

Abstract
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), a potent AhR ligand, is an environmental contaminant that is known for mediating toxicity across generations. However, whether TCDD can induce multigenerational changes in the expression of microRNAs (miRs) has not been previously studied. In the current study, we investigated the effect of administration of TCDD in pregnant mice (F0) on gestational day 14, on the expression of miRs in the thymus of F0 and subsequent generations (F1 and F2). Of the 3200 miRs screened, 160 miRs were dysregulated similarly in F0, F1, and F2 generations, while 46 miRs were differentially altered in F0 to F2 generations. Pathway analysis revealed that the changes in miR signature profile mediated by TCDD affected the genes that regulate cell signaling, apoptosis, thymic atrophy, cancer, immunosuppression, and other physiological pathways. A significant number of miRs that showed altered expression exhibited dioxin response elements (DRE) on their promoters. Focusing on one such miR, namely miR-203 that expressed DREs and was induced across F0 to F2 by TCDD, promoter analysis showed that one of the DREs expressed by miR-203 was functional to TCDD-mediated upregulation. Also, the histone methylation status of H3K4me3 in the miR-203 promoter was significantly increased near the transcriptional start site in TCDD-treated thymocytes across F0 to F2 generations. Genome-wide chromatin immunoprecipitation sequencing study suggested that TCDD may cause alterations in histone methylation in certain genes across the three generations. Together, the current study demonstrates that gestational exposure to TCDD can alter the expression of miRs in F0 through direct activation of DREs as well as across F0, F1, and F2 generations through epigenetic pathways.
Keywords: TCDD, multigenerational, epigenetics, microRNA, histone
Significance Statement.
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is an environmental contaminant that was found in Agent Orange used during the Vietnam war. The US National Academy of Sciences reported that there was evidence demonstrating that TCDD exposure caused birth defects. Subsequent studies confirmed the deleterious effects of TCDD on reproductive health across multiple generations. The current study demonstrates for the first time that perinatal exposure to TCDD alters the expression of a large number of miRNAs in a similar fashion in F0, F1, and F2 generations. We also provide evidence that such alterations in the miRNAs may result from epigenetic changes involving histone methylation. Because miRNAs play a critical role in the epigenetic regulation of gene expression, the current study provides new insights into the multigenerational impact of TCDD.
Introduction
The environmental factors affect and alter developmental programming leading to the onset of diseases, including diabetes, cancer, reproductive disorders, and cardiovascular disease in adult animals and humans (1–6). To this end, exposure to endocrine disruptors, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), is of great significance and concern as they play important roles in regulating transient and irreversible developmental processes. There is evidence suggesting that gestational exposure to chemicals, including TCDD, results in influencing the disease status not only in the F0 maternal life but also in subsequent generations (7–10). Among the susceptibility to such diseases, the most well characterized include reproductive disorders.
TCDD has been well characterized for causing a wide range of toxicity, including immunotoxicity (10–18). The fetal basis of adult disease hypothesis suggests that exposure to environmental factors, stress, or malnutrition during pregnancy, can have a long-lasting impact on the developing fetus, leading to increased susceptibility to a wide range of diseases, including autoimmune diseases, cardiovascular, cancer, and hypertension later in life (19, 20). While the impact of toxic chemicals such as TCDD during gestation on fetal health can be explained because such chemicals can cross the placenta and can directly affect the fetus, as in the case of induction of thymic atrophy (21, 22), any effect on subsequent generations such as F3 and beyond can be explained primarily by the ability of the chemicals to induce epigenetic changes in the germ-line. This phenomenon is called transgenerational epigenetic inheritance, a process that involves the transmission of an altered epigenome and its impact on the phenotype across generations through the germline even when the chemical is not present to directly cause the toxicity [reviewed in (23)]. The pathways through which transgenerational epigenetic inheritance is mediated include primarily DNA methylation, histone modifications, and noncoding RNAs, including microRNA (miR) (23).
miRs are a class of noncoding RNAs that have been shown to play a key role in the regulation of gene expression. In most instances, miRs interact with the 3′ untranslated regions (3′ UTR) of target mRNAs to cause the degradation of mRNA and translational repression. However, miRs can also interact with other regions such as the 5′ UTR, coding sequence, and gene promoters, as well as activate translation or regulate transcription (24). Sperm miRs are very sensitive to environmental changes, which can alter the sperm miR profile (25). Thus, after entering the zygote, these miRs can induce epigenetic modifications in the embryo, thereby causing changes in the physiological functions and impacting health (25).
We have previously shown that TCDD causes thymic atrophy in adult mice, which is associated with significant changes in miR expression in the thymocytes (10). Interestingly, we and others have shown that many miRs express dioxin response elements (DREs) in their 3′ UTR, thereby directly regulating their expression (26, 27). Thus, the alterations in the expression of such miRs in the thymi of pregnant mice (F0) and the fetus (F1) following gestational exposure to TCDD can be explained by direct exposure of the thymus to TCDD and activation of DREs on miRs. However, whether gestational exposure to TCDD alters the expression of miRs even in the F2 generation, which would be indicative of TCDD not acting directly on DREs of miRs in the thymus, but potentially through epigenetic modulations involving germ cells, has not been previously investigated.
Therefore, in the current study, we investigated the effect of gestational exposure to TCDD on miRs in thymocytes from F0, F1, and F2 generations and found that a significant number of miRs were similarly up- or downregulated across these generations. Such changes could be associated with histone modifications. For example, TCDD treatment led to an increased expression of miR-203, an important regulator of the immune response, in all three generations, which correlated with its H3K4me3 signal level in the promoter.
Results
TCDD triggers multigenerational alterations in the miR profile
In this study, we first determined the multigenerational effect of TCDD (Fig. 1) on miR expression in the thymi of mice following gestational exposure of pregnant female mice using miR arrays. The cluster analysis of variable miRs in VEH- and TCDD-treated samples were performed using Ward’s method. Also, the measure of miRs in the VEH and TCDD-treated groups in three generations (F0, F1, and F2) were performed using the Half Square Euclidean Distance method. Similarly, the ordering function of miRs was performed based on Input rank. The visualization of cluster analysis of miRs has been shown as a dendrogram based on the similarity between them (Fig. 2A) and their expression pattern was reflected in a range from +326.5 to −71.9 (Fig. 2A). The data presented in Fig. 2B demonstrated the heatmap of altered expression of miRs in the three generations of TCDD-treated groups when compared to their respective vehicle control groups.
Fig. 1.
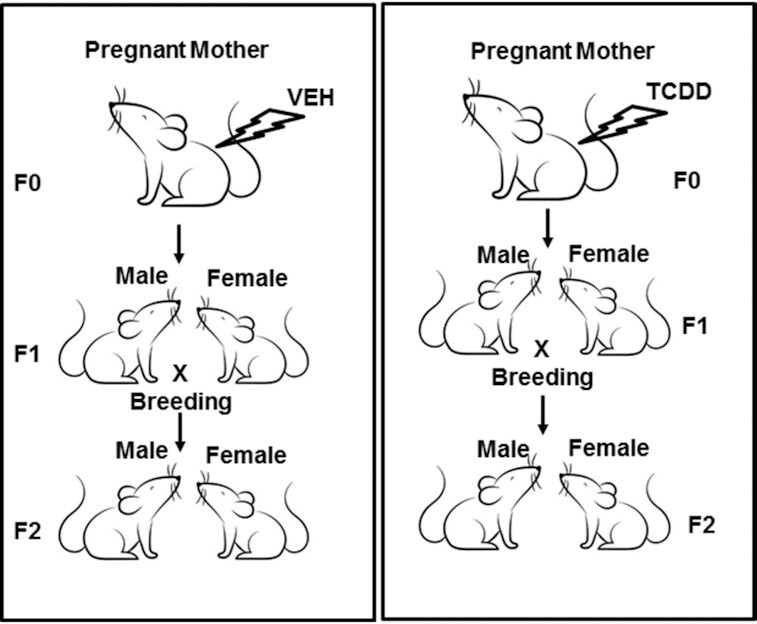
Showing a graphical sketch of the breeding protocol after gestational exposure to TCDD or vehicle control.
Fig. 2.

