scholar]
- 208.Legouis D, Ricksten SE, Faivre A, Verissimo T, Gariani K, Verney C, Galichon P, Berchtold L, Feraille E, Fernandez M, Placier S, Koppitch K, Hertig A, Martin PY, Naesens M, Pugin J, McMahon AP, Cippa PE, de Seigneux S. Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat Metab 2: 732–743, 2020. [DOI] [PubMed] [Google Scholar]
- 209.Li J, Liu H, Takagi S, Nitta K, Kitada M, Srivastava SP, Takagaki Y, Kanasaki K, Koya D. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight 5: e129034, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Li J, Neal B, Perkovic V, de Zeeuw D, Neuen BL, Arnott C, Simpson R, Oh R, Mahaffey KW, Heerspink HJL. Mediators of the effects of canagliflozin on kidney protection in patients with type 2 diabetes. Kidney Int 98: 769–777, 2020. [DOI] [PubMed] [Google Scholar]
- 211.Li J, Woodward M, Perkovic V, Figtree GA, Heerspink HJL, Mahaffey KW, de Zeeuw D, Vercruysse F, Shaw W, Matthews DR, Neal B. Mediators of the effects of canagliflozin on heart failure in patients with type 2 diabetes. JACC Heart Fail 8: 57–66, 2020. [DOI] [PubMed] [Google Scholar]
- 212.Li Z, Agrawal V, Ramratnam M, Sharma RK, D’Auria S, Sincoular A, Jakubiak M, Music ML, Kutschke WJ, Huang XN, Gifford L, Ahmad F. Cardiac sodium-glucose co-transporter 1 (SGLT1) is a novel mediator of ischemia/reperfusion injury. Cardiovasc Res 115: 1646–1658, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Limbutara K, Chou CL, Knepper MA. Quantitative proteomics of all 14 renal tubule segments in rat. J Am Soc Nephrol, 2020. DOI: 10.1681/ASN.2020010071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lin J, Curhan GC. Associations of sugar and artificially sweetened soda with albuminuria and kidney function decline in women. Clin J Am Soc Nephrol 6: 160–166, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Linden KC, DeHaan CL, Zhang Y, Glowacka S, Cox AJ, Kelly DJ, Rogers S. Renal expression and localization of the facilitative glucose transporters GLUT1 and GLUT12 in animal models of hypertension and diabetic nephropathy. Am J Physiol Renal Physiol 290: F205–F213, 2006. [DOI] [PubMed] [Google Scholar]
- 216.Litvinov D, Selvarajan K, Garelnabi M, Brophy L, Parthasarathy S. Anti-atherosclerotic actions of azelaic acid, an end product of linoleic acid peroxidation, in mice. Atherosclerosis 209: 449–454, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Liu H, Takagaki Y, Kumagai A, Kanasaki K, Koya D. The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1alpha accumulation. J Diabetes Investig 12: 697–709, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Liu R, Carretero OA, Ren Y, Garvin JL. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int 67: 1837–1843, 2005. [DOI] [PubMed] [Google Scholar]
- 219.Locatelli M, Zoja C, Zanchi C, Corna D, Villa S, Bolognini S, Novelli R, Perico L, Remuzzi G, Benigni A, Cassis P. Manipulating Sirtuin 3 pathway ameliorates renal damage in experimental diabetes. Sci Rep 10: 8418, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Loeschke K, Baumann K, Renschler H, Ullrich KJ. Differentiation of the active and passive components of D-glucose transport in the proximal tubule of rat kidney. Pflugers Arch 305: 118–138, 1969. [DOI] [PubMed] [Google Scholar]
- 221.Lu YH, Chang YP, Li T, Han F, Li CJ, Li XY, Xue M, Cheng Y, Meng ZY, Han Z, Sun B, Chen LM. Empagliflozin attenuates hyperuricemia by upregulation of ABCG2 via AMPK/AKT/CREB signaling pathway in type 2 diabetic mice. Int J Biol Sci 16: 529–542, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ly JP, Onay T, Sison K, Sivaskandarajah G, Sabbisetti V, Li L, Bonventre JV, Flenniken A, Paragas N, Barasch JM, Adamson SL, Osborne L, Rossant J, Schnermann J, Quaggin SE. The sweet pee model for Sglt2 mutation. J Am Soc Nephrol 22: 113–123, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lynch MR, Tran MT, Parikh SM. PGC1alpha in the kidney. Am J Physiol Renal Physiol 314: F1–F8, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lytvyn Y, Bjornstad P, Katz A, Singh SK, Godoy LC, Chung LT, Vinovskis CL, Pyle L, Roussel R, Perkins BA, Cherney D. SGLT2 inhibition increases serum copeptin in young adults with type 1 diabetes. Diabetes Metab, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lytvyn Y, Skrtic M, Yang GK, Yip PM, Perkins BA, Cherney DZ. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am J Physiol Renal Physiol 308: F77–F83, 2015. [DOI] [PubMed] [Google Scholar]
- 226.Macdonald FR, Peel JE, Jones HB, Mayers RM, Westgate L, Whaley JM, Poucher SM. The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes Metab 12: 1004–1012, 2010. [DOI] [PubMed] [Google Scholar]
- 227.Macdonald I, Keyser A, Pacy D. Some effects, in man, of varying the load of glucose, sucrose, fructose, or sorbitol on various metabolites in blood. Am J Clin Nutr 31: 1305–1311, 1978. [DOI] [PubMed] [Google Scholar]
- 228.Madunic IV, Breljak D, Karaica D, Koepsell H, Sabolic I. Expression profiling and immunolocalization of Na(+)-D-glucose-cotransporter 1 in mice employing knockout mice as specificity control indicate novel locations and differences between mice and rats. Pflugers Arch 469: 1545–1565, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Maenpaa PH, Raivio KO, Kekomaki MP. Liver adenine nucleotides: Fructose-induced depletion and its effect on protein synthesis. Science 161: 1253–1254, 1968. [DOI] [PubMed] [Google Scholar]
- 230.Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 52: 691–697, 2009. [DOI] [PubMed] [Google Scholar]
- 231.Malek M, Nematbakhsh M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J Renal Inj Prev 4: 20–27, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Marks J, Carvou NJ, Debnam ES, Srai SK, Unwin RJ. Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J Physiol 553: 137–145, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Martin MG, Turk E, Lostao MP, Kerner C, Wright EM. Defects in Na+/glucose cotransporter (SGLT1) trafficking and function cause glucose-galactose malabsorption. Nat Genet 12: 216–220, 1996. [DOI] [PubMed] [Google Scholar]
