

Abstract
Over 40% of veterans from the Persian Gulf War (GW) (1990-1991) suffer from Gulf War Illness (GWI). Thirty years since the GW, the exposure and mechanism contributing to GWI remain unclear. One possible exposure that has been attributed to GWI are chemical warfare agents (CWA). While there are treatments for isolated symptoms of GWI, the number of respiratory and cognitive/neurological issues continues to rise with minimum treatment options. This issue doesn’t only affect veterans of the GW, importantly these chronic multisymptom illnesses are also growing amongst veterans who have served in the Afghanistan-Iraq war. What both wars have in common are their regions and inhaled exposures. In this review, we will describe the CWA exposures such as sarin, cyclosarin, and mustard gas in both wars and discuss the various respiratory and neurocognitive issues experienced by veterans. We will bridge the respiratory and neurological symptoms experienced to the various potential mechanisms described for each CWA provided with the most up-to-date models and hypotheses.
Keywords: Chemical Warfare Agents, Sarin, Cyclosarin, Sulfur Mustard, Gulf War Illness, Chronic Multisymptom Illness, Respiratory, Neurological effects, Organophosphorus compounds, Nitrogen Mustard, CEES
Introduction
In August 1990, over 700,000 U.S. troops were deployed for the Persian Gulf War (GW) to serve in combat missions: Operation Desert Shield and Desert Storm1. Upon returning from the GW, veterans experienced a collection of chronic multisymptomatic health complications within a year2 . These symptoms included fatigue, headache, memory problems, muscle/joint pain, diarrhea, dyspepsia/indigestion, terminal tumors, skin conditions, arthritis/ joint issues, gastrointestinal problems, respiratory complications, post-traumatic stress disorder, and chronic fatigue syndrome2. The combination of these symptoms is better known today as Gulf War Illness (GWI) or Chronic Multisymptom Illness (CMI).
Diagnosis Criteria for Gulf War Illness
GWI is diagnosed using either the Center for Disease Control’s (CDC) CMI criteria or the Kansas GWI Criteria. The CDC CMI criteria require that veterans have two of the following chronic symptoms, lasting for more than six months: pain, fatigue, mood swings, and alterations to cognition3,4. The Kansas GWI criteria is the stricter of the two, defined as having at least one chronic symptom in three of the following classifications: fatigue/sleep problems, pain, neurologic/cognitive/mood symptoms, gastrointestinal symptoms, respiratory symptoms, and skin symptoms3,4. The U.S Department of Veteran Affairs (VA) and the Department of Defense (DoD) are working together to merge both these criteria to create uniform guidelines for clinical and research practices. Currently, the VA guidelines for diagnosing GWI are based on the time and location of service during exposure, types of exposure, potential exposures contributing to symptoms, and symptoms persisting for at least six months resulting in a disability rating of 10% or more5.
Prevalence of Chronic Respiratory and Neurological Symptoms of GWI
Approximately 40% of veterans who served in the GW have been diagnosed with GWI3,4. GWI progression has been well documented in the Ft. Devens Cohort (FDC), one of the longest studied GWI cohorts (1991)6. This longitudinal study compared the prevalence of nine chronic medical conditions in 2013 and 20146. The study results found a significant association between self-reported chemical/biological weapons exposure and chronic diseases such as asthma and bronchitis6,7. FDC men were approximately two times more likely (OR=1.987) to report having asthma compared to the general population (National Health and Nutrition Examination Survey; NHANES) and over three times more likely (OR = 3.175) to report chronic bronchitis compared to the general population as of 20136.
In addition to respiratory issues, veterans also reported an array of neurological problems that have been associated with exposures during the GW 8–10. GW veterans also have a higher incidence of amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), brain cancer, repeated seizures, neuritis, chronic migraine headaches, chronic cognitive disorders, and mood-related problems compared to the general population11–14. Thirty- years since the GW, the VA has seen a rise in GWI diagnosis (25% to ~35%), and a rise in veterans experiencing various respiratory and neurological complications2,4,5,15.
Sex differences and sex-specific effects have been observed in GWI sufferers. Male GW veterans have reported higher rates of sexual dysfunction and genital and bladder problems than non-GW veterans 16 . Female veterans’ health outcomes have been examined using the GW veterans from the VA Cooperative Studies Program 585 GW Era Cohort and Biorepository (GWECB)6,17,18. In 2016, the GWECB found that GW-deployed women were significantly more likely to report cognitive (17% excess prevalence rate), neurological (15% excess prevalence rate), mood (20% excess prevalence rate), and respiratory issues (19% excess prevalence rate) compared to non-deployed GW-era women veterans6,17. Women GW veterans also have higher rates of birth defects in their children and other reproductive issues than their non-GW veteran counterparts 19. There is evidence that sex differences exist in GWI pathology and in the mechanisms driving it. GW-deployed women had a higher prevalence of most GWI symptoms than GW-deployed men, along with having higher rates of mild-to-moderate or severe GWI 17. Plasma lipid profiles differ significantly between male and female veterans with GWI 20. Plasma levels of central nervous system (CNS) autoantibodies were more elevated in men with GWI than in women with GWI 21. Homeostatic modeling using data from veterans with GWI indicated that men and women with GWI have different homeostatic states, with men having hypercortisolism, low testosterone, and a disposition towards Th1 immune activation while women had hypocortisolism, high estrogen, and a disposition towards Th2 immune activation 22 . Sex differences also appear in animal models of GWI. A mouse model of GWI saw more cognitive impairment and increased CNS immune cell activation and inflammation in females compared to males 23. Interestingly, this same study saw more greatly increased markers of peripheral inflammation in males than in females 23. Another study using mice found that pyridostigmine bromide (PB), a reversible acetylcholinesterase inhibitor used in the Gulf War as an anti-nerve agent drug 24, increased fecal pellet production and delayed colonic transit in females, but not males 25. This study also found that prior exposure to PB altered the effects of later treatment with palmitoylethanolamide, an anti-inflammatory, in sex-specific ways, with females experiencing an increase in myenteric neurons and muscarinic M3 receptors while males did not 25. The neuroimmune effects of PB exposure were found to be more immediate in males, but delayed in females in a mouse model of GWI 26. These animal studies have been summarized in Table 1..
Table 1:
Studies on the toxic effect of pyridostigmine bromide.
| Pyridostigmine Bromide | |||
|---|---|---|---|
| Study (Ref #) | Model | Exposure | Results |
| Bryant et al., 2021 (23) | Male and female C57BL/6 mice (WT, NLRP3−/−, and STINGgt/gt) | 0.7 mg/kg PB and 200 mg/kg Per in 50 mL DMSO daily for 10 days | Greater neuroinflammation and cognitive impairment in female mice; greater peripheral inflammation in males |
| Hernandez et al., 2020 (25) | Male and female C57BL/6 mice | 90 mg/mL PB in drinking water for 7 days; 0.07 mg/mL PEA in drinking water for one month | PB alone increased fecal pellet production and colonic transit time only in females, PB+PEA increased myenteric neurons and muscarinic M3 receptors only in females |
| Hernandez et al., 2019 (26) | Male and female C57BL/6 mice | 9 or 90 mg/mL PB in drinking water for 7 days | PB-induced neuroimmune effects seen in males immediately after treatment, but not in females until 30 days after beginning of treatment |
Over 30 years after the GW, scientists have yet to understand the etiology of GWI. Multiple exposures have been implicated, particularly inhaled toxicants, such as depleted uranium (DU), pesticides (organophosphates and carbamates), fine particulate matter (PM) (building fires, oil well fires, and desert dust), and chemical warfare agents (sarin and sulfur mustard)24. Other non-respiratory exposures that have been suspected of contributing to the disease etiology include microbiological exposures, immunizations against anthrax, plague, botulism, and physiological stress components (i.e., sleep deprivation, extreme temperature, and combat stress)31–33. This review will focus on the etiology of GWI/CMI, focusing on chemical warfare agent (CWA) inhalation exposures suggested in disease pathology. For these exposures, this review will summarize the prevalence and likelihood of veterans being exposed, mechanistic studies, and the environmental factors that can cause neurological and lung inflammation resulting from inhaling CWAs.
The following terms were used during the literature search: “gulf war illness sex”, “gulf war children”, “gulf war illness reproductive”, “gulf war illness reproduction”,“GWI reproductive”, “gulf war illness”, “civilians chemical weapons gulf war”, “civilian exposure chemical weapons gulf war”, “civilian exposure chemical weapons”, “sarin Afghanistan”, “chemical warfare Afghanistan”, “chemical weapons Afghanistan”, “chemical exposure Afghanistan”, and “organophosphate inflammation”.
Chemical Warfare Agents
Soldiers were exposed to CWAs throughout both the Gulf War. CWA exposures included organophosphate (OP) compounds such as sarin (GB), cyclosarin (GF), diisopropyl fluorophosphate (DFP), and mustard gas (sulfur mustard; SM)28,34,35. During the time of Desert Storm, Iraq had an active chemical warfare program with vast amounts of munitions containing CWAs and bulk reservoirs located throughout the country35. Surprisingly, most exposures to these chemicals did not come from active combat; instead, after the war ended in March 1991, when U.S. soldiers destroyed the stockpiles of the Iraqi CWAs at the Khamisiyah munitions depot34,35. During this time, U.S troops also bombed various CWA research, storage, and mass production sites34.
Much of the evidence suggests that troops were exposed, and many of the civilians who lived nearby were also affected by CWAs in the atmosphere. Shortly after known CWA exposures, military personnel and civilians reported respiratory complications and neurological symptoms due to the inhalation exposures from previously mentioned incidents27,30,35.
Inhalation Exposure to Sarin (GB) and Cyclosarin (GF)
An estimated 99,000 U.S troops were potentially exposed to a mixture of GB and GF34. It is believed that the long-term effects of GB and GF inhalation could play a contributing role in the respiratory and neurological issues manifested in GWI. The munition dump filled with GB and GF, located in Khamisiyah, Iraq, was destroyed at a time when the highest number of U.S. troops were in the area and likely exposed to varying levels of the CWA mixtures10,34. Five years after the initial event, the DoD developed weather and wind schematics of the affected area to determine the number of troops affected and the potential concentration of the exposure37. Considering the size of the plumes from the destruction of the munition dump paired with the initial weather mapping, new maps were created seven years since the exposure and suggested that over 100,000 U.S. troops were exposed38.
Organophosphorus Chemical Warfare Agent Mechanism
GB and GF are OP compounds known for inhibiting acetylcholinesterase (AChE)39,40. AChE’s primary purpose is to hydrolyze acetylcholine (ACh) at the synapse of neuromuscular junctions41. Inhibition occurs when GB and GF phosphorylate the serine hydroxyl group on the active site of AChE. The inhibition of AChE will result in rapid accumulation of ACh, producing overstimulation at cholinergic synapses, often referred to as cholinergic crisis40,42. Two major classes of cholinergic receptors are the nicotinic and muscarinic receptors. Nicotinic receptors are found at the neuromuscular junctions, autonomic ganglia, and some parts of the CNS43. In contrast, muscarinic receptors are found on myocardial muscle, throughout the lungs, and in regions of the CNS44.
Like with most organophosphorus compounds, the process of “aging”, or the dealkylation of AChE when inhibited by OP compounds and subsequently becoming unable to be directly reactivated by pyridinium oximes (one of the main treatments for OP-poisoning), will occur with GB and GF if left untreated45. Once phosphorylated, the active site of AChE loses an alkyl group, making it resistant to hydrolysis, blocking dephosphorylation. This aging process after GB and GF exposures can occur within 5 h of exposure34. GB and GF’s mild to moderate toxicity results in pinpoint pupils, blurry vision, rhinorrhea, shortness of breath, chest tightness, and muscular weakness10,34,39,40. In comparison, severe symptoms can lead to seizures, bronchoconstriction, lung edema, and respiratory failure/pulmonary collapse leading to death due to the overload of ACh on the autonomic nervous system39.
Many GW veterans experienced acute cholinergic syndrome, which was noticeable seconds to hours after initial exposure, presenting with previously mentioned mild to moderate toxicity symptoms34,46. Many low-level exposures can lead to acute cholinergic syndrome and should resolve within days. There is also evidence indicating a non-cholinergic pathway by which OP compounds exert their neurotoxic effects. Studies with organophosphate exposure in various models have been summarized in Table 2. Terry Jr et al. found that DFP exposures that did not produce cholinergic distress could still result in cognitive impairment in rats49. Locker et al. studied the effects of multiple GW-relevant AChE inhibitors, including the OP compounds DFP and chlorpyrifos-oxon (CPO), on neuroinflammation under stress conditions50. In this study, it was observed that only the OP AChE inhibitors caused significant neuroinflammation under either stressed or on-stressed conditions. Even more interestingly, it was found that the stressed condition both exacerbated neuroinflammation and reduced AChE inhibition in the OP-treated mice 50. While the authors were not able to point out a specific non-AChE pathway responsible for the observed neuroinflammation, they did reject the role of AChE and postulated that phosphorylation by OP compounds may be a potential cause 50. Miller et al. found similar results, with stress conditions exacerbating OP compound-induced neuroinflammation and markers of neuroinflammation generally not correlating with ACh activity in multiple regions of the brain51. The possibility that phosphorylation by OP compounds could be a causal factor of neuroinflammation was also mentioned by O’Callaghan et al. 52. Yates et al. found that DFP plus cortisol in human-induced pluripotent stem cells (hiPSC’s) resulted in alterations in tau levels, microtubule acetylation, and mitochondrial function. The authors also found that DFP plus cortisol in rats increased total tau in the CA3 region of the hippocampus, but nowhere else, and reduced total cell number without altering cell morphology in the CA3 region of the hippocampus53. Lein et al. treated guinea pigs with OP pesticides at doses that did not result in cholinergic intoxication, but did exacerbate vagally induced bronchoconstriction and bradycardia, leading the authors to conclude that OP compounds were acting directly on the muscarinic receptors. These effects were seen at doses that did not inhibit AChE activity, which, combined with the inability of the OP compounds at the doses given to exacerbate ACh-induced bronchoconstriction, indicates that OP compounds may exert their effects on the respiratory system in a manner independent of AChE54. Further evidence that OP compounds can act directly on muscarinic receptors is provided by Katz and Marquis, who found that paraoxon inhibits tritiated quinuclidinyl benzilate, [3H]QNB, an antagonist of cholinergic muscarinic receptors55. While some OP compounds can act directly on muscarinic receptors at doses that do not inhibit AChE, OP compound that act directly on nicotinic receptors only do so at doses that also inhibit AChE56. Other possible mechanisms of OP-induced toxicity include inhibition of neurotoxic esterase and inhibition of ACh synthesis via reduced cAMP resulting in reduced choline uptake56.