Showing analysis of thymic miRs of F0, F1, and F2 generations exposed to VEH or TCDD. Total miRs from groups of five pooled mouse thymi of F0, F1, and F2 generations of control or TCDD groups were analyzed. (A) Heat map depicting miR expression in thymi of VEH- or TCDD-exposed F0, F1, and F2 generations. The spectrum of downregulated to the upregulated expression pattern of miRs expression is shown from green to red. (B) Showing fold change (>1.5) expression profile of miRs of F0, F1, and F2 generations between the two groups (VEH- or TCDD-exposed). (C) Showing upregulation or downregulation (>1.5-fold change) of miRs regulated by TCDD, when compared to miRs of VEH-treated groups.
Next, dysregulated (upregulated or downregulated) expression of miRs was analyzed using a two-sample t-test, and the significance of analysis of microarrays was performed using the Kaplan–Meier method. A P-value of <0.05 in the t-test was considered significant. Of the >3200 miRs screened, 160 miRs showed altered expression by >1.5-fold in a similar fashion in TCDD groups of all three generations, when compared to vehicle-treated groups of the three respective generations (Table S1). We also observed that miRs were differentially dysregulated in the three generations (Table S2).
Linear Discriminant Analysis (LDA) to identify TCDD-specific biomarkers and the role of TCDD-mediated miRs in the regulation of various pathways and diseases
Next, we performed LDA of miRs in TCDD vs VEH groups (Fig. 3A). The VEH group included miRs profiles from VEH-treated F0, F1, and F2 generations and the TCDD group included miRs profiles from TCDD-treated F0, F1, and F2 generations. LDA analysis, which was set at a threshold of 2.0 led to the identification of 31 miRs as possible biomarkers, unique to the TCDD group. All the miRs depicted are miR-biomarkers enriched in mice treated with TCDD in F0, F1, and F2 generations. Together, these data demonstrated that the TCDD group expressed unique sets of miRs.
Fig. 3.

Pathway analysis of TCDD-regulated miRs and their association with functional networks. (A) LDA of miRs in the TCDD group. TCDD group included miR profiles from TCDD-treated F0, F1, and F2 generations. LDA analysis, which was set at a threshold of 2.0 led to the identification of 31 miRs as possible biomarkers, unique to the TCDD group. (B) TCDD-mediated changes in miRs by 1.5-fold or higher were analyzed using IPA software and their database. The bars in the graph demonstrate various pathways regulated by TCDD-induced miRs. The –log (P-value) on Y-axis represents the significance of function by random chance (IPA software, Qiagen Inc.). (C) Showing miRs involved in a pathway with various functions. Pink color represents upregulated mature miRs and green color represents downregulated miRs in TCDD vs VEH-treated groups.
To analyze the role of miRs whose expression was altered by TCDD, we next selected those expressing more than 1.5-fold (upregulated or downregulated) in F0, F1, and F2 generations, which yielded 160 dysregulated miRs. These miRs were analyzed using IPA software from Qiagen and the database of the company (Qiagen Inc., Hilden, Germany). Upon analysis, we observed that there were as many as 27 pathways that might be affected by various miRs altered by TCDD in the thymi of three generations (Fig. 3B). There were at least six miRs involved in the regulation of endocrine system disorders, six miRs in gastrointestinal diseases, and six miRs in metabolic diseases (Fig. 3B). Similarly, there were at least four sets of different miRs involved in cellular development, infectious diseases, organismal development, and hepatic system disease (Fig. 3B). There were more than three different sets of miRs involved in 10 different pathways/diseases including cell death and survival, cell to cell signaling and interaction, cellular assembly and organization, cellular functions and maintenance, embryonic development, hair and skin development, nervous system development and functions, organ development, tissue development, tissue morphology, cardiovascular system development, cell morphology, and skeletal and muscular system development and function (Fig. 3B). We also observed that there were at least three or more miRs involved in cancer, respiratory diseases, and tumor morphology, and at least one miR or more in drug metabolism, lipid metabolism, molecular transport, and small molecule biochemistry (Fig. 3B). Figure 3C shows the role of up- and downregulated miRs in signaling pathways involved in various molecular and physiological mechanisms. For example, the downregulation of miR-134 by TCDD may increase the expression of FoxP3, a transcription factor expressed by the regulatory T cells (Tregs) as shown in earlier studies (28). Taken together, the analysis revealed that TCDD affects a large number of miRs multigenerationally that play a role in regulating several signaling pathways/diseases.
Real-time PCR (RT-qPCR) to validate the expression of a few select miRs
To validate the data on the expression of TCDD-induced miRs obtained from miR arrays, we performed RT-qPCR on a few select miRs that were immunologically relevant and either upregulated or downregulated in the thymi of mice of all generations. To that end, we selected upregulated miRs (miR-146a and miR203) and downregulated miRs (miR-30a, miR-31, miR134, miR-155, miR-182, and miR-499). RT-qPCR was performed using cDNAs converted from total RNA from thymocytes of mice treated with TCDD or VEH (F0, F1, and F2 generations) as described in the section “Materials and methods.” RT-qPCR analysis confirmed the expression of all the selected miRs. There was upregulated expression of miR-146a (Fig. 4A) and miR-203 (Fig. 4B) in thymocytes from mice treated with TCDD when compared to vehicle-treated thymocytes (Fig. 4A). Similarly, we observed downregulation of miR-30a, miR-31, and miR-155 (Fig. 4A), miR-134 (Fig. 4B), and miR-182 and miR-499 (Fig. 4C) in TCDD-treated thymocytes when compared to vehicle controls (Fig. 4A to C). The RT-qPCR data on the expression profile of select miRs validated their expression in thymi of mice of all three generations (F0, F1, and F2) obtained from the miR arrays.
Fig. 4.

Validation of miRs expression in thymocytes of F0, F1, and F2 generations. Mice were treated with VEH- or TCDD as described in Fig. 1 legend from groups of five pooled mouse thymi that were analyzed by quantitative real-time PCR (qRT-PCR). Total miRs of F0, F1, and F2 generations of control or TCDD groups were analyzed. (A) Thymi exposed to VEH or TCDD of F0, F1, and F2 generations were analyzed for the expression of Th1 (IFN-γ), Th17 (IL-17), and Tregs (FoxP3)-specific miRs (miR-30a, -31, -155, and -146a). (B) Expression of CYP1A1-specific miRs (miR-134 and miR-203). (C) Expression of AhR-specific miRs (miR-182 and miR-499). SNORD96A was used as an internal control for miRs. Data are depicted as mean ± SEM of technical triplicates from pooled thymi of five mice per group. Asterisk (*) in (A) to (C) indicates statistically significant (P < 0.05) differences between the groups compared.
Determination of select miR-specific gene expression in the thymus
We investigated the expression of the target genes of the select miRs using RT-qPCR. To that end, we determined the expression of CYP1A1 (miR-134-specific), AhR (miR-182 and -499-specific), FoxP3 (miR-31-specific), IL-17 (miR-203-specific), and IFN-γ (miR-146a-specific) genes in the thymocytes of TCDD- and VEH-exposed mice of F0, F1, and F2 generations. The expression of CYP1A1, AhR, and FoxP3 was significantly upregulated in thymocytes of TCDD-exposed mice when compared to VEH-treated mice in all F0, F1, and F2 generations (Fig. 5A and B). Also, the expression of IL-17 and IFN-γ was significantly downregulated in thymocytes of F0, F1, and F2 groups treated with TCDD when compared to those exposed to the vehicle (Fig. 5B). When we studied the thymic cellularity, we found that TCDD-exposed mice showed a slight decrease in thymic cellularity in F0 mice, while the F1 and F2 generations failed to exhibit significant change (Fig. 5C).
Fig. 5.

Determination of select miR-specific gene expression in thymocytes of F0, F1, and F2 generations of VEH- or TCDD-treated groups using qRT-PCR. (A) Thymi exposed to VEH or TCDD of F0, F1, and F2 generations were analyzed for the expression of CYP1A1 (miR-134-specific). (B) Expression of AhR (miR-182 and -499-specific), FoxP3 (miR-31-specific), IL-17 (miR-203-specific), and IFN-γ (miR-146-specific). A housekeeping gene, 18S, was used as an internal control. (C) Thymic cellularity shows data expressed as mean ± SEM of five individual mice per group. The qRT-PCR data are depicted as mean ± SEM of technical triplicates from pooled thymi of five mice per group. Asterisk (*) in panels (A) and (B) indicates statistically significant (P < 0.05) difference between the groups compared.