- 234.Masuda T, Muto S, Fukuda K, Watanabe M, Ohara K, Koepsell H, Vallon V, Nagata D. Osmotic diuresis by SGLT2 inhibition stimulates vasopressin-induced water reabsorption to maintain body fluid volume. Physiol Rep 8: e14360, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Masuda T, Watanabe Y, Fukuda K, Watanabe M, Onishi A, Ohara K, Imai T, Koepsell H, Muto S, Vallon V, Nagata D. Unmasking a sustained negative effect of SGLT2 inhibition on body fluid volume in the rat. Am J Physiol Renal Physiol 315: F653–F664, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Mate A, de la Hermosa MA, Barfull A, Planas JM, Vazquez CM. Characterization of D-fructose transport by rat kidney brush-border membrane vesicles: Changes in hypertensive rats. Cell Mol Life Sci 58: 1961–1967, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25: 4777–4786, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, Wiriyasermkul P, Kikuchi Y, Oda T, Nishiyama J, Nakamura T, Morimoto Y, Kamakura K, Sakurai Y, Nonoyama S, Kanai Y, Shinomiya N. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet 83: 744–751, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Mavrias DA, Mayer RJ. Metabolism of fructose in the small intestine. 1. The effect of fructose feeding on fructose transport and metabolism in rat small intestine. Biochim Biophys Acta 291: 531–537, 1973. [DOI] [PubMed] [Google Scholar]
- 240.McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O’Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjostrand M, Langkilde AM. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 381: 1995–2008, 2019. [DOI] [PubMed] [Google Scholar]
- 241.Mellanby K. Metabolic water and desiccation. Nature 150: 21–21, 1942. [Google Scholar]
- 242.Merovci A, Solis-Herrera C, Daniele G, Eldor R, Fiorentino TV, Tripathy D, Xiong J, Perez Z, Norton L, Abdul-Ghani MA, DeFronzo RA. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest 124: 509–514, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Meyer C, Stumvoll M, Nadkarni V, Dostou J, Mitrakou A, Gerich J. Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest 102: 619–624, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99: 989–994, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mirtschink P, Krishnan J, Grimm F, Sarre A, Horl M, Kayikci M, Fankhauser N, Christinat Y, Cortijo C, Feehan O, Vukolic A, Sossalla S, Stehr SN, Ule J, Zamboni N, Pedrazzini T, Krek W. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 522: 444–449, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Moen MF, Zhan M, Hsu VD, Walker LD, Einhorn LM, Seliger SL, Fink JC. Frequency of hypoglycemia and its significance in chronic kidney disease. Clin J Am Soc Nephrol 4: 1121–1127, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mogensen CE. Maximum tubular reabsorption capacity for glucose and renal hemodynamics during rapid hypertonic glucose infusion in normal and diabetic subjects. Scand J Clin Lab Invest 28: 101–109, 1971. [DOI] [PubMed] [Google Scholar]
- 248.Molitoris BA, Kinne R. Ischemia induces surface membrane dysfunction. Mechanism of altered Na+-dependent glucose transport. J Clin Invest 80: 647–654, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Monami M, Nardini C, Mannucci E. Efficacy and safety of sodium glucose co-transport-2 inhibitors in type 2 diabetes: A meta-analysis of randomized clinical trials. Diabetes Obes Metab 16: 457–466, 2014. [DOI] [PubMed] [Google Scholar]
- 250.Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. diabetes care 39: 1115–1122, 2016. [DOI] [PubMed] [Google Scholar]
- 251.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34: 121–138, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Mulder S, Heerspink HJL, Darshi M, Kim JJ, Laverman GD, Sharma K, Pena MJ. Effects of dapagliflozin on urinary metabolites in people with type 2 diabetes. Diabetes Obes Metab, 2019. [DOI] [PubMed] [Google Scholar]
- 253.Nakagawa T. Uncoupling of the VEGF-endothelial nitric oxide axis in diabetic nephropathy: An explanation for the paradoxical effects of VEGF in renal disease. Am J Physiol Renal Physiol 292: F1665–F1672, 2007. [DOI] [PubMed] [Google Scholar]
- 254.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera-Acosta J, Patel JM, Johnson RJ. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 290: F625–F631, 2006. [DOI] [PubMed] [Google Scholar]
- 255.Nakagawa T, Johnson RJ, Andres-Hernando A, Roncal-Jimenez C, Sanchez-Lozada LG, Tolan DR, Lanaspa MA. Fructose production and metabolism in the kidney. J Am Soc Nephrol 31: 898–906, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Nakagawa T, Lanaspa MA, Johnson RJ. The effects of fruit consumption in patients with hyperuricaemia or gout. Rheumatology (Oxford) 58: 1133–1141, 2019. [DOI] [PubMed] [Google Scholar]
- 257.Nakagawa T, Lanaspa MA, Millan IS, Fini M, Rivard CJ, Sanchez-Lozada LG, Andres-Hernando A, Tolan DR, Johnson RJ. Fructose contributes to the Warburg effect for cancer growth. Cancer Metab 8: 16, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Nakagawa T, Tanabe K, Croker BP, Johnson RJ, Grant MB, Kosugi T, Li Q. Endothelial dysfunction as a potential contributor in diabetic nephropathy. Nat Rev Nephrol 7: 36–44, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Nakagawa T, Tuttle KR, Short RA, Johnson RJ. Hypothesis: Fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol 1: 80–86, 2005. [DOI] [PubMed] [Google Scholar]
- 260.Nakayama T, Kosugi T, Gersch M, Connor T, Sanchez-Lozada LG, Lanaspa MA, Roncal C, Perez-Pozo SE, Johnson RJ, Nakagawa T. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am J Physiol Renal Physiol 298: F712–F720, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Nangaku M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J Am Soc Nephrol 17: 17–25, 2006. [DOI] [PubMed] [Google Scholar]
- 262.Nannipieri M, Lanfranchi A, Santerini D, Catalano C, Van de Werve G, Ferrannini E. Influence of long-term diabetes on renal glycogen metabolism in the rat. Nephron 87: 50–57, 2001. [DOI] [PubMed] [Google Scholar]
- 263.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377: 644–657, 2017. [DOI] [PubMed] [Google Scholar]
- 264.Neill O, Fasching A, Pihl L, Patinha D, Franzen S, Palm F. Acute SGLT inhibition normalizes oxygen tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am J Physiol Renal Physiol 309: F227–F234, 2015. [DOI] [PubMed] [Google Scholar]
- 265.Neilson EG. The fructose nation. J Am Soc Nephrol 18: 2619–2621, 2007. [DOI] [PubMed] [Google Scholar]
- 266.Nespoux J, Patel R, Hudkins KL, Huang W, Freeman B, Kim Y, Koepsell H, Alpers CE, Vallon V. Gene deletion of the Na-glucose cotransporter SGLT1 ameliorates kidney recovery in a murine model of acute kidney injury induced by ischemia-reperfusion. Am J Physiol Renal Physiol 316: F1201–F1210, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Nespoux J, Patel R, Zhang H, Huang W, Freeman B, Sanders PW, Kim YC, Vallon V. Gene knockout of the Na(+)-glucose cotransporter SGLT2 in a murine model of acute kidney injury induced by ischemia-reperfusion. Am J Physiol Renal Physiol 318: F1100–F1112, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Nespoux J, Vallon V. Renal effects of SGLT2 inhibitors: An update. Curr Opin Nephrol Hypertens 29: 190–198, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Nespoux J, Vallon V. SGLT2 inhibition and kidney protection. Clin Sci (Lond) 132: 1329–1339, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Neuen BL, Young T, Heerspink HJL, Neal B, Perkovic V, Billot L, Mahaffey KW, Charytan DM, Wheeler DC, Arnott C, Bompoint S, Levin A, Jardine MJ. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol 7: 845–854, 2019. [DOI] [PubMed] [Google Scholar]