Table 2:
Studies on the toxic effect of organophosphates.
| Organophosphates | |||
|---|---|---|---|
| Study (Ref #) | Model | Exposure | Results |
| Maxwell et al., 2013 (40) | Recombinant human AChE | AChE incubated with OP compounds, including sarin and tabun | OP compounds deactivated AChE |
| Terry et al., 2011 (49) | Male albino Wistar rats | 0.25, 0.5, 0.75, or 1.0 mg/kg DFP, CAS 55-91-4 every other day for 30 days | DFP could cause cognitive impairment in doses that do not cause cholinergic distress |
| Locker et al., 2017 (50) | Male C57BL/6 mice | 8 mg/kg CPO, 4 mg/kg DFP, 3 mg/kg PB, or 0.5 mg/kg PHY; 400 mg/L CORT in drinking water for 4 days before treatment with AChE inhibitor | Only OP AChE inhibitors cause neuroinflammation; stress increases neuroinflammation and decreases AChE inhibition; Importance of AChE in OP-induced neuroinflammation rejected |
| Miller at al., 2018 (51) | Male C57BL/6 mice | 8 mg/kg CPO, 4 mg/kg DFP, 3 mg/kg PB, or 0.5 mg/kg PHY; 400 mg/L CORT in drinking water for 4 days before treatment with AChE inhibitor | Stress exacerbates OP-induced neuroinflammation; Markers of neuroinflammation do not correlate with AChE activity in the brain |
| O’Callaghan et al., 2015 (52) | Male C57BL/6 mice | 2 mg/kg PB and 30 mg/kg DEET daily on days 1-14 (100 mg/kg MINO on days 1-15 for MINO experiment); 200 mg/L CORT in drinking water on days 8-15, 4 mg/kg DFP on day 15 | CORT exacerbated DFP-induced neuroinflammation and removed protective effects of PB/DEET; MINO suppressed CORT+DFP-induced neuroinflammation; Organophosphorylation implicated in neuroinflammation |
| Yates et al., 2021 (53) | hiPSC cells generated from PBMCs from GW veterans; male albino Wistar rats | Cells exposed to 2 mM cortisol and either 200 nM, 1 mM, or 2.5 mM DFP; Rats exposed to 0.4 mg/kg CORT in drinking water for 7 days and then given 1.5 mg/kg DFP on day 7 | Altered tau levels, microtubule acetylation, and mitochondrial function in hiPSC cells; increased total tau, decreased total cell numbers,but unaltered cell morphology in CA3 region of hippocampus |
| Lein, P.J. & Fryer, A.D., 2005 (54) | Male guinea pigs | 0.1-10 mg/kg parathion, 0.75-75 mg/kg diazinon, or 150 mg/kg permethrin | Exacerbated vagally induced bronchoconstriction and bradycardia without causing cholinergic distress; no exacerbation of Ach-induced bronchoconstriction |
| Katz, L.S. & Marquis, J.K., 1989 (55) | Caudate nuclei harvested from calf brains | Displacement of [3H]QNB by muscarinic antagonists was measured in the presence or absence of paraoxon (dose not given) | Paraoxon, an OP compound inhibits [3H]QNB |
| Guignet et al., 2020 (57) | Male Sprague Dawley rats | 0.1 mg/kg PB and either 4 mg/kg DFP or vehicle | Reactive astrocytes, microgliosis, and increased oxidative stress in brains of OP-treated rats |
| Zhang et al., 2021 (59) | Male C57BL/6 mice | 0.01, 0.1, 1, or 10 mg/kg CPF for five days per week for 12 weeks | Liver inflammation and altered gut microbiota |
| Meng et al., 2022 (60) | Female BALB/c mice | 10 or 100 mg/kg TnBP or 10 or 100 mg/kg TBOEP each day for three weeks | Lung inflammation and exacerbated allergic response |
| Pena-Philippides et al., 2007 (67) | Male Fischer 344 rats | 10 mg/kg CHL 7 days before sarin exposure; 0.4 mg/m3/h/day sarin for 1 or 5 days | Significantly increased gene expression of TNF-a, IL-1b, and IL-6; effects did not persist beyond two weeks |
| Pena-Philippides et al., 2007 (67) | Male Fischer 344 rats | 10 mg/kg CHL 7 days before sarin exposure; 0.4 mg/m3/h/day sarin for 1 or 5 days | Significantly increased gene expression of TNF-a, IL-1b, and IL-6; effects did not persist beyond two weeks |
| Kassa et al., 2004 (68) | Female BALB/c mice | Clinically asymptomatic concentration of sarin given either once or three times each other day via inhalation | Inhibition of T-cell function |
| Gundavarapu et al., 2014 (71) | Male F344 rats | 13-15 mg/m3 sarin for 10 min | Severe bronchoconstriction |
| Shi et al., 2021 (76) | Male guinea pigs | 16.8 mg/kg sarin daily for 14 days | Disrupted lipid metabolism in hippocampus |
| Genovese et al., 2006 (83) | Male Sprague-Dawley rats | 16-5.2 mg/m3 cyclosarin | Impaired learning; recovered months later |
| Phillips and Deshpande, 2016 (85) | Male Sprague-Dawley rats | 400 mg/kg DFP daily for 5 days | Severe cognitive impairment and neuronal cell death in hippocampus three months after exposure |
| Valiyaveettil et al., 2011 (89) | Male Hartley guinea pigs | 846 mg/m3 sarin or 841 mg/m3 soman; 1 mg purified human or 0.5 mg purified rabbit serum PON1 | PON1 protected against sarin and soman |
| Raveh et al., 1993 (92) | Male albino ICR mice and male Sprague-Dawley rats | 12-45 nmol HuBChE and various doses of OP compounds | HuBChE protects against OP compound-induced toxicity |
| Worek et al., 2014 (96) | Male Dunkin-Hartley guinea pigs | 0.2 or 1.0 mg/kg IlG1 and 100 mg/kg cyclosarin | Prevented cyclosarin toxicity |
| Melzer et al., 2012 (97) | Male Wistar rats | 270 mg/kg soman; 12.2, 35.8, or 71.0 mg/kg WT DFPase; 35.8 mg/kg PEGylated DFPase | High dose of DFPase prevented soman-induced mortality |
| Kuruba et al., 2018 (98) | Male Sprague-Dawley rats | 3.2 mg/kg DFP; 5 mg/kg Diazepam at 10 minute, 60 minute, or 120 minutes post-DFP exposure | Diazepam effective when given at 10 minutes, but completely ineffective at later time points |
| Reddy et al., 2020 (99) | Male Sprague-Dawley rats | 3.2 mg/kg DFP;30, 60, or 100 mg/kg phenobarbital | Highest dose of phenobarbital protected against DFP-induced seizures and neuronal damage, but could also cause coma or death |
| Rojas et al., 2018 (100) | Sprague-Dawley rats (sex unspecified) | 9.5 mg/kg i.p. or 5 mg/kg s.c. DFP; 10 mg/kg diazepam; 0.8 g/kg urethane | Urethane ended DFP-induced seizures and prevented further seizures |
| Rojas et al., 2020 (101) | Male Sprague-Dawley rats | 5 mg/kg DFP; 0.8 g/kg urethane | Urethane suppresses neurodegeneration and inflammatory mediators |
| Lumley et al., 2019 (102) | Male Sprague-Dawley rats | 453 mg-min/m3 of GB for 60 minutes; 10 mg/kg diazepam and either vehicle or 4 mg/kg pregnanolone | Diazepam plus pregnanolone reduced seizures and neurodegeneration |
| Zhu et al., 2020 (105) | Male Sprague-Dawley rats | 0.5 mg/kg DFP daily for 5 days; various doses of ketamine, R-ketamine, or S-ketamine | Ketamine improved behavior |
Inflammation has been seen in both animal and human models of OP exposure. Increased expression of proinflammatory cytokines in the CNS has been observed upon OP exposure in mice50,52. Guignet et al. found evidence of reactive astrocytes, microgliosis, and increased oxidative stress in the brains of rats exposed to OP compounds57. An observational study of agricultural workers exposed to OP pesticides found elevated levels of TNF-α (tumor necrosis factor), interleukin-6 (IL-6), and C-reactive protein in their blood58. Oral exposure to chlorpyrifos in a mouse model resulted in inflammation in the liver and altered gut microbiota59. OP compound exposure in a mouse model was also seen to produce lung inflammation as well as an exacerbated allergic response in ovalbumin (OVA)-sensitized mice60.
Inhalation effects of GB and GF on the Respiratory System
The primary route of exposure causing the most toxic effects to GB and GF is via inhalation, but victims can also be exposed via dermal and ocular routes61. GB and GF trigger airway hyperreactivity through the inhibition of AChE, which in turn decreases the hydrolysis of ACh that is readily available62. By decreasing hydrolysis, ACh will accumulate on the cholinergic receptors, such as the muscarinic receptors. In the lung, muscarinic receptors are present throughout the airways and on epithelial cells63. In addition, the parasympathetic nerves that are innervated throughout the lung are known for releasing ACh onto the muscarinic receptors64. Given that the lung is one of the primary routes of exposure to GB and GF, victims of such exposure will experience respiratory symptoms. Still, it is unclear if exposure contributes to long-term effects observed in GWI.
Stemming from the vagus nerve, parasympathetic nerves are responsible for most of the lung’s autonomic functions, such as airway contraction and relaxation. Normal contraction and bronchoconstriction result from the parasympathetic nerves releasing ACh onto the muscarinic receptors65. The severity of GB and GF exposures is highly dependent on the exposure dose. Less severe, acute toxicity to GB (ECt50=1.0 mg-min/m3) and GF (ECt50=0.5 mg-min/ m3) leads to increased nasal discharge, bronchoconstriction, wheezing, and increased mucus secretions64. As a result of the GB/GF ECt50’s mentioned above, the overload of ACh will cause bronchoconstriction through the persistent activation of specific muscarinic receptors such as the M3 receptors on smooth airway muscles64. In severe cases, respiratory distress is likely to occur at concentrations higher than the ECt50 but below the LCt50 for GB (35 mg-min/m3) and GF (35 mg-min/m3)64. In more severe cases, respiratory distress can lead to death associated with the prolonged inhibition of AChE and cholinergic overload in the autonomic nervous system. This overload causes continual muscle contraction in essential organs such as the lungs (bronchospasms) and the heart (tachycardia) to the point of paralysis in the diaphragm and intercostal muscles64. Minimum information is available on long-term damage to lungs in humans due to the toxic nature of these compounds and their ability to predominantly affect the CNS.
Research is sparse in addressing the long-term respiratory effects of GB and GF exposure. It has been reported in rodent models that a one-time high dose inhalation exposure to GB (51.2 mg/m3 for 15 min) did not produce lasting inflammatory effects in the lung and was cleared 2-3 weeks after initial exposure66. In another study, examining whether subclinical doses had lasting respiratory impacts that closely resemble GW exposures, a 5-day repeated exposure to a subclinical dose of 0.4 mg/m3/h/day (nose-only exposure) of GB was administered to rats to determine inflammation in the lung67. While changes in gene expression of proinflammatory cytokines TNF-α, interleukin-1β (IL-1β), and IL-6 were significantly increased as well as inhibition of T-cell function, however, these inflammatory findings did not persist beyond two weeks67,68. Despite the lack of evidence in humans, much of the animal model data indicates acute short-term respiratory damage but fails to confirm long-term damage. This topic merits further exploration to determine potential long-term synergistic effects of acute injury experienced from exposure to GB/GF in addition to other exposures that potentially contribute to the respiratory impacts observed in GWI.
Inhalation Effects of GB and GF on the Central Nervous System
Though neurocognitive symptoms and brain alterations don’t occur until months or even years after initial exposure to GB and GF, acute neurological effects follow immediately after initial exposure, such as seizures, paralysis, and in severe cases, death69. The enzyme AChE is essential in function of the autonomic nervous system and within the autonomic nervous system reside the parasympathetic nerves, as previously mentioned. By inhibiting AChE, nerve signaling occurs indefinitely, overloading cholinergic receptors with ACh62. The continuous firing of these nerve signals leads to continual muscle contractions throughout the body, causing paralysis, respiratory failure, tachycardia, and death if left untreated or exposed to a high concentration70,71. Once AChE inhibition subsides, the neurocognitive decline becomes more apparent in affected veterans, such as attention, memory, and learning10,72–74. In addition to neurocognitive decline, there have been several reports of brain tissue changes, predominantly in the hippocampus, resulting from GB and GF exposures that are detected months to years after exposure75,76.
Many studies have focused on how GB and GF exposure affects the brain structures and their potential involvement in neurocognitive decline experienced by veterans with GWI. The brain structure affected by inhalation of GB and GF is the hippocampus region of the brain75,76. The hippocampus is located between the temporal lobes and comprises of the dentate gyrus and what is known as Ammon’s horn (cornu ammonis). The role of the hippocampus involves receiving and processing information that contributes to learning and memory formation along with spatial navigation77. Given the hippocampus function, veterans who disclosed being near the plumes of CWA have reported a decline in dexterity, visuospatial functions, and memory78–80. Similarly, reduction in white matter and hippocampal volume was also found in veterans, which was associated with the decline in neurocognition as mentioned earlier81. Fourteen to nineteen years after the GW, veterans took part in GWI research at the San Francisco Veterans Affairs Medical center between 2005 and 2010. These Veterans had a significant reduction in hippocampal volume and gray matter volume (observed through 1.5T and 4T magnetic resonance imaging) as well as notable changes on the continuous performance test (measuring attention) (n=56 exposed/unexposed)75. These findings correlate with known GB attacks victims such as the 1995 Tokyo terrorist attacks, who also notably had smaller hippocampal volume than those unexposed82.
In further understanding the mechanism behind GB/GF exposure in the brain, animal models have been used to replicate the neurological symptoms in GWI. In rodent models, a one-time exposure to GF (1.6-5.2 mg/m3) caused a drastic decrease in learned behavioral tasks but was recovered months after83. In characterizing the effect of GB on the hippocampus, a metabolomic study examined the impact of a low dose exposure of GB [0.4 x LD50= 16.8 μg/kg via subcutaneously (s.c.)] and found that phospholipid and sphingolipid metabolism was disrupted in the hippocampi of guinea pigs after 14 days76. In studying the effects of GB on the brain, an OP surrogate known as DFP has been routinely used84. DFP has made it possible to recapitulate many neurological effects observed in GWI in rat models. In a study done in rats, 400 mg/kg of DFP, a dose authors said to be comparable to what GW soldiers experienced in the field, was administered s.c. over five days85. Three months after DFP exposure, rats had several cognitive issues such as anxiety, chronic depression, and memory problems that persisted along with neuronal cell death in the hippocampus85. Although different neuronal populations within the hippocampus have been attributed to memory issues, it has also been hypothesized that Ca2+ fluctuations may contribute to synaptic plasticity leading to the persistent memory issues exhibited in GWI86. The further evaluation of the Ca2+ hypothesis in neurological morbidities can provide a relevant therapeutic target. While the information provided is advantageous in elucidating a mechanism of GB/GF in the brain, most studies lack an appropriate route of exposure (s.c. injections versus inhalation exposure) which is critical in understanding the manifestation of GWI.
The Progression of Therapeutics for GB and GF Exposures
Some of the most commonly used therapeutics for OP poisoning are atropine, oxime, and diazepam. As an anticholinergic, atropine blocks the effects of the ACh overload on muscarinic receptor regions62. In addition to atropine, oximes are often used to reactivate AChE, aiding in regaining respiratory functions40. The usage of oximes such as pralidoxime as a treatment is controversial due to timing and mechanism in alleviating respiratory distress87. There are a couple of conditions where oxime treatments are ineffective and detrimental to victims, such as the aging of AChE and the rate of AChE reactivation in response to the oxime is slower than the rate of bond formation between OP and AChE40,87. Paired with atropine and oxime usage, the anticonvulsant diazepam alleviates victims of seizures following such exposure88. Although these therapeutics are effective when the exposure is known, and the victim can seek medical attention immediately, timing is a huge factor in their effectiveness. Due to the above limitations of veterans not knowing when and what they were exposed to, alternative therapies are being explored for OP poisoning concerning GB/GF exposures that encompass catalytic bio-scavengers, phenobarbital, urethane, neurosteroids, and ketamine.