TCDD regulates miR-203 expression through DREs present on its promoter
Our earlier studies demonstrated that TCDD downregulates the expression of IL-17 (29). Also, in the current study, we noted that miR-203, which was upregulated in F0, F1, and F2 generations (Fig. 4B) was found to target the IL-17 gene (Fig. 6A). We, therefore, undertook additional studies on this miR to address if miR-203 expressed DREs and if TCDD-mediated induction of miR-203 was dependent on DREs. We found that the promoter of miR-203 expressed four DREs (Fig. 5B). The role of DREs in the regulation of the expression of miR-203 by TCDD was determined by performing luciferase assay. As described in the section “Materials and methods,” the four DREs present in the miR-203 promoter were cloned in pGL4 vector. The pGL4 constructs with or without the DRE regions, as detailed in Fig. 6B, were transfected into mouse liver cell line Hep1a. Twenty-four hours post-transfection, the cells were treated with various concentrations of TCDD and incubated at 37°C, 5% CO2 in an incubator. Dual luciferase assays were performed 24 h after the treatment using a kit from Promega and following the protocol of the company. The data obtained from luciferase assays demonstrated that the DRE domain located near 430 bp upstream of transcriptional start site (TSS) of miR-203 responded to TCDD in a dose-dependent manner (Fig. 6), suggesting that TCDD may induce miR-203 through binding to this DRE motif and not the other three motifs present on the promoter of miR-203.
Fig. 6.
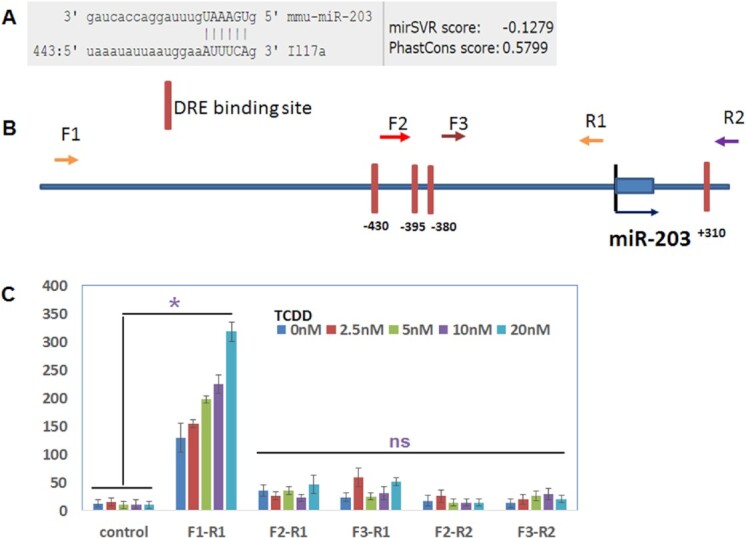
TCDD regulates the expression of miR-203 through DRE present in its promoter. (A) Showing binding affinity of miR-203 with IL-17 (microRNA.org). (B) Schematic diagram of the promoter and DRE sites in the promoter of miR-203. Arrows indicate the PCR primers and their relative locations (F, forward primer; R, reverse primer). (C) The PCR products generated with the combination of different forward and reverse primers were cloned into a luciferase reporter vector. Luciferase activities were measured after the addition of the indicated amounts of TCDD. The empty luciferase vector was used as the control. Data presented in panel (C) are depicted areas mean ± SEM of three technical replicates from pooled thymi of five mice per group. Asterisks (*) in panel (C) indicate a statistically significant (P < 0.05) difference between the groups compared.
TCDD promotes histone methylation patterns in the promoters that might have multigenerational effects in mice
Because the histone methylation signals in the promoter region affect gene expression, we determined whether TCDD affects the overall histone methylation near the TSS. The genomic region between 3 kb upstream and 3 kb downstream of TSS was defined as the promoter region. H3K4me3 was significantly increased near the TSS of genes in the vehicle and TCDD-treated samples of all three generations (Fig. 7). On the other hand, the methylation signal of H3K9me3 and H3K27me3 (Fig. 7) decreased significantly within 1 kb upstream and downstream of TSS in all samples. These patterns were consistent with the notion that H3K4me3 is associated with gene activation, while H3K27me3 and H3K9me3 are associated with gene expression repression. Overall, the treatment with TCDD did not significantly affect the histone methylation patterns in the promoter region. However, those histone marks may differ significantly in individual genes. As seen in Fig. 8, TCDD treatment altered the distribution of histone marks as demonstrated by correlation analysis and visualization in a genome browser (Fig. 8A to F).
Fig. 7.

Analysis of H3K4me3, H3k9me3, and H3K27me3 in TCDD-treated and VEH-treated samples. Showing signals for H3K4me3, H3K27me3, and H3K9me3 F0, F1, and F2 generations. The data presented are representative of one of the three experiments.
Fig. 8.

Analysis of H3K4me3, H3k9me3, and HK27me3 by performing chromatin immunoprecipitation sequencing (ChIP-seq). VEH- or TCDD-treated thymocyte samples from F0, F1, and F2 generations were analyzed. A representative of histones mark, H3K4me3 (A), H3k9me3 (B), and HK27me3 (C) ChIP-seq signal obtained after visualization of the sequencing data in Integrated Genome Browser (IGB) for mouse chromosome 1 are shown. The data presented depict a representative experiment out of three.
Next, we examined the histone methylation status of miR-203 and found that H3K4me3 was significantly increased near the TSS of miR-203 in TCDD-treated samples when compared to vehicle-treated samples from all three generations (Fig. 9A). We also examined DNA methylation levels in the CpG island near the TSS of miR-203. The TCDD-treated sample had less DNA methylation in the CpG island when compared to the vehicle-treated sample (Fig. 9B). Together, these data suggested that epigenetic regulations such as histone methylation and DNA methylation of miR-203 may be responsible for its overexpression multigenerationally.
Fig. 9.
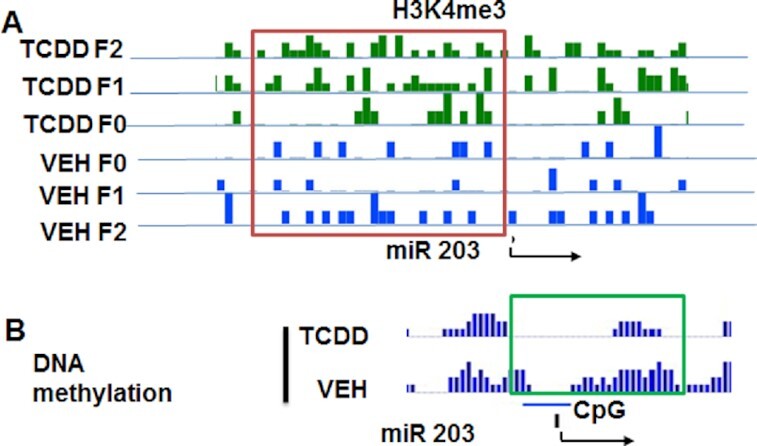
Histone methylation of H3K4me3 in the promoter of miR-203. (A) Showing the status of H3K4me3 in thymocyte samples exposed to TCDD or VEH from F0, F1, and F2 generations. (B) Showing DNA methylation status of CpG islands present in miR-203 promoter in thymocyte samples treated with TCDD or VEH. The data presented depict a representative experiment out of three.