- 271.Nguyen S, Choi HK, Lustig RH, Hsu CY. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J Pediatr 154: 807–813, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Niculescu L, Veiga-da-Cunha M, Van Schaftingen E. Investigation on the mechanism by which fructose, hexitols and other compounds regulate the translocation of glucokinase in rat hepatocytes. Biochem J 321 (Pt 1): 239–246, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Norton L, Shannon CE, Fourcaudot M, Hu C, Wang N, Ren W, Song J, Abdul-Ghani M, DeFronzo RA, Ren J, Jia W. Sodium-glucose co-transporter (SGLT) and glucose transporter (GLUT) expression in the kidney of type 2 diabetic subjects. Diabetes Obes Metab 19: 1322–1326, 2017. [DOI] [PubMed] [Google Scholar]
- 274.Nosadini R, Semplicini A, Fioretto P, Lusiani L, Trevisan R, Donadon V, Zanette G, Nicolosi GL, Dall’Aglio V, Zanuttini D, et al. Sodium-lithium countertransport and cardiorenal abnormalities in essential hypertension. Hypertension 18: 191–198, 1991. [DOI] [PubMed] [Google Scholar]
- 275.Novikov A, Fu Y, Huang W, Freeman B, Patel R, van Ginkel C, Koepsell H, Busslinger M, Onishi A, Nespoux J, Vallon V. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am J Physiol Renal Physiol 316: F173–F185, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ohmura C, Watada H, Azuma K, Shimizu T, Kanazawa A, Ikeda F, Yoshihara T, Fujitani Y, Hirose T, Tanaka Y, Kawamori R. Aldose reductase inhibitor, epalrestat, reduces lipid hydroperoxides in type 2 diabetes. Endocr J 56: 149–156, 2009. [DOI] [PubMed] [Google Scholar]
- 277.Onishi A, Fu Y, Darshi M, Crespo-Masip M, Huang W, Song P, Patel R, Kim YC, Nespoux J, Freeman B, Soleimani M, Thomson SC, Sharma K, Vallon V. Effect of renal tubule-specific knockdown of the Na(+)/H(+) exchanger NHE3 in Akita diabetic mice. Am J Physiol Renal Physiol 317: F419–F434, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Onishi A, Fu Y, Patel R, Darshi M, Crespo-Masip M, Huang W, Song P, Freeman B, Kim YC, Soleimani M, Sharma K, Thomson SC, Vallon V. A role for tubular Na(+)/H(+) exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol 319: F712–F728, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Ono S, Kume S, Yasuda-Yamahara M, Yamahara K, Takeda N, Chin-Kanasaki M, Araki H, Sekine O, Yokoi H, Mukoyama M, Uzu T, Araki SI, Maegawa H. O-linked beta-N-acetylglucosamine modification of proteins is essential for foot process maturation and survival in podocytes. Nephrol Dial Transplant 32: 1477–1487, 2017. [DOI] [PubMed] [Google Scholar]
- 280.Osorio H, Bautista R, Rios A, Franco M, Santamaria J, Escalante B. Effect of treatment with losartan on salt sensitivity and SGLT2 expression in hypertensive diabeticrats. Diabetes Res Clin Pract 86: e46–e49, 2009. [DOI] [PubMed] [Google Scholar]
- 281.Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF Jr. Liver and kidney metabolism during prolonged starvation. J Clin Invest 48: 574–583, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277: 30409–30412, 2002. [DOI] [PubMed] [Google Scholar]
- 283.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, Januzzi J, Verma S, Tsutsui H, Brueckmann M, Jamal W, Kimura K, Schnee J, Zeller C, Cotton D, Bocchi E, Bohm M, Choi DJ, Chopra V, Chuquiure E, Giannetti N, Janssens S, Zhang J, Gonzalez Juanatey JR, Kaul S, Brunner-La Rocca HP, Merkely B, Nicholls SJ, Perrone S, Pina I, Ponikowski P, Sattar N, Senni M, Seronde MF, Spinar J, Squire I, Taddei S, Wanner C, Zannad F, Investigators EM-RT. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med 383: 1413–1424, 2020. [DOI] [PubMed] [Google Scholar]
- 284.Palatini P, Mormino P, Dorigatti F, Santonastaso M, Mos L, De Toni R, Winnicki M, Dal Follo M, Biasion T, Garavelli G, Pessina AC, Group HS. Glomerular hyperfiltration predicts the development of microalbuminuria in stage 1 hypertension: The HARVEST. Kidney Int 70: 578–584, 2006. [DOI] [PubMed] [Google Scholar]
- 285.Panchapakesan U, Pegg K, Gross S, Komala MG, Mudaliar H, Forbes J, Pollock C, Mather A. Effects of SGLT2 inhibition in human kidney proximal tubular cells–renoprotection in diabetic nephropathy? PLoS One 8: e54442, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Papademetriou V, Lovato L, Doumas M, Nylen E, Mottl A, Cohen RM, Applegate WB, Puntakee Z, Yale JF, Cushman WC. Chronic kidney disease and intensive glycemic control increase cardiovascular risk in patients with type 2 diabetes. Kidney Int 87: 649–659, 2015. [DOI] [PubMed] [Google Scholar]
- 287.Park TJ, Reznick J, Peterson BL, Blass G, Omerbasic D, Bennett NC, Kuich P, Zasada C, Browe BM, Hamann W, Applegate DT, Radke MH, Kosten T, Lutermann H, Gavaghan V, Eigenbrod O, Begay V, Amoroso VG, Govind V, Minshall RD, Smith ESJ, Larson J, Gotthardt M, Kempa S, Lewin GR. Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science 356: 307–311, 2017. [DOI] [PubMed] [Google Scholar]
- 288.Pascual JM, Wang D, Lecumberri B, Yang H, Mao X, Yang R, De Vivo DC. GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol 150: 627–633, 2004. [DOI] [PubMed] [Google Scholar]
- 289.Pereira-Moreira R, Muscelli E. Effect of Insulin on Proximal Tubules Handling of Glucose: A Systematic Review. J Diabetes Res 2020: 8492467, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Perkins BA, Cherney DZ, Partridge H, Soleymanlou N, Tschirhart H, Zinman B, Fagan NM, Kaspers S, Woerle HJ, Broedl UC, Johansen OE. Sodium-Glucose Cotransporter 2 Inhibition and Glycemic Control in Type 1 Diabetes: Results of an 8-Week Open-Label Proof-of-Concept Trial. Diabetes Care 37: 1480–1483, 2014. [DOI] [PubMed] [Google Scholar]
- 291.Perkovic V, Jardine M, Vijapurkar U, Meininger G. Renal effects of canagliflozin in type 2 diabetes mellitus. Curr Med Res Opin 31: 2219–2231, 2015. [DOI] [PubMed] [Google Scholar]
- 292.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de ZD, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, and Mahaffey KW. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med 380: 2295–2306, 2019. [DOI] [PubMed] [Google Scholar]
- 293.Persson P, Hansell P, Palm F. Reduced adenosine A2a receptor-mediated efferent arteriolar vasodilation contributes to diabetes-induced glomerular hyperfiltration. Kidney Int 87: 109–115, 2015. [DOI] [PubMed] [Google Scholar]
- 294.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol 25: 2028–2039, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Pfaff IL, Vallon V. Protein kinase C beta isoenzymes in diabetic kidneys and their relation to nephroprotective actions of the ACE inhibitor lisinopril. Kidney Blood Press Res 25: 329–340, 2002. [DOI] [PubMed] [Google Scholar]