An alternative therapeutic currently being studied in animals are catalytic bioscavengers. Bioscavengers are a novel approach, with their ability to efficiently eliminate nerve agents such as GB/GF by catalyzing their hydrolysis, entirely inhibiting toxicity89,90. Bioscavengers would be used prophylactically and could render benefits if soldiers anticipate an attack89,90. Its limitation resides in the fact that administration after exposure would be useless. There needs to be enough bioscavengers in the body before an exposure to eradicate GB/GF efficiently. A bioscavenger of interest is human butyrylcholinesterase (BChE)91,92. Human butyrylcholinesterase is a non-specific cholinesterase enzyme made in the liver and is found in plasma92. Large doses of BChE must be readily available in the blood to catalyze lethal concentrations of nerve agents, making it a less viable option91,92. However, it is possible to improve efficacy, as site-directed mutagenesis of BChE has shown promising results as a viable prophylactic option. The G117H mutation has yielded exciting results against GB, catalyzing the reaction 174-fold higher than wild-type BChE, but this remains insufficient to implement as a treatment91,93. This is a significant limitation of this bioscavenger as a therapeutic, and the search for a bioscavenger with a higher catalytic reaction rate is underway.
Since the discovery of BChE, other bioscavengers have been identified, including mammalian paraoxonase (PON1) and diisopropylfluorophosphatase (DFPase) from Loligo vulgaris89,94,95. PON1 is a lipoprotein secreted by the liver, but local synthesis is said to occur in most tissues in the body. Intravenous (i.v.) injection of a chimeric PON1 mutant, IIG1, at 0.2 and 1.0 mg/kg, 60 min before s.c. injection of GF (100 μg/kg), completely prevented signs of poisoning and lethality in guinea pigs96. While the PON1 mutant, IIG1, shows some promise as a prophylactic treatment in in vitro and in vivo studies, more research is needed to determine efficacy in humans.
DFPase, originating from squid (Loligo vulgaris), is another potential bioscavenger. Unlike previously mentioned bioscavengers, the DFPase enzyme’s non-human origin causes it to readily clear the body faster and poses a potential risk for unwanted immune responses97. To make this enzyme more favorable, polyethylene glycol (PEG) can be added to solve the issues mentioned. This concept was tested using a similar OP nerve agent, Soman. It was found that dosing rats with 71 mg/kg PEGylated DFPase (considered extremely high) 5 minutes before Soman (3 x LD50) s.c. kept the rats alive while lower doses did not97. While the higher dose of PEGylated DFPase kept the rats alive, the doses are not biologically relevant unless an increase in substrate affinity allows for the dosage to be substantially lowered.
A prominent effect of OP’s (GB, GF, and DFP) are the seizures and refractory status epilepticus (SE) following exposure. While diazepam is one of the main treatments to treat seizures and SE after OP exposure, victims are sometimes unresponsive88,98. Due to the failure of diazepam, drugs such as phenobarbital and urethane have been studied for therapeutic consideration. In rats, SE was induced by a s.c. administration of 3.2 mg/kg DFP99. Forty minutes after DFP exposure, phenobarbital was administered at 30,60, and 100 mg/kg99. At 100 mg/kg of phenobarbital, seizure activity was terminated entirely, and there was significant neuroprotection in the brain regions associated with SE99. However, there are benefits and inherent risks in using phenobarbital, such as an unconscious state or death.
Urethane, which was formerly used as an anesthetic, is presently an anticonvulsant. Electroencephalography on DFP exposed rats (0.8 urethane administered 1 h after a 5mg/kg DFP) showed that urethane treatment terminated SE and prevented future seizures100. Urethane has also been shown to robustly attenuate neurodegeneration and inflammatory mediators (IL-1β and TNFα) at 24 h after DFP administration in rats101. Another drug under investigation for treating seizures and SE after OP exposure is the neurosteroid pregnenolone. Dual therapy with pregnanolone (4 mg/kg) and diazepam (10 mg/kg) has been shown to reduce experienced SE time in rats exposed to GB and decreased neuronal degeneration months after a 60-min one-time exposure of 3.0 LCt50 of GB102.
While seizures and SE remain significant OP symptoms of poisoning, another considerable concern is the effects OP has on mood and the chronic fatigue experienced by veterans103,104. GB/GF are thought to be underlying factors that play a role in mood dysfunction seen in veterans. A study utilizing a DFP exposed rat model showed that when treated with ketamine (3, 5, and 10 mg/kg, i.p.), rats exhibited significantly improved behavioral signs as early as 1 h post-administration, persisting to 24h105. Many of these treatment strategies have yielded promising results that need further investigation. GWI remains a significant threat to countless veterans’ health and quality of life. While many of the discussed treatment strategies here have yielded promising results, we must direct additional resources to elucidate these mechanisms to understand and counteract the progression of these disorders.
Inhalation Exposure to Sulfur Mustard
Along with GB and GF, Sulfur mustard (bis(2-chloroethyl) sulfide; SM) is a CWA identified in destroyed munitions in Khamisiyah, Iraq, as well as Muammadiyat Ammunition Storage Site46,106. The DoD recorded over 15 metric tons of SM at the Muammadiyat Ammunition destruction site alone46,106. In Khamisiyah, the amount of mustard gas was reported to be far less than the nerve agents, GB and GF, but troops near the site still ran the risk of exposure35. DoD estimates over 4,000 soldiers have been acutely exposed to high concentrations of mustard gas. In contrast, the remaining troops in neighboring areas could have been exposed to lower concentrations due to wind patterns35,46. This estimation is expected to be much higher according to government documents, but an exact number remains unknown46. SM is thought to cause long-term respiratory and neurological damage, as seen in veterans throughout different wars that experienced this exposure. An overview of the symptoms experienced following SM exposure is detailed in Figure 1 and the animal studies utilizing SM and analogues have been summarized in Table 3..
Figure 1. Overview of SM exposure.
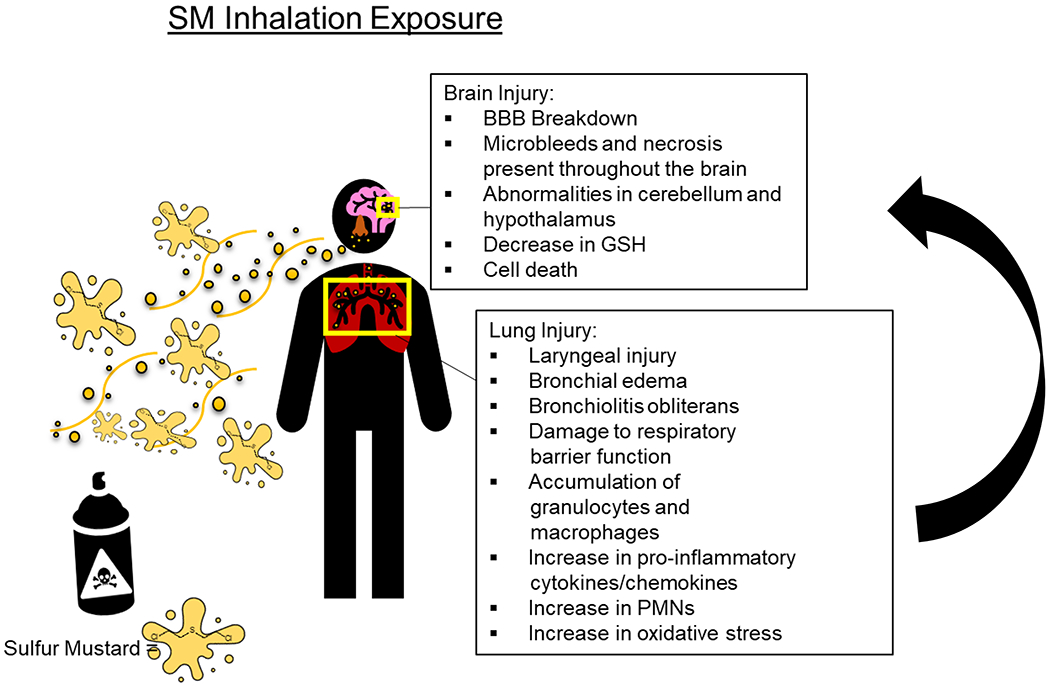
Inhalation to SM has been attributed to lung and brain injury. After exposure to SM, soldiers and civilians develop respiratory complications, such as asthma-like pathologies, chronic bronchitis, and pulmonary fibrosis. While the lungs are known for their susceptibility to SM toxicity, the brain’s effects remain primarily unknown. Figure created with biorender.com
Table 3:
Studies on the toxic effect of vesicants (sulfur mustard, nitrogen mustard, and CEES).
| Pyridostigmine Bromide | |||
|---|---|---|---|
| Study (Ref #) | Model | Exposure | Results |
| Bryant et al., 2021 (23) | Male and female C57BL/6 mice (WT, NLRP3−/−, and STINGgt/gt) | 0.7 mg/kg PB and 200 mg/kg Per in 50 mL DMSO daily for 10 days | Greater neuroinflammation and cognitive impairment in female mice; greater peripheral inflammation in males |
| Hernandez et al., 2020 (25) | Male and female C57BL/6 mice | 90 mg/mL PB in drinking water for 7 days; 0.07 mg/mL PEA in drinking water for one month | PB alone increased fecal pellet production and colonic transit time only in females, PB+PEA increased myenteric neurons and muscarinic M3 receptors only in females |
| Sulfur Mustard | |||
| Study (Ref #) | Model | Exposure | Results |
| Inturi et al., 2014 (117) | JB6 cells | 0.75 mM nitrogen mustard | Decreased cell growth; cell cycle arrest |
| Byers et al, 2000 (120) | Hairless guinea pigs | 7 minutes exposure to 10 mL sulfur mustard in vapor cups | Sulfur mustard depletes NAD+, leading to ATP deficiency and cell death |
| Malaviya et al., 2010 (124) | Male Crl:CD (SD) rats | 0.7, 1.0, or 1.4 mg/kg sulfur mustard via inhalation | Damage, cell death, and inflammation in the lungs |
| Chevillard et al., 1992 (127) | Respiratory epithelial (tracheae of male New Zealand white rabbits) | 0.1 mM sulfur mustard | Cessation of ciliary beating |
| Malaviya et al., 2020 (129) | Male Wistar rats | 0.06-0.6 mg/kg sulfur mustard via inhalation | Increased TNF-α in lungs |
| Feng et al., 2019 (130) | Male ICR mice | 40 mg/kg SM for survival and respiratory function experiments; 30 mg/kg SM for other experiments | Increased pro-inflammatory cytokine expression in lungs |
| Sunil et al., 2011 (133) | Male Wistar rats | 0.25 mg/kg nitrogen mustard | Alveolar macrophages increased in size and number; increased neutrophil numbers |
| Gray et al., 2010 (145) | MLE-15 murine lung epithelial cells | 100 mg/mL CEES | NADPH cytochrome P450 reductase inhibited in CEES-treated cells |
| Xiaoji et al., 2016 (157) | Male Sprague-Dawley rats | 2 mg/kg SM | Tissue damage and inflammatory cell infiltration in the lungs |
| McGraw et al., 2018 (158) | Male Sprague-Dawley rats | 1.0 +/− 0.05 mg/kg SM | Decreased lung function; development of bronchiolitis obliterans and pulmonary fibrosis |
| Jugg et al., 2016 (159) | Female large white pigs | 60 or 100 mg/kg SM | Lung damage and alterations in pathways such as the Nrf2 pathway |
| Rana et al., 2020 (162) | Male Sprague-Dawley rats | 15 minutes exposure to CEES vapor | Epithelial damage, decreased microRNA-140-5p, increased pro-inflammatory cytokines, and increased superoxide dismutase |
| Sunil et al., 2020 (163) | Male and female C57BL6/J mice | 0.8 mg/kg NM | Tissue damage and inflammation in the lung |
| Cruz-Hernandez et al., 2021 (164) | Male C57BL/6J and KitW-sh mice; BMMCs from C57BL/6J mice | 0.125 mg/kg NM | Inflammation and lung injury occurred in C57BL/6 mice, but not in the mast cell-deficient mice |
| Malaviya et al., 2015 (165) | Male Wistar rats | 0.125 mg/kg NM; 15 mg/kg anti-TNFa antibody every nine days after NM exposure | Anti-TNFa antibody reduced M1 macrophage numbers and TGF-b levels |
| Solopov et al., 2020 (167) | Male C57BL/6J mice | 0.312 or 0.625 mg/kg NM | Increased pro-fibrotic biomarkers and collagen expression at 10 and 30-days post-exposure |
| Tekiner et al., 2009 (176) | Male brown rats | 800 mg/m3/min NM for 10 minutes; 100 mg/kg/day proanthocyanin | Vascular edema, abnormal neuronal and glial processes, and altered cellular components in the brain; proanthocyanin alleviated NM-induced oxidative stress |
| Gao et al., 2011 (177) | Male Crl:CD SD BR rats | 0.25 mg SM in 100 mL inhaled over 50 minutes; 10, 20, or 40 mg/kg roxithromycin | Roxithromycin improved lung function in SM-exposed rats |
| Calvet et al., 1994 (178) | Male Hartley guinea pigs | 0.3 mL/kg SM injected into trachea; trachea incubated with 10−5 M phosphoramidon | Phosphoramidon inhibited SM-induced sensitivity to substance P |
| Calvet et al., 1996 (179) | Male Hartley guinea pigs | 0.3 mL/kg SM (stored 100 mg/mL in EtOH and then diluted 1:100 in 0.9% NaCl) given intratracheally; 3 mg/kg betamethasone in drinking water for seven days | Betamethasone repaired airway damage caused by SM |
| Wigenstam et al., 2012 (180) | Female C57BL/6 mice | 1 mg/kg melphalan; 10 mg/kg dexamethasone given 1, 2, or 6 hours after melphalan exposure | Dexamethasone reduced inflammatory cell infiltration in lungs of SM-exposed mice |
| O’Neill et al., 2010 (181) | Male Sprague-Dawley rats | 5% CEES in ethanol at 12.7 cc/hour for 15 minutes; 5 mg/kg AEOL 10150 injected at 1 or 9-hours post-exposure | AEOL 10150 decreased markers of oxidative stress in SM-exposed rats |
| Sharma et al., 2010 (186) | Female Swiss mice | 11.9 mg/kg HN-1, 20.0 mg/kg HN-2, 7.1 mg/kg HN-3, or 8.1 mg/kg SM | Differences in toxic effects and efficacy of antidotes seen between mustard compounds |
Mechanism of Action for Sulfur Mustard
SM is a bifunctional vesicating agent known for causing severe blistering and burns on the skin, eyes, and most notably, the respiratory tract107,108. Despite popular belief, SM is not a gas; it is an oily liquid aerosolized when used on the battlefield, giving it a “gas-like” appearance109. For this reason, one of the most common modes of exposure is inhalation. Many mechanisms of toxicity have been proposed, but to date, no mechanism fully encapsulates the systemic impairment and damage experienced as a result of SM inhalation. The lack of a particular mechanism has made developing a viable treatment for SM exposure challenging. Below we will discuss some of the significant mechanisms highlighted in the literature that contribute to SM-mediated toxicity.