Discussion
TCDD is an environmental pollutant and a very potent toxicant, known to cause tumorigenesis, immunological dysfunction, and teratogenesis (10–12, 14–18, 30–38). This has raised questions on whether gestational exposure to TCDD would impact the health of the fetus as well as the subsequent generations. Because TCDD can cross the placental barrier (39), the direct effect of TCDD on the thymus in F1 generation can be expected, however, whether the toxic effects can be preserved even in the F2 and subsequent generations remains an intriguing possibility. To that end, several studies have focused on the transgenerational effects of TCDD on the reproductive functions [reviewed in (40, 41)] and shown that F1 mice exposed to TCDD in utero exhibit reduced fertility and an increased incidence of premature birth, an effect that is maintained for the next three generations (42). Similarly, female C57BL/6 mice (F1), exposed in utero to TCDD (10 μg/kg) by oral gavage at gestational day (GD) 15 exhibited endometriosis-like uterine phenotype in adult life (43). Also, even in the absence of direct TCDD exposure, the three subsequent generations (F2 to F4) presented adenomyosis [60] Such effects of TCDD on F2 and subsequent generations even in the absence of direct TCDD exposure on target organs suggest TCDD-mediated alterations occurring in the germline (41) through potential epigenetic pathways.
Several epigenetic pathways can regulate gene expression without changing the DNA sequence in the genome. Such pathways include DNA methylation, histone modifications, noncoding RNAs, chromatin structure, and RNA methylations (44). miRs are small endogenous noncoding RNAs that negatively regulate the protein production of their mRNA targets. There is significant interaction between miR and epigenetic regulators (45), and miR can also be controlled by epigenetic factors (44). The effects of TCDD on miR across generations have not been studied thus far. The current study demonstrated that gestational exposure to TCDD leads to alterations in the miR expression in the thymus of F0, F1, and F2 generations.
The data obtained from miR arrays showed that TCDD caused dysregulation of a large number of miRs. More than 160 miRs were >1.5-fold similarly up- or downregulated in all three generations (F0, F1, and F2). Furthermore, based on the LDA analysis of dysregulated miRs amongst all the generations (F0, F1, and F2), we identified 31 miRs that were altered and were unique to the TCDD group. Moreover, we also identified miRs that were specific to AhR (miR-182 and miR-499), CYP1A1 (miR-134 and miR-203), FoxP3 (miR-31), IFN-γ (miR-146a), IL-17 (miR-203), and the like. The common dysregulation of certain miRs across all three generations (F0, F1, and F2) is intriguing and suggested epigenetic regulation of the expression of such miRs. It is noteworthy that the expression of miRs reciprocally correlated with the target genes expressed in the thymi of all three generations. It is also interesting that while CYPIA1 was targeted by two miRs, miR-134, which was downregulated and miR-203, which was upregulated following TCDD exposure, the expression of CYPIA1 was upregulated. This can be explained by the fact that multiple miRs can target the same gene, and that the combination of all these activities ultimately determines the expression of miR target genes (46).
Several studies have shown that TCDD triggers thymic atrophy (34, 47, 48). In the current multigenerational study, we found that while F0 showed slight decrease in thymic cellularity, the F1 and F2 generations did not show any decrease in thymic cellularity. This can be explained by the fact that F0 mice were directly exposed to TCDD, whereas F1 was exposed as a fetus and as a neonate, and by the time we harvested the thymi of F1 at the age of 6 to 8 weeks, the effect of TCDD may have waned, consistent with previous studies that the effect of TCDD on thymic atrophy is reversible (34). Also, the F2 generation thymus was not directly exposed to TCDD. The failure of TCDD-exposed F1 and F2 generation thymi to show atrophy also suggested that the changes in miR expression caused by TCDD in F1 and F2 did not directly affect the thymic cellularity.
It should be noted that because the F0 and F1 generations were directly exposed to TCDD, it is also likely that TCDD caused a direct effect on the thymus thereby altering the expression of some miRs through the regulation of DREs expressed on their promoters. DREs play an important role in the TCDD-mediated regulation of genes (16, 38). Upon analysis of select eight miRs for the presence DREs in their promoter, we observed that all the miRs that we analyzed had DREs in their respective promoters (Table 4). To further confirm that TCDD acts through DREs, we performed a luciferase assay, which demonstrated that the DRE domain located near 430 bp upstream of TSS of miR-203 responded to TCDD in a dose-dependent manner. This suggested that TCDD induces miR-203 through binding to this DRE motif present in the promoter of miR-203. While TCDD may directly alter the expression of miRs in the thymi of F0 and F1 generations, because the thymi in F2 generation were not directly exposed to TCDD, any effect on F2 thymi is suggestive of the involvement of epigenetic pathways, which may regulate such miRs through the action on germ cells. It should be noted that some of the miRs that expressed DREs were downregulated by TCDD. In this context, it is noteworthy that AhR signaling can occur through canonical and noncanonical pathways (49). Also, some of the miRs may be downregulated by TCDD through indirect pathways by enhancing the negative regulators of miRs or through epigenetic regulation as shown in this study.
Table 4.
Showing DRE sites in the promoters of selected miRs.
| miRs | DREs in the promoter of miRs | Sequences |
|---|---|---|
| miR-30a | 4 | TCAGGCGTGTT, AAATGCGTGA, CTGGGCGTGC, and GCGCGCGTGAGCTC |
| miR-31 | 2 | CCGCGTGAAA and ACGTGCGTGCAATT |
| miR-134 | 2 | GGAGCGTGGTTAG and CCCGCGTGAGCA |
| miR-146a | 3 | CTAGCGTGAT, TCAGCGTGGGG, and TTTGCGTGTCAA |
| miR-155 | 3 | GCAGCGTGACAAT, TGTGTGCGTGTGTG, and CTGCTCTGCGTGACC |
| miR-182 | 4 | GGTGGCGTGC, GTGCGTGCGT, GCGTGCGTGTGTG, and GCTGGCGTGTAC |
| miR-203 | 4 | TGAAGCGTGGTTC, CATGCGTGTGCCT, AGCGCGTGCCC, and TGCATGCGTGCCTT |
| miR-499 | 4 | GCCGTGCGTGCCA, CCTCGCGTGTGCC, GACTGCGTGTATA, and GTGTGCGTGTGCAT |
Histone modification is an important factor of epigenetic regulation leading to alteration in gene expression and long-term effects. We also investigated the multigenerational effect of TCDD on histone modifications and observed that TCDD caused histone changes, especially in H3K4me3, H3K9me3, and H3K27me3. H3K4me3 is a histone marker responsible for the upregulation of genes (50). A higher H3K4me3 signal indicates that this histone modification could lead to upregulated multigenerational expression of many genes by TCDD. In our study, we observed an increased H3K4me3 signal near TSS (Fig. 7). In contrast, there were opposite modifications in H3K9me3 and H3K27me3 histones in all three generations examined (Fig. 7). Analysis of H3K4me3 signal in miR-203 in TCDD- and vehicle-treated samples, showed a significantly higher level of H3K4me3 in TCDD-treated samples when compared to vehicle-treated samples. Furthermore, analysis of the DNA methylation status of CpG islands in the promoter of miR-203, showed that there was significantly less methylation of CpG islands in TCDD-treated sample when compared to vehicle-treated sample (Fig. 8B). Both indicated an increased expression of miR-203 after TCDD treatment.
For the epigenetic changes to be maintained across multiple generations, such changes need to occur at the germ cells and previous studies suggested that TCDD may exert true transgenerational effects. For instance, exposure to TCDD in F0 leads to a decreased fertility in F1 to F3 (23, 41). A recent epigenome-wide association study in rats to identify potential sperm DNA methylation biomarkers for specific multigenerational toxicity showed disease-specific epimutation in DNA methylation regions, which could account for TCDD-mediated multigenerational susceptibility to certain diseases (48). Similarly, exposure of F0 females to TCDD led to reproductive disorders in F1 and F3 generations that was associated with DNA methylation in gene promoters in F3 epigenome (49). Transgenerational effects caused by histone methylation have also been demonstrated. Overexpression of KDM1A, a histone K4 demethylase during mouse spermatogenesis in F0 disrupts histone methylation causing reduced survivability and increased abnormal development in F1 to F3 generations, which do not have overexpressed KDM1A (51). Although the mechanisms of transgenerational epigenetic inheritance are not clear, such effects are likely gene specific. In Caenorhabditis elegans, high fat diet is associated with inheritable changes of H3K4me3 at lipid metabolism related genes. Further study indicated that wdr-5.1, a component of H3K4me3 modifier is required for transmitting high fat diet-induced histone mark transgenerationally (52), suggesting that inheritable epigenetic marks are gene-specific and dependent on certain regulatory factors.