- 296.Pfaff IL, Wagner HJ, Vallon V. Immunolocalization of protein kinase C isoenzymes alpha, beta1 and betaII in rat kidney. J Am Soc Nephrol 10: 1861–1873, 1999. [DOI] [PubMed] [Google Scholar]
- 297.Phillips AO, Steadman R, Morrisey K, Williams JD. Polarity of stimulation and secretion of transforming growth factor-beta 1 by cultured proximal tubular cells. Am J Pathol 150: 1101–1111, 1997. [PMC free article] [PubMed] [Google Scholar]
- 298.Powell DR, DaCosta CM, Gay J, Ding ZM, Smith M, Greer J, Doree D, Jeter-Jones S, Mseeh F, Rodriguez LA, Harris A, Buhring L, Platt KA, Vogel P, Brommage R, Shadoan MK, Sands AT, Zambrowicz B. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab 304: E117–E130, 2013. [DOI] [PubMed] [Google Scholar]
- 299.Powell DR, DaCosta CM, Smith M, Doree D, Harris A, Buhring L, Heydorn W, Nouraldeen A, Xiong W, Yalamanchili P, Mseeh F, Wilson A, Shadoan M, Zambrowicz B, Ding ZM. Effect of LX4211 on glucose homeostasis and body composition in preclinical models. J Pharmacol Exp Ther 350: 232–242, 2014. [DOI] [PubMed] [Google Scholar]
- 300.Pruijm M, Milani B, Pivin E, Podhajska A, Vogt B, Stuber M, Burnier M. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int 93: 932–940, 2018. [DOI] [PubMed] [Google Scholar]
- 301.Qiu H, Novikov A, Vallon V. Ketosis and diabetic ketoacidosis in response to SGLT2 inhibitors: Basic mechanisms and therapeutic perspectives. Diabetes Metab Res Rev 33: 5, 2017. [DOI] [PubMed] [Google Scholar]
- 302.Quamme GA, Freeman HJ. Evidence for a high-affinity sodium-dependent D-glucose transport system in the kidney. Am J Physiol 253: F151–F157, 1987. [DOI] [PubMed] [Google Scholar]
- 303.Quinn PG, Yeagley D. Insulin regulation of PEPCK gene expression: A model for rapid and reversible modulation. Curr Drug Targets Immune Endocr Metabol Disord 5: 423–437, 2005. [DOI] [PubMed] [Google Scholar]
- 304.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54: 3427–3434, 2005. [DOI] [PubMed] [Google Scholar]
- 305.Raivio KO, Kekomaki MP, Maenpaa PH. Depletion of liver adenine nucleotides induced by D-fructose. Dose-dependence and specificity of the fructose effect. Biochem Pharmacol 18: 2615–2624, 1969. [DOI] [PubMed] [Google Scholar]
- 306.Rajasekeran H, Lytvyn Y, Bozovic A, Lovshin JA, Diamandis E, Cattran D, Husain M, Perkins BA, Advani A, Reich HN, Kulasingam V, Cherney DZI. Urinary adenosine excretion in type 1 diabetes. Am J Physiol Renal Physiol 313: F184–F191, 2017. [DOI] [PubMed] [Google Scholar]
- 307.Rand EB, Depaoli AM, Davidson NO, Bell GI, Burant CF. Sequence, tissue distribution, and functional characterization of the rat fructose transporter GLUT5. Am J Physiol 264: G1169–G1176, 1993. [DOI] [PubMed] [Google Scholar]
- 308.Rasch R. Tubular lesions in streptozotocin-diabetic rats. Diabetologia 27: 32–37, 1984. [DOI] [PubMed] [Google Scholar]
- 309.Rebholz CM, Young BA, Katz R, Tucker KL, Carithers TC, Norwood AF, Correa A. Patterns of beverages consumed and risk of incident kidney disease. Clin J Am Soc Nephrol 14: 49–56, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Reinecke RM, Hauser PJ. Arterio venous blood sugar studies on the kidney of the eviscerated dog. Fed Proc 7: 99, 1948. [PubMed] [Google Scholar]
- 311.Ren Y, Garvin JL, Liu R, Carretero OA. Possible mechanism of efferent arteriole (Ef-Art) tubuloglomerular feedback. Kidney Int 71: 861–866, 2007. [DOI] [PubMed] [Google Scholar]
- 312.Rieg T, Masuda T, Gerasimova M, Mayoux E, Platt K, Powell DR, Thomson SC, Koepsell H, Vallon V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol 306: F188–F193, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Ritchie S, Waugh D. The pathology of Armanni-Ebstein diabetic nephropathy. Am J Pathol 33: 1035–1057, 1957. [PMC free article] [PubMed] [Google Scholar]
- 314.Rohde R, Deetjen P. Glucose reabsorption in the rat kidney. Micropuncture analysis of tubular glucose concentration during free flow. Pflugers Arch 302: 219–232, 1968. [DOI] [PubMed] [Google Scholar]
- 315.Roncal-Jimenez CA, Ishimoto T, Lanaspa MA, Milagres T, Hernando AA, Jensen T, Miyazaki M, Doke T, Hayasaki T, Nakagawa T, Marumaya S, Long DA, Garcia GE, Kuwabara M, Sanchez-Lozada LG, Kang DH, Johnson RJ. Aging-associated renal disease in mice is fructokinase dependent. Am J Physiol Renal Physiol 311: F722–F730, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Roncal Jimenez CA, Ishimoto T, Lanaspa MA, Rivard CJ, Nakagawa T, Ejaz AA, Cicerchi C, Inaba S, Le M, Miyazaki M, Glaser J, Correa-Rotter R, Gonzalez MA, Aragon A, Wesseling C, Sanchez-Lozada LG, Johnson RJ. Fructokinase activity mediates dehydration-induced renal injury. Kidney Int 86: 294–302, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Rosen S, Epstein FH, Brezis M. Determinants of intrarenal oxygenation: Factors in acute renal failure. Ren Fail 14: 321–325, 1992. [DOI] [PubMed] [Google Scholar]
- 318.Saab KR, Kendrick J, Yracheta JM, Lanaspa MA, Pollard M, Johnson RJ. New insights on the risk for cardiovascular disease in African Americans: The role of added sugars. J Am Soc Nephrol 26: 247–257, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Sabolic I, Vrhovac I, Eror DB, Gerasimova M, Rose M, Breljak D, Ljubojevic M, Brzica H, Sebastiani A, Thal SC, Sauvant C, Kipp H, Vallon V, Koepsell H. Expression of Na+-D-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am J Physiol Cell Physiol 302: C1174–C1188, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Sala-Rabanal M, Hirayama BA, Ghezzi C, Liu J, Huang SC, Kepe V, Koepsell H, Yu A, Powell DR, Thorens B, Wright EM, Barrio JR. Revisiting the physiological roles of SGLTs and GLUTs using positron emission tomography in mice. J Physiol 594: 4425–4438, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Saldana TM, Basso O, Darden R, Sandler DP. Carbonated beverages and chronic kidney disease. Epidemiology 18: 501–506, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Salomon LL, Lanza FL, Smith DE. Renal conversion of fructose to glucose. Am J Physiol 200: 871–877, 1961. [DOI] [PubMed] [Google Scholar]
- 323.Sanchez-Lozada LG, Andres-Hernando A, Garcia-Arroyo FE, Cicerchi C, Li N, Kuwabara M, Roncal-Jimenez CA, Johnson RJ, Lanaspa MA. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J Biol Chem 294: 4272–4281, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia M, Garcia-Arroyo F, Soto V, Cruz-Robles D, Nakagawa T, Yu MA, Kang DH, Johnson RJ. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Experimental nephrology 121: e71–e78, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Sanchez-Lozada LG, Tapia E, Jimenez A, Bautista P, Cristobal M, Nepomuceno T, Soto V, Avila-Casado C, Nakagawa T, Johnson RJ, Herrera-Acosta J, Franco M. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am J Physiol Renal Physiol 292: F423–F429, 2007. [DOI] [PubMed] [Google Scholar]