Alkylation Events:
One of SM’s first and most studied cytotoxic chemical reactions is DNA alkylation. Biologically, SM will form sulfonium ions that will rapidly undergo another reaction, the opening of the sulfonium ring, forming carbenium ions109. Carbenium ions are then readily available to interact with DNA107. Approximately 61% of alkylation events occur on the guanine at the N-7 position, and 8% of alkylation events occur on an adenine at the N-1 position forming adducts110,111. SM can react with other nucleotides at various positions, but these are less common110. Once these alkylation events occur on one side of the DNA strand, due to SM’s bifunctional characteristics, it can react once more, forming a cross-linkage (Figure 2)110,111. This cross-linkage reaction between guanines/adenines of the double helix molecule disrupts cellular metabolism and alterations in the DNA replication mechanism110. It has been estimated that about 17% of these alkylation events will form inter-strand links causing DNA strand breaks, in turn inhibiting cellular function such as replication, and ultimately causing mutations (cancer formation) or cell death110,112. This is especially detrimental to fast-dividing cells such as precursor immune cells found in the bone marrow. The death of these cells will directly affect immune responses to infection, as an example. SM can form adducts with other molecules aside from DNA, such as RNA, proteins, and lipids (Figure 2)113. SM’s ability to alkylate different biological components is voluminous when diving into the damage and cellular impairment SM causes. Still, the double-strand cross-linkage only occurs in about 17% of events which doesn’t account for the remaining damage SM causes. It is essential to understand that DNA alkylation, either single or double strand cross-linkage event, activates downstream mechanisms that are believed pivotal to SM-induced injury.
Figure 2. Overview of Proposed Mechanisms of Toxicity for Sulfur Mustard.
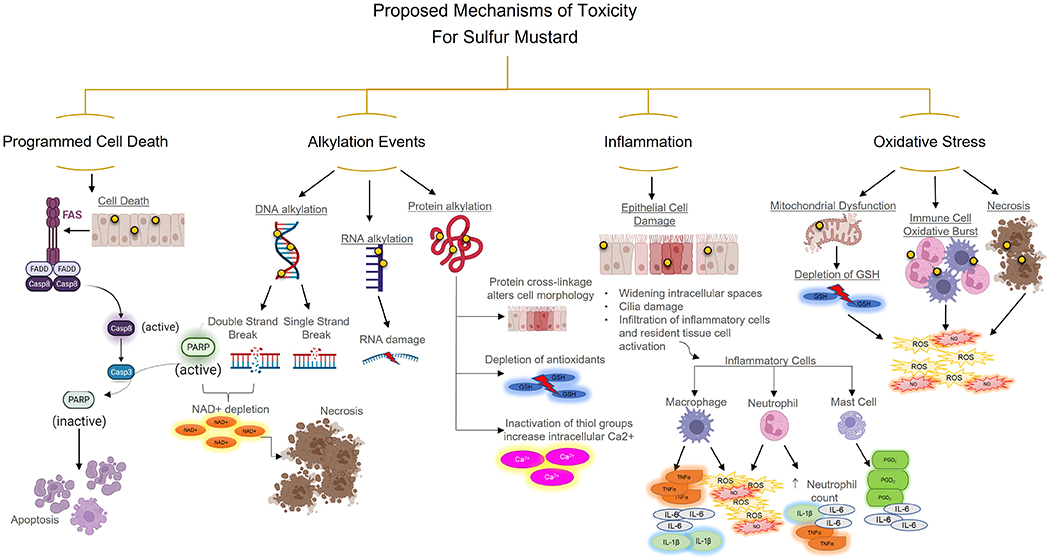
(A) SM can cause apoptosis via the FAS/FASL pathway. (B) SM alkylates DNA, RNA, and proteins. DNA alkylation leads to single and double-strand breaks, which leads to the activation of PARP. Overactivation of PARP leads to NAD+ depletion and necrosis. While RNA and protein alkylation don’t lead directly to cell death, lack of antioxidants increases intracellular Ca2+ and alters cell morphology. (C) SM’s damage and toxicity causes epithelial damage leading to a robust inflammatory response through macrophages, neutrophils, and mast cells. (D) SM causes mitochondrial dysfunction, immune cell accumulation (macrophage and neutrophils), leading to a respiratory burst of reactive oxygen species (ROS), nitric oxide (NO), and necrotic cell death. Figure created with biorender.com
In the formation of DNA adducts, poly(ADP-ribose)polymerase type1 (PARP-1) is activated. PARP-1 is an essential protein in the DNA base excision repair mechanism113–115. In the event SM has formed an adduct, PARP-1 is recruited and serves as a biological platform that utilizes nicotine adenine dinucleotide (NAD+) as a substrate to covalently bond poly-ADP- ribose (PAR) and other proteins to PARP-1 itself (known as automodification)114,116. Excision repair by PARP-1 in mild SM-DNA adduct formation will preserve the cellular integrity and viability and hinder mutations from occurring within the DNA chain114,116. Issues arise when the body is exposed to high or prolonged amounts of SM, leading to necrotic cellular damage117,118. The overwhelming demand for PARP-1 will lead to rapid NAD+ and adenosine triphosphate (ATP) depletion causing cellular rupture113,114,119. Depleting the NAD+ pools in the cell will lead to a deficiency in ATP, critical to cellular energetics and ultimately the cell’s fate120. Severe ATP deficiency will result in PARP not being cleaved by caspase 3, thus potentially resulting in necrosis (Figure 2)121. It is thought that PARP has a regulatory function if it may need to shift from apoptosis to necrosis121,122. SM does cause necrosis as well as apoptosis at the site of injury via the Fas/FasL pathway123. When this pathway is activated, PARP is cleaved by caspase 3, and cells undergo apoptosis (Figure 2)123. The cell’s inability to produce or use energy for vital functions for its survival leads to cell death, either via apoptosis or necrosis, resulting in extensive tissue damage. Extensive cell death leads to immune activation and the formation of oxidative stress.
Inflammatory cell activation:
The inflammatory process is complex. Many pro-inflammatory and anti-inflammatory mediators play a critical role in the various inflammatory mechanisms that initiate inflammation and repair at the injury site. When SM is inhaled, it’s been shown that significant necrosis occurs amongst epithelial cells and, in turn, releases various mediators such as IL-1α to activate immune cells to the site of injury124–126. Aside from necrosis, ciliated cells become damaged and no longer have cilia which become critical in mucus clearance (Figure 2)127. In in vitro experiments, it has been shown that SM increases the production Muc5Ac increasing mucus128. When the epithelial cells become damaged, cellular matrix adhesion is lost, and the membrane becomes leaky (Figure 2)128. Aside from resident tissue immune cells such as mast cells and macrophages, the leaky membrane and pro-inflammatory mediators released from the necrotic cells and resident inflammatory cells signal more inflammatory cells to the site of injury. In the bronchioalveolar lavage (BAL) rodent models, there is an increase in IL-6 and TNFα, which has been attributed to macrophages (Figure 2)129,130.
Furthermore, in the BAL, patients who exhibited SM-induced pulmonary fibrosis had elevated macrophage inflammatory protein 1α (MIP-1α) and monocyte chemoattractant protein −1 (MCP-1, CCL-2)131. The release of MIP-1α is known to attract neutrophils to the site of injury (Figure 2)131. Various animal models depict the accumulation of neutrophils following inhaled SM. Neutrophils are known to accumulate within minutes to hours of SM exposure132. Macrophages release various proinflammatory cytokines such as IL-1β, TNFα, IL-6, and MIP1α to recruit neutrophils to the site of injury129,133. Once at the affected tissue, neutrophils also release pro-inflammatory cytokines such as IL-1β, TNFα, IL-6, and IL-8132,134.
Oxidative Stress Generation and Cellular Response:
As the internal cellular homeostasis is disturbed by DNA damage, overactivation of PARP-1, and reduction of the NAD+ and ATP cellular pools, the cells located at the site of injury form an abundance of reactive oxygen species (ROS)135. A sign of early toxicity is inflammation, which can be triggered in response to ROS formation directly/indirectly from cell death and resident tissue cells signaling the initiation of an immune response. After inhalation of SM, macrophages neutrophils become a major source of ROS and nitric oxide (NO) production at the site of injury136,137. It has been hypothesized that these immune effector cells can produce ROS in what is known as a respiratory burst or oxidative burst138–140. As the name implies, these cells can rapidly release ROS to mediate the damage and initiate repair140. However, if the excess ROS is not scavenged, it causes oxidative stress to neighbor cells in the affected region, resulting in more damage before repair, including mitochondrial dysfunction140,141. While macrophages and neutrophils remain primary extracellular sources for NO and ROS, mitochondria within epithelial cells represent intracellular sources of oxidative stress122. Mitochondrial dysfunction leads to the reduction of detoxifying thiols such as glutathione (GSH)142,143. The decrease in GSH paired with the overproduction of ROS will lead to an imbalance in redox factors and changes in cellular metabolism142,144. These changes directly impact DNA base oxidation, lipid peroxidation, and protein oxidation113,142. As oxidative stress lingers, extensive damage to the cell membrane also occurs and contributes to secondary damage induced by SM, leading to such conditions as bronchiolitis obliterans, asthma, and chronic obstructive pulmonary disease (COPD). Overall, ROS is one of the major causes of chronic toxicity to SM exposure. Much debate remains around whether ROS initiates inflammation and mitochondrial dysfunction or does DNA alkylation events and damage trigger an inflammatory response, leading to ROS increase.
Inhibition of NADPH-cytochrome P450 reductase:
Another mechanism that contributes to SM toxicity is the inhibition of NADPH-cytochrome P450 reductase145. Cytochrome P450 are enzymes that are abundantly found in the liver but are also located in many extrahepatic cells, including type II epithelial cells in the lung146. Additionally, the enzyme NADPH- cytochrome P450 reductase undergoes oxidation to donate an electron to cytochrome P450 to detoxify SM and its metabolites145. While this mechanism isn’t as extensively studied as previously mentioned, it has been shown in the literature that SM is a potent inhibitor of cytochrome P450 and NADPH-cytochrome P450 reductase145,147. If SM exposure is persistent and overloads the body, the inhibition of NADPH-cytochrome P450 reductase cannot detoxify SM and its metabolites. The excess of SM and its metabolites are readily available to react with other biomolecules.
Since many of the previously mentioned mechanisms have been individually studied based on the effects of SM, there is a clear connection of how each mechanism plays a critical role within each other. What remains unclear is what mechanism is initiated first, and how SM primarily exerts its toxicity. Once this is determined, a viable treatment option can be developed.
Respiratory Effects After Sulfur Mustard Inhalation
Veterans and civilians exposed to acute low dose SM will experience symptoms within 12-24 h such as rhinitis, sneezing, bloody nose, burning sensation of the nose and throat, shortness of breath, and a dry cough148. Those who have been exposed to a much higher concentrations experience these symptoms within 2-4 h according to the CDC (LCt50 = 3000mg-min/m3)35,148. Long-term effects from a one-time single exposure result in dyspnea, bronchitis, asthma, pulmonary fibrosis, bronchiolitis obliterans, and COPD148,149. While the mortality rate is low, most veterans experience chronic toxicity originating from the lung, attributed to the robust immune response35. This immune response is thought to impact many other organ systems further negatively throughout the body years after the exposure150.
While respiratory effects are less studied than effects in the skin and the eyes, it is thought that inflammatory processes occurring in the lung play a major role in long-term disease. One of the most studied cohorts are veterans who served in the Iraq-Iran war (1986). This cohort had known direct SM exposures while veterans who served in the GW and Iraq-Afghanistan war were thought to be exposed to a mixture of chemical warfare agents. The in-depth analysis of the Iran-Iraq cohort has allowed us to identify similarities between the veterans who have served in the GW and the Iraq-Afghanistan war. Veterans from all three wars have reported similar disease pathologies such as increased asthma, COPD, difficulty breathing, and bronchiolitis obliterans, years after exposure30,151,152. Several studies regarding the Iranian cohort of veterans determined that 10 to 20 years after SM exposures, the most affected organ is the lung (42.5-95%) compared to the skin and eyes153,154. In further assessing lung damage years after exposure in Iranian veterans, chest X-ray findings and spirometry tests were used.
Findings confirm approximately 5% of veterans have increased pleural thickening, 41% have increased vascular markings, and 37.3% have pathology consistent with bronchiectasis155. The spirometry test revealed obstructive breathing patterns in half of the veterans (110/220 cases)155. Moreover, studies have looked at inflammatory markers in exposed Iranian veterans’ BAL fluid. Analysis of the BAL fluid revealed a significant elevation in various chemokines such as MCP-1, MIP-1α, MIP-1β together with elevated neutrophils, lymphocytes, and eosinophils compared to control initial exposure in 1986, confirming the presence of persistent inflammation131. Similarly, there have been reports of elevated proinflammatory cytokines such as IL-6, TNF-α, and IL-1β in the same cohort of veterans131,134.. There has been a notable increase in bronchiolitis obliterans and evidence of pulmonary fibrosis amongst other respiratory issues such as chronic pulmonary obstruction, chronic bronchitis, and acute respiratory distress syndrome (ARDS)15,151,156. A recent study looked at the 10-year prevalence rate for fifteen respiratory diseases affecting veterans who actively served in the GW and veterans who were not deployed. Out of the 15 respiratory illnesses that were considered, this study found that per 100,000 veterans; there was an increase in the prevalence of chronic bronchitis (PR 1.19; 95% CI 1.10, 1.28), emphysema (PR 1.11; 95% CI 1.01, 1.21), chronic airway obstruction (PR 1.09; 95% CI 1.07, 1.12), and chronic obstructive pulmonary disease (PR 1.09; 1.07, 1.11)151. Many of these respiratory illnesses in this study overlap with the respiratory diseases experienced by Iranian veterans. While the literature fails to link GW veterans to SM-only exposures due to the many other exposures that occurred during the GW, it is essential to note that the similar lung pathology and respiratory symptoms experienced by Iranian veterans exposed to SM are analogous to the respiratory symptoms that comprise GWI. In understanding the mechanism of action of SM, there have been many advances in in vitro and in vivo research supporting the respiratory pathology in GWI.
Rat models have been extensively used in looking at the damage and acute inflammatory responses within the lung. Rats exposed to a high dose of 2 mg/kg of SM via intratracheal instillation had ulcerations, shedding of the epithelium, pulmonary fibrosis, airway remodeling, and inflammatory cell infiltration within 6 h persisting at 72 h157. The results described are comparable to what soldiers and civilians have experienced after a high exposure. In characterizing lung inflammation in rats following SM exposure, there was an increase in T and B lymphocytes, increased macrophages, and decreased glutathione peroxidase (GPx) by 72 h in the BAL157. The inflammatory cell infiltration at 6 h of SM exposure combined with an increase in GPx further strengthens the hypothesis that oxidative stress and immune response contribute to lung damage. SM exposure is known to cause pulmonary fibrosis and bronchiolitis obliterans in humans exposed, as previously mentioned. A study looking at rats exposed to 1.0 mg/kg of SM further demonstrated that bronchiolitis obliterans and pulmonary fibrosis gradually intensified while lung function decreased158. By 28 days, TGF-β, platelet derived growth factor (PDGF), and plasminogen activator inhibitor type 1 (PAI-1) elevated in the BAL, suggesting airway remodeling and activation of pro-fibrotic pathways contributing to the collagen formation is observed158. While rodent models are typical when assessing the toxicity of SM, swine models have also been used. In female swine exposed to 60 (sublethal SM dose) and 100 μg/kg (LD40) doses for 10 min showed an increase in damage measured by lung pathology scoring as well as various transcriptional changes159. This study utilized canonical pathway analysis and saw changes in various pathways such as aryl hydrocarbon receptor signaling, Nrf-2 mediated oxidative stress, differences in immune response, and effects on cell signaling metabolism159.