During the generation of sperms or eggs, primordial germ cells undergo genome-wide DNA demethylation and erasure of histone modification marks, a process called epigenetic reprogramming, which is critical for the embryo development (53). However, increasing evidence suggests that not all epigenetic marks are erased during reprogramming. There are some fractions of genomic loci including the imprint control regions, differentially methylated regions, intracisternal A particles, transposons, and some enhancer elements, which are more resistant to the global methylation reprogramming that is seen during early development (53). While it is currently unclear why and how these regions are refractory to methylation reprogramming, such sites may facilitate methylation patterns to be transferred transgenerationally. We speculate that such factors may help explain the epigenetic regulation of miR-203 multigenerationally from F0 to F2 following TCDD exposure.
There are some limitations in the current study. First, we included only female mice for all three generations, and not the male mice. It is well established that males and females of the same species can display significant differences in the sensitivity to a wide range of TCDD-mediated toxicity (54, 55). There are several sex-specific TCDD-responsive genes that account for such sex-differences. Additionally, there are also complex interactions between the AhR and sex hormone receptors that contribute to the observable differences seen in the sexes (56). It is for this reason that we felt that the inclusion of male mice in this study may further complicate the interpretation of the complex epigenetic data. We currently plan to pursue studies on male mice as well as compare sex differences as it relates to multigenerational epigenetic changes. Secondly, we used one of the miRs (miR-203) comprehensively for the epigenetic marks and used these data to suggest that such a pathway may lead to altered expression of miRs across generations. Clearly, additional studies are necessary to test if other miRs are similarly regulated by histone modifications. Lastly, while we used some well-established marks like H3K4me3 and H3K27me3, there are other marks such as H3K27ac, which could be significant marks of active promoter. In the future, we will examine other histone marks including acetylation and mono-, di-, and tri-methylation. Besides the promoters, we will also examine histone modification in other genomic regions.
In summary, in this study, we demonstrate for the first time that perinatal exposure to TCDD can alter the expression of miRs across F0 to F2 generations. Some of these changes in miRs are identical across the F0 to F2 generations, while others are unique to the specific generations. The impact of TCDD on miRs in F0 and F1 can be explained in part by the direct action of TCDD on the thymus through the DREs expressed on miRs. However, the continued TCDD-mediated alterations in the expression of certain miRs even in F2 generation in the absence of a direct effect on the thymus suggest epigenetic changes induced by TCDD in the germ cells.
Materials and methods
Mice
C57BL/6 pregnant mice were purchased from the Jackson Laboratories (Bar Harbor, ME, USA). All mice were housed in the animal facility of the School of Medicine (SOM), University of South Carolina. Care and maintenance of animals were carried out in accordance with the guidelines for the care and use of laboratory animals as adopted by Institutional and NIH guidelines. The protocol for this research was approved by the University of South Carolina Animal Care and Use Committee.
TCDD
Dr Stephen Safe, Institute of Biosciences and Technology, Texas A&M Health Sciences Center, Houston, Texas, USA, kindly provided TCDD. Acetone was first used to dissolve TCDD and DMSO was used to dilute TCDD for in-vitro studies and TCDD diluted in corn oil was used for in-vivo studies, as described previously (10).
Chemical reagents and antibodies
The following reagents (RPMI 1640, HEPES, L-Glutamine, Penn/Strep, FBS, and PBS) were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). RNeasy Mini kit, iScript cDNA synthesis kit, miScript primer assays kit, and miScript SYBR Green PCR kit were purchased from Qiagen (Valencia, CA, USA). MicroChIP and MicroPlex Library Preparation Package, MicroPlex Library kits, and antibodies against mouse H3K4me3, H3k9me, and H3K27me were purchased from Diagenode Inc. (Diagenode Inc., Denville, NJ, USA).
Gestational exposure to TCDD and breeding of mice
Groups of five pregnant mice on GD 14 were injected intraperitoneally (i.p.) with the vehicle (VEH; corn oil) or with TCDD dissolved in acetone and suspended in corn oil (10 µg/kg body weight), as described previously (10).
The pups (F1) from the pregnant mothers (F0) exposed to VEH or TCDD, were bred to obtain F2 generation in the SOM Animal facility. The details of the breeding procedure are described in the graphical sketch (Fig. 1). Female F1 and F2 mice at the age of 6 to 8 weeks were used for studying the miR expression in the thymus. We used only female mice for all three generations (F0 to F2) to avoid any differences between the generations based on sex.
Multigenerational effect of TCDD on thymi
We have previously shown that TCDD causes thymic atrophy (10, 12). In this study also, we determined the effect of TCDD on thymi of multigeneration (F0, F1, and F2). To this end, thymi from VEH- and TCDD-treated mice were harvested and single-cell suspensions were prepared. Thymic cellularity was measured as described earlier (11, 12).
miR arrays and analysis
To evaluate the multigenerational effects of TCDD, we performed miR arrays as described earlier (27, 57). In brief, total RNA, including miR, was isolated from groups of five pooled thymi of VEH- or TCDD-treated F0, F1, and F2 generations using miRNeasy kit from Qiagen and following their protocol. miR array consisting of 3200 miRs was performed using the Affymetrix instrument (GCS2000; version 4.1). MicroRNA array data has been submitted to NIH Geo database (accession number: GSE220204) and DOI: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE220204. Analysis of obtained miR data was performed using Transcriptome Analysis Console (TAC) software. Change in miR expression in the TCDD group when compared to the control group with 1.5-fold or higher was considered for further analysis. Altered miRs were then analyzed using Ingenuity Pathway Analysis (IPA) software (Qiagen) for their role in various pathways, as described previously (10, 58). We used several databases including MicroRNA.org, miRwalk, TargetScan, Tools4miRs, etc. to identify the sequence alignment regions between miRs and their target genes.
RT-qPCR to confirm the expression of miRs in the thymocytes of VEH- or TCDD-treated mice
We performed RT-qPCR on thymocytes from groups of five F0, F1, and F2 mice to validate the expression of a few significantly altered miRs identified from miR array data, as described previously (10, 58). Because TCDD is well known for its immunotoxicity, we chose Th1 (IFNγ-specific miR-146a), Th17 (IL17-specific miR-203), Tregs (FoxP3-specific miR-31), and additionally, CYP1A1-specific miRs (miR-134 and miR-203), and AhR-specific miRs (miR-182, miR-203, and miR-499). We also chose miR-155 and miR-30a because of their role in inflammation. To that end, cDNA was generated from total RNAs including miRs isolated from thymocytes of mice (F0, F1, and F2 generations) exposed to VEH or TCDD. We used miScript primer assay kit (details in Table 1) and miScript SYBR Green PCR kit from Qiagen and followed the protocol of the company (Qiagen, Valencia, CA, USA).
Table 1.
Details of miRs targeted for RT-qPCR.
| miRBase ID | Target sequences | Qiagen Cat number |
|---|---|---|
| Mmu-miR-30a_st | UGUAAACAUCCUCGACUGGAAG | MS00011704 |
| Mmu-miR-31_st | AGGCAAGAUGCUGGCAUAGCUG | MS00001407 |
| Mmu-miR-134a_st | UGUGACUGGUUGACCAGAGGGG | MS00001568 |
| Mmu-miR-146a_st | UGAGAACUGAAUUCCAUGGGUU | MIMAT0000158 |
| Mmu-miR-155_st | UUAAUGCUAAUUGUGAUAGGGGU | MIMAT0000165 |
| Mmu-miR-182_st | GUGGUUCUAGACUUGCCAACU | MIMAT0016995 |
| Mmu-miR-203_st | GUGAAAUGUUUAGGACCACUAG | MS00001848 |
| Mmu-miR-499_st | UUAAGACUUGCAGUGAUGUUU | MS00002576 |
RT-qPCR was performed on StepOnePlus system V2.1 (Applied Biosystems, Carlsbad, CA, USA). The following conditions were used: 40 cycles using the following conditions: 15 min at 95°C (initial activation step), 15 s at 94°C (denaturing temperature), 30 s at 55°C (annealing temperature), and 30 s at 70°C (extension temperature and fluorescence data collection) were used. We calculated normalized expression (NE) of miRs using NE ¼ 2_DDCt, where Ct is the threshold cycle to detect fluorescence. The data of the above miRs were normalized against internal control miR (SNORD96) and fold change of miRs was calculated against the control miR, and the treatment group (TCDD) was compared with the vehicle group. To define significant differences in the expression miRs, ANOVA was performed using GraphPad version 6.0 (GraphPad Software, Inc., San Diego, CA, USA). Differences between treatment groups were considered significant when P < 0.05.