- 326.Santer R, Calado J. Familial renal glucosuria and SGLT2: From a Mendelian trait to a therapeutic target. Clin J Am Soc Nephrol 5: 133–141, 2010. [DOI] [PubMed] [Google Scholar]
- 327.Santer R, Groth S, Kinner M, Dombrowski A, Berry GT, Brodehl J, Leonard JV, Moses S, Norgren S, Skovby F, Schneppenheim R, Steinmann B, Schaub J. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi-Bickel syndrome. Hum Genet 110: 21–29, 2002. [DOI] [PubMed] [Google Scholar]
- 328.Santer R, Schneppenheim R, Suter D, Schaub J, Steinmann B. Fanconi-Bickel syndrome–the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature. Eur J Pediatr 157: 783–797, 1998. [DOI] [PubMed] [Google Scholar]
- 329.Saponaro C, Muhlemann M, Acosta-Montalvo A, Piron A, Gmyr V, Delalleau N, Moerman E, Thevenet J, Pasquetti G, Coddeville A, Cnop M, Kerr-Conte J, Staels B, Pattou F, Bonner C. Inter-individual heterogeneity of SGLT2 expression and function in human pancreatic islets. Diabetes, 2020. [DOI] [PubMed] [Google Scholar]
- 330.Sasaki M, Sasako T, Kubota N, Sakurai Y, Takamoto I, Kubota T, Inagi R, Seki G, Goto M, Ueki K, Nangaku M, Jomori T, Kadowaki T. Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubules of the kidney. Diabetes 66: 2339–2350, 2017. [DOI] [PubMed] [Google Scholar]
- 331.Satirapoj B, Korkiatpitak P, Supasyndh O. Effect of sodium-glucose cotransporter 2 inhibitor on proximal tubular function and injury in patients with type 2 diabetes: A randomized controlled trial. Clin Kidney J 12: 326–332, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Sato Y, Feig DI, Stack AG, Kang DH, Lanaspa MA, Ejaz AA, Sanchez-Lozada LG, Kuwabara M, Borghi C, Johnson RJ. The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nat Rev Nephrol 15: 767–775, 2019. [DOI] [PubMed] [Google Scholar]
- 333.Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl 77: S13–S18, 2000. [DOI] [PubMed] [Google Scholar]
- 334.Schmidt C, Hocherl K, Schweda F, Kurtz A, Bucher M. Regulation of renal sodium transporters during severe inflammation. J Am Soc Nephrol 18: 1072–1083, 2007. [DOI] [PubMed] [Google Scholar]
- 335.Schmidt U, Guder WG. Sites of enzyme activity along the nephron. Kidney Int 9: 233–242, 1976. [DOI] [PubMed] [Google Scholar]
- 336.Schmidt U, Marosvari I, Dubach UC. Renal metabolism of glucose: Anatomical sites of hexokinase activity in the rat nephron. FEBS Lett 53: 26–28, 1975. [DOI] [PubMed] [Google Scholar]
- 337.Schnermann J, Briggs JP. Single nephron comparison of the effect of loop of Henle flow on filtration rate and pressure in control and angiotensin II-infused rats. Miner Electrolyte Metab 15: 103–107, 1989. [PubMed] [Google Scholar]
- 338.Schulte K, Kunter U, Moeller MJ. The evolution of blood pressure and the rise of mankind. Nephrol Dial Transplant 30: 713–723, 2015. [DOI] [PubMed] [Google Scholar]
- 339.Schurek HJ, Johns O. Is tubuloglomerular feedback a tool to prevent nephron oxygen deficiency? Kidney Int 51: 386–392, 1997. [DOI] [PubMed] [Google Scholar]
- 340.Scott TW, Setchell BP, Bassett JM. Characterization and metabolism of ovine foetal lipids. Biochem J 104: 1040–1047, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Seidner G, Alvarez MG, Yeh JI, O’Driscoll KR, Klepper J, Stump TS, Wang D, Spinner NB, Birnbaum MJ, De Vivo DC. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet 18: 188–191, 1998. [DOI] [PubMed] [Google Scholar]
- 342.Sha S, Devineni D, Ghosh A, Polidori D, Chien S, Wexler D, Shalayda K, Demarest K, Rothenberg P. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab 13: 669–672, 2011. [DOI] [PubMed] [Google Scholar]
- 343.Shanley PF, Brezis M, Spokes K, Silva P, Epstein FH, Rosen S. Transport-dependent cell injury in the S3 segment of the proximal tubule. Kidney Int 29: 1033–1037, 1986. [DOI] [PubMed] [Google Scholar]
- 344.Shen L, Kristensen SL, Bengtsson O, Bohm M, de Boer RA, Docherty KF, Inzucchi SE, Katova T, Kober L, Kosiborod MN, Langkilde AM, Lindholm D, Martinez MFA, O’Meara E, Nicolau JC, Petrie MC, Ponikowski P, Sabatine MS, Schou M, Sjostrand M, Solomon SD, Jhund PS, McMurray JJV. Dapagliflozin in HFrEF patients treated with mineralocorticoid receptor antagonists: An analysis of DAPA-HF. JACC Heart Fail 9: 254–264, 2021. [DOI] [PubMed] [Google Scholar]
- 345.Shepard BD, Pluznick JL. Saving the sweetness: Renal glucose handling in health and disease. Am J Physiol Renal Physiol 313: F55–F61, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Shinozaki K, Kashiwagi A, Nishio Y, Okamura T, Yoshida Y, Masada M, Toda N, Kikkawa R. Abnormal biopterin metabolism is a major cause of impaired endothelium-dependent relaxation through nitric oxide/O2- imbalance in insulin-resistant rat aorta. Diabetes 48: 2437–2445, 1999. [DOI] [PubMed] [Google Scholar]
- 347.Shiota M, Galassetti P, Monohan M, Neal DW, Cherrington AD. Small amounts of fructose markedly augment net hepatic glucose uptake in the conscious dog. Diabetes 47: 867–873, 1998. [DOI] [PubMed] [Google Scholar]
- 348.Shoham DA, Durazo-Arvizu R, Kramer H, Luke A, Vupputuri S, Kshirsagar A, Cooper RS. Sugary soda consumption and albuminuria: Results from the National Health and Nutrition Examination Survey, 1999–2004. PLoS One 3: e3431, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Singh AK, Amlal H, Haas PJ, Dringenberg U, Fussell S, Barone SL, Engelhardt R, Zuo J, Seidler U, Soleimani M. Fructose-induced hypertension: Essential role of chloride and fructose absorbing transporters PAT1 and Glut5. Kidney Int 74: 438–447, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.So A, Thorens B. Uric acid transport and disease. J Clin Invest 120: 1791–1799, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, Fujisaka S, Serra D, Herrero L, Willoughby J, Fitzgerald K, Ilkayeva O, Newgard CB, Gibson BW, Schilling B, Cohen DE, Kahn CR. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab 30: 735–753 e734, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Solini A, Rossi C, Mazzanti CM, Proietti A, Koepsell H, Ferrannini E. Sodium-glucose co-transporter (SGLT)2 and SGLT1 renal expression in patients with type 2 diabetes. Diabetes Obes Metab 19: 1289–1294, 2017. [DOI] [PubMed] [Google Scholar]
- 353.Soncrant T, Komnenov D, Beierwaltes WH, Chen H, Wu M, Rossi NF. Bilateral renal cryodenervation decreases arterial pressure and improves insulin sensitivity in fructose-fed Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol 315: R529–R538, 2018. [DOI] [PubMed] [Google Scholar]