In studying SM exposure, a few surrogates are often employed due to SM’s restricted use and toxic nature. The two most used surrogates that elicit comparable immune response and damage in the lung are 2-chloroethyl ethyl sulfide (CEES, half mustard) and mechlorethamine (HN2, Nitrogen Mustard; NM). For most in vivo studies using CEES, the liquid is aerosolized at concentrations of 7.5-10% CEES in 90-92.5% ethanol and delivered via a nose-only system160,161. After 12- 18 h of CEES exposure in rats, there was evidence of epithelial damage due to increases in tissue factor activity in BAL, decreases in microRNA −140-5p (a marker for acute lung injury in tissue), and an increase in superoxide dismutase (SOD)162. Paired with the damage observed, there was an increase in proinflammatory cytokines IL-6, TNF-α, and IL-1α/β162. The comparable inflammatory responses of SM and CEES that occur as early as 12 h post-exposure raises the question of whether the inflammatory processes are initiating damage or if the damage caused is causing the inflammatory responses.
NM has been extensively used in rat and mice models via intratracheal instillation and oropharyngeal aspiration to determine if inflammation is responsible for generating the bulk of the damage in the lung. NM exposure is associated with increases in proinflammatory cytokines such as IL-6, TNF-α, IL-1β, and growth factors (TGF-β) at doses 0.08 mg/kg – 0.25 mg/kg133,163. First responding cells of the immune system are commonly macrophages and mast cells. Both cells are believed to play a critical role in initiating inflammation163–165. From 12- 72 h after NM treatment, macrophages [M1 (cytotoxic) and M2 (pro-resolving)] at the site of injury generate oxidative stress, eicosanoid production, and cytokines (TNF-α and IL6) as well as chemokines, all contributing to the altered lung function in rats and mice133,163. In understanding the attenuation of lung injury in rats by using an anti-TNF-α antibody (15 mg/kg TNF-α antibody 30 min after initial intratracheal instillation and then every nine days for 28 days), there was a decrease in M1 macrophage, but M2 remained mostly unchanged165. In this study, TGF-β, a profibrotic mediator, was also significantly reduced165. In looking at such exposures in mice, 14 days after 0.08 mg/kg of NM delivered via oropharyngeal aspirations, there was an increased expression of high mobility group protein 1 (HMGB1) and TNF-α associated with macrophage accumulation in tissue as well as high numbers of M1 macrophages correlating with TGF-β and fibrosis formation163.
In addition to macrophages, mast cells have very similar roles in the initiation and resolution of inflammatory responses due to NM exposure164. Mast cells are critical in the formation of fibrosis and are known for rapidly releasing granules with preformed mediators recruiting macrophages and neutrophils to the site of injury166. Within 72 h, SM exposure (0.2-0.6 mg/kg) correlates with an increase in TNF-α followed by infiltration of macrophages and neutrophils following a 3-day exposure129. This increase in macrophages and neutrophils three days after initial exposure may be due to the various mediators that mast cells release. In a recent study looking at the role of mast cells in C57BL/6J mice (Wildtype; WT) versus mast cell-deficient mice (Kitw-sh) 3 days after NM exposure (0.125 mg/kg via oropharyngeal aspiration), there was significant damage and immune cell infiltration in the WT that was absent in the mast cell deficient mice164. Additionally, there were increases in mRNA gene expression for proinflammatory mediators IL-6 and TNF-α in WT that were absent in mast cell deficient mice as well as the increase in eicosanoid formation (PGD2) in WT compared to the mast cell deficient mice164. Furthermore, it’s been reported while inflammatory responses are robust early on, 10 days and 30 days after initial exposure (0.625 and 0.312 mg/kg NM) in mice revealed an upregulation in pro-fibrotic biomarkers such as serine/threonine – selective protein kinases (p-ERK) and heat shock protein 90 in addition to an increase in collagen expression167. Mast cells play a prominent role in fibrosis and could be contributing to the collagen formation by producing TGF-β which is responsible for fibroblast proliferation and migration168. In identifying treatment targets, stabilizing mast cells could be a viable option for treatment immediately after SM exposure. While further research is needed to determine if mast cells or macrophages are viable treatment targets to reduce inflammation in the lung, inhalation to SM has also been implicated in damage in the brain169–171.
Inhalation of Sulfur Mustard Impacts the Central Nervous System
Inhalation of SM also results in neurological symptoms such as impaired cognition, severe headaches, lethargy, sleep disturbances, and increased depression 172,173. While SM-induced damage is known to target organs such as the skin, eyes, and the lung, the mechanism(s) by which SM exposure results in cognitive impairment has not been fully elucidated. In Iranian veterans, SM mustard exposure contributes to the breakdown of the blood-brain barrier (BBB), abnormalities in the cerebellum and hypothalamus, and the many microbleeds in areas where necrosis is present throughout the brain contributing to cognitive dysfunction 174. Due to its lipophilic properties, the accumulation of SM in the brain of post-mortem veterans gives us reason to believe that SM toxicity impacts regions of the brain responsible for cognition171. Interestingly, cognitive deficits are prominent features associated with GWI and include cognitive and memory problems, mood dysfunction, sleep disorders, and chronic fatigue 80. In addition to sharing similar cognitive deficits as seen in exposure to SM, GWI primarily affects those veterans deployed in wars mentioned previously where SM exposure has been documented 175.
Few studies have looked at the inhalation effects of SM on the brain, but it has been reported that significant neuronal and structural changes occur. Tekiner et al. exposed rats to a toxic dose of vaporized NM for 10 min (800 mg/m3/min), resulting in significant vascular oedema around major blood vessels and capillaries, abnormalities in neuronal and glial processes, and substantial changes in cellular components such as evidence of nuclear and mitochondrial degeneration176. Further analyzing whole brain tissue homogenates showed a significant increase in thiobarbituric acid reactive substances (TBARS), SOD activity, and GPx activity after NM exposure176. The same group wanted to determine if introducing the antioxidant proanthocyanin (PC) in the feed (100 mg/kg/day) would inhibit effects caused by NM exposure176. Their findings confirmed that the administration of an antioxidant such as PC could alleviate oxidative stress and lipid peroxidation in various brain regions and inhibit cellular degeneration176. Other studies have looked at the effects of SM, NM, and CEES on the brain through different modes of exposure, such as intraperitoneal (i.p.) and diffusion through the skin. Although the routes of exposure are different, there are comparable results of neuronal degeneration and ROS formation in the brain.
Potential Therapeutics and Therapeutic Targets
Considering there are no therapeutic options, much of the current research focuses on identifying potential targets to develop therapeutics to treat individuals exposed through inhalation of SM. Many anti-inflammatory drugs have been used in vivo. In acute studies involving inhalation exposure to SM, NM, or CEES, they have shown promises, such as macrolides, glucocorticoid steroids, antioxidants, and anti-TNF-α antibodies. Macrolides are antibiotics typically used in the treatment of bacterial infections. In rats exposed to 0.25 mg of SM delivered via an intratracheal tube, roxithromycin (commonly used macrolide) given 1 h prior to exposure and therein 24 h after for 1, 3, and 7 days found that at three days there was a significant improvement in lung function177.
Additionally, lung scores and survival at all doses of roxithromycin (10, 20, 40 mg/kg) improved in a dose-dependent manner177. Glucocorticoid steroids such as betamethasone and dexamethasone have been around for a few decades and have been previously used in guinea pigs exposed to SM178,179. Administration of both glucocorticoid steroids decreased airway hyperresponsiveness and repaired parts of the airway epithelium in SM exposed guinea pigs179. In addition to this study, C57BL/6J mice were exposed to 10 mg/kg of NM, and dexamethasone was administered (i.p.) once after either 1, 2, or 6 h post-exposure180. Dexamethasone protected the airways of mice significantly at all time points through the reduction of inflammatory cell infiltration180.
Interestingly at 1 h of dexamethasone administration, there was better inhibition of collagen formation after 14 days180. Early treatment with glucocorticoid steroids in both SM and NM models (guinea pigs and mice) shows a significant reduction in inflammatory cells at different concentrations, making it a viable prophylactic179,180. As previously mentioned, ROS is a substantial contributor to injury. Metalloporphyrin catalytic antioxidant AEOL 10150 (after 1 and 9 h of 5mg/kg s.c.) showed significant improvement in the lungs of mice that were exposed to 5% of CEES for 15 minutes181. Oxidative stress markers 8-OHdG and 4-HNE were significantly decreased in the lung, which demonstrates AEOL10150 is an efficient antioxidant agent181. Other studies have shown the elevation of TNF-α after NM or SM inhalation exposures in rodent models. Treatment with anti-TNF-α was associated with a reduction in collagen deposition and suppression of proinflammatory cytotoxic macrophages in NM exposed lung165. Another therapeutics target being considered is an extracellular nucleic acid (eNA), which are essential mediators in the inflammatory process and damage161,182. With eNA’s being a potential therapeutic target, this study utilizes eNA scavenger hexadimethrine bromide161. Twelve hours after a 10% CEES inhalation exposure in rats, there was an increase in eNA in the BAL of the lungs161. Upregulation in eNA in the BAL can lead to inflammation and barrier dysfunction within the lungs161. Hexadimethrine bromide is an extracellular nucleic acid scavenger161,183. Hexadimethrine bromide (10 mg/kg) was administered to rats 2 h post-exposure and resulted in improved blood oxygenation, reversal of ARDS, and little to no alveolar-capillary barrier damage161. While there aren’t any approved treatment options to prevent the pathophysiological effects of SM exposure, various cellular targets have been identified for potential therapeutics. There is a clear need to identify therapeutics further potentially targeting vital immune cells such as mast cells and macrophages or oxidative stress based on our knowledge of mechanisms of toxicity.
Iraq and Afghanistan Wars
Many of the exposures experienced in the GW were also experienced in the Iraq-Afghanistan war but over a longer time frame. Soldiers during the Iraq War were exposed to aging chemical weapons left over from the Iran-Iraq War36. Thus far, in the Iraq war, there were reports of CWA exposure in both operations Iraqi Freedom and New Dawn36. Little is known about the chemical exposure experienced during the Afghanistan war since it ended very recently in August 2021. Although many veterans’ self-reported exposures to GB, there is difficulty in corroborating the reports with military records28. This lack of corroboration is a significant issue as researchers cannot confirm that soldiers self-reporting exposures to chemical weapons were actually near any confirmed sites of chemical attack or weapons storage and/or demolition28. In one study using veteran self-reports, many veterans reported receiving PB, even though they were not supposed to have been administered PB 28. In recent military operations, there have been reports of soldiers being exposed to SM through the handling and destruction of explosives during the Iraq- Afghanistan conflicts between 2003 to 201528. While this information has been reported in the media, the exact number of U.S. soldiers that were exposed remains unconfirmed.
As a result, veterans who served in the Iraq-Afghanistan war (2001-2021) are experiencing similar symptoms27–29. Amidst the 2.5 million men and women deployed to the Iraq-Afghanistan war, also known as the “War on Terror” (post 9/11/2001 terrorist attacks), many veterans who have returned experienced symptoms related to CMI. One year after returning from deployment, 49.5% of veterans met the criteria for CMI, similar to that seen in GW veterans27. Respiratory issues were one of the many reported symptoms expressed by veterans27,30. There was a 28% increase in veterans experiencing allergies and a 19% increase in asthma after soldiers returned from deployment30. To further assess military exposure and long-term health outcomes, the Millennium cohort group was established in 2001. This prospective study conducted by the DoD reported an increase in mental and respiratory illness months after arriving from deployment27. Veterans in the Millennium cohort had trouble sleeping (51.4%), were moody/irritable (50.8%), had joint pain (46.0%), fatigue (39.5%), difficulty remembering (39.8%), concentration issues, headaches (36.1%), and sinus problems (30.0%)27. Veterans who served in the GW and Iraq- Afghanistan war have reported lung disease pathologies similar to those in SM-exposed veterans of the Iran-Iraq War.
Due to the nature of the recently concluded conflict in Afghanistan (2021) and the exposures troops experienced that parallel those in the GW, there is more to be learned on the factors potentially contributing to the CMI observed in the Iraq-Afghanistan veterans.
Conclusion
Over 30% of veterans who served in the Persian Gulf War have suffered respiratory and cognitive effects due to various exposures, including CWAs35. While there isn’t an exact number of veterans affected from the Afghanistan- Iraq war that recently ended (2021), there is a rising number of reports of respiratory and cognitive effects28.
With GWI/CMI recognized in both veteran populations, and exposures to CWAs occurring during both wars, it is suggested that CWAs such as GB, GF, and SM have critical roles in the manifestation of this illness5,29,46,75. While there are few reports of the lungs being affected in GB and GF exposures, the mode by which many of these animal models are exposed is not what is typically experienced during the war (i.p. or s.c.) when studying respiratory effects. Due to GB and GF being an OP compound, the vast knowledge in the literature revolves around neurological effects, and very little explores the respiratory damage experienced by veterans as a result of inhaling these compounds. This needs to be addressed because pulmonary injury and inflammation are critical factors in GWI/CMI15,30. The known neurocognitive effects resulting from GB/GF exposure are thought to involve hippocampal injury, neuroinflammation, and AChE inhibition74,75. It remains unclear what mechanisms contribute to neuroinflammation and hippocampal injury and why veterans experience memory loss years after exposure74,75. Long-term animal studies could help bridge this gap in the literature. Although GB, GF, and SM are highly toxic chemicals, many veterans exposed do not die from the exposure itself, rather experience various symptoms years after exposure35,64,75,148. Long-term animal studies will help further identify mechanisms and biomarkers that may contribute to both lung and brain pathology encountered in veterans.
Sulfur mustard is also believed to play a critical role in the manifestation of GWI. Many of the symptoms manifested in GWI have also been observed in Iranian veterans from the Iran-Iraq war who had known SM exposures150,151,174. While surrogates are valuable in understanding the potential mechanisms that SM is implicated in, many of these studies need to be carried out further using the actual compound SM. It has been shown that CEES and NM compared to SM have similar immune effects and damage on the lung and various other organs, but there are still differences in toxicity amongst these compounds184–186. In addition to using the actual compound SM, the literature lacks exploring the neurocognitive effects of SM exposure and its role in neuronal damage. There is evidence that the parent compound, SM, accumulates in post-mortem patients, and such exposure can contribute to cognition decline experienced by veterans171,173,174. These findings have been shown in Iranian veterans, but very few animal studies have addressed mechanisms.
Both wars previously mentioned had various other exposures such as depleted uranium (DU), pesticides (organophosphates and carbamates), fine particulate matter (PM) (building fires, oil well fires, and desert dust), and CWAs were not exclusive to either war. With what is already known about CWA’s and the injury it has inflicted on various military personnel, it does appear that CWA’s may play a critical role in the disease pathology seen in GWI/CMI.