Validation of target gene expression in F0, F1, and F2 generations by RT-qPCR.
To confirm the role of miRs in the regulation of their target genes, we performed RT-qPCR. We chose miR-146a-specific IFNγ, miR-203-specific IL-17, miR-31-specific FoxP3, miR-134-specific CYP1A1, and miR-182- and miR-499-specific AhR genes for their expression in thymocytes of F0, F1, and F2 generations. Details of miRs and their binding target are described in Table 2. To this end, RT-qPCR on cDNAs generated from total RNAs isolated from thymocytes of F0, F1, and F2 mice were performed as described previously (10, 58). The details of the primers used are presented in Table 3.
Table 2.
Details of miRs and their target gene with binding affinity.
| Predicted pairing of target gene and miR | Weighted score |
| 3′ CCACUGAUCCACCGGGUGUC mmu-miR-134 | | | | | | | | 724: 5′. . .CUGGGCACAGCAGAGGCCCACAG CYP1A1 |
−0.16 |
| 3′ AUUGGCUAAAGUUUACCACGAU mmu-miR-499 | | | | | | | | 511:5′. . .UUAAAGUAAAUGGUUUGGUGCUA AhR |
−031 |
| 3′gucgauacggUC-GUAGAACGGa 5′ mmu-miR-31 | | | | | | | | | | 1088:5′ gguugcucaaAGUCUUCUUGCCc 3′ Foxp3 |
−0.18 |
| 3′ gaucaccaggauuugUAAAGUg5′ mmu-miR-203 | | | | | | 443:5′ uaaauauuaauggaaAUUUCAg 3′ IL17A |
−0.09 |
| 3′ UGGUCUUGACUCAGGGAUCCCG mmu-miR-146a | | | | | | | | 524: 5′. . .ACUUGACACCUGGUCCUAGGGCA IFN-gamma |
−0.12 |
Table 3.
Primer pairs used for RT-qPCR.
| Genes | Forward primer | Reverse primer |
|---|---|---|
| AhR | 5′-GCGGCCGCAGGAAGTGA G-3′ | 5′-GTGCCGTTGATTTGCGTGTGCT-3′ |
| CYP1A1 | 5′-CCACAGCACCACAAGAGATA-3′ | 5′-AAGTAGGAGGCAGGCACAATGTC-3′ |
| FOXP3 | 5′-CCCATCCCCAGGAGTCTTG-3′ | 5′-ACCATGACTAGGGGCACTGTA-3′ |
| IL-17A | 5′-TTTAACTCCCTTGGCGCAAAA-3 | 5′-CTTTCCCTCCGCATTGACAC-3′ |
| IFN-γ | 5′-GAGTGTGGAGACCATGGCAAG-3′ | 5′-TGCTTTGCGTTGGACATTCAAGTC-3′ |
| 18S | 5′-GCCCGAGCCGCCTGGATAC-3′ | 5′-CCGGCGGGTCATGGGAATAAC-3′ |
RT-qPCR was performed on the following conditions: 40 cycles: 5 min at 95°C (initial activation step), 15 s at 94°C, 30 s at 60°C, and 45 s at 72°C were used. The value of genes was normalized against the housekeeping gene (18S) and fold-change of genes was calculated against 18S and the TCDD group was compared with the vehicle group. To define significant differences in the expression of genes, ANOVA was performed using GraphPad version 6.0 (GraphPad Software, Inc., San Diego, CA, USA), and differences between the groups were considered significant when P < 0.05.
Identification of DRE on the promoter of select miR-203
We selected miR-203 for further analysis. The selection of miR-203 was due to its role in the regulation of IL-17A expressed by Th17 cells. To this end, we first analyzed the promoter of miR-203 for the presence of DREs as described previously (38). The details of DREs in the promoter of miR-203 are described in Table 4.
Generation of constructs containing miR-203 promoter DRE regions
To test the regulation of miR-203 by TCDD through DRE, we generated a construct using pGL4 vector and regions containing DRE in the promoter of miR-203. We selected miR-203 based on its 3′UTR’s high binding affinity with the IL-17 gene (Fig. 5A). In brief, within 1 kb upstream and downstream of the TSS of miR-203, as shown in Table 4, there are four DRE-binding sites (GCGTG). They are located near −430, −395, −380, and +310 of the miR-203 promoter. To determine which DREs would respond to TCDD, we designed several sets of primers (Table 5). Various promoter fragments of miR-203 containing different regions of the promoter with or without DRE motifs were generated by high-fidelity PCR and subcloned into luciferase reporter pGL4 vector, as described previously (38). After verification of the construct, the transformation was performed using competent Escherichia coli. The positive clones were selected and cultured in a liquid growth medium. Plasmids were isolated using a Qiagen kit (Qiagen, Valencia, CA, USA). After determining the quality and quantity of the isolated pGL4 plasmid, aliquots were prepared and stored at a temperature of −80°C for future use.
Table 5.
Showing primers used to clone miR-203 promoter regions containing DRE region into luciferase vector.
| Primer F1: 5′-GGGGGTACCAAACCCTACACTGTTCACCCTACAA-3′ |
| Primer F2: 5′-GGGGGTACCGCGCCGCCCGTCGGGCTCGG-3′ |
| Primer F3: 5′-GGGGGTACCGCCCCTTGGCGAGCATTGCAAA-3′ |
| Primer R1: 5′-GGGCTCGAGGCGCCTGCACACAGAAGTTCT-3′ |
| Primer R2: 5′-GGGCTCGAGAGATGACTACCAAGGGCTCGGC-3′ |
Luciferase assays to determine the role of various DREs of miR-203
To determine the role of TCDD in the regulation of miRs (miR-203), a luciferase assay was performed, as described (59). In brief, the mouse liver cell line, Hep1a, was transfected with various pGL4 constructs containing DRE regions present in the promoter of miR-203. We used the Effectane Transfection reagents kit from Qiagen and followed the protocol of the company (Qiagen Inc., Hilden, Germany). After transfection, the cells were treated with various concentrations (0, 2.5, 5, 10, and 20 nM) of TCDD and were incubated at 37°C, 5% CO2. Dual luciferase assays were performed 24 h after the treatment using a Promega kit (Promega, Madison, WI, USA).
Chromatin immunoprecipitation sequencing
To determine TCDD-induced multigenerational effects on histones modifications, we performed ChIP-seq as described previously using True MicroChIP & MicroPlex Library Preparation Package and MicroPlex Library Preparation Kit v2 from Diagenode (Denville, NJ, USA) (50). In brief, thymocytes from F0, F1, and F2 generations of VEH- or TCDD-exposed mice were isolated, and the histone and DNA were cross-linked using 1% formaldehyde by incubating for 10 min at room temperature with gentle shaking followed by quenching with 0.5 M glycine, and the cells were then collected after washing twice with cold PBS. Next, nuclear membrane disruption and chromatin shearing were performed in lysis buffer and by sonicating the samples at 4°C in a temperature controlled Bioruptor sonicator (Diagenode Inc. Denville, NJ, USA). The samples were centrifuged at 13,000 rpm for 10 min and the supernatant was used for chromatin immunoprecipitation (ChIP) with antibodies against mouse H3K4me3 or H3K9me3 or H3K27me3 purchased from Abcam (Waltham, MA, USA). After overnight immunoprecipitation at 4°C with gentle rotation, protein G beads were added and incubated further for 2 h. Chromatin was re-suspended in elution buffer and the crosslink was reversed by treating the immunoprecipitated chromatin with proteinase K at 65°C for 45 min with constant vortexing. Size selection of the fragments was performed using SPRI beads from Beckman Coulter (Brea, CA, USA) and DNA fragments of the desired size were then re-suspended in water and quantified. The sequencing library was then constructed using MicroPlex Library Preparation Kit v2 (Diagenode Inc.). Sequencing was performed using Illumina NextSeq550 at the University of South Carolina School of Medicine.