- 354.Song P, Huang W, Onishi A, Patel R, Kim Y, van Ginkel C, Fu Y, Freeman B, Koepsell H, Thomson SC, Liu R, Vallon V. Knockout of Na-glucose-cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase-1 in macula densa and glomerular hyperfiltration. Am J Physiol Renal Physiol 317: F207–F217, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Song Z, Roncal-Jimenez CA, Lanaspa-Garcia MA, Oppelt SA, Kuwabara M, Jensen T, Milagres T, Andres-Hernando A, Ishimoto T, Garcia GE, Johnson G, MacLean PS, Sanchez-Lozada LG, Tolan DR, Johnson RJ. Role of fructose and fructokinase in acute dehydration-induced vasopressin gene expression and secretion in mice. J Neurophysiol 117: 646–654, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Sotak M, Marks J, Unwin RJ. Putative tissue location and function of the SLC5 family member SGLT3. Exp Physiol 102: 5–13, 2017. [DOI] [PubMed] [Google Scholar]
- 357.Soty M, Penhoat A, Amigo-Correig M, Vinera J, Sardella A, Vullin-Bouilloux F, Zitoun C, Houberdon I, Mithieux G. A gut-brain neural circuit controlled by intestinal gluconeogenesis is crucial in metabolic health. Mol Metab 4: 106–117, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Srivastava SP, Goodwin JE, Kanasaki K, Koya D. Metabolic reprogramming by N-acetyl-seryl-aspartyl-lysyl-proline protects against diabetic kidney disease. Br J Pharmacol 177: 3691–3711, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Srivastava SP, Li J, Kitada M, Fujita H, Yamada Y, Goodwin JE, Kanasaki K, Koya D. SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis 9: 997, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Stenvinkel P, Jani AH, Johnson RJ. Hibernating bears (Ursidae): Metabolic magicians of definite interest for the nephrologist. Kidney Int 83: 207–212, 2013. [DOI] [PubMed] [Google Scholar]
- 361.Suga T, Kikuchi O, Kobayashi M, Matsui S, Yokota-Hashimoto H, Wada E, Kohno D, Sasaki T, Takeuchi K, Kakizaki S, Yamada M, Kitamura T. SGLT1 in pancreatic alpha cells regulates glucagon secretion in mice, possibly explaining the distinct effects of SGLT2 inhibitors on plasma glucagon levels. Mol Metab 19: 1–12, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Sugawara-Yokoo M, Suzuki T, Matsuzaki T, Naruse T, Takata K. Presence of fructose transporter GLUT5 in the S3 proximal tubules in the rat kidney. Kidney Int 56: 1022–1028, 1999. [DOI] [PubMed] [Google Scholar]
- 363.Sullivan MA, Forbes JM. Glucose and glycogen in the diabetic kidney: Heroes or villains? EBioMedicine 47: 590–597, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Sun S, Ning X, Zhang Y, Lu Y, Nie Y, Han S, Liu L, Du R, Xia L, He L, Fan D. Hypoxia-inducible factor-1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int 75: 1278–1287, 2009. [DOI] [PubMed] [Google Scholar]
- 365.Sundborn G, Thornley S, Merriman TR, Lang B, King C, Lanaspa MA, Johnson RJ. Are liquid sugars different from solid sugar in their ability to cause metabolic syndrome? Obesity (Silver Spring) 27: 879–887, 2019. [DOI] [PubMed] [Google Scholar]
- 366.Swe MT, Pongchaidecha A, Chatsudthipong V, Chattipakorn N, Lungkaphin A. Molecular signaling mechanisms of renal gluconeogenesis in nondiabetic and diabetic conditions. J Cell Physiol 234: 8134–8151, 2019. [DOI] [PubMed] [Google Scholar]
- 367.Szablewski L. Distribution of glucose transporters in renal diseases. J Biomed Sci 24: 64, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Takagi S, Li J, Takagaki Y, Kitada M, Nitta K, Takasu T, Kanasaki K, Koya D. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J Diabetes Investig 9: 1025–1032, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Tazawa S, Yamato T, Fujikura H, Hiratochi M, Itoh F, Tomae M, Takemura Y, Maruyama H, Sugiyama T, Wakamatsu A, Isogai T, Isaji M. SLC5A9/SGLT4, a new Na+-dependent glucose transporter, is an essential transporter for mannose, 1, 5-anhydro-D-glucitol, and fructose. Life Sci 76: 1039–1050, 2005. [DOI] [PubMed] [Google Scholar]
- 370.Thach TT, Wu C, Hwang KY, Lee SJ. Azelaic acid induces mitochondrial biogenesis in skeletal muscle by activation of olfactory receptor 544. Front Physiol 11: 329, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Thomson S, Vallon V, Blantz RC. Asymmetry of tubuloglomerular feedback effector mechanism with respect to ambient tubular flow. Am J Physiol 271: F1123–F1130, 1996. [DOI] [PubMed] [Google Scholar]
- 372.Thomson SC, Deng A, Komine N, Hammes JS, Blantz RC, Gabbai FB. Early diabetes as a model for testing the regulation of juxtaglomerular NOS I. Am J Physiol Renal Physiol 287: F732–F738, 2004. [DOI] [PubMed] [Google Scholar]
- 373.Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, Singh P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol 302: R75–R83, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Thomson SC, Vallon V. Effects of SGLT2 inhibitor and dietary NaCl on glomerular hemodynamics assessed by micropuncture in diabetic rats. Am J Physiol Renal Physiol 320: F761–F777, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Thomson SC, Vallon V. Renal Effects of Sodium-Glucose Co-Transporter Inhibitors. Am J Cardiol 124 (Suppl 1): S28–S35, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Thorens B, Lodish HF, Brown D. Differential localization of two glucose transporter isoforms in rat kidney. Am J Physiol 259: C286–C294, 1990. [DOI] [PubMed] [Google Scholar]
- 377.Tiku A, Johnson DW, Badve SV. Recent evidence on the effect of urate-lowering treatment on the progression of kidney disease. Curr Opin Nephrol Hypertens 30: 346–352, 2021. [DOI] [PubMed] [Google Scholar]
- 378.Tiwari S, Riazi S, Ecelbarger CA. Insulin’s impact on renal sodium transport and blood pressure in health, obesity, and diabetes. Am J Physiol Renal Physiol 293: F974–F984, 2007. [DOI] [PubMed] [Google Scholar]
- 379.Tojo A, Onozato ML, Fujita T. Role of macula densa neuronal nitric oxide synthase in renal diseases. Med Mol Morphol 39: 2–7, 2006. [DOI] [PubMed] [Google Scholar]
- 380.Tolins JP, Shultz PJ, Raij L, Brown DM, Mauer SM. Abnormal renal hemodynamic response to reduced renal perfusion pressure in diabetic rats: Role of NO. Am J Physiol 265: F886–F895, 1993. [DOI] [PubMed] [Google Scholar]
- 381.Tomita I, Kume S, Sugahara S, Osawa N, Yamahara K, Yasuda-Yamahara M, Takeda N, Chin-Kanasaki M, Kaneko T, Mayoux E, Mark M, Yanagita M, Ogita H, Araki SI, Maegawa H. SGLT2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mTORC1 inhibition. Cell Metab 32: 404–419, 2020. [DOI] [PubMed] [Google Scholar]
- 382.Toyoki D, Shibata S, Kuribayashi-Okuma E, Xu N, Ishizawa K, Hosoyamada M, Uchida S. Insulin stimulates uric acid reabsorption via regulating urate transporter 1 and ATP-binding cassette subfamily G member 2. Am J Physiol Renal Physiol 313: F826–F834, 2017. [DOI] [PubMed] [Google Scholar]