Funding:
This research was supported by the National Institute of Environmental Health Sciences R01 ES019311S1, T32 ES029074 and U.S. Army Medical Research Grant W81XWH1810169.
List of abbreviations:
- ACh
Acetylcholine
- AChE
Acetylcholinesterase
- ALS
Amyotrophic lateral sclerosis
- ARDS
Acute respiratory distress syndrome
- ATP
Adenosine triphosphate
- BAL
Bronchioalveolar lavage
- BBB
Blood-brain barrier
- BChE
Butyrylcholinesterase
- CDC
Center for Disease Control
- CEES
2-chloroethyl ethyl sulfide
- CMI
Chronic multisymptom illness
- CNS
Central nervous system
- COPD
Chronic obstructive pulmonary disease
- CPO
Chlorpyrifos-oxon
- CWA
Chemical warfare agent
- DFP
Diisopropyl fluorophosphate
- DFPase
Diisopropyl fluorophosphatase
- DoD
Department of Defense
- DU
Depleted uranium
- eNA
extracellular nucleic acid
- FDC
Ft. Devens cohort
- GB
Sarin
- GF
Cyclosarin
- GPx
Glutathione peroxidase
- GSH
Glutathione
- GW
Gulf war
- GWECB
Gulf war era cohort and biorepository
- GWI
Gulf war illness
- HMGB1
High mobility group box 1 protein
- MCP-1
Monocyte chemoattractant protein-1
- MIP-1α
Macrophage inflammatory protein 1
- NAD
Nicotine adenine dinucleotide
- NHANES
National Health and Nutrition Examination Survey
- NM
Nitrogen mustard
- NO
Nitric oxide
- OP
Organophosphate
- PAI-1
Plasminogen activator inhibitor type 1
- PAR
Poly-ADP-ribose
- PARP-1
Poly (ADP-ribose) polymerase type1
- PB
Pyridostigmine bromide
- PC
Proanthocyanin
- PD
Parkinson’s disease
- PDGF
Platelet derived growth factor
- PEG
Polyethylene glycol
- PM
Particulate matter
- PON1
Paraoxonase 1
- ROS
Reactive oxygen species
- SE
Status epilepticus
- SM
Sulfur mustard
- SOD
Superoxide dismutase
- TBARS
Thiobarbituric acid reactive substances
- VA
Veteran affairs
Bibliography
- 1.Institute of Medicine Committee on Gulf, W. & Health: Health Effects of Serving in the Gulf War, U. in Gulf War and Health: Volume 8: Update of Health Effects of Serving in the Gulf War (National Academies Press (US)Copyright 2010 by the National Academy of Sciences. All rights reserved., 2010). [Google Scholar]
- 2.Murphy FM Gulf war syndrome. BMJ 318, 274–275, doi: 10.1136/bmj.318.7179.274 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaimal G & Dieterich-Hartwell R Grappling with Gulf War Illness: Perspectives of Gulf War Providers. Int J Environ Res Public Health 17, doi: 10.3390/ijerph17228574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maule AL et al. Meta-analysis of self-reported health symptoms in 1990–1991 Gulf War and Gulf War-era veterans. BMJ Open 8, e016086, doi: 10.1136/bmjopen-2017-016086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Affairs, U. S. D. o. V. Gulf War Illnesses linked to Southwest Asia service, <https://www.va.gov/disability/eligibility/hazardous-materials-exposure/gulf-war-illness-southwest-asia/> (2021).
- 6.Zundel CG et al. Rates of Chronic Medical Conditions in 1991 Gulf War Veterans Compared to the General Population. Int J Environ Res Public Health 16, doi: 10.3390/ijerph16060949 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steele L Prevalence and patterns of Gulf War illness in Kansas veterans: association of symptoms with characteristics of person, place, and time of military service. Am J Epidemiol 152, 992–1002, doi: 10.1093/aje/152.10.992 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Barth SK, Kang HK, Bullman TA & Wallin MT Neurological mortality among U.S. veterans of the Persian Gulf War: 13-year follow-up. Am J Ind Med 52, 663–670, doi: 10.1002/ajim.20718 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Heaton KJ et al. Quantitative magnetic resonance brain imaging in US army veterans of the 1991 Gulf War potentially exposed to sarin and cyclosarin. Neurotoxicology 28, 761–769, doi: 10.1016/j.neuro.2007.03.006 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Proctor SP, Heaton KJ, Heeren T & White RF Effects of sarin and cyclosarin exposure during the 1991 Gulf War on neurobehavioral functioning in US army veterans. Neurotoxicology 27, 931–939, doi: 10.1016/j.neuro.2006.08.001 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Horner RD et al. Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology 61, 742–749, doi: 10.1212/01.wnl.0000069922.32557.ca (2003). [DOI] [PubMed] [Google Scholar]
- 12.Chao L Do Gulf War veterans with high levels of deployment-related exposures display symptoms suggestive of Parkinson’s disease? Int J Occup Med Environ Health 32, 503–526, doi: 10.13075/ijomeh.1896.01346 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barth SK, Dursa EK, Bossarte RM & Schneiderman AI Trends in brain cancer mortality among U.S. Gulf War veterans: 21 year follow-up. Cancer Epidemiol 50, 22–29, doi: 10.1016/j.canep.2017.07.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christova P et al. Subcortical brain atrophy in Gulf War Illness. Exp Brain Res 235, 2777–2786, doi: 10.1007/s00221-017-5010-8 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Santos Charles J., M.D. , & Stevens Rachael R., B.A. Gulf War Syndrome: Respiratory and Psychiatric Distress in a Gulf War Veteran. American Journal of Psychiatry Residents’ Journal 15, 26–28, doi: 10.1176/appi.ajp-rj.2020.150415 (2020). [DOI] [Google Scholar]
- 16.Simmons R, Maconochie N & Doyle P Self-reported ill health in male UK Gulf War veterans: a retrospective cohort study. BMC Public Health 4, 27, doi: 10.1186/1471-2458-4-27 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heboyan V et al. Sex Differences in Gulf War Illness: A Reanalysis of Data From the CDC Air Force Study Using CDC and Modified Kansas Case Definitions. J Occup Environ Med 61, 610–616, doi: 10.1097/JOM.0000000000001620 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Krengel M et al. Neurotoxicant exposures and rates of Chronic Multisymptom Illness and Kansas Gulf War Illness criteria in Gulf War deployed women veterans. Life Sci 280, 119623, doi: 10.1016/j.lfs.2021.119623 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Coughlin SS et al. A Review of Epidemiologic Studies of the Health of Gulf War Women Veterans. J Environ Health Sci 3, doi: 10.15436/2378-6841.17.1551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oberlin S et al. Sex-specific differences in plasma lipid profiles are associated with Gulf War Illness. J Transl Med 20, 73, doi: 10.1186/s12967-022-03272-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abou-Donia MB et al. Sex-Based Differences in Plasma Autoantibodies to Central Nervous System Proteins in Gulf War Veterans versus Healthy and Symptomatic Controls. Brain Sci 11, doi: 10.3390/brainsci11020148 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Craddock TJ et al. A role for homeostatic drive in the perpetuation of complex chronic illness: Gulf War Illness and chronic fatigue syndrome. PLoS One 9, e84839, doi: 10.1371/journal.pone.0084839 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryant JD et al. Neuroimmune mechanisms of cognitive impairment in a mouse model of Gulf War illness. Brain Behav Immun 97, 204–218, doi: 10.1016/j.bbi.2021.07.015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binns J et al. Research advisory committee on Gulf War veterans’ illnesses. Gulf War illness and the health of gulf war veterans. Washington, DC: Department of Veterans Affairs; (2008). [Google Scholar]
- 25.Hernandez S, Morales-Soto W, Grubisic V, Fried D & Gulbransen BD Pyridostigmine bromide exposure creates chronic, underlying neuroimmune disruption in the gastrointestinal tract and brain that alters responses to palmitoylethanolamide in a mouse model of Gulf War Illness. Neuropharmacology 179, 108264, doi: 10.1016/j.neuropharm.2020.108264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez S, Fried DE, Grubisic V, McClain JL & Gulbransen BD Gastrointestinal neuroimmune disruption in a mouse model of Gulf War illness. FASEB J 33, 6168–6184, doi: 10.1096/fj.201802572R (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McAndrew LM et al. Iraq and Afghanistan Veterans report symptoms consistent with chronic multisymptom illness one year after deployment. J Rehabil Res Dev 53, 59–70, doi: 10.1682/JRRD.2014.10.0255 (2016). [DOI] [PubMed] [Google Scholar]
- 28.DeBeer BB et al. The Association Between Toxic Exposures and Chronic Multisymptom Illness in Veterans of the Wars of Iraq and Afghanistan. J Occup Environ Med 59, 54–60, doi: 10.1097/JOM.0000000000000922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brower V Gulf War syndrome revisited. EMBO Rep 4, 551–553, doi: 10.1038/sj.embor.embor874 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garshick E et al. Respiratory Health after Military Service in Southwest Asia and Afghanistan. An Official American Thoracic Society Workshop Report. Ann Am Thorac Soc 16, e1–e16, doi: 10.1513/AnnalsATS.201904-344WS (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peakman M, Skowera A & Hotopf M Immunological dysfunction, vaccination and Gulf War illness. Philos Trans R Soc Lond B Biol Sci 361, 681–687, doi: 10.1098/rstb.2006.1826 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyams KC, Riddle J, Trump DH & Graham JT Endemic infectious diseases and biological warfare during the Gulf War: a decade of analysis and final concerns. Am J Trop Med Hyg 65, 664–670, doi: 10.4269/ajtmh.2001.65.664 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Haley RW Is Gulf War syndrome due to stress? The evidence reexamined. Am J Epidemiol 146, 695–703, doi: 10.1093/oxfordjournals.aje.a009343 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Health, I. o. M. U. C. o. G. W. a. in Gulf War and Health: Updated Literature Review of Sarin (2004). [PubMed] [Google Scholar]
- 35.National Academies of Sciences, E., and Medicine; Health and Medicine Division; Board on Population Health and Public Health Practice. in Gulf War and Health: Volume 11: Generational Health Effects of Serving in the Gulf War Vol. 11 (National Academies Press (US), 2018). [PubMed] [Google Scholar]
- 36.Baird C et al. Chemical Weapons Exposures in Iraq: Challenges of a Public Health Response a Decade Later. US Army Med Dep J, 75–84 (2016). [PubMed] [Google Scholar]
- 37.Stewart RW War in the Persian Gulf: Operations Desert Shield and Desert Storm: August 1990-March 1991. Vol. 70 (Government Printing Office, 2010). [Google Scholar]
- 38.Committee, H. (Washington, DC United States Congress, 105th Congress, 1st Session). [Google Scholar]
- 39.Abou-Donia MB, Siracuse B, Gupta N & Sobel Sokol A Sarin (GB, O-isopropyl methylphosphonofluoridate) neurotoxicity: critical review. Crit Rev Toxicol 46, 845–875, doi: 10.1080/10408444.2016.1220916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maxwell DM, Brecht KM & Sweeney RE A common mechanism for resistance to oxime reactivation of acetylcholinesterase inhibited by organophosphorus compounds. Chem Biol Interact 203, 72–76, doi: 10.1016/j.cbi.2012.08.024 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Colovic MB, Krstic DZ, Lazarevic-Pasti TD, Bondzic AM & Vasic VM Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol 11, 315–335, doi: 10.2174/1570159X11311030006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor P Anticholinesterase agents. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. Twelfth edition. New York, Macmillan, 239–254 (2011). [Google Scholar]
- 43.Hogg RC, Raggenbass M & Bertrand D Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol 147, 1–46, doi: 10.1007/s10254-003-0005-1 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Weinstock M Activation of central muscarinic receptors causes respiratory stimulation in conscious animals. Br J Pharmacol 74, 587–592, doi: 10.1111/j.1476-5381.1981.tb10468.x (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhuang Q et al. Efforts toward treatments against aging of organophosphorus-inhibited acetylcholinesterase. Ann N Y Acad Sci 1374, 94–104, doi: 10.1111/nyas.13124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brimfield AA Chemicals of military deployments: revisiting Gulf War Syndrome in light of new information. Prog Mol Biol Transl Sci 112, 209–230, doi: 10.1016/B978-0-12-415813-9.00007-6 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Mahan CM, Page WF, Bullman TA & Kang HK Health effects in Army Gulf War veterans possibly exposed to chemical munitions destruction at Khamisiyah, Iraq: Part I. Morbidity associated with potential exposure. Mil Med 170, 935–944, doi: 10.7205/milmed.170.11.935 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Haley RW et al. Cholinergic autonomic dysfunction in veterans with Gulf War illness: confirmation in a population-based sample. JAMA Neurol 70, 191–200, doi: 10.1001/jamaneurol.2013.596 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Terry AV Jr. et al. Repeated, intermittent exposures to diisopropylfluorophosphate in rats: protracted effects on cholinergic markers, nerve growth factor-related proteins, and cognitive function. Neuroscience 176, 237–253, doi: 10.1016/j.neuroscience.2010.12.031 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Locker AR et al. Corticosterone primes the neuroinflammatory response to Gulf War Illness-relevant organophosphates independently of acetylcholinesterase inhibition. J Neurochem 142, 444–455, doi: 10.1111/jnc.14071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller JV et al. The Neuroinflammatory Phenotype in a Mouse Model of Gulf War Illness is Unrelated to Brain Regional Levels of Acetylcholine as Measured by Quantitative HILIC-UPLC-MS/MS. Toxicol Sci 165, 302–313, doi: 10.1093/toxsci/kfy130 (2018). [DOI] [PubMed] [Google Scholar]
- 52.O’Callaghan JP, Kelly KA, Locker AR, Miller DB & Lasley SM Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J Neurochem 133, 708–721, doi: 10.1111/jnc.13088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yates PL et al. A cellular approach to understanding and treating Gulf War Illness. Cell Mol Life Sci 78, 6941–6961, doi: 10.1007/s00018-021-03942-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lein PJ & Fryer AD Organophosphorus insecticides induce airway hyperreactivity by decreasing neuronal M2 muscarinic receptor function independent of acetylcholinesterase inhibition. Toxicol Sci 83, 166–176, doi: 10.1093/toxsci/kfi001 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Katz LS & Marquis JK Modulation of central muscarinic receptor binding in vitro by ultralow levels of the organophosphate paraoxon. Toxicol Appl Pharmacol 101, 114–123, doi: 10.1016/0041-008x(89)90217-2 (1989). [DOI] [PubMed] [Google Scholar]