ChIP-seq data analysis
The sequencing reads were aligned to mouse genome build mm9 using Bowtie (60). Uniquely mapped reads were used for peak calling by SICER with the statistic threshold value (E value) set as 0.01 (61–64). The peaks in WIG file format were visualized in the UCSC genome browser. The heat map sample-to-sample correlation of the overall histone methylation signal was generated using DiffBind software (65). The signal distribution pattern within 3 bp upstream and downstream of the TSS was calculated using CEAS software (66–68).
Statistical analysis
We used groups of five randomized female pregnant mice treated with vehicle or TCDD (F0). In subsequent generations (F1 and F2), we also included five randomized female mice for each group. The thymi from each group were pooled to study the miR profile. This was necessary because of the small size of the thymus. The details on the number of mice used and the replicates have been provided under each figure legend. We used GraphPad Prism version 6.01 to generate graphs and statistical analyses. We used Student’s t-test for paired observations if data followed a normal distribution to compare between two groups, while two-factor ANOVA variance was used to compare more than two groups. P-value of ≤ 0.05 was considered statistically significant.
Supplementary Material
Notes
Competing Interest: The authors declare no competing interest.
Contributor Information
Narendra P Singh, Department of Pathology, Microbiology and Immunology, University of South Carolina School of Medicine, Columbia, SC 29208, USA.
Xiaoming Yang, Department of Pathology, Microbiology and Immunology, University of South Carolina School of Medicine, Columbia, SC 29208, USA.
Marpe Bam, Department of Pathology, Microbiology and Immunology, University of South Carolina School of Medicine, Columbia, SC 29208, USA.
Mitzi Nagarkatti, Department of Pathology, Microbiology and Immunology, University of South Carolina School of Medicine, Columbia, SC 29208, USA.
Prakash Nagarkatti, Department of Pathology, Microbiology and Immunology, University of South Carolina School of Medicine, Columbia, SC 29208, USA.
Funding
This work was funded in part by the National Institutes of Health Grants R01ES030144, P01AT003961, P20GM103641, R01AI123947, and R01AI160896.
Authors' Contributions
N.P.S. designed and performed some of the experiments, as well as wrote the manuscript, X.Y. performed some of the experiments (cloning of DRE regions and luciferase assays) and reviewed the manuscript, M.B. performed some of the experiments (ChIP assays), M.N. designed the experiments and reviewed the manuscript, and provided the funding, and P.N. conceived, designed the experiments, reviewed the manuscript, and provided the funding.
References
- 1. Gluckman PD, Cutfield W, Hofman P, Hanson MA. 2005. The fetal, neonatal, and infant environments—the long-term consequences for disease risk. Early Hum Dev. 81(1):51–59. [DOI] [PubMed] [Google Scholar]
- 2. Gluckman PD, Hanson MA, Pinal C. 2005. The developmental origins of adult disease. Matern Child Nutr. 1(3):130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gluckman PD, Hanson MA, Spencer HG, Bateson P. 2005. Environmental influences during development and their later consequences for health and disease: implications for the interpretation of empirical studies. Proc Biol Sci. 272(1564):671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heindel JJ. 2005. The fetal basis of adult disease: role of environmental exposures—introduction. Birth Defects Res A Clin Mol Teratol. 73(3):131–132. [DOI] [PubMed] [Google Scholar]
- 5. Heindel JJ, Levin E. 2005. Developmental origins and environmental influences—introduction. Birth Defects Res A Clin Mol Teratol. 73(7):469. [DOI] [PubMed] [Google Scholar]
- 6. Lau C, Rogers JM. 2004. Embryonic and fetal programming of physiological disorders in adulthood. Birth Defects Res C Embryo Today Rev. 72(4):300–312. [DOI] [PubMed] [Google Scholar]
- 7. Dong W, Teraoka H, Kondo S, Hiraga T. 2001. 2,3,7,8-tetrachlorodibenzo-p-dioxin induces apoptosis in the dorsal midbrain of zebrafish embryos by activation of arylhydrocarbon receptor. Neurosci Lett. 303(3):169–172. [DOI] [PubMed] [Google Scholar]
- 8. Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM, Skinner MK. 2012. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS One. 7(2):e31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. 2012. Pesticide and insect repellent mixture (permethrin and DEET) induces epigenetic transgenerational inheritance of disease and sperm epimutations. Reprod Toxicol. 34(4):708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh NP, Singh UP, Guan H, Nagarkatti P, Nagarkatti M. 2012. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS One. 7(9):e45054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Camacho IA, Nagarkatti M, Nagarkatti PS. 2002. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces Fas-dependent activation-induced cell death in superantigen-primed T cells. Arch Toxicol. 76(10):570–580. [DOI] [PubMed] [Google Scholar]
- 12. Camacho IA, Nagarkatti M, Nagarkatti PS. 2004. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on maternal immune response during pregnancy. Arch Toxicol. 78(5):290–300. [DOI] [PubMed] [Google Scholar]
- 13. Camacho IA, Singh N, Hegde VL, Nagarkatti M, Nagarkatti PS. 2005. Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. J Immunol. 175(1):90–103. [DOI] [PubMed] [Google Scholar]
- 14. Durrin LK, Jones PB, Fisher JM, Galeazzi DR, Whitlock JP Jr.. 1987. 2,3,7,8-Tetrachlorodibenzo-p-dioxin receptors regulate transcription of the cytochrome P1-450 gene. J Cell Biochem. 35(2):153–160. [DOI] [PubMed] [Google Scholar]
- 15. Ehrlich AK, Pennington JM, Bisson WH, Kolluri SK, Kerkvliet NI. 2018. TCDD, FICZ, and other high affinity AhR ligands dose-dependently determine the fate of CD4+ T cell differentiation. Toxicol Sci. 161(2):310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fisher MT, Nagarkatti M, Nagarkatti PS. 2005. 2,3,7,8-tetrachlorodibenzo-p-dioxin enhances negative selection of T cells in the thymus but allows autoreactive T cells to escape deletion and migrate to the periphery. Mol Pharmacol. 67(1):327–335. [DOI] [PubMed] [Google Scholar]
- 17. Nagarkatti PS, Sweeney GD, Gauldie J, Clark DA. 1984. Sensitivity to suppression of cytotoxic T cell generation by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is dependent on the Ah genotype of the murine host. Toxicol Appl Pharmacol. 72(1):169–176. [DOI] [PubMed] [Google Scholar]
- 18. Singh NP, Nagarkatti M, Nagarkatti P. 2008. Primary peripheral T cells become susceptible to 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated apoptosis in vitro upon activation and in the presence of dendritic cells. Mol Pharmacol. 73(6):1722–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barker DJ, Eriksson JG, Forsen T, Osmond C. 2002. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 31(6):1235–1239. [DOI] [PubMed] [Google Scholar]
- 20. Phillips DI. 2006. External influences on the fetus and their long-term consequences. Lupus. 15(11):794–800. [DOI] [PubMed] [Google Scholar]
- 21. Blaylock BL, Holladay SD, Comment CE, Heindel JJ, Luster MI. 1992. Exposure to tetrachlorodibenzo-p-dioxin (TCDD) alters fetal thymocyte maturation. Toxicol Appl Pharmacol. 112(2):207–213. [DOI] [PubMed] [Google Scholar]
- 22. Singh NP, et al. 2011. Resveratrol (trans-3,5,4'-trihydroxystilbene) suppresses EL4 tumor growth by induction of apoptosis involving reciprocal regulation of SIRT1 and NF-κB. Mol Nutr Food Res. 55(8):1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaspari L, et al. 2021. Experimental evidence of 2,3,7,8-tetrachlordibenzo-p-dioxin (TCDD) transgenerational effects on reproductive health. Int J Mol Sci. 22(16):9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O’Brien J, Hayder H, Zayed Y, Peng C. 2018. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. 9:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dupont C, Kappeler L, Saget S, Grandjean V, Levy R. 2019. Role of miRNA in the transmission of metabolic diseases associated with paternal diet-induced obesity. Front Genet. 10:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Disner GR, Lopes-Ferreira M, Lima C. 2021. Where the aryl hydrocarbon receptor meets the microRNAs: literature review of the last 10 years. Front Mol Biosci. 8:725044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neamah WH, et al. 2019. AhR activation leads to massive mobilization of myeloid-derived suppressor cells with immunosuppressive activity through regulation of CXCR2 and microRNA miR-150-5p and miR-543-3p that target anti-inflammatory genes. J Immunol. 203(7):1830–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sultan M, et al. 2021. The endocannabinoid anandamide attenuates acute respiratory distress syndrome by downregulating miRNA that target inflammatory pathways. Front Pharmacol. 12:644281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Al-Ghezi ZZ, et al. 2019. AhR activation by TCDD (2,3,7,8-Tetrachlorodibenzo-p-dioxin) attenuates pertussis toxin-Induced inflammatory responses by differential regulation of tregs and Th17 cells through specific targeting by microRNA. Front Microbiol. 10:2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Faith RE, Luster MI. 1979. Investigations on the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on parameters of various immune functions. Ann NY Acad Sci. 320:564–571. [DOI] [PubMed] [Google Scholar]