- 383.Turner RJ, Moran A. Heterogeneity of sodium-dependent D-glucose transport sites along the proximal tubule: Evidence from vesicle studies. Am J Physiol 242: F406–F414, 1982. [DOI] [PubMed] [Google Scholar]
- 384.Uchida S, Endou H. Substrate specificity to maintain cellular ATP along the mouse nephron. Am J Physiol 255: F977–F983, 1988. [DOI] [PubMed] [Google Scholar]
- 385.Umino H, Hasegawa K, Minakuchi H, Muraoka H, Kawaguchi T, Kanda T, Tokuyama H, Wakino S, Itoh H. High basolateral glucose increases sodium-glucose cotransporter 2 and reduces Sirtuin-1 in renal tubules through glucose transporter-2 detection. Sci Rep 8: 6791, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, Jancev M, Hollmann MW, Weber NC, Coronel R, Zuurbier CJ. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 61: 722–726, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Uthman L, Nederlof R, Eerbeek O, Baartscheer A, Schumacher C, Buchholtz N, Hollmann MW, Coronel R, Weber NC, Zuurbier CJ. Delayed ischaemic contracture onset by empagliflozin associates with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc Res 115: 1533–1545, 2019. [DOI] [PubMed] [Google Scholar]
- 388.Vallon V. Glucose transporters in the kidney in health and disease. Pflugers Arch 472: 1345–1370, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Vallon V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu Rev Med 66: 255–270, 2015. [DOI] [PubMed] [Google Scholar]
- 390.Vallon V. Molecular determinants of renal glucose transport. Am J Physiol Cell Physiol 300: C6–C8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300: R1009–R1022, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Vallon V. Tubular transport in acute kidney injury: relevance for diagnosis. Prognosis and Intervention. Nephron 134: 160–166, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Vallon V, Blantz RC, Thomson S. Glomerular hyperfiltration and the salt paradox in early type 1 diabetes mellitus: A tubulo-centric view. J Am Soc Nephrol 14: 530–537, 2003. [DOI] [PubMed] [Google Scholar]
- 394.Vallon V, Blantz RC, Thomson S. Homeostatic efficiency of tubuloglomerular feedback is reduced in established diabetes mellitus in rats. Am J Physiol 269: F876–F883, 1995. [DOI] [PubMed] [Google Scholar]
- 395.Vallon V, Broer S, Nigam SK. Renal handling of organic solutes. In: Yu AS, Chertow G, Luyckx V, Marsden PA, Skorecki K, Taal MW, editors. Brenner & Rector’s The Kidney. Philadelphia: Elsevier, 2020, p. 218–246. [Google Scholar]
- 396.Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, Koepsell H, Thomson SC, Rieg T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 306: F194–F204, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Vallon V, Grahammer F, Richter K, Bleich M, Lang F, Barhanin J, Volkl H, Warth R. Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J Am Soc Nephrol 12: 2003–2011, 2001. [DOI] [PubMed] [Google Scholar]
- 398.Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K, Lang F. KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci U S A 102: 17864–17869, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Vallon V, Komers R. Pathophysiology of the diabetic kidney. Compr Physiol 1: 1175–1232, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev 86: 901–940, 2006. [DOI] [PubMed] [Google Scholar]
- 401.Vallon V, Osswald H. Dipyridamole prevents diabetes-induced alterations of kidney function in rats. Naunyn Schmiedebergs Arch Pharmacol 349: 217–222, 1994. [DOI] [PubMed] [Google Scholar]
- 402.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J Am Soc Nephrol 10: 2569–2576, 1999. [DOI] [PubMed] [Google Scholar]
- 404.Vallon V, Richter K, Huang DY, Rieg T, Schnermann J. Functional consequences at the single-nephron level of the lack of adenosine A1 receptors and tubuloglomerular feedback in mice. Pflugers Arch 448: 214–221, 2004. [DOI] [PubMed] [Google Scholar]
- 405.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Vallon V, Schwark JR, Richter K, Hropot M. Role of Na(+)/H(+) exchanger NHE3 in nephron function: Micropuncture studies with S3226, an inhibitor of NHE3. Am J Physiol Renal Physiol 278: F375–F379, 2000. [DOI] [PubMed] [Google Scholar]
- 407.Vallon V, Thomson S. Inhibition of local nitric oxide synthase increases homeostatic efficiency of tubuloglomerular feedback. Am J Physiol 269: F892–F899, 1995. [DOI] [PubMed] [Google Scholar]
- 408.Vallon V, Thomson SC. Renal function in diabetic disease models: The tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol 74: 351–375, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 60: 215–225, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol 16: 317–336, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Vallon V, Traynor T, Barajas L, Huang YG, Briggs JP, Schnermann J. Feedback control of glomerular vascular tone in neuronal nitric oxide synthase knockout mice. J Am Soc Nephrol 12: 1599–1606, 2001. [DOI] [PubMed] [Google Scholar]
- 412.Vallon V, Unwin R, Inscho EW, Leipziger J, Kishore BK. Extracellular nucleotides and P2 receptors in renal function. Physiol Rev 100: 211–269, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Vallon V, Verma S. Effects of SGLT2 inhibitors on kidney and cardiovascular function. Annu Rev Physiol 83: 503–528, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.van Bommel EJM, Muskiet MHA, van Baar MJB, Tonneijck L, Smits MM, Emanuel AL, Bozovic A, Danser AHJ, Geurts F, Hoorn EJ, Touw DJ, Larsen EL, Poulsen HE, Kramer MHH, Nieuwdorp M, Joles JA, van Raalte DH. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int 97: 202–212, 2020. [DOI] [PubMed] [Google Scholar]
- 415.Van Schaftingen E, Detheux M, Veiga da Cunha M. Short-term control of glucokinase activity: Role of a regulatory protein. FASEB J 8: 414–419, 1994. [DOI] [PubMed] [Google Scholar]
- 416.Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, Castelo-Branco RC, Pastor-Soler N, Arranz CT, Yu ASL, McDonough AA. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, Dyck JE, Uddin GM, Oudit GY, Mayoux E, Lehrke M, Marx N, Lopaschuk GD. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl Sci 3: 575–587, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Vrhovac I, Balen ED, Klessen D, Burger C, Breljak D, Kraus O, Radovic N, Jadrijevic S, Aleksic I, Walles T, Sauvant C, Sabolic I, Koepsell H. Localizations of Na-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 467: 1881–1898, 2015. [DOI] [PubMed] [Google Scholar]
- 419.Walker RW, Goran MI. Laboratory determined sugar content and composition of commerical infant formulas, baby foods and common grocery items targeted to children. Nutrients 7: 5850–5867, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Wang J, Shibayama Y, Kobori H, Liu Y, Kobara H, Masaki T, Wang Z, Nishiyama A. High glucose augments angiotensinogen in human renal proximal tubular cells through hepatocyte nuclear factor-5. PLoS One 12: e0185600, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Wang XX, Levi J, Luo Y, Myakala K, Herman-Edelstein M, Qiu L, Wang D, Peng Y, Grenz A, Lucia S, Dobrinskikh E, D’Agati VD, Koepsell H, Kopp JB, Rosenberg A, Levi M. SGLT2 expression is increased in human diabetic nephropathy: SGLT2 inhibition decreases renal lipid accumulation, inflammation and the development of nephropathy in diabetic mice. J Biol Chem 292: 5335–5348, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Mattheus EM, Johansen OE, Woerle HJ, Broedl UC, Zinman B. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 375: 323–334, 2016. [DOI] [PubMed] [Google Scholar]
- 423.Warburg O. On respiratory impairment in cancer cells. Science 124: 269–270, 1956. [PubMed] [Google Scholar]
- 424.Warburg O. On the origin of cancer cells. Science 123: 309–314, 1956. [DOI] [PubMed] [Google Scholar]
- 425.Watanabe S, Kang DH, Feng L, Nakagawa T, Kanellis J, Lan H, Mazzali M, Johnson RJ. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 40: 355–360, 2002. [DOI] [PubMed] [Google Scholar]
- 426.Wei Q, Su J, Dong G, Zhang M, Huo Y, Dong Z. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am J Physiol Renal Physiol 316: F1162–F1172, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Weinberg JM, Molitoris BA. Illuminating mitochondrial function and dysfunction using multiphoton technology. J Am Soc Nephrol 20: 1164–1166, 2009. [DOI] [PubMed] [Google Scholar]
- 428.Wells RG, Pajor AM, Kanai Y, Turk E, Wright EM, Hediger MA. Cloning of a human kidney cDNA with similarity to the sodium-glucose cotransporter. Am J Physiol 263: F459–F465, 1992. [DOI] [PubMed] [Google Scholar]
- 429.Wen SF. Micropuncture studies of glucose transport in the dog: Mechanism of renal glucosuria. Am J Physiol 231: 468–475, 1976. [DOI] [PubMed] [Google Scholar]
- 430.White CE, Piper EL, Noland PR, Daniels LB. Fructose utilization for nucleic acid synthesis in the fetal pig. J Anim Sci 55: 73–76, 1982. [DOI] [PubMed] [Google Scholar]
- 431.Wilcox CS, Welch WJ, Murad F, Gross SS, Taylor G, Levi R, Schmidt HH. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci U S A 89: 11993–11997, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Witkowska K, Smith KM, Yao SY, Ng AM, O’Neill D, Karpinski E, Young JD, Cheeseman CI. Human SLC2A9a and SLC2A9b isoforms mediate electrogenic transport of urate with different characteristics in the presence of hexoses. Am J Physiol Renal Physiol 303: F527–F539, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): Expanded families of sugar transport proteins. Br J Nutr 89: 3–9, 2003. [DOI] [PubMed] [Google Scholar]
- 434.Woods HF, Eggleston LV, Krebs HA. The cause of hepatic accumulation of fructose 1-phosphate on fructose loading. Biochem J 119: 501–510, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes 60: 2354–2369, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Wright EM. Glucose transport families SLC5 and SLC50. Mol Aspects Med 34: 183–196, 2013. [DOI] [PubMed] [Google Scholar]
- 437.Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol 280: F10–F18, 2001. [DOI] [PubMed] [Google Scholar]
- 438.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 91: 733–794, 2011. [DOI] [PubMed] [Google Scholar]
- 439.Wright EM, Turk E. The sodium/glucose cotransport family SLC5. Pflugers Arch 447: 510–518, 2004. [DOI] [PubMed] [Google Scholar]
- 440.Wu C, Hwang SH, Jia Y, Choi J, Kim YJ, Choi D, Pathiraja D, ChoiI G, Koo SH, Lee SJ. Olfactory receptor 544 reduces adiposity by steering fuel preference toward fats. J Clin Invest 127: 4118–4123, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Yale JF, Bakris G, Cariou B, Yue D, David-Neto E, Xi L, Figueroa K, Wajs E, Usiskin K, Meininger G. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab 15: 463–473, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Yamaoka T, Nishimura C, Yamashita K, Itakura M, Yamada T, Fujimoto J, Kokai Y. Acute onset of diabetic pathological changes in transgenic mice with human aldose reductase cDNA. Diabetologia 38: 255–261, 1995. [DOI] [PubMed] [Google Scholar]
- 443.Yin XN, Wang J, Cui LF, Fan WX. Enhanced glycolysis in the process of renal fibrosis aggravated the development of chronic kidney disease. Eur Rev Med Pharmacol Sci 22: 4243–4251, 2018. [DOI] [PubMed] [Google Scholar]
- 444.You G, Lee WS, Barros EJ, Kanai Y, Huo TL, Khawaja S, Wells RG, Nigam SK, Hediger MA. Molecular characteristics of Na(+)-coupled glucose transporters in adult and embryonic rat kidney. J Biol Chem 270: 29365–29371, 1995. [DOI] [PubMed] [Google Scholar]
- 445.Zapata-Morales JR, Galicia-Cruz OG, Franco M, Martinez YM. Hypoxia-inducible factor-1alpha (HIF-1alpha) protein diminishes sodium glucose transport 1 (SGLT1) and SGLT2 protein expression in renal epithelial tubular cells (LLC-PK1) under hypoxia. J Biol Chem 289: 346–357, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RHM, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Sabatine MS. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 393: 31–39, 2019. [DOI] [PubMed] [Google Scholar]
- 447.Zenner ZP, Gordish KL, Beierwaltes WH. Free radical scavenging reverses fructose-induced salt-sensitive hypertension. Integr Blood Press Control 11: 1–9, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Zhang G, Darshi M, Sharma K. The Warburg effect in diabetic kidney disease. Semin Nephrol 38: 111–120, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Zhang J, Wei J, Jiang S, Xu L, Wang L, Cheng F, Buggs J, Koepsell H, Vallon V, Liu R. Macula densa SGLT1-NOS1-TGF pathway – a new mechanism for glomerular hyperfiltration during hyperglycemia. J Am Soc Nephrol 30: 578–593, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Zhang SL, Filep JG, Hohman TC, Tang SS, Ingelfinger JR, Chan JS. Molecular mechanisms of glucose action on angiotensinogen gene expression in rat proximal tubular cells. Kidney Int 55: 454–464, 1999. [DOI] [PubMed] [Google Scholar]
- 451.Zhang Y, Nakano D, Guan Y, Hitomi H, Uemura A, Masaki T, Kobara H, Sugaya T, Nishiyama A. A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor-dependent pathway after renal injury in mice. Kidney Int 94: 524–535, 2018. [DOI] [PubMed] [Google Scholar]
- 452.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng X, Trefely S, Fernandez S, Carrer A, Miller KD, Schug ZT, Snyder NW, Gade TP, Titchenell PM, Rabinowitz JD, Wellen KE. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579: 586–591, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Zierler K Whole body glucose metabolism. Am J Physiol 276: E409–E426, 1999. [DOI] [PubMed] [Google Scholar]
- 454.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373: 2117–2128, 2015. [DOI] [PubMed] [Google Scholar]