- 56.Pope CN Organophosphorus pesticides: do they all have the same mechanism of toxicity? J Toxicol Environ Health B Crit Rev 2, 161–181, doi: 10.1080/109374099281205 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Guignet M et al. Persistent behavior deficits, neuroinflammation, and oxidative stress in a rat model of acute organophosphate intoxication. Neurobiol Dis 133, 104431, doi: 10.1016/j.nbd.2019.03.019 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leonel Javeres MN et al. Analysis of PON1 gene polymorphisms (rs662 and rs854560) and inflammatory markers in organophosphate pesticides exposed cohorts from two distinct populations. Environ Res 191, 110210, doi: 10.1016/j.envres.2020.110210 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y et al. Effects of chlorpyrifos exposure on liver inflammation and intestinal flora structure in mice. Toxicol Res (Camb) 10, 141–149, doi: 10.1093/toxres/tfaa108 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meng Y et al. Alkyl organophosphate flame retardants (OPFRs) induce lung inflammation and aggravate OVA-simulated asthmatic response via the NF-small ka, CyrillicB signaling pathway. Environ Int 163, 107209, doi: 10.1016/j.envint.2022.107209 (2022). [DOI] [PubMed] [Google Scholar]
- 61.Zhuang J et al. Inhalation of the nerve gas sarin impairs ventilatory responses to hypercapnia and hypoxia in rats. Toxicol Appl Pharmacol 232, 440–447, doi: 10.1016/j.taap.2008.07.016 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stewart W The effects of sarin and atropine on the respiratory center and neuromuscular junctions of the rat. Canadian journal of biochemistry and physiology 37, 651–660 (1959). [PubMed] [Google Scholar]
- 63.Ikeda T et al. Regional quantification of muscarinic acetylcholine receptors and beta-adrenoceptors in human airways. Br J Pharmacol 166, 1804–1814, doi: 10.1111/j.1476-5381.2012.01881.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Willis K, Salem H & Sidell FR in Encyclopedia of Toxicology (Third Edition) (ed Philip Wexler) 213–217 (Academic Press, 2014). [Google Scholar]
- 65.Buels KS & Fryer AD Muscarinic receptor antagonists: effects on pulmonary function. Handb Exp Pharmacol, 317–341, doi: 10.1007/978-3-642-23274-9_14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pant SC, Vijayaraghavan R & Das Gupta S Sarin induced lung pathology and protection by standard therapy regime. Biomed Environ Sci 6, 103–111 (1993). [PubMed] [Google Scholar]
- 67.Pena-Philippides JC et al. Long- and short-term changes in the neuroimmune-endocrine parameters following inhalation exposures of F344 rats to low-dose sarin. Toxicol Sci 97, 181–188, doi: 10.1093/toxsci/kfm017 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Kassa J et al. The influence of single or repeated low-level sarin exposure on immune functions of inbred BALB/c mice. Basic Clin Pharmacol Toxicol 94, 139–143, doi: 10.1111/j.1742-7843.2004.pto940307.x (2004). [DOI] [PubMed] [Google Scholar]
- 69.Program NT NTP monograph on the systematic review of long-term neurological effects following acute exposure to sarin. Ntp Monograph (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rickett DL, Glenn JF & Beers ET Central respiratory effects versus neuromuscular actions of nerve agents. Neurotoxicology 7, 225–236 (1986). [PubMed] [Google Scholar]
- 71.Gundavarapu S et al. A critical role of acute bronchoconstriction in the mortality associated with high-dose sarin inhalation: effects of epinephrine and oxygen therapies. Toxicol Appl Pharmacol 274, 200–208, doi: 10.1016/j.taap.2013.11.007 (2014). [DOI] [PubMed] [Google Scholar]
- 72.White RF et al. Neuropsychological function in Gulf War veterans: relationships to self-reported toxicant exposures. Am J Ind Med 40, 42–54, doi: 10.1002/ajim.1070 (2001). [DOI] [PubMed] [Google Scholar]
- 73.Toomey R et al. Neuropsychological functioning of U.S. Gulf War veterans 10 years after the war. J Int Neuropsychol Soc 15, 717–729, doi: 10.1017/S1355617709990294 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Chao LL Evidence of Objective Memory Impairments in Deployed Gulf War Veterans With Subjective Memory Complaints. Mil Med 182, e1625–e1631, doi: 10.7205/MILMED-D-16-00309 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Chao LL, Kriger S, Buckley S, Ng P & Mueller SG Effects of low-level sarin and cyclosarin exposure on hippocampal subfields in Gulf War Veterans. Neurotoxicology 44, 263–269, doi: 10.1016/j.neuro.2014.07.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi M et al. Repeated low-dose exposures to sarin disrupted the homeostasis of phospholipid and sphingolipid metabolism in guinea pig hippocampus. Toxicol Lett 338, 32–39, doi: 10.1016/j.toxlet.2020.11.020 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Lisman J et al. Viewpoints: how the hippocampus contributes to memory, navigation and cognition. Nat Neurosci 20, 1434–1447, doi: 10.1038/nn.4661 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jeffrey MG et al. Neuropsychological Findings in Gulf War Illness: A Review. Front Psychol 10, 2088, doi: 10.3389/fpsyg.2019.02088 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hom J, Haley RW & Kurt TL Neuropsychological correlates of Gulf War syndrome. Arch Clin Neuropsychol 12, 531–544 (1997). [PubMed] [Google Scholar]
- 80.Janulewicz PA et al. Neuropsychological characteristics of Gulf War illness: A meta-analysis. PLoS One 12, e0177121, doi: 10.1371/journal.pone.0177121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Christova P et al. Gulf War Illness: C-reactive protein is associated with reduction of the volume of hippocampus and decreased fractional anisotropy of the fornix. Journal of Neurology & Neuromedicine 5 (2020). [Google Scholar]
- 82.Yamasue H et al. Human brain structural change related to acute single exposure to sarin. Ann Neurol 61, 37–46, doi: 10.1002/ana.21024 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Genovese RF, Benton BJ, Shippee SJ, Jakubowski EM & Bonnell JC Effects of low-level inhalation exposure to cyclosarin on learned behaviors in Sprague-Dawley rats. J Toxicol Environ Health A 69, 2167–2180, doi: 10.1080/15287390600748153 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Chaubey K et al. Differential proteome analysis of rat plasma after diisopropyl fluorophosphate (DFP) intoxication, a surrogate of nerve agent sarin. Chem Biol Interact 298, 66–71, doi: 10.1016/j.cbi.2018.10.026 (2019). [DOI] [PubMed] [Google Scholar]
- 85.Phillips KF & Deshpande LS Repeated low-dose organophosphate DFP exposure leads to the development of depression and cognitive impairment in a rat model of Gulf War Illness. Neurotoxicology 52, 127–133, doi: 10.1016/j.neuro.2015.11.014 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Phillips KF & Deshpande LS Calcium Hypothesis of Gulf War Illness: Role of Calcium Ions in Neurological Morbidities in a DFP-Based Rat Model for Gulf War Illness. Neurosci Insights 15, 2633105520979841, doi: 10.1177/2633105520979841 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eyer P The role of oximes in the management of organophosphorus pesticide poisoning. Toxicol Rev 22, 165–190, doi: 10.2165/00139709-200322030-00004 (2003). [DOI] [PubMed] [Google Scholar]
- 88.Marrs TC Diazepam in the treatment of organophosphorus ester pesticide poisoning. Toxicol Rev 22, 75–81, doi: 10.2165/00139709-200322020-00002 (2003). [DOI] [PubMed] [Google Scholar]
- 89.Valiyaveettil M et al. Protective efficacy of catalytic bioscavenger, paraoxonase 1 against sarin and soman exposure in guinea pigs. Biochem Pharmacol 81, 800–809, doi: 10.1016/j.bcp.2010.12.024 (2011). [DOI] [PubMed] [Google Scholar]
- 90.Trovaslet-Leroy M et al. Organophosphate hydrolases as catalytic bioscavengers of organophosphorus nerve agents. Toxicol Lett 206, 14–23, doi: 10.1016/j.toxlet.2011.05.1041 (2011). [DOI] [PubMed] [Google Scholar]
- 91.McGarry KG et al. A Novel, Modified Human Butyrylcholinesterase Catalytically Degrades the Chemical Warfare Nerve Agent, Sarin. Toxicol Sci 174, 133–146, doi: 10.1093/toxsci/kfz251 (2020). [DOI] [PubMed] [Google Scholar]
- 92.Raveh L et al. Human butyrylcholinesterase as a general prophylactic antidote for nerve agent toxicity. In vitro and in vivo quantitative characterization. Biochem Pharmacol 45, 2465–2474, doi: 10.1016/0006-2952(93)90228-o (1993). [DOI] [PubMed] [Google Scholar]
- 93.Yao Y, Liu J & Zhan CG Why does the G117H mutation considerably improve the activity of human butyrylcholinesterase against sarin? Insights from quantum mechanical/molecular mechanical free energy calculations. Biochemistry 51, 8980–8992, doi: 10.1021/bi3009246 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wymore T, Field MJ, Langan P, Smith JC & Parks JM Hydrolysis of DFP and the nerve agent (S)-sarin by DFPase proceeds along two different reaction pathways: implications for engineering bioscavengers. J Phys Chem B 118, 4479–4489, doi: 10.1021/jp410422c (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Allahyari H & Latifi AM Diisopropyl-fluorophosphatase as a catalytic bioscavenger. Journal of Applied Biotechnology Reports 3, 477–482 (2016). [Google Scholar]
- 96.Worek F et al. Efficacy of the rePON1 mutant IIG1 to prevent cyclosarin toxicity in vivo and to detoxify structurally different nerve agents in vitro. Arch Toxicol 88, 1257–1266, doi: 10.1007/s00204-014-1204-z (2014). [DOI] [PubMed] [Google Scholar]
- 97.Melzer M et al. In vitro and in vivo efficacy of PEGylated diisopropyl fluorophosphatase (DFPase). Drug Test Anal 4, 262–270, doi: 10.1002/dta.363 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Kuruba R, Wu X & Reddy DS Benzodiazepine-refractory status epilepticus, neuroinflammation, and interneuron neurodegeneration after acute organophosphate intoxication. Biochim Biophys Acta Mol Basis Dis 1864, 2845–2858, doi: 10.1016/j.bbadis.2018.05.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reddy DS et al. Phenobarbital as alternate anticonvulsant for organophosphate-induced benzodiazepine-refractory status epilepticus and neuronal injury. Epilepsia Open 5, 198–212, doi: 10.1002/epi4.12389 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rojas A et al. Beneficial Outcome of Urethane Treatment Following Status Epilepticus in a Rat Organophosphorus Toxicity Model. eNeuro 5, doi: 10.1523/ENEURO.0070-18.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rojas A, Wang J, Glover A & Dingledine R Urethane attenuates early neuropathology of diisopropylfluorophosphate-induced status epilepticus in rats. Neurobiol Dis 140, 104863, doi: 10.1016/j.nbd.2020.104863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lumley L et al. Neurosteroid and benzodiazepine combination therapy reduces status epilepticus and long-term effects of whole-body sarin exposure in rats. Epilepsia Open 4, 382–396, doi: 10.1002/epi4.12344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davies R, Ahmed G & Freer T Chronic exposure to organophosphates: background and clinical picture. Advances in Psychiatric Treatment 6, 187–192 (2000). [Google Scholar]
- 104.McCauley LA et al. Chronic fatigue in a population-based study of Gulf War veterans. Arch Environ Health 57, 340–348, doi: 10.1080/00039890209601419 (2002). [DOI] [PubMed] [Google Scholar]
- 105.Zhu J, Hawkins E, Phillips K & Deshpande LS Assessment of Ketamine and Its Enantiomers in an Organophosphate-Based Rat Model for Features of Gulf War Illness. Int J Environ Res Public Health 17, doi: 10.3390/ijerph17134710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Illnesses GW (GAO-04-821T. Washington, DC: US GAO, June 1, 2004. Gulf War Illnesses; …, 2004). [Google Scholar]
- 107.Shakarjian MP et al. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol Sci 114, 5–19, doi: 10.1093/toxsci/kfp253 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kehe K et al. Sulfur mustard research--strategies for the development of improved medical therapy. Eplasty 8, e32 (2008). [PMC free article] [PubMed] [Google Scholar]
- 109.Kortepeter MG & Newmark J Sulfur mustard: a liquid, not a gas. Clin Infect Dis 54, 885–886, doi: 10.1093/cid/cir962 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ludlum DB, Austin-Ritchie P, Hagopian M, Niu TQ & Yu D Detection of sulfur mustard-induced DNA modifications. Chem Biol Interact 91, 39–49, doi: 10.1016/0009-2797(94)90005-1 (1994). [DOI] [PubMed] [Google Scholar]
- 111.Bielmann A et al. Synthesis of different glutathione–sulfur mustard adducts of verified and potential biomarkers. RSC advances 8, 23881–23890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ludlum DB, Kent S & Mehta JR Formation of O6-ethylthioethylguanine in DNA by reaction with the sulfur mustard, chloroethyl sulfide, and its apparent lack of repair by O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 7, 1203–1206, doi: 10.1093/carcin/7.7.1203 (1986). [DOI] [PubMed] [Google Scholar]
- 113.Tewari-Singh N & Agarwal R Mustard vesicating agent-induced toxicity in the skin tissue and silibinin as a potential countermeasure. Ann N Y Acad Sci 1374, 184–192, doi: 10.1111/nyas.13099 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bhat KR, Benton BJ, Rosenthal DS, Smulson ME & Ray R Role of poly(ADP-ribose) polymerase (PARP) in DNA repair in sulfur mustard-exposed normal human epidermal keratinocytes (NHEK). J Appl Toxicol 20 Suppl 1, S13–17, doi: (2000). [DOI] [PubMed] [Google Scholar]
- 115.Rosenthal DS et al. Sulfur mustard induces markers of terminal differentiation and apoptosis in keratinocytes via a Ca2+-calmodulin and caspase-dependent pathway. Journal of Investigative Dermatology 111, 64–71 (1998). [DOI] [PubMed] [Google Scholar]
- 116.Shieh WM et al. Poly (ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. Journal of Biological Chemistry 273, 30069–30072 (1998). [DOI] [PubMed] [Google Scholar]
- 117.Inturi S, Tewari-Singh N, Agarwal C, White CW & Agarwal R Activation of DNA damage repair pathways in response to nitrogen mustard-induced DNA damage and toxicity in skin keratinocytes. Mutat Res 763-764, 53–63, doi: 10.1016/j.mrfmmm.2014.04.002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kehe K, Balszuweit F, Steinritz D & Thiermann H Molecular toxicology of sulfur mustard-induced cutaneous inflammation and blistering. Toxicology 263, 12–19 (2009). [DOI] [PubMed] [Google Scholar]
- 119.Papirmeister B, Gross CL, Meier HL, Petrali JP & Johnson JB Molecular basis for mustard-induced vesication. Fundam Appl Toxicol 5, S134–149 (1985). [PubMed] [Google Scholar]
- 120.Byers S, Anderson D, Brobst D & Cowan F Automated assay for nicotinamide adenine dinucleotide (NAD+). J Appl Toxicol 20 Suppl 1, S19–22, doi: (2000). [DOI] [PubMed] [Google Scholar]
- 121.Chiarugi A & Moskowitz MA Cell biology. PARP-1--a perpetrator of apoptotic cell death? Science 297, 200–201, doi: 10.1126/science.1074592 (2002). [DOI] [PubMed] [Google Scholar]
- 122.Nourani MR, Mahmoodzadeh Hosseini H, Azimzadeh Jamalkandi S & Imani Fooladi AA Cellular and molecular mechanisms of acute exposure to sulfur mustard: a systematic review. J Recept Signal Transduct Res 37, 200–216, doi: 10.1080/10799893.2016.1212374 (2017). [DOI] [PubMed] [Google Scholar]
- 123.Pirzad G et al. The Role of Fas-FasL Signaling Pathway in Induction of Apoptosis in Patients with Sulfur Mustard-Induced Chronic Bronchiolitis. J Toxicol 2010, 373612, doi: 10.1155/2010/373612 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Malaviya R et al. Inflammatory effects of inhaled sulfur mustard in rat lung. Toxicol Appl Pharmacol 248, 89–99, doi: 10.1016/j.taap.2010.07.018 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yaraee R et al. Alterations in serum levels of inflammatory cytokines (TNF, IL-1alpha, IL-1beta and IL-1Ra) 20 years after sulfur mustard exposure: Sardasht-Iran cohort study. Int Immunopharmacol 9, 1466–1470, doi: 10.1016/j.intimp.2009.09.001 (2009). [DOI] [PubMed] [Google Scholar]
- 126.Suwara MI et al. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol 7, 684–693, doi: 10.1038/mi.2013.87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chevillard M, Lainee P, Robineau P & Puchelle E Toxic effects of sulfur mustard on respiratory epithelial cells in culture. Cell Biol Toxicol 8, 171–181, doi: 10.1007/BF00260566 (1992). [DOI] [PubMed] [Google Scholar]
- 128.Pohl C et al. Acute morphological and toxicological effects in a human bronchial coculture model after sulfur mustard exposure. Toxicological sciences 112, 482–489 (2009). [DOI] [PubMed] [Google Scholar]
- 129.Malaviya R et al. Progressive Lung Injury, Inflammation, and Fibrosis in Rats Following Inhalation of Sulfur Mustard. Toxicol Sci 178, 358–374, doi: 10.1093/toxsci/kfaa150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Feng Y et al. The therapeutic effects of bone marrow-derived mesenchymal stromal cells in the acute lung injury induced by sulfur mustard. Stem Cell Res Ther 10, 90, doi: 10.1186/s13287-019-1189-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Emad A & Emad V Elevated levels of MCP-1, MIP-alpha and MIP-1 beta in the bronchoalveolar lavage (BAL) fluid of patients with mustard gas-induced pulmonary fibrosis. Toxicology 240, 60–69, doi: 10.1016/j.tox.2007.07.014 (2007). [DOI] [PubMed] [Google Scholar]
- 132.Ham HY et al. Sulfur mustard primes human neutrophils for increased degranulation and stimulates cytokine release via TRPM2/p38 MAPK signaling. Toxicol Appl Pharmacol 258, 82–88, doi: 10.1016/j.taap.2011.10.010 (2012). [DOI] [PubMed] [Google Scholar]
- 133.Sunil VR et al. Functional and inflammatory alterations in the lung following exposure of rats to nitrogen mustard. Toxicol Appl Pharmacol 250, 10–18, doi: 10.1016/j.taap.2010.09.016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Emad A & Emad Y Levels of cytokine in bronchoalveolar lavage (BAL) fluid in patients with pulmonary fibrosis due to sulfur mustard gas inhalation. J Interferon Cytokine Res 27, 38–43, doi: 10.1089/jir.2006.0084 (2007). [DOI] [PubMed] [Google Scholar]
- 135.Laskin JD et al. Oxidants and antioxidants in sulfur mustard–induced injury. Annals of the New York Academy of Sciences 1203, 92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang SC, Tsai YF, Pan YL & Hwang TL Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed J 44, 439–446, doi: 10.1016/j.bj.2020.09.001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Laskin DL, Sunil VR, Gardner CR & Laskin JD Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol 51, 267–288, doi: 10.1146/annurev.pharmtox.010909.105812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Iles KE & Forman HJ Macrophage signaling and respiratory burst. Immunol Res 26, 95–105, doi: 10.1385/IR:26:1-3:095 (2002). [DOI] [PubMed] [Google Scholar]
- 139.Tung JP, Fraser JF, Wood P & Fung YL Respiratory burst function of ovine neutrophils. BMC Immunol 10, 25, doi: 10.1186/1471-2172-10-25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang P, Huang S & Xu A in Amphioxus Immunity 153–165 (Elsevier, 2016). [Google Scholar]
- 141.Jezek J, Cooper KF & Strich R Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants (Basel) 7, doi: 10.3390/antiox7010013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mari M et al. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants (Basel) 9, doi: 10.3390/antiox9100909 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gould NS, White CW & Day BJ A role for mitochondrial oxidative stress in sulfur mustard analog 2-chloroethyl ethyl sulfide-induced lung cell injury and antioxidant protection. Journal of Pharmacology and Experimental Therapeutics 328, 732–739 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Beigi Harchegani A et al. Free Radical Production and Oxidative Stress in Lung Tissue of Patients Exposed to Sulfur Mustard: An Overview of Cellular and Molecular Mechanisms. Chem Res Toxicol 31, 211–222, doi: 10.1021/acs.chemrestox.7b00315 (2018). [DOI] [PubMed] [Google Scholar]
- 145.Gray JP, Mishin V, Heck DE, Laskin DL & Laskin JD Inhibition of NADPH cytochrome P450 reductase by the model sulfur mustard vesicant 2-chloroethyl ethyl sulfide is associated with increased production of reactive oxygen species. Toxicol Appl Pharmacol 247, 76–82, doi: 10.1016/j.taap.2010.05.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kamata S et al. Expression of cytochrome P450 mRNAs in Type II alveolar cells from subjects with chronic obstructive pulmonary disease. Pharmacol Res Perspect 6, e00405, doi: 10.1002/prp2.405 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brimfield AA et al. Free radical production from the interaction of 2-chloroethyl vesicants (mustard gas) with pyridine nucleotide-driven flavoprotein electron transport systems. Toxicol Appl Pharmacol 234, 128–134, doi: 10.1016/j.taap.2008.10.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goverman J et al. Sulfur mustard gas exposure: case report and review of the literature. Ann Burns Fire Disasters 27, 146–150 (2014). [PMC free article] [PubMed] [Google Scholar]
- 149.White CW, Rancourt RC & Veress LA Sulfur mustard inhalation: mechanisms of injury, alteration of coagulation, and fibrinolytic therapy. Ann N Y Acad Sci 1378, 87–95, doi: 10.1111/nyas.13130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shoeibi N et al. Long-term complications of sulfur mustard poisoning: retinal electrophysiological assessment in 40 severely intoxicated Iranian veterans. International journal of retina and vitreous 3, 1–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dursa EK et al. Respiratory illness among Gulf War and Gulf War era veterans who use the Department of Veterans Affairs for healthcare. American Journal of Industrial Medicine 63, 980–987 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Razavi SM, Ghanei M, Salamati P & Safiabadi M Long-term effects of mustard gas on respiratory system of Iranian veterans after Iraq-Iran war: a review. Chinese Journal of Traumatology 16, 163–168 (2013). [PubMed] [Google Scholar]
- 153.Khateri S, Ghanei M, Keshavarz S, Soroush M & Haines D Incidence of lung, eye, and skin lesions as late complications in 34,000 Iranians with wartime exposure to mustard agent. Journal of occupational and environmental medicine 45, 1136–1143 (2003). [DOI] [PubMed] [Google Scholar]
- 154.Balali‐Mood M et al. Long‐term complications of sulphur mustard poisoning in severely intoxicated Iranian veterans. Fundamental & clinical pharmacology 19, 713–721 (2005). [DOI] [PubMed] [Google Scholar]
- 155.Bijani K & Moghadamnia A Long-term effects of chemical weapons on respiratory tract in Iraq–Iran war victims living in Babol (North of Iran). Ecotoxicology and environmental safety 53, 422–424 (2002). [DOI] [PubMed] [Google Scholar]
- 156.Pugh MJ et al. Increasing Prevalence of Chronic Lung Disease in Veterans of the Wars in Iraq and Afghanistan. Mil Med 181, 476–481, doi: 10.7205/MILMED-D-15-00035 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Xiaoji Z et al. Mechanism underlying acute lung injury due to sulfur mustard exposure in rats. Toxicol Ind Health 32, 1345–1357, doi: 10.1177/0748233714560603 (2016). [DOI] [PubMed] [Google Scholar]
- 158.McGraw MD et al. Bronchiolitis Obliterans and Pulmonary Fibrosis after Sulfur Mustard Inhalation in Rats. Am J Respir Cell Mol Biol 58, 696–705, doi: 10.1165/rcmb.2017-0168OC (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jugg BJ et al. Acute Gene Expression Profile of Lung Tissue Following Sulfur Mustard Inhalation Exposure in Large Anesthetized Swine. Chem Res Toxicol 29, 1602–1610, doi: 10.1021/acs.chemrestox.6b00069 (2016). [DOI] [PubMed] [Google Scholar]
- 160.Rancourt RC et al. Airway tissue factor-dependent coagulation activity in response to sulfur mustard analog 2-chloroethyl ethyl sulfide. Am J Physiol Lung Cell Mol Physiol 302, L82–92, doi: 10.1152/ajplung.00306.2010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mariappan N et al. Extracellular nucleic acid scavenging rescues rats from sulfur mustard analog-induced lung injury and mortality. Arch Toxicol 94, 1321–1334, doi: 10.1007/s00204-020-02699-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rana T et al. MicroRNA-mediated inflammation and coagulation effects in rats exposed to an inhaled analog of sulfur mustard. Ann N Y Acad Sci 1479, 148–158, doi: 10.1111/nyas.14416 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sunil VR et al. Lung injury, oxidative stress and fibrosis in mice following exposure to nitrogen mustard. Toxicol Appl Pharmacol 387, 114798, doi: 10.1016/j.taap.2019.114798 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cruz-Hernandez A et al. Mast Cells Promote Nitrogen Mustard-Mediated Toxicity in the Lung Associated With Proinflammatory Cytokine and Bioactive Lipid Mediator Production. Toxicol Sci 184, 127–141, doi: 10.1093/toxsci/kfab107 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Malaviya R et al. Attenuation of nitrogen mustard-induced pulmonary injury and fibrosis by anti-tumor necrosis factor-α antibody. Toxicological sciences 148, 71–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Krystel-Whittemore M, Dileepan KN & Wood JG Mast Cell: A Multi-Functional Master Cell. Front Immunol 6, 620, doi: 10.3389/fimmu.2015.00620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Solopov P, Marinova M, Dimitropoulou C, Colunga Biancatelli RML & Catravas JD Development of chronic lung injury and pulmonary fibrosis in mice following acute exposure to nitrogen mustard. Inhal Toxicol 32, 141–154, doi: 10.1080/08958378.2020.1757791 (2020). [DOI] [PubMed] [Google Scholar]
- 168.Conti P et al. Critical role of inflammatory mast cell in fibrosis: Potential therapeutic effect of IL-37. Cell Prolif 51, e12475, doi: 10.1111/cpr.12475 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pesonen M et al. Capsaicinoids, chloropicrin and sulfur mustard: possibilities for exposure biomarkers. Front Pharmacol 1, 140, doi: 10.3389/fphar.2010.00140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Roshan R et al. Long-term effects of sulfur mustard on civilians’ mental health 20 years after exposure (The Sardasht-Iran Cohort Study). Health and quality of life outcomes 11, 1–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Drasch G, Kretschmer E, Kauert G & von Meyer L Concentrations of mustard gas [bis(2-chloroethyl)sulfide] in the tissues of a victim of a vesicant exposure. J Forensic Sci 32, 1788–1793 (1987). [PubMed] [Google Scholar]
- 172.Thomsen AB, Eriksen J & Smidt-Nielsen K Chronic neuropathic symptoms after exposure to mustard gas: a long-term investigation. J Am Acad Dermatol 39, 187–190, doi: 10.1016/s0190-9622(98)70072-6 (1998). [DOI] [PubMed] [Google Scholar]
- 173.Isono O et al. Long-term neurological and neuropsychological complications of sulfur mustard and Lewisite mixture poisoning in Chinese victims exposed to chemical warfare agents abandoned at the end of WWII. Toxicol Lett 293, 9–15, doi: 10.1016/j.toxlet.2018.04.017 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Darchini-Maragheh E, Nemati-Karimooy H, Hasanabadi H & Balali-Mood M Delayed neurological complications of sulphur mustard and tabun poisoning in 43 Iranian veterans. Basic Clin Pharmacol Toxicol 111, 426–432, doi: 10.1111/j.1742-7843.2012.00922.x (2012). [DOI] [PubMed] [Google Scholar]
- 175.Gupta RC 1,199 pages (Elsevier : AP,, London, England, 2015). [Google Scholar]
- 176.Tekiner A et al. The effect of nitrogen mustard on the rat brain and the therapeutic value of proanthocyanidin. Turk Neurosurg 19, 360–366 (2009). [PubMed] [Google Scholar]
- 177.Gao X et al. Pathological studies on the protective effect of a macrolide antibiotic, roxithromycin, against sulfur mustard inhalation toxicity in a rat model. Toxicologic pathology 39, 1056–1064 (2011). [DOI] [PubMed] [Google Scholar]
- 178.Calvet J et al. Glucocorticoids inhibit sulfur mustard-induced airway muscle hyperresponsiveness to substance P. Journal of Applied Physiology 77, 2325–2332 (1994). [DOI] [PubMed] [Google Scholar]
- 179.Calvet J et al. Airway epithelial damage induced by sulfur mustard in uinea pigs, effects of glucocorticoids. Human & experimental toxicology 15, 964–971 (1996). [DOI] [PubMed] [Google Scholar]
- 180.Wigenstam E, Jonasson S, Koch B & Bucht A Corticosteroid treatment inhibits airway hyperresponsiveness and lung injury in a murine model of chemical-induced airway inflammation. Toxicology 301, 66–71, doi: 10.1016/j.tox.2012.06.020 (2012). [DOI] [PubMed] [Google Scholar]
- 181.O’Neill HC et al. Treatment with the catalytic metalloporphyrin AEOL 10150 reduces inflammation and oxidative stress due to inhalation of the sulfur mustard analog 2-chloroethyl ethyl sulfide. Free Radic Biol Med 48, 1188–1196, doi: 10.1016/j.freeradbiomed.2010.01.039 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Preissner KT & Herwald H Extracellular nucleic acids in immunity and cardiovascular responses: between alert and disease. Thromb Haemost 117, 1272–1282, doi: 10.1160/TH-16-11-0858 (2017). [DOI] [PubMed] [Google Scholar]
- 183.Aswani A et al. Scavenging Circulating Mitochondrial DNA as a Potential Therapeutic Option for Multiple Organ Dysfunction in Trauma Hemorrhage. Front Immunol 9, 891, doi: 10.3389/fimmu.2018.00891 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang Q-Q, Begum RA, Day VW & Bowman-James K Sulfur, oxygen, and nitrogen mustards: stability and reactivity. Organic & biomolecular chemistry 10, 8786–8793 (2012). [DOI] [PubMed] [Google Scholar]
- 185.Tsoutsoulopoulos A, Brockmoller S, Thiermann H & Steinritz D Comparison of the toxicity of sulfur mustard and its oxidation products in vitro. Toxicol Lett 321, 69–72, doi: 10.1016/j.toxlet.2019.12.015 (2020). [DOI] [PubMed] [Google Scholar]
- 186.Sharma M, Vijayaraghavan R & Agrawal OP Comparative toxic effect of nitrogen mustards (HN-1, HN-2, and HN-3) and sulfur mustard on hematological and biochemical variables and their protection by DRDE-07 and its analogues. International journal of toxicology 29, 391–401 (2010). [DOI] [PubMed] [Google Scholar]