- 31. Golor G, Yamashita K, Koner W, Hagenmaier H, Neubert D. 2001. Kinetics and inductive potency of 1,2,3,4,6,7,8-heptachlorodibenzo-p-dioxin (H7CDD) in rats. Life Sci. 69(5):493–508. [DOI] [PubMed] [Google Scholar]
- 32. Kamath AB, Camacho I, Nagarkatti PS, Nagarkatti M. 1999. Role of Fas–Fas ligand interactions in 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD)-induced immunotoxicity: increased resistance of thymocytes from Fas-deficient (lpr) and Fas ligand-defective (gld) mice to TCDD-induced toxicity. Toxicol Appl Pharmacol. 160(2):141–155. [DOI] [PubMed] [Google Scholar]
- 33. Kamath AB, Nagarkatti PS, Nagarkatti M. 1998. Characterization of phenotypic alterations induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin on thymocytes in vivo and its effect on apoptosis. Toxicol Appl Pharmacol. 150(1):117–124. [DOI] [PubMed] [Google Scholar]
- 34. Kamath AB, Xu H, Nagarkatti PS, Nagarkatti M. 1997. Evidence for the induction of apoptosis in thymocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in vivo. Toxicol Appl Pharmacol. 142(2):367–377. [DOI] [PubMed] [Google Scholar]
- 35. Kerkvliet NI. 2002. Recent advances in understanding the mechanisms of TCDD immunotoxicity. Int Immunopharmacol. 2(2-3):277–291. [DOI] [PubMed] [Google Scholar]
- 36. Lundberg K, Dencker L, Gronvik KO. 1992. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) inhibits the activation of antigen-specific T-cells in mice. Int J Immunopharmacol. 14(4):699–705. [DOI] [PubMed] [Google Scholar]
- 37. McConkey DJ, Hartzell P, Duddy SK, Hakansson H, Orrenius S. 1988. 2,3,7,8-Tetrachlorodibenzo-p-dioxin kills immature thymocytes by Ca2+-mediated endonuclease activation. Science. 242(4876):256–259. [DOI] [PubMed] [Google Scholar]
- 38. Singh NP, Nagarkatti M, Nagarkatti PS. 2007. Role of dioxin response element and nuclear factor-κB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression. Mol Pharmacol. 71(1):145–157. [DOI] [PubMed] [Google Scholar]
- 39. Gogal RM Jr., Holladay SD. 2008. Perinatal TCDD exposure and the adult onset of autoimmune disease. J Immunotoxicol. 5(4):413–418. [DOI] [PubMed] [Google Scholar]
- 40. Lee YH, Wu MR, Hsiao JK. 2021. Organic anion transporting polypeptide 1B1 is a potential reporter for dual MR and optical imaging. Int J Mol Sci. 22(16):8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Viluksela M, Pohjanvirta R. 2019. Multigenerational and transgenerational effects of dioxins. Int J Mol Sci. 20(12):2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bruner-Tran KL, Osteen KG. 2011. Developmental exposure to TCDD reduces fertility and negatively affects pregnancy outcomes across multiple generations. Reprod Toxicol. 31(3):344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang ZY, et al. 2006. Induction of dendritic cell maturation by pertussis toxin and its B subunit differentially initiate Toll-like receptor 4-dependent signal transduction pathways. Exp Hematol. 34(8):1115–1124. [DOI] [PubMed] [Google Scholar]
- 44. Bianchi M, Renzini A, Adamo S, Moresi V. 2017. Coordinated actions of microRNAs with other epigenetic factors regulate skeletal muscle development and adaptation. Int J Mol Sci. 18(4):840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Osella M, Riba A, Testori A, Cora D, Caselle M. 2014. Interplay of microRNA and epigenetic regulation in the human regulatory network. Front Genet. 5:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu S, et al. 2010. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene. 29(15):2302–2308. [DOI] [PubMed] [Google Scholar]
- 47. De Heer C, et al. 1994. Time course of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced thymic atrophy in the Wistar rat. Toxicol Appl Pharmacol. 128(1):97–104. [DOI] [PubMed] [Google Scholar]
- 48. McConkey DJ, Orrenius S. 1989. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) kills glucocorticoid-sensitive thymocytes in vivo. Biochem Biophys Res Commun. 160(3):1003–1008. [DOI] [PubMed] [Google Scholar]
- 49. Wright EJ, De Castro KP, Joshi AD, Elferink CJ. 2017. Canonical and non-canonical aryl hydrocarbon receptor signaling pathways. Curr Opin Toxicol. 2:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bam M, et al. 2020. Increased H3K4me3 methylation and decreased miR-7113-5p expression lead to enhanced Wnt/β-catenin signaling in immune cells from PTSD patients leading to inflammatory phenotype. Mol Med. 26(1):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Siklenka K, et al. 2015. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science. 350(6261):aab2006. [DOI] [PubMed] [Google Scholar]
- 52. Wan QL, et al. 2022. Histone H3K4me3 modification is a transgenerational epigenetic signal for lipid metabolism in Caenorhabditis elegans. Nat Commun. 13(1):768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tuscher JJ, Day JJ. 2019. Multigenerational epigenetic inheritance: one step forward, two generations back. Neurobiol Dis. 132:104591. [DOI] [PubMed] [Google Scholar]
- 54. Lee J, et al. 2015. Male and female mice show significant differences in hepatic transcriptomic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BMC Genomics. 16:625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pohjanvirta R, Miettinen H, Sankari S, Hegde N, Linden J. 2012. Unexpected gender difference in sensitivity to the acute toxicity of dioxin in mice. Toxicol Appl Pharmacol. 262(2):167–176. [DOI] [PubMed] [Google Scholar]
- 56. Ohtake F, Fujii-Kuriyama Y, Kawajiri K, Kato S. 2011. Cross-talk of dioxin and estrogen receptor signals through the ubiquitin system. J Steroid Biochem Mol Biol. 127(1-2):102–107. [DOI] [PubMed] [Google Scholar]
- 57. Alharris E, et al. 2018. Resveratrol attenuates allergic asthma and associated inflammation in the lungs through regulation of miRNA-34a that targets FoxP3 in mice. Front Immunol. 9:2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Singh NP, et al. 2011. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One. 6(8):e23522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Becker W, Nagarkatti M, Nagarkatti PS. 2018. miR-466a targeting of TGF-β2 contributes to FoxP3+ regulatory T cell differentiation in a murine model of allogeneic transplantation. Front Immunol. 9:688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Z, et al. 2009. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 138(5):1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Araki Y, et al. 2009. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 30(6):912–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zang C, et al. 2009. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 25(15):1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wei G, et al. 2009. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 30(1):155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ross-Innes CS, et al. 2012. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 481(7381):389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu T, et al. 2011. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12(8):R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu T, et al. 2011. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 21(2):227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shin H, Liu T, Duan X, Zhang Y, Liu XS. 2013. Computational methodology for ChIP-seq analysis. Quant Biol. 1(1):54–